intrinsic platelet defects in hereditary thrombocytopenia
TRANSCRIPT

INTRINSIC PLATELET DEFECTS IN HEREDITARY THROMBOCYTOPENIA*
Scott Murphy Presbyterian-University of Pennsylvania Medical Center
Philadelphia, Pennsylvania 19104
In the past four years, we have had the opportunity to study 24 patients from 1 1 families with hereditary thrombocytopenia. In this paper, the data that they have provided will be presented and a classification for these syndromes will be proposed. At the outset, I should like to emphasize the clinical importance of recognizing these patients. For the clinician, the patient presents with thrombo- cytopenia, no abnormality of leukocytes or erythrocytes, a normal marrow aspirate with at least adequate numbers of megakaryocytes, and no findings to suggest drug purpura, systemic lupus erythematosus, or lymphoma. The diagnosis of idiopathic thrombocytopenia purpura (ITP) is irresistable. In many of our patients, that diagnosis had been made and treatment for ITP had been pre- scribed. In most instances, it was not until hemorrhagic symptoms developed in family members that the correct diagnosis was appreciated. One cannot anticipate the same response to splenectomy as one sees in ITP. Twelve of our patients have undergone splenectomy; many responded with a modest increase in platelet count, but only two had permanent elevations into the normal range. Further- more, the risk of overwhelming, fatal, postsplenectomy infection' is very high in the Wiskott-Aldrich Syndrome (WAS) and its variants; the confusion of this syndrome with ITP can have tragic consequences.
ASSAY FOR ANTIPLATELET FACTOR The separation of the hereditary and acquired varieties of hemolytic anemia
has been facilitated by the Coombs test and assays for cold agglutinins. In the separation of hereditary and acquired thrombocytopenic states, we have used a modification of an assay for circulating antiplatelet factor first described by Karpatkin and In this technique, a globulin fraction of test serum reacts with normal platelets to release platelet phospholipid into the supernatant. This release is detected by the shortening of the recalcification time of supefnatant plasma in the presence of contact product. To date, we have obtained positive tests in 38 of 54 patients with ITP, 2 of 29 thrombocytopenic patients with marrow aplasia or malignancy, and none of 15 patients with hereditary thrombo- cytopenia. Therefore, the history of chronic thrombocytopenia resembling ITP, particularly if it dates back to early childhood, and a negative assay for anti- platelet factor should initiate a vigorous family study to search for other members who are thrombocytopenic.
CLASSIFICATION : LABORATORY PROCEDURES We have found many varieties of hereditary thrombocytopenia; one of our
major concerns has been to develop a classification for them. The two laboratory
Supported in part by Clinical Research Center grants (SMO1-PR40, PR75) from the Division of Research Facilities and Resources, National Institutes of Health, by grants (HE 11047-04, PH 43-67-1384) from the United States Public Health Service, and by a grant from the John A. Hartford Foundation.
42 1
422 Annals New York Academy of Sciences
procedures that have been most helpful in this regard have been measurements of platelet survival and electronic determinations of platelet size.
