interaction of metal-free maleic and fumaric acids with...
TRANSCRIPT

362
Chapter X
Interaction of metal-free maleic and fumaric acids with alkyl
diamines and metallation of chiral zwitter ions leading to the
formation of some novel products
Abstract: This chapter deals with the study of the interaction of maleic and fumaric acids with various
Lewis bases (en and dap) in metal-free state and probe the possibility of having any cis-trans
isomerization. Besides the interaction can also lead to acid:diamine adducts. Since both maleic and
fumaric acids are dicarboxylic acids the diamines ’en’ and ‘dap’ are expected to form 1:1 type
adducts/salts even if they don’t interact to give any chiral zwitterions. Interestingly, we find 2:1 type
adduct for maleic acid:diamine while the fumaric acid gives the anticipated 1:1 adduct whatever may be
the ratio in which we try the reaction. Detailed crystal and molecular structure features of 4 different
adducts (65, 66, 67, 68) formed could be carried out which showed several interesting features. Since the
zwitterions we have generated through insertion reaction are essentially amino acids we attempted to
derivatize them using metal salts as they can then be relevant in bio-systems and as enzyme mimics. We
were able to synthesize a Ca2+
derivative 69 and structurally characterize it through single crystal XRD.
The structure reveals some novel characteristics. We were also able to generate a novel ternary type 1D
polymer 70 made up of Cu+2
, zwitterion 16 and fumaric acid, the fumaric acid being generated in situ by
dissociation of part of the zwitter ion used. The 1D polymer has a unique paddle-wheel type
configuration in which the zwitter ions act as capping ligands and fumarate moiety as ‘connectors’. The
molecular and packing features of 70 is unique and unprecedented. The structural features of all the
products (65-70) are presented in detail in the chapter.

363
10.1 Introduction
Having observed the unprecedented reaction of [M(Hmal)2(H2O)4] with alkyl
amines generating novel cyclically interconnected one-dimensional coordination
polymers made up of [M(mal)2]2-
motifs we have been interested in probing how these
two isomeric dicarboxylic acids react in their natural form with various alkyl amines.
Since these two acids are structurally and chemically different because of their cis and
tras configurations we thought that they would react differently with amines forming
structurally dissimilar products. We have considered only alkyl diamines because both
maleic and fumaric acids are dicarboxylic acids. Further, there are a few scattered
reports available on these two acids reacting with aromatic diamines like 4,4’-
bipyridine.96,114
We have confined our studies on such a perspective by taking into
consideration only 1,2-diaminoethane (en) and 1,3-diaminopropane (dap) as diamines
which can be considered as almost similar but having difference only in the nature of -
(CH2)- spacer lengths. In this chapter we try to consolidate all the relevant details on
the nature of reaction and also the type of products obtained through the reaction of
H2mal and H2fum with both en and dap and present crystal and molecular structures of
some interesting adducts formed (65, 66, 67 and 68).
Since the chiral zwitterions (like 16) generated during our reactions discussed
earlier (Chapters IV and V) are similar to some of the important amino acids, we have
also attempted to synthesize their metal derivatives to look at their structural and
conformational features. An interesting Ca complex 69 formed from 23 containing two
coordinated chiral zwitter ions could be made and its structural features studied through
single crystal XRD. Presented in the chapter are also some details on a novel and
unprecedented metal-zwitterion derivative 70 including its unique structural features.
We have made use of a wide variety of experimental techniques like CHN analysis, TG,
DTA, FTIR, 1H NMR,
13C NMR, EPR, PXRD and single crystal X-ray diffraction

364
studies rather extensively for the detailed characterization of all the products obtained
during the transformation reactions.
10.2 Experimental
Materials
The diamines 1,2-diaminoethane and 1,3-diaminopropane were purchased from
Merck KgaA. Maleic acid and fumaric acid were E. Merck (India) Limited products.
All chemicals were used as received.
Analytical Methods
Elemental analyses (C, H and N) were performed using an Elementar Vario EL III
elemental analyzer. IR spectra (4000-400 cm-1
) were measured on a Shimadzu FTIR-
8400S spectrophotometer, where KBr was used as the dispersal medium. Thermo
gravimetric analyses were carried out on a Shimadzu DTG-60 simultaneous DAT-TG
apparatus. EPR spectrum was recorded in solution state in methanol on a Varian E-112
EPR spectrometer operating in X-band using DPPH as the ‘g’ marker. Single crystal X-
ray diffraction data were collected at 293 ± 2K on an automated Bruker axs (Kappa
apex2) CCD diffractometer.
Synthesis
Considering the possibility of formation of different types of adducts/compounds
the reaction of diamines with both maleic and fumaric acids were carried out both in 1:1
and 2:1 (acid: diamine) molar ratios. We could see that irrespective of these two acid:
diamine molar ratios we start with, the composition and nature of products obtained was
seen to be always dependent on the nature of the acid (maleic or fumaric acid) we have
employed. In the case of maleic acid the acid: diamine ratio in the product was always
2:1 while in the case of fumaric acid it was 1:1. Therefore we have carried out the acid-
diamine reaction by maintaining the respective optimum molar ratios. The specific
synthetic conditions/details for each case are given below.

365
Maleic acid-en adduct/salt, 65
An aqueous solution (10mL) of maleic acid (0.232g, 2mmol) was mixed with an
aqueous solution of (10mL) 1,2-diaminoethane (0.07mL, 1mmol). The solution was
warmed on a water bath and kept aside for slow evaporation. Colourless block crystals,
65 were obtained after three days. The crystals were washed with water and dried.
Yield: 80%
Maleic acid-dap adduct/salt, 66
The experimental procedure employed for 66 was almost the same as above. To a
10mL of aqueous solution of maleic acid (0.232g, 2mmol) a solution of 1,3-
diaminopropane (0.08mL, 1mmol, 10mL water) was added, stirred and kept for slow
evaporation. Colourless block crystals of 66 obtained were collected and washed with
water and dried in air. Yield: 75%
Fumaric acid -en adduct/salt, 67
A methanolic solution (10mL) of fumaric acid (0.116g, 1mmol) was mixed with
an aqueous solution (10mL) of 1,2-diaminoethane (0.07mL, 1mmol) under mild stirring
and heating. The clear solution yielded colourless block crystals after a three days.
Yield: 85%
Fumaric acid - dap adduct/salt, 68
The experimental procedure was similar to the above one. In this case a
methanolic solution (10mL) of fumaric acid (0.116g, 1mmol) was mixed with 10mL of
aqueous solution of 1,3-diaminopropane (0.08mL, 1mmol) under heating. Colourless
block crystals of 68 obtained were collected, washed with water and dried. Yield: 80%
[Ca(3-pic.zwitterion)2(H2O)2].4H2O, 69
The 3-picolinium succinate zwitterion, 23 (0.209g, 1mmol) which was obtained as
a clear product through the reaction mentioned in Section 5.2.3 was dissolved in water

366
by continous heating and stirring. To this aqueous solution CaCO3 was added slowly.
Brisk effervescence was seen observed in the initial stages of addition indicating that
the Ca salt is reacting with the zwitterionic acid. Addition of CaCO3 was continued till
there was no effervescence. The solution was filtered and kept for crystallisation.
Colourless block crystals of 69 were formed after two days. Yield: 80%
[Cu2(fum)(zwitterion)2(H2O)2], 70
To an aqueous solution (20mL) of pyridinium succinate zwitterion, 16 (0.195g,
1mmol) obtained as a pure product from the reaction discussed in Section 4.3.1, solid
CuCO3 was added slowly with heating and stirring till there was no evolution of
effervescence. Heating was continued for half an hour and the resultant blue solution
was filtered and kept for slow evaporation. Tiny prismatic blue crystals of 70 were
formed after four days which were collected and washed with water and dried. Yield:
35%
10.3 Results and Discussion
Since the studies covered in this chapter belong to two different aspects the results
and discussion are presented under the following two main headings: (1) maleic
/fumaric acid: aliphatic amine adducts/salts and (2) metal derivatives of chiral
zwitterions.
10.3.1 Maleic /fumaric acid:aliphatic diamine adducts/salts
We have considered the reaction of maleic acid and fumaric acids with two alkyl
diamines, 1,2-diaminoethane (en) and 1,3-diaminopropane (dap) having varying lengths
of -(CH2)- spacer moieties between the –NH2 ends to look at the nature of products
formed. We wanted to probe whether there is any chance of –NH2 moiety getting
inserted in the -HC=CH- double bond to form a chiral amino acid or if the diamine acts
only as a base the nature and structure of the adducts formed. There could be significant
difference in the nature of salt/adduct formed with maleic and fumaric acids by these
diamines because of their wide structural difference. As mentioned in the preparative

