international journal of pharmaceutics - chemlabcorp.com · (ivivc) by the simultaneous ... dosage...
TRANSCRIPT

International Journal of Pharmaceutics 491 (2015) 180–189
In vitro dissolution–permeation evaluation of an electrospuncyclodextrin-based formulation of aripiprazole using mFluxTM
EnikÅ Borbása, Attila Balogha, Katalin Bocza, Judit Müllera, Éva Kiserdeia, Tamás Vigha,Bálint Sinkób, Attila Marosic, Attila Halászd, Zoltán Dohányosd, Lajos Szentee,György T. Baloghf,**, Zsombor K. Nagya,*aBudapest University of Technology and Economics, Department of Organic Chemistry and Technology, Hungaryb PION Inc., USAc Semmelweis University, Department of Pharmaceutical Chemistry, HungarydGedeon Richter Plc., Department of Discovery Pharmacokinetics, HungaryeCYCLOLAB, Cyclodextrin Research and Development Laboratory Ltd., HungaryfGedeon Richter Plc., Compound Profiling Laboratory, Hungary
A R T I C L E I N F O
Article history:Received 26 April 2015Received in revised form 12 June 2015Accepted 14 June 2015Available online 24 June 2015
Keywords:ElectrospinningNanofiberDissolution-permeationMicrofluxPermeationCyclodextrinAripiprazole
A B S T R A C T
Since it is a well-known fact that among the newly discovered active pharmaceutical ingredients thenumber of poorly water soluble candidates is continually increasing, dissolution enhancement of poorlywater soluble drugs has become one of the central challenges of pharmaceutical studies. So far thepreclinical studies have been mainly focused on formulation methods to enhance the dissolution ofactive compounds, in many cases disregarding the fact that the formulation matrix not only affectsdissolution but also has an effect on the transport through biological membranes, changing permeationof the drug molecules. The aim of this study was to test an electrospun cyclodextrin-based formulation ofaripiprazole with the novel mFlux apparatus, which monitors permeation together with dissolution, andby this means better in vitro–in vivo correlation is achieved. It was evinced that a cyclodextrin-basedelectrospun formulation of aripiprazole has the potential to ensure fast drug delivery through the oralmucosa owing to the ultrafast dissolution of the drug from the formulation and the enhanced flux acrossmembranes as shown by the result of the novel in vitro dissolution and permeation test.
ã 2015 Elsevier B.V. All rights reserved.
Contents lists available at ScienceDirect
International Journal of Pharmaceutics
journa l home page : www.e l sev ier .com/ loca te / i jpharm
1. Introduction
As most of the newly discovered active pharmaceuticalingredients (APIs) have poor water solubility, dissolution enhance-ment of the Biopharmaceutical Classification System (BCS) II type(poorly water-soluble, but highly permeable) drugs is one of themost pressing pharmaceutical challenges (Keserü and Makara,2009; Lipinski, 2000). For the evaluation of dissolution enhance-ment, dissolution tests are used in the industrial protocol.However, the results of these tests do not always correlate withthe bioavailability. One common reason for the lack of correlationis that studying dissolution without permeability can be deceptive,since both of them have an impact on in vivo bioavailability (Dahanand Miller, 2012). In addition, the solubility-permeability interplay
* Corresponding author at: 1111 Budapest, Máegyetem rkp. 3, Hungary.** Corresponding author at: 1103 Budapest, GyömrÅi út 19-21, Hungary.
E-mail addresses: [email protected] (G.T. Balogh), [email protected](Z.K. Nagy).
http://dx.doi.org/10.1016/j.ijpharm.2015.06.0190378-5173/ã 2015 Elsevier B.V. All rights reserved.
cannot be overlooked in cases where the API is formulated toenhance its dissolution, because additives, such as surfactants,polymers and cyclodextrins, can have an effect on the permeationof the drug molecules through the biological membranes (Loftssonand Brewster, 2011; Dahan et al., 2010; Fenyvesi et al., 2011). Forexample, when the API has pH-dependent solubility, microenvi-ronmental pH-modification is a possible way to improve dissolu-tion behavior (Tran et al., 2008; Tran et al., 2010; Taniguchi et al.,2014; Loftsson and Brewster, 2012). However, when usingformulation techniques like this former one, it is important tonote that modification of pH might though enhance API solubilityby ionization, it can hinder absorption, which can cause that thebioavailability cannot be predicted from only dissolution testresults. In order to permeate, the ionizable molecule must be in itsuncharged form at the membrane surface to permeate by passivediffusion most efficiently (Avdeef, 2012; Balogh et al., 2013).Moreover, when an API dissolves from its amorphous soliddispersion, in most cases a supersaturated solution is created,and according to a recent study reported by Frank et al., such a

E. Borbás et al. / International Journal of Pharmaceutics 491 (2015) 180–189 181
lasting supersaturation can be the key for enhancing not only thedissolution but the absorption rate of the API as well (Frank et al.,2014). In the case of in vitro dissolution tests the concentration ofthe API is not lowered by transports like in biological systems,meaning that the probability of precipitation from a supersaturat-ed solution is higher in vitro then in vivo. Therefore, dissolution–permeability tests have the possibility to be more predictive as toin vivo results if precipitation occurs.
There have been many attempts to improve the in vitro–in vivocorrelation (IVIVC) by the simultaneous testing of dissolution andpermeability (Grundy et al.,1997; Kobayashi et al., 2001; Dickinsonet al., 2012), e.g. by connecting dissolution cells with Caco-2 tests(Ginski and Polli, 1999). Due to the fact that Caco-2 permeabilityassay is a living cell-based method, its time and cost effectivenessis low compared to non-cell-based tests like Parallel ArtificialMembrane Permeability Assay (PAMPA). Although PAMPA has theadvantage that it is a high throughput screening method based onsimple analytical detection, the small volume of the donor andacceptor compartments in which the experiments are carried outmakes the application of this assay more difficult in the case offormulations. Therefore, it is proposed that a scaled up version ofPAMPA could be an eligible way of combining dissolution andpermeability testing. mFlux is a novel technique, that (bysimultaneously measuring dissolution and permeability) has thepotential to improve IVIVC and to reduce the number of experi-ments needed on animals. In the case of formulation screening, itcould be a reproducible and cost-effective way of testing comparedto cell-based or animal tests. Dosage forms produced by variousformulation strategies could be investigated with this newanalytical method.
Creating amorphous solid dispersions is one of the mostpromising strategies to improve the bioavailability of poorly water-soluble drugs. This is largely due to the amorphous state of the API(Seif et al., 2015), by the administration of which the achievableconcentration can be significantly higher than the solubility of anyof the crystalline forms (Hancock and Parks, 2000), and to thehydrophilic nature of the surrounding matrix material that can aidwetting once exposed to the aqueous biological media (Shah et al.,2014). A relatively new and emerging way of generating soliddispersions is electrospinning, a process that can create polymericfibers at nano- and micro-scale from a viscous polymer solutioncontaining an API, using the drawing force of an electrostatic field.With the help of this promising continuous technology fastdissolution of the drug can be reached owing to the high surfacearea, the wettability of the polymer mat and the amorphisation ofthe API (Nagy et al., 2012). Although electrospun drug-loadednanofibers have been widely investigated for potential applica-tions to provide fast release (Yu et al., 2009), sustained release (Yuet al., 2012) and biphasic controlled release profiles (Yu et al.,2013), to exploited as solid dosage forms (Yu et al., 2013a,b; Nagyet al., 2013; Balogh et al., 2014; Nagy et al., 2015; Vrbata et al.,2014), wound dressings (Sridhar et al., 2014; Pelipenko et al., 2013),topical drug delivery systems (Huang et al., 2012), transdermalpatches (Kataria et al., 2014), vaginal dosage forms (Nagy et al.,2014; Huang et al., 2014) and mucoadhesive orally dissolving drugdelivery system for dental care (Samprasit et al., 2015), few studieshave reported about their dissolution–absorption properties.
