improvingheterogeneouscatalyst stabilityforliquid ... · sccerbiosweet–the swiss competence...
TRANSCRIPT

582 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
doi102533chimia2015582 Chimia 69 (2015) 582ndash591 copy Schweizerische Chemische Gesellschaft
Correspondence Prof Dr J S LuterbacherLaboratory of Sustainable and Catalytic ProcessingInstitute of Chemical Sciences and EngineeringEacutecole Polytechnique Feacutedeacuterale de Lausanne (EPFL)CH-1015 Lausanne SwitzerlandE-mail jeremyluterbacherepflch
Improving Heterogeneous CatalystStability for Liquid-phase BiomassConversion and Reforming
Florent Heacuteroguel Bartosz Rozmysłowicz and Jeremy S Luterbacher
Abstract Biomass is a possible renewable alternative to fossil carbon sources Today many bio-resourcescan be converted to direct substitutes or suitable alternatives to fossil-based fuels and chemicals Howevercatalyst deactivation under the harsh often liquid-phase reaction conditions required for biomass treatment is amajor obstacle to developing processes that can compete with the petrochemical industry This review presentsrecently developed strategies to limit reversible and irreversible catalyst deactivation such as metal sintering andleaching metal poisoning and support collapse Methods aiming to increase catalyst lifetime include passivationof low-stability atoms by overcoating creation of microenvironments hostile to poisons improvement of metalstability or reduction of deactivation by process engineering
Keywords Biomass conversion middot Catalyst stabilization middot Deactivation middot Heterogeneous catalysis middotProcess engineering
1 Introduction
Production of fuels and chemicals inthe 20th century has largely been focusedon conversion of fossil resources Crudeoil was easily available at a relatively lowextraction cost and constituted a feedstockcontaining molecules of similar chemicalproperties[1] This led to the developmentof robust catalytic processes in the pet-rochemical industry Hydrocarbon-basedfeeds with low oxygen content can be eas-ily converted to transportation fuels orcatalytically cracked to elemental chemi-cal building blocks and selectively oxi-dized to valuable chemicals Most of thoseprocesses are solvent-free and are oftenconducted in the gas phase at elevatedtemperatures[2] These features have madeit possible for petrochemical conversion tooperate at very large scale and to be veryprofitable[3]
However despite the presence of thesewell-established and robust commercialprocesses for producing carbon-basedproducts there is increasing interest infinding renewable carbon sources to re-place fossil resources The geological tim-escales at which these fossil resources arecreated are much too long with respect tothe rates at which they are consumed Forthis reason resource depletion appears in-evitable In addition the global communityis increasingly acknowledging the effect offossil-based carbon emissions to the atmo-sphere which have severe consequenceswith respect to climate change This hasled to new regulations[4] forcing usage ofbiofuels and a general increased interestin biomass conversion in the past two de-cades[5]
Biomass consists of three main groupsof polymeric compounds cellulose andhemicellulose formed by carbohydratemonomers and lignin which is a complexpolymer with aromatic functionalities[6]We have recently reviewed the targeteddeconstruction of lignocellulosic mate-rial and the initial biomass deconstructionsteps that generally produce the carbohy-drate monomers of the aforementionedpolysaccharides (mainly glucose and xy-lose) or the dehydration products of thesemonomers (including furans or levulinicacid)[7] Lignin deconstruction processesare much less mature and most explor-atory processes include the hydrogenoly-sis or oxidation to syringyl and guaicylderivatives[78] These main routes for thedeconstruction of biomass into platformmolecules usually occur in water usingvarious combinations of acids[910] bas-
es[11] solvents[12ndash14] or ionic liquids[1516]All these methods typically produce sol-uble biomass-derived molecules at fairlylow concentrations (lt20 gL) in aqueousor solvent solutions[7] though some carbo-hydrate solutions can reach 100ndash220 gLusing solvent pretreatment and enzymatichydrolysis[17ndash19] or solvent systems[1314]Therefore biomass-derived platform mol-ecules are often produced in solution atfairly low concentrations
Biomass-derived carbohydrates canalso be converted to many other moleculesby fermentation Though combining chem-ical catalysis with biological conversioncan dramatically increase the number ofmolecules that can be produced from bio-mass these molecules also tend to be pro-duced in fairly dilute aqueous streams[20]In addition biological processes tend tointroduce biogenic impurities includingproteins and inorganic salts that can poisonchemical catalysts[20] Finally even pyroly-sis oils ndash a third major source of biomass-derived molecules ndash which are much moreconcentrated ndash usually contain significantamounts of water and nonvolatile com-pounds Therefore platform molecules de-rived from biomass are often produced indilute liquid and often aqueous streamsTheir high oxygen content makes themnonvolatile and vulnerable to degradationTherefore these molecules are usually fur-ther upgraded in the aqueous phase[21ndash24]with reaction operation conditions limitedto low temperatures by the low stability ofthe reactants especially compared to pet-rochemical processes[25] (Fig 1)
While high catalytic activity is an im-portant parameter for developing more
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 583
a regeneration cycle regeneration cyclesincrease process costs and can themselvesbe problematic by modifying or destroyingsupports notably during high temperaturecalcination In the third part we discussmethods to improve stability of the sup-port as opposed to just the nanoparticlesFinally we discuss methods besides cata-lyst design including tuning the processparameters to increase catalyst stabilityand process lifetime The main methodsreviewed here as well as their commonali-ties are summarized in Table 1
2 Irreversible Deactivation viaSintering andor Leaching ofSupported NPs
Sintering is one of the main deac-tivation pathways for supported metalnanoparticles especially for catalytic pro-cesses performed at high temperaturesUnderstanding sintering is a prerequisitefor the development of stable materials andhas been the subject of several reviews[30]Sintering can occur via two distinct mech-anisms i) particle migration and coales-cence where particles statically move onthe support surface and coalesce to formlarger particles and ii) Ostwald ripeningwhere particles size and geometry evolveto minimize free energy via adatom mi-gration In situ studies and Monte-Carlosimulations have helped to clarify the con-tribution of these two mechanisms Initialrapid catalyst deactivation due to loss ofmetal surface area is attributed to Ostwaldripening while particle migration and co-alescence becomes preponderant in a sec-ond phase once small metal particles havedisappeared[47]
Leaching is also a major issue for liq-uid phase processes as it leads to both adecrease in activity and pollution of theproduct stream Leaching occurs by ex-traction of individual metal atoms fromparticles into the solution via formation offree metal ions
Many efforts have been dedicatedto the stabilization of supported metalnanoparticles[48] and recent techniquesinvolving deposition of a protective layerto prevent sintering and leaching of metalnanoparticles appear especially promis-ing Overcoating was first reported byLu et al for the stabilization of PdAl
2O
3during high-temperature dehydrogenationof ethane to ethylene[39] More recentlyOrsquoNeill et al used the same strategy for thestabilization of alumina-supported cop-per nanoparticles by the deposition of anAl
2O
3overcoat[33] Stabilization of copper
was especially noteworthy because whileprecious metal catalysts are generally pre-ferred due to their great stability base met-als such as copper suffer from irreversible
competitive processes catalyst lifetime isof prime importance under the harsh oftenhydrothermal conditions used for biomassconversion processes Liquid-phase condi-tions can often increase the reactivity ofcatalyst surfaces Furthermore the waterdissociation equilibrium is modified underhydrothermal conditions during which theionization constant increases from 10ndash14 at25 degC to 10ndash11 at 250 degC[26] This results inhigher ion concentrations that can perturbcatalyst stability[27] In addition as men-tioned above biomass-derived feeds oftencontain high quantities of biogenic impuri-ties that can act as catalyst poisons such assulfur- or nitrogen-containing compoundswhich can deactivate catalysts This prob-lem is often even more pronounced whena fermentation step is used Given the ex-tent to which biomass-derived feeds dif-fer from petroleum-based feeds shiftingcatalyst properties and robustness to meetthese new demands is an important area ofresearch This review aims to address themain developments in the design of morestable catalysts for biomass conversion andreforming reactions (Fig 2)
Supported metal particles are amongthe main catalyst families and often dis-play a limited stability independent of
their preparation method[2829] The maindeactivation paths include sintering whichleads to a decrease in metal dispersion[30]leaching ie metal dissolution and pollu-tion of surface metal with byproducts ascoke These phenomena dramatically de-crease the amount of active metallic sitesin the catalysts by reducing the ratio ofactive surface sites to inactive bulk metalin the case of sintering removing activesites by leaching or blocking active sitesin the case of poisoning This is especiallyproblematic because the metallic particlesare often the most expensive element of thecatalyst
The first section of this review reportson the major advances in catalyst designthat aim to stabilize supported nanopar-ticles against sintering and leachingSintering and leaching reduce the amountof active sites and are irreversible deactiva-tion pathways ie initial properties cannotbe recovered by regeneration such as cal-cination or reduction In the second part ofthe review we address catalyst poisoningand methods aiming to reduce it duringwhich an impurity or reaction byproduct(eg coke) blocks the active site Eventhough this is a reversible deactivationsince the poison can be removed during
0 100 200 300 400 500 600 700 800 9000
50
100
150
200
T (degC)
p(a
tm)
PetrochemicalProcessesH
ydro
gena
tion
Hyd
roge
noly
sis
Aqueous-Phase
Reforming
DehydratationHydrolysis
IsomerizationAldol Condensation
Oxidation
Aqueous phase Vapor
Vapor-liquidequilibrium
Fig 1 Approximatereaction conditionranges for catalyticbiomass conversionversus those forpetroleum processesAdapted fromref [25]
leaching
sintering
support dissolution
cokedeposition
overcoating
time on stream
activity
time on stream
activity
severe biomassconversion conditions
alloying
support doping
stabilization strategiesprocess engineering
classical catalystdeactivation
limited deactivationstable metal surface
hydrothermal stability
Fig 2 Main deactivation pathways for solid catalysts for conversion of biomass and major cata-lysts stabilization concepts
584 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
area increase to 39 m2g and re-exposureof the Cu0 sites (23 vs 86 mmolg beforecoating) Stability of the catalysts was in-vestigated in the hydrogenation of furfuralto furfuryl alcohol in 1-butanol After 15days in stream the catalyst had deactivat-ed significantly and was regenerated by insitu calcination and reduction and catalyticactivity was further tested (Fig 4c and d)After four successive cycles initial activitycould not be recovered with the uncoatedcatalyst (Fig 4c) which showed irrevers-ible deactivation In contrast the over-coated CuAl
2O
3led to identical turnover
frequencies over the course of several reac-tionregeneration cycles in 1-butanol (Fig4c) and water (Fig 4d) Irreversible deac-tivation was attributed to the decrease ofmetal surface area due to metal sintering asobserved by TEM which showed a notableparticle size increase for the uncoated cata-lyst (Fig 4b) Moreover operando X-rayabsorption spectroscopy studies confirmedthat the overcoat provided stabilization bylimiting sintering most likely because of
deactivation through leaching and sinteringunder severe liquid-phase conditions Thelatter catalyst was prepared by first form-ing Al
2O
3-supported copper oxide parti-
cles by incipient wetness impregnation ofCu(NO
3)2followed by reduction and pas-
sivation The overcoat was then depositedby 45 successive cycles of Atomic LayerDeposition (ALD) Among various cata-lytic design techniques[49] ALD has beenextensively used to tune properties at themolecular level[50]ALD allows depositionof thin layers (lt 100 nm) with high levelof conformality In comparison state-of-the-art catalyst preparation methods thatinclude wet impregnation and depositionprecipitation can lead to a mixture of siteswhich result in side reactions Thereforedespite the high price of ALD equipmentand the limits of its applicability for indus-trial-scale preparation ALD is a promis-ing technique for catalyst modification andstabilization In the case of Al
2O
3-ALD
deposition was achieved under vacuum at200 degC by alternating cycles consisting of
exposure of the substrate to an aluminumprecursor (trimethyl aluminium or TMA)and water as depicted on Fig 3b
Conventional ALD reactors designedfor wafers cannot be employed for ALDon powders due to their high surface ar-ea Fluidized bed reactors equipped withmechanical stirring or rotating chamberswhich allow long contact times with pre-cursors (gt 30 s compared with 30 ms ex-posure for wafers substrates) were usedin this and other work to uniformly coatsupported catalyst and other high surfacematerials while preventing particle ag-gregation[51ndash53] When treating the Al
2O
3-
supported copper particles a continuousovercoat was formed as shown by TEMpictures (Fig 4a) In addition both a sig-nificant surface area decrease (from 190 to16 m2g) and the absence of accessible Cu0
sites detected by N2O chemisorption after
reduction were observed[33] Subsequentcalcination at 700 degC under air flow al-lowed for pore formation within the over-coat (Fig 3a) as attested by the surface
Table 1 Summary of the main catalyst stabilization strategies
Deactivation pathway Catalyst Strategies Method Reaction ref
Metal leaching andsintering
CuAl2O
3
Overcoat to stabilizeundercoordinated atoms
Al2O
3ALD
Furfural hydrogenation [31ndash33]
CuCr2O
4-
CuO Furfural hydrogenation [3435]
CoTiO2
TiO2ALD Aqueous phase hydro-
genation [36]
NiCNF Avoid acidic conditionsIncrease pH byaddition of KOHand hydrogen
Ethylene glycol conversion [37]
Metal leaching andsinteringCoke deposition
CoHZSM-5 Embedment into supportstructure
Growth of HZSM-5onto Co
3O
3SiO
2
Conversion of levulinic acidto valeric biofuel [38]
Coke poisoningCuAl
2O
3
Mixed Al2O
3-MgO
xovercoat to decreasesupport acidy
ALD Furfural hydrogenation [31]
PdAl2O
3
Overcoat to passivateundercoordinated atoms Al
2O
3ALD Dehydrogenation of ethane
to ethylene [39]
Poisoning by aminoacids PdAuAl
2O
3
Microenvironment hostileto biogenic impurities
Polymer cross-linking
Hydrogenation of triaceticacid lactone [40]
Support dissolution
PdSiO2
PdAl2O
3 Carbon overcoat toincrease hydrophobicity
Sucrose pyrolysisto form thin carbonfilm
Hydrogenation of acetylene [41]
PtAl2O
3RuAl
2O
3
Formation ofgraphitic carbon byCLD
Ethylene glycol reforming [42]
HY zeolite Hydrophobic micro-environment
Grafting silanefunctionalities Refining of pyrolysis oil [43]
Pdniobia Formation of mixed oxide Doping by additionof 5 SiO
2 GVL to pentanoic acid[44]
PdSBAsilica
Deposition of hydrother-mally stable overcoat Niobia ALD [45]
Coke deposition anddealumination
MicroporousHZSM-5 Change reaction media Methanol co-feed Bio-oil to hydrocarbons [46]
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 585
a preferential interaction of the overcoatwith undercoordinated Cu atomswhich aremore exposed to sintering and leaching[32]However access to Cu sites decreasedover time on stream due to coke deposit inthe small pores in the overcoat leading todeactivation reversible upon calcinationFinally the overcoat did not influence se-lectivity as furfuryl alcohol remained theonly hydrogenation product that was ob-served
Al2O
3overcoating by ALD could also
be used to increase the stability in gas-phase reactions Zhang et al reported en-hanced stability for the hydrogenation offurfural by ALD overcoating using copperchromite catalysts in the gas phase[3435]Similarly to the CuAl
2O
3system described
above the overcoat preferentially blockedundercoordinated Cu atoms which werepresent in the form of step edges and sur-face defects The overcoat led to increasedstability by decreasing three deactivationpathways sintering of Cu coke formationand coverage of active Cu sites by chromitemigration In addition secondary reactionssuch as further dehydration of furfuryl al-cohol to 2-methyl furan were disfavoredwhen covering undercoordinated atoms re-sulting in higher furfuryl alcohol selectiv-ity ThinALD overcoat (10 cycles) showed
high activity but low stability while thickALD overcoat (45 cycles) provided highstability but lower activities The high ac-tivity of coated materials was attributed toan increased reduction temperature whichdecreased the activation energy of furfuralon Cu thanks to a modified ratio of Cu0Cu+ and Cu2+species Prior FTIR studies ofadsorbed CO on Pd catalysts ruled out theexistence of electronic effects between theALD layer and metal nanoparticles[3954]
Rather the authors suggested that the over-coat influences the transport of gas to andfrom the copper surface
Besides Al2O
3 successful TiO
2ALD
overcoating was used to stabilize CoTiO
2during aqueous phase hydrogenation
(APH) reactions[36] The analogous Al2O
3
Coγ-Al2O
3was inactive for the APH of
furfural due to the formation of cobalt alu-minate after calcination treatment whichwas unreducible up to 800 degC Hencethe authors investigated the performancesof TiO
2CoTiO
2prepared by ALD from
TiCl4on TiO
2-supported Co particles H
2-
temperature programmed reduction (TPR)revealed that switching fromAl
2O
3to TiO
2allowed reduction of Co at 600 degC lead-ing to furfural conversionWhile the parentcatalyst suffered fromcobalt leaching (10Co lost over 2100minutes) the metal load-ing of overcoated material remained con-stant even after regeneration Finally TEMstudy showed that the overcoat prevented
Fig 3 a) Strategy for the synthesis of stable CuAl2O3 catalysts formation of Al2O3 supportedcopper particles overcoating by ALD and cracking of overcoat by calcination b) Mechanismof Al2O3 ALD on Al2O3 (red O purple Al black C blue H) c) Embedment of Co onto HZSM-5matrix Reproduced with permission from ref [38] Copyright 2014 American Chemical Society
c)
d)
a)
b)
Fig 4 STEM image of 45ALDCuAl2O3 (a) Particle size distribution of fresh and used catalysts(b) TOF in furfural hydrogenation for 45ALDCuAl2O3 (solid) and CuAl2O3 (hollow) in 1-butanol (c)and water (d) (fresh one regeneration O two regenerations loz three regenerations Δ) Adaptedwith permission from Wiley-VCH Verlag from ref [33]
586 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
the nanoparticles from sintering whereassintering was observed during calcinationin the absence of overcoat
The methods above stabilized metalnanoparticles by adding a separate protec-tive layer over selected parts of the supportand metal particle surface Sintering andleaching of base metal catalysts could alsobe limited by embedding the metal into astructure[55] For instance Co nanoparti-cles were loaded into an HZSM-5 zeoliteproviding a bifunctional catalyst for theconversion of levulinic acid into valericbiofuel where cobalt performed hydroge-nation while the acidic support catalyzedγ-valerolactone (GVL) ring opening (Fig3c)[38] In contrast to the incipient wetnessimpregnation approach cobalt was em-bedded into HZSM-5 by nucleation andgrowth of HZSM-5 crystals onto Co
3O
4
SiO2whereas SiO
2was progressively dis-
solved thanks to the basicity of the systemH
2-TPR showed a lower temperature for
the reduction of cobalt oxide for the im-pregnated (CoHZSM-5) compared to theembedded (CoHZSM-5) catalyst dem-onstrating a higher metal-support interac-tion for the latter The strong interactionhad a limited influence on the Co acces-sibility as demonstrated by a similar H
2up-
take for both materials Structural charac-terization was performed on CoHZSM-5and CoHZSM-5 before and after cataly-sis to identify stability differences FirstTEM revealed a stable Co NPs size forCoHZSM-5 while for CoHZSM-5 theaverage Co NP size increased from 25 to32 nm Second coke formation was onlydetected for CoHZSM-5 by thermogravi-metric analysis (TGA) as well as N
2physi-
sorption (decrease of surface area and porevolume) Finally XRD patterns showedthat both materials maintained their crys-tal structure
Similar to Cu and Co supported Niparticles are relevant systems for the low-temperature conversion of biomass-de-rived compounds ndash notably for lignin hy-drogenolysis[56ndash58] However as for manybase metals the Ni particles suffer fromsintering Nanoconfinement of Ni par-ticles by self-assembly of Ni atoms insidethe nanoribbons of hydrotalcite-derivedmixed oxide provided increased catalyticstability for methane steam reformingcompared to Ni supported on commercialsupport surface[59] Metal embedment hasalso proven to be an efficient stabilizationmethod for lactic acid production fromglycerol Morales et al showed that MFIconstituted a stabilizing matrix for Sn andsignificantly decreased leaching of said Snwhile slowing down deactivation in com-parison to bifunctional carbon-silica com-posites and BEA zeolites[60]
Above we discussed various methodsdemonstrating that controlling local nano-
structures around the active metal catalystcould increase stability Abdelrahman etal recently showed that a simple judiciouschoice of the support could limit particlesintering during the hydrogenation oflevulinic acid[61] In fact fast deactivationwas observed for ruthenium particles sup-ported on SiO
2while increased stability
was achieved by using carbon and TiO2as
supports Al2O
3was the support that led to
the most stable catalyst which the authorsattributed to its lowest electronegativity
Another strategy is to tune the com-position of the metal itself Formation ofalloys for improved catalyst performanceshas often been used including in reactionsrelevant to biomass conversion[62] In thecase of SBA-15 the use of supported Ru-Fe catalysts led to stable conversion andselectivities over 300 h with no aggrega-tion of bimetallic particles no structurecollapse and improved selectivity for hy-drogenation of acetic acid to ethanol com-pared to monometallic catalysts[63]
Finally while many efforts have beendedicated to the control of individual char-acteristics of supported metal nanopar-ticles and their immediate environmentPrieto et al showed that controlling thecollective properties of supported metalnanoparticles led to increased stability[64]By achieving near-maximum interpar-ticle spacing for silica-supported coppernanoparticles deactivation during metha-nol synthesis was limited with deactivationrates which were an order of magnitudelower than those observed with a referencecatalyst that had a non-uniform spatial dis-tribution of particles
