introduction center for medical genetics staff (81) clinicians and psychologists laboratory...
TRANSCRIPT

IntroductionCenter for Medical Genetics
Staff (81)
Clinicians and psychologistsLaboratory supervisors ResearchersLaboratory techniciansSecretary

IntroductionCenter for Medical Genetics
Diagnosis of genetic disorders
Clinical assesmentLaboratory investigations (three labs)Counseling

Cursus Human Molecular Genetics
Les 1 : From human cytogenetics to molecular cytogenetics
Les 2 : Monogenic disorders
Les 3 : Familial cancer
Les 4 : Multifactorial genetic disorders
Les 5 : Diagnosis and Research in Human Genetics

From human cytogenetics to molecular cytogenetics
• introduction
• historical overview (the birth of human cytogenetics)
• progress in (molecular) cytogenetics
• general aspects of (molecular) cytogenetics
• molecular mechanisms for constitutional chromosomal rearrangements in humans

The birth of human cytogenetics
• 1956: Tjio and Levan count the full complement of 46 human chromosomes

The birth of human cytogenetics
• 1956: Tjio and Levan count the full complement of 46 human chromosomes
• serendipitous addition of water to a suspension of fixed cells
• 3 years after description of DNA structure • 30 years after count of 48 chromosomes by Thomas Painter

The birth of human cytogenetics
• human chromosomes have a morphology which allows classification

The birth of human cytogenetics
rapidly associations were found between human diseases (syndromes) and specific chromosome abnormalities 1959 Lejeune et al : +21 in Down syndrome Ford et al. : 45,X in Turner syndrome Jacobs et al : 47,XXY in Klinefelter syndrome
1960 Nowel and Hungerford Philadelphia chromosome in CML 1973 Rowley: t(9;22)(q34;q11) in CML


The birth of human cytogenetics
rapidly associations were found between human diseases (syndromes) and specific chromosome abnormalities 1963 chromosome 5 short arm partial deletion in Cri du Chat syndrome
1963 D-chromosome deletion in patient with bilateral retinoblastoma

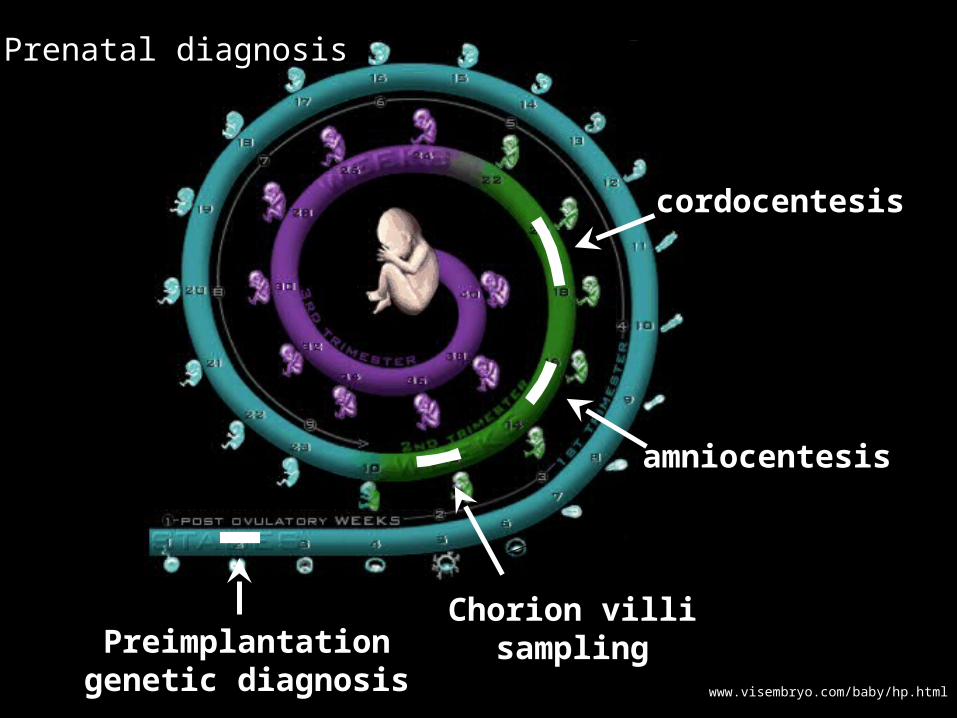
www.visembryo.com/baby/hp.html
Preimplantationgenetic diagnosis
Chorion villisampling
cordocentesis
amniocentesis
Prenatal diagnosis