Excipients such as cyclodextrins are widely used in thepharmaceutical industry, because these cyclic oligosaccharidescan function as solubilizing and stabilizing agents owing to theinclusion complex formation with lipophilic API substances asguest molecules (Challa et al., 2005; Loftsson, 2014; Vigh et al.,2013). In case of aripiprazole the use of HPbCD and SBEbCD wasreported for the solubility enhancement of the drug (Mihajlovicet al., 2012; Nerurkar and Naringrekar, 2009).
There are already several products on the market, such asinjections or tablets (e.g. Sugammadex), but recent studies statethat cyclodextrins could be used even for drug delivery throughalternative routes like buccal, nasal or pulmonary administration(Jansook et al., 2011; Loftsson and Masson, 2001; Schipper et al.,1992; Marques et al., 1991).
In this work we investigated the in vitro dissolution–perme-ation properties of a complex formulation matrix using mFlux.Various formulation strategies were combined to create amor-phous solid dispersions using electrospinning, sulfobutylether-b-cyclodextrin as solubilizing agent and citric acid as micro-enviromental pH modifying agent to enhance the dissolution andthe bioavailabilty of a poorly soluble antipsychotic drug, aripipra-zole (Fig. 1). This API has not only therapeutic, but economicalpotential as well, especially in the USA, where it was the marketleader in 2013. After formulating the API by creating an orallydisintegrating and fast dissolving drug delivery system, it has thepossibility to ensure proper and painless delivery of an antipsy-chotic drug even in the case of poor patient compliance (Patel et al.,2011).
2. Materials and methods
2.1. Materials
Aripiprazole (ARP, 448.385 g/mol), citric acid and buffercomponents (KH2PO4, NaOH) were purchased from Sigma–AldrichCo., LLC. (St. Louis, MO, The United States), sulfobuthylether-b-cyclodextrin (SBEbCD, (1135.0 + n � 158.2) g/mol, degree ofsubstitution 6.2–6.9) was kindly supplied by Cyclolab Ltd. (Buda-pest, Hungary), poly(ethylene oxide) grades (PEOs, with averagemolar weights of 1 �106, 2 � 106, 4 �106, 7 � 106 g/mol) werereceived from Colorcon Ltd. (Dartford Kent, United Kingdom).
2.2. Phase solubility study
The solubility of aripiprazole in the presence of SBEbCD wasdetermined in 3.33 mg/mL citric acid solution. Specifically, excessamounts of ARP were added in the citric acid solution withincreasing concentration of SBEbCD (0–100 mM). Suspensionswere stirred at 30 �C for 24 h. The suspensions were filteredthrough a 0.45 mm membrane filter, diluted and analyzedspectrofotometrically (HP 8452A UV–vis spectrophotometer) at248 nm.
2.3. Electrospinning
The polymer, the ARP, the cyclodextrin and the citric acid ofcalculated amounts were added into 10 mL of the solvent (ethanoland water 1:1) and stirred magnetically (VELP Scientifica, Usmate,Italy) at 600 rpm until the complete dissolution. The electricalpotential applied on the spinneret electrode was 40 kV (NT-35 highvoltage DC supply, MA2000 device, Unitronik Ltd., Nagykanizsa,Hungary). A grounded aluminum plate covered with aluminum foilwas used as collector (50 cm from the spinneret). Polymersolutions were dosed with 2 mL/h at room temperature (25 �C)with an SEP-10S Plus type syringe pump (Aitecs, Vilnius,Lithuania).
Fig. 1. Chemical structure of aripiprazole.

182 E. Borbás et al. / International Journal of Pharmaceutics 491 (2015) 180–189
2.4. Scanning electron microscopy
Morphology of the samples was investigated with a JEOL JSM-6380LA (JEOL Ltd., Tokyo, Japan) type scanning electron micro-scope. Each specimen was fixed by conductive double-sided carbonadhesive tape and coated with gold–palladium alloy beforeexamination. The applied accelerating voltage and workingdistance were between 15 and 30 kV and 10 and 12 mm,respectively.
2.5. Differential scanning calorimetry
Differential scanning calorimetry (DSC) measurements werecarried out using a Setaram DSC 92 apparatus (Caluire, France).(Sample weight: 10–15 mg, open aluminum pan, nitrogen flush.)The temperature program consisted of an isothermal period(1 min) at room temperature, with subsequent linear heating from25 �C to 200 �C at the rate of 10 �C/min.
2.6. X-ray powder diffraction
X-ray diffraction patterns of the samples were recorded bymeans of a PANalytical (Amelo, the Netherlands) X’pert ProMDP X-ray diffractometer using Cu-K” a radiation (1.542 Å) and a Ni filter.The applied voltage was 40 kV, while the current was 30 mA. Thesamples were analyzed between 4� and 42� 2u.
2.7. Titration of phosphate buffer and human saliva with citric acid
Human saliva samples from 3 healthy man volunteers (agedbetween 20 and 30 years) were collected before meal. 5 mL of eachsample were then titrated with citric acid at room temperature,immediately after collection. Series of pH 6.8 phosphate bufferswith different buffer capacities were prepared by diluting a0.05 mol/L KH2PO4 buffer with distilled water. 5 mL of humansaliva (from healthy men volunteers, n = 3) and pH 6.8 phosphatebuffers were measured, then in each step the weighted amount ofcitric acid was added to the solution and stirred for 2–3 min to get ahomogenous solution, then the pH of the solution was measured(Metler Toledo MP220, Hungary) at room temperature (25 �C � 1�C).
2.8. Measurement of the thermodynamic solubility
For measuring the thermodynamic solubility 300 mL of distilledwater or citric acid solution (3.33 mg/L, pH 2.5) were added tocrystalline ARP (�1 mg) (n = 5). The resulting mixtures wereshaken at room temperature for 24 h (to the solution equilibrium)and were filtered on a 0.45 mm Millipore Multiscreen Filter Plate.The concentrations of the filtrates were measured by LC–MS(Agilent 1200 coupled with an Agilent 6410 triple quadrupole massspectrometer equipped with an ESI ion source) in the case of the
Table 1Lipid compositions of membranes used for PAMPA.
Gastrointestinal lipid mixturea (mg lipids/mL n-dode
Phosphatidylcholine 26.7
Cholesterol 13.3
Palmitic acid –
Glucosylceramide –
Sphingomyelin –
Phosphatidylinositol –
Phosphatidylethanolamine –
a Modified Kansy method (Kansy et al., 1998).
distilled water, and by LC-UV (Agilent 1200 LC-DAD, 220 nm) in thecase of the citric acid solution, based on calibration (n = 3).
2.9. Small-volume “in syringe” dissolution testing
Small-volume dissolution studies were performed using aprototype of “in-syringe” dissolution testing apparatus in pH6.8 phosphate buffer (0.025 mol/dm3 KH2PO4) at 25 �C. Theapparatus consisted of a disposable 10 mL plastic syringe, a0.45 mm syringe filter and a magnetic stirrer bar (10 mm long witha diameter of 3 mm). The whole dissolution experiment wasconducted inside of the barrel of the syringe, while samples weretaken from the stirred medium through the syringe filter placed onthe adaptor of the syringe. For dissolution testing, the preparedphysical mixtures or the electrospun samples equivalent to 5 mg ofaripiprazole, or the pure crystalline form of the API (5 mg) wereweighted into the syringe. Then, the stirring bar was placed on topof the powder, and the plunger was reinstated. 5 mL of pH6.8 phosphate buffer was poured into the syringe through theadaptor hole using automatic pipette. Then the syringe filter wasreinstated, and the apparatus was immediately placed onto amagnetic stirrer (Heidolph MR Hei-Tec, Nuremberg, Germany)without heating and stirred at (digitally controlled) 150 rpm, atroom temperature (25 �C � 1 �C). Dissolution was followed for60 min and two drops of dissolution medium were carefully takenas samples through the syringe filter at predetermined timeintervals. From the two drops of the sample 50 mL were taken usinga micropipette, diluted to 3 mL and analyzed spectrophotometri-cally at a wavelength of 248 nm (HP 8452A UV–vis spectropho-tometer).
