inhibition of histone demethylase jmjd1a improves anti...
TRANSCRIPT

Molecular and Cellular Pathobiology
Inhibition of Histone Demethylase JMJD1A Improves Anti-Angiogenic Therapy and Reduces Tumor-AssociatedMacrophages
Tsuyoshi Osawa1,2,5, Rika Tsuchida5, Masashi Muramatsu5,6, Teppei Shimamura4, Feng Wang5,Jun-ichi Suehiro1, Yasuharu Kanki2, Youichiro Wada2, Yasuhito Yuasa5, Hiroyuki Aburatani3,Satoru Miyano4, Takashi Minami1, Tatsuhiko Kodama2, and Masabumi Shibuya5,7
AbstractAntiangiogenic strategies can be effective for cancer therapy, but like all therapies resistance poses a major
clinical challenge. Hypoxia and nutrient starvation select for aggressive qualities thatmay render tumors resistantto antiangiogenic attack. Here, we show that hypoxia and nutrient starvation cooperate to drive tumoraggressiveness through epigenetic regulation of the histone demethylase JMJD1A (JHDM2A; KDM3A). In cancercells rendered resistant to long-term hypoxia and nutrient starvation, we documented a stimulation of AKTphosphorylation, cell morphologic changes, cell migration, invasion, and anchorage-independent growth inculture. These qualities associated in vivowith increased angiogenesis and infiltration ofmacrophages into tumortissues. Through expression microarray analysis, we identified a cluster of functional drivers such as VEGFA,FGF18, and JMJD1A, the latter whichwas upregulated in vitro under conditions of hypoxia and nutrient starvationand in vivo before activation of the angiogenic switch or the prerefractory phase of antiangiogenic therapy.JMJD1A inhibition suppressed tumor growth by downregulating angiogenesis and macrophage infiltration, bysuppressing expression of FGF2, HGF, and ANG2. Notably, JMJD1A inhibition enhanced the antitumor effects ofthe anti-VEGF compound bevacizumab and the VEGFR/KDR inhibitor sunitinib. Our results form the foundationof a strategy to attack hypoxia- and nutrient starvation–resistant cancer cells as an approach to leverageantiangiogenic treatments and limit resistance to them. Cancer Res; 73(10); 3019–28. �2013 AACR.
IntroductionAngiogenesis is essential for tumor growth and metastasis
(1, 2). VEGFs and their receptors (VEGFR) are key regulatoryfactors of angiogenesis (3, 4). Therefore, targeting the VEGF/VEGFR pathways by antiangiogenesis is a clinically validatedanticancer treatment (5). Antiangiogenic therapies are, how-ever, limited to certain types of cancer andmay elicitmalignant
progression and metastasis (6, 7). Thus, the molecular mech-anism of eliminating resistant and refractory cancer cells byusing such antiangiogenic treatments needs to be elucidated.Under such treatments tumor cells may be exposed to bothhypoxia and nutrient starvation. We previously reported thathypoxic and nutrient-starved tumor microenvironment mayplay a critical role in tumor aggression and the refractoriness tothe antiangiogenic treatments (8) and that affected host cellsresulted in secondary leukemia in murine cancer models (9) Inaddition, we found that an epigenetic regulator, histonedemethylase JHDM1D, suppressed tumor growth by regulatingangiogenesis under nutrient starvation (10). Epigenetic regu-lation of angiogenesis under hypoxia and nutrient starvationcan be important for tumor progression and aggression, but itsmechanism has not yet been elucidated. The present study wasconducted to investigate the mechanism of tumor aggres-siveness, and our findings show that histone demethylaseJMJD1A plays an important role in tumor progression underhypoxia and nutrient starvation.
Materials and MethodsCell culture, hypoxia, and nutrient starvation double-deprivation
Human cervix HeLa, hepatic HepG2, epidermoid A431carcinomas, glioblastoma T98G, rabdomyosarcoma A673,and murine melanoma B16 cell lines were purchased from
Authors' Affiliations: 1Laboratory for Vascular Biology, 2Laboratory forSystems Biology and Medicine, 3Division of Genome Science, RCAST,4Laboratory of DNA Information Analysis, Genome Center, Institute ofMedical Science, The University of Tokyo; 5Department of MolecularOncology, Graduate School of Medicine and Dentistry, Tokyo MedicalandDental University, Tokyo, Japan; 6CancerGenetics Center for Geneticsand Pharmacology, Roswell Park Cancer Institute, Buffalo, New York; and7Department of Research and Education, Jobu University, Gunma, Japan
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
R. Tsuchida and M. Muramatsu contributed equally to this work.
Corresponding Authors: Masabumi Shibuya, Department of Researchand Education, Jobu University, 634-1 Toyazuka-machi, Isesaki, Gunma372-8588, Japan. Phone: 813-58035086; Fax: 011-813-5803-0125;E-mail: [email protected]; and Tsuyoshi Osawa, Laboratory forSystems Biology and Medicine, RCAST, The University of Tokyo, Tokyo,Japan. E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-12-3231
�2013 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 3019
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231

the American Type Culture Collection. The murine uterinecancer cell line, HSML, was kindly provided by Dr. Kudoh(Hirosaki University, Hirosaki, Japan). Cells were maintainedin Dulbecco's Modified Eagle Medium (DMEM) (NacalaiTesque) supplemented with 10% FBS at 37�C in a 5% CO2
atmosphere, except HSML cells, which were maintained inRPMI-1640 medium (Nacalai Tesque). Nutrient deprivationwas conducted as previously described under hypoxia (Fig.1A; refs. 8, 9).
Cell proliferation assayCell proliferation wasmeasured using the sulforhodamine B
(SRB) assay as previously described (8).
Phase-contrast microscopyCells were plated on Petri dishes and images were captured
using the �10 to �20 objective lenses of an Olympus invertedmicroscope. Phase-contrast imageswere acquired usingKinet-ic Imaging software connected to a Hamamatsu CCD camera(Hamamatsu Photnics K.K).
Wound-healing assayCells were seeded onto 12-well plates at a density of 1� 106
cells. Following attachment, 3 to 6 hours after seeding,scratches were made within the monolayer by using asterile pipette tip. The same spots were photographed underphase-contrast microscopy at 12 hours post-"wounding." The
A
B
Wonded
fill
ed
(%
)
0
10
20
30
40
50
HeLa-
DDS10
HeLa0
20
40
60
T98G-
DDS10
T98G
Wonde
d fill
ed
(%
)
Invasio
n (
count)
Invasio
n (
count)
*
0
20
40
60
80
100
120 **
0
20
40
60
80 **
**
Colo
ny (
count)
Colo
ny (
count)
0
20
40
60
80*
020406080
100120
*
D
E
F
p-Akt
Akt
β-Actin
HeL
a
HeL
a-DDS10
T98G
T98G-D
DS10
473
C
HeLa-
DDS10
HeLa T98G-
DDS10
T98G
T98G-
DDS10
T98GHeLa-
DDS10
HeLa
0
0.5
1
1.5
2
2.5
00.20.40.60.8
11.21.41.6** **
Ce
ll g
row
th
Ce
ll g
row
th
HeLa
T98G
Cont DDS10
G
T98G-
DDS10
T98GHeLa-
DDS10
HeLa
0 1 2 3 4 5Day
Oxygen (%)
Nutrition (ratio)
1 21
100301
Death RecoveryCell
Figure 1. Hypoxia and nutrient starvation double-deprivation increases Akt phosphorylation, cellular migration and invasion, and anchorage-independenttumor cell growth in vitro. A, schematic diagramof ahypoxia andnutrient starvationDDScycle.Humancancer cellswere cultured under hypoxia (pO2, 1%) andnutrient starvation (1:100 ratio) for approximately 3 days and recovered by culturing under normoxia in 30% nutrition for 1 day and then 100% nutritionfor more than a day until 80% to 90% confluence was achieved. This hypoxia and nutrient starvation cycle was conducted for 10 or more cycles. B,ten cyclesofDDS (DDS10) inHeLa andT98Gcells induced acellmorphologic change fromepithelial cobblestone–like cells tomesenchymal-like spindle cells.C, DDS stimulated Akt phosphorylation in HeLa and T98G cells. D, DDS increased the cellular migration, which was examined using a wound-healingassay. E, DDS increased cellular invasion into Matrigel migration chambers. F, DDS increased anchorage-independent tumor cell growth in HeLa andT98G cells. G, HeLa-DDS10 and T98G-DDS10 cells become less sensitive to pacritaxel. Cells were treated with 10 mmol/L pacritaxel for 3 days. �, P < 0.05;��, P < 0.01, determined by the Student t test.