367
section the composition of the acid-diamine products formed was dependent on the type
of acid used. So we have carried out synthesis using the optimum composition needed
in each case. Given in Table 10.1 are the analytical data of each of the product obtained
which agree with the composition indicated. It is seen that maleic acid forms 2:1
adducts with both en and dap (65 and 66) while the composition of the fumaric acid:
diamine products have 1:1 composition for both en and dap (67 and 68).
Table10.1 Elemental analytical data of the maleic/fumaric acid: aliphatic amine
adducts
Compound (Emp.formula) Formula
weight
Elemental content (%)
Found (Calcd.) Colour and nature
(solubility in water) C H N
maleic acid-en salt, 65
(Hmal-1
)2 (enH22+
).H2O
(C10 H18 N2 O9)
310.26
38.67
38.35
5.80
5.62
9.02
8.93
colourless crystals
(soluble)
maleic acid-dap salt, 66
(Hmal-1
)6 (dapH22+
)3.H2O
C33 H56 N6 O25
936.84 41.99
(42.26)
6.49
(5.97)
9.00
(8.96)
colourless crystals
(soluble)
fumaric acid-en salt, 67
(fum-2
) (enH22+
)
C6H12N2O4
176.18 40.53
(40.86)
6.72
(6.81)
15.83
(15.89)
colourless crystals
(soluble)
fumaric acid - dap salt, 68
(fum-2
) (dapH22+
)
C7 H14 N2 O4
190.20
44.54
(44.16)
7.42
(7.36)
14.51
(14.72)
colourless crystals
(soluble)

368
FTIR spectral data of 65, 66, 67 and 68
Given in Table 10.2 are some of the important IR absorption peaks of the four phase
pure forms of acid-amine adducts obtained. In both 65 and 66 we could observe the presence
of strong bands around 1701cm-1
indiacting the presence of free –COOH groups in the maleic
acid adducts. In contrast we could not find any such peaks for the fumaric acid adducts.
Besides the –COOH peaks in 65 and 66 the strong absorptions due to νas(COO-) and νs(COO
-
) were seen with a Δν value of around 200 cm-1
indicating the presence of ionic type –COO-
moiety.82
In the case of fumaric acid adducts with both en and dap we could notice νas(COO-)
and νs(COO-) specific vibrations around 1600 and 1400 cm
-1 with a Δν value around 200 cm
-
1 which also suggests the ionic nature of –COO
- moieties. We were able to notice ν(NH3
+)
specific vibration peaks as a broad band below 3100 cm-1 . No peaks characteristic to NH2
group are seen in all the samples indicating that all the NH2 moieties are in the protonated
form. The broad nature and low value of 3100 cm-1
indicates that the NH3+ groups are
involved in strong H-bonding. The simultaneous presence of the characteristic peak of free –
COOH group at 1701cm-1
and the characteristic peaks for νas(COO-) and νs(COO-) show
that only one carboxyl group is deprotonated in the maleic acid salts/adducts 65 and 66. This
is also clear from the molecular structures (Fig.10.1 and 10.3).This accounts for the fact that
the maleic acid :diamine composition is always 2:1. In the case of fumaric acid salts (67 and
68) we could not see any –COOH specific peaks which suggests that both of the carboxylate
moieties are in the deprotonated form. Consequently the fumaric acid: diamine composition
expected is 1:1 which is what we find from the analytical data of both 67 and 68. Further, we
find νOH(H2O) peaks only for the maleic acid salts (65 and 66) which are due to the H2O
molecules present in them.

369
Table 10.2 IR spectral data of the acid: amine adducts (in cm-1
)
65 66 67 68
ν OH(H2O) 3494 3490 --- ---
ν (COOH) 1701 1701 --- ---
νas(COO) 1573 1573 1600 1600
νs(COO) 1369 1366 1393 1407
Δν 204 207 207 193
ν(N-H)
3093
3058
3031
3058
3008
3062
3020
2974
3058
3004
ν(C-H) aliphatic 2893 2908 2812 2893
Thermal decomposition features
TGA show that only the maleic acid salts/adducts (65 and 66) have water of
crystallization while the adducts of fumaric acid with both en and dap (67 and 68) are
water free. In the first thermal stage guest water is lost by about 63°C. The remaining
portion of the thermograms is similar for all the four salts. The next stage is a strong
endothermic loss of the amine. It is observed that the heat of loss of amine is higher for
en salts (3.5 and 3.6kJ/g for 65 and 67) compared to that of dap salts (2.57 and 2.58kJ/g
for 66 and 68) which shows that en may be more strongly hydrogen bonded. The last
step is the decomposition of the acid which begins at about 250°C and ends at around
600°C with a broad exothermic peak in the DTA curve, with a heat of decomposition of
about 20 kJ/g.
Structural details of the amine adducts
Regardless of whether the reacting molar ratios of acid to diamine are 1:1 or 2:1,
maleic acid adducts always crystallize with a 2:1 ratio of acid to diamine (for both en and
dap) in the final product. However, the fumaric acid adducts have a 1:1 stoichiometry for
both en and dap. We could carry out the single crystal XRD studies of all the four
maleic/fumaric acid: amine salts. Discussed below are the crystal structure details in brief in