Further progress in human cytogeneticsis fueled by technical innovations (I)
• 1968 Caspersson et al differential staining of chromosomes produces a recognizable banding pattern (chromosomal barcode) along the length of the chromosomes
• chromosome bands are related to differences in base pair composition, gene density, repetitive elements, chromatin packaging but molecular basis is not understood
• greatly facilitates classification and recognition of structural aberations

general aspects of (molecular) cytogenetics


ISCN 1995International System for Human Cytogenetic Nomenclature
groep A (1-3)
groep B (4-5)
groep C (6-12, X)
groep D (13-15)
groep E (16-18)
groep F (19-20)

general aspects of (molecular) cytogenetics

general aspects of (molecular) cytogenetics
chromosomal rearrangements
• numerical chromosome changes/aneuploidyresult from errors occurring during meiotic or mitotic segregation
• structural chromosome changestranslocationsinversionsinsertionsdeletionsduplications

CYTOGENETICA EN MOLECULAIRE CYTOGENETICA:CONSTITUTIONELE EN VERWORVEN
CHROMOSOMALE DEFECTEN

general aspects of (molecular) cytogenetics

general aspects of (molecular) cytogenetics

general aspects of (molecular) cytogenetics

reciprocal translocation
general aspects of (molecular) cytogenetics

ISCN 1995International System for Human Cytogenetic Nomenclature
Reciprocal translocation
http://www.waisman.wisc.edu/cytogenetics/abnormalities/abnormalities.html
45,XX,der(13;14)(q10;q10)
Robertsonian translocation
46,XY,t(6;9)(q24;p23)

ISCN 1995International System for Human Cytogenetic Nomenclature
Reciprocal translocation (unbalanced)
http://www.waisman.wisc.edu/cytogenetics/abnormalities/abnormalities.html
46,XY,t(6;9)(q24;p23)
46,XY,der(6)t(6;9)(q24;p23)

ISCN 1995International System for Human Cytogenetic Nomenclature
inversion
46,XX,inv(9)(p13q13)
insertion
46,XY,ins(5;2)(p14;q22q32)

ISCN 1995International System for Human Cytogenetic Nomenclature
duplication deletion
46,X,dup(X)(p11.2p22.1) del(18)(pterp11.2)del(18)(p11.2)

Further progress in human cytogeneticsis fueled by technical innovations (II)
methods for mapping (disease) genes basedupon chromosomal rearrangements
• Somatic cell hybrids• flow sorted chromosomes• FISH

Further progress in human cytogeneticsis fueled by technical innovations (II)
Somatic cell hybrids

Further progress in human cytogeneticsis fueled by technical innovations (II)

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
Positional cloning of t(1;17) breakpoints
• constitutional (1;17)(p36.2;q11.2) in patient with neuroblastoma• 1p36 region is frequently lost in NB• association of a translocation with a particular disease phenotype may point at the chromosomal localisation of the disease gene• additional evidence from eg LOH, linkage,mouse,…• positional cloning: cloning of disease gene based upon the assumption of the chromosomal localisation• physical mapping, identification of candidate genes mutation analysis, expression studies, functional evidence

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
Positional cloning of t(1;17) breakpoints


Further progress in human cytogeneticsis fueled by technical innovations
late ’80ies introduction of FISH
• significant increase of sensitivity (10.000x)• new possiblities eg interphase• various applications eg gene mapping, genetic diagnosis, research• “the FISH have spawned”
CGHM-FISH/SKYFICTIONfibre FISH

FISH (fluorescentie in situ hybridisatie)

Fluorescence in situ hybridisation
Labeling: nick translation
DNA
Denaturation and incubation at 37°C
Mix
Denaturation
Hybridisatie o/n
Commercial Cot1 DNA
Wash, detectionand counterstain
10ml culture

2 X chromosome 13
2 X chromosome 18
2 X chromosome 21
1 X chromosome Y
Controllymphocytes
(FISH 952-35)



CGH part I

CGH part I

CGH
advantages
whole genome in 1 experiment
no need to culture tumor cells
sensitive detection of gene amplification
disadvantages
limited resolution (~10 Mb del/dup)
laborious
only gains and losses / no balanced rearrangementsno information on the nature of the aberrations
part I
retrospective analysis


Molecular mechanisms for constitutionalchromosomal rearrangements in humans
• chromosomal rearrangements require the formation of double strand breaks (DSBs) and subsequent rejoining of the broken ends between two (or more) breakpoints
• exogenous causes of structural aberrations X-rays, -rays, -particles and other forms of ionizing radiation
cause formation of oxidants which are powerful clastogens
duration of exposure

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
exogenous causes of structural aberrations
• chemicals: alkylating agents, purine and pyrimidine analoges, alkyl epoxides, aromatic amines, nitroso compounds and heavy metals
most often generation of breaks at G2
• viral infections
• lesions may undergo repair or misrepair by a wide range of DNA repair systems