Osawa et al.
Cancer Res; 73(10) May 15, 2013 Cancer Research3020
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231

migrated distance was assessed using 9 measurements at timepoint. Wounded area was calculated using the followingequation: Wounded filled (%)¼ [(wound width at 12 hours�wound width at 0 hours)/wounded width at 0 hours] � 100.The experiments were carried out in triplicate. SEM wascalculated and analyzed using the unpaired, 2-sided Studentt test.
Invasion assayTwenty microliters of Matrigel (BD Biosciences) was added
to aBoyden chamber insert containing 8-mmpores (BDFalcon)and solidified for 2 hours in 37�C. Then, 200 mL of DMEMsupplemented with 0.5% FBS containing 5 � 104 cells wereplated on the upper chamber, and 300 mL DMEM supplemen-ted with 10% FBS was added in the lower chamber. Cells werecultured for 8 to 48 hours depending on the cell types, and thenumber of invasive cells was counted following staining withcrystal violet.
Colony formation assayCells were suspended in DMEM containing 0.6% methylcel-
lulose (Wako) supplemented with 10% FBS and seeded into 6-well plates at a density of 1 � 104 to 2.5 � 104 cells per well intriplicate. After 7 to 10 days, anchorage-independent colonieswere counted using a microscope.
Western blottingCell lysates were applied to a 7.5% to 15% PAGE and
transferred to a polyvinylidene difluoride membrane (Invitro-gen). The membrane was incubated with rabbit polyclonalanti-JHDM2A antibody, anti-histone H3K9me1, anti-histoneH3K9me3, anti-histone H3, anti-Akt, anti-phosphorylated Akt,or mouse monoclonal anti-histone H3K9me2 and anti-b-actinantibody (1:1,000; Millipore), followed by incubation withhorseradish peroxidase–conjugated secondary antibodies (GEHealthcare). Signals were detected using enhanced chemilu-minescence detection reagents (GE Healthcare) and imageswere acquired using a luminescent image analyzer (LAS3000,FujiFilm).
Gene expression analysis using real-time PCRTotal RNA was extracted from cells using the Isogen
reagent (Nippon Gene), converted to cDNA by using thePrime Script reverse transcriptase (Takara) as per the man-ufacturer's instructions, and used for quantitative real-timePCR amplification using SYBR Green (Takara; Supplemen-tary Table S1).
Gene expression profiling and statistical analysisTotal RNA (5 mg) was converted to aminoallyl-modified
cDNA by random 6-mer and oligo(dT)-primed polymerizationusingPrimeScript reverse transcriptase (Takara), coupled toN-hydroxysuccinimidyl esters of Cy3 (Agilent), and then hybrid-ized to a human Oligo Microarray slide (human G4110F,Agilent) as per the manufacturer's instructions. Arrays werescanned using a G2565BA Microarray Scanner System andanalyzed using the GeneSpring GX software (Tomy DigitalBiology).
Animal studies and tumor xenograftMurine B16 or human HeLa or A673 cells (1 � 107) were
subcutaneously injected into C57BL/6J or C.B17/Icr-scidJclscid/scid mice, respectively. Mice were treated with i.v. injec-tion of 1 to 10 mg/kg/wk intraperitoneally (i.p.) of eitherhuman IgG or anti-VEGF antibody (Chugai-Roche) every 7days starting on day 2. VEGFR inhibitor (sunitinib) wassolubilized in methylselrose and administrated orally at theconcentration of 40mg/kg/d. To visualize the hypoxic region inthe tumor tissue, 60mg/kg of pimonidazole was administrated(i.p./i.v.) at 30 to 45 minutes before perfusion fixation. Tumorvolume was measured, and the data were analyzed using theStudent t test. All animal care procedures were in accordancewith institutional guidelines approved by the University ofTokyo and Tokyo Medical and Dental University.
Histological analysis and immunohistochemistryTumor tissues were fixed in 4% paraformaldehyde (PFA),
embedded in paraffin, and subjected to hematoxylin and eosin(HE) and periodic acid-Schiff (PAS) staining.
Immunofluorescence stainingFreshly frozen or 4% PFA-fixed tumor tissues were cut 14-
mm thick by using a Cryostat (Leica) and stained with hamsteranti-CD31 (BD Biosciences), rat anti-CD11b (BD Biosciences),rat anti-F4/80 (AbD Serotec), mouse monoclonal anti-aSMA1(Sigma), and mouse monoclonal anti-pimonidazole (Cosmobio). The sections were incubated with the appropriate sec-ondary antibody and the nuclear-staining dye To-Pro-3 (Invi-trogen Life Technologies) and then analyzed using a confocalmicroscope (Radiance 2000; Bio-Rad). We quantified the num-ber of CD31þ/aSMA1þblood vessels/pericytes in the total areaof the tumor. We analyzed 5 to 10 fields per sample (n¼ 5) forquantification of blood vessel density (k pixels). We alsocounted the number of CD11bþ and F4/80þ cells in the totalarea of the tumor in 5 to 10 fields for each sample (n ¼ 5).
siRNAsiRNAs were designed against human JMJD1A and obtained
commercially (Invitrogen Life Technologies). In this study, 2sequences of siRNAswere used against hJMJD1A, designated assiRNA1 (AGAAGAAUUCAAGAGAUUCCGGAGG) and siRNA2(AACACAUUCCAGUUGCUCUUAUUGU). Cells were trans-fected with JMJD1A siRNAs or negative (scramble) controlsiRNA by using the Lipofectamine RNAi MAX transfectionreagent according to the manufacturer's instructions (Invitro-gen Life Technologies). The ability of the siRNA to inhibitJMJD1A expression was assessed at 48 hours after transfection.