370
each case. Crystallographic data and details of structure refinements of the salts 65, 66, 67
and 68 are consolidated in Table 10.3.
Maleic acid-en salt, (Hmal-)2 (enH2
2+)H2O, 65
The colorless needle like crystals of 65 is seen to be crystallising in monoclinic
form with space group Pc. The crystal data and refinement parameters are presented in
Table 10.3. ORTEP plot of compound 65 is given in Fig. 10.1 which clearly shows that
only one of the carboxyl groups of the maleic acid is deprotoaned while the other
remains in the –COOH form itself. The packing features of monodeprotonated maleic
acid (Hmal) and the relative orientations of successive Hmal moieties in the crystal are
interesting which are depicted in Fig 10.1. The C1-O1 bond length is shorter than C1-
O2 bond length showing that the former is a double bond and hence one carboxyl group
of the maleate anion is not deprotonated. C4-O3 bond is longer than C4-O4 because O3
is involved in intra-molecular hydrogen bond with O2-H. The H(1A)-N(1)-H(1C) bond
angle of 109.5° shows that the ammonium N is sp3
hybridised as expected. There exist
extensive H-bonding interactions among the carboxylate O, -NH3+ and H2O molecules
which is clear from Fig. 10.2. Selected bond lengths and bond angles are given in Table
10.4. The extensive H-bonds are evident from Table 10.5.
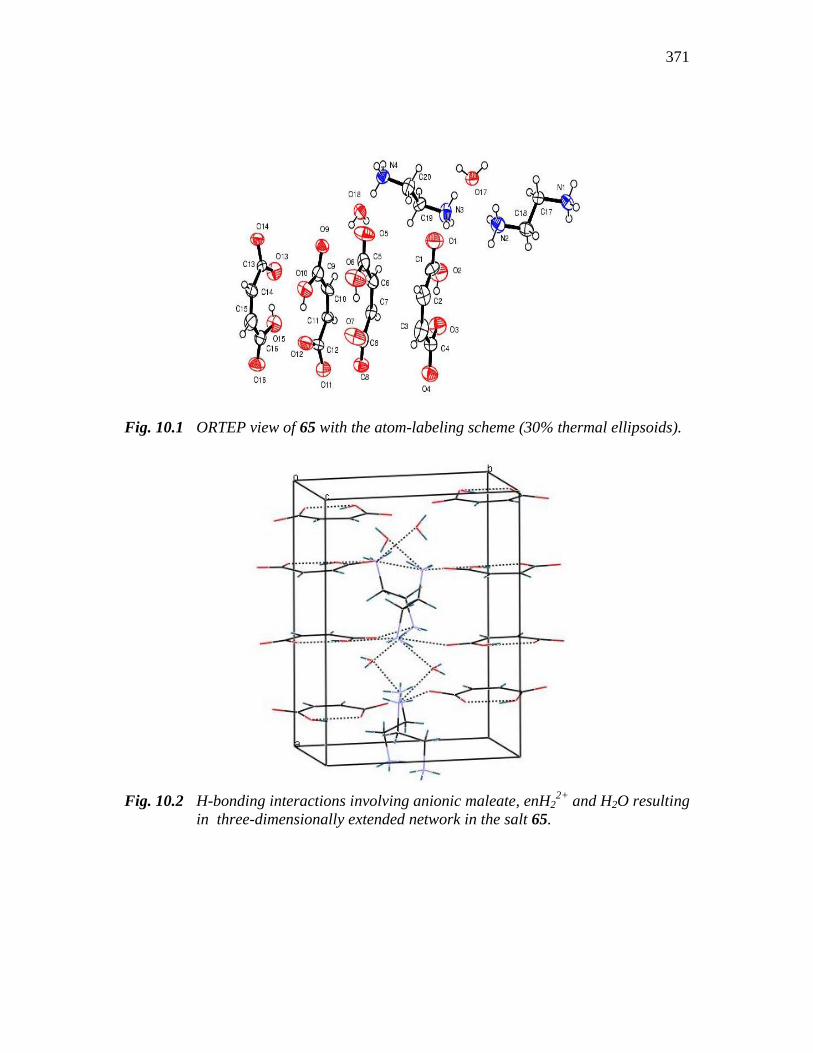
371
Fig. 10.1 ORTEP view of 65 with the atom-labeling scheme (30% thermal ellipsoids).
Fig. 10.2 H-bonding interactions involving anionic maleate, enH22+
and H2O resulting
in three-dimensionally extended network in the salt 65.
372
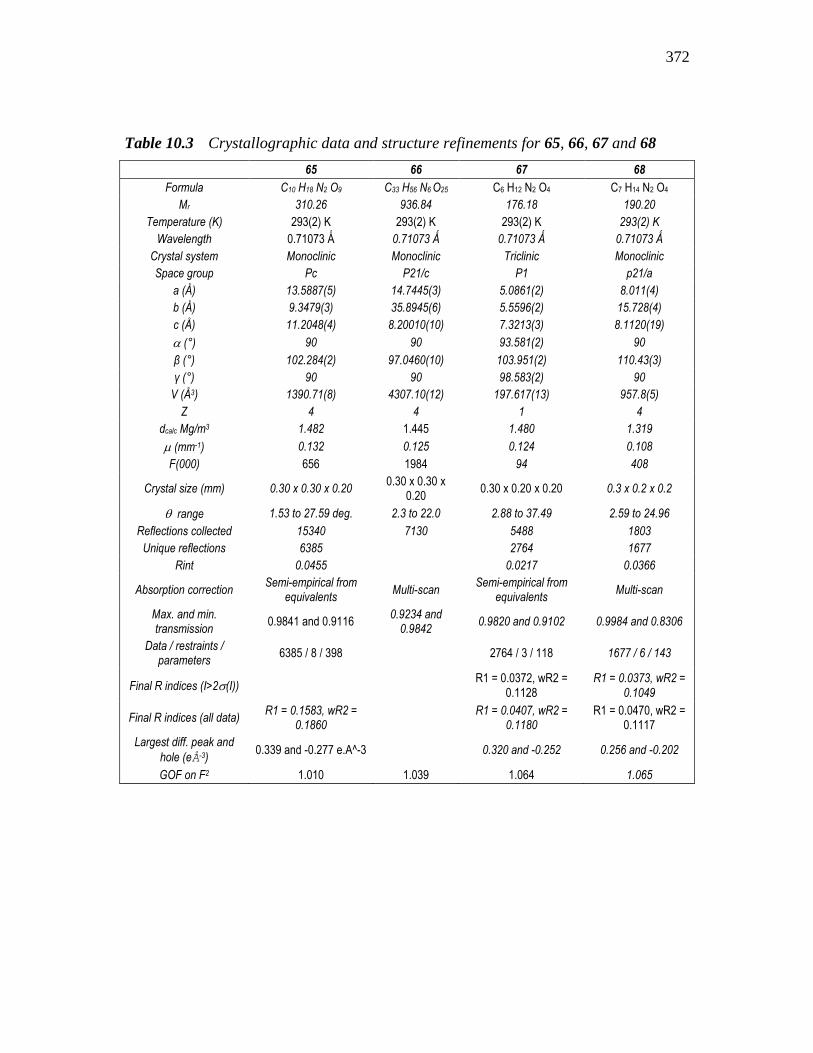
Table 10.3 Crystallographic data and structure refinements for 65, 66, 67 and 68
65 66 67 68
Formula C10 H18 N2 O9 C33 H56 N6 O25 C6 H12 N2 O4 C7 H14 N2 O4
Mr 310.26 936.84 176.18 190.20
Temperature (K) 293(2) K 293(2) K 293(2) K 293(2) K
Wavelength 0.71073 Ǻ 0.71073 Ǻ 0.71073 Ǻ 0.71073 Ǻ
Crystal system Monoclinic Monoclinic Triclinic Monoclinic
Space group Pc P21/c P1 p21/a
a (Å) 13.5887(5) 14.7445(3) 5.0861(2) 8.011(4)
b (Å) 9.3479(3) 35.8945(6) 5.5596(2) 15.728(4)
c (Å) 11.2048(4) 8.20010(10) 7.3213(3) 8.1120(19)
(°) 90 90 93.581(2) 90
β (°) 102.284(2) 97.0460(10) 103.951(2) 110.43(3)
γ (°) 90 90 98.583(2) 90
V (Å3) 1390.71(8) 4307.10(12) 197.617(13) 957.8(5)
Z 4 4 1 4
dcalc Mg/m3 1.482 1.445 1.480 1.319
(mm-1) 0.132 0.125 0.124 0.108
F(000) 656 1984 94 408
Crystal size (mm) 0.30 x 0.30 x 0.20 0.30 x 0.30 x
0.20 0.30 x 0.20 x 0.20 0.3 x 0.2 x 0.2
range 1.53 to 27.59 deg. 2.3 to 22.0 2.88 to 37.49 2.59 to 24.96
Reflections collected 15340 7130 5488 1803
Unique reflections 6385 2764 1677
Rint 0.0455 0.0217 0.0366
Absorption correction Semi-empirical from
equivalents Multi-scan
Semi-empirical from equivalents
Multi-scan
Max. and min. transmission
0.9841 and 0.9116 0.9234 and
0.9842 0.9820 and 0.9102 0.9984 and 0.8306
Data / restraints / parameters
6385 / 8 / 398 2764 / 3 / 118 1677 / 6 / 143
Final R indices (I>2(I))
R1 = 0.0372, wR2 = 0.1128
R1 = 0.0373, wR2 = 0.1049
Final R indices (all data) R1 = 0.1583, wR2 =
0.1860
R1 = 0.0407, wR2 = 0.1180
R1 = 0.0470, wR2 = 0.1117
Largest diff. peak and
hole (eÅ-3) 0.339 and -0.277 e.A^-3 0.320 and -0.252 0.256 and -0.202
GOF on F2 1.010 1.039 1.064 1.065