3 Poisoning of the Active Sites
In addition to the reduction of the num-ber of active sites by sintering and leach-ing competitive adsorption of undesirable
compounds such as coke or sulfur is a ma-jor catalyst deactivation pathway becauseit leads to the blocking of active sitesSome poisons including coke can oftenbe removed by regenerating the catalystunder oxidative or reductive media to formvolatile species However these methodsare sometimes incompatible with certainsupports such as carbon which has lowthermal stability and will degrade duringthese regeneration treatments In additionprocesses not requiring periodic catalystregeneration are generally favored foreconomic and practical reasons Henceseveral paths have been explored to limitcatalyst poisoning
Catalyst poisoning is often describedas especially problematic in the contextof fermentation-derived media which hasbeen proposed as an important source ofbiomass-derived platform molecules[65] Inparticular sulfur-containing amino acidsseem to be particularly detrimental to cat-alyst activity and are commonly found infermentation broths[65] To alleviate deacti-vation by coke deposition of alumina-sup-ported Pd catalysts during hydrogenationof fermentation-derived triacetic acid lac-tone (TAL) Schwartz et al formed equi-molar bimetallic PdAu particles Howeverthe alloy still suffered from sulfur poison-ing similar to the parent monometalliccatalyst when co-feeding model biogenicimpurities such as methionine The au-thors reasoned that a microenvironmentunfavorable to polar species would limitdeactivation by biogenic impurities whilethe reaction with the less polar substratewould still go forward[40] TAL is highlysoluble in alcohols whereas amino acidsare not hence a poly(vinyl alcohol) (PVA)overcoat was selected to form a polymericpseudo-solvent microenvironment (Fig5a) The PVA-overcoated PdAuAl
2O
3catalyst displayed a deactivation rateof 002 hndash1 vs 012 hndash1 for the parent Pd
CuAl2O3sintering and leaching
coke deposition
Al2O3CuAl2O3low sintering and leaching
coke deposition
MgOxCuAl2O3sintering and leachinglow coke deposition
Al2O3MgOxAl2O3CuAl2O3low sintering and leaching
low coke deposition
OH OH OH
OH OH OH OH
OH
OO
OH
S OH
O
NH2
substrate poison
a) b)
Fig 5 a) Creation of microenvironment unfavorable to poisons Adapted with permission fromWiley-VCH Verlag from ref [40] b) Stabilization of Cu(black)Al2O3 via ALD of Al2O3 (violet) andMgOx (blue)
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 587
Al2O
3for initial conversions between 50
and 70 Stabilization by PVA was alsohighly efficient when using TAL producedby fermentation without any feed purifi-cation Comparison of 13C NMR spectraof 13C-enriched methionine adsorbed onPVA-covered and non-covered PdAl
2O
3showed that the polymer acts as a lsquosolidsolventrsquo by limiting methionine adsorptionon Pd
The ALD overcoat strategy describedin the previous section for metal sinteringand leaching prevention has also provenuseful for limiting coke deposition Lu etal reported low reversible deactivation ofovercoated PdAl
2O
3for the dehydroge-
nation of ethane to ethylene at 650 degC[39]After using 45ALDcycles to form anAl
2O
3thin film the amount of coke deposited asdetermined by thermogravimetric analy-sis was more than 16 times lower than theamount measured for the parent PdAl
2O
3catalyst The reduction of the amount ofcoke formed was attributed to the blockingof the low-coordination Pd surface siteswhich were preferentially coated In factthe later sites favored CndashC bond scissionand hydrogen stripping resulting in the un-desirable formation of coke CH
4 CO and
CO2 Hence in addition to a lower catalyst
deactivation coating led to higher ethyleneyields
The Al2O
3overcoated CuAl
2O
3de-
scribed earlier suffered from reversiblecoke deposition In a separate study theauthors managed to limit this phenomenonby finely tuning the overcoat In addition totheAl
2O
3overcoat which provided particle
stabilization basic MgOxwas deposited to
reduce the overcoatrsquos acidity which wasassumed to catalyze the formation of coke(Fig 5b)[31] Interestingly pure MgO
xdid
not provide stabilization against sinteringThe local structure of the active site can
also reduce poisoning by affecting localmass transfer To suppress deactivation byfouling due to coke deposition during theconversion of propanal to hydrocarbons onAl-MFI Luo et al used an open mesopo-rous structure based on nanosheets to limitthe effects of fouling[66] This strategy re-sulted in a five-fold decrease of deactiva-tion rates In addition to an increase in thenumber of pore openings improved sta-bility was attributed to the short diffusionpath for reagents in the single unit-cell-thick nanosheet material which limitedcoke formation that was favored by longercontact times with the reaction products
4 Increasing Support Stability inHydrothermal Conditions
In addition to deactivation due to metalleaching and sintering loss of surface areaand pore collapsing in oxide supports is a
major source of irreversible deactivationespecially in aqueous phase conditionsFor biomass conversion processes whichoften require liquid phase andor aqueousmedia at low pH hydrothermal stabilityis a major concern Metal oxides are of-ten used as supports due to their thermalstability and high surface area Howeverthey tend to have low stability under theaforementioned conditions[27] Such ox-ides can sometimes be substituted by morestable support such as carbon but someprocesses require specific surface func-tionalities such as the acidic sites presenton alumina In addition contrary to car-bon most oxides have excellent thermalstability which is necessary during certaincatalyst regeneration processes Prominentsupports such as alumina and silica-basedmaterials undergo dissolution by hydroly-sis of aluminoxane and siloxane bonds Inaddition to loss of surface area hydrolysisof oxide network can lead to modificationof chemical composition For instance hy-drolysis of the Si-O-Al bond in zeolites fa-vors dealumination This section describesthe main techniques used to stabilize metaloxides supports in the area of biomass con-version
Formation of mixed oxides or supportdoping can be used to limit this phenome-non For instance Pham et al reported thatthe stability of Pdniobia during transfor-mation of γ-valerolactone (GVL) to pen-tanoic acid could be improved by additionof 5 SiO
2into niobia[44] Niobia could
not be entirely substituted by silica be-cause acid sites are required for GVL ringopening in addition to metal sites for thehydrogenation reaction However the ad-dition of a small portion of SiO
2improves
stability while retaining activity The com-parison of the N
2physisorption isotherms
of a Pdniobia HY commercial referencecatalyst before and after the GVL reactionshowed a dramatic surface area loss from150 to 8 m2g while the porous structurewas affected to a lesser extent for PdNb-Sioxide with a decrease from 81 to 31 m2gSimilarly HAADF-STEM and HRTEMrevealed a lower Pd particle growth uponreaction for the stabilized system than forthe PdHY reference catalyst SimilarlyMazumder et al reported the promotionof Niγ-Al
2O
3by 5 wt La
2O
3leading to
dramatic improvement of activity and sta-bility for the catalytic gasification of glu-cose compared to the parent catalyst whichsuffered from metal sintering[67]
The stabilizing oxide can also be in-troduced by ALD similar to the thin over-coats deposited for the stabilization ofsupported metal particles as described inSection 2 For instance hydrothermallystable Nb-Si oxide catalysts were preparedusing ALD of niobia within the pores ofmesoporous silica SBA-15 using between10 and 30 cycles which alternated expo-sure of the Nb(OCH
2CH
3)4precursor and
H2O to the SBA-15 surface (Fig 6a)[45]
The consumption of silanol group on thesurface was complete after 19 cycles as
a)
b)
c)
Fig 6 a) Strategyfor the synthesis ofhydrothermally stablehighly ordered niobiamesoporous materialsby ALD Reproducedwith permission fromref [45] Copyright2011 AmericanChemical Societyb) Strategy for theformation of niobiacarbon compositecatalysts Adaptedwith permission ofElsevier from ref [68]c) Deposition of gra-phitic carbon layer onalumina powder andpellets by chemicallayer deposition fromCH4 Reproducedwith permission fromWiley-VCH Verlagfrom ref [42]
588 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
demonstrated by the disappearance of theSiOH band at 3735 cmndash1 in the FTIR spec-tra Hydrated amorphous niobia was iden-tified as a major species by Raman spec-troscopy for samples presenting 10 19 and30 ALD layers N
2physisorption showed
a decrease of surface area pore volumeand pore diameter with increasing niobialoadings The presence of acid sites formedupon niobia deposition was verified byNH
3-TPD and hydrothermal stability was
tested by treating the materials in liquidwater for 12 h at 200 degC and 28 bar argonThe structure of the coated materials aftertreatment was confirmed by X-ray diffrac-tion with the persistence of the hexagonalmesoporous structure pattern while the lat-ter pattern was suppressed after hydrother-mal treatment of the parent SBA-15 TEManalysis confirmed the stability differencesobserved by XRD A bifunctional catalystfor the transformation of GVL to pentanoicacid was also formed by subsequent depo-sition of Pd nanoparticles As for the sup-port this catalyst showed higher stabilitythan the conventional PdHY-340
Improved hydrothermal stability ofa niobia-supported Pd catalyst was alsoachieved by deposition of the latter ona more stable surface such as carbonHighly dispersed spherical particles of aniobiacarbon composite were formed bythe reaction of d-glucose and ammoniumniobium oxalate in an autoclave at 200degC (pH was controlled by urea decom-position) followed by pyrolysis at 400degC under N
2(Fig 6b)[68] Hydrothermal
stability was investigated during liquid-phase butanol dehydration at 240 degC and51 bar While initial activity of the com-posite bifunctional catalyst was lower thanthe commercial HY-340 catalyst the lat-ter quickly deactivated during the first 20h of reaction while the modified catalystsremained very stable and surpassed HY-340 after 8 h Interestingly the analogousmaterial obtained by incipient wetness im-pregnation of niobia led to the formation oflarge niobia crystallites underscoring theimportance of strong interaction betweenniobia and carbon[69] Finally bifunctionalcatalysts were prepared by incipient wet-ness impregnation of Pd(NO
3)2and tested
during the conversion of GVL to pentanoicacid leading to improved hydrothermalstability for the niobiacarbon compositecatalysts just like the metal-free system
For silica and alumina improved hy-drothermal stability above 100 degC wasachieved by deposition of a thin film of car-bon formed from simple sugars Pham et aldeposited carbon by pyrolysis of adsorbedsucrose at 400 degC under N
2flow on SBA-
15 silica gel and fumed alumina leadingto improved stability after treatment in liq-uid water at 100 degC for 12 h[41] MoreoverPd was deposited on the carbon-coated
oxides to test their ability as supports forthe catalytic hydrogenation of acetylenein the presence of excess ethylene For acomparable Pd particle size (1ndash15 nm)the authors observed formation of ethaneat high acetylene conversion on both SiO
2andAl
2O
3 while carbon-coating prevented
over-hydrogenation to ethane The FTIRstudy showed a low hydration potential forthe coated materials compared to SiO
2and
Al2O
3 Therefore the higher selectivity to
ethylene for the coated materials was at-tributed to their increased hydrophobicity
More recently Xiong et al reported onthe deposition of a graphitic carbon layerover alumina by chemical vapor deposition(CVD) from CH
4(Fig 6c)[42] The graphit-
ic carbon layer provided higher stabilitythan the carbon deposited from pyrolysisof sugars as demonstrated by the higherstability achieved under flowing air (500vs 250 degC) As monitored by the growthof the characteristic graphitic peak at 24deg2θ on the XRD pattern the amount of de-posited carbon was controlled by the CH
4deposition time (from 05 to 6 h) whereasother peaks attributed to γ-Al
2O
3were un-
touched TEM and energy-filtered TEM at-tested that the carbon coating was uniformon the oxide support Raman spectros-copy mapping confirmed that depositionwas uniform over the entire surface of thespheres Furthermore the relatively highratio (071) between the peak intensitiesfor G and D bands (at 1590 and 1350 cmndash1respectively) in the Raman spectrum of gc-Al
2O
3revealed a high degree of graphitiza-
tion on the entire surface No such featureswere observed for blank alumina and pyro-lyzed carbon on alumina
The hydrothermal stability of the cata-lyst was investigated using a treatmentunder static conditions at 220 degC underautogenic pressure in an autoclave usingwater After treatment the XRD pattern ofγ-Al
2O
3displayed the well-defined diffrac-
tion peaks of the boehmite phase attestingto a reaction with water Simultaneouslyno significant change was observed forgraphitic-carbon covered γ-Al
2O
3(gc-
Al2O
3) Improved stability for gc-Al
2O
3was confirmed by N
2physisorption results
which showed that no significant decreaseof surface area pore volume and poresize occurred during hydrothermal treat-ment while γ-Al
2O
3saw its surface area
decrease by 80 during the same treat-ment Stability of gc-Al
2O
3as a support
was tested in the aqueous phase reforming(APR) of ethylene glycol at 250 degC and70 bar after deposition of platinum par-ticles Conversion using Ptgc-Al
2O
3was
twice as high as for Ptγ-Al2O
3 This result
was consistent with the leached Al and Ptdetected in the liquid phase after reactionby ICP-OES for the uncoated materialIn addition to full conversion of γ-Al
2O
3
to boehmite observed by XRD STEMshowed Pt particle size increase for Ptγ-Al
2O
3 Hence deactivation of Ptγ-Al
2O
3was also attributed to Pt particle sinteringin addition to leaching whereas the use ofgraphitic carbon overcoat prevented bothphenomena under APR conditions Thisapproach was also successful when usingRugc-Al
2O
3for lactic acid hydrogenation
Overall graphitic carbon layers depositedthrough vapor phase route provided higherhydrothermal stability than pyrolytic car-bon-coatings and seems to be a promisingstabilization method for a wide range ofapplications
In addition to the deposition of thinfilm precise surface functionalization canalso be used to improve catalyst stabilityFor instance the generation of hydropho-bic surfaces by grafting of octadecyltri-chlorosilane groups onHY zeolite prevent-ed the support structure from collapsingduring stability tests which mimicked theupgrading of biomass-derived liquid com-pounds in biphasic systems at 200 degC[43]Stabilization was attributed to a limitedcontact between the zeolitersquos hydrophobicsurface and the aqueous phase The sameauthors also reported a systematic studywith various alkylsilanes from C
2to C
18[70]
Hydrothermal stability of zeolites hasbeen studied in great detail as zeolites arethemost widely used solid acid catalysts[71]and are often proposed for biomass con-version[7273] As shown by detailed char-acterization using 27Al and 29Si solid-stateNMR and NH
3-TPD siloxane bridge hy-
drolysis was the main deactivation routein the presence of hot liquid water whilezeolites treated under steam suffered fromdealumination[74] After a treatment in hotwater at 200 degC mimicking APR condi-tions ZSM-5 was stable independent ofits SiAl ratio while degradation of zeoliteY increased with increasing SiAl ratioas opposed to the better stability of zeo-lites with higher SiAl in the presence ofsteam Finally Al-rich zeolite Y could bestabilized by incorporation of lanthanumcations[74]
Recently Gardner et al reported thattesting the hydrothermal stability of zeo-lites in distilled water was not completelyrelevant to biomass conversion reactionconditions[75] In fact dry biomass con-tains about 15 wt of inorganic saltssuch as NaCl and the dissolution rate ofmetal oxides could be dramatically en-hanced by cations in solution[76] Catalytictests showed that the presence of NaCl hadno influence on glucose conversion duringthe liquid-phase transformation of glucoseto HMF on ZSM-5 However zeolite hy-drothermal stability was significantly per-turbed by the presence of NaCl leading tothe hydrolytic attack of extra-frameworkspecies andor defects sites Interestingly
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 589
leached Al species were active in the con-version of glucose to fructose
Together with metal particles zeolitesconstitute an important class of bifunc-tional catalysts While studies mentionedabove aimed at adding a stabilizing ele-ment to an existing material Ennaert et alassessed the stability of Ruzeolite H-USYin hot liquid water for the hydrolytic hy-drogenation of cellulose using variousSiAl ratios in order to select the optimalzeolite composition[77] They identifiedthat the lattice was stabilized by frame-work aluminum atoms which preventedhydrolysis of framework bonds while theextra-framework aluminum atoms locatedat the external surface prevented zeolitesolubilization With a SiAl ratio greaterthan 3 a stable quantitative hexitol yieldwas obtained
5 Improving Catalyst Stability byProcess Design
Previous sections demonstrated howimportant smart catalyst design can be forprolonged catalyst lifetime In additionimproved stability for a given catalyst canbe achieved by tuning process parametersIn fact proper reactions conditions such asthe choice of gas atmosphere (eg hydro-gen versus an inert gas) or concentrationof reagents and solvents can have dra-matic effects on processes As mentionedpreviously leaching and sintering of metalnanoparticles tends to be more pronouncedat low pH Nickel at pH values below 7 canbe oxidized by water or acids[78] to formNi2+ species[79] To counterbalance this ef-fect van Haasterecht et al used additionof KOH in ethylene glycol conversion overNiCNF catalyst with KOH concentrationbetween 0ndash12 M to increase the pH andimprove catalyst stability (Fig 7)[37] Niparticles were less stable in hydrothermalconditions in comparison to ethylene gly-col In both cases increasing the pH hada significant impact though it was not aspronounced in ethylene glycol Moreoverat very high pH (gt13) sintering occurredagain which was probably due to a for-mation of highly mobile hydroxide nickelspecies
A reductive gas atmosphere can lead toincreased catalyst stability A typical ex-ample is the aqueous phase reforming ofethylene glycol conducted over Ni-basedcatalysts[37] Introduction of hydrogen inthe gas atmosphere suppressed the growthof nickel nanoparticles The average par-ticle size after reaction in a hydrogen andan inert atmosphere was 58 and 32 nmrespectively Moreover leaching of thenickel decreased leading to 026 catalystmass loss under inert atmosphere com-pared to 015 mass loss in hydrogen
However the use of hydrogen is notalways beneficial It was reported that forplatinum supported on graphite at highpH under a hydrogen-rich atmosphereand at an elevated temperature agglom-eration of platinum could occur[80] Thereason was that removal of the graphiteoxygen functional groups by hydrogenlead to lower adhesion and thereforehigher mobility of the nanoparticles onthe surface
Coke formation over the catalyst canalso be inhibited by co-feeding other sub-stances to the reactor For instance in cata-lytic conversion of bio-oil into hydrocar-bons a stream of bio-oil was diluted witha ratio 12 with water which decreased themass of carbon deposits on microporousZSM-5[46] Unfortunately this treatmentled to irreversible catalyst deactivation dueto dealumination of the zeolite caused bywater at high temperature[8182] Howeverthe same authors found that the use of a70 wt methanol with a bio-oil mixturecould decrease coke formation up to 70and prevent dealumination and thereforemaintain activity of the catalyst
Preventing deactivation can also beachieved by controlling the reaction me-dia During the process of producing fur-fural from xylans high deactivation ofthe GaUSY catalyst was observed whenusing water as a solvent XRD analysis ofthe spent catalyst confirmed the collapseof the zeolite structure during reactionMoreover gallium content dramaticallydecreased after 24 hours of reaction from59 wt to 027 wt To avoid the nega-tive effects of water on the catalyst thebiphasic system waterndashmethyl isobutyl
ketone (MIBK) was used with a ratio of2080 Gallium leaching decreased duringthe first few hours (36 wt Ga) and con-tinued but at a lower rate to reach a metalcontent of 19 wt at the end of the reac-tion Furthermore XRD confirmed that thezeolite retained its structure at the end ofthe experiment confirming the importanceof using a co-solvent[83]
6 Conclusions and Outlook
Deactivation of catalysts under theharsh conditions used for biomass conver-sion is a bottleneck in the developmentof competitive processes for bioresourcevalorization to renewable alternatives tofossil-based fuels and chemicals Biomass-derived molecules that must be further up-graded are usually produced in relativelydilute liquid (often aqueous) streamswhich contain biogenic and inorganic im-purities This introduces significant chal-lenges for heterogeneous catalysts whichhave often been developed in the context ofreforming fairly pure gas-phase hydrocar-bons An important category of catalystsfor catalytic biomass upgrading are oxide-supported metal particles due to their usein hydrogenation and hydrodeoxygenationreactions are essential for removing oxy-gen from bio-based molecules Several re-searchers have sought to reduce the maindeactivation pathways for these catalystsnotably their irreversible deactivation viametal leaching and sintering in the liquidphase as well as their reversible deactiva-tion by poisoning Leaching and sinteringoccur preferentially due to the instability
Fig 7 Approximation based on experimental data between nickelnanoparticles size of the spent NiCNF catalysts and the pH of the finalreaction mixture in aqueous phase reforming of ethylene glycol ThepH of the feed solutions was adjusted by addition of KOH (0-12 M) toaqueous solutions of 1 wt ethylene glycol (green line) and to water(blue line) Reactions were performed at 230 degC for 6 h under inert gasAdapted with permission of Elsevier from ref [37]
590 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
of undercoordinated metal atoms Suchatoms can also favor byproduct formationdue to their high reactivity These byprod-ucts include coke in which case these re-active atoms contribute to poisoning theother active sites by initiating the forma-tion of carbon deposits For these reasonsseveral authors have reported methods topassivate these undercoordinated atomsusing overcoats deposited either by ALDsugar pyrolysis or CVD thereby stabiliz-ing the catalyst of interest Another issueis the leaching and structure modificationsof oxide supports due to the poor hydro-thermal stability of these solids whichinclude silica and zeolites In this caseseveral authors have turned to fine-tuningthe local environment of these catalystsagain by physically covering the materialof interest The addition of hydrophobicsurface functionalities or overcoats hasbeen shown to reduce support hydrolysisbywater molecules Similarly depositing apolymeric layer over metal hydrogenationsites was shown to act as an organic pseu-do-solvent which reduced the presence ofpolar biogenic catalyst poisons and thuscatalyst deactivation Finally increasedcatalyst lifetime was also improved byprocess engineering In fact tuning thereaction conditions including by loweringpH or co-feeding organic solvents and hy-drogen proved to be efficient for limitingcatalyst deactivation