ResultsHypoxia and nutrient starvation accelerate tumoraggressiveness and resistance to chemotherapy
To clarify whether the hypoxia and nutrient starvationdouble-deprivation stress (DDS) stimulates tumor aggres-siveness in human cancer cells, we used thewell-knownhumancancer cell lines HeLa (human cervical carcinoma) and T98G(human glioblastoma). As previously reported, we developeda cell culture system for maintaining these cells under DDS
Inhibition of JMJD1A Improves Antiangiogenesis
www.aacrjournals.org Cancer Res; 73(10) May 15, 2013 3021
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231
(Fig. 1A; refs. 8, 9). HeLa and T98G cells were continuouslyexposed to 10 cycles of DDS (HeLa-DDS10 and T98G-DDS10cells). HeLa-DDS10 and T98G-DDS10 cells showed cell mor-phologic changes (Fig. 1B), increased Akt phosphorylation (Fig.1C), cellularmigration (Fig. 1D), invasion (Fig. 1E), and anchor-age-independent cell growth (Fig. 1F) in vitro. To investigatewhether increased invasion is due, in part, to the high prolif-eration rate of DDS cells, we compared the cell proliferationrates of untreated HeLa and T98G cells to the proliferationrates of HeLa- and T98G-DDS10 cells. We found no significant
difference in the rate of cell proliferation in HeLa and T98Gcells (Supplementary Fig. S1), suggesting that cell proliferationrate was not responsible for the increased migration andinvasiveness of DDS cells (Fig. 1D and E). Notably, HeLa-DDS10and T98G-DDS10 cells were less sensitive to conventionalchemotherapeutic agents such as paclitaxel (Fig. 1G, Supple-mentary Fig. S2; ref. 8). This finding suggests that hypoxia- andnutrient starvation–resistant human cancer cells developtumor aggressiveness and resistance to chemotherapeuticagents.
A
B16 B16DDS10
HeLa HeLaDDS10
B16 B16DDS10
HeLa HeLaDDS10
* *
* *HeLa-cont
B16-cont
HeLa-DDS10
B16-DDS10
0
200
400
600
800
B16 B16DDS10
Tum
or
volu
me (
mm
)
CD
31 C
D11
b n
ucle
us
CD
31 C
D11
b n
ucle
us
0
50
100
150
200
250
HeLa HeLaDDS10
Tum
or
volu
me (
mm
)
B
C
anti-VEGF Ab
*
*D E F
G H I J
H
H
33
Control IgG
Hypoxia
Nutr
itio
n
0
Cont IgG 10mg/kg
anti-VEGF 1mg/kg
anti-VEGF 10mg/kg
Nutrition (PAS)
Oxygen (HP)
Vessels
Nutrition (PAS)
Oxygen (HP)
Vessels
Cont IgG
anti-VEGF
+++
+++
+
+
-
-
Tum
or
volu
me (
mm
3) 2
1
x1000
0
5
10
15
20
25
30
35
0
5
10
15
20
25
30
35
40
0
1
2
3
4
5
6
0
2
4
6
8
10
12
14x10
9
x109
x107
x107
CD
11b+
cells
(counts
in tota
l are
a)
Blo
od v
essel density
(pix
els
in tota
are
a)
Blo
od v
essel density
(pix
els
in tota
are
a)
CD
11b+
cells
(counts
in tota
l are
a)
1.21.41.61.8
00.20.40.60.8
0 5 10 15 20
Days after injection
P
P
V
*
*
**
****
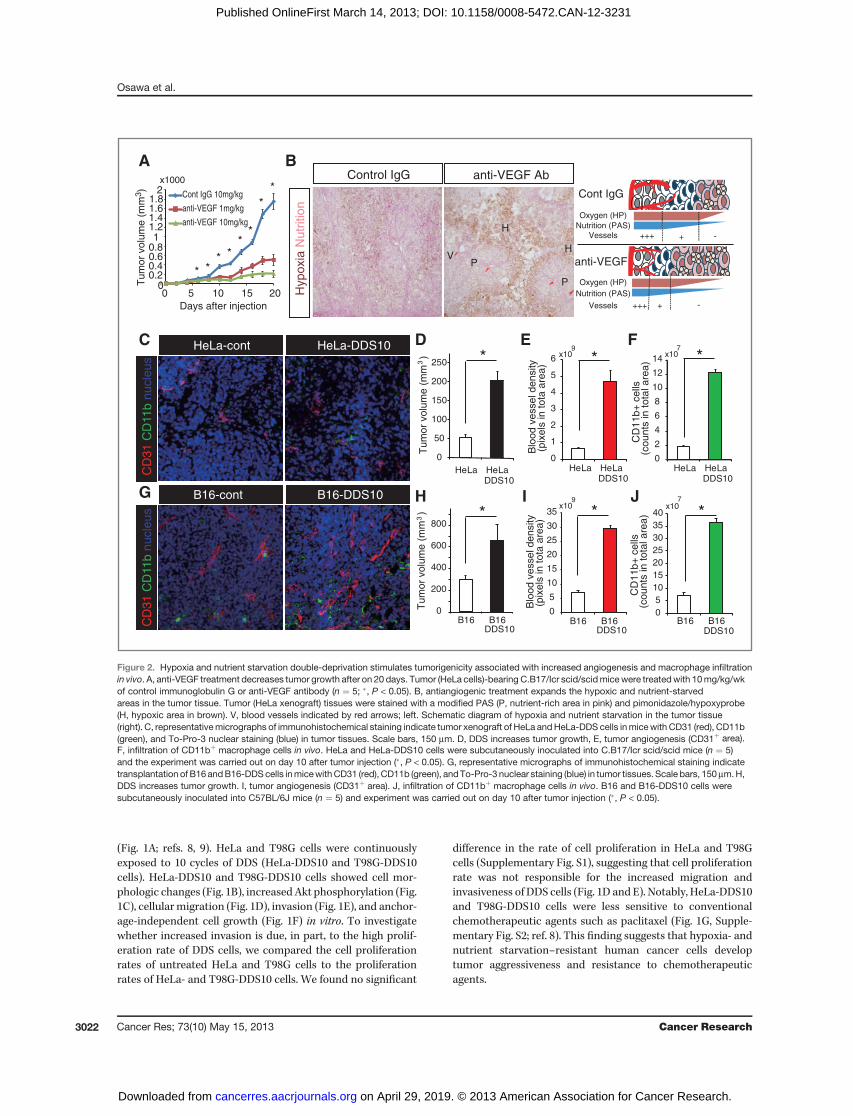
Figure 2. Hypoxia and nutrient starvation double-deprivation stimulates tumorigenicity associated with increased angiogenesis and macrophage infiltrationin vivo. A, anti-VEGF treatment decreases tumor growth after on 20 days. Tumor (HeLa cells)-bearing C.B17/Icr scid/scidmicewere treatedwith 10mg/kg/wkof control immunoglobulin G or anti-VEGF antibody (n ¼ 5; �, P < 0.05). B, antiangiogenic treatment expands the hypoxic and nutrient-starvedareas in the tumor tissue. Tumor (HeLa xenograft) tissues were stained with a modified PAS (P, nutrient-rich area in pink) and pimonidazole/hypoxyprobe(H, hypoxic area in brown). V, blood vessels indicated by red arrows; left. Schematic diagram of hypoxia and nutrient starvation in the tumor tissue(right). C, representativemicrographs of immunohistochemical staining indicate tumor xenograft of HeLa andHeLa-DDS cells inmicewith CD31 (red), CD11b(green), and To-Pro-3 nuclear staining (blue) in tumor tissues. Scale bars, 150 mm. D, DDS increases tumor growth, E, tumor angiogenesis (CD31þ area).F, infiltration of CD11bþ macrophage cells in vivo. HeLa and HeLa-DDS10 cells were subcutaneously inoculated into C.B17/Icr scid/scid mice (n ¼ 5)and the experiment was carried out on day 10 after tumor injection (�, P < 0.05). G, representative micrographs of immunohistochemical staining indicatetransplantation of B16andB16-DDScells inmicewithCD31 (red), CD11b (green), andTo-Pro-3 nuclear staining (blue) in tumor tissues. Scale bars, 150mm.H,DDS increases tumor growth. I, tumor angiogenesis (CD31þ area). J, infiltration of CD11bþ macrophage cells in vivo. B16 and B16-DDS10 cells weresubcutaneously inoculated into C57BL/6J mice (n ¼ 5) and experiment was carried out on day 10 after tumor injection (�, P < 0.05).