2.10. Paralell artificial membrane permeability assay (PAMPA)
2.10.1. PAMPA methodSolutions of each compound were prepared in dimethyl
sulfoxide (DMSO) at 10 mM and then diluted with phosphatebuffer of a pH of 6.8 to obtain the donor drug solution with the finalconcentration of 100 mM. This solution was shaken for an hour inroom temperature in a 96-well polypropylene plate (Agilent,Waldbronn, Germany).
A slightly modified version of PAMPA (Kansy et al., 1998) wasused to determine the effective permeability (Pe, cm/s). Each wellof donor plates (MultiscreenTM-IP, MAIPN4510, pore size 0.45 mm;Millipore) was coated with 5 mL of n-dodecane or lipid solutiondissolved in n-dodecane (the applied lipid compositions are shownin Table 1). Then 150 mL (100 mM) of the API solution was put onthe membrane. The bottom plate (96-well PTFE acceptor plates(Multiscreen Acceptor Plate, MSSACCEPTOR; Millipore) was filledwith 300 mL of the buffer solution (0.01 M PBS, pH 7.4). The donorand acceptor plates were fit, and the donor plate was coated with asheet of wet paper tissue and a plate lid to avoid the evaporation ofthe solvent. The plates were incubated (37 �C, 4 h, HeidolphTitramax 1000). After that the PAMPA sandwich plates were
cane) Buccal lipid mixture (Kokate et al., 2009) (mg lipids/mL n-dodecane)
8.010.34.06.03.30.87.5

E. Borbás et al. / International Journal of Pharmaceutics 491 (2015) 180–189 183
separated, and the concentrations of the API (at zero time pointand after incubation) in the donor and acceptor solutions weredetermined by HPLC-DAD. Test solutions, originated from PAMPAexperiments were prepared in 96-well polypropylene plates(Agilent, Waldbronn, Germany) and sealed before injection.
The effective permeability and the membrane retention ofdrugs were calculated using the following equation (Avdeef, 2012):
Pe ¼ �2:303A � ðt � tssÞ �
11 þ ra
� �
� 1g �ra þ 1 þ ra1 � MR
� �� CDðtÞCDð0Þ
� �; (1)
where Pe is the effective permeability coefficient (cm/s), A is thefilter area (0.3 cm2), VD and VA are the volumes in the donor(0.15 cm3) and acceptor phase (0.3 cm3), t is the incubation time (s),tSS is the time (s) to reach steady-state, CD(t) is the concentration(mol/cm3) of the compound in the donor phase at time point t,CD(0) is the concentration (mol/cm3) of the compound in the donorphase at time 0, MR is the membrane retention factor, ra is the sinkasymmetry ratio (gradient-pH-induced), defined as
ra ¼ VD
VA
� �PðA!DÞe
PðD!AÞe
; (2)
MR ¼ 1 � CDðtÞCDð0Þ �
VA
VD
CAðtÞCDð0Þ
� �; (3)
The effective permeability value of each compound was measuredwith 3 replicates.
Quantitative chromatographic analyses were performed using aSHIMADZU Prominence Modular HPLC system (Shimadzu Corpo-ration, Japan) at 40 �C on a Kinetex 2.6 mm XB-C18 100A(30 � 3 mm) column with a mobile phase flow rate of 1.1 mL/min. Composition of mobile phase: A (0.1% (v/v) formic acid inwater), B (acetonitrile/water 95/5 (v/v) with 0.1% (v/v) formic acid).The applied two-step linear gradient program: 0–15% B (0–0.3 min), 15–100% B (0.3–1.8 min), 100% B (1.8–2.4 min), 0% B(2.41 min), equilibration (for 1.2 min). The chromatograms wererecorded at 200–500 nm (UVmax) and the applied injection volumewas 6 mL.
Fig. 2. Schematic figure of the m
2.11. In vitro dissolution–permeation study
In vitro dissolution–permeation studies were carried out usingmFLUX (pION Inc., Boston, USA) (Fig. 2). The apparatus consists of adonor and an acceptor chamber (20 mL volumes) separated by anartificial membrane (PVDF, polyvinylidenfluoride, 0.45 mm,8.55 cm2). In the case of buccal drug delivery, the donor chamberrepresents the oral cavity, the artificial membrane the oral mucosa,while the acceptor chamber represents the blood circulation. Atfirst 75 mL of n-dodecane was dribbled on the membrane surface,then 20 mL of pH 6.8 phosphate buffer was added into the donorchamber, and 20 mL of sink buffer was added into the acceptorchamber. The solubility of ARP is 30 mg/L in the acceptor buffer,which meets the requirement for sink condition, i.e. the volume ofmedium in the acceptor chamber is at least three times greaterthan required to form a saturated solution of the drug substance(FIP Guidelines, 1981; USP 23, 1995). Samples equivalent to 20 mgof ARP were placed in the donor chamber. Both chambers werestirred at 200 rpm at room temperature. In both chambers the APIconcentration was followed UV-spectrofotometrically at 252 nm.
The flux across the membrane was calculated using thefollowing equation:
JðtÞ ¼ DnA � Dt
where the flux (J) of a drug through the membrane is defined as theamount (n) of drug crossing a unit area (A) perpendicular to its flowper unit time (t).
3. Results and discussions
Firstly the optimization of the formulation matrix, secondly themorphology of the electrospun amorphous solid dispersion aredescribed. Then, results of small-volume “in syringe” dissolutiontests and dissolution–permeability tests using mFlux technique areshown and discussed.
3.1. Optimization of the formulation matrix
Taking into account that sulfobutylether-b-cyclodextrin sodi-um salt is approved in 6 marketed pharmaceutical productsaccording to USP-NF, it was selected and used to enhance theapparent solubility of ARP, the thermodynamic solubility of which
FLUX (pION inc.) apparatus.

Table 2Compositions of the electrospun solutions.a
Nr. PEO (mg) Mw (106 g/mol) Electropun fibers
ES1 50 1 noneES2 50 2 noneES3 50 4 noneES4 50 7 noneES5 75 1 beadedES6 75 2 beadlessES7 75 4 noneES8 75 7 none
a 75 mg ARP, 1500 mg SBEbCD, 33.3 mg citric acid, solvent: 5 mL ethanol, 5 mLwater.
184 E. Borbás et al. / International Journal of Pharmaceutics 491 (2015) 180–189
is under the quantification limit (4.93 �10�3mg/L) at 25 �C indistilled water. The use of 150 mg/mL SBEbCD enabled to reach anAPI concentration in water as high as 148 mg/L, which meansapproximately a 30,000 times greater achievable concentrationthan in the case of the pure ARP.
ARP is a weak base (cpKa = 7.46 ChemAxon, DrugBank) andconsequently has pH dependent solubility. The solubility of ARPincreases with decreasing pH; therefore, with micro-environmen-tal pH-lowering the solubility of ARP increases. By adding citricacid to the mixture of SBEbCD and the API, a concentration of4464 mg/L was reached at a pH of 4.0, and could be used forpreparing a stable polymer solution from ARP and this is a 30 timesgreater concentration than without citric acid (thermodynamicsolubility of ARP is 430 mg/L in 3.33 mg/L citric acid solution,25 �C).
In Fig. 3 phase solubility of ARP in 3.33 mg/ml citric acidsolution with different SBEbCD contents is presented. From theresults the apparent stability constant was calculated and found tobe 1822 dm3/mol.