373
Table 10.4 Selected bond lengths [Å] and angles [°] for compound 65
___________________________________________________________
C(1)-O(1) 1.257(9) O(4)-C(4)-O(3) 121.2(6)
C(1)-O(2) 1.299(10) O(1)-C(1)-C(2) 119.6(8)
C(4)-O(3) 1.319(9) N(1)-C(17)-C(18) 110.9(4)
C(4)-O(4) 1.230(6) N(1)-C(17)-H(17A) 109.3
O(1)-C(1)-O(2) 118.2(9) C(17)-N(1)-H(1A) 109.6
H(1A)-N(1)-H(1C) 109.5 H(1B)-N(1)-H(1C) 109.5
Table 10.5 Selected H-Bonds [Å] and angles [°] for compound 66
D-H...A d(D-H) d(H...A) d(D...A) <(DHA)
N(1)-H(1A)...O(9)#1 0.89 1.89 2.755(8) 163.1
N(1)-H(1B)...O(11)#2 0.89 2.03 2.850(9) 153.3
N(1)-H(1C)...O(18)#3 0.89 2.15 2.884(9) 139.6
N(2)-H(2A)...O(14)#3 0.89 1.91 2.773(8) 162.4
N(2)-H(2B)...O(17) 0.89 2.15 2.887(9) 140.0
N(2)-H(2C)...O(16)#4 0.89 2.02 2.847(9) 154.5
N(3)-H(3A)...O(4)#5 0.89 1.91 2.791(8) 172.8
N(3)-H(3B)...O(2) 0.89 2.18 2.967(9) 147.2
N(3)-H(3C)...O(17) 0.89 2.18 2.812(8) 127.7
N(4)-H(4A)...O(8)#6 0.89 1.91 2.799(8) 174.6
N(4)-H(4B)...O(18) 0.89 2.12 2.797(9) 132.0
O(2)-H(2O)...O(3) 0.82 1.52 2.333(9) 169.7
O(6)-H(6O)...O(7) 0.82 1.64 2.453(8) 174.8
O(10)-H(10O)...O(12) 0.82 1.62 2.424(8) 167.8
O(18)-H(18C)...O(9) 0.855(10) 1.95(2) 2.767(8) 159(5)
O(17)-H(17D)...O(14)#1 0.846(10) 1.97(3) 2.740(8) 151(5)
O(17)-H(17C)...O(4)#6 0.848(10) 2.06(3) 2.850(8) 154(5)
O(18)-H(18D)...O(8)#5 0.847(10) 2.06(2) 2.836(8) 153(4)
O(15)-H(13O)...O(13) 0.76(3) 1.68(4) 2.416(9) 164(4)
___________________________________________________________
Symmetry transformations used to generate equivalent atoms: #1 x-1,-y+1,z-1/2 #2 x-1,-y+2,z-1/2 #3 x-1,y,z
#4 x-1,y-1,z #5 x,-y+2,z-1/2 #6 x,y-1,z
Maleic acid-dap salt, (Hmal-1
)6 (dapH22+
)3.H2O, 66

374
Just as 65 the maleic acid: dap adduct 66 also crystallize in monoclinic form but
with a different space group P21/c. The crystal data are presented in Table 10.3 along
with that of other derivatives. Both the maleic acid salts (65 and 66) have four
molecular units per unit cell but the cell volume of 66 is about three times that of 65.
This is evident because one molecular unit of 66 has six Hmal- moieties where as that of
65 has only two Hmal- ions (as clear from the chemical composition). Given in Fig.
10.3 is the ORTEP of compound 66 with atom label. The mono-deprotonated form of
the maleic acid in 66 is also clear from the molecular plot. C11-C12 bond length of
1.330(3)Ǻ is shorter than C10-C11 bond length of 1.476(3)Ǻ because the former is the
double bond of the maleate ion. C13-O4 of 1.231(3)Ǻ is shorter than C12-C13 of
1.473(3)Ǻ because the former is a double bond while latter is a single bond, which
indicates that one of the carboxyl groups in the maleate ion is not deprotonated. O3- H3
of 1.13(4)Ǻ shows strong intra-molecular hydrogen bond. C1 - N1 - H1A bond angle of
109.5° shows that the –NH2 is protonated and the N atom is converted to sp3 hybridised.
As in the case of compound 65 there exist extensive H-bonding interactions in 66 which
are depicted in Fig. 10.4. Relevant bond lengths and angles are given in Table 10.6. The
extensive H-bonds are clear from Table 10.7.
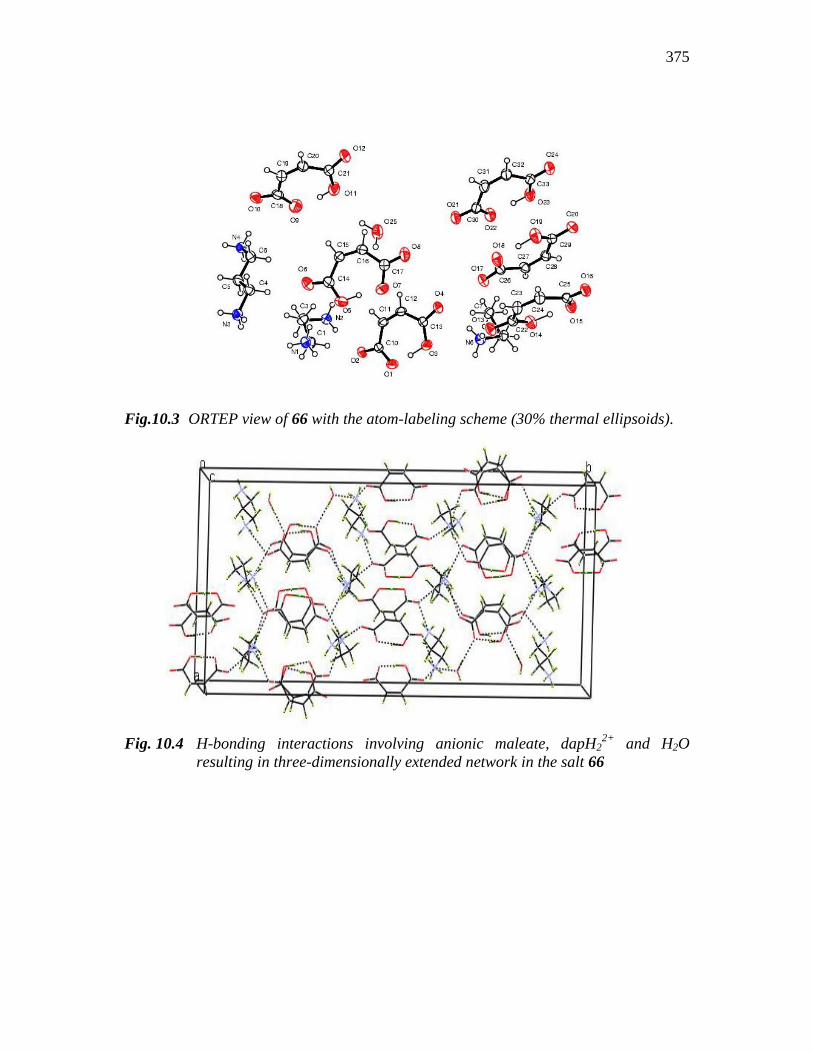
375
Fig.10.3 ORTEP view of 66 with the atom-labeling scheme (30% thermal ellipsoids).
Fig. 10.4 H-bonding interactions involving anionic maleate, dapH22+
and H2O
resulting in three-dimensionally extended network in the salt 66