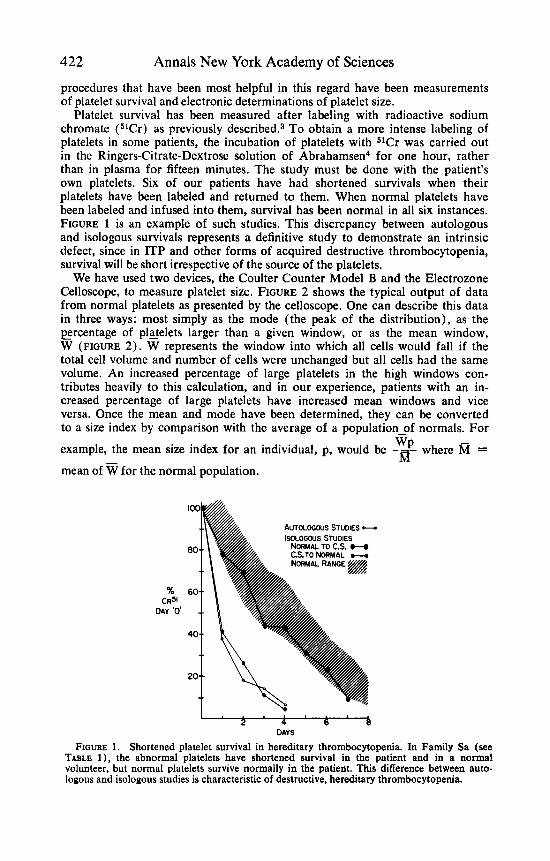
Platelet survival has been measured after labeling with radioactive sodium chromate (51Cr) as previously de~cr ibed .~ To obtain a more intense labeling of platelets in some patients, the incubation of platelets with 51Cr was carried out in the Ringers-Citrate-Dextrose solution of Abrahamsen4 for one hour, rather than in plasma for fifteen minutes. The study must be done with the patient’s own platelets. Six of our patients have had shortened survivals when their platelets have been labeled and returned to them. When normal platelets have been labeled and infused into them, survival has been normal in all six instances. FIGURE 1 is an example of such studies. This discrepancy between autologous and isologous survivals represents a definitive study to demonstrate an intrinsic defect, since in ITP and other forms of acquired destructive thrombocytopenia, survival will be short irrespective of the source of the platelets.
We have used two devices, the Coulter Counter Model B and the Electrozone Celloscope, to measure platelet size. FIGURE 2 shows the typical output of data from normal platelets as presented by the celloscope. One can describe this data in three ways: most simply as the mode (the peak of the distribution), as the ercentage of platelets larger than a given window, or as the mean window, W (FIGURE 2). W represents the window into which all cells would fall if the total cell volume and number of cells were unchanged but all cells had the same volume. An increased percentage of large platelets in the high windows con- tributes heavily to this calculation, and in our experience, patients with an in- creased percentage of large platelets have increased mean windows and vice versa. Once the mean and mode have been determined, they can be converted to a size index by comparison with the average of a population-of normals. For
example, the mean size index for an individual, p, would be where R =
mean of
WP
for the normal population.
h AUTOLOGOUS STUDIES -
% CR5’
DAY ‘0’
DAYS
FIGURE 1. Shortened platelet survival in hereditary thrombocytopenia. In Family Sa (see TABLE l ) , the abnormal platelets have shortened survival in the patient and in a normal volunteer, but normal platelets survive normally in the patient. This difference between auto- logous and isologous studies is characteristic of destructive, hereditary thrombocytopenia.

Murphy: Hereditary Thrombocytopenia 423
WINDOW NUMBERS FIGURE 2. Platelet size distribution determined by the celloscope. A polyvinyltoluene
particle is used as a standard. The results in the first ten windows are ignored, since they represent largely electrical background. The ascending limb of the distribution is extrapolated to baseline and the "mean window" ( W ) is calculated:
X, + 2X, + . . . + nx. XI + x, + . . . + X"
- W =- -0.5
where x,, is the number of cells counted in window n.
We emphasize this because of our findings in acquired disease (FIGURE 3) . One would hope to be able to distinguish between thrombocytopenic patients with shortened and normal platelet survivals; that is, between ITP and Aplasia. In fact, an index based on the mean window does not allow such a distinction, but an index based on modes does. These patients with aplasia have increased mean windows because they have an increased percentage of large platelets, so that one cannot discriminate on that basis either. In hereditary patients, the mode and mean windows are either raised or lowered in concert, allowing a clear-cut identification of patients with small, normal, or large platelet size.