Osawa et al.
Cancer Res; 73(10) May 15, 2013 Cancer Research3022
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231

Cancer cells resistant to hypoxia and nutrient starvationincreased tumor growth and tumor angiogenesis in vivoThe extent of hypoxia and nutrient starvation in tumor
tissues following treatment with antiangiogenic agents wasexamined by treating tumor-bearing immunodeficient micewith VEGF-neutralizing antibody (bevacizumab; 0–10 mg/kg/wk i.p.), and the hypoxic- and nutrient-rich areas were visu-alized using anti-pimonidazol and a modified PAS, respective-ly. While tumor growth was significantly suppressed after anti-VEGF treatment (Fig. 2A, Supplementary Fig. S3A), hypoxic(pimonidazolþ) and nutrient-starved (PAS�) areas wereincreased in tumor tissues in HeLa and A673 cells (Fig. 2B,Supplementary Fig. S3B). In conclusion, hypoxia alone does notaffect cell proliferation, but hypoxia and nutrient starvationcooperatively suppressed the cell proliferation of cancer cellsin vitro (Supplementary Fig. S4).To investigate whether cancer cells resistant to hypoxia and
nutrient starvation stimulate tumorigenicity, we subcutane-ously inoculated 1� 107 of originalHeLa andHeLa-DDS10 cellsin scid/scid mice. HeLa-DDS10 cells showed increased initialtumor growth (Fig. 2C and D), increased CD31þ vascularformation (Fig. 2E), and infiltration of CD11bþ monocytes(Fig. 2F) and F4/80þ macrophages (Supplementary Fig. S5),suggesting a contribution of tumor-associated monocytes/macrophages (TAM; refs. (11, 12). In addition, orthotopictransplantation of B16-DDS10 cells in immunoproficient miceshowed a significant increase in the initial tumor growth (Fig.
2G and H), angiogenesis (Fig. 2I), and monocyte (Fig. 2J) andmacrophage infiltration (Supplementary Fig. S5) comparedwith implantation with original B16 cells (11, 13). Notably,blood vessel functionality and coverage of pericytes(a-SMA1þ) on CD31þ blood vessels were not significantlydifferent between control andDDS cells in vivo (SupplementaryFig. S6). In addition, HeLa-DDS10 cells were still sensitive totreatment with anti-VEGF antibody (bevacizumab; 5 mg/kg/wk�1 i.p.) in vivo (Supplementary Fig. S7). Thus, cancer cellssurvived under the DDS-stimulated tumorigenicity by regula-tion of tumor angiogenesis.
Hypoxia and nutrient starvation cooperatively inducethe epigenetic regulator JMJD1A
To determine the molecular targets against oxygen andnutrition DDS–resistant cancer cells, we conducted a micro-array analysis of 4 different human cancer cells (HeLa, HepG2,A431, and T98G) that were subjected to hypoxia, nutritionstarvation, and DDS. We found that a cluster of DDS-respon-sive genes such as VEGF-A, FGF18, and jumonji domain-con-taining protein 1A (JMJD1A) was upregulated in cancer cells(Fig. 3A–C, Supplementary Table S2 and Supplementary Fig.S8). Histone demethylase JMJD1A was upregulated under DDSin both murine and human cancer cells (Fig. 3D, Supplemen-tary Table S3) and the upregulation prominently increasedfollowing long-term exposure to DDS (48–72 hours; Fig. 3E). Inaddition, JMJD1A expression was normalized by returning the
Hyp
oxia
Nu
trie
nt
Sta
rva
tio
n
DD
S
Hypoxia inducible
Nutrient starvation
inducible
DDSinducible
Hypoxia/NS high
DDS
down regulation
He
La
He
pG
2
A4
31
T9
8G
FGF18C1orf51C8orf58
JMJD1AHK2
FLJ32679GOLGA8B
SAMD4A
DKFZp667E0512
FLJ23834GPR146LAPR2
VEGFA
IL27
ZNF33A
TBC1D3TCP11L2
He
La
He
pG
2
A4
31
T9
8G
He
La
He
pG
2
A4
31
T9
8G
HypoxiaNutrient
StarvationDDS
updown
3-3 1 2-1-2Fold-change
A B
C D
E
0
2
4
6
8
10
12
0 24 48 720
4
8
12
16
20
0 24 48 72
mJM
JD1A
exp
ress
ion
Time (h)
Control
Hypoxia
Nutrient starvation
DDS
HeLa B16
0
2
4
6
8
10
12
0
2
4
6
8
0
4
8
12
16
20
0
1
2
3
4
hJM
JD1A
exp
ress
ion
hJM
JD1A
exp
ress
ion
HeLa HepG2 A431 T98G
C H N DS C H N DS C H N DS C H N DS
C DS C DS C DS C DS
HeLa HepG2 A431 T98G
42 kDa147 kDa
17 kDa17 kDa17 kDa
JMJD1Ab-act
H3K9me2
H3K9me3H3
Mouse
DDS genesHuman
DDS genes
***
57035128
JMJD1A
*
Figure 3. Hypoxia and nutrient starvation DDS induces DDS-responsible gene clusters including JMJD1A. A, cluster of DDS-inducible genes following DDS inhuman cancer cells (HeLa, HepG2, A431, and T98G) under hypoxia, nutrient starvation, and DDS. B, expression of JMJD1A mRNA was cooperativelyupregulated under hypoxia and nutrient starvation (C, control; H, hypoxia, NS, nutrient starvation; DDS, hypoxia and nutrient starvation double stress).�,P < 0.05. C, JMJD1Aexpressionwas upregulated underDDSat the protein level. D, Venn diagramofDDS-responsible genes common to humanandmousecancer cells. E, JMJD1A mRNA expression was especially increased in human and murine cancer cells in response to long-term exposure to DDS.