SBEbCD does not have the ability to form fibers without using apolymeric matrix because of the repulsive forces between theSBEbCD molecules caused by their ionization. Therefore, poly(ethylene oxide) (PEO) was selected as a fiber forming polymer. It isable to stabilize supersaturated systems during dissolution, but hasa retard effect on dissolution (Campbell et al., 2008) as well, whichis unfavorable when aiming at creating a fast dissolving drugdelivery system. In order to avoid this retard effect, the polymerconcentration was kept at minimum. The concentration of the APIdissolved in the polymer solution (containing SBEbCD and citricacid) was kept constant at 7500 mg/L, because this concentration isclose to the achievable maximum, but could be reproducibly andquickly reached.
At first, distilled water was used to prepare the polymersolution, but adding ethanol to the solvent improved theprocessability through lowering the surface tension of the polymersolution (Kostakova et al., 2014); therefore, ethanol and water in a1:1 ratio was used afterwards.
The composition of the ES solutions were changed systemati-cally (Table 2) until homogeneous, non-beaded, thin, and discretepoly(ethylene oxide) fibers were obtained. PEO fibers wereexpected to form when sufficient entanglements of the linearpolymer were achieved in the solution (Shenoy et al., 2005).
During the optimization of the process, the polymer concen-tration was studied on two levels. It was found that the PEO with an
Fig. 3. Phase solubility diagram of ARP in 3.33 mg/L citric acid solution withdifferent SBEbCD contents.
average molecular weight (Mw) of 2 � 106 g/mol was able toproduce beadless fibers at 0.75% (w/v) polymer concentration(ES6), the diameters of these electrospun fibers ranged from 900 to1500 nm (Figure 4). At lower concentration (0.5%) no fibers wereformed, only electrospraying occured. When applying 0.75% (w/v)PEO of a lower Mw (1 �106 g/mol), many beads were found in theproduct, while at higher Mw (4 �106 g/mol, 7 � 106 g/mol) thepolymer solution became too viscous, making the ES processinefficient (Table 2).
3.2. Characterisation of the electrospun samples
3.2.1. X-ray powder diffractionX-ray powder diffraction was used to characterize the
morphological changes of ARP induced by electrospinning. Fig. 5shows the diffractogram of ES6 compared to those of the individualcomponents. In the case of the electropsun fibers (e), no diffractionpeaks are observed but only a diffuse background scattering as aproof of the amorphous nature of the sample, No signs ofcrystallization were observed even after 3 months, either (g),indicating the stability of the product No signs of crystallizationwere observed even after 3 months, either (g), indicating thestability of the product.
3.2.2. Differential scanning calorimetryDifferential scanning calorimetry analyses were also performed
in order to study the physical state of the components in thesamples (Fig. 6). The absence of a melting peak of ARP in theelectrospun fibers (ES 6) leads to the conclusion that the drug is inamorphous form. No signs of crystallization were observed evenafter 3 months, either (g), indicating the stability of the product.The melting peak of PEO is also absent in the case of thenanofibrous product. In the thermograms of SBEbCD and thenanofibers, a broad endotherm, characteristic of water lossbetween room temperature and 150 �C, is observed.
Based on the DSC curves and the X-ray diffractograms the pureAPI is present in form X, which is the thermodynamically stableform of the API at room temperature. Characteristic peaks at 9.94,12.55, 21.77 and 22.21 2 theta was found, which indicatesaccording to Braun et al., (2009) that it is form X. On the DSCcurve there is small peak at 128 �C and a larger one at 150 �C whichalso suggest that form X is present.
Based on the above-mentioned results, drug dissolutionenhancement should be expected because of the high surfacearea and huge porosity of the medicated nanofibers and theamorphous state of drug presenting in them.
3.3.1. Selection of the dissolution mediumThe dissolution medium needed to be optimized because of the
pH dependency of the drug dissolution and the micro-environ-mental pH lowering agent, citric acid, used in the formulationmatrix. Human saliva has the approximate pH of 6.8 (Marques
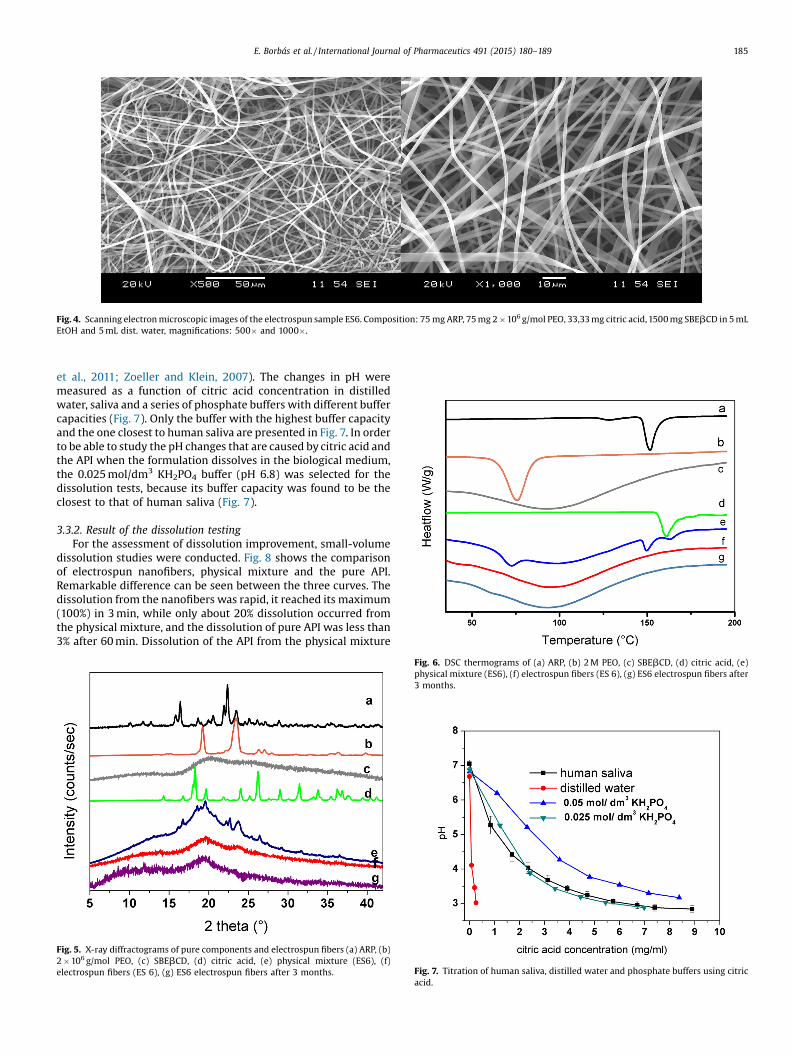
Fig. 4. Scanning electron microscopic images of the electrospun sample ES6. Composition: 75 mg ARP, 75 mg 2 � 106 g/mol PEO, 33,33 mg citric acid, 1500 mg SBEbCD in 5 mLEtOH and 5 mL dist. water, magnifications: 500� and 1000�.
E. Borbás et al. / International Journal of Pharmaceutics 491 (2015) 180–189 185
et al., 2011; Zoeller and Klein, 2007). The changes in pH weremeasured as a function of citric acid concentration in distilledwater, saliva and a series of phosphate buffers with different buffercapacities (Fig. 7). Only the buffer with the highest buffer capacityand the one closest to human saliva are presented in Fig. 7. In orderto be able to study the pH changes that are caused by citric acid andthe API when the formulation dissolves in the biological medium,the 0.025 mol/dm3 KH2PO4 buffer (pH 6.8) was selected for thedissolution tests, because its buffer capacity was found to be theclosest to that of human saliva (Fig. 7).