Many laboratory procedures have not been helpful in distinguishing one family from another. The families to be presented have normal or increased numbers of megakaryocytes in marrow smears, even when platelet survival is normal. The megakaryocytes do not have distinctive morphologic features. Electron micro- copic preparations have been prepared in all families except Family Au (see TABLE 1 ) . We have looked particularly for abnormalities of the normal disc shape, the marginal bundle of microtubules, the dense granules, mitochondria, and glycogen, and we have found none even when platelet size has been abnormal. In five families studied (Ra, Ri, Sa, Sh, Ep), we have not found reduced levels of 14 glycolytic and hexose monophosphate shunt enzymes when measured as previously de~cr ibed .~ Surface charge, measured as electrophoretic mobility by Dr. R. Leonardi and Dr. B. Alexander, using a modification of the method of Fuhrman and Ruhenstroth-Ba~er,~ has been normal in five families studied (Ri, Sa, Sh, Ep, Ha) .
PATIENTS TABLE 1 presents data on 22 patients from nine families indicating the mode of
inheritance, the presence or absence of features of the WAS (decreased re-

424 Annals New York Academy of Sciences
M A N INDEX
8
MODE INDEX
' s s NORMAL
RANGE
..
FIGURE 3. Size index determinations in ITP and aplasia. Indices based on the mean window overlap, but those based on the modal window do not.
sistance to infection and eczema), the 51Cr survival t%, and platelet size. To facilitate examination of the data, it is indicated that the first ten patients have small platelets, while the next seven have normal-sized platelets, and the last five have large platelets. In the first five families, platelet survival has been shown to be short in four families and assumed to be short in the fifth, since the typical features of the WAS are present in Family Br. In the next three families, platelet survival is normal; it has not been measured in Family Ha.
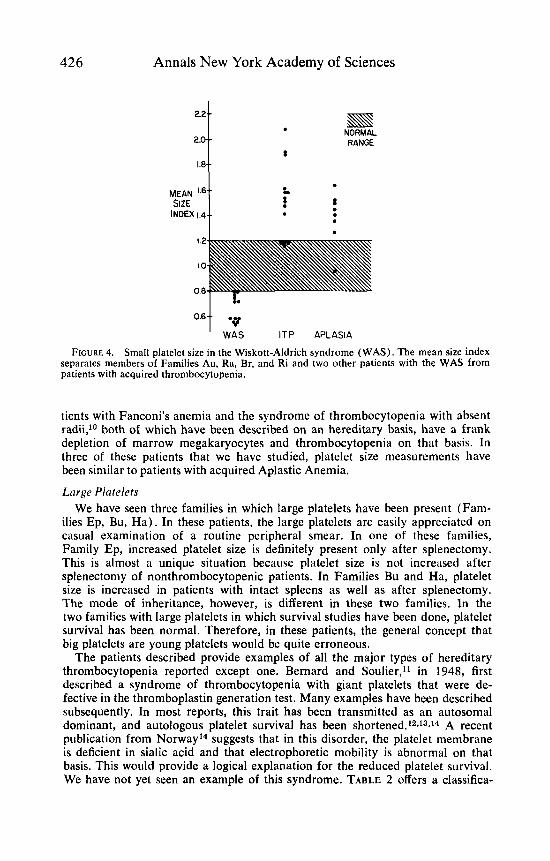
Small Platelets The first four families represent examples of the WAS or its variants. As others
have reported,E we have found that platelets are small in this syndrome. As shown in FIGURE 4, the mean size index provides a crisp differential with other forms of thrombocytopenia. We believe that this test will be most helpful in suggesting or confirming this diagnosis, which is often not straightforwxd. Several investigators6J have reported that autologous platelet survival is short in this syndrome, but normal platelets survive normally in the pstients. In fact, Family Au is the same family in which Baldini and coworkers' first demonstrated this fact. We can confirm this in patient E. Ra., whose family has the typical features of the syndrome, and in Family Ri, where platelet survival is short, normal platelets survive normally, platelet size is small, and the inheritance pattern is typical for the WAS but the characteristic dermotologic and immunologic features are lacking. In this family, platelet size and survival studies have confirmed a relationship to the Wiskott-Aldrich Syndrome that would not have been apparent on a clinical basis.