Further research opportunities exist tocombine the principle strategies mentionedabove to further improve catalyst stabiliza-tion Tailored overcoats could be used bothto stabilize undercoordinated atoms and atthe same time to provide favorable micro-environments that could reduce hydrolysisandor concentrations of catalyst poisonsSuch an approach was used in part by add-ingMgO
xto reduce the acidity of an alumi-
na overcoat which was thought to catalyzecoke formation while the overcoat wasused to stabilize undercoordinated atomsHowever this approach could be furtherstrengthened by adding functionality to theovercoat and further tailoring themoleculardesign of the catalystrsquos local environmentFurthermore controlling pore size open-ings within these overcoats could intro-duce shape selectivity and be used to sieveout undesired reactants as was recentlydemonstrated for an alumina overcoat on atitania catalyst used for photocatalysis[84]Finally it will become increasingly impor-tant to use real biomass-derived streamsfor testing catalytic stability The negativeeffects of using liquid or aqueous condi-tions can be compounded by the presenceof numerous perhaps yet unidentified in-organic or biogenic impurities This wasnotably demonstrated by the acceleratedstructure collapse of zeolite structures inthe presence of inorganic salts
In summary current synthetic tech-niques can lead to unprecedented controlof the nanostructures surrounding a cata-lystrsquos active site but harsh conditions pres-ent for biomass conversion are equally un-precedented in industrial catalytic process-ing As society comes under increasingpressure to develop routes to sustainablecarbon-based molecules these syntheticmethods will likely prove essential in ad-dressing the challenges posed by catalyti-cally upgrading renewable resources suchas biomass
AcknowledgementsThe authors acknowledge financial support
from the Swiss National Science Foundationthrough grant number 407040_153866 as partof the National Research Program 70 and grantnumber PYAPP2_154281 and Benjamin LeMonnier for help with the design of figures
Received June 17 2015
[1] K Sugiura M Ishihara T Shimauchi SHarayama Environ Sci Technol 1997 31 45
[2] S Matar M J Mirbach HA Tayim lsquoCatalysisin Petrochemical Processesrsquo Springer Scienceamp Business Media 1989
[3] O S Abdul-Hamid Monthly Oil MarketReport - May 2015 OPEC 2015
[4] The European Parliament and the Council of theEuropean Union OJ 2009 L140 16
[5] R Rinaldi F Schuumlth Energy Environ Sci2009 2 610
[6] A A Peterson F Vogel R P Lachance MFroumlling J Michael JAntal JW Tester EnergyEnviron Sci 2008 1 32
[7] J S Luterbacher D M Alonso J A DumesicGreen Chem 2014 16 4816
[8] A Rahimi A Ulbrich J J Coon S S StahlNature 2014 515 249
[9] A Eyal R Jansen R Mali A Vitner lsquoSugarMixtures and Methods for Production and UseThereofrsquo 2012 WO 2011161685 A3
[10] D V Kazachkin M Colakyan F J MoeslerlsquoSupercritical Hydrolysis of Biomassrsquo 2015US 8999065 B2
[11] D L Sills J M Gossett Bioresour Technol2011 102 1389
[12] D M Alonso S G Wettstein M A MellmerE I Gurbuz J A Dumesic Energy EnvironSci 2012 6 76
[13] J S Luterbacher J M Rand D M Alonso JHan J T Youngquist C T Maravelias B FPfleger J A Dumesic Science 2014 343 277
[14] J S Luterbacher D M Alonso J M Rand YM Questell-Santiago J H Yeap B F PflegerJ A Dumesic ChemSusChem 2015 8 1317
[15] H Tadesse R Luque Energy Environ Sci2011 4 3913
[16] J B Binder R T Raines J Am Chem Soc2009 131 1979
[17] J S Luterbacher J W Tester L P WalkerBiotechnol Bioeng 2010 107 451
[18] D B Hodge M N Karim D J Schell J DMcMillan Bioresour Technol 2008 99 8940
[19] J S Luterbacher Q Chew Y Li J W TesterL P Walker Energy Environ Sci 2012 56990
[20] T J Schwartz B J OrsquoNeill B H Shanks J ADumesic ACS Catal 2014 4 2060
[21] G W Huber J N Chheda C J Barrett J ADumesic Science 2005 308 1446
[22] G W Huber R D Cortright J A DumesicAngew Chem Int Ed 2004 43 1549
[23] J N Chheda Y Romaacuten-Leshkov J ADumesic Green Chem 2007 9 342
[24] D W Rackemann W O Doherty BiofuelsBioprod Biorefining 2011 5 198
[25] J N Chheda G W Huber J A DumesicAngew Chem Int Ed 2007 46 7164
[26] A V Bandura S N Lvov J Phys Chem RefData 2006 35 15
[27] H Xiong H N Pham A K Datye GreenChem 2014 16 4627
[28] K P De Jong lsquoSynthesis Of Solid CatalystsrsquoWiley-VCHVerlag GmbH amp Co KGaA 2009
[29] F Heacuteroguel G Siddiqi M D Detwiler DY Zemlyanov O V Safonova C Copeacuteret JCatal 2015 321 81
[30] T W Hansen A T DeLaRiva S R Challa AK Datye Acc Chem Res 2013 46 1720
[31] B J OrsquoNeill C Sener D H K Jackson T FKuech J A Dumesic ChemSusChem 2014 73247
[32] B J OrsquoNeill J T Miller P J Dietrich FG Sollberger F H Ribeiro J A DumesicChemCatChem 2014 6 2493
[33] B J OrsquoNeill D H K Jackson A J Crisci CA Farberow F Shi A C Alba-Rubio J Lu PJ Dietrich X Gu C L Marshall et al AngewChem 2013 125 14053
[34] H Zhang Y Lei A J Kropf G Zhang J WElam J T Miller F Sollberger F Ribeiro MC Akatay E A Stach J A Dumesic C LMarshall J Catal 2014 317 284
[35] H ZhangY Lei A Jeremy Kropf G Zhang JW Elam J T Miller F Sollberger F RibeiroM Cem Akatay E A Stach J A Dumesic CL Marshall J Catal 2015 323 165
[36] J Lee D H K Jackson T Li R E Winans JA Dumesic T F Kuech G W Huber EnergyEnviron Sci 2014 7 1657
[37] T van Haasterecht C C I Ludding K P deJong J H Bitter J Catal 2014 319 27
[38] P Sun G Gao Z Zhao C Xia F Li ACSCatal 2014 4 4136
[39] J Lu B Fu M C Kung G Xiao J W ElamH H Kung P C Stair Science 2012 3351205
[40] T J Schwartz R L Johnson J Cardenas AOkerlund N A Da Silva K Schmidt-Rohr JA Dumesic Angew Chem Int Ed 2014 5312718
[41] H N Pham A E Anderson R L Johnson KSchmidt-Rohr A K Datye Angew Chem IntEd 2012 51 13163
[42] H Xiong T J Schwartz N I Andersen J ADumesic A K Datye Angew Chem Int Ed2015 early view DOI 101002anie201502206
[43] P A Zapata J Faria M P Ruiz R E JentoftD E Resasco J Am Chem Soc 2012 1348570
[44] H N Pham Y J Pagan-Torres J C Serrano-Ruiz D Wang J A Dumesic A K DatyeAppl Catal Gen 2011 397 153
[45] Y J Pagaacuten-Torres J M R Gallo D WangH N Pham J A Libera C L Marshall J WElam A K Datye J A Dumesic ACS Catal2011 1 1234
[46] A G Gayubo B Valle A T Aguayo MOlazar J Bilbao Energy Fuels 2009 23 4129
[47] S R Challa A T Delariva T W Hansen SHelveg J Sehested P L Hansen F Garzon AK Datye J Am Chem Soc 2011 133 20672
[48] J A Moulijn A E van Diepen F KapteijnAppl Catal Gen 2001 212 3
[49] S L Wegener T J Marks P C Stair AccChem Res 2012 45 206
[50] B J OrsquoNeill D H K Jackson J Lee CCanlas P C Stair C L Marshall J W ElamT F Kuech J A Dumesic G W Huber ACSCatal 2015 5 1804
[51] D M King J A Spencer II X Liang L FHakim A W Weimer Surf Coat Technol2007 201 9163
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 591
[52] J A McCormick B L Cloutier AWWeimerS M George J Vac Sci Technol A 2007 2567
[53] D Longrie D Deduytsche C Detavernier JVac Sci Technol A 2014 32 010802
[54] J Lu J W Elam P C Stair Acc Chem Res2013 46 1806
[55] L De Rogatis M Cargnello V GombacB Lorenzut T Montini P FornasieroChemSusChem 2010 3 24
[56] A G Sergeev J D Webb J F Hartwig J AmChem Soc 2012 134 20226
[57] J He C Zhao J A Lercher J Am Chem Soc2012 134 20768
[58] Q Song F Wang J CaiY Wang J Zhang WYu J Xu Energy Environ Sci 2013 6 994
[59] R Dehghan-Niri J C Walmsley A HolmenP A Midgley E Rytter A H Dam A BHungria J C Hernandez-Garrido D ChenCatal Sci Technol 2012 2 2476
[60] M Morales P Y Dapsens I Giovinazzo JWitte C Mondelli S PapadokonstantakisK Hungerbuumlhler J Peacuterez-Ramiacuterez EnergyEnviron Sci 2015 8 558
[61] O A Abdelrahman H Y Luo A Heyden YRomaacuten-Leshkov J Q Bond J Catal 2015329 10
[62] D M Alonso S G Wettstein J A DumesicChem Soc Rev 2012 41 8075
[63] W Li L Ye P Long J Chen H Ariga KAsakura YYuan RSC Adv 2014 4 29072
[64] G Prieto J Zecevic H Friedrich K P deJong P E de Jongh Nat Mater 2013 12 34
[65] Z Zhang J E Jackson D J Miller BioresourTechnol 2008 99 5873
[66] H Luo T Prasomsri Y Romaacuten-Leshkov TopCatal 2015 394
[67] J Mazumder H I de Lasa Catal Today 2014237 100
[68] H Xiong H N Pham A K Datye J Catal2013 302 93
[69] H Xiong M Nolan B H ShanksA K DatyeAppl Catal Gen 2014 471 165
[70] P A Zapata Y Huang M A Gonzalez-BorjaD E Resasco J Catal 2013 308 82
[71] A Corma Chem RevWash DC U S 1995 95559
[72] P A Jacobs M Dusselier B F Sels AngewChem Int Ed 2014 53 8621
[73] M Milina S Mitchell J Peacuterez-Ramiacuterez CatalToday 2014 235 176
[74] R M Ravenelle F Schuumlssler A DrsquoAmico NDanilina J A van Bokhoven J A Lercher CW Jones C Sievers J Phys Chem C 2010114 19582
[75] D W Gardner J Huo T C Hoff R LJohnson B H Shanks J-P Tessonnier ACSCatal 2015 DOI 101021acscatal5b00888
[76] P M Dove N Han J J D Yoreo Proc NatlAcad Sci 2005 102 15357
[77] T Ennaert J Geboers E Gobechiya C MCourtin M Kurttepeli K Houthoofd C E AKirschhock P C M M Magusin S Bals P AJacobs B F Sels ACS Catal 2015 5 754
[78] B Beverskog I Puigdomenech Corros Sci1997 39 969
[79] D A Palmer P Beacuteneacutezeth C Xiao D JWesolowski L M Anovitz J Solut Chem2011 40 680
[80] J H Vleeming B F M Kuster G B Marin FOudet P Courtine J Catal 1997 166 148
[81] A G Gayubo A T Aguayo M Olazar RVivanco J Bilbao Chem Eng Sci 2003 585239
[82] A de Lucas P CanizaresA DuraacutenA CarreroAppl Catal Gen 1997 154 221
[83] CAellig D Scholz PY Dapsens C MondelliJ Peacuterez-Ramiacuterez Catal Sci Technol 2014 5142
[84] C P Canlas J Lu N A Ray N A Grosso-Giordano S Lee J W Elam R E Winans RP Van Duyne P C Stair J M Notestein NatChem 2012 4 1030

SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 583
a regeneration cycle regeneration cyclesincrease process costs and can themselvesbe problematic by modifying or destroyingsupports notably during high temperaturecalcination In the third part we discussmethods to improve stability of the sup-port as opposed to just the nanoparticlesFinally we discuss methods besides cata-lyst design including tuning the processparameters to increase catalyst stabilityand process lifetime The main methodsreviewed here as well as their commonali-ties are summarized in Table 1
2 Irreversible Deactivation viaSintering andor Leaching ofSupported NPs
Sintering is one of the main deac-tivation pathways for supported metalnanoparticles especially for catalytic pro-cesses performed at high temperaturesUnderstanding sintering is a prerequisitefor the development of stable materials andhas been the subject of several reviews[30]Sintering can occur via two distinct mech-anisms i) particle migration and coales-cence where particles statically move onthe support surface and coalesce to formlarger particles and ii) Ostwald ripeningwhere particles size and geometry evolveto minimize free energy via adatom mi-gration In situ studies and Monte-Carlosimulations have helped to clarify the con-tribution of these two mechanisms Initialrapid catalyst deactivation due to loss ofmetal surface area is attributed to Ostwaldripening while particle migration and co-alescence becomes preponderant in a sec-ond phase once small metal particles havedisappeared[47]
Leaching is also a major issue for liq-uid phase processes as it leads to both adecrease in activity and pollution of theproduct stream Leaching occurs by ex-traction of individual metal atoms fromparticles into the solution via formation offree metal ions
Many efforts have been dedicatedto the stabilization of supported metalnanoparticles[48] and recent techniquesinvolving deposition of a protective layerto prevent sintering and leaching of metalnanoparticles appear especially promis-ing Overcoating was first reported byLu et al for the stabilization of PdAl
2O
3during high-temperature dehydrogenationof ethane to ethylene[39] More recentlyOrsquoNeill et al used the same strategy for thestabilization of alumina-supported cop-per nanoparticles by the deposition of anAl
2O
3overcoat[33] Stabilization of copper
was especially noteworthy because whileprecious metal catalysts are generally pre-ferred due to their great stability base met-als such as copper suffer from irreversible
competitive processes catalyst lifetime isof prime importance under the harsh oftenhydrothermal conditions used for biomassconversion processes Liquid-phase condi-tions can often increase the reactivity ofcatalyst surfaces Furthermore the waterdissociation equilibrium is modified underhydrothermal conditions during which theionization constant increases from 10ndash14 at25 degC to 10ndash11 at 250 degC[26] This results inhigher ion concentrations that can perturbcatalyst stability[27] In addition as men-tioned above biomass-derived feeds oftencontain high quantities of biogenic impuri-ties that can act as catalyst poisons such assulfur- or nitrogen-containing compoundswhich can deactivate catalysts This prob-lem is often even more pronounced whena fermentation step is used Given the ex-tent to which biomass-derived feeds dif-fer from petroleum-based feeds shiftingcatalyst properties and robustness to meetthese new demands is an important area ofresearch This review aims to address themain developments in the design of morestable catalysts for biomass conversion andreforming reactions (Fig 2)
Supported metal particles are amongthe main catalyst families and often dis-play a limited stability independent of
their preparation method[2829] The maindeactivation paths include sintering whichleads to a decrease in metal dispersion[30]leaching ie metal dissolution and pollu-tion of surface metal with byproducts ascoke These phenomena dramatically de-crease the amount of active metallic sitesin the catalysts by reducing the ratio ofactive surface sites to inactive bulk metalin the case of sintering removing activesites by leaching or blocking active sitesin the case of poisoning This is especiallyproblematic because the metallic particlesare often the most expensive element of thecatalyst
The first section of this review reportson the major advances in catalyst designthat aim to stabilize supported nanopar-ticles against sintering and leachingSintering and leaching reduce the amountof active sites and are irreversible deactiva-tion pathways ie initial properties cannotbe recovered by regeneration such as cal-cination or reduction In the second part ofthe review we address catalyst poisoningand methods aiming to reduce it duringwhich an impurity or reaction byproduct(eg coke) blocks the active site Eventhough this is a reversible deactivationsince the poison can be removed during
0 100 200 300 400 500 600 700 800 9000
50
100
150
200
T (degC)
p(a
tm)
PetrochemicalProcessesH
ydro
gena
tion
Hyd
roge
noly
sis
Aqueous-Phase
Reforming
DehydratationHydrolysis
IsomerizationAldol Condensation
Oxidation
Aqueous phase Vapor
Vapor-liquidequilibrium
Fig 1 Approximatereaction conditionranges for catalyticbiomass conversionversus those forpetroleum processesAdapted fromref [25]
leaching
sintering
support dissolution
cokedeposition
overcoating
time on stream
activity
time on stream
activity
severe biomassconversion conditions
alloying
support doping
stabilization strategiesprocess engineering
classical catalystdeactivation
limited deactivationstable metal surface
hydrothermal stability
Fig 2 Main deactivation pathways for solid catalysts for conversion of biomass and major cata-lysts stabilization concepts
584 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
area increase to 39 m2g and re-exposureof the Cu0 sites (23 vs 86 mmolg beforecoating) Stability of the catalysts was in-vestigated in the hydrogenation of furfuralto furfuryl alcohol in 1-butanol After 15days in stream the catalyst had deactivat-ed significantly and was regenerated by insitu calcination and reduction and catalyticactivity was further tested (Fig 4c and d)After four successive cycles initial activitycould not be recovered with the uncoatedcatalyst (Fig 4c) which showed irrevers-ible deactivation In contrast the over-coated CuAl
2O
3led to identical turnover
frequencies over the course of several reac-tionregeneration cycles in 1-butanol (Fig4c) and water (Fig 4d) Irreversible deac-tivation was attributed to the decrease ofmetal surface area due to metal sintering asobserved by TEM which showed a notableparticle size increase for the uncoated cata-lyst (Fig 4b) Moreover operando X-rayabsorption spectroscopy studies confirmedthat the overcoat provided stabilization bylimiting sintering most likely because of
deactivation through leaching and sinteringunder severe liquid-phase conditions Thelatter catalyst was prepared by first form-ing Al
2O
3-supported copper oxide parti-
cles by incipient wetness impregnation ofCu(NO
3)2followed by reduction and pas-
sivation The overcoat was then depositedby 45 successive cycles of Atomic LayerDeposition (ALD) Among various cata-lytic design techniques[49] ALD has beenextensively used to tune properties at themolecular level[50]ALD allows depositionof thin layers (lt 100 nm) with high levelof conformality In comparison state-of-the-art catalyst preparation methods thatinclude wet impregnation and depositionprecipitation can lead to a mixture of siteswhich result in side reactions Thereforedespite the high price of ALD equipmentand the limits of its applicability for indus-trial-scale preparation ALD is a promis-ing technique for catalyst modification andstabilization In the case of Al
2O
3-ALD
deposition was achieved under vacuum at200 degC by alternating cycles consisting of
exposure of the substrate to an aluminumprecursor (trimethyl aluminium or TMA)and water as depicted on Fig 3b
Conventional ALD reactors designedfor wafers cannot be employed for ALDon powders due to their high surface ar-ea Fluidized bed reactors equipped withmechanical stirring or rotating chamberswhich allow long contact times with pre-cursors (gt 30 s compared with 30 ms ex-posure for wafers substrates) were usedin this and other work to uniformly coatsupported catalyst and other high surfacematerials while preventing particle ag-gregation[51ndash53] When treating the Al
2O
3-
supported copper particles a continuousovercoat was formed as shown by TEMpictures (Fig 4a) In addition both a sig-nificant surface area decrease (from 190 to16 m2g) and the absence of accessible Cu0
sites detected by N2O chemisorption after
reduction were observed[33] Subsequentcalcination at 700 degC under air flow al-lowed for pore formation within the over-coat (Fig 3a) as attested by the surface
Table 1 Summary of the main catalyst stabilization strategies
Deactivation pathway Catalyst Strategies Method Reaction ref
Metal leaching andsintering
CuAl2O
3
Overcoat to stabilizeundercoordinated atoms
Al2O
3ALD
Furfural hydrogenation [31ndash33]
CuCr2O
4-
CuO Furfural hydrogenation [3435]
CoTiO2
TiO2ALD Aqueous phase hydro-
genation [36]
NiCNF Avoid acidic conditionsIncrease pH byaddition of KOHand hydrogen
Ethylene glycol conversion [37]
Metal leaching andsinteringCoke deposition
CoHZSM-5 Embedment into supportstructure
Growth of HZSM-5onto Co
3O
3SiO
2
Conversion of levulinic acidto valeric biofuel [38]
Coke poisoningCuAl
2O
3
Mixed Al2O
3-MgO
xovercoat to decreasesupport acidy
ALD Furfural hydrogenation [31]
PdAl2O
3
Overcoat to passivateundercoordinated atoms Al
2O
3ALD Dehydrogenation of ethane
to ethylene [39]
Poisoning by aminoacids PdAuAl
2O
3
Microenvironment hostileto biogenic impurities
Polymer cross-linking
Hydrogenation of triaceticacid lactone [40]
Support dissolution
PdSiO2
PdAl2O
3 Carbon overcoat toincrease hydrophobicity
Sucrose pyrolysisto form thin carbonfilm
Hydrogenation of acetylene [41]
PtAl2O
3RuAl
2O
3
Formation ofgraphitic carbon byCLD
Ethylene glycol reforming [42]
HY zeolite Hydrophobic micro-environment
Grafting silanefunctionalities Refining of pyrolysis oil [43]
Pdniobia Formation of mixed oxide Doping by additionof 5 SiO
2 GVL to pentanoic acid[44]
PdSBAsilica
Deposition of hydrother-mally stable overcoat Niobia ALD [45]
Coke deposition anddealumination
MicroporousHZSM-5 Change reaction media Methanol co-feed Bio-oil to hydrocarbons [46]
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 585
a preferential interaction of the overcoatwith undercoordinated Cu atomswhich aremore exposed to sintering and leaching[32]However access to Cu sites decreasedover time on stream due to coke deposit inthe small pores in the overcoat leading todeactivation reversible upon calcinationFinally the overcoat did not influence se-lectivity as furfuryl alcohol remained theonly hydrogenation product that was ob-served
Al2O
3overcoating by ALD could also
be used to increase the stability in gas-phase reactions Zhang et al reported en-hanced stability for the hydrogenation offurfural by ALD overcoating using copperchromite catalysts in the gas phase[3435]Similarly to the CuAl
2O
3system described
above the overcoat preferentially blockedundercoordinated Cu atoms which werepresent in the form of step edges and sur-face defects The overcoat led to increasedstability by decreasing three deactivationpathways sintering of Cu coke formationand coverage of active Cu sites by chromitemigration In addition secondary reactionssuch as further dehydration of furfuryl al-cohol to 2-methyl furan were disfavoredwhen covering undercoordinated atoms re-sulting in higher furfuryl alcohol selectiv-ity ThinALD overcoat (10 cycles) showed
high activity but low stability while thickALD overcoat (45 cycles) provided highstability but lower activities The high ac-tivity of coated materials was attributed toan increased reduction temperature whichdecreased the activation energy of furfuralon Cu thanks to a modified ratio of Cu0Cu+ and Cu2+species Prior FTIR studies ofadsorbed CO on Pd catalysts ruled out theexistence of electronic effects between theALD layer and metal nanoparticles[3954]
Rather the authors suggested that the over-coat influences the transport of gas to andfrom the copper surface
Besides Al2O
3 successful TiO
2ALD
overcoating was used to stabilize CoTiO
2during aqueous phase hydrogenation
(APH) reactions[36] The analogous Al2O
3
Coγ-Al2O
3was inactive for the APH of
furfural due to the formation of cobalt alu-minate after calcination treatment whichwas unreducible up to 800 degC Hencethe authors investigated the performancesof TiO
2CoTiO
2prepared by ALD from
TiCl4on TiO
2-supported Co particles H
2-
temperature programmed reduction (TPR)revealed that switching fromAl
2O
3to TiO
2allowed reduction of Co at 600 degC lead-ing to furfural conversionWhile the parentcatalyst suffered fromcobalt leaching (10Co lost over 2100minutes) the metal load-ing of overcoated material remained con-stant even after regeneration Finally TEMstudy showed that the overcoat prevented
Fig 3 a) Strategy for the synthesis of stable CuAl2O3 catalysts formation of Al2O3 supportedcopper particles overcoating by ALD and cracking of overcoat by calcination b) Mechanismof Al2O3 ALD on Al2O3 (red O purple Al black C blue H) c) Embedment of Co onto HZSM-5matrix Reproduced with permission from ref [38] Copyright 2014 American Chemical Society
c)
d)
a)
b)
Fig 4 STEM image of 45ALDCuAl2O3 (a) Particle size distribution of fresh and used catalysts(b) TOF in furfural hydrogenation for 45ALDCuAl2O3 (solid) and CuAl2O3 (hollow) in 1-butanol (c)and water (d) (fresh one regeneration O two regenerations loz three regenerations Δ) Adaptedwith permission from Wiley-VCH Verlag from ref [33]