Inhibition of JMJD1A Improves Antiangiogenesis
www.aacrjournals.org Cancer Res; 73(10) May 15, 2013 3023
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231
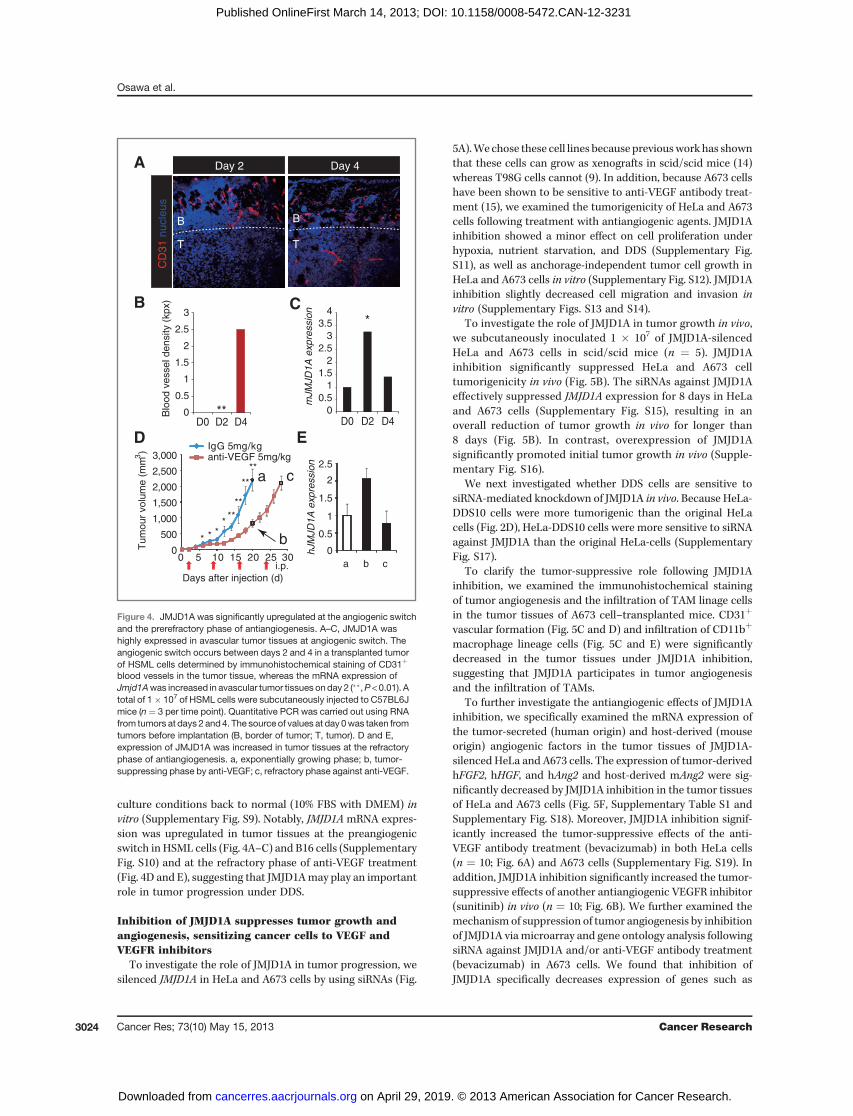
culture conditions back to normal (10% FBS with DMEM) invitro (Supplementary Fig. S9). Notably, JMJD1A mRNA expres-sion was upregulated in tumor tissues at the preangiogenicswitch in HSML cells (Fig. 4A–C) and B16 cells (SupplementaryFig. S10) and at the refractory phase of anti-VEGF treatment(Fig. 4D and E), suggesting that JMJD1Amay play an importantrole in tumor progression under DDS.
Inhibition of JMJD1A suppresses tumor growth andangiogenesis, sensitizing cancer cells to VEGF andVEGFR inhibitors
To investigate the role of JMJD1A in tumor progression, wesilenced JMJD1A in HeLa and A673 cells by using siRNAs (Fig.
5A).We chose these cell lines because previouswork has shownthat these cells can grow as xenografts in scid/scid mice (14)whereas T98G cells cannot (9). In addition, because A673 cellshave been shown to be sensitive to anti-VEGF antibody treat-ment (15), we examined the tumorigenicity of HeLa and A673cells following treatment with antiangiogenic agents. JMJD1Ainhibition showed a minor effect on cell proliferation underhypoxia, nutrient starvation, and DDS (Supplementary Fig.S11), as well as anchorage-independent tumor cell growth inHeLa and A673 cells in vitro (Supplementary Fig. S12). JMJD1Ainhibition slightly decreased cell migration and invasion invitro (Supplementary Figs. S13 and S14).
To investigate the role of JMJD1A in tumor growth in vivo,we subcutaneously inoculated 1 � 107 of JMJD1A-silencedHeLa and A673 cells in scid/scid mice (n ¼ 5). JMJD1Ainhibition significantly suppressed HeLa and A673 celltumorigenicity in vivo (Fig. 5B). The siRNAs against JMJD1Aeffectively suppressed JMJD1A expression for 8 days in HeLaand A673 cells (Supplementary Fig. S15), resulting in anoverall reduction of tumor growth in vivo for longer than8 days (Fig. 5B). In contrast, overexpression of JMJD1Asignificantly promoted initial tumor growth in vivo (Supple-mentary Fig. S16).
We next investigated whether DDS cells are sensitive tosiRNA-mediated knockdown of JMJD1A in vivo. Because HeLa-DDS10 cells were more tumorigenic than the original HeLacells (Fig. 2D), HeLa-DDS10 cells were more sensitive to siRNAagainst JMJD1A than the original HeLa-cells (SupplementaryFig. S17).
To clarify the tumor-suppressive role following JMJD1Ainhibition, we examined the immunohistochemical stainingof tumor angiogenesis and the infiltration of TAM linage cellsin the tumor tissues of A673 cell–transplanted mice. CD31þ
vascular formation (Fig. 5C and D) and infiltration of CD11bþ
macrophage lineage cells (Fig. 5C and E) were significantlydecreased in the tumor tissues under JMJD1A inhibition,suggesting that JMJD1A participates in tumor angiogenesisand the infiltration of TAMs.
To further investigate the antiangiogenic effects of JMJD1Ainhibition, we specifically examined the mRNA expression ofthe tumor-secreted (human origin) and host-derived (mouseorigin) angiogenic factors in the tumor tissues of JMJD1A-silenced HeLa and A673 cells. The expression of tumor-derivedhFGF2, hHGF, and hAng2 and host-derived mAng2 were sig-nificantly decreased by JMJD1A inhibition in the tumor tissuesof HeLa and A673 cells (Fig. 5F, Supplementary Table S1 andSupplementary Fig. S18). Moreover, JMJD1A inhibition signif-icantly increased the tumor-suppressive effects of the anti-VEGF antibody treatment (bevacizumab) in both HeLa cells(n ¼ 10; Fig. 6A) and A673 cells (Supplementary Fig. S19). Inaddition, JMJD1A inhibition significantly increased the tumor-suppressive effects of another antiangiogenic VEGFR inhibitor(sunitinib) in vivo (n ¼ 10; Fig. 6B). We further examined themechanism of suppression of tumor angiogenesis by inhibitionof JMJD1A viamicroarray and gene ontology analysis followingsiRNA against JMJD1A and/or anti-VEGF antibody treatment(bevacizumab) in A673 cells. We found that inhibition ofJMJD1A specifically decreases expression of genes such as
a b c
B
D E
0
500
1,000
1,500
2,000
2,500
3,000
0 5 10 15 20 25 30
IgG 5mg/kganti-VEGF 5mg/kg
Days after injection (d)
Tu
mo
ur
vo
lum
e (
mm
)
i.p.
* * ****
**
**
**
3
hJM
JD1A
exp
ress
ion
0
0.5
1
1.5
2
2.5
0
0.5
1
1.5
2
2.5
3
3.5
4
D0 D2 D40
0.5
1
1.5
2
2.5
3
D0 D2 D4
mJM
JD1A
exp
ress
ion
Blo
od
ve
sse
l d
en
sity (
kp
x)
**
*
A
T
B
T
B
a
b
c
C
Day 2 Day 4C
D3
1 n
ucle
us
Figure 4. JMJD1A was significantly upregulated at the angiogenic switchand the prerefractory phase of antiangiogenesis. A–C, JMJD1A washighly expressed in avascular tumor tissues at angiogenic switch. Theangiogenic switch occurs between days 2 and 4 in a transplanted tumorof HSML cells determined by immunohistochemical staining of CD31þ
blood vessels in the tumor tissue, whereas the mRNA expression ofJmjd1Awas increased in avascular tumor tissues onday 2 (��,P <0.01). Atotal of 1� 107 of HSML cells were subcutaneously injected to C57BL6Jmice (n ¼ 3 per time point). Quantitative PCR was carried out using RNAfrom tumors at days 2 and4. The source of values at day 0was taken fromtumors before implantation (B, border of tumor; T, tumor). D and E,expression of JMJD1A was increased in tumor tissues at the refractoryphase of antiangiogenesis. a, exponentially growing phase; b, tumor-suppressing phase by anti-VEGF; c, refractory phase against anti-VEGF.