Only two other conditions associated with small platelets have been dzscribed. Harker6 reported that platelets were small in iron deficiency; we h:ve not been able to confirm this in three patients studied. Weiss and colleagues9 have reported a family with thrombocytopathy and small platelets; in that family, thrombo- cytopenia was minimal or absent, the functional defect was most prominent, and
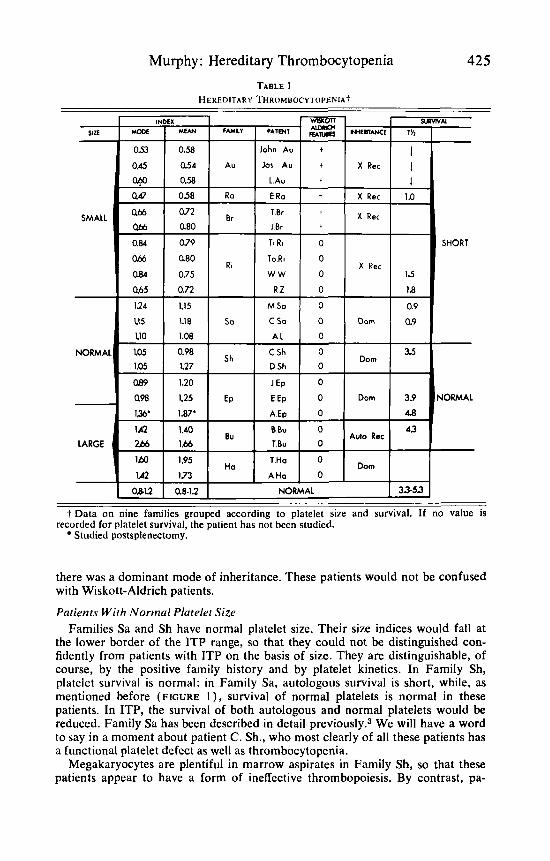
Murphy: Hereditary Thrombocytopenia 425 TABLE 1
HEREDITARY THROMBOCYTOPENIA~
NORMAL I 33-53 t Data on nine families grouped according to platelet size and survival. If no value is
Studied postsplenectomy. recorded for platelet survival, the patient has not been studied.
there was a dominant mode of inheritance. These patients would not be confused with Wiskott-Aldrich patients.
Patients With Normal Platelet Size Families Sa and Sh have normal platelet size. Their size indices would fall at
the lower border of the ITP range, so that they could not be distinguished con- fidently from patients with ITP on the basis of size. They are distinguishable, of course, by the positive family history and by platelet kinetics. In Family Sh, platelet survival is normal: in Family Sa, autologous survival is short, while, as mentioned before (FIGURE l ) , survival of normal platelets is normal in these patients. In ITP, the survival of both autologous and normal platelets would be reduced. Family Sa has been described in detail p rev io~s ly .~ We will have a word to say in a moment about patient C. Sh., who most clearly of all these patients has a functional platelet defect as well as thrombocytopenia.
Megakaryocytes are plentiful in marrow aspirates in Family Sh, so that these patients appear to have a form of ineffective thrombopoiesis. By contrast, pa-
426
22.-
2.0-
1.8-
MEAN ‘6- SIZE
INDEX 1.4-
Annals New York Academy of Sciences
. s s NORMAL RANGE
8
f t : . .
I
I T P APLASIA 0.6t WAS
FIGURE 4. Small platelet size in the Wiskott-Aldrich syndrome (WAS). The mean size index separates members of Families Au, Ra, Br, and Ri and two other patients with the WAS from patients with acquired thrombocytopenia.
tients with Fanconi’s anemia and the syndrome of thrombocytopenia with absent radii,1° both of which have been described on an hereditary basis, have a frank depletion of marrow megakaryocytes and thrombocytopenia on that basis. In three of these patients that we have studied, platelet size measurements have been similar to patients with acquired Aplastic Anemia.