586 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
the nanoparticles from sintering whereassintering was observed during calcinationin the absence of overcoat
The methods above stabilized metalnanoparticles by adding a separate protec-tive layer over selected parts of the supportand metal particle surface Sintering andleaching of base metal catalysts could alsobe limited by embedding the metal into astructure[55] For instance Co nanoparti-cles were loaded into an HZSM-5 zeoliteproviding a bifunctional catalyst for theconversion of levulinic acid into valericbiofuel where cobalt performed hydroge-nation while the acidic support catalyzedγ-valerolactone (GVL) ring opening (Fig3c)[38] In contrast to the incipient wetnessimpregnation approach cobalt was em-bedded into HZSM-5 by nucleation andgrowth of HZSM-5 crystals onto Co
3O
4
SiO2whereas SiO
2was progressively dis-
solved thanks to the basicity of the systemH
2-TPR showed a lower temperature for
the reduction of cobalt oxide for the im-pregnated (CoHZSM-5) compared to theembedded (CoHZSM-5) catalyst dem-onstrating a higher metal-support interac-tion for the latter The strong interactionhad a limited influence on the Co acces-sibility as demonstrated by a similar H
2up-
take for both materials Structural charac-terization was performed on CoHZSM-5and CoHZSM-5 before and after cataly-sis to identify stability differences FirstTEM revealed a stable Co NPs size forCoHZSM-5 while for CoHZSM-5 theaverage Co NP size increased from 25 to32 nm Second coke formation was onlydetected for CoHZSM-5 by thermogravi-metric analysis (TGA) as well as N
2physi-
sorption (decrease of surface area and porevolume) Finally XRD patterns showedthat both materials maintained their crys-tal structure
Similar to Cu and Co supported Niparticles are relevant systems for the low-temperature conversion of biomass-de-rived compounds ndash notably for lignin hy-drogenolysis[56ndash58] However as for manybase metals the Ni particles suffer fromsintering Nanoconfinement of Ni par-ticles by self-assembly of Ni atoms insidethe nanoribbons of hydrotalcite-derivedmixed oxide provided increased catalyticstability for methane steam reformingcompared to Ni supported on commercialsupport surface[59] Metal embedment hasalso proven to be an efficient stabilizationmethod for lactic acid production fromglycerol Morales et al showed that MFIconstituted a stabilizing matrix for Sn andsignificantly decreased leaching of said Snwhile slowing down deactivation in com-parison to bifunctional carbon-silica com-posites and BEA zeolites[60]
Above we discussed various methodsdemonstrating that controlling local nano-
structures around the active metal catalystcould increase stability Abdelrahman etal recently showed that a simple judiciouschoice of the support could limit particlesintering during the hydrogenation oflevulinic acid[61] In fact fast deactivationwas observed for ruthenium particles sup-ported on SiO
2while increased stability
was achieved by using carbon and TiO2as
supports Al2O
3was the support that led to
the most stable catalyst which the authorsattributed to its lowest electronegativity
Another strategy is to tune the com-position of the metal itself Formation ofalloys for improved catalyst performanceshas often been used including in reactionsrelevant to biomass conversion[62] In thecase of SBA-15 the use of supported Ru-Fe catalysts led to stable conversion andselectivities over 300 h with no aggrega-tion of bimetallic particles no structurecollapse and improved selectivity for hy-drogenation of acetic acid to ethanol com-pared to monometallic catalysts[63]
Finally while many efforts have beendedicated to the control of individual char-acteristics of supported metal nanopar-ticles and their immediate environmentPrieto et al showed that controlling thecollective properties of supported metalnanoparticles led to increased stability[64]By achieving near-maximum interpar-ticle spacing for silica-supported coppernanoparticles deactivation during metha-nol synthesis was limited with deactivationrates which were an order of magnitudelower than those observed with a referencecatalyst that had a non-uniform spatial dis-tribution of particles
3 Poisoning of the Active Sites
In addition to the reduction of the num-ber of active sites by sintering and leach-ing competitive adsorption of undesirable
compounds such as coke or sulfur is a ma-jor catalyst deactivation pathway becauseit leads to the blocking of active sitesSome poisons including coke can oftenbe removed by regenerating the catalystunder oxidative or reductive media to formvolatile species However these methodsare sometimes incompatible with certainsupports such as carbon which has lowthermal stability and will degrade duringthese regeneration treatments In additionprocesses not requiring periodic catalystregeneration are generally favored foreconomic and practical reasons Henceseveral paths have been explored to limitcatalyst poisoning
Catalyst poisoning is often describedas especially problematic in the contextof fermentation-derived media which hasbeen proposed as an important source ofbiomass-derived platform molecules[65] Inparticular sulfur-containing amino acidsseem to be particularly detrimental to cat-alyst activity and are commonly found infermentation broths[65] To alleviate deacti-vation by coke deposition of alumina-sup-ported Pd catalysts during hydrogenationof fermentation-derived triacetic acid lac-tone (TAL) Schwartz et al formed equi-molar bimetallic PdAu particles Howeverthe alloy still suffered from sulfur poison-ing similar to the parent monometalliccatalyst when co-feeding model biogenicimpurities such as methionine The au-thors reasoned that a microenvironmentunfavorable to polar species would limitdeactivation by biogenic impurities whilethe reaction with the less polar substratewould still go forward[40] TAL is highlysoluble in alcohols whereas amino acidsare not hence a poly(vinyl alcohol) (PVA)overcoat was selected to form a polymericpseudo-solvent microenvironment (Fig5a) The PVA-overcoated PdAuAl
2O
3catalyst displayed a deactivation rateof 002 hndash1 vs 012 hndash1 for the parent Pd
CuAl2O3sintering and leaching
coke deposition
Al2O3CuAl2O3low sintering and leaching
coke deposition
MgOxCuAl2O3sintering and leachinglow coke deposition
Al2O3MgOxAl2O3CuAl2O3low sintering and leaching
low coke deposition
OH OH OH
OH OH OH OH
OH
OO
OH
S OH
O
NH2
substrate poison
a) b)
Fig 5 a) Creation of microenvironment unfavorable to poisons Adapted with permission fromWiley-VCH Verlag from ref [40] b) Stabilization of Cu(black)Al2O3 via ALD of Al2O3 (violet) andMgOx (blue)
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 587
Al2O
3for initial conversions between 50
and 70 Stabilization by PVA was alsohighly efficient when using TAL producedby fermentation without any feed purifi-cation Comparison of 13C NMR spectraof 13C-enriched methionine adsorbed onPVA-covered and non-covered PdAl
2O
3showed that the polymer acts as a lsquosolidsolventrsquo by limiting methionine adsorptionon Pd
The ALD overcoat strategy describedin the previous section for metal sinteringand leaching prevention has also provenuseful for limiting coke deposition Lu etal reported low reversible deactivation ofovercoated PdAl
2O
3for the dehydroge-
nation of ethane to ethylene at 650 degC[39]After using 45ALDcycles to form anAl
2O
3thin film the amount of coke deposited asdetermined by thermogravimetric analy-sis was more than 16 times lower than theamount measured for the parent PdAl
2O
3catalyst The reduction of the amount ofcoke formed was attributed to the blockingof the low-coordination Pd surface siteswhich were preferentially coated In factthe later sites favored CndashC bond scissionand hydrogen stripping resulting in the un-desirable formation of coke CH
4 CO and
CO2 Hence in addition to a lower catalyst
deactivation coating led to higher ethyleneyields
The Al2O
3overcoated CuAl
2O
3de-
scribed earlier suffered from reversiblecoke deposition In a separate study theauthors managed to limit this phenomenonby finely tuning the overcoat In addition totheAl
2O
3overcoat which provided particle
stabilization basic MgOxwas deposited to
reduce the overcoatrsquos acidity which wasassumed to catalyze the formation of coke(Fig 5b)[31] Interestingly pure MgO
xdid
not provide stabilization against sinteringThe local structure of the active site can
also reduce poisoning by affecting localmass transfer To suppress deactivation byfouling due to coke deposition during theconversion of propanal to hydrocarbons onAl-MFI Luo et al used an open mesopo-rous structure based on nanosheets to limitthe effects of fouling[66] This strategy re-sulted in a five-fold decrease of deactiva-tion rates In addition to an increase in thenumber of pore openings improved sta-bility was attributed to the short diffusionpath for reagents in the single unit-cell-thick nanosheet material which limitedcoke formation that was favored by longercontact times with the reaction products
4 Increasing Support Stability inHydrothermal Conditions
In addition to deactivation due to metalleaching and sintering loss of surface areaand pore collapsing in oxide supports is a
major source of irreversible deactivationespecially in aqueous phase conditionsFor biomass conversion processes whichoften require liquid phase andor aqueousmedia at low pH hydrothermal stabilityis a major concern Metal oxides are of-ten used as supports due to their thermalstability and high surface area Howeverthey tend to have low stability under theaforementioned conditions[27] Such ox-ides can sometimes be substituted by morestable support such as carbon but someprocesses require specific surface func-tionalities such as the acidic sites presenton alumina In addition contrary to car-bon most oxides have excellent thermalstability which is necessary during certaincatalyst regeneration processes Prominentsupports such as alumina and silica-basedmaterials undergo dissolution by hydroly-sis of aluminoxane and siloxane bonds Inaddition to loss of surface area hydrolysisof oxide network can lead to modificationof chemical composition For instance hy-drolysis of the Si-O-Al bond in zeolites fa-vors dealumination This section describesthe main techniques used to stabilize metaloxides supports in the area of biomass con-version
Formation of mixed oxides or supportdoping can be used to limit this phenome-non For instance Pham et al reported thatthe stability of Pdniobia during transfor-mation of γ-valerolactone (GVL) to pen-tanoic acid could be improved by additionof 5 SiO
2into niobia[44] Niobia could
not be entirely substituted by silica be-cause acid sites are required for GVL ringopening in addition to metal sites for thehydrogenation reaction However the ad-dition of a small portion of SiO
2improves
stability while retaining activity The com-parison of the N
2physisorption isotherms
of a Pdniobia HY commercial referencecatalyst before and after the GVL reactionshowed a dramatic surface area loss from150 to 8 m2g while the porous structurewas affected to a lesser extent for PdNb-Sioxide with a decrease from 81 to 31 m2gSimilarly HAADF-STEM and HRTEMrevealed a lower Pd particle growth uponreaction for the stabilized system than forthe PdHY reference catalyst SimilarlyMazumder et al reported the promotionof Niγ-Al
2O
3by 5 wt La
2O
3leading to
dramatic improvement of activity and sta-bility for the catalytic gasification of glu-cose compared to the parent catalyst whichsuffered from metal sintering[67]
The stabilizing oxide can also be in-troduced by ALD similar to the thin over-coats deposited for the stabilization ofsupported metal particles as described inSection 2 For instance hydrothermallystable Nb-Si oxide catalysts were preparedusing ALD of niobia within the pores ofmesoporous silica SBA-15 using between10 and 30 cycles which alternated expo-sure of the Nb(OCH
2CH
3)4precursor and
H2O to the SBA-15 surface (Fig 6a)[45]
The consumption of silanol group on thesurface was complete after 19 cycles as
a)
b)
c)
Fig 6 a) Strategyfor the synthesis ofhydrothermally stablehighly ordered niobiamesoporous materialsby ALD Reproducedwith permission fromref [45] Copyright2011 AmericanChemical Societyb) Strategy for theformation of niobiacarbon compositecatalysts Adaptedwith permission ofElsevier from ref [68]c) Deposition of gra-phitic carbon layer onalumina powder andpellets by chemicallayer deposition fromCH4 Reproducedwith permission fromWiley-VCH Verlagfrom ref [42]
588 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
demonstrated by the disappearance of theSiOH band at 3735 cmndash1 in the FTIR spec-tra Hydrated amorphous niobia was iden-tified as a major species by Raman spec-troscopy for samples presenting 10 19 and30 ALD layers N
2physisorption showed
a decrease of surface area pore volumeand pore diameter with increasing niobialoadings The presence of acid sites formedupon niobia deposition was verified byNH
3-TPD and hydrothermal stability was
tested by treating the materials in liquidwater for 12 h at 200 degC and 28 bar argonThe structure of the coated materials aftertreatment was confirmed by X-ray diffrac-tion with the persistence of the hexagonalmesoporous structure pattern while the lat-ter pattern was suppressed after hydrother-mal treatment of the parent SBA-15 TEManalysis confirmed the stability differencesobserved by XRD A bifunctional catalystfor the transformation of GVL to pentanoicacid was also formed by subsequent depo-sition of Pd nanoparticles As for the sup-port this catalyst showed higher stabilitythan the conventional PdHY-340
Improved hydrothermal stability ofa niobia-supported Pd catalyst was alsoachieved by deposition of the latter ona more stable surface such as carbonHighly dispersed spherical particles of aniobiacarbon composite were formed bythe reaction of d-glucose and ammoniumniobium oxalate in an autoclave at 200degC (pH was controlled by urea decom-position) followed by pyrolysis at 400degC under N
2(Fig 6b)[68] Hydrothermal
stability was investigated during liquid-phase butanol dehydration at 240 degC and51 bar While initial activity of the com-posite bifunctional catalyst was lower thanthe commercial HY-340 catalyst the lat-ter quickly deactivated during the first 20h of reaction while the modified catalystsremained very stable and surpassed HY-340 after 8 h Interestingly the analogousmaterial obtained by incipient wetness im-pregnation of niobia led to the formation oflarge niobia crystallites underscoring theimportance of strong interaction betweenniobia and carbon[69] Finally bifunctionalcatalysts were prepared by incipient wet-ness impregnation of Pd(NO
3)2and tested
during the conversion of GVL to pentanoicacid leading to improved hydrothermalstability for the niobiacarbon compositecatalysts just like the metal-free system
For silica and alumina improved hy-drothermal stability above 100 degC wasachieved by deposition of a thin film of car-bon formed from simple sugars Pham et aldeposited carbon by pyrolysis of adsorbedsucrose at 400 degC under N
2flow on SBA-
15 silica gel and fumed alumina leadingto improved stability after treatment in liq-uid water at 100 degC for 12 h[41] MoreoverPd was deposited on the carbon-coated
oxides to test their ability as supports forthe catalytic hydrogenation of acetylenein the presence of excess ethylene For acomparable Pd particle size (1ndash15 nm)the authors observed formation of ethaneat high acetylene conversion on both SiO
2andAl
2O
3 while carbon-coating prevented
over-hydrogenation to ethane The FTIRstudy showed a low hydration potential forthe coated materials compared to SiO
2and
Al2O
3 Therefore the higher selectivity to
ethylene for the coated materials was at-tributed to their increased hydrophobicity
More recently Xiong et al reported onthe deposition of a graphitic carbon layerover alumina by chemical vapor deposition(CVD) from CH
4(Fig 6c)[42] The graphit-
ic carbon layer provided higher stabilitythan the carbon deposited from pyrolysisof sugars as demonstrated by the higherstability achieved under flowing air (500vs 250 degC) As monitored by the growthof the characteristic graphitic peak at 24deg2θ on the XRD pattern the amount of de-posited carbon was controlled by the CH
4deposition time (from 05 to 6 h) whereasother peaks attributed to γ-Al
2O
3were un-
touched TEM and energy-filtered TEM at-tested that the carbon coating was uniformon the oxide support Raman spectros-copy mapping confirmed that depositionwas uniform over the entire surface of thespheres Furthermore the relatively highratio (071) between the peak intensitiesfor G and D bands (at 1590 and 1350 cmndash1respectively) in the Raman spectrum of gc-Al
2O
3revealed a high degree of graphitiza-
tion on the entire surface No such featureswere observed for blank alumina and pyro-lyzed carbon on alumina
The hydrothermal stability of the cata-lyst was investigated using a treatmentunder static conditions at 220 degC underautogenic pressure in an autoclave usingwater After treatment the XRD pattern ofγ-Al
2O
3displayed the well-defined diffrac-
tion peaks of the boehmite phase attestingto a reaction with water Simultaneouslyno significant change was observed forgraphitic-carbon covered γ-Al
2O
3(gc-
Al2O
3) Improved stability for gc-Al
2O
3was confirmed by N
2physisorption results
which showed that no significant decreaseof surface area pore volume and poresize occurred during hydrothermal treat-ment while γ-Al
2O
3saw its surface area
decrease by 80 during the same treat-ment Stability of gc-Al
2O
3as a support
was tested in the aqueous phase reforming(APR) of ethylene glycol at 250 degC and70 bar after deposition of platinum par-ticles Conversion using Ptgc-Al
2O
3was
twice as high as for Ptγ-Al2O
3 This result
was consistent with the leached Al and Ptdetected in the liquid phase after reactionby ICP-OES for the uncoated materialIn addition to full conversion of γ-Al
2O
3
to boehmite observed by XRD STEMshowed Pt particle size increase for Ptγ-Al
2O
3 Hence deactivation of Ptγ-Al
2O
3was also attributed to Pt particle sinteringin addition to leaching whereas the use ofgraphitic carbon overcoat prevented bothphenomena under APR conditions Thisapproach was also successful when usingRugc-Al
2O
3for lactic acid hydrogenation
Overall graphitic carbon layers depositedthrough vapor phase route provided higherhydrothermal stability than pyrolytic car-bon-coatings and seems to be a promisingstabilization method for a wide range ofapplications
In addition to the deposition of thinfilm precise surface functionalization canalso be used to improve catalyst stabilityFor instance the generation of hydropho-bic surfaces by grafting of octadecyltri-chlorosilane groups onHY zeolite prevent-ed the support structure from collapsingduring stability tests which mimicked theupgrading of biomass-derived liquid com-pounds in biphasic systems at 200 degC[43]Stabilization was attributed to a limitedcontact between the zeolitersquos hydrophobicsurface and the aqueous phase The sameauthors also reported a systematic studywith various alkylsilanes from C
2to C
18[70]
Hydrothermal stability of zeolites hasbeen studied in great detail as zeolites arethemost widely used solid acid catalysts[71]and are often proposed for biomass con-version[7273] As shown by detailed char-acterization using 27Al and 29Si solid-stateNMR and NH
3-TPD siloxane bridge hy-
drolysis was the main deactivation routein the presence of hot liquid water whilezeolites treated under steam suffered fromdealumination[74] After a treatment in hotwater at 200 degC mimicking APR condi-tions ZSM-5 was stable independent ofits SiAl ratio while degradation of zeoliteY increased with increasing SiAl ratioas opposed to the better stability of zeo-lites with higher SiAl in the presence ofsteam Finally Al-rich zeolite Y could bestabilized by incorporation of lanthanumcations[74]
Recently Gardner et al reported thattesting the hydrothermal stability of zeo-lites in distilled water was not completelyrelevant to biomass conversion reactionconditions[75] In fact dry biomass con-tains about 15 wt of inorganic saltssuch as NaCl and the dissolution rate ofmetal oxides could be dramatically en-hanced by cations in solution[76] Catalytictests showed that the presence of NaCl hadno influence on glucose conversion duringthe liquid-phase transformation of glucoseto HMF on ZSM-5 However zeolite hy-drothermal stability was significantly per-turbed by the presence of NaCl leading tothe hydrolytic attack of extra-frameworkspecies andor defects sites Interestingly
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 589
leached Al species were active in the con-version of glucose to fructose
Together with metal particles zeolitesconstitute an important class of bifunc-tional catalysts While studies mentionedabove aimed at adding a stabilizing ele-ment to an existing material Ennaert et alassessed the stability of Ruzeolite H-USYin hot liquid water for the hydrolytic hy-drogenation of cellulose using variousSiAl ratios in order to select the optimalzeolite composition[77] They identifiedthat the lattice was stabilized by frame-work aluminum atoms which preventedhydrolysis of framework bonds while theextra-framework aluminum atoms locatedat the external surface prevented zeolitesolubilization With a SiAl ratio greaterthan 3 a stable quantitative hexitol yieldwas obtained
5 Improving Catalyst Stability byProcess Design
Previous sections demonstrated howimportant smart catalyst design can be forprolonged catalyst lifetime In additionimproved stability for a given catalyst canbe achieved by tuning process parametersIn fact proper reactions conditions such asthe choice of gas atmosphere (eg hydro-gen versus an inert gas) or concentrationof reagents and solvents can have dra-matic effects on processes As mentionedpreviously leaching and sintering of metalnanoparticles tends to be more pronouncedat low pH Nickel at pH values below 7 canbe oxidized by water or acids[78] to formNi2+ species[79] To counterbalance this ef-fect van Haasterecht et al used additionof KOH in ethylene glycol conversion overNiCNF catalyst with KOH concentrationbetween 0ndash12 M to increase the pH andimprove catalyst stability (Fig 7)[37] Niparticles were less stable in hydrothermalconditions in comparison to ethylene gly-col In both cases increasing the pH hada significant impact though it was not aspronounced in ethylene glycol Moreoverat very high pH (gt13) sintering occurredagain which was probably due to a for-mation of highly mobile hydroxide nickelspecies
A reductive gas atmosphere can lead toincreased catalyst stability A typical ex-ample is the aqueous phase reforming ofethylene glycol conducted over Ni-basedcatalysts[37] Introduction of hydrogen inthe gas atmosphere suppressed the growthof nickel nanoparticles The average par-ticle size after reaction in a hydrogen andan inert atmosphere was 58 and 32 nmrespectively Moreover leaching of thenickel decreased leading to 026 catalystmass loss under inert atmosphere com-pared to 015 mass loss in hydrogen