376
Table 10.6 Selected bond lengths [Å] and angles [°] for compound 66
___________________________________________________________
C1 N1 1.475(3) N1 H1B 0.8900
C1 C2 1.521(3) N1 H1C 0.8900
C2 C3 1.492(3) O25 H25A 0.90(2)
C3 N2 1.478(3) O25 H25B 0.90(3)
C10 O2 1.235(2) O1 H3 1.31(4)
C10 O1 1.270(3) O3 H3 1.13(4)
C10 C11 1.476(3) N1 C1 C2 109.6(2)
C11 C12 1.330(3) C3 C2 C1 113.8(2)
C12 C13 1.473(3) C1 N1 H1A 109.5
C13 O4 1.231(3) C1 N1 H1B 109.5
C13 O3 1.277(3) H1A N1 H1B 109.5
N1 H1A 0.8900 C1 N1 H1C 109.5
___________________________________________________________
Table 10.7 Selected H-Bonds [Å] and angles [°] for compound 66
___________________________________________________________
D-H...A d(D-H) d(H...A) d(D...A) <(DHA)
N1 H1C O5 0.89 2.31 3.077(3) 144.8
N1 H1C O6 0.89 2.32 3.150(3) 155.6
N2 H2A O2 0.89 2.14 3.018(2) 170.6
O3 H3 O1 1.13(4) 1.31(4) 2.433(2) 171(4)
O5 H7 O7 1.04(5) 1.38(5) 2.415(3) 174(4)
O11 H11 O9 0.96(3) 1.49(3) 2.443(3) 170(3)
N1 H1A O4 0.89 2.15 3.037(2) 171.74
N1 H1B O21 0.89 1.96 2.822(2) 163.04
N2 H2C O17 0.89 1.95 2.842(2) 178.24
N3 H3A O25 0.89 1.94 2.799(3) 160.74
O25 H25A O9 0.90(2) 1.841(8) 2.726(3) 167(3)4
O25 H25B O6 0.90(3) 2.041(4) 2.939(3) 176(3)4
_________________________________________________________________
Fumaric acid - en salt, (fum-2
) (enH22+
), 67

377
Crystal data and structure refinement parameters of 67 are consolidated in Table
10.3. It is seen that while the two maleic acid adducts (65 and 66) crystallise in
monoclinic system the fumaric acid-en salt is formed in triclinic system with P1 space
group. It is also interesting to see that this compound has the smallest cell volume
compared to the other three acid: amine salts. This is because only one molecular unit is
present per unit cell. Contrary to the maleic acid salts, the fumaric acid salt has an acid:
amine ratio 1:1 which is clear from the ORTEP of 67 as given in Fig.10.5. This is
expected because in the salts of fumaric acid both carboxyl groups can be deprotonated
since there is no intra-molecular hydrogen bond due to the trans-
configuration. The en entity adopts a trans configuration in order to facilitate H-bonding
interactions with the fumarate dianion, which is also clear from the ORTEP. The
difference in the C-O bond lengths is due to the difference in the number of hydrogen
bonds attached to the O atoms of the carboxyl groups. H-bonds are represented in Fig.
10.6. C2-C3 distance of 1.32Ǻ is the characteristic double bond length. All the
ammonium hydrogens are equivalent. H(1A)-N(1)-H(1C) bond angle of 109.5° is
consistent with the tetrahedral geometry of the -NH3 group. Relevant bond lenghts and
angles are given in Table 10.8 and H-bonds are presented in Table 10.9. Packing
features of 67 are illustrated in Figs. 10.7, 10.8 and 10.9.

378
Fig.10.5 ORTEP view of 67 with the atom-labeling scheme (30% thermal
ellipsoids).
Fig.10.6 H-bonding interactions involving anionic fumarate, and (enH22+
), resulting
in the three-dimensionally extended network in 67.

379
Fig. 10.7 Linear chain of fumarate units and the sandwiched ethylene diamonium
cations. (view along c axis).
Fig.10.8 Network of H-bonds. (only some H-bonded cites are shown
for clarity). View along a axis.

380
Fig. 10.9 The 3D network due to H-bonding interactions in 67
Table 10.8 Bond lengths [Ǻ] and angles [°] for 67.
_____________________________________________________________
C(1)-O(2) 1.251(3) N(1)-H(1B) 0.8900
C(1)-O(1) 1.258(3) N(1)-H(1C) 0.8900
C(4)-O(4) 1.234(3) O(2)-C(1)-O(1) 123.0(3)
C(4)-O(3) 1.275(2) C(2)-C(3)-C(4) 123.63(12)
C(1)-C(2) 1.496(3) O(4)-C(4)-O(3) 124.4(3)
C(2)-C(3) 1.3253(11)
N(1)-H(1A) 0.8900
N(1)-C(5)-H(5A) 109.6
N(1)-C(5)-C(6) 110.13(16)
H(1A)-N(1)-H(1C) 109.5
_________________________________________________________
381
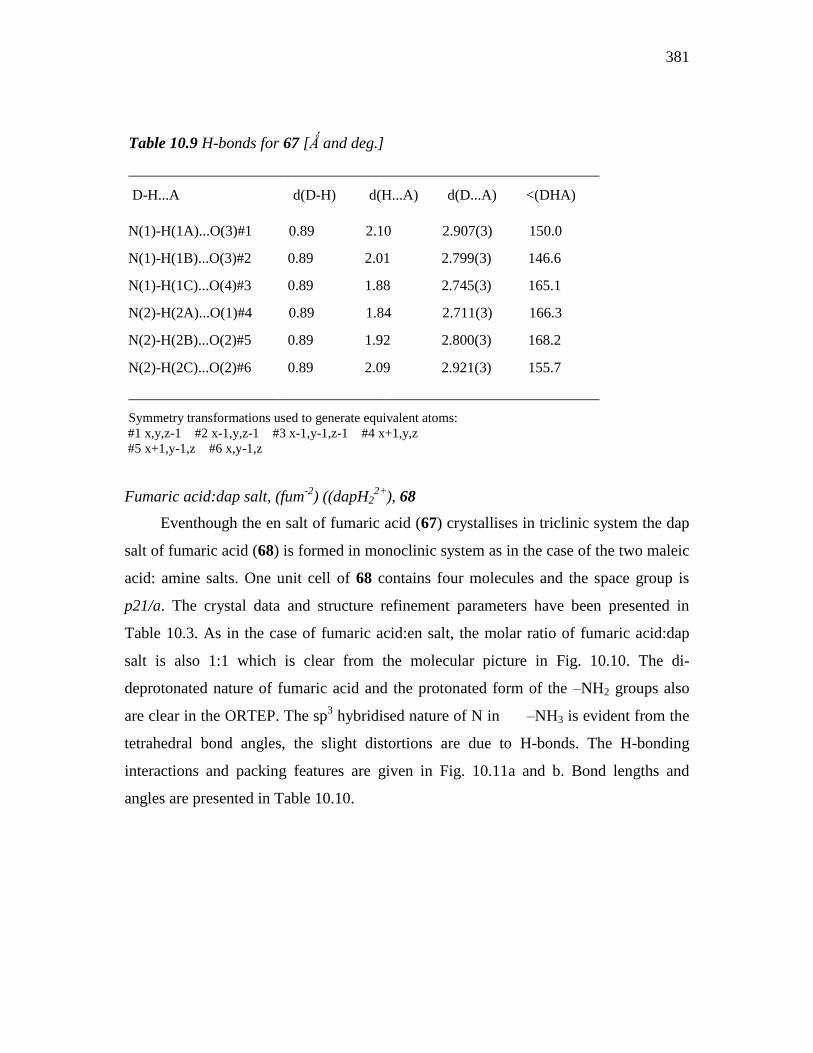
Table 10.9 H-bonds for 67 [Ǻ and deg.]
___________________________________________________________
D-H...A d(D-H) d(H...A) d(D...A) <(DHA)
N(1)-H(1A)...O(3)#1 0.89 2.10 2.907(3) 150.0
N(1)-H(1B)...O(3)#2 0.89 2.01 2.799(3) 146.6
N(1)-H(1C)...O(4)#3 0.89 1.88 2.745(3) 165.1
N(2)-H(2A)...O(1)#4 0.89 1.84 2.711(3) 166.3
N(2)-H(2B)...O(2)#5 0.89 1.92 2.800(3) 168.2
N(2)-H(2C)...O(2)#6 0.89 2.09 2.921(3) 155.7
___________________________________________________________
Symmetry transformations used to generate equivalent atoms: #1 x,y,z-1 #2 x-1,y,z-1 #3 x-1,y-1,z-1 #4 x+1,y,z
#5 x+1,y-1,z #6 x,y-1,z
Fumaric acid:dap salt, (fum-2
) ((dapH22+
), 68
Eventhough the en salt of fumaric acid (67) crystallises in triclinic system the dap
salt of fumaric acid (68) is formed in monoclinic system as in the case of the two maleic
acid: amine salts. One unit cell of 68 contains four molecules and the space group is
p21/a. The crystal data and structure refinement parameters have been presented in
Table 10.3. As in the case of fumaric acid:en salt, the molar ratio of fumaric acid:dap
salt is also 1:1 which is clear from the molecular picture in Fig. 10.10. The di-
deprotonated nature of fumaric acid and the protonated form of the –NH2 groups also
are clear in the ORTEP. The sp3 hybridised nature of N in –NH3 is evident from the
tetrahedral bond angles, the slight distortions are due to H-bonds. The H-bonding
interactions and packing features are given in Fig. 10.11a and b. Bond lengths and
angles are presented in Table 10.10.