3.3.2. Result of the dissolution testingFor the assessment of dissolution improvement, small-volume
dissolution studies were conducted. Fig. 8 shows the comparisonof electrospun nanofibers, physical mixture and the pure API.Remarkable difference can be seen between the three curves. Thedissolution from the nanofibers was rapid, it reached its maximum(100%) in 3 min, while only about 20% dissolution occurred fromthe physical mixture, and the dissolution of pure API was less than3% after 60 min. Dissolution of the API from the physical mixture
Fig. 5. X-ray diffractograms of pure components and electrospun fibers (a) ARP, (b)2 � 106 g/mol PEO, (c) SBEbCD, (d) citric acid, (e) physical mixture (ES6), (f)electrospun fibers (ES 6), (g) ES6 electrospun fibers after 3 months. Fig. 7. Titration of human saliva, distilled water and phosphate buffers using citric
acid.
Fig. 6. DSC thermograms of (a) ARP, (b) 2 M PEO, (c) SBEbCD, (d) citric acid, (e)physical mixture (ES6), (f) electrospun fibers (ES 6), (g) ES6 electrospun fibers after3 months.
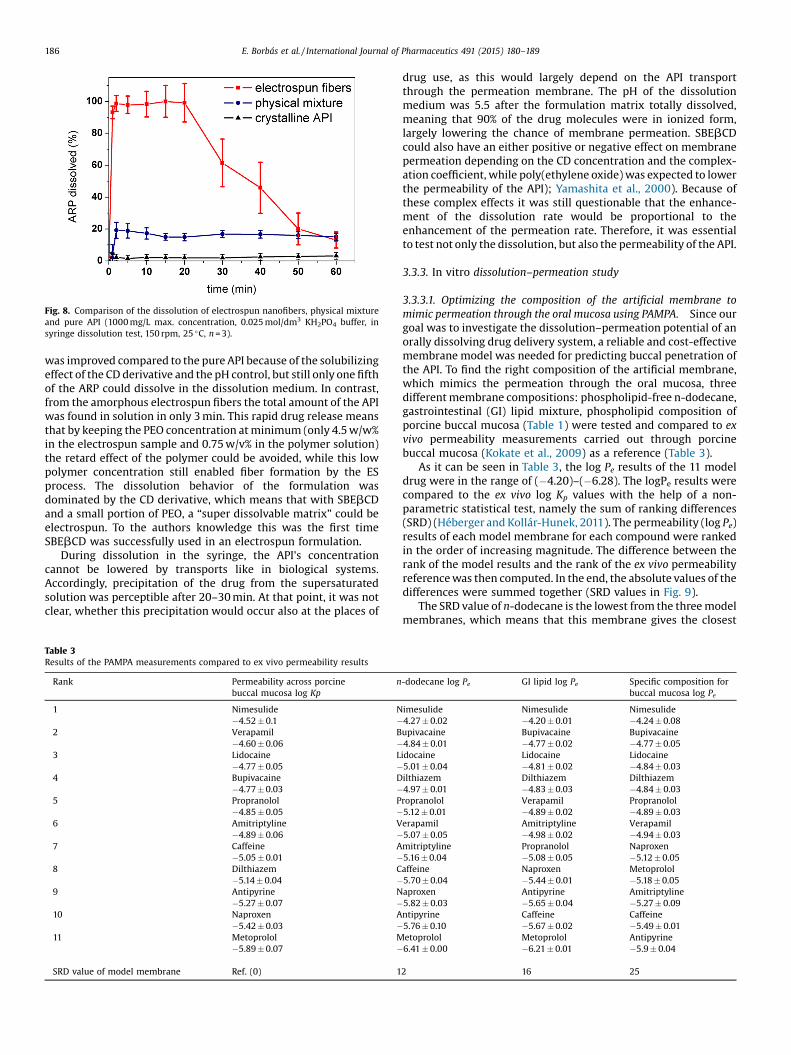
Fig. 8. Comparison of the dissolution of electrospun nanofibers, physical mixtureand pure API (1000 mg/L max. concentration, 0.025 mol/dm3 KH2PO4 buffer, insyringe dissolution test, 150 rpm, 25 �C, n = 3).
186 E. Borbás et al. / International Journal of Pharmaceutics 491 (2015) 180–189
was improved compared to the pure API because of the solubilizingeffect of the CD derivative and the pH control, but still only one fifthof the ARP could dissolve in the dissolution medium. In contrast,from the amorphous electrospun fibers the total amount of the APIwas found in solution in only 3 min. This rapid drug release meansthat by keeping the PEO concentration at minimum (only 4.5 w/w%in the electrospun sample and 0.75 w/v% in the polymer solution)the retard effect of the polymer could be avoided, while this lowpolymer concentration still enabled fiber formation by the ESprocess. The dissolution behavior of the formulation wasdominated by the CD derivative, which means that with SBEbCDand a small portion of PEO, a “super dissolvable matrix” could beelectrospun. To the authors knowledge this was the first timeSBEbCD was successfully used in an electrospun formulation.
During dissolution in the syringe, the API's concentrationcannot be lowered by transports like in biological systems.Accordingly, precipitation of the drug from the supersaturatedsolution was perceptible after 20–30 min. At that point, it was notclear, whether this precipitation would occur also at the places of
Table 3Results of the PAMPA measurements compared to ex vivo permeability results
Rank Permeability across porcinebuccal mucosa log Kp
n
1 Nimesulide N�4.52 � 0.1 �
2 Verapamil B�4.60 � 0.06 �
3 Lidocaine L�4.77 � 0.05 �
4 Bupivacaine D�4.77 � 0.03 �
5 Propranolol P�4.85 � 0.05 �
6 Amitriptyline V�4.89 � 0.06 �
7 Caffeine A�5.05 � 0.01 �
8 Dilthiazem C�5.14 � 0.04 �
9 Antipyrine N�5.27 � 0.07 �
10 Naproxen A�5.42 � 0.03 �
11 Metoprolol M�5.89 � 0.07 �
SRD value of model membrane Ref. (0) 1
drug use, as this would largely depend on the API transportthrough the permeation membrane. The pH of the dissolutionmedium was 5.5 after the formulation matrix totally dissolved,meaning that 90% of the drug molecules were in ionized form,largely lowering the chance of membrane permeation. SBEbCDcould also have an either positive or negative effect on membranepermeation depending on the CD concentration and the complex-ation coefficient, while poly(ethylene oxide) was expected to lowerthe permeability of the API); Yamashita et al., 2000). Because ofthese complex effects it was still questionable that the enhance-ment of the dissolution rate would be proportional to theenhancement of the permeation rate. Therefore, it was essentialto test not only the dissolution, but also the permeability of the API.
3.3.3. In vitro dissolution–permeation study
3.3.3.1. Optimizing the composition of the artificial membrane tomimic permeation through the oral mucosa using PAMPA. Since ourgoal was to investigate the dissolution–permeation potential of anorally dissolving drug delivery system, a reliable and cost-effectivemembrane model was needed for predicting buccal penetration ofthe API. To find the right composition of the artificial membrane,which mimics the permeation through the oral mucosa, threedifferent membrane compositions: phospholipid-free n-dodecane,gastrointestinal (GI) lipid mixture, phospholipid composition ofporcine buccal mucosa (Table 1) were tested and compared to exvivo permeability measurements carried out through porcinebuccal mucosa (Kokate et al., 2009) as a reference (Table 3).
As it can be seen in Table 3, the log Pe results of the 11 modeldrug were in the range of (�4.20)–(�6.28). The logPe results werecompared to the ex vivo log Kp values with the help of a non-parametric statistical test, namely the sum of ranking differences(SRD) (Héberger and Kollár-Hunek, 2011). The permeability (log Pe)results of each model membrane for each compound were rankedin the order of increasing magnitude. The difference between therank of the model results and the rank of the ex vivo permeabilityreference was then computed. In the end, the absolute values of thedifferences were summed together (SRD values in Fig. 9).