However the use of hydrogen is notalways beneficial It was reported that forplatinum supported on graphite at highpH under a hydrogen-rich atmosphereand at an elevated temperature agglom-eration of platinum could occur[80] Thereason was that removal of the graphiteoxygen functional groups by hydrogenlead to lower adhesion and thereforehigher mobility of the nanoparticles onthe surface
Coke formation over the catalyst canalso be inhibited by co-feeding other sub-stances to the reactor For instance in cata-lytic conversion of bio-oil into hydrocar-bons a stream of bio-oil was diluted witha ratio 12 with water which decreased themass of carbon deposits on microporousZSM-5[46] Unfortunately this treatmentled to irreversible catalyst deactivation dueto dealumination of the zeolite caused bywater at high temperature[8182] Howeverthe same authors found that the use of a70 wt methanol with a bio-oil mixturecould decrease coke formation up to 70and prevent dealumination and thereforemaintain activity of the catalyst
Preventing deactivation can also beachieved by controlling the reaction me-dia During the process of producing fur-fural from xylans high deactivation ofthe GaUSY catalyst was observed whenusing water as a solvent XRD analysis ofthe spent catalyst confirmed the collapseof the zeolite structure during reactionMoreover gallium content dramaticallydecreased after 24 hours of reaction from59 wt to 027 wt To avoid the nega-tive effects of water on the catalyst thebiphasic system waterndashmethyl isobutyl
ketone (MIBK) was used with a ratio of2080 Gallium leaching decreased duringthe first few hours (36 wt Ga) and con-tinued but at a lower rate to reach a metalcontent of 19 wt at the end of the reac-tion Furthermore XRD confirmed that thezeolite retained its structure at the end ofthe experiment confirming the importanceof using a co-solvent[83]
6 Conclusions and Outlook
Deactivation of catalysts under theharsh conditions used for biomass conver-sion is a bottleneck in the developmentof competitive processes for bioresourcevalorization to renewable alternatives tofossil-based fuels and chemicals Biomass-derived molecules that must be further up-graded are usually produced in relativelydilute liquid (often aqueous) streamswhich contain biogenic and inorganic im-purities This introduces significant chal-lenges for heterogeneous catalysts whichhave often been developed in the context ofreforming fairly pure gas-phase hydrocar-bons An important category of catalystsfor catalytic biomass upgrading are oxide-supported metal particles due to their usein hydrogenation and hydrodeoxygenationreactions are essential for removing oxy-gen from bio-based molecules Several re-searchers have sought to reduce the maindeactivation pathways for these catalystsnotably their irreversible deactivation viametal leaching and sintering in the liquidphase as well as their reversible deactiva-tion by poisoning Leaching and sinteringoccur preferentially due to the instability
Fig 7 Approximation based on experimental data between nickelnanoparticles size of the spent NiCNF catalysts and the pH of the finalreaction mixture in aqueous phase reforming of ethylene glycol ThepH of the feed solutions was adjusted by addition of KOH (0-12 M) toaqueous solutions of 1 wt ethylene glycol (green line) and to water(blue line) Reactions were performed at 230 degC for 6 h under inert gasAdapted with permission of Elsevier from ref [37]
590 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
of undercoordinated metal atoms Suchatoms can also favor byproduct formationdue to their high reactivity These byprod-ucts include coke in which case these re-active atoms contribute to poisoning theother active sites by initiating the forma-tion of carbon deposits For these reasonsseveral authors have reported methods topassivate these undercoordinated atomsusing overcoats deposited either by ALDsugar pyrolysis or CVD thereby stabiliz-ing the catalyst of interest Another issueis the leaching and structure modificationsof oxide supports due to the poor hydro-thermal stability of these solids whichinclude silica and zeolites In this caseseveral authors have turned to fine-tuningthe local environment of these catalystsagain by physically covering the materialof interest The addition of hydrophobicsurface functionalities or overcoats hasbeen shown to reduce support hydrolysisbywater molecules Similarly depositing apolymeric layer over metal hydrogenationsites was shown to act as an organic pseu-do-solvent which reduced the presence ofpolar biogenic catalyst poisons and thuscatalyst deactivation Finally increasedcatalyst lifetime was also improved byprocess engineering In fact tuning thereaction conditions including by loweringpH or co-feeding organic solvents and hy-drogen proved to be efficient for limitingcatalyst deactivation
Further research opportunities exist tocombine the principle strategies mentionedabove to further improve catalyst stabiliza-tion Tailored overcoats could be used bothto stabilize undercoordinated atoms and atthe same time to provide favorable micro-environments that could reduce hydrolysisandor concentrations of catalyst poisonsSuch an approach was used in part by add-ingMgO
xto reduce the acidity of an alumi-
na overcoat which was thought to catalyzecoke formation while the overcoat wasused to stabilize undercoordinated atomsHowever this approach could be furtherstrengthened by adding functionality to theovercoat and further tailoring themoleculardesign of the catalystrsquos local environmentFurthermore controlling pore size open-ings within these overcoats could intro-duce shape selectivity and be used to sieveout undesired reactants as was recentlydemonstrated for an alumina overcoat on atitania catalyst used for photocatalysis[84]Finally it will become increasingly impor-tant to use real biomass-derived streamsfor testing catalytic stability The negativeeffects of using liquid or aqueous condi-tions can be compounded by the presenceof numerous perhaps yet unidentified in-organic or biogenic impurities This wasnotably demonstrated by the acceleratedstructure collapse of zeolite structures inthe presence of inorganic salts
In summary current synthetic tech-niques can lead to unprecedented controlof the nanostructures surrounding a cata-lystrsquos active site but harsh conditions pres-ent for biomass conversion are equally un-precedented in industrial catalytic process-ing As society comes under increasingpressure to develop routes to sustainablecarbon-based molecules these syntheticmethods will likely prove essential in ad-dressing the challenges posed by catalyti-cally upgrading renewable resources suchas biomass
AcknowledgementsThe authors acknowledge financial support
from the Swiss National Science Foundationthrough grant number 407040_153866 as partof the National Research Program 70 and grantnumber PYAPP2_154281 and Benjamin LeMonnier for help with the design of figures
Received June 17 2015
[1] K Sugiura M Ishihara T Shimauchi SHarayama Environ Sci Technol 1997 31 45
[2] S Matar M J Mirbach HA Tayim lsquoCatalysisin Petrochemical Processesrsquo Springer Scienceamp Business Media 1989
[3] O S Abdul-Hamid Monthly Oil MarketReport - May 2015 OPEC 2015
[4] The European Parliament and the Council of theEuropean Union OJ 2009 L140 16
[5] R Rinaldi F Schuumlth Energy Environ Sci2009 2 610
[6] A A Peterson F Vogel R P Lachance MFroumlling J Michael JAntal JW Tester EnergyEnviron Sci 2008 1 32
[7] J S Luterbacher D M Alonso J A DumesicGreen Chem 2014 16 4816
[8] A Rahimi A Ulbrich J J Coon S S StahlNature 2014 515 249
[9] A Eyal R Jansen R Mali A Vitner lsquoSugarMixtures and Methods for Production and UseThereofrsquo 2012 WO 2011161685 A3
[10] D V Kazachkin M Colakyan F J MoeslerlsquoSupercritical Hydrolysis of Biomassrsquo 2015US 8999065 B2
[11] D L Sills J M Gossett Bioresour Technol2011 102 1389
[12] D M Alonso S G Wettstein M A MellmerE I Gurbuz J A Dumesic Energy EnvironSci 2012 6 76
[13] J S Luterbacher J M Rand D M Alonso JHan J T Youngquist C T Maravelias B FPfleger J A Dumesic Science 2014 343 277
[14] J S Luterbacher D M Alonso J M Rand YM Questell-Santiago J H Yeap B F PflegerJ A Dumesic ChemSusChem 2015 8 1317
[15] H Tadesse R Luque Energy Environ Sci2011 4 3913
[16] J B Binder R T Raines J Am Chem Soc2009 131 1979
[17] J S Luterbacher J W Tester L P WalkerBiotechnol Bioeng 2010 107 451
[18] D B Hodge M N Karim D J Schell J DMcMillan Bioresour Technol 2008 99 8940
[19] J S Luterbacher Q Chew Y Li J W TesterL P Walker Energy Environ Sci 2012 56990
[20] T J Schwartz B J OrsquoNeill B H Shanks J ADumesic ACS Catal 2014 4 2060
[21] G W Huber J N Chheda C J Barrett J ADumesic Science 2005 308 1446
[22] G W Huber R D Cortright J A DumesicAngew Chem Int Ed 2004 43 1549
[23] J N Chheda Y Romaacuten-Leshkov J ADumesic Green Chem 2007 9 342
[24] D W Rackemann W O Doherty BiofuelsBioprod Biorefining 2011 5 198
[25] J N Chheda G W Huber J A DumesicAngew Chem Int Ed 2007 46 7164
[26] A V Bandura S N Lvov J Phys Chem RefData 2006 35 15
[27] H Xiong H N Pham A K Datye GreenChem 2014 16 4627
[28] K P De Jong lsquoSynthesis Of Solid CatalystsrsquoWiley-VCHVerlag GmbH amp Co KGaA 2009
[29] F Heacuteroguel G Siddiqi M D Detwiler DY Zemlyanov O V Safonova C Copeacuteret JCatal 2015 321 81
[30] T W Hansen A T DeLaRiva S R Challa AK Datye Acc Chem Res 2013 46 1720
[31] B J OrsquoNeill C Sener D H K Jackson T FKuech J A Dumesic ChemSusChem 2014 73247
[32] B J OrsquoNeill J T Miller P J Dietrich FG Sollberger F H Ribeiro J A DumesicChemCatChem 2014 6 2493
[33] B J OrsquoNeill D H K Jackson A J Crisci CA Farberow F Shi A C Alba-Rubio J Lu PJ Dietrich X Gu C L Marshall et al AngewChem 2013 125 14053
[34] H Zhang Y Lei A J Kropf G Zhang J WElam J T Miller F Sollberger F Ribeiro MC Akatay E A Stach J A Dumesic C LMarshall J Catal 2014 317 284
[35] H ZhangY Lei A Jeremy Kropf G Zhang JW Elam J T Miller F Sollberger F RibeiroM Cem Akatay E A Stach J A Dumesic CL Marshall J Catal 2015 323 165
[36] J Lee D H K Jackson T Li R E Winans JA Dumesic T F Kuech G W Huber EnergyEnviron Sci 2014 7 1657
[37] T van Haasterecht C C I Ludding K P deJong J H Bitter J Catal 2014 319 27
[38] P Sun G Gao Z Zhao C Xia F Li ACSCatal 2014 4 4136
[39] J Lu B Fu M C Kung G Xiao J W ElamH H Kung P C Stair Science 2012 3351205
[40] T J Schwartz R L Johnson J Cardenas AOkerlund N A Da Silva K Schmidt-Rohr JA Dumesic Angew Chem Int Ed 2014 5312718
[41] H N Pham A E Anderson R L Johnson KSchmidt-Rohr A K Datye Angew Chem IntEd 2012 51 13163
[42] H Xiong T J Schwartz N I Andersen J ADumesic A K Datye Angew Chem Int Ed2015 early view DOI 101002anie201502206
[43] P A Zapata J Faria M P Ruiz R E JentoftD E Resasco J Am Chem Soc 2012 1348570
[44] H N Pham Y J Pagan-Torres J C Serrano-Ruiz D Wang J A Dumesic A K DatyeAppl Catal Gen 2011 397 153
[45] Y J Pagaacuten-Torres J M R Gallo D WangH N Pham J A Libera C L Marshall J WElam A K Datye J A Dumesic ACS Catal2011 1 1234
[46] A G Gayubo B Valle A T Aguayo MOlazar J Bilbao Energy Fuels 2009 23 4129
[47] S R Challa A T Delariva T W Hansen SHelveg J Sehested P L Hansen F Garzon AK Datye J Am Chem Soc 2011 133 20672
[48] J A Moulijn A E van Diepen F KapteijnAppl Catal Gen 2001 212 3
[49] S L Wegener T J Marks P C Stair AccChem Res 2012 45 206
[50] B J OrsquoNeill D H K Jackson J Lee CCanlas P C Stair C L Marshall J W ElamT F Kuech J A Dumesic G W Huber ACSCatal 2015 5 1804
[51] D M King J A Spencer II X Liang L FHakim A W Weimer Surf Coat Technol2007 201 9163
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 591
[52] J A McCormick B L Cloutier AWWeimerS M George J Vac Sci Technol A 2007 2567
[53] D Longrie D Deduytsche C Detavernier JVac Sci Technol A 2014 32 010802
[54] J Lu J W Elam P C Stair Acc Chem Res2013 46 1806
[55] L De Rogatis M Cargnello V GombacB Lorenzut T Montini P FornasieroChemSusChem 2010 3 24
[56] A G Sergeev J D Webb J F Hartwig J AmChem Soc 2012 134 20226
[57] J He C Zhao J A Lercher J Am Chem Soc2012 134 20768
[58] Q Song F Wang J CaiY Wang J Zhang WYu J Xu Energy Environ Sci 2013 6 994
[59] R Dehghan-Niri J C Walmsley A HolmenP A Midgley E Rytter A H Dam A BHungria J C Hernandez-Garrido D ChenCatal Sci Technol 2012 2 2476
[60] M Morales P Y Dapsens I Giovinazzo JWitte C Mondelli S PapadokonstantakisK Hungerbuumlhler J Peacuterez-Ramiacuterez EnergyEnviron Sci 2015 8 558
[61] O A Abdelrahman H Y Luo A Heyden YRomaacuten-Leshkov J Q Bond J Catal 2015329 10
[62] D M Alonso S G Wettstein J A DumesicChem Soc Rev 2012 41 8075
[63] W Li L Ye P Long J Chen H Ariga KAsakura YYuan RSC Adv 2014 4 29072
[64] G Prieto J Zecevic H Friedrich K P deJong P E de Jongh Nat Mater 2013 12 34
[65] Z Zhang J E Jackson D J Miller BioresourTechnol 2008 99 5873
[66] H Luo T Prasomsri Y Romaacuten-Leshkov TopCatal 2015 394
[67] J Mazumder H I de Lasa Catal Today 2014237 100
[68] H Xiong H N Pham A K Datye J Catal2013 302 93
[69] H Xiong M Nolan B H ShanksA K DatyeAppl Catal Gen 2014 471 165
[70] P A Zapata Y Huang M A Gonzalez-BorjaD E Resasco J Catal 2013 308 82
[71] A Corma Chem RevWash DC U S 1995 95559
[72] P A Jacobs M Dusselier B F Sels AngewChem Int Ed 2014 53 8621
[73] M Milina S Mitchell J Peacuterez-Ramiacuterez CatalToday 2014 235 176
[74] R M Ravenelle F Schuumlssler A DrsquoAmico NDanilina J A van Bokhoven J A Lercher CW Jones C Sievers J Phys Chem C 2010114 19582
[75] D W Gardner J Huo T C Hoff R LJohnson B H Shanks J-P Tessonnier ACSCatal 2015 DOI 101021acscatal5b00888
[76] P M Dove N Han J J D Yoreo Proc NatlAcad Sci 2005 102 15357
[77] T Ennaert J Geboers E Gobechiya C MCourtin M Kurttepeli K Houthoofd C E AKirschhock P C M M Magusin S Bals P AJacobs B F Sels ACS Catal 2015 5 754
[78] B Beverskog I Puigdomenech Corros Sci1997 39 969
[79] D A Palmer P Beacuteneacutezeth C Xiao D JWesolowski L M Anovitz J Solut Chem2011 40 680
[80] J H Vleeming B F M Kuster G B Marin FOudet P Courtine J Catal 1997 166 148
[81] A G Gayubo A T Aguayo M Olazar RVivanco J Bilbao Chem Eng Sci 2003 585239
[82] A de Lucas P CanizaresA DuraacutenA CarreroAppl Catal Gen 1997 154 221
[83] CAellig D Scholz PY Dapsens C MondelliJ Peacuterez-Ramiacuterez Catal Sci Technol 2014 5142
[84] C P Canlas J Lu N A Ray N A Grosso-Giordano S Lee J W Elam R E Winans RP Van Duyne P C Stair J M Notestein NatChem 2012 4 1030

584 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
area increase to 39 m2g and re-exposureof the Cu0 sites (23 vs 86 mmolg beforecoating) Stability of the catalysts was in-vestigated in the hydrogenation of furfuralto furfuryl alcohol in 1-butanol After 15days in stream the catalyst had deactivat-ed significantly and was regenerated by insitu calcination and reduction and catalyticactivity was further tested (Fig 4c and d)After four successive cycles initial activitycould not be recovered with the uncoatedcatalyst (Fig 4c) which showed irrevers-ible deactivation In contrast the over-coated CuAl
2O
3led to identical turnover
frequencies over the course of several reac-tionregeneration cycles in 1-butanol (Fig4c) and water (Fig 4d) Irreversible deac-tivation was attributed to the decrease ofmetal surface area due to metal sintering asobserved by TEM which showed a notableparticle size increase for the uncoated cata-lyst (Fig 4b) Moreover operando X-rayabsorption spectroscopy studies confirmedthat the overcoat provided stabilization bylimiting sintering most likely because of
deactivation through leaching and sinteringunder severe liquid-phase conditions Thelatter catalyst was prepared by first form-ing Al
2O
3-supported copper oxide parti-
cles by incipient wetness impregnation ofCu(NO
3)2followed by reduction and pas-
sivation The overcoat was then depositedby 45 successive cycles of Atomic LayerDeposition (ALD) Among various cata-lytic design techniques[49] ALD has beenextensively used to tune properties at themolecular level[50]ALD allows depositionof thin layers (lt 100 nm) with high levelof conformality In comparison state-of-the-art catalyst preparation methods thatinclude wet impregnation and depositionprecipitation can lead to a mixture of siteswhich result in side reactions Thereforedespite the high price of ALD equipmentand the limits of its applicability for indus-trial-scale preparation ALD is a promis-ing technique for catalyst modification andstabilization In the case of Al
2O
3-ALD
deposition was achieved under vacuum at200 degC by alternating cycles consisting of
exposure of the substrate to an aluminumprecursor (trimethyl aluminium or TMA)and water as depicted on Fig 3b
Conventional ALD reactors designedfor wafers cannot be employed for ALDon powders due to their high surface ar-ea Fluidized bed reactors equipped withmechanical stirring or rotating chamberswhich allow long contact times with pre-cursors (gt 30 s compared with 30 ms ex-posure for wafers substrates) were usedin this and other work to uniformly coatsupported catalyst and other high surfacematerials while preventing particle ag-gregation[51ndash53] When treating the Al
2O
3-
supported copper particles a continuousovercoat was formed as shown by TEMpictures (Fig 4a) In addition both a sig-nificant surface area decrease (from 190 to16 m2g) and the absence of accessible Cu0
sites detected by N2O chemisorption after
reduction were observed[33] Subsequentcalcination at 700 degC under air flow al-lowed for pore formation within the over-coat (Fig 3a) as attested by the surface
Table 1 Summary of the main catalyst stabilization strategies
Deactivation pathway Catalyst Strategies Method Reaction ref
Metal leaching andsintering
CuAl2O
3
Overcoat to stabilizeundercoordinated atoms
Al2O
3ALD
Furfural hydrogenation [31ndash33]
CuCr2O
4-
CuO Furfural hydrogenation [3435]
CoTiO2
TiO2ALD Aqueous phase hydro-
genation [36]
NiCNF Avoid acidic conditionsIncrease pH byaddition of KOHand hydrogen
Ethylene glycol conversion [37]
Metal leaching andsinteringCoke deposition
CoHZSM-5 Embedment into supportstructure
Growth of HZSM-5onto Co
3O
3SiO
2
Conversion of levulinic acidto valeric biofuel [38]
Coke poisoningCuAl
2O
3
Mixed Al2O
3-MgO
xovercoat to decreasesupport acidy
ALD Furfural hydrogenation [31]
PdAl2O
3
Overcoat to passivateundercoordinated atoms Al
2O
3ALD Dehydrogenation of ethane
to ethylene [39]
Poisoning by aminoacids PdAuAl
2O
3
Microenvironment hostileto biogenic impurities
Polymer cross-linking
Hydrogenation of triaceticacid lactone [40]
Support dissolution
PdSiO2
PdAl2O
3 Carbon overcoat toincrease hydrophobicity
Sucrose pyrolysisto form thin carbonfilm
Hydrogenation of acetylene [41]
PtAl2O
3RuAl
2O
3
Formation ofgraphitic carbon byCLD
Ethylene glycol reforming [42]
HY zeolite Hydrophobic micro-environment
Grafting silanefunctionalities Refining of pyrolysis oil [43]
Pdniobia Formation of mixed oxide Doping by additionof 5 SiO
2 GVL to pentanoic acid[44]
PdSBAsilica
Deposition of hydrother-mally stable overcoat Niobia ALD [45]
Coke deposition anddealumination
MicroporousHZSM-5 Change reaction media Methanol co-feed Bio-oil to hydrocarbons [46]
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 585
a preferential interaction of the overcoatwith undercoordinated Cu atomswhich aremore exposed to sintering and leaching[32]However access to Cu sites decreasedover time on stream due to coke deposit inthe small pores in the overcoat leading todeactivation reversible upon calcinationFinally the overcoat did not influence se-lectivity as furfuryl alcohol remained theonly hydrogenation product that was ob-served
Al2O
3overcoating by ALD could also
be used to increase the stability in gas-phase reactions Zhang et al reported en-hanced stability for the hydrogenation offurfural by ALD overcoating using copperchromite catalysts in the gas phase[3435]Similarly to the CuAl
2O
3system described
above the overcoat preferentially blockedundercoordinated Cu atoms which werepresent in the form of step edges and sur-face defects The overcoat led to increasedstability by decreasing three deactivationpathways sintering of Cu coke formationand coverage of active Cu sites by chromitemigration In addition secondary reactionssuch as further dehydration of furfuryl al-cohol to 2-methyl furan were disfavoredwhen covering undercoordinated atoms re-sulting in higher furfuryl alcohol selectiv-ity ThinALD overcoat (10 cycles) showed
high activity but low stability while thickALD overcoat (45 cycles) provided highstability but lower activities The high ac-tivity of coated materials was attributed toan increased reduction temperature whichdecreased the activation energy of furfuralon Cu thanks to a modified ratio of Cu0Cu+ and Cu2+species Prior FTIR studies ofadsorbed CO on Pd catalysts ruled out theexistence of electronic effects between theALD layer and metal nanoparticles[3954]
Rather the authors suggested that the over-coat influences the transport of gas to andfrom the copper surface
Besides Al2O
3 successful TiO
2ALD
overcoating was used to stabilize CoTiO
2during aqueous phase hydrogenation
(APH) reactions[36] The analogous Al2O
3
Coγ-Al2O
3was inactive for the APH of
furfural due to the formation of cobalt alu-minate after calcination treatment whichwas unreducible up to 800 degC Hencethe authors investigated the performancesof TiO
2CoTiO
2prepared by ALD from
TiCl4on TiO
2-supported Co particles H
2-
temperature programmed reduction (TPR)revealed that switching fromAl
2O
3to TiO
2allowed reduction of Co at 600 degC lead-ing to furfural conversionWhile the parentcatalyst suffered fromcobalt leaching (10Co lost over 2100minutes) the metal load-ing of overcoated material remained con-stant even after regeneration Finally TEMstudy showed that the overcoat prevented
Fig 3 a) Strategy for the synthesis of stable CuAl2O3 catalysts formation of Al2O3 supportedcopper particles overcoating by ALD and cracking of overcoat by calcination b) Mechanismof Al2O3 ALD on Al2O3 (red O purple Al black C blue H) c) Embedment of Co onto HZSM-5matrix Reproduced with permission from ref [38] Copyright 2014 American Chemical Society
c)
d)
a)
b)
Fig 4 STEM image of 45ALDCuAl2O3 (a) Particle size distribution of fresh and used catalysts(b) TOF in furfural hydrogenation for 45ALDCuAl2O3 (solid) and CuAl2O3 (hollow) in 1-butanol (c)and water (d) (fresh one regeneration O two regenerations loz three regenerations Δ) Adaptedwith permission from Wiley-VCH Verlag from ref [33]
586 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
the nanoparticles from sintering whereassintering was observed during calcinationin the absence of overcoat
The methods above stabilized metalnanoparticles by adding a separate protec-tive layer over selected parts of the supportand metal particle surface Sintering andleaching of base metal catalysts could alsobe limited by embedding the metal into astructure[55] For instance Co nanoparti-cles were loaded into an HZSM-5 zeoliteproviding a bifunctional catalyst for theconversion of levulinic acid into valericbiofuel where cobalt performed hydroge-nation while the acidic support catalyzedγ-valerolactone (GVL) ring opening (Fig3c)[38] In contrast to the incipient wetnessimpregnation approach cobalt was em-bedded into HZSM-5 by nucleation andgrowth of HZSM-5 crystals onto Co
3O
4
SiO2whereas SiO
2was progressively dis-
solved thanks to the basicity of the systemH
2-TPR showed a lower temperature for
the reduction of cobalt oxide for the im-pregnated (CoHZSM-5) compared to theembedded (CoHZSM-5) catalyst dem-onstrating a higher metal-support interac-tion for the latter The strong interactionhad a limited influence on the Co acces-sibility as demonstrated by a similar H
2up-