Osawa et al.
Cancer Res; 73(10) May 15, 2013 Cancer Research3024
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231

FGF2, CSF2, and S100A9 (Supplementary Table S4) related tocell movement, leukocyte and myeloid cell migration, angio-genesis, and lymphangiogenesis (Supplementary Fig. S20). Thisexplains why an inhibition of JMJD1A by siRNAs can preventthe infiltration of immune cells such as monocytes/macro-phages into tumor tissue, resulting in decreased tumor angio-genesis (Fig. 5). Introducing epigenetic modifications in hyp-oxic and nutrient-starved cancer cells, such as by JMJD1Ainhibition, is an effective anticancer strategy for improvingantiangiogenesis (Fig. 7).
DiscussionAntiangiogenesis is a clinically validated cancer treatment
(5). However, the emergence of drug resistance and tumoraggressiveness, partly due to an extreme tumor microenviron-ment, can be major clinical problems (6–9). In this study, weinvestigated the mechanism underlying tumor aggressivenessby using a model system of antiangiogenesis (8, 9). Oxygen andnutrition deficiency stimulated cellmigration, invasion (Fig. 1),and tumorigenicity associated with increased tumor angio-
genesis and infiltration of TAMs (Fig. 2). We have shown thathistone demethylase JMJD1A, an epigenetic regulator, is highlyexpressed in cancer cells in response to hypoxia and nutrientstarvation (Fig. 3) and that JMJD1A inhibition suppressedtumor angiogenesis by downregulating fibroblast growth fac-tor (FGF)2, hepatocyte growth factor (HGF), and Ang2 andresulted in significant suppression of tumor growth andimprovement of anti-VEGF antibody (bevacizumab) andVEGFR inhibitor (sunitinib) treatment (Figs. 5 and 6). Takentogether, our results indicate that targeting cancer cell resis-tance to hypoxia and nutrient starvation can be a strategy forovercoming resistance to antiangiogenic treatments.
Inhibition of angiogenesis suppresses the formation ofnew tumor vasculature (5) andmay normalize the vasculature,resulting in increased efficacy of the combination chemother-apy (16). We found that antiangiogenic treatments expandedthe hypoxic and nutrient-starved areas within the tumortissue (Fig. 2B) and that hypoxia- and nutrient starvation–resistant cancer cells showed tumor aggressiveness such asincreased migration, invasion (Fig. 1), tumorigenicity, tumor
CD31 CD11b nucleus
JMJD
1A s
c co
nt
JMJD
1A s
iRN
A
*
***
**
**
** *
*
A B
C
12
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
HeLa sc
HeLa siRNA
0
0.2
0.4
0.6
0.8
1
1.2
1.4
A673 sc
A673 siRNA
0
500
1,000
1,500
2,000
2,500
3,000
3,500
4,000
4,500
0 5 10 15 20
A673 + control siRNA
A673 + JMJD1A siRNA1
A673 + JMJD1A siRNA2
Days after injection (d)
*
**
*
**
0
200
400
600
800
1,000
1,200
1,400
1,600
1,800
0 5 10 15 20
HeLa + control siRNA
HeLa + JMJD1A siRNA1
HeLa + JMJD1A siRNA2
Tu
mo
r vo
lum
e (
mm
3)
Days after injection (d)
**
*
*
*****
HeLa A673
sc scsi1 si1si2 si2
42 kDa
147 kDa
17 kDa
17 kDa
17 kDa
JMJD1A
H3K9me2
H3K9me3
b-act
H3
D
E
F
hAng
2 e
xp
ressio
nhA
ng2
exp
ressio
n
sc si1 si2
CD
11
b+
ce
llsB
loo
d v
esse
l d
en
sity
(pix
els
in
to
tal a
rea
)
0
2
4
6
8
10
12
14
16x10
8
(co
un
ts in
to
tal a
rea
)
0
2
4
6
8
10
12
14
16
18
sc si1 si2
x1010
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
0
0.2
0.4
0.6
0.8
1
1.2
HeLa sc
HeLa siRNA
hHG
F e
xp
ressio
n
HeLa sc
HeLa siRNA
hFG
F2
exp
ressio
n
0
0.2
0.4
0.6
0.8
1
1.2
1.4
A673 sc
A673 siRNA
0
0.2
0.4
0.6
0.8
1
1.2
1.4
A673 sc
A673 siRNA
hHG
F e
xp
ressio
n
hFG
F2
exp
ressio
n
0
0.2
0.4
0.6
0.8
1
1.2
0
0.2
0.4
0.6
0.8
1
1.2
A673 sc
A673 siRNA
mA
ng2
exp
ressio
nm
Ang
2 e
xp
ressio
n
HeLa sc
HeLa siRNA
Tu
mo
r vo
lum
e (
mm
3)
Figure 5. JMJD1A inhibition suppressed tumor growth via downregulating angiogenesis in vivo. A and B, JMJD1A inhibition by siRNAs suppressed in vivotumor growth. Tumor volumes of the xenografts of JMJD1A-silencedHeLa andA673 cells weremeasured and comparedwith that of the controls (�,P < 0.05).C–E, inhibition of JMJD1A decreased tumor angiogenesis and infiltration of macrophage cells and showed additive effects in combination withantiangiogenesis. Immunohistochemical staining revealed that JMJD1A inhibition decreased formation of CD31þ blood vessels and infiltration ofCD11bþ cells in tumors (�,P<0.05). F, expressionof proangiogenic factors (Ang1,Ang2, andHgf) in tumor tissueswere decreased in siJMJD1A-treated tumorexamined by quantitative real-time PCR analysis (�, P < 0.05).
Inhibition of JMJD1A Improves Antiangiogenesis
www.aacrjournals.org Cancer Res; 73(10) May 15, 2013 3025
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231

angiogenesis, and infiltration of tumor-associated monocytesand macrophage cells, suggesting that DDS10 cells activelyrecruit monocytes and macrophages (Fig. 2, SupplementaryFig. S5). In addition, hypoxia- and nutrient starvation–resistantcells became less sensitive to conventional chemotherapy (Fig.1G; ref. 8), suggesting that cancer cell refractoriness followingantiangiogenic treatment (6, 7, 17, 18) could be due to resis-tance to the combinational chemotherapy, in addition toactivation of both tumor cells and associated cells (19). Thus,combination chemotherapeutic regimens should be consid-ered to perturb the DDS-resistant cancer cells and tumor-associated cells in combinationwith antiangiogenic treatment.