382
Fig. 10.10 ORTEP view of 68 with the atom-labeling scheme (30% thermal ellipsoids).
a b
Fig.10.11 (a) and (b) H-bonding interactions involving anionic fumarate, and dapH22+
resulting in the 3D extended network in salt 68.

383
Table 10.10 Bond lengths [Ǻ] and angles [deg] for 68
_______________________________________________________
C(1)-N(1) 1.480(2) N(2)-H(4N) 0.8403(11)
C(1)-C(2) 1.506(2) C(4)#1-C(4)-C(5) 123.49(19)
C(4)-C(4)#1 1.316(3) O(2)-C(5)-C(4) 119.95(15)
C(4)-C(5) 1.495(2) H(1N)-N(1)-H(2N) 108(2)
C(5)-O(2) 1.243(2) H(1N)-N(1)-H(3N) 112(2)
C(5)-O(1) 1.272(2) H(2N)-N(1)-H(3N) 107(2)
___________________________________________________________
Symmetry transformations used to generate equivalent atoms:
#1 -x+3,-y,-z+2 #2 -x+3,-y,-z+1
10.3.2 Metal derivatives of zwitterions
As mentioned in Chapter IV and V the pyridinium zwitterions, which contain an
active –COOH, can be utilized for coordination to metal centers to generate MOCNs or
coordination compounds. Since the zwitterions are essentially amino acids their
metallated forms can be of relevance in biological context and also as useful bio-
mimics. Even though we have tried to synthesise the metal derivatives of the three
zwitterions (16, 23 and 25) with various metals like Mn, Co, Ni, Cu and Zn we could
not get totally phase pure form of the derivative through conventional route. However
we were successful to get one of the Ca derivatives (69) in phase pure form and as good
quality crystals. Similarly yet another ternary type zwitterion product 70 incorporating a
chiral zwitterion and fumarate ion coordinated to Cu ion could be successfully
synthesized. Given below are the synthetic and structural details of the compounds
including their crystal and molecular structures.
[Ca(3-pic zwitterion)2(H2O)2].4H2O, 69
The preparative details of the compound are discussed already. While the
zwitterion itself and its other metal derivatives are not easy to crystallize we were able

384
to get the Ca product 69 as good quality colorless needle like crystals rather easily.
Elemental analytical data of 69 are consistent with a composition [Ca(3-pic
zwitterion)2(H2O)2].4H2O. The CHN data and the nature of the metal-zwitterion
derivative 69 is given in Table 10.11
Table 10.11 Elemental analytical data of 69 and 70
Compound (Emp. formula) Formula
weight
Elemental content (%)
Found (calcd.) Colour and nature
(solubility in water)
C H N
[Ca(3-piczwitterion)2(H2O)2].4H2O, 69
(Ca C20 H32 N2 O14) 564.56
42.12
(42.51)
5.43
(5.67)
4.21
(4.25)
colourless crystals
(soluble)
[Cu2(fum)(zwitterion)2(H2O)2], 70
(Cu2 C22 H22 N2 O14) 665.50
39.32
39.67
3.05
3.31
3.53
3.61
blue crystals
(sparingly soluble)
FTIR spectral data of 69 and 70
Given in Table 10.12 are the IR stretching vibrations seen for various
characteristic groups in 69. The Ca derivative has its νas(COO-), νs(COO
-) and Δν values
1566, 1380, 186 cm-1
respectively which suggests the mono-dentate coordination mode
of the carboxylate group of both of its zwitterion moieties. There are also H2O specific
vibrations seen for the compound at 3232 and 3402 cm-1
indicating the presence of both
coordinated and guest type H2O molecules. This could be confirmed from both TGA
and also crystal structure studies.

385
Table10.12 FTIR spectral data of the metal derivatives of the zwitterions, 69 and 70
compounds νOH(H2O) νas(COO) νs(COO) Δν ν(M-O) ν(C-H)
aromatic ν(C-H)
aliphatic
ν(C-C), ν(C-N)
ring stretch
Ring deformation of pyridine
69 3402
3232 1566 1380 186 563 3066
2966
2935
1508
1434 667
70 3290 1635
1625
1427
1396
208
229 474
3128
3055
2970
2939
1500
1481 698
10.3.2.1 Structural characterization of [Ca(3-pic zwitterion)2 (H2O)2].4H2O, 69
Structural details:
Compound 69 has the composition [Ca(3-pic zwitterion)2(H2O)2].4H2O. The
compound crystallizes in monoclinic form with space group P2/n. The crystal data along
with structural refinement details are given in Table 10.13. Given in Fig. 10.12 is the
ORTEP plot of 69 which clearly shows the η1 mode of the carboxylate groups of the
zwitterion to the Ca2+
ion. Interestingly the Ca2+
ion is seen in CaO6 octahedral
environment. A closer look at the coordinating moieties on the Ca2+
ion (Fig 10.13a)
shows that there are in fact four carboxylate moieties coordinated to a single metal ion
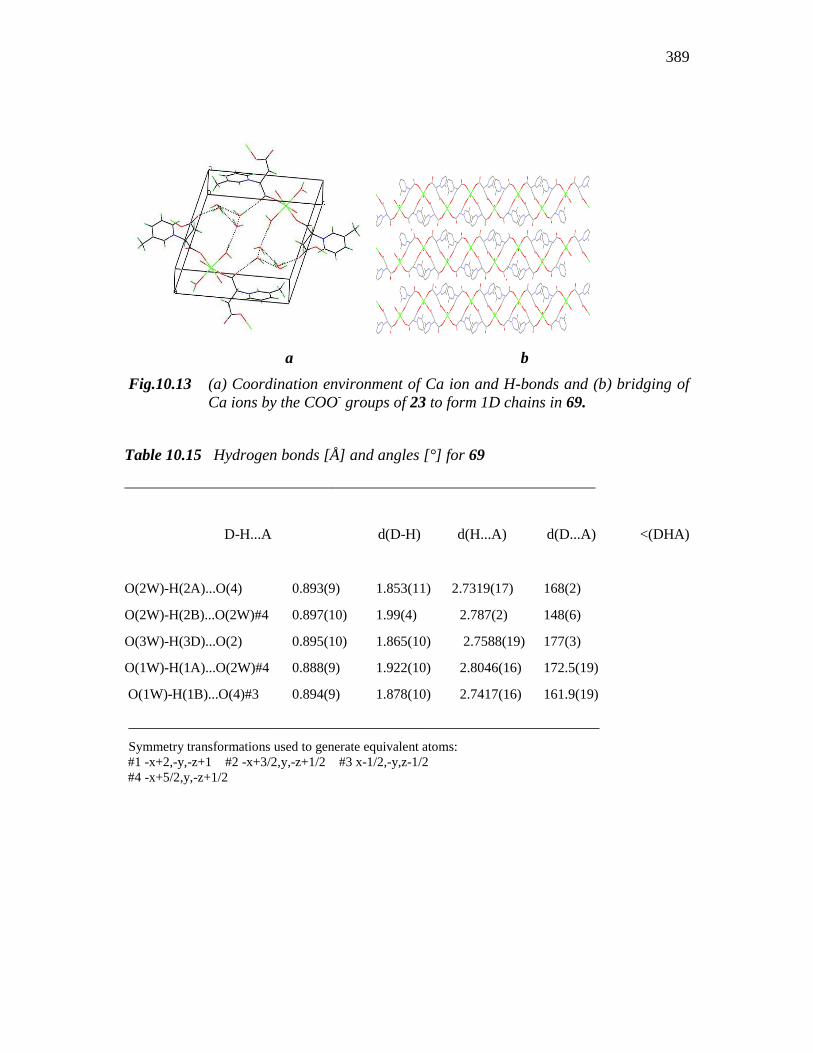
along with two H2O molecules. Further, all the four carboxylate moieties coordinated to
a single metal uion come from for different zwitterions. The resulting geometry of CaO6
is distorted octahedron. The two oxygens situated in the opposite corners of the