take for both materials Structural charac-terization was performed on CoHZSM-5and CoHZSM-5 before and after cataly-sis to identify stability differences FirstTEM revealed a stable Co NPs size forCoHZSM-5 while for CoHZSM-5 theaverage Co NP size increased from 25 to32 nm Second coke formation was onlydetected for CoHZSM-5 by thermogravi-metric analysis (TGA) as well as N
2physi-
sorption (decrease of surface area and porevolume) Finally XRD patterns showedthat both materials maintained their crys-tal structure
Similar to Cu and Co supported Niparticles are relevant systems for the low-temperature conversion of biomass-de-rived compounds ndash notably for lignin hy-drogenolysis[56ndash58] However as for manybase metals the Ni particles suffer fromsintering Nanoconfinement of Ni par-ticles by self-assembly of Ni atoms insidethe nanoribbons of hydrotalcite-derivedmixed oxide provided increased catalyticstability for methane steam reformingcompared to Ni supported on commercialsupport surface[59] Metal embedment hasalso proven to be an efficient stabilizationmethod for lactic acid production fromglycerol Morales et al showed that MFIconstituted a stabilizing matrix for Sn andsignificantly decreased leaching of said Snwhile slowing down deactivation in com-parison to bifunctional carbon-silica com-posites and BEA zeolites[60]
Above we discussed various methodsdemonstrating that controlling local nano-
structures around the active metal catalystcould increase stability Abdelrahman etal recently showed that a simple judiciouschoice of the support could limit particlesintering during the hydrogenation oflevulinic acid[61] In fact fast deactivationwas observed for ruthenium particles sup-ported on SiO
2while increased stability
was achieved by using carbon and TiO2as
supports Al2O
3was the support that led to
the most stable catalyst which the authorsattributed to its lowest electronegativity
Another strategy is to tune the com-position of the metal itself Formation ofalloys for improved catalyst performanceshas often been used including in reactionsrelevant to biomass conversion[62] In thecase of SBA-15 the use of supported Ru-Fe catalysts led to stable conversion andselectivities over 300 h with no aggrega-tion of bimetallic particles no structurecollapse and improved selectivity for hy-drogenation of acetic acid to ethanol com-pared to monometallic catalysts[63]
Finally while many efforts have beendedicated to the control of individual char-acteristics of supported metal nanopar-ticles and their immediate environmentPrieto et al showed that controlling thecollective properties of supported metalnanoparticles led to increased stability[64]By achieving near-maximum interpar-ticle spacing for silica-supported coppernanoparticles deactivation during metha-nol synthesis was limited with deactivationrates which were an order of magnitudelower than those observed with a referencecatalyst that had a non-uniform spatial dis-tribution of particles
3 Poisoning of the Active Sites
In addition to the reduction of the num-ber of active sites by sintering and leach-ing competitive adsorption of undesirable
compounds such as coke or sulfur is a ma-jor catalyst deactivation pathway becauseit leads to the blocking of active sitesSome poisons including coke can oftenbe removed by regenerating the catalystunder oxidative or reductive media to formvolatile species However these methodsare sometimes incompatible with certainsupports such as carbon which has lowthermal stability and will degrade duringthese regeneration treatments In additionprocesses not requiring periodic catalystregeneration are generally favored foreconomic and practical reasons Henceseveral paths have been explored to limitcatalyst poisoning
Catalyst poisoning is often describedas especially problematic in the contextof fermentation-derived media which hasbeen proposed as an important source ofbiomass-derived platform molecules[65] Inparticular sulfur-containing amino acidsseem to be particularly detrimental to cat-alyst activity and are commonly found infermentation broths[65] To alleviate deacti-vation by coke deposition of alumina-sup-ported Pd catalysts during hydrogenationof fermentation-derived triacetic acid lac-tone (TAL) Schwartz et al formed equi-molar bimetallic PdAu particles Howeverthe alloy still suffered from sulfur poison-ing similar to the parent monometalliccatalyst when co-feeding model biogenicimpurities such as methionine The au-thors reasoned that a microenvironmentunfavorable to polar species would limitdeactivation by biogenic impurities whilethe reaction with the less polar substratewould still go forward[40] TAL is highlysoluble in alcohols whereas amino acidsare not hence a poly(vinyl alcohol) (PVA)overcoat was selected to form a polymericpseudo-solvent microenvironment (Fig5a) The PVA-overcoated PdAuAl
2O
3catalyst displayed a deactivation rateof 002 hndash1 vs 012 hndash1 for the parent Pd
CuAl2O3sintering and leaching
coke deposition
Al2O3CuAl2O3low sintering and leaching
coke deposition
MgOxCuAl2O3sintering and leachinglow coke deposition
Al2O3MgOxAl2O3CuAl2O3low sintering and leaching
low coke deposition
OH OH OH
OH OH OH OH
OH
OO
OH
S OH
O
NH2
substrate poison
a) b)
Fig 5 a) Creation of microenvironment unfavorable to poisons Adapted with permission fromWiley-VCH Verlag from ref [40] b) Stabilization of Cu(black)Al2O3 via ALD of Al2O3 (violet) andMgOx (blue)
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 587
Al2O
3for initial conversions between 50
and 70 Stabilization by PVA was alsohighly efficient when using TAL producedby fermentation without any feed purifi-cation Comparison of 13C NMR spectraof 13C-enriched methionine adsorbed onPVA-covered and non-covered PdAl
2O
3showed that the polymer acts as a lsquosolidsolventrsquo by limiting methionine adsorptionon Pd
The ALD overcoat strategy describedin the previous section for metal sinteringand leaching prevention has also provenuseful for limiting coke deposition Lu etal reported low reversible deactivation ofovercoated PdAl
2O
3for the dehydroge-
nation of ethane to ethylene at 650 degC[39]After using 45ALDcycles to form anAl
2O
3thin film the amount of coke deposited asdetermined by thermogravimetric analy-sis was more than 16 times lower than theamount measured for the parent PdAl
2O
3catalyst The reduction of the amount ofcoke formed was attributed to the blockingof the low-coordination Pd surface siteswhich were preferentially coated In factthe later sites favored CndashC bond scissionand hydrogen stripping resulting in the un-desirable formation of coke CH
4 CO and
CO2 Hence in addition to a lower catalyst
deactivation coating led to higher ethyleneyields
The Al2O
3overcoated CuAl
2O
3de-
scribed earlier suffered from reversiblecoke deposition In a separate study theauthors managed to limit this phenomenonby finely tuning the overcoat In addition totheAl
2O
3overcoat which provided particle
stabilization basic MgOxwas deposited to
reduce the overcoatrsquos acidity which wasassumed to catalyze the formation of coke(Fig 5b)[31] Interestingly pure MgO
xdid
not provide stabilization against sinteringThe local structure of the active site can
also reduce poisoning by affecting localmass transfer To suppress deactivation byfouling due to coke deposition during theconversion of propanal to hydrocarbons onAl-MFI Luo et al used an open mesopo-rous structure based on nanosheets to limitthe effects of fouling[66] This strategy re-sulted in a five-fold decrease of deactiva-tion rates In addition to an increase in thenumber of pore openings improved sta-bility was attributed to the short diffusionpath for reagents in the single unit-cell-thick nanosheet material which limitedcoke formation that was favored by longercontact times with the reaction products
4 Increasing Support Stability inHydrothermal Conditions
In addition to deactivation due to metalleaching and sintering loss of surface areaand pore collapsing in oxide supports is a
major source of irreversible deactivationespecially in aqueous phase conditionsFor biomass conversion processes whichoften require liquid phase andor aqueousmedia at low pH hydrothermal stabilityis a major concern Metal oxides are of-ten used as supports due to their thermalstability and high surface area Howeverthey tend to have low stability under theaforementioned conditions[27] Such ox-ides can sometimes be substituted by morestable support such as carbon but someprocesses require specific surface func-tionalities such as the acidic sites presenton alumina In addition contrary to car-bon most oxides have excellent thermalstability which is necessary during certaincatalyst regeneration processes Prominentsupports such as alumina and silica-basedmaterials undergo dissolution by hydroly-sis of aluminoxane and siloxane bonds Inaddition to loss of surface area hydrolysisof oxide network can lead to modificationof chemical composition For instance hy-drolysis of the Si-O-Al bond in zeolites fa-vors dealumination This section describesthe main techniques used to stabilize metaloxides supports in the area of biomass con-version
Formation of mixed oxides or supportdoping can be used to limit this phenome-non For instance Pham et al reported thatthe stability of Pdniobia during transfor-mation of γ-valerolactone (GVL) to pen-tanoic acid could be improved by additionof 5 SiO
2into niobia[44] Niobia could
not be entirely substituted by silica be-cause acid sites are required for GVL ringopening in addition to metal sites for thehydrogenation reaction However the ad-dition of a small portion of SiO
2improves
stability while retaining activity The com-parison of the N
2physisorption isotherms
of a Pdniobia HY commercial referencecatalyst before and after the GVL reactionshowed a dramatic surface area loss from150 to 8 m2g while the porous structurewas affected to a lesser extent for PdNb-Sioxide with a decrease from 81 to 31 m2gSimilarly HAADF-STEM and HRTEMrevealed a lower Pd particle growth uponreaction for the stabilized system than forthe PdHY reference catalyst SimilarlyMazumder et al reported the promotionof Niγ-Al
2O
3by 5 wt La
2O
3leading to
dramatic improvement of activity and sta-bility for the catalytic gasification of glu-cose compared to the parent catalyst whichsuffered from metal sintering[67]
The stabilizing oxide can also be in-troduced by ALD similar to the thin over-coats deposited for the stabilization ofsupported metal particles as described inSection 2 For instance hydrothermallystable Nb-Si oxide catalysts were preparedusing ALD of niobia within the pores ofmesoporous silica SBA-15 using between10 and 30 cycles which alternated expo-sure of the Nb(OCH
2CH
3)4precursor and
H2O to the SBA-15 surface (Fig 6a)[45]
The consumption of silanol group on thesurface was complete after 19 cycles as
a)
b)
c)
Fig 6 a) Strategyfor the synthesis ofhydrothermally stablehighly ordered niobiamesoporous materialsby ALD Reproducedwith permission fromref [45] Copyright2011 AmericanChemical Societyb) Strategy for theformation of niobiacarbon compositecatalysts Adaptedwith permission ofElsevier from ref [68]c) Deposition of gra-phitic carbon layer onalumina powder andpellets by chemicallayer deposition fromCH4 Reproducedwith permission fromWiley-VCH Verlagfrom ref [42]
588 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
demonstrated by the disappearance of theSiOH band at 3735 cmndash1 in the FTIR spec-tra Hydrated amorphous niobia was iden-tified as a major species by Raman spec-troscopy for samples presenting 10 19 and30 ALD layers N
2physisorption showed
a decrease of surface area pore volumeand pore diameter with increasing niobialoadings The presence of acid sites formedupon niobia deposition was verified byNH
3-TPD and hydrothermal stability was
tested by treating the materials in liquidwater for 12 h at 200 degC and 28 bar argonThe structure of the coated materials aftertreatment was confirmed by X-ray diffrac-tion with the persistence of the hexagonalmesoporous structure pattern while the lat-ter pattern was suppressed after hydrother-mal treatment of the parent SBA-15 TEManalysis confirmed the stability differencesobserved by XRD A bifunctional catalystfor the transformation of GVL to pentanoicacid was also formed by subsequent depo-sition of Pd nanoparticles As for the sup-port this catalyst showed higher stabilitythan the conventional PdHY-340
Improved hydrothermal stability ofa niobia-supported Pd catalyst was alsoachieved by deposition of the latter ona more stable surface such as carbonHighly dispersed spherical particles of aniobiacarbon composite were formed bythe reaction of d-glucose and ammoniumniobium oxalate in an autoclave at 200degC (pH was controlled by urea decom-position) followed by pyrolysis at 400degC under N
2(Fig 6b)[68] Hydrothermal
stability was investigated during liquid-phase butanol dehydration at 240 degC and51 bar While initial activity of the com-posite bifunctional catalyst was lower thanthe commercial HY-340 catalyst the lat-ter quickly deactivated during the first 20h of reaction while the modified catalystsremained very stable and surpassed HY-340 after 8 h Interestingly the analogousmaterial obtained by incipient wetness im-pregnation of niobia led to the formation oflarge niobia crystallites underscoring theimportance of strong interaction betweenniobia and carbon[69] Finally bifunctionalcatalysts were prepared by incipient wet-ness impregnation of Pd(NO
3)2and tested
during the conversion of GVL to pentanoicacid leading to improved hydrothermalstability for the niobiacarbon compositecatalysts just like the metal-free system
For silica and alumina improved hy-drothermal stability above 100 degC wasachieved by deposition of a thin film of car-bon formed from simple sugars Pham et aldeposited carbon by pyrolysis of adsorbedsucrose at 400 degC under N
2flow on SBA-
15 silica gel and fumed alumina leadingto improved stability after treatment in liq-uid water at 100 degC for 12 h[41] MoreoverPd was deposited on the carbon-coated
oxides to test their ability as supports forthe catalytic hydrogenation of acetylenein the presence of excess ethylene For acomparable Pd particle size (1ndash15 nm)the authors observed formation of ethaneat high acetylene conversion on both SiO
2andAl
2O
3 while carbon-coating prevented
over-hydrogenation to ethane The FTIRstudy showed a low hydration potential forthe coated materials compared to SiO
2and
Al2O
3 Therefore the higher selectivity to
ethylene for the coated materials was at-tributed to their increased hydrophobicity
More recently Xiong et al reported onthe deposition of a graphitic carbon layerover alumina by chemical vapor deposition(CVD) from CH
4(Fig 6c)[42] The graphit-
ic carbon layer provided higher stabilitythan the carbon deposited from pyrolysisof sugars as demonstrated by the higherstability achieved under flowing air (500vs 250 degC) As monitored by the growthof the characteristic graphitic peak at 24deg2θ on the XRD pattern the amount of de-posited carbon was controlled by the CH
4deposition time (from 05 to 6 h) whereasother peaks attributed to γ-Al
2O
3were un-
touched TEM and energy-filtered TEM at-tested that the carbon coating was uniformon the oxide support Raman spectros-copy mapping confirmed that depositionwas uniform over the entire surface of thespheres Furthermore the relatively highratio (071) between the peak intensitiesfor G and D bands (at 1590 and 1350 cmndash1respectively) in the Raman spectrum of gc-Al
2O
3revealed a high degree of graphitiza-
tion on the entire surface No such featureswere observed for blank alumina and pyro-lyzed carbon on alumina
The hydrothermal stability of the cata-lyst was investigated using a treatmentunder static conditions at 220 degC underautogenic pressure in an autoclave usingwater After treatment the XRD pattern ofγ-Al
2O
3displayed the well-defined diffrac-
tion peaks of the boehmite phase attestingto a reaction with water Simultaneouslyno significant change was observed forgraphitic-carbon covered γ-Al
2O
3(gc-
Al2O
3) Improved stability for gc-Al
2O
3was confirmed by N
2physisorption results
which showed that no significant decreaseof surface area pore volume and poresize occurred during hydrothermal treat-ment while γ-Al
2O
3saw its surface area
decrease by 80 during the same treat-ment Stability of gc-Al
2O
3as a support
was tested in the aqueous phase reforming(APR) of ethylene glycol at 250 degC and70 bar after deposition of platinum par-ticles Conversion using Ptgc-Al
2O
3was
twice as high as for Ptγ-Al2O
3 This result
was consistent with the leached Al and Ptdetected in the liquid phase after reactionby ICP-OES for the uncoated materialIn addition to full conversion of γ-Al
2O
3
to boehmite observed by XRD STEMshowed Pt particle size increase for Ptγ-Al
2O
3 Hence deactivation of Ptγ-Al
2O
3was also attributed to Pt particle sinteringin addition to leaching whereas the use ofgraphitic carbon overcoat prevented bothphenomena under APR conditions Thisapproach was also successful when usingRugc-Al
2O
3for lactic acid hydrogenation
Overall graphitic carbon layers depositedthrough vapor phase route provided higherhydrothermal stability than pyrolytic car-bon-coatings and seems to be a promisingstabilization method for a wide range ofapplications
In addition to the deposition of thinfilm precise surface functionalization canalso be used to improve catalyst stabilityFor instance the generation of hydropho-bic surfaces by grafting of octadecyltri-chlorosilane groups onHY zeolite prevent-ed the support structure from collapsingduring stability tests which mimicked theupgrading of biomass-derived liquid com-pounds in biphasic systems at 200 degC[43]Stabilization was attributed to a limitedcontact between the zeolitersquos hydrophobicsurface and the aqueous phase The sameauthors also reported a systematic studywith various alkylsilanes from C
2to C
18[70]
Hydrothermal stability of zeolites hasbeen studied in great detail as zeolites arethemost widely used solid acid catalysts[71]and are often proposed for biomass con-version[7273] As shown by detailed char-acterization using 27Al and 29Si solid-stateNMR and NH
3-TPD siloxane bridge hy-
drolysis was the main deactivation routein the presence of hot liquid water whilezeolites treated under steam suffered fromdealumination[74] After a treatment in hotwater at 200 degC mimicking APR condi-tions ZSM-5 was stable independent ofits SiAl ratio while degradation of zeoliteY increased with increasing SiAl ratioas opposed to the better stability of zeo-lites with higher SiAl in the presence ofsteam Finally Al-rich zeolite Y could bestabilized by incorporation of lanthanumcations[74]
Recently Gardner et al reported thattesting the hydrothermal stability of zeo-lites in distilled water was not completelyrelevant to biomass conversion reactionconditions[75] In fact dry biomass con-tains about 15 wt of inorganic saltssuch as NaCl and the dissolution rate ofmetal oxides could be dramatically en-hanced by cations in solution[76] Catalytictests showed that the presence of NaCl hadno influence on glucose conversion duringthe liquid-phase transformation of glucoseto HMF on ZSM-5 However zeolite hy-drothermal stability was significantly per-turbed by the presence of NaCl leading tothe hydrolytic attack of extra-frameworkspecies andor defects sites Interestingly
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 589
leached Al species were active in the con-version of glucose to fructose
Together with metal particles zeolitesconstitute an important class of bifunc-tional catalysts While studies mentionedabove aimed at adding a stabilizing ele-ment to an existing material Ennaert et alassessed the stability of Ruzeolite H-USYin hot liquid water for the hydrolytic hy-drogenation of cellulose using variousSiAl ratios in order to select the optimalzeolite composition[77] They identifiedthat the lattice was stabilized by frame-work aluminum atoms which preventedhydrolysis of framework bonds while theextra-framework aluminum atoms locatedat the external surface prevented zeolitesolubilization With a SiAl ratio greaterthan 3 a stable quantitative hexitol yieldwas obtained
5 Improving Catalyst Stability byProcess Design
Previous sections demonstrated howimportant smart catalyst design can be forprolonged catalyst lifetime In additionimproved stability for a given catalyst canbe achieved by tuning process parametersIn fact proper reactions conditions such asthe choice of gas atmosphere (eg hydro-gen versus an inert gas) or concentrationof reagents and solvents can have dra-matic effects on processes As mentionedpreviously leaching and sintering of metalnanoparticles tends to be more pronouncedat low pH Nickel at pH values below 7 canbe oxidized by water or acids[78] to formNi2+ species[79] To counterbalance this ef-fect van Haasterecht et al used additionof KOH in ethylene glycol conversion overNiCNF catalyst with KOH concentrationbetween 0ndash12 M to increase the pH andimprove catalyst stability (Fig 7)[37] Niparticles were less stable in hydrothermalconditions in comparison to ethylene gly-col In both cases increasing the pH hada significant impact though it was not aspronounced in ethylene glycol Moreoverat very high pH (gt13) sintering occurredagain which was probably due to a for-mation of highly mobile hydroxide nickelspecies
A reductive gas atmosphere can lead toincreased catalyst stability A typical ex-ample is the aqueous phase reforming ofethylene glycol conducted over Ni-basedcatalysts[37] Introduction of hydrogen inthe gas atmosphere suppressed the growthof nickel nanoparticles The average par-ticle size after reaction in a hydrogen andan inert atmosphere was 58 and 32 nmrespectively Moreover leaching of thenickel decreased leading to 026 catalystmass loss under inert atmosphere com-pared to 015 mass loss in hydrogen
However the use of hydrogen is notalways beneficial It was reported that forplatinum supported on graphite at highpH under a hydrogen-rich atmosphereand at an elevated temperature agglom-eration of platinum could occur[80] Thereason was that removal of the graphiteoxygen functional groups by hydrogenlead to lower adhesion and thereforehigher mobility of the nanoparticles onthe surface
Coke formation over the catalyst canalso be inhibited by co-feeding other sub-stances to the reactor For instance in cata-lytic conversion of bio-oil into hydrocar-bons a stream of bio-oil was diluted witha ratio 12 with water which decreased themass of carbon deposits on microporousZSM-5[46] Unfortunately this treatmentled to irreversible catalyst deactivation dueto dealumination of the zeolite caused bywater at high temperature[8182] Howeverthe same authors found that the use of a70 wt methanol with a bio-oil mixturecould decrease coke formation up to 70and prevent dealumination and thereforemaintain activity of the catalyst
Preventing deactivation can also beachieved by controlling the reaction me-dia During the process of producing fur-fural from xylans high deactivation ofthe GaUSY catalyst was observed whenusing water as a solvent XRD analysis ofthe spent catalyst confirmed the collapseof the zeolite structure during reactionMoreover gallium content dramaticallydecreased after 24 hours of reaction from59 wt to 027 wt To avoid the nega-tive effects of water on the catalyst thebiphasic system waterndashmethyl isobutyl