The adaptation of cancer cells to tumor microenvironmentsinvolving oxygen or nutrient deficiencies, acidosis, and reactiveoxygen species (ROS) is required for tumor progression andmetastasis (20, 21). The oxygen or nutrient deficiency andacidosis can be adapted by metabolic alterations to glycolysis(Warburg effect), gluconeogenesis, and other pathways (22–24). Although prokaryotic cells use a "stringent control" under
nutrient deficiencies by terminating cell proliferation whilesynthesizing defective nutrient components (25, 26), the met-abolic alteration can be achieved in mammalian cells viatranscriptional and epigenetic regulation (27). Epigenetic reg-ulation of metastasis-related genes can also lead to resistanceto ROS (28). The "stringent control" of mammalian cells mayplay important roles during nutrient starvation, but to datethere are no studies showing this. We previously reported thathistone demethylase JHDM1D is an essential epigenetic reg-ulator in tumor progression under nutrient starvation byangiogenesis downregulation (10). Interestingly, JMJD1A actsas a pro-angiogenic regulator that promotes initiation of tumorgrowth, whereas JHDM1D suppresses tumor growth, possiblyplaying a role during the adaptation to both hypoxia andnutrient starvation. Thus, epigenetic regulation under hypoxiaand nutrient starvation could also be essential for both tumoraggressiveness and the shift in metabolism, but the controlmechanism underlying this has not yet been identified.
We identified that the histone demethylase JMJD1A is anessential epigenetic regulator of tumor progression underhypoxia and nutrient starvation. JMJD1A is involved in sper-matogenic development (29), metabolism in obesity (30), andhypoxic cancer cells underHIF1a regulation (31). Expression ofJMJD1A can be a poor prognostic maker in patients withcancer (32). Loss of JMJD1A resulted in reduced proliferationand invasiveness of human colon cancer cells in vitro, whichmay lead to decreased tumor growth in vivo (32). In this study,while JMJD1A significantly decreased tumor growth in vivo(Fig. 5), inhibition of JMJD1A showed only a minor effect onproliferation and invasion of HeLa and A673 cells in vitro(Supplementary Figs. S11 and S12). Therefore, we suggest thatJMJD1A alters the tumormicroenvironment by affecting angio-genesis and TAMs.
However, the exact role of JMJD1A in tumor progression andaggression has not been elucidated to date. JMJD1A inhibitionsignificantly decreased tumor angiogenesis and macrophageinfiltration into tumor tissues (Fig. 5C–E). Although Ang1 andAng2 conduct antagonistic as well as agonistic functions onthe regulation of angiogenesis (33), expression of several
Growth rich
Aggressive
Dormant
JMJD1AHypoxia&Starvation
Novel Anti cancer Therapy
Anti angiogenesisChemotherapy
ReccurenceResistance
Figure 7. Epigenetic modification of cancer cells resistant to hypoxia andnutrient starvation improves antiangiogenic therapy. Schematic diagramof targeting aggressive cancer cells that survived under DDS by theinhibition of DDS-responsive genes such as histone demethylaseJMJD1A.
0
500
1,000
1,500
2,000
2,500
3,000
0 5 10 15 20 25 30
0
500
1,000
1,500
2,000
2,500
0 5 10 15 20 25
Tum
or
volu
me (
mm
3)
Tum
or
volu
me (
mm
3)
Days after injection (d) Days after injection (d)
Placebo + scRNA
Sunitinib + scRNA
Sunitinib + siRNA
IgG + scRNA
Bevacizmab + scRNA
Bevacizmab + siRNA
siRNA
A B
anti-VEGF Sunitinib**
********** *
*********
Anti-VEGF
VEGFR inhibition
siJ
MJD
1A
siJ
MJD
1A
Figure 6. JMJD1A inhibition enhances antitumor effects of anti-VEGF and anti-VEGF receptor treatments using HeLa cells in vivo. A, JMJD1A inhibitionenhances effect of anti-VEGF antibody treatment (bevacizumab; 5 mg/kg/wk, i.p.). A one-way ANOVA was used to detect mean differences of tumorvolumes following 3 treatments for each timepoint (�,P < 0.05). B, JMJD1A inhibition enhances effect of VEGFRs inhibitor (sunitinib; 40mg/kg/d, administeredorally). A one-way ANOVA was used to detect mean differences of tumor volumes following 3 treatments for each time point (�, P < 0.05).
Osawa et al.
Cancer Res; 73(10) May 15, 2013 Cancer Research3026
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231

pro-angiogenic factors such as angiopoietins, FGFs. and HGFwas downregulated by JMJD1A inhibition within the tumortissues of HeLa andA673 cells (Fig. 5F, Supplementary Table S4and Supplementary Fig. S18). Moreover, JMJD1A was signifi-cantly upregulated at the angiogenic switch and the prere-fractory phase of antiangiogenesis (Fig. 4, Supplementary Fig.S10), suggesting the importance of JMJD1A at these stages.Strikingly, JMJD1A inhibition delayed tumor growth in com-bination with anti-VEGF (bevacizumab) and anti-VEGFRtreatment (sunitinib); possibly, by affecting the refractoryphase of the tumor against antiangiogenesis (Fig. 6, Supple-mentary Fig. S19), suggesting that modification of epigeneticregulators can influence both cancer cells and tumor-associ-ated cells.Recently, small-molecule inhibitors targeting epigenetic
modulators have garneredmuch attention in the developmentof cancer chemotherapy drugs. For example, DNAdemethylaseinhibitors, 5-azacytidine and 5-aza-20-deoxycytidine, common-ly used in clinical cancer therapy (34). Many inhibitors ofhistone modifiers such as Ezh2 are now in clinical trials. Thus,it is possible that epigenetic modification through inhibitionof JMJD1A can be a novel way to overcome the refractorinessof tumors to antiangiogenic therapy. On the basis of ourresults, therapies targeting cancer cells that are resistant tohypoxic and nutrient-starved tumor microenvironmentscan comprise an anticancer strategy for sensitizing cells anti-angiogenic treatments and preventing tumor recurrence andaggressiveness.
Disclosure of Potential Conflict of InterestM. Shibuya has Honoraria from Speakers Bureau as the symposium speaker
and is on the Consultant/Advisory Board as a research advisor. No potentialconflicts of interest were disclosed by the other authors.
Authors' ContributionsConception and design: T. Osawa, M. ShibuyaDevelopment of methodology: T. Osawa, R. Tsuchida, M. Muramatsu, TeppeiShimamura, F. WangAcquisition of data (provided animals, acquired managed patients,provided facilities, etc.): Y. Yuasa, H. Aburatani, T. Minami, T. KodamaAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): T. Osawa, M. Muramatsu, T. Shimamura, J. Suehiro,Y. Kanki, Y. Wada, S. MiyanoWriting, review, and/or revision of the manuscript: T. Osawa, R. Tsuchida,M. ShibuyaStudy supervision: M. Shibuya
AcknowledgmentsThe authors thank the members of the Department of Molecular Oncology,
Tokyo Dental and Medical University, and Laboratory for Systems Biology andMedicine, the RCAST, University of Tokyo. They also thank A. Izumi, K. Kaneki,M. Miura (LSBM, RCAST, University of Tokyo), Dr. A. Kamio (Genome ScienceDivision, RCAST, University of Tokyo), Dr. S. Hiratsuka and Y.Maru (Departmentof Pharmacology, Tokyo Women's Medical University), and Dr. S. Yamamoto(Department of Molecular and Medical Pharmacology, Graduate School ofMedicine and Pharmaceutical Sciences, University of Toyama) for helpfuldiscussions and supports.