386
Table 10.13 Crystallographic data and structure refinements for 69 and 70
69 70
Empirical formula Ca C20 H32 N2 O14 Cu2 C22 H22 N2 O14
Formula weight 564.56 665.50
Temperature 293(2) K 293(2) K
Wavelength 0.71073 Ǻ 0.71073 Ǻ
Crystal system Monoclinic Triclinic
space group P2/n P-1
a (Å) 10.6243(4) 7.8181(5)
b (Å) 9.8887(4) 8.7831(6)
c (Å) 13.0621(5) 9.6464(6)
(°) 90 79.008(4)
β (°) 106.537(2) 77.974(3)
γ (°) 90 73.723(3)
Volume 1315.55(9) 615.68(7) A^3
Z 2 1
Calculated density 1.425 Mg/m^3 1.795 Mg/m^3
Absorption coefficient 0.309 1.806 mm^-1
F(000) 338
Theta range for data collection 2.18 to 31.42 deg.
Reflections collected 15782 16351
unique 3505 7282
R(int) 0.0321 0.0261
Absorption correction Semi-empirical from
equivalents
Max. and min. transmission 0.9143 and 0.7823
Refinement method Full-matrix least-
squares on F^2
Data / restraints / parameters 7282 / 12 / 373
Goodness-of-fit on F^2 1.034
Final R indices [I>2sigma(I)] R1 = 0.0361, wR2 = 0.1006 R1 = 0.0346, wR2 =
0.0925
R indices (all data) R1 = 0.0461, wR2 = 0.1084 R1 = 0.0465, wR2 =
0.0995
Largest diff. peak and hole 0.533 and -0.550 e.A^-3

387
octahedron are from two coordinated water molecules (Fig. 10.12 and 10.13a). Even
though there are four carboxylate groups coordinated to a Ca2+
ion the overall charge
appears to be compensated because one of the COO- moieties of the zwitterions 23 is
always with a -1 charge (which is internally charge compensated by +1 charge on the
pyridinium moiety) and it is this –COO- moiety which is coordinated to the Ca2+
ion
along with the deprotonated –COOH group. Thus there are two deprotonated –COOH
and two originally present –COO- moieties coordinated to each Ca2+
ion along with two
H2O molecules. Because four different zwitter ionic moieties are coordinated to Ca2+
ion the bonding and crystal packing is such that they form neatly arranged 2D layers
(Fig 10.13b). There are four lattice water molecules also per molecule of 69 which
participate in inter-chain hydrogen bonding interactions through both coordinated and
lattice H2O molecules or between the water and the carboxylate ions (Fig. 10.13a). The
Ca-O bond distances in 69 are in the range 2.2911(9)-2.3509(11)Å. Crystal parameters,
selected bond lengths and bond angles are listed in Table 10.13, 10.14 and 10.15
respectively.
Fig.10.12 ORTEP of 69 with atom label.

388
Table 10.14 Selected bond lengths [Å] and angles [°] for 69
___________________________________________________________
C(1)-O(2) 1.2303(16) C(8)-C(9) 1.3738(19)
C(1)-O(1) 1.2506(15) C(8)-C(10) 1.500(2)
C(1)-C(2) 1.5504(16) C(9)-N(1) 1.3453(17)
C(2)-N(1) 1.4821(15) O(1)-Ca(1) 2.2911(9)
C(2)-C(3) 1.5202(18) O(1W)-Ca(1) 2.3509(11)
C(3)-C(4) 1.5183(17) O(3)-Ca(1)#1 2.3407(9)
C(4)-O(3) 1.2394(16) Ca(1)-O(1)#2 2.2911(9)
C(4)-O(4) 1.2534(17) Ca(1)-O(3)#3 2.3407(9)
C(5)-N(1) 1.3422(16) Ca(1)-O(3)#1 2.3407(9)
C(5)-C(6) 1.375(2) Ca(1)-O(1W)#2 2.3509(11)
C(7)-C(8) 1.391(2)
O(2)-C(1)-C(2) 119.44(11) O(1)-Ca(1)-O(1)#2 88.24(5)
O(1)-C(1)-C(2) 114.01(11) O(1)-Ca(1)-O(3)#3 178.40(4)
N(1)-C(2)-C(3) 113.09(10) O(1)#2-Ca(1)-O(3)#3 90.46(4)
N(1)-C(2)-C(1) 110.11(10) O(1)-Ca(1)-O(3)#1 90.46(4)
C(3)-C(2)-C(1) 111.58(10) O(1)#2-Ca(1)-O(3)#1 178.40(4)
C(4)-C(3)-C(2) 114.36(11) O(3)#3-Ca(1)-O(3)#1 90.86(6)
O(3)-C(4)-O(4) 124.70(12) O(1)-Ca(1)-O(1W)#2 90.31(5)
C(5)-N(1)-C(2) 119.44(10) O(1)#2-Ca(1)-O(1W)#2 98.91(4)
C(9)-N(1)-C(2) 119.48(10) O(3)#3-Ca(1)-O(1W)#2 88.98(4)
C(1)-O(1)-Ca(1) 145.81(9) O(3)#1-Ca(1)-O(1W)#2 82.02(4)
C(4)-O(3)-Ca(1)#1 135.67(9) O(1)-Ca(1)-O(1W) 98.91(4)
___________________________________________________________
Symmetry transformations used to generate equivalent atoms: #1 -x+2,-y,-z+1 #2 -x+3/2,y,-z+1/2
#3 x-1/2,-y,z-1/2
389
a b
Fig.10.13 (a) Coordination environment of Ca ion and H-bonds and (b) bridging of
Ca ions by the COO- groups of 23 to form 1D chains in 69.
Table 10.15 Hydrogen bonds [Å] and angles [°] for 69
___________________________________________________________
D-H...A d(D-H) d(H...A) d(D...A) <(DHA)
O(2W)-H(2A)...O(4) 0.893(9) 1.853(11) 2.7319(17) 168(2)
O(2W)-H(2B)...O(2W)#4 0.897(10) 1.99(4) 2.787(2) 148(6)
O(3W)-H(3D)...O(2) 0.895(10) 1.865(10) 2.7588(19) 177(3)
O(1W)-H(1A)...O(2W)#4 0.888(9) 1.922(10) 2.8046(16) 172.5(19)
O(1W)-H(1B)...O(4)#3 0.894(9) 1.878(10) 2.7417(16) 161.9(19)
___________________________________________________________
Symmetry transformations used to generate equivalent atoms: #1 -x+2,-y,-z+1 #2 -x+3/2,y,-z+1/2 #3 x-1/2,-y,z-1/2
#4 -x+5/2,y,-z+1/2