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
endogenous causes of structural aberrations
• rare autosomal recessive chromosome breakage syndromes caused by defective DNA repair enzymes (AT, ATM; BS, BLM; NBS, NBS1)
• transposable elements
• short and long interspersed elements (LINE, SINE)300 bp Alu (every 4 kb, gene rich), longer LINE (gene poor)
• segmental duplication, gene duplication
• fragile sites

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
Segmental duplications: an ‘expanding’ role in genomicinstability and disease. Emanuel and Shaikh, Nature ReviewsGenetics, Volume 2, October 2001, 791-800.
• Segmental duplications = region or chromosome specific low-copy repeats, new class of repetitive DNA elements recently identified • resulting genetic aberrations
deletionsinterstitial duplicationstranslocationsinversionsmarker chromosomes



Molecular mechanisms for constitutionalchromosomal rearrangements in humans
DiGeorge/velo-cardio-facial syndromerecurrent reciprocal translocation t(11;22)cat eye syndrome (CES)

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
red blocks: low copy repeatsADU: DGS patient with translocationTDR: common 3 Mb typically deleted regiona-f: unusual deletions

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
DiGeorge syndromevelo-cardio-facial syndrome

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
DiGeorge syndromevelo-cardio-facial syndrome
• 1/4000 live births• gene haploinsufficiency syndrome• 90% de novo, 10% inherited• deletion encompasses ~30 genes• clinical features are highly variable (table), variable expressivity and incomplete penetrance• affect pharyngeal and neurobehavioural development• which genes are critically involved ???? mouse models: candidate TBX1, T-box family of genes highly expressed in pharyngeal arches, TBX1 KO

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
VCFS: CP, velopharyngeal insufficiency, small mouth, retrognathia, bulbous nasal tip, microcephaly, concotruncal heart defects, MR, learning disabilities, short stature,
DGS: parathyroid hypoplasia, thymic hypoplasia and immune defect due to T cell deficit

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
DiGeorge syndromevelo-cardio-facial syndrome

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
DiGeorge syndromevelo-cardio-facial syndrome

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
DiGeorge syndromevelo-cardio-facial syndrome

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
cat eye syndrome t(11;22)
carriers have normal phenotypeare at risk for unbalanced progeny
1:3 segregation leading to 47,XX,+der(22)
MR, multipel malformation syndrome including characteristic eye abnormalities

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
CMT1A/HNPP
• Charcot-Marie-Tooth disease 1A
• duplication within 17p12
• peripheral myelin protein 22
• most common inherited peripheral neuropathy
• 70% of CMT1 inherited demyelating neuropathy

CMT1A/HNPP
• most common inherited peripheral neuropathy
• 70% of CMT1 inherited demyelating neuropathy
First described in 1886 by Charcot and Marie in Paris, France and Tooth in Cambridge, England. Most common inherited disorder of the peripheral nerves affecting 1 in 2500 individuals in their 20s and 30s. Characterised by distal muscle atrophy and weakness, first involving the legs and particularly the peritoneal muscles. Sensory loss may be present but is always less pronounced than muscle weakness, and tendon reflexes are absent or diminished. High arched feet (pes cavus) are often present. Also described as Hereditary and Motor Sensory Neuropathy (HMSN).
Molecular mechanisms for constitutionalchromosomal rearrangements in humans

CMT1A/HNPP
• hereditary neuropathy with liability the pressure palsies
• idem inherited peripheral neuropathy but with episodic and milder manifestations
Hereditary Neuropathy with liability to Pressure Palsies or HNPP is a slowly progressive, hereditary, neuromuscular disorder which makes an individual very susceptible to nerve injury from pressure, stretch or repetitive use. When injured, the nerves demyelinate or lose their insulating covering. This causes episodes of numbness and weakness in the injured area, which are referred to as the ‘pressure palsies'. These episodes can be mild and more of a nuisance than anything, or so severe almost all movement in the affected limb is impossible. They may last several minutes to months. Because the symptoms can come and go, and most neurologists have not yet heard of or seen a case of HNPP, it can be very difficult and lengthy process to be diagnosed.
Molecular mechanisms for constitutionalchromosomal rearrangements in humans

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
CMT1A/HNPP
• hereditary neuropathy with liability the pressure palsies
• idem inherited peripheral neuropathy but with episodic and milder manifestations
SMS Smith-Magenis syndrome
• mental retardation/malformation syndrome • ~5 Mb deletion