ketone (MIBK) was used with a ratio of2080 Gallium leaching decreased duringthe first few hours (36 wt Ga) and con-tinued but at a lower rate to reach a metalcontent of 19 wt at the end of the reac-tion Furthermore XRD confirmed that thezeolite retained its structure at the end ofthe experiment confirming the importanceof using a co-solvent[83]
6 Conclusions and Outlook
Deactivation of catalysts under theharsh conditions used for biomass conver-sion is a bottleneck in the developmentof competitive processes for bioresourcevalorization to renewable alternatives tofossil-based fuels and chemicals Biomass-derived molecules that must be further up-graded are usually produced in relativelydilute liquid (often aqueous) streamswhich contain biogenic and inorganic im-purities This introduces significant chal-lenges for heterogeneous catalysts whichhave often been developed in the context ofreforming fairly pure gas-phase hydrocar-bons An important category of catalystsfor catalytic biomass upgrading are oxide-supported metal particles due to their usein hydrogenation and hydrodeoxygenationreactions are essential for removing oxy-gen from bio-based molecules Several re-searchers have sought to reduce the maindeactivation pathways for these catalystsnotably their irreversible deactivation viametal leaching and sintering in the liquidphase as well as their reversible deactiva-tion by poisoning Leaching and sinteringoccur preferentially due to the instability
Fig 7 Approximation based on experimental data between nickelnanoparticles size of the spent NiCNF catalysts and the pH of the finalreaction mixture in aqueous phase reforming of ethylene glycol ThepH of the feed solutions was adjusted by addition of KOH (0-12 M) toaqueous solutions of 1 wt ethylene glycol (green line) and to water(blue line) Reactions were performed at 230 degC for 6 h under inert gasAdapted with permission of Elsevier from ref [37]
590 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
of undercoordinated metal atoms Suchatoms can also favor byproduct formationdue to their high reactivity These byprod-ucts include coke in which case these re-active atoms contribute to poisoning theother active sites by initiating the forma-tion of carbon deposits For these reasonsseveral authors have reported methods topassivate these undercoordinated atomsusing overcoats deposited either by ALDsugar pyrolysis or CVD thereby stabiliz-ing the catalyst of interest Another issueis the leaching and structure modificationsof oxide supports due to the poor hydro-thermal stability of these solids whichinclude silica and zeolites In this caseseveral authors have turned to fine-tuningthe local environment of these catalystsagain by physically covering the materialof interest The addition of hydrophobicsurface functionalities or overcoats hasbeen shown to reduce support hydrolysisbywater molecules Similarly depositing apolymeric layer over metal hydrogenationsites was shown to act as an organic pseu-do-solvent which reduced the presence ofpolar biogenic catalyst poisons and thuscatalyst deactivation Finally increasedcatalyst lifetime was also improved byprocess engineering In fact tuning thereaction conditions including by loweringpH or co-feeding organic solvents and hy-drogen proved to be efficient for limitingcatalyst deactivation
Further research opportunities exist tocombine the principle strategies mentionedabove to further improve catalyst stabiliza-tion Tailored overcoats could be used bothto stabilize undercoordinated atoms and atthe same time to provide favorable micro-environments that could reduce hydrolysisandor concentrations of catalyst poisonsSuch an approach was used in part by add-ingMgO
xto reduce the acidity of an alumi-
na overcoat which was thought to catalyzecoke formation while the overcoat wasused to stabilize undercoordinated atomsHowever this approach could be furtherstrengthened by adding functionality to theovercoat and further tailoring themoleculardesign of the catalystrsquos local environmentFurthermore controlling pore size open-ings within these overcoats could intro-duce shape selectivity and be used to sieveout undesired reactants as was recentlydemonstrated for an alumina overcoat on atitania catalyst used for photocatalysis[84]Finally it will become increasingly impor-tant to use real biomass-derived streamsfor testing catalytic stability The negativeeffects of using liquid or aqueous condi-tions can be compounded by the presenceof numerous perhaps yet unidentified in-organic or biogenic impurities This wasnotably demonstrated by the acceleratedstructure collapse of zeolite structures inthe presence of inorganic salts
In summary current synthetic tech-niques can lead to unprecedented controlof the nanostructures surrounding a cata-lystrsquos active site but harsh conditions pres-ent for biomass conversion are equally un-precedented in industrial catalytic process-ing As society comes under increasingpressure to develop routes to sustainablecarbon-based molecules these syntheticmethods will likely prove essential in ad-dressing the challenges posed by catalyti-cally upgrading renewable resources suchas biomass
AcknowledgementsThe authors acknowledge financial support
from the Swiss National Science Foundationthrough grant number 407040_153866 as partof the National Research Program 70 and grantnumber PYAPP2_154281 and Benjamin LeMonnier for help with the design of figures
Received June 17 2015
[1] K Sugiura M Ishihara T Shimauchi SHarayama Environ Sci Technol 1997 31 45
[2] S Matar M J Mirbach HA Tayim lsquoCatalysisin Petrochemical Processesrsquo Springer Scienceamp Business Media 1989
[3] O S Abdul-Hamid Monthly Oil MarketReport - May 2015 OPEC 2015
[4] The European Parliament and the Council of theEuropean Union OJ 2009 L140 16
[5] R Rinaldi F Schuumlth Energy Environ Sci2009 2 610
[6] A A Peterson F Vogel R P Lachance MFroumlling J Michael JAntal JW Tester EnergyEnviron Sci 2008 1 32
[7] J S Luterbacher D M Alonso J A DumesicGreen Chem 2014 16 4816
[8] A Rahimi A Ulbrich J J Coon S S StahlNature 2014 515 249
[9] A Eyal R Jansen R Mali A Vitner lsquoSugarMixtures and Methods for Production and UseThereofrsquo 2012 WO 2011161685 A3
[10] D V Kazachkin M Colakyan F J MoeslerlsquoSupercritical Hydrolysis of Biomassrsquo 2015US 8999065 B2
[11] D L Sills J M Gossett Bioresour Technol2011 102 1389
[12] D M Alonso S G Wettstein M A MellmerE I Gurbuz J A Dumesic Energy EnvironSci 2012 6 76
[13] J S Luterbacher J M Rand D M Alonso JHan J T Youngquist C T Maravelias B FPfleger J A Dumesic Science 2014 343 277
[14] J S Luterbacher D M Alonso J M Rand YM Questell-Santiago J H Yeap B F PflegerJ A Dumesic ChemSusChem 2015 8 1317
[15] H Tadesse R Luque Energy Environ Sci2011 4 3913
[16] J B Binder R T Raines J Am Chem Soc2009 131 1979
[17] J S Luterbacher J W Tester L P WalkerBiotechnol Bioeng 2010 107 451
[18] D B Hodge M N Karim D J Schell J DMcMillan Bioresour Technol 2008 99 8940
[19] J S Luterbacher Q Chew Y Li J W TesterL P Walker Energy Environ Sci 2012 56990
[20] T J Schwartz B J OrsquoNeill B H Shanks J ADumesic ACS Catal 2014 4 2060
[21] G W Huber J N Chheda C J Barrett J ADumesic Science 2005 308 1446
[22] G W Huber R D Cortright J A DumesicAngew Chem Int Ed 2004 43 1549
[23] J N Chheda Y Romaacuten-Leshkov J ADumesic Green Chem 2007 9 342
[24] D W Rackemann W O Doherty BiofuelsBioprod Biorefining 2011 5 198
[25] J N Chheda G W Huber J A DumesicAngew Chem Int Ed 2007 46 7164
[26] A V Bandura S N Lvov J Phys Chem RefData 2006 35 15
[27] H Xiong H N Pham A K Datye GreenChem 2014 16 4627
[28] K P De Jong lsquoSynthesis Of Solid CatalystsrsquoWiley-VCHVerlag GmbH amp Co KGaA 2009
[29] F Heacuteroguel G Siddiqi M D Detwiler DY Zemlyanov O V Safonova C Copeacuteret JCatal 2015 321 81
[30] T W Hansen A T DeLaRiva S R Challa AK Datye Acc Chem Res 2013 46 1720
[31] B J OrsquoNeill C Sener D H K Jackson T FKuech J A Dumesic ChemSusChem 2014 73247
[32] B J OrsquoNeill J T Miller P J Dietrich FG Sollberger F H Ribeiro J A DumesicChemCatChem 2014 6 2493
[33] B J OrsquoNeill D H K Jackson A J Crisci CA Farberow F Shi A C Alba-Rubio J Lu PJ Dietrich X Gu C L Marshall et al AngewChem 2013 125 14053
[34] H Zhang Y Lei A J Kropf G Zhang J WElam J T Miller F Sollberger F Ribeiro MC Akatay E A Stach J A Dumesic C LMarshall J Catal 2014 317 284
[35] H ZhangY Lei A Jeremy Kropf G Zhang JW Elam J T Miller F Sollberger F RibeiroM Cem Akatay E A Stach J A Dumesic CL Marshall J Catal 2015 323 165
[36] J Lee D H K Jackson T Li R E Winans JA Dumesic T F Kuech G W Huber EnergyEnviron Sci 2014 7 1657
[37] T van Haasterecht C C I Ludding K P deJong J H Bitter J Catal 2014 319 27
[38] P Sun G Gao Z Zhao C Xia F Li ACSCatal 2014 4 4136
[39] J Lu B Fu M C Kung G Xiao J W ElamH H Kung P C Stair Science 2012 3351205
[40] T J Schwartz R L Johnson J Cardenas AOkerlund N A Da Silva K Schmidt-Rohr JA Dumesic Angew Chem Int Ed 2014 5312718
[41] H N Pham A E Anderson R L Johnson KSchmidt-Rohr A K Datye Angew Chem IntEd 2012 51 13163
[42] H Xiong T J Schwartz N I Andersen J ADumesic A K Datye Angew Chem Int Ed2015 early view DOI 101002anie201502206
[43] P A Zapata J Faria M P Ruiz R E JentoftD E Resasco J Am Chem Soc 2012 1348570
[44] H N Pham Y J Pagan-Torres J C Serrano-Ruiz D Wang J A Dumesic A K DatyeAppl Catal Gen 2011 397 153
[45] Y J Pagaacuten-Torres J M R Gallo D WangH N Pham J A Libera C L Marshall J WElam A K Datye J A Dumesic ACS Catal2011 1 1234
[46] A G Gayubo B Valle A T Aguayo MOlazar J Bilbao Energy Fuels 2009 23 4129
[47] S R Challa A T Delariva T W Hansen SHelveg J Sehested P L Hansen F Garzon AK Datye J Am Chem Soc 2011 133 20672
[48] J A Moulijn A E van Diepen F KapteijnAppl Catal Gen 2001 212 3
[49] S L Wegener T J Marks P C Stair AccChem Res 2012 45 206
[50] B J OrsquoNeill D H K Jackson J Lee CCanlas P C Stair C L Marshall J W ElamT F Kuech J A Dumesic G W Huber ACSCatal 2015 5 1804
[51] D M King J A Spencer II X Liang L FHakim A W Weimer Surf Coat Technol2007 201 9163
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 591
[52] J A McCormick B L Cloutier AWWeimerS M George J Vac Sci Technol A 2007 2567
[53] D Longrie D Deduytsche C Detavernier JVac Sci Technol A 2014 32 010802
[54] J Lu J W Elam P C Stair Acc Chem Res2013 46 1806
[55] L De Rogatis M Cargnello V GombacB Lorenzut T Montini P FornasieroChemSusChem 2010 3 24
[56] A G Sergeev J D Webb J F Hartwig J AmChem Soc 2012 134 20226
[57] J He C Zhao J A Lercher J Am Chem Soc2012 134 20768
[58] Q Song F Wang J CaiY Wang J Zhang WYu J Xu Energy Environ Sci 2013 6 994
[59] R Dehghan-Niri J C Walmsley A HolmenP A Midgley E Rytter A H Dam A BHungria J C Hernandez-Garrido D ChenCatal Sci Technol 2012 2 2476
[60] M Morales P Y Dapsens I Giovinazzo JWitte C Mondelli S PapadokonstantakisK Hungerbuumlhler J Peacuterez-Ramiacuterez EnergyEnviron Sci 2015 8 558
[61] O A Abdelrahman H Y Luo A Heyden YRomaacuten-Leshkov J Q Bond J Catal 2015329 10
[62] D M Alonso S G Wettstein J A DumesicChem Soc Rev 2012 41 8075
[63] W Li L Ye P Long J Chen H Ariga KAsakura YYuan RSC Adv 2014 4 29072
[64] G Prieto J Zecevic H Friedrich K P deJong P E de Jongh Nat Mater 2013 12 34
[65] Z Zhang J E Jackson D J Miller BioresourTechnol 2008 99 5873
[66] H Luo T Prasomsri Y Romaacuten-Leshkov TopCatal 2015 394
[67] J Mazumder H I de Lasa Catal Today 2014237 100
[68] H Xiong H N Pham A K Datye J Catal2013 302 93
[69] H Xiong M Nolan B H ShanksA K DatyeAppl Catal Gen 2014 471 165
[70] P A Zapata Y Huang M A Gonzalez-BorjaD E Resasco J Catal 2013 308 82
[71] A Corma Chem RevWash DC U S 1995 95559
[72] P A Jacobs M Dusselier B F Sels AngewChem Int Ed 2014 53 8621
[73] M Milina S Mitchell J Peacuterez-Ramiacuterez CatalToday 2014 235 176
[74] R M Ravenelle F Schuumlssler A DrsquoAmico NDanilina J A van Bokhoven J A Lercher CW Jones C Sievers J Phys Chem C 2010114 19582
[75] D W Gardner J Huo T C Hoff R LJohnson B H Shanks J-P Tessonnier ACSCatal 2015 DOI 101021acscatal5b00888
[76] P M Dove N Han J J D Yoreo Proc NatlAcad Sci 2005 102 15357
[77] T Ennaert J Geboers E Gobechiya C MCourtin M Kurttepeli K Houthoofd C E AKirschhock P C M M Magusin S Bals P AJacobs B F Sels ACS Catal 2015 5 754
[78] B Beverskog I Puigdomenech Corros Sci1997 39 969
[79] D A Palmer P Beacuteneacutezeth C Xiao D JWesolowski L M Anovitz J Solut Chem2011 40 680
[80] J H Vleeming B F M Kuster G B Marin FOudet P Courtine J Catal 1997 166 148
[81] A G Gayubo A T Aguayo M Olazar RVivanco J Bilbao Chem Eng Sci 2003 585239
[82] A de Lucas P CanizaresA DuraacutenA CarreroAppl Catal Gen 1997 154 221
[83] CAellig D Scholz PY Dapsens C MondelliJ Peacuterez-Ramiacuterez Catal Sci Technol 2014 5142
[84] C P Canlas J Lu N A Ray N A Grosso-Giordano S Lee J W Elam R E Winans RP Van Duyne P C Stair J M Notestein NatChem 2012 4 1030

SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 585
a preferential interaction of the overcoatwith undercoordinated Cu atomswhich aremore exposed to sintering and leaching[32]However access to Cu sites decreasedover time on stream due to coke deposit inthe small pores in the overcoat leading todeactivation reversible upon calcinationFinally the overcoat did not influence se-lectivity as furfuryl alcohol remained theonly hydrogenation product that was ob-served
Al2O
3overcoating by ALD could also
be used to increase the stability in gas-phase reactions Zhang et al reported en-hanced stability for the hydrogenation offurfural by ALD overcoating using copperchromite catalysts in the gas phase[3435]Similarly to the CuAl
2O
3system described
above the overcoat preferentially blockedundercoordinated Cu atoms which werepresent in the form of step edges and sur-face defects The overcoat led to increasedstability by decreasing three deactivationpathways sintering of Cu coke formationand coverage of active Cu sites by chromitemigration In addition secondary reactionssuch as further dehydration of furfuryl al-cohol to 2-methyl furan were disfavoredwhen covering undercoordinated atoms re-sulting in higher furfuryl alcohol selectiv-ity ThinALD overcoat (10 cycles) showed
high activity but low stability while thickALD overcoat (45 cycles) provided highstability but lower activities The high ac-tivity of coated materials was attributed toan increased reduction temperature whichdecreased the activation energy of furfuralon Cu thanks to a modified ratio of Cu0Cu+ and Cu2+species Prior FTIR studies ofadsorbed CO on Pd catalysts ruled out theexistence of electronic effects between theALD layer and metal nanoparticles[3954]
Rather the authors suggested that the over-coat influences the transport of gas to andfrom the copper surface
Besides Al2O
3 successful TiO
2ALD
overcoating was used to stabilize CoTiO
2during aqueous phase hydrogenation
(APH) reactions[36] The analogous Al2O
3
Coγ-Al2O
3was inactive for the APH of
furfural due to the formation of cobalt alu-minate after calcination treatment whichwas unreducible up to 800 degC Hencethe authors investigated the performancesof TiO
2CoTiO
2prepared by ALD from
TiCl4on TiO
2-supported Co particles H
2-
temperature programmed reduction (TPR)revealed that switching fromAl
2O
3to TiO
2allowed reduction of Co at 600 degC lead-ing to furfural conversionWhile the parentcatalyst suffered fromcobalt leaching (10Co lost over 2100minutes) the metal load-ing of overcoated material remained con-stant even after regeneration Finally TEMstudy showed that the overcoat prevented
Fig 3 a) Strategy for the synthesis of stable CuAl2O3 catalysts formation of Al2O3 supportedcopper particles overcoating by ALD and cracking of overcoat by calcination b) Mechanismof Al2O3 ALD on Al2O3 (red O purple Al black C blue H) c) Embedment of Co onto HZSM-5matrix Reproduced with permission from ref [38] Copyright 2014 American Chemical Society
c)
d)
a)
b)
Fig 4 STEM image of 45ALDCuAl2O3 (a) Particle size distribution of fresh and used catalysts(b) TOF in furfural hydrogenation for 45ALDCuAl2O3 (solid) and CuAl2O3 (hollow) in 1-butanol (c)and water (d) (fresh one regeneration O two regenerations loz three regenerations Δ) Adaptedwith permission from Wiley-VCH Verlag from ref [33]
586 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
the nanoparticles from sintering whereassintering was observed during calcinationin the absence of overcoat
The methods above stabilized metalnanoparticles by adding a separate protec-tive layer over selected parts of the supportand metal particle surface Sintering andleaching of base metal catalysts could alsobe limited by embedding the metal into astructure[55] For instance Co nanoparti-cles were loaded into an HZSM-5 zeoliteproviding a bifunctional catalyst for theconversion of levulinic acid into valericbiofuel where cobalt performed hydroge-nation while the acidic support catalyzedγ-valerolactone (GVL) ring opening (Fig3c)[38] In contrast to the incipient wetnessimpregnation approach cobalt was em-bedded into HZSM-5 by nucleation andgrowth of HZSM-5 crystals onto Co
3O
4
SiO2whereas SiO
2was progressively dis-
solved thanks to the basicity of the systemH
2-TPR showed a lower temperature for
the reduction of cobalt oxide for the im-pregnated (CoHZSM-5) compared to theembedded (CoHZSM-5) catalyst dem-onstrating a higher metal-support interac-tion for the latter The strong interactionhad a limited influence on the Co acces-sibility as demonstrated by a similar H
2up-
take for both materials Structural charac-terization was performed on CoHZSM-5and CoHZSM-5 before and after cataly-sis to identify stability differences FirstTEM revealed a stable Co NPs size forCoHZSM-5 while for CoHZSM-5 theaverage Co NP size increased from 25 to32 nm Second coke formation was onlydetected for CoHZSM-5 by thermogravi-metric analysis (TGA) as well as N
2physi-
sorption (decrease of surface area and porevolume) Finally XRD patterns showedthat both materials maintained their crys-tal structure
Similar to Cu and Co supported Niparticles are relevant systems for the low-temperature conversion of biomass-de-rived compounds ndash notably for lignin hy-drogenolysis[56ndash58] However as for manybase metals the Ni particles suffer fromsintering Nanoconfinement of Ni par-ticles by self-assembly of Ni atoms insidethe nanoribbons of hydrotalcite-derivedmixed oxide provided increased catalyticstability for methane steam reformingcompared to Ni supported on commercialsupport surface[59] Metal embedment hasalso proven to be an efficient stabilizationmethod for lactic acid production fromglycerol Morales et al showed that MFIconstituted a stabilizing matrix for Sn andsignificantly decreased leaching of said Snwhile slowing down deactivation in com-parison to bifunctional carbon-silica com-posites and BEA zeolites[60]
Above we discussed various methodsdemonstrating that controlling local nano-
structures around the active metal catalystcould increase stability Abdelrahman etal recently showed that a simple judiciouschoice of the support could limit particlesintering during the hydrogenation oflevulinic acid[61] In fact fast deactivationwas observed for ruthenium particles sup-ported on SiO
2while increased stability
was achieved by using carbon and TiO2as
supports Al2O
3was the support that led to
the most stable catalyst which the authorsattributed to its lowest electronegativity
Another strategy is to tune the com-position of the metal itself Formation ofalloys for improved catalyst performanceshas often been used including in reactionsrelevant to biomass conversion[62] In thecase of SBA-15 the use of supported Ru-Fe catalysts led to stable conversion andselectivities over 300 h with no aggrega-tion of bimetallic particles no structurecollapse and improved selectivity for hy-drogenation of acetic acid to ethanol com-pared to monometallic catalysts[63]
Finally while many efforts have beendedicated to the control of individual char-acteristics of supported metal nanopar-ticles and their immediate environmentPrieto et al showed that controlling thecollective properties of supported metalnanoparticles led to increased stability[64]By achieving near-maximum interpar-ticle spacing for silica-supported coppernanoparticles deactivation during metha-nol synthesis was limited with deactivationrates which were an order of magnitudelower than those observed with a referencecatalyst that had a non-uniform spatial dis-tribution of particles
3 Poisoning of the Active Sites
In addition to the reduction of the num-ber of active sites by sintering and leach-ing competitive adsorption of undesirable
compounds such as coke or sulfur is a ma-jor catalyst deactivation pathway becauseit leads to the blocking of active sitesSome poisons including coke can oftenbe removed by regenerating the catalystunder oxidative or reductive media to formvolatile species However these methodsare sometimes incompatible with certainsupports such as carbon which has lowthermal stability and will degrade duringthese regeneration treatments In additionprocesses not requiring periodic catalystregeneration are generally favored foreconomic and practical reasons Henceseveral paths have been explored to limitcatalyst poisoning
Catalyst poisoning is often describedas especially problematic in the contextof fermentation-derived media which hasbeen proposed as an important source ofbiomass-derived platform molecules[65] Inparticular sulfur-containing amino acidsseem to be particularly detrimental to cat-alyst activity and are commonly found infermentation broths[65] To alleviate deacti-vation by coke deposition of alumina-sup-ported Pd catalysts during hydrogenationof fermentation-derived triacetic acid lac-tone (TAL) Schwartz et al formed equi-molar bimetallic PdAu particles Howeverthe alloy still suffered from sulfur poison-ing similar to the parent monometalliccatalyst when co-feeding model biogenicimpurities such as methionine The au-thors reasoned that a microenvironmentunfavorable to polar species would limitdeactivation by biogenic impurities whilethe reaction with the less polar substratewould still go forward[40] TAL is highlysoluble in alcohols whereas amino acidsare not hence a poly(vinyl alcohol) (PVA)overcoat was selected to form a polymericpseudo-solvent microenvironment (Fig5a) The PVA-overcoated PdAuAl
2O
3catalyst displayed a deactivation rateof 002 hndash1 vs 012 hndash1 for the parent Pd
CuAl2O3sintering and leaching
coke deposition
Al2O3CuAl2O3low sintering and leaching
coke deposition
MgOxCuAl2O3sintering and leachinglow coke deposition
Al2O3MgOxAl2O3CuAl2O3low sintering and leaching
low coke deposition
OH OH OH
OH OH OH OH
OH
OO
OH
S OH
O
NH2
substrate poison
a) b)
Fig 5 a) Creation of microenvironment unfavorable to poisons Adapted with permission fromWiley-VCH Verlag from ref [40] b) Stabilization of Cu(black)Al2O3 via ALD of Al2O3 (violet) andMgOx (blue)
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 587
Al2O
3for initial conversions between 50
and 70 Stabilization by PVA was alsohighly efficient when using TAL producedby fermentation without any feed purifi-cation Comparison of 13C NMR spectraof 13C-enriched methionine adsorbed onPVA-covered and non-covered PdAl
2O
3showed that the polymer acts as a lsquosolidsolventrsquo by limiting methionine adsorptionon Pd
The ALD overcoat strategy describedin the previous section for metal sinteringand leaching prevention has also provenuseful for limiting coke deposition Lu etal reported low reversible deactivation ofovercoated PdAl
2O
3for the dehydroge-
nation of ethane to ethylene at 650 degC[39]After using 45ALDcycles to form anAl
2O
3thin film the amount of coke deposited asdetermined by thermogravimetric analy-sis was more than 16 times lower than theamount measured for the parent PdAl