390
10.3.2.2 Structural characterization of [Cu2(fum)(zwitterion)2(H2O)2, 70
It is interesting to note that even though we carried out reactions of Cu2+
salt
(CuCO3) with zwitter ion 16 in hot aqueous condition in almost 1:2 ratio both analytical,
spectral and crystal structure data clearly indicated the ternary nature of the compound
with fumarate ion as an additional and surprising constituent. The analytical data (Table
10.11.) and FTIR spectra clearly showed the presence of the fumarate moiety along with
the zwitterions in the compound. The νas(COO-), νs(COO
-) and Δν values of the
compound also indicated that carboxylate moieties are acting as bridging bidentate
moieties characteristic of paddle-wheel type structure for the compound.19
The important
vibrations are listed in Table 10.12.
We were able to get good quality blue needle like single crystals for the
compound 70 which is seen crsyallising in triclinic form with P-1 space group. Crystal
data and structure refinement parameters are given in Table 10.13. Given in Fig 10.14 is
the ORTEP plot of the compound which shows the presence of both zwitter ion and
fumarate moiety. The copper atoms have a five-coordinate square pyramidal
environment (Fig. 10.14). The basal plane is defined by four oxygen atoms from two
distinct fumarate carboxylato groups and two pyridinium succinate zwitterions. The
apical position is occupied by H2O molecule. The coordination environment consists of
tetracarboxylate-bridged dimetallic paddle-wheel secondary building units (SBUs). The
asymmetric unit consists of one copper atom, one pyridinium succinate zwitterion, half
of fumarate ligand and one water molecule. The copper atom is in a distorted square
pyramidal environment [Cu1-O 1.950(6)-2.095(6)Ǻ, Cu…Cu 2.6606(4)Ǻ]. The
fumarate ligands bridge the copper ions to form a 1D chain (Fig.10.15). These chains
are connected by hydrogen bonds between the coordinated water molecules and the free
carboxylate groups of the zwitterions to form a 3D network (Fig. 10.16). Bond lengths
and angles are given in Table 10.16. Selected H-bonds are summarized in Table 10.17.

391
It has been very surprising matter for us how a fumarate moiety was incorporated
in 70 in addition to 16 while we carried out reaction of Cu2+
salt with only chiral
zwitterion 16. In fact we carried out the complexation reaction in hot aqueous
condition. Since fumarate moiety in 70 can come only from the zwitterions, we heated a
suspension of zwitter ion 16 in aqueous condition for a few hours and checked whether
the zwitter ion remains still stable. Analysis of the product obtained after heating in
aqueous condition showed that the compound obtained after heating has turned to a
product mixture containing the original zwitter ion ad fumaric acid. We could also
notice the evolution of pyridine during the reaction. This clearly indicates the partial
instability of the zwitter ion in hot aqueous condition. Since the zwitter ion can be
considered as a product formed by the insertion of pyridine in either fumaric acid or
maleic acid, heating it in aqueous condition may be causing the reverse reaction thereby
generating fumaric acid in solution. Therefore we believe that fumarate moiety which is
contained in 70 must have been generated through pyridine expulsion from part of the
zwitterions taken for the reaction. In any case we believe that compound 70 is very
novel and unprecedented ternary type paddle-wheel type compound the design of which
has not been done or reported so far.

392
Fig.10.14 ORTEP view of 70 with the atom-labeling scheme (30% thermal
ellipsoids).
a
b
Fig. 10.15 (a) and (b) The 1D chain of 70 showing the paddle wheel arrangement of
the ligating atoms.

393
a b
Fig. 10.16 (a) and (b) The 3D network of hydrogen bonds in 70
Table 10.16 Bond lengths [Ǻ] and angles [deg] for 70
___________________________________________________________
Cu(1)-Cu(2) 2.6606(4) O(9)-Cu(1)-O(3) 167.5(3)
O(9)-Cu(1) 1.970(7) O(11)-Cu(1)-O(5) 96.2(3)
O(3)-Cu(1) 1.985(6) O(12)-Cu(2)-O(6) 99.8(3)
O(10)-Cu(2) 1.965(6) O(10)-C(18)-O(9) 126.9(8)
O(4)-Cu(2) 1.963(6) O(3)-C(9)-O(4) 124.7(7)
O(5)-Cu(1) 2.095(6) O(13)#2-Cu(1)-O(11) 167.6(3)
O(6)-Cu(2) 2.145(7) O(13)#2-Cu(1)-O(9) 91.9(3)
O(11)-Cu(1) 1.950(6) O(5)-Cu(1)-Cu(2) 174.47(16)
O(12)-Cu(2) 1.950(7) O(12)-Cu(2)-O(14)#2 167.1(3)
O(4)-Cu(2)-O(10) 166.8(3) O(12)-Cu(2)-Cu(1) 83.7(2)
__________________________________________________________
Symmetry transformations used to generate equivalent atoms: #1 x,y-1,z #2 x,y+1,z

394
Table 10.17 H-bonds for 70 [Ǻ and deg.].
___________________________________________________________
D-H...A d(D-H) d(H...A) d(D...A) <(DHA)
O(5)-H(5A)...O(2)#3 0.852(9) 2.14(2) 2.924(10) 153(3)
O(5)-H(5B)...O(7)#4 0.846(9) 2.039(15) 2.871(9) 168(4)
O(6)-H(6A)...O(7)#5 0.853(10) 2.16(6) 2.808(9) 133(6)
O(6)-H(6B)...O(2)#6 0.856(10) 1.84(4) 2.595(9) 146(7)
O(6)-H(6B)...O(14)#2 0.856(10) 2.796(18) 2.993(10) 94.9(11)
___________________________________________________________
Symmetry transformations used to generate equivalent atoms: #1 x,y-1,z #2 x,y+1,z #3 x+1,y,z-1 #4 x-1,y,z
#5 x-1,y,z+1 #6 x+1,y,z
EPR spectrum of [Cu2(fum)(zwitterion)2(H2O)2, 70
We have made some attempt to look at the bonding features of 70 by measuring
EPR spectrum in methanolic solution at liquid nitrogen temperature. The spectrum
shows four aniosotropic hyperfine splittings which is characteristic of Cu2+
ion (Fig.
10.17).
The EPR spin-Hamiltonian parameters of the compound are evaluated assuming
axial symmetry and using DPPH as the ‘g’markerthe. The values are :A║ = 9mT, H║ =
309.5mT, A┴ = 5.66mT, H┴ = 320.7mT, g║ = 2.14, g┴ = 2.06, giso= 2.08, G=2.38, α2=
0.4524. The low A║ value of 9mT shows that Cu is not octahedrally coordinated by the
ligands. g║> g┴ > 2 indicates tetragonally elongated Cu(II) complex and also indicates
that the unpaired electron resides in the dx2-y
2 orbital. The fact that g║< 2.3 indicates the
covalent nature of the complex. G value less than 4, indicates a strong ligand field.
Similarly the low value (0.4524) of inplane σ covalency parameter α2 shows that the
metal- ligand bond has a high covalent character.

395
3000 3500-0.10
-0.05
0.00
0.05
0.10
30502960
Inte
ns
ity
GAUSS
Fig. 10.17 X-band EPR spectrum of a methanolic solution of 70 in LNT.
Summary and conclusion
Relevant details on the nature of reaction and also the type of products obtained through
the reaction of maleic acid and fumaric acid with both en and dap have been presented. Crystal
and molecular structures of some interesting adducts also have been discussed. An interesting
Ca complex formed from 3-picoline zwitterion containing two coordinated chiral zwitter ions
could be made and its structural features studied through single crystal XRD. Presented in the
chapter are also some details on a novel and unprecedented metal-zwitterion derivative 70
including its unique structural features. We have made use of a wide variety of experimental
techniques like CHN analysis, TG, DTA, FTIR, 1H NMR,
13C NMR, EPR, PXRD and single
crystal X-ray diffraction studies rather extensively for the detailed characterization of all the
products obtained during the transformation reactions.