Lorge Platelets We have seen three families in which large platelets have been present (Fam-
ilies Ep, Bu, Ha) . In these patients, the large platelets are easily appreciated on casual examination of a routine peripheral smear. In one of these families, Family Ep, increased platelet size is definitely present only after splenectomy. This i s almost a unique situation because platelet size is not increased after splenectomy of nonthrombocytopenic patients. In Families Bu and Ha, platelet size is increased in patients with intact spleens as well as after splenectomy. The mode of inheritance, however, is different in these two families. In the two families with large platelets in which survival studies have been done, platelet survival has been normal. Therefore, in these patients, the general concept that big platelets are young platelets would be quite erroneous.
The patients described provide examples of all the major types of hereditary thrombocytopenia reported except one. Bernard and Soulier,ll in 1948, first described a syndrome of thrombocytopenia with giant platelets that were de- fective in the thromboplastin generation test. Many examples have been described subsequently. In most reports, this trait has been transmitted as an autosomal dominant, and autologous platelet survival has been shortened.12J3J4 A recent publication from Norway14 suggests that in this disorder, the platelet membrane is deficient in sialic acid and that electrophoretic mobility is abnormal on that basis. This would provide a logical explanation for the reduced platelet survival. We have not yet seen an example of this syndrome. TABLE 2 offers a classifica-

Murphy: Hereditary Thrombocytopenia TABLE 2
427
HEREDITARY THROMBOCYTOPENIA'
I. Microthrombocytes A. Sex-linked recessive inheritance and shortened lifespan
1. Wiskott-Aldrich Syndrome (Au. Br, Ra) 2. Incomplete WAS (Ri) Dominant inheritance - functional defect predominates, only minimal thrombocytopenia ( 9 )
B.
11. Normal Platelet Size A. Dominant inheritance
1. Normal lifespan (Sh) 2. Shortened lifespan (Sa)
B. Autosomal recessive inheritance (10)
111. Macrothrombocytes A. Normal lifespan
1. Dominant inheritance (Ep) 2. Recessive inheritance (Bu) Shortened lifespan - Dominant inheritance (1 1-14) B.
* Letters or numbers in parentheses refer to one of the families listed in TABLE 1 or a pertinent publication in the reference section.
tion of hereditary thrombocytopenia based on platelet size, platelet lifespan, and mode of inheritance.
PLATELET FUNCTION Nothing has yet been said about platelet function in these patients. Certainly
we can anticipate that the intrinsic defects responsible for thrombocytopenia might compromise platelet function as well. We have not felt secure interpreting aggregation studies in these patients because of the low platelet counts in platelet- rich plasma (PRP) obtained from them. Aggregation with epinephrine and collagen become abnormal as normal PRP is diluted to thrombocytopenic levels. We have studied aggregation with adenosine diphosphate (ADP) in six of these families; results have been normal in five (Ra, Br, Ri, Sa, Ep) . In patient C.Sh.,
0-
a OD 4- / PLATELETS-70,000/mm3
I 2 i M MINUTES 0 ADP
FIGURE 5. Defective platelet function in Patient C.Sh. On three separate occasions when the patient was receiving no medications, the addition of ADP to citrated PRP (final con- centration, 1-5pM) produced little or no change in the optical density of the solution. In each instance, control PRP adjusted to the same platelet count demonstrated a normal aggregation curve.

428 Annals New York Academy of Sciences
however, we have repeatedly found abnormal aggregation with ADP (FIGURE 5 ) . The proposed defect in aggregation was confirmed by an Ivy bleeding time of greater than 20 minutes at a time when his platelet count was 145,000/mms. We would expect a normal bleeding time at this level. Another family, similar to Family Sh, in which inherited thrombocytopenia was associated with a functional defect similar to that seen in thrombasthenia, has been previously r e~0r t ed . I~ In our view, better definition of the functional defects in these patients awaits better techniques for studying platelet function in thrombocytopenic patients.