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
Williams-Beuren syndrome
• deletion of the elastin gene, responsable for supravalvular aortic stenosis
• ~1.6 Mb deletion at 7q11.23
• heart defects, facial dysmorphy, mental retardation, behavioural abn
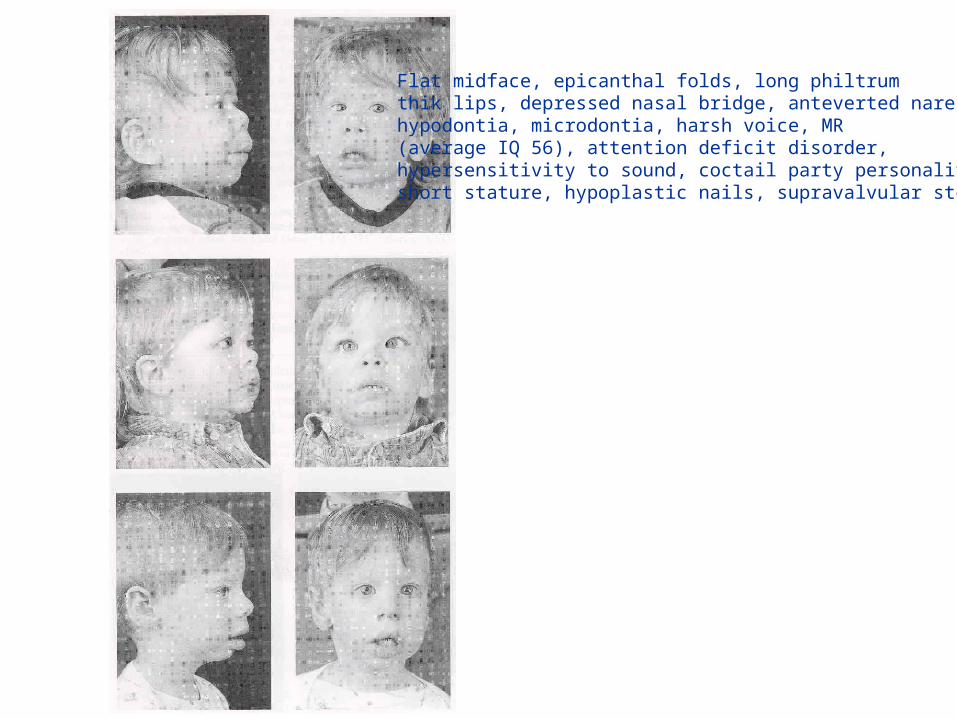
Molecular mechanisms for constitutionalchromosomal rearrangements in humans
Flat midface, epicanthal folds, long philtrumthik lips, depressed nasal bridge, anteverted nares,hypodontia, microdontia, harsh voice, MR(average IQ 56), attention deficit disorder, hypersensitivity to sound, coctail party personality, short stature, hypoplastic nails, supravalvular stenosis

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
PWS/AS
• ~4 Mb deletion of imprinted region on 15q12
• maternal deletion or paternal disomy leads to AS (profound MR, no speach development, uncontrolled laughter), deletion of UBE3A (mouse KO)
• paternal deletion or maternal disomy leads to PWS (MR, obesity, dysmorphic) SNRP associated imprinting center

Failure to thrive in infancy, obesity, dolichocephaly,narrow bitemporal diameter, almond-shaped eyes,strabismus, thin upper lip, small appearing mouth,down turned corners of the mouth, hypogonadismsmall hands, hypopigmentation, learning disabilities,behavioural problems

Microbrachycephaly, prognathia, protruding tongue, macrostomia, widely spaced teeth,severe MR, paroxysmal laughter, absent speech, ataxia with jerky arm movements,seizures, hypopigmentation

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
X-linked ichtyosis
• deletion of the steroid sulphatase gene
haemophilia A
• inversion that disrupts factor VIII gene int22h in intron 22 and two inverted int22h at ~500 kb telomeric
Emery-Dreifuss muscular dystrophy
• inversion in the emerin genetel tel
x
tel

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
simple segmental duplication
• CMT1A-REPtwo copies that flank the region24 kb in size98.7% identity
• S323 elements on Xp22, separated by 1.9 Mb
• two 11.3-kb inverted repeats that mediate the inversion in the emerin gene, >99% identity
• int22h (intron homologous region) sequence which mediates the inversion in the factor VIII gene

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
complex segmental duplication
• 22q11 repeatdifferences in size, content, organisationtruncated gene segments and pseudogenespotentially recombinogenic sequences including palindromic (A+T) rich repeats (PATRR) and VNTRs
• SMS at least four genes or pseudogenes
• PWS/AS duplications of HERC2
• BWS several (pseudo)genes in complex configurations
• evidence for presence recombinational hot spots

Molecular mechanisms for constitutionalchromosomal rearrangements in humans

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
Mechanistic models for rearrangements

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
Mechanistic models for rearrangements

Molecular mechanisms for constitutionalchromosomal rearrangements in humans
Mechanistic models for rearrangements