The SRD value of n-dodecane is the lowest from the three modelmembranes, which means that this membrane gives the closest
-dodecane log Pe GI lipid log Pe Specific composition forbuccal mucosa log Pe
imesulide Nimesulide Nimesulide4.27 � 0.02 �4.20 � 0.01 �4.24 � 0.08upivacaine Bupivacaine Bupivacaine4.84 � 0.01 �4.77 � 0.02 �4.77 � 0.05idocaine Lidocaine Lidocaine5.01 � 0.04 �4.81 � 0.02 �4.84 � 0.03ilthiazem Dilthiazem Dilthiazem4.97 � 0.01 �4.83 � 0.03 �4.84 � 0.03ropranolol Verapamil Propranolol5.12 � 0.01 �4.89 � 0.02 �4.89 � 0.03erapamil Amitriptyline Verapamil5.07 � 0.05 �4.98 � 0.02 �4.94 � 0.03mitriptyline Propranolol Naproxen5.16 � 0.04 �5.08 � 0.05 �5.12 � 0.05affeine Naproxen Metoprolol5.70 � 0.04 �5.44 � 0.01 �5.18 � 0.05aproxen Antipyrine Amitriptyline5.82 � 0.03 �5.65 � 0.04 �5.27 � 0.09ntipyrine Caffeine Caffeine5.76 � 0.10 �5.67 � 0.02 �5.49 � 0.01etoprolol Metoprolol Antipyrine6.41 � 0.00 �6.21 � 0.01 �5.9 � 0.04
2 16 25
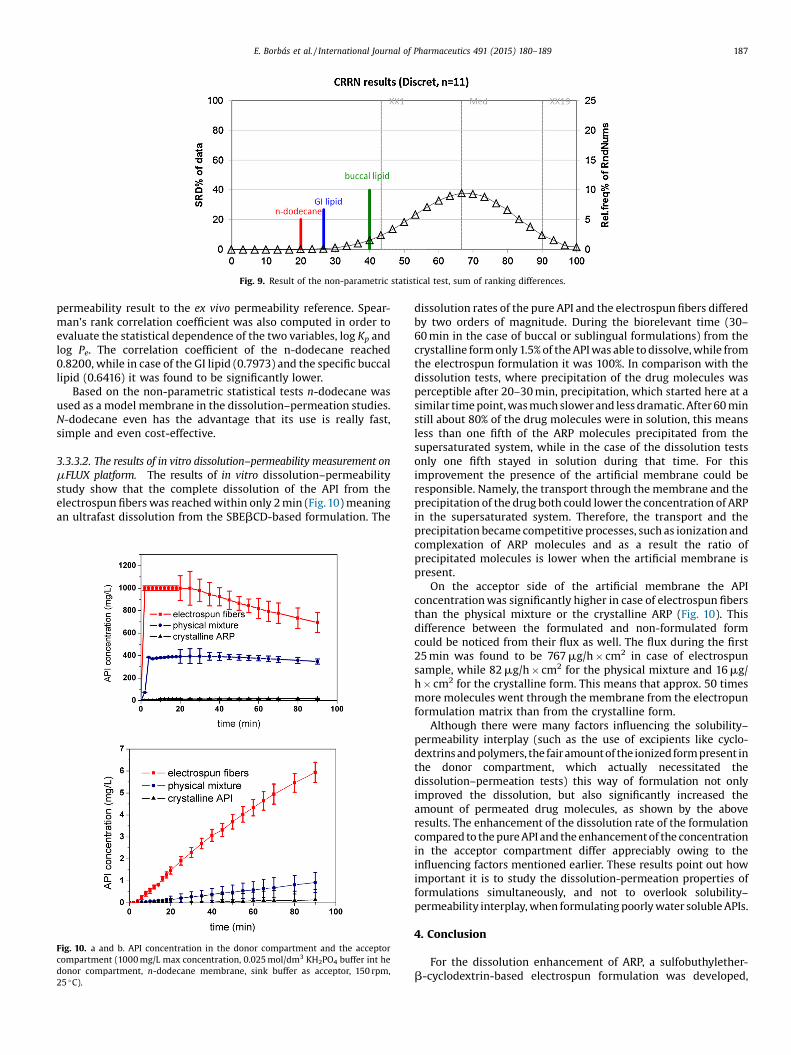
Fig. 9. Result of the non-parametric statistical test, sum of ranking differences.
E. Borbás et al. / International Journal of Pharmaceutics 491 (2015) 180–189 187
permeability result to the ex vivo permeability reference. Spear-man’s rank correlation coefficient was also computed in order toevaluate the statistical dependence of the two variables, log Kp andlog Pe. The correlation coefficient of the n-dodecane reached0.8200, while in case of the GI lipid (0.7973) and the specific buccallipid (0.6416) it was found to be significantly lower.
Based on the non-parametric statistical tests n-dodecane wasused as a model membrane in the dissolution–permeation studies.N-dodecane even has the advantage that its use is really fast,simple and even cost-effective.
3.3.3.2. The results of in vitro dissolution–permeability measurement onmFLUX platform. The results of in vitro dissolution–permeabilitystudy show that the complete dissolution of the API from theelectrospun fibers was reached within only 2 min (Fig.10) meaningan ultrafast dissolution from the SBEbCD-based formulation. The
Fig. 10. a and b. API concentration in the donor compartment and the acceptorcompartment (1000 mg/L max concentration, 0.025 mol/dm3 KH2PO4 buffer int hedonor compartment, n-dodecane membrane, sink buffer as acceptor, 150 rpm,25 �C).
dissolution rates of the pure API and the electrospun fibers differedby two orders of magnitude. During the biorelevant time (30–60 min in the case of buccal or sublingual formulations) from thecrystalline form only 1.5% of the API was able to dissolve, while fromthe electrospun formulation it was 100%. In comparison with thedissolution tests, where precipitation of the drug molecules wasperceptible after 20–30 min, precipitation, which started here at asimilar time point, was much slower and less dramatic. After 60 minstill about 80% of the drug molecules were in solution, this meansless than one fifth of the ARP molecules precipitated from thesupersaturated system, while in the case of the dissolution testsonly one fifth stayed in solution during that time. For thisimprovement the presence of the artificial membrane could beresponsible. Namely, the transport through the membrane and theprecipitation of the drug both could lower the concentration of ARPin the supersaturated system. Therefore, the transport and theprecipitation became competitive processes, such as ionization andcomplexation of ARP molecules and as a result the ratio ofprecipitated molecules is lower when the artificial membrane ispresent.
On the acceptor side of the artificial membrane the APIconcentration was significantly higher in case of electrospun fibersthan the physical mixture or the crystalline ARP (Fig. 10). Thisdifference between the formulated and non-formulated formcould be noticed from their flux as well. The flux during the first25 min was found to be 767 mg/h � cm2 in case of electrospunsample, while 82 mg/h � cm2 for the physical mixture and 16 mg/h � cm2 for the crystalline form. This means that approx. 50 timesmore molecules went through the membrane from the electropunformulation matrix than from the crystalline form.
Although there were many factors influencing the solubility–permeability interplay (such as the use of excipients like cyclo-dextrins and polymers, the fairamount of the ionized form present inthe donor compartment, which actually necessitated thedissolution–permeation tests) this way of formulation not onlyimproved the dissolution, but also significantly increased theamount of permeated drug molecules, as shown by the aboveresults. The enhancement of the dissolution rate of the formulationcompared to the pure API and the enhancement of the concentrationin the acceptor compartment differ appreciably owing to theinfluencing factors mentioned earlier. These results point out howimportant it is to study the dissolution-permeation properties offormulations simultaneously, and not to overlook solubility–permeability interplay, when formulating poorly water soluble APIs.
4. Conclusion
For the dissolution enhancement of ARP, a sulfobuthylether-b-cyclodextrin-based electrospun formulation was developed,

188 E. Borbás et al. / International Journal of Pharmaceutics 491 (2015) 180–189
which was the first SBEbCD-containing electrospun formulation ofthe API. Despite that PEO was used as fiber forming polymer, whichhas a retard effect on dissolution, the creation of a fast dissolvingdrug delivery system was possible by using only 0.75 w/v% PEO(2 �106 g/mol) for the polymer solution. Even that low concentra-tion of PEO was enough to create fine, beadless fibers by a solvent-based electrospinning process. The API in the electrospun fiberswas found to be in amorphous state and no crystallization wasobserved even after 3 months.