SUMMARY Nine families with hereditary thrombocytopenia have been described. They
have been discussed and classified according to mode of inheritance, the presence or absence of clinical features of the Wiskott-Aldrich Syndrome, platelet size, and platelet lifespan studies. These features and an assay for serum Antiplatelet Factor help to distinguish hereditary disease from acquired thrombocytopenias such as idiopathic thrombocytopenic purpura. This distinction is important, since response to therapy, particularly splenectomy, is clearly different for hereditary and acquired disease.
ACKNOWLEDGMENTS These studies were carried out with the assistance of many colleagues, including
Dr. John Egan, Dr. Margaret Johnson, Dr. Charles Lusch, Dr. Michael Miller, Dr. Lawrence Naiman, Dr. Frank Oski, Dr. Arnold Rubin, Dr. Richard Smalley, Dr. Harvey Weiss, and Dr. James Wolff.
REFERENCES 1. DIAMOND, L. K. 1969. Splenectomy in childhood and the hazard of overwhelming
infection. Pediatrics 43: 886. 2. KARPATKIN, S. & G. W. SISKIND. 1969. I n vitro detection of platelet antibody in patients
with idiopathic thrombocytopenic purpura and systemic lupus erythematosus. Blood 33: 795.
3. MURPHY, S., F. A. OSKI & F. H. GARDNER. 1969. Hereditary thrombocytopenia with an intrinsic platelet defect. New. Eng. J. Med. 281: 857.
4. ABRAHAMSEN, A. F. 1968. A modification of the technique for 51Cr-labelling of blood platelets giving increased circulating platelet radioactivity. Scand. J. Haematol. 5: 53.
5. FUHRMAN, G. F. & G. RUHANSTROTH-BAUER. 1965. In Cell Electrophoresis. E. J. Ambrose, Ed. : 22. J. A. Churchill. London, England.
6. GROTTUM, K. A., T. HOVIG, H. HOLMSEN, A. F. ABRAHAMSEN, M. JEREMIC & M. SEIP. 1969. Wiskott-Aldrich syndrome: qualitative platelet defects and short platelet survival Brit. J. Haematol. 17: 373.
7. BALDINI, M., B. KIM, M. STEINER, A. KURAMATO, M. OKUMA & B. W. OTRIDGE. 1969. Metabolic platelet defect in the Wiskott-Aldrich syndrome. Proceedings of the 39th Annual Meeting of the Society for Pediatric Research : p. 48.
8. HARKER, L. A. 1968. Significance of platelet volume alterations. (Abstr.) 22-7. XI1 Congress of the International Society of Hematology. New York, N. Y.
9. WEISS, H. J., P. A. CHERVENICK, R. ZALUSKY & A. FACTOR. 1969. A fami!ial defect in platelet function associated with impaired release of adenosine diphosphate. New Eng. J. Med. 281: 1264.
10. HALL, J. G., J. LEVIN, J. P. KUHN, E. J. OTTENHEIMER, K. A. PETER VAN BERKUM & V. A. MCKUSICK. 1969. Thrombocytopenia with absent radius (TAR). Medicine 48: 41 1.
11. BERNARD, J. & J. P. SOULIER. 1948. Sur une nouvelle varidte de dystrophie thrombo- cytaire hdmorragipare congtnitale. Sem. HBp. Paris 24: 3217.
12. NAJEAN, Y., N. ARDAILLOU. J. CAEN, M.-J. LARRIEU & J. BERNARD. 1963. Survival of radiochromium-labeled platelets in thrombocytopenias. Blood 22: 718.
13. CULLUM, C., D. P. COONEY & S. L. SCHRIER. 1967. Familial thrombocytopenic thrombo- cytopathy. Brit. J. Haematol. 13: 147.
14. GROTNM, K. A. & N. 0. SOLUM. 1969. Congenital thrombocytopenia with giant platelets; a defect in the platelet membrane. Brit. J. Haematol. 16: 277.
IS. SHETH, N. K. &A. J. PRANKERD. 1968. Inherited thrombocytopenia with thrombasthenia. J. Clin. Path. 21: 154.