Grant SupportThis work was supported by a Grant-in-Aid Special Project Research on
Cancer-Bioscience (17014020, M. Shibuya), Grant-in-Aid for Young Scientist(23701047, T. Osawa), and Grant-in-Aid for Scientific Research on InnovativeAreas (24116509, T. Osawa) from the Ministry of Education, Culture, Sports,Science and Technology of Japan, the Takeda Science Foundation (T. Osawa), theResearch Fellowships of the Japan Society for the Promotion of Science (JSPS) forYoung Scientists (22-40024, R. Tsuchida), and the "JSPS Funding Program forNext GenerationWorld-Leading Researchers, initiated by theCouncil for Scienceand Technology Policy (JS038, T. Minami).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicate thisfact.
Received August 14, 2012; revised January 23, 2013; accepted February 24, 2013;published OnlineFirst March 14, 2013.
References1. Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature
2005;438:967–74.2. Carmeliet P. Angiogenesis in health and disease. Nature Med
2003;9:653–60.3. Risaw W. Mechanism of angiogenesis. Nature 1997;386:671–4.4. ShibuyaM, Claesson-Welsh L. Signal transduction by VEGF receptors
in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res2006;312:549–60.
5. Folkman J. Angiogenesis: an organizing principle for drug discovery?Nat Rev Drug Discov 2007;6:273–86.
6. Paez-RibesM,Allen E,Hudock J, TakedaT,OkuyamaH,Vi~nals F. Anti-angiogenic therapy elicits malignant progression of tumors toincreased local invasion and distant metastasis. Cancer Cell 2009;15:220–31.
7. Ebos JML, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG,Kerbel RS. Accelerated metastasis after short-term treatment with apatient inhibitor of tumor angiogenesis. Cancer Cell 2009;15:232–9.
8. Osawa T, Muramatsu M, Watanabe M, Shibuya M. Hypoxia and lownutrition double stress induces aggressiveness in a murine model ofmelanoma. Cancer Sci 2009;100:844–51.
9. Osawa T, Tsuchida R, Muramatsu M, Yuasa Y, Shibuya M. Humanglioblastoma cells exposed to long-term hypoxia and nutrient starva-tion stimulated induction of secondary T cell leukemia in mice. BloodCancer J 2011;1:e6.
10. Osawa T, Muramatsu M, Wang F, Tsuchida R, Kodama T, Minami T,et al. Increased expression of histone demethylase JHDM1D under
nutrient starvation suppresses tumor growth via downregulatingangiogenesis. Proc Natl Acad Sci USA 2011;108:20725–9.
11. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progres-sion and metastasis. Cell 2010;141:39–51.
12. MuramatsuM, Yamamoto S, Osawa T, ShibuyaM. VEGFR-1 signalingpromotes mobilization of macrophage-linage cells from bone marrowand stimulates solid tumor growth. Cancer Res 2010;70:8211–21.
13. HiratsukaS,Goel S, KamounWS,MaruY, FukumuraD,DudaDG, et al.MMP9 induction by vascular endothelial growth factor receptor-1 isinvolved in lung-specific metastasis. Cancer Cell 2002;2:289–300.
14. WangF,OsawaT, TsuchidaR, YuasaY, ShibuyaM.Downregulation ofreceptor for activated C-kinase 1 (RACK1) suppresses tumor growthby inhibiting tumor cell proliferation and tumor-associated angiogen-esis. Cancer Sci 2011;102:2007–13.
15. Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibitionof vascular endothelial growth factor-induced angiogenesis sup-presses tumour growth in vivo. Nature 1993;362:841–4.
16. Jain RK. Normalization of tumor vasculature: an emerging concept inantiangiogenic therapy. Science 2005;307:58–62.
17. Shojaei F, Wu X, Qu X, KowanetzM, Yu L, TanM, et al. G-CSF initiatedmyeloid cell mobilization and angiogenesis mediate tumor refractori-ness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci USA2009;106:6742–7.
18. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance byevasion of antiangiogenic targeting of VEGF signaling in late-stagepancreatic islet tumors. Cancer Cell 2005;8:299–309.
Inhibition of JMJD1A Improves Antiangiogenesis
www.aacrjournals.org Cancer Res; 73(10) May 15, 2013 3027
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231

19. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, SampaolesiM, et al. Tie2 identifies a hematopoietic lineage of proangiogenicmonocytes required for tumor vessel formation and a mesenchymalpopulation of pericyte progenitors. Cancer Cell 2005;8:211–26.
20. Warburg O. On the origin of cancer cells. Science 1956;123:309–14.21. Gupta GP,Massague J. Cancermetastasis: building a framework. Cell
2006;127:679–95.22. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the
Warburg effect: the metabolic requirements of cell proliferation.Science 2009;324:1029–33.
23. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis?Nat Rev Cancer 2004;4:891–9.
24. DeBerardinis RJ, Thompson CB. Cellular metabolism and disease:what do metabolic outliers teach us? Cell 2012;148:1132–44.
25. Sokawa Y, Sokawa J, Kaziro Y. Role of rel gene in translationduring amino acid starvation in Escherichia coli. Nature 1974;249:59–62.
26. ShibuyaM, Kaziro Y. Studies on stringent control in a cell-free system.Regulation by guanosine-50-diphosphate-30-diphosphate of the syn-thesis of elongation factor Tu. J Biochem 1979;86:403–11.
27. Baylin SB, Jones PA. A decade of exploring the cancer epigenome -biological and translational implications. Nat Rev Cancer 2011;11:726–34.
28. Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M,Yoshida GJ, et al. Alternative splicing of CD44 mRNA by ESRP1enhances lung colonization of metastatic cancer cell. Nat Commun2012;3:883.
29. Okada Y, Scott G, Ray MK, Mishina Y, Zhang Y. Histone demethylaseJHDM2A is critical for Tnp1 and Prm1 transcription and spermato-genesis. Nature 2007;450:119–23.
30. Tateishi K, Okada Y, Kallin EM, Zhang Y. Role of Jhdm2a in regulatingmetabolic gene expression and obesity resistance. Nature 2009;458:757–61.
31. Krieg AJ, Rankin EB, Chan D, RazorenovaO, Fernandez S, Giaccia AJ.Regulation of the histone demethylase JMJD1A by hypoxia-induciblefactor 1 alpha enhances hypoxic gene expression and tumor growth.Mol Cell Biol 2010;30:344–53.
32. UemuraM, Yamamoto H, Takemasa I, Mimori K, Hemmi H,MizushimaT, et al. Jumonji domain containing 1A is a novel prognostic marker forcolorectal cancer: in vivo identification from hypoxic tumor cells. ClinCancer Res 2010;16:4636–46.
33. Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascularmorphogenesis and homeostasis through the angiopoietin–Tie sys-tem. Nat Rev Mol Cell Biol 2009;10:165–77.
34. Yoo PB, Jones PA. Epigenetic therapy of cancer: past, present andfuture. Nat Rev Drug Discov 2006;5:37–50.
Osawa et al.
Cancer Res; 73(10) May 15, 2013 Cancer Research3028
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231

2013;73:3019-3028. Published OnlineFirst March 14, 2013.Cancer Res Tsuyoshi Osawa, Rika Tsuchida, Masashi Muramatsu, et al. MacrophagesAnti-Angiogenic Therapy and Reduces Tumor-Associated Inhibition of Histone Demethylase JMJD1A Improves
Updated version
10.1158/0008-5472.CAN-12-3231doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2013/03/14/0008-5472.CAN-12-3231.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/73/10/3019.full#ref-list-1
This article cites 34 articles, 8 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/73/10/3019.full#related-urls
This article has been cited by 7 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/73/10/3019To request permission to re-use all or part of this article, use this link
on April 29, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 14, 2013; DOI: 10.1158/0008-5472.CAN-12-3231