An electrospun cyclodextrin-based formulation of aripiprazolehas the potential to ensure fast drug delivery through the oralmucosa owing to the ultrafast dissolution of the drug from thenanofibrous dosage form even in a small volume of thedissolution medium and the enhanced flux across biologicalmembranes, as shown by the results of the novel in vitrodissolution and permeation test. For the dissolution test of theformulation, the saliva-modeling buffer composition and for thedissolution–permeation test the artificial membrane compositionwas optimized. These test results predict a significantly enhancedbioavailability for the electrospun formulation. The difference inthe results of dissolution and dissolution- permeation experi-ments exemplify well that with dissolution-permeation testscarried out with mFlux apparatus it is possible to predictbioavailability more precisely, because with these results notonly the dissolution but the interplay between solubility andpermeability is also considered.
Acknowledgements
This work was financially supported by the New SzéchenyiDevelopment Plan (TÁMOP-4.2.1/B-09/1/KMR-2010-0002), OTKAresearch fund (grant number K112644 and PD108975), MedInProtSynergy Program, Gedeon Richter Talentum Foundation and theJános Bolyai Research Scholarship of the Hungarian Academy ofSciences. The authors would like to express their special thanks toAnna E. Káncz for the thermodynamic solubility measurments andfor János Madarász for the XRPD measurments.
References
Avdeef, A., 2012. Absorption and Drug Development: Solubility, Permeability, andCharge State. John Wiley & Sons.
Balogh, G.T., Müller, J., Könczöl, Á., 2013. pH-Gradient PAMPA-based in vitro modelassay for drug-induced phospholipidosis in early stage of drug discovery. Eur. J.Pharm. Sci. 49 (1), 81–89.
Balogh, A., Drávavölgyi, G., Faragó, K., Farkas, A., Vigh, T., Sóti, P.L., Nagy, Z.K., 2014.Plasticized drug-loaded melt electrospun polymer mats: characterization,thermal degradation, and release kinetics. J. Pharm. Sci. 103 (4), 1278–1287.
Braun, D.E., Gelbrich, T., Kahlenberg, V., Tessadri, R., Wieser, J., Griesser, J., 2009.Conformational polymorphism in aripiprazole: preparation, stability andstructure of five modifications. J. Pharm. Sci. 98 (6), 2010–2026.
Campbell, K., Craig, D.Q., McNally, T., 2008. Poly (ethylene glycol) layered silicatenanocomposites for retarded drug release prepared by hot-melt extrusion. Int. J.Pharm. 363 (1), 126–131.
Challa, R., Ahuja, A., Ali, J., Khar, R.K., 2005. Cyclodextrins in drug delivery: anupdated review. Aaps Pharmscitech 6 (2), E329–E357.
Dahan, A., Miller, J.M., 2012. The solubility–permeability interplay and itsimplications in formulation design and development for poorly soluble drugs.AAPS J. 14 (2), 244–251.
Dahan, A., Miller, J.M., Hoffman, A., Amidon, G.E., Amidon, G.L., 2010. The solubility–permeability interplay in using cyclodextrins as pharmaceutical solubilizers:mechanistic modeling and application to progesterone. J. Pharm. Sci. 99 (6),2739–2749.
Dickinson, P.A., Rmaileh, R.A., Ashworth, L., Barker, R.A., Burke, W.M., Patterson, C.M., Yasin, M., 2012. An investigation into the utility of a multi-compartmental,dynamic, system of the upper gastrointestinal tract to support formulationdevelopment and establish bioequivalence of poorly soluble drugs. AAPS J. 14(2), 196–205.
FIP Guidelines for Dissolution Testing of Solid Oral Products. 1981, Pharm. Ind. 43,334–343.
Fenyvesi, F., Kiss, T., Fenyvesi É, Szente, L., Veszelka, S., Deli, M.A., Bácskay, I., 2011.Randomly methylated Éácyclodextrin derivatives enhance taxol permeabilitythrough human intestinal epithelial Caco-2 cell monolayer. J. Pharm. Sci. 100(11), 4734–4744.
Frank, K.J., Westedt, U., Rosenblatt, K.M., Hölig, P., Rosenberg, J., Mägerlein, M.,Brandl, M., 2014. What is the mechanism behind increased permeation rate of apoorly soluble drug from aqueous dispersions of an amorphous solid dispersion.J. Pharm. Sci. 103 (6), 1779–1786.
Ginski, M.J., Polli, J.E., 1999. Prediction of dissolution–absorption relationships froma dissolution/Caco-2 system. Int. J. Pharm. 177 (1), 117–125.
Grundy, J.S., Anderson, K.E., Rogers, J.A., Foster, R.T., 1997. Studies on dissolutiontesting of the nifedipine gastrointestinal therapeutic system. I. Description of atwo-phase in vitro dissolution test. J. Control. Release 48 (1), 1–8.
Héberger, K., Kollár-Hunek, K., 2011. Sum of ranking differences for methoddiscrimination and its validation: comparison of ranks with random numbers. J.Chemometrics 25 (4), 151–158.
Hancock, B.C., Parks, M., 2000. What is the true solubility advantage for amorphouspharmaceuticals? Pharm. Res. 17 (4), 397–404.
Huang, C., Soenen, S.J., Rejman, J., Trekker, J., Chengxun, L., Lagae, L., De Smedt, S.C.,2012. Magnetic electrospun fibers for cancer therapy. Adv. Funct. Mater. 22 (12),2479–2486.
Huang, C., Soenen, S.J., Gulck, E., Rejman, J., Vanham, G., Lucas, B., De Smedt, S.C.,2014. Electrospun polystyrene fibers for HIV entrapment. Polym. Adv. Technol.25 (8), 827–834.
Jansook, P., Brewster, M.E., Loftsson, T., 2011. Cyclodextrin-enhanced drug deliverythrough mucous membranes. Cyclodextrins in pharmaceutics, cosmetics, andbiomedicine. Curr. Future Ind. Appl. 145–158.
Kansy, M., Senner, F., Gubernator, K., 1998. Physicochemical high throughput screening:parallel artificial membrane permeation assay in the description of passiveabsorption processes. J. Med. Chem. 41 (7), 1007–1010.
Kataria, K., Gupta, A., Rath, G., Mathur, R.B., Dhakate, S.R., 2014. In vivo woundhealing performance of drug loaded electrospun composite nanofiberstransdermal patch. Int. J. Pharm. 469 (1), 102–110.
Keserü, G.M., Makara, G.M., 2009. The influence of lead discovery strategies on theproperties of drug candidates. Nat. Rev. Drug Discov. 8 (3), 203–212.
Kobayashi, M., Sada, N., Sugawara, M., Iseki, K., Miyazaki, K., 2001. Development of anew system for prediction of drug absorption that takes into account drugdissolution and pH change in the gastro-intestinal tract. Int. J. Pharm. 221 (1),87–94.
Kokate, A., Li, X., Williams, P.J., Singh, P., Jasti, B.R., 2009. In silico prediction of drugpermeability across buccal mucosa. Pharm. Res. 26 (5), 1130–1139.
Kostakova, E., Seps, M., Pokorny, P., Lukas, D., 2014. Study of polycaprolactone wetelectrospinning process. Exp. Polym. Lett. 8 (8) .
Lipinski, C.A., 2000. Drug-like properties and the causes of poor solubility and poorpermeability. J. Pharmacol. Toxicol. Methods 44 (1), 235–249.
Loftsson, T., Brewster, M.E., 2011. Pharmaceutical applications of cyclodextrins:effects on drug permeation through biological membranes. J. Pharm.Pharmacol. 63 (9), 1119–1135.
Loftsson, T., Brewster, M.E., 2012. Cyclodextrins as functional excipients: methods toenhance complexation efficiency. J. Pharm. Sci. 101 (9), 3019–3032.
Loftsson, T., Masson, M., 2001. Cyclodextrins in topical drug formulations: theoryand practice. Int. J. Pharm. 225 (1), 15–30.
Loftsson, T., 2014. Self-assembled cyclodextrin nanoparticles and drug delivery. J.Inclusion Phenomena Macrocyclic Chem. 80 (1–2), 1–7.
Marques, H.M.C., Hadgraft, J., Kellaway, I.W., Taylor, G., 1991. Studies of cyclodextrininclusion complexes. IV. The pulmonary absorption of salbutamol from a complexwith 2-hydroxypropyl-b-cyclodextrin in rabbits. Int. J. Pharm. 77 (2), 303–307.
Marques, M.R., Loebenberg, R., Almukainzi, M., 2011. Simulated biological fluidswith possible application in dissolution testing. Dissol. Technol. 18 (3), 15–28.
Mihajlovic, T., Kachrimanis, K., Graovac, A., Djuric, Z., Ibric, S., 2012. Improvement ofaripiprazole solubility by complexation with (2-hydroxy) propyl-b-cyclodextrinusing spray drying technique. AAPS Pharmscitech 13 (2), 623–631.
Nagy, Z.K., Balogh, A., Vajna, B., Farkas, A., Patyi, G., Kramarics, Á., Marosi, G., 2012.Comparison of electrospun and extruded soluplus1-based solid dosage forms ofimproved dissolution. J. Pharm. Sci. 101 (1), 322–332.
Nagy, Z.K., Balogh, A., Drávavölgyi, G., Ferguson, J., Pataki, H., Vajna, B., Marosi, G.,2013. Solvent-free melt electrospinning for preparation of fast dissolving drugdelivery system and comparison with solvent-based electrospun and meltextruded systems. J. Pharm. Sci. 102 (2), 508–517.
Nagy, Z.K., Wagner, I., Suhajda, Á., Tobak, T., Harasztos, Á.H., Vigh, T., Marosi, G., 2014.Nanofibrous solid dosage form of living bacteria prepared by electrospinning.Exp. Polym. Lett. 8 (5) .
Nagy, Z.K., Balogh, A., Démuth, B., Pataki, H., Vigh, T., Szabó, B., Brewster, M.E., 2015.High speed electrospinning for scaled-up production of amorphous soliddispersion of itraconazole. Int. J. Pharm..
Nerurkar, M., Naringrekar, V., 2009. U.S. Patent Application 12/417,067.Patel, V.F., Liu, F., Brown, M.B., 2011. Advances in oral transmucosal drug delivery. J.
Control. Release 153 (2), 106–116.Pelipenko, J., Kocbek, P., Govedarica, B., Rošic, R., Baumgartner, S., Kristl, J., 2013. The
topography of electrospun nanofibers and its impact on the growth andmobility of keratinocytes. Eur. J. Pharm. Biopharm. 84 (2), 401–411.
Samprasit, W., Kaomongkolgit, R., Sukma, M., Rojanarata, T., Ngawhirunpat, T.,Opanasopit, P., 2015. Mucoadhesive electrospun chitosan-based nanofibre matsfor dental caries prevention. Carbohydr. Polym. 117, 933–940.
Schipper, N.G.M., Verhoef, J., Romeijn, S.G., Merkus, F.W.H.M., 1992. Absorptionenhancers in nasal insulin delivery and their influence on nasal ciliaryfunctioning. J. Control. Release 21, 173–185 1).
Seif, S., Franzen, L., Windbergs, M., 2015. Overcoming drug crystallization inelectrospun fibers—elucidating key parameters and developing strategies fordrug delivery. Int. J. Pharm. 478 (1), 390–397.

E. Borbás et al. / International Journal of Pharmaceutics 491 (2015) 180–189 189
Shah, N., Sandhu, H., Choi, D.S., Chokshi, H., Malick, A.W., 2014. Amorphous SolidDispersions. Springer-Verlag, New York.
Shenoy, S.L., Bates, W.D., Frisch, H.L., Wnek, G.E., 2005. Role of chain entanglementson fiber formation during electrospinning of polymer solutions: good solvent,non-specific polymer–polymer interaction limit. Polymer 46 (10), 3372–3384.
Sridhar, R., Sundarrajan, S., Vanangamudi, A., Singh, G., Matsuura, T., Ramakrishna,S., 2014. Green processing mediated novel polyelectrolyte nanofibers and theirantimicrobial evaluation. Macromol. Mater. Eng. 299 (3), 283–289.
Taniguchi, C., Kawabata, Y., Wada, K., Yamada, S., Onoue, S., 2014.Microenvironmental pH-modification to improve dissolution behavior and oralabsorption for drugs with pH-dependent solubility. Exp. Opin. Drug Deliv.11 (4),505–516.
Tran, P.H.L., Tran, H.T.T., Lee, B.J., 2008. Modulation of microenvironmental pH andcrystallinity of ionizable telmisartan using alkalizers in solid dispersions forcontrolled release. J. Control. Release 129 (1), 59–65.
Tran, P.H.L., Tran, T.T.D., Lee, K.H., Kim, D.J., Lee, B.J., 2010. Dissolution-modulatingmechanism of pH modifiers in solid dispersion containing weakly acidic or basicdrugs with poor water solubility. Exp. Opin. Drug Deliv. 7 (5), 647–661.
Absorption and drug development: solubility, permeability, and charge state. JohnWiley & Sons <1088> In-vitro and in-vivo evaluation of dosage forms (1995)USP 23.
Vigh, T., Horváthová, T., Balogh, A., Sóti, P.L., Drávavölgyi, G., Nagy, Z.K., Marosi, G.,2013. Polymer-free and polyvinylpirrolidone-based electrospun solid dosageforms for drug dissolution enhancement. Eur. J. Pharm. Sci. 49 (4), 595–602.
Vrbata, P., Berka, P., Stránská, D., Doležal, P., Lázní9cek, M., 2014. Electrospinning ofdiosmin from aqueous solutions for improved dissolution and oral absorption.Int. J. Pharm. 473 (1), 407–413.
Yamashita, S., Furubayashi, T., Kataoka, M., Sakane, T., Sezaki, H., Tokuda, H., 2000.Optimized conditions for prediction of intestinal drug permeability using Caco-2 cells. Eur. J. Pharm. Sci. 10 (3), 195–204.
Yu, D.G., Shen, X.X., Branford-White, C., White, K., Zhu, L.M., Bligh, S.A., 2009. Oralfast-dissolving drug delivery membranes prepared from electrospunpolyvinylpyrrolidone ultrafine fibers. Nanotechnology 20 (5), 055104.
Yu, D.G., Yu, J.H., Chen, L., Williams, G.R., Wang, X., 2012. Modified coaxialelectrospinning for the preparation of high-quality ketoprofen-loaded celluloseacetate nanofibers. Carbohydr. Polym. 90 (2), 1016–1023.
Yu, D.G., Chian, W., Wang, X., Li, X.Y., Li, Y., Liao, Y.Z., 2013a. Linear drug releasemembrane prepared by a modified coaxial electrospinning process. J. Membr.Sci. 428, 150–156.
Yu, D.G., Wang, X., Li, X.Y., Chian, W., Li, Y., Liao, Y.Z., 2013b. Electrospun biphasicdrug release polyvinylpyrrolidone/ethyl cellulose core/sheath nanofibers. ActaBiomater. 9 (3), 5665–5672.
Zoeller, T., Klein, S., 2007. Simplified biorelevant media for screening dissolutionperformance of poorly soluble drugs. Dissol. Technol 14 (4), 8–13.