2O
3catalyst The reduction of the amount ofcoke formed was attributed to the blockingof the low-coordination Pd surface siteswhich were preferentially coated In factthe later sites favored CndashC bond scissionand hydrogen stripping resulting in the un-desirable formation of coke CH
4 CO and
CO2 Hence in addition to a lower catalyst
deactivation coating led to higher ethyleneyields
The Al2O
3overcoated CuAl
2O
3de-
scribed earlier suffered from reversiblecoke deposition In a separate study theauthors managed to limit this phenomenonby finely tuning the overcoat In addition totheAl
2O
3overcoat which provided particle
stabilization basic MgOxwas deposited to
reduce the overcoatrsquos acidity which wasassumed to catalyze the formation of coke(Fig 5b)[31] Interestingly pure MgO
xdid
not provide stabilization against sinteringThe local structure of the active site can
also reduce poisoning by affecting localmass transfer To suppress deactivation byfouling due to coke deposition during theconversion of propanal to hydrocarbons onAl-MFI Luo et al used an open mesopo-rous structure based on nanosheets to limitthe effects of fouling[66] This strategy re-sulted in a five-fold decrease of deactiva-tion rates In addition to an increase in thenumber of pore openings improved sta-bility was attributed to the short diffusionpath for reagents in the single unit-cell-thick nanosheet material which limitedcoke formation that was favored by longercontact times with the reaction products
4 Increasing Support Stability inHydrothermal Conditions
In addition to deactivation due to metalleaching and sintering loss of surface areaand pore collapsing in oxide supports is a
major source of irreversible deactivationespecially in aqueous phase conditionsFor biomass conversion processes whichoften require liquid phase andor aqueousmedia at low pH hydrothermal stabilityis a major concern Metal oxides are of-ten used as supports due to their thermalstability and high surface area Howeverthey tend to have low stability under theaforementioned conditions[27] Such ox-ides can sometimes be substituted by morestable support such as carbon but someprocesses require specific surface func-tionalities such as the acidic sites presenton alumina In addition contrary to car-bon most oxides have excellent thermalstability which is necessary during certaincatalyst regeneration processes Prominentsupports such as alumina and silica-basedmaterials undergo dissolution by hydroly-sis of aluminoxane and siloxane bonds Inaddition to loss of surface area hydrolysisof oxide network can lead to modificationof chemical composition For instance hy-drolysis of the Si-O-Al bond in zeolites fa-vors dealumination This section describesthe main techniques used to stabilize metaloxides supports in the area of biomass con-version
Formation of mixed oxides or supportdoping can be used to limit this phenome-non For instance Pham et al reported thatthe stability of Pdniobia during transfor-mation of γ-valerolactone (GVL) to pen-tanoic acid could be improved by additionof 5 SiO
2into niobia[44] Niobia could
not be entirely substituted by silica be-cause acid sites are required for GVL ringopening in addition to metal sites for thehydrogenation reaction However the ad-dition of a small portion of SiO
2improves
stability while retaining activity The com-parison of the N
2physisorption isotherms
of a Pdniobia HY commercial referencecatalyst before and after the GVL reactionshowed a dramatic surface area loss from150 to 8 m2g while the porous structurewas affected to a lesser extent for PdNb-Sioxide with a decrease from 81 to 31 m2gSimilarly HAADF-STEM and HRTEMrevealed a lower Pd particle growth uponreaction for the stabilized system than forthe PdHY reference catalyst SimilarlyMazumder et al reported the promotionof Niγ-Al
2O
3by 5 wt La
2O
3leading to
dramatic improvement of activity and sta-bility for the catalytic gasification of glu-cose compared to the parent catalyst whichsuffered from metal sintering[67]
The stabilizing oxide can also be in-troduced by ALD similar to the thin over-coats deposited for the stabilization ofsupported metal particles as described inSection 2 For instance hydrothermallystable Nb-Si oxide catalysts were preparedusing ALD of niobia within the pores ofmesoporous silica SBA-15 using between10 and 30 cycles which alternated expo-sure of the Nb(OCH
2CH
3)4precursor and
H2O to the SBA-15 surface (Fig 6a)[45]
The consumption of silanol group on thesurface was complete after 19 cycles as
a)
b)
c)
Fig 6 a) Strategyfor the synthesis ofhydrothermally stablehighly ordered niobiamesoporous materialsby ALD Reproducedwith permission fromref [45] Copyright2011 AmericanChemical Societyb) Strategy for theformation of niobiacarbon compositecatalysts Adaptedwith permission ofElsevier from ref [68]c) Deposition of gra-phitic carbon layer onalumina powder andpellets by chemicallayer deposition fromCH4 Reproducedwith permission fromWiley-VCH Verlagfrom ref [42]
588 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
demonstrated by the disappearance of theSiOH band at 3735 cmndash1 in the FTIR spec-tra Hydrated amorphous niobia was iden-tified as a major species by Raman spec-troscopy for samples presenting 10 19 and30 ALD layers N
2physisorption showed
a decrease of surface area pore volumeand pore diameter with increasing niobialoadings The presence of acid sites formedupon niobia deposition was verified byNH
3-TPD and hydrothermal stability was
tested by treating the materials in liquidwater for 12 h at 200 degC and 28 bar argonThe structure of the coated materials aftertreatment was confirmed by X-ray diffrac-tion with the persistence of the hexagonalmesoporous structure pattern while the lat-ter pattern was suppressed after hydrother-mal treatment of the parent SBA-15 TEManalysis confirmed the stability differencesobserved by XRD A bifunctional catalystfor the transformation of GVL to pentanoicacid was also formed by subsequent depo-sition of Pd nanoparticles As for the sup-port this catalyst showed higher stabilitythan the conventional PdHY-340
Improved hydrothermal stability ofa niobia-supported Pd catalyst was alsoachieved by deposition of the latter ona more stable surface such as carbonHighly dispersed spherical particles of aniobiacarbon composite were formed bythe reaction of d-glucose and ammoniumniobium oxalate in an autoclave at 200degC (pH was controlled by urea decom-position) followed by pyrolysis at 400degC under N
2(Fig 6b)[68] Hydrothermal
stability was investigated during liquid-phase butanol dehydration at 240 degC and51 bar While initial activity of the com-posite bifunctional catalyst was lower thanthe commercial HY-340 catalyst the lat-ter quickly deactivated during the first 20h of reaction while the modified catalystsremained very stable and surpassed HY-340 after 8 h Interestingly the analogousmaterial obtained by incipient wetness im-pregnation of niobia led to the formation oflarge niobia crystallites underscoring theimportance of strong interaction betweenniobia and carbon[69] Finally bifunctionalcatalysts were prepared by incipient wet-ness impregnation of Pd(NO
3)2and tested
during the conversion of GVL to pentanoicacid leading to improved hydrothermalstability for the niobiacarbon compositecatalysts just like the metal-free system
For silica and alumina improved hy-drothermal stability above 100 degC wasachieved by deposition of a thin film of car-bon formed from simple sugars Pham et aldeposited carbon by pyrolysis of adsorbedsucrose at 400 degC under N
2flow on SBA-
15 silica gel and fumed alumina leadingto improved stability after treatment in liq-uid water at 100 degC for 12 h[41] MoreoverPd was deposited on the carbon-coated
oxides to test their ability as supports forthe catalytic hydrogenation of acetylenein the presence of excess ethylene For acomparable Pd particle size (1ndash15 nm)the authors observed formation of ethaneat high acetylene conversion on both SiO
2andAl
2O
3 while carbon-coating prevented
over-hydrogenation to ethane The FTIRstudy showed a low hydration potential forthe coated materials compared to SiO
2and
Al2O
3 Therefore the higher selectivity to
ethylene for the coated materials was at-tributed to their increased hydrophobicity
More recently Xiong et al reported onthe deposition of a graphitic carbon layerover alumina by chemical vapor deposition(CVD) from CH
4(Fig 6c)[42] The graphit-
ic carbon layer provided higher stabilitythan the carbon deposited from pyrolysisof sugars as demonstrated by the higherstability achieved under flowing air (500vs 250 degC) As monitored by the growthof the characteristic graphitic peak at 24deg2θ on the XRD pattern the amount of de-posited carbon was controlled by the CH
4deposition time (from 05 to 6 h) whereasother peaks attributed to γ-Al
2O
3were un-
touched TEM and energy-filtered TEM at-tested that the carbon coating was uniformon the oxide support Raman spectros-copy mapping confirmed that depositionwas uniform over the entire surface of thespheres Furthermore the relatively highratio (071) between the peak intensitiesfor G and D bands (at 1590 and 1350 cmndash1respectively) in the Raman spectrum of gc-Al
2O
3revealed a high degree of graphitiza-
tion on the entire surface No such featureswere observed for blank alumina and pyro-lyzed carbon on alumina
The hydrothermal stability of the cata-lyst was investigated using a treatmentunder static conditions at 220 degC underautogenic pressure in an autoclave usingwater After treatment the XRD pattern ofγ-Al
2O
3displayed the well-defined diffrac-
tion peaks of the boehmite phase attestingto a reaction with water Simultaneouslyno significant change was observed forgraphitic-carbon covered γ-Al
2O
3(gc-
Al2O
3) Improved stability for gc-Al
2O
3was confirmed by N
2physisorption results
which showed that no significant decreaseof surface area pore volume and poresize occurred during hydrothermal treat-ment while γ-Al
2O
3saw its surface area
decrease by 80 during the same treat-ment Stability of gc-Al
2O
3as a support
was tested in the aqueous phase reforming(APR) of ethylene glycol at 250 degC and70 bar after deposition of platinum par-ticles Conversion using Ptgc-Al
2O
3was
twice as high as for Ptγ-Al2O
3 This result
was consistent with the leached Al and Ptdetected in the liquid phase after reactionby ICP-OES for the uncoated materialIn addition to full conversion of γ-Al
2O
3
to boehmite observed by XRD STEMshowed Pt particle size increase for Ptγ-Al
2O
3 Hence deactivation of Ptγ-Al
2O
3was also attributed to Pt particle sinteringin addition to leaching whereas the use ofgraphitic carbon overcoat prevented bothphenomena under APR conditions Thisapproach was also successful when usingRugc-Al
2O
3for lactic acid hydrogenation
Overall graphitic carbon layers depositedthrough vapor phase route provided higherhydrothermal stability than pyrolytic car-bon-coatings and seems to be a promisingstabilization method for a wide range ofapplications
In addition to the deposition of thinfilm precise surface functionalization canalso be used to improve catalyst stabilityFor instance the generation of hydropho-bic surfaces by grafting of octadecyltri-chlorosilane groups onHY zeolite prevent-ed the support structure from collapsingduring stability tests which mimicked theupgrading of biomass-derived liquid com-pounds in biphasic systems at 200 degC[43]Stabilization was attributed to a limitedcontact between the zeolitersquos hydrophobicsurface and the aqueous phase The sameauthors also reported a systematic studywith various alkylsilanes from C
2to C
18[70]
Hydrothermal stability of zeolites hasbeen studied in great detail as zeolites arethemost widely used solid acid catalysts[71]and are often proposed for biomass con-version[7273] As shown by detailed char-acterization using 27Al and 29Si solid-stateNMR and NH
3-TPD siloxane bridge hy-
drolysis was the main deactivation routein the presence of hot liquid water whilezeolites treated under steam suffered fromdealumination[74] After a treatment in hotwater at 200 degC mimicking APR condi-tions ZSM-5 was stable independent ofits SiAl ratio while degradation of zeoliteY increased with increasing SiAl ratioas opposed to the better stability of zeo-lites with higher SiAl in the presence ofsteam Finally Al-rich zeolite Y could bestabilized by incorporation of lanthanumcations[74]
Recently Gardner et al reported thattesting the hydrothermal stability of zeo-lites in distilled water was not completelyrelevant to biomass conversion reactionconditions[75] In fact dry biomass con-tains about 15 wt of inorganic saltssuch as NaCl and the dissolution rate ofmetal oxides could be dramatically en-hanced by cations in solution[76] Catalytictests showed that the presence of NaCl hadno influence on glucose conversion duringthe liquid-phase transformation of glucoseto HMF on ZSM-5 However zeolite hy-drothermal stability was significantly per-turbed by the presence of NaCl leading tothe hydrolytic attack of extra-frameworkspecies andor defects sites Interestingly
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 589
leached Al species were active in the con-version of glucose to fructose
Together with metal particles zeolitesconstitute an important class of bifunc-tional catalysts While studies mentionedabove aimed at adding a stabilizing ele-ment to an existing material Ennaert et alassessed the stability of Ruzeolite H-USYin hot liquid water for the hydrolytic hy-drogenation of cellulose using variousSiAl ratios in order to select the optimalzeolite composition[77] They identifiedthat the lattice was stabilized by frame-work aluminum atoms which preventedhydrolysis of framework bonds while theextra-framework aluminum atoms locatedat the external surface prevented zeolitesolubilization With a SiAl ratio greaterthan 3 a stable quantitative hexitol yieldwas obtained
5 Improving Catalyst Stability byProcess Design
Previous sections demonstrated howimportant smart catalyst design can be forprolonged catalyst lifetime In additionimproved stability for a given catalyst canbe achieved by tuning process parametersIn fact proper reactions conditions such asthe choice of gas atmosphere (eg hydro-gen versus an inert gas) or concentrationof reagents and solvents can have dra-matic effects on processes As mentionedpreviously leaching and sintering of metalnanoparticles tends to be more pronouncedat low pH Nickel at pH values below 7 canbe oxidized by water or acids[78] to formNi2+ species[79] To counterbalance this ef-fect van Haasterecht et al used additionof KOH in ethylene glycol conversion overNiCNF catalyst with KOH concentrationbetween 0ndash12 M to increase the pH andimprove catalyst stability (Fig 7)[37] Niparticles were less stable in hydrothermalconditions in comparison to ethylene gly-col In both cases increasing the pH hada significant impact though it was not aspronounced in ethylene glycol Moreoverat very high pH (gt13) sintering occurredagain which was probably due to a for-mation of highly mobile hydroxide nickelspecies
A reductive gas atmosphere can lead toincreased catalyst stability A typical ex-ample is the aqueous phase reforming ofethylene glycol conducted over Ni-basedcatalysts[37] Introduction of hydrogen inthe gas atmosphere suppressed the growthof nickel nanoparticles The average par-ticle size after reaction in a hydrogen andan inert atmosphere was 58 and 32 nmrespectively Moreover leaching of thenickel decreased leading to 026 catalystmass loss under inert atmosphere com-pared to 015 mass loss in hydrogen
However the use of hydrogen is notalways beneficial It was reported that forplatinum supported on graphite at highpH under a hydrogen-rich atmosphereand at an elevated temperature agglom-eration of platinum could occur[80] Thereason was that removal of the graphiteoxygen functional groups by hydrogenlead to lower adhesion and thereforehigher mobility of the nanoparticles onthe surface
Coke formation over the catalyst canalso be inhibited by co-feeding other sub-stances to the reactor For instance in cata-lytic conversion of bio-oil into hydrocar-bons a stream of bio-oil was diluted witha ratio 12 with water which decreased themass of carbon deposits on microporousZSM-5[46] Unfortunately this treatmentled to irreversible catalyst deactivation dueto dealumination of the zeolite caused bywater at high temperature[8182] Howeverthe same authors found that the use of a70 wt methanol with a bio-oil mixturecould decrease coke formation up to 70and prevent dealumination and thereforemaintain activity of the catalyst
Preventing deactivation can also beachieved by controlling the reaction me-dia During the process of producing fur-fural from xylans high deactivation ofthe GaUSY catalyst was observed whenusing water as a solvent XRD analysis ofthe spent catalyst confirmed the collapseof the zeolite structure during reactionMoreover gallium content dramaticallydecreased after 24 hours of reaction from59 wt to 027 wt To avoid the nega-tive effects of water on the catalyst thebiphasic system waterndashmethyl isobutyl
ketone (MIBK) was used with a ratio of2080 Gallium leaching decreased duringthe first few hours (36 wt Ga) and con-tinued but at a lower rate to reach a metalcontent of 19 wt at the end of the reac-tion Furthermore XRD confirmed that thezeolite retained its structure at the end ofthe experiment confirming the importanceof using a co-solvent[83]
6 Conclusions and Outlook
Deactivation of catalysts under theharsh conditions used for biomass conver-sion is a bottleneck in the developmentof competitive processes for bioresourcevalorization to renewable alternatives tofossil-based fuels and chemicals Biomass-derived molecules that must be further up-graded are usually produced in relativelydilute liquid (often aqueous) streamswhich contain biogenic and inorganic im-purities This introduces significant chal-lenges for heterogeneous catalysts whichhave often been developed in the context ofreforming fairly pure gas-phase hydrocar-bons An important category of catalystsfor catalytic biomass upgrading are oxide-supported metal particles due to their usein hydrogenation and hydrodeoxygenationreactions are essential for removing oxy-gen from bio-based molecules Several re-searchers have sought to reduce the maindeactivation pathways for these catalystsnotably their irreversible deactivation viametal leaching and sintering in the liquidphase as well as their reversible deactiva-tion by poisoning Leaching and sinteringoccur preferentially due to the instability
Fig 7 Approximation based on experimental data between nickelnanoparticles size of the spent NiCNF catalysts and the pH of the finalreaction mixture in aqueous phase reforming of ethylene glycol ThepH of the feed solutions was adjusted by addition of KOH (0-12 M) toaqueous solutions of 1 wt ethylene glycol (green line) and to water(blue line) Reactions were performed at 230 degC for 6 h under inert gasAdapted with permission of Elsevier from ref [37]
590 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
of undercoordinated metal atoms Suchatoms can also favor byproduct formationdue to their high reactivity These byprod-ucts include coke in which case these re-active atoms contribute to poisoning theother active sites by initiating the forma-tion of carbon deposits For these reasonsseveral authors have reported methods topassivate these undercoordinated atomsusing overcoats deposited either by ALDsugar pyrolysis or CVD thereby stabiliz-ing the catalyst of interest Another issueis the leaching and structure modificationsof oxide supports due to the poor hydro-thermal stability of these solids whichinclude silica and zeolites In this caseseveral authors have turned to fine-tuningthe local environment of these catalystsagain by physically covering the materialof interest The addition of hydrophobicsurface functionalities or overcoats hasbeen shown to reduce support hydrolysisbywater molecules Similarly depositing apolymeric layer over metal hydrogenationsites was shown to act as an organic pseu-do-solvent which reduced the presence ofpolar biogenic catalyst poisons and thuscatalyst deactivation Finally increasedcatalyst lifetime was also improved byprocess engineering In fact tuning thereaction conditions including by loweringpH or co-feeding organic solvents and hy-drogen proved to be efficient for limitingcatalyst deactivation
Further research opportunities exist tocombine the principle strategies mentionedabove to further improve catalyst stabiliza-tion Tailored overcoats could be used bothto stabilize undercoordinated atoms and atthe same time to provide favorable micro-environments that could reduce hydrolysisandor concentrations of catalyst poisonsSuch an approach was used in part by add-ingMgO
xto reduce the acidity of an alumi-
na overcoat which was thought to catalyzecoke formation while the overcoat wasused to stabilize undercoordinated atomsHowever this approach could be furtherstrengthened by adding functionality to theovercoat and further tailoring themoleculardesign of the catalystrsquos local environmentFurthermore controlling pore size open-ings within these overcoats could intro-duce shape selectivity and be used to sieveout undesired reactants as was recentlydemonstrated for an alumina overcoat on atitania catalyst used for photocatalysis[84]Finally it will become increasingly impor-tant to use real biomass-derived streamsfor testing catalytic stability The negativeeffects of using liquid or aqueous condi-tions can be compounded by the presenceof numerous perhaps yet unidentified in-organic or biogenic impurities This wasnotably demonstrated by the acceleratedstructure collapse of zeolite structures inthe presence of inorganic salts
In summary current synthetic tech-niques can lead to unprecedented controlof the nanostructures surrounding a cata-lystrsquos active site but harsh conditions pres-ent for biomass conversion are equally un-precedented in industrial catalytic process-ing As society comes under increasingpressure to develop routes to sustainablecarbon-based molecules these syntheticmethods will likely prove essential in ad-dressing the challenges posed by catalyti-cally upgrading renewable resources suchas biomass
AcknowledgementsThe authors acknowledge financial support
from the Swiss National Science Foundationthrough grant number 407040_153866 as partof the National Research Program 70 and grantnumber PYAPP2_154281 and Benjamin LeMonnier for help with the design of figures
Received June 17 2015
[1] K Sugiura M Ishihara T Shimauchi SHarayama Environ Sci Technol 1997 31 45
[2] S Matar M J Mirbach HA Tayim lsquoCatalysisin Petrochemical Processesrsquo Springer Scienceamp Business Media 1989
[3] O S Abdul-Hamid Monthly Oil MarketReport - May 2015 OPEC 2015
[4] The European Parliament and the Council of theEuropean Union OJ 2009 L140 16
[5] R Rinaldi F Schuumlth Energy Environ Sci2009 2 610
[6] A A Peterson F Vogel R P Lachance MFroumlling J Michael JAntal JW Tester EnergyEnviron Sci 2008 1 32
[7] J S Luterbacher D M Alonso J A DumesicGreen Chem 2014 16 4816
[8] A Rahimi A Ulbrich J J Coon S S StahlNature 2014 515 249
[9] A Eyal R Jansen R Mali A Vitner lsquoSugarMixtures and Methods for Production and UseThereofrsquo 2012 WO 2011161685 A3
[10] D V Kazachkin M Colakyan F J MoeslerlsquoSupercritical Hydrolysis of Biomassrsquo 2015US 8999065 B2
[11] D L Sills J M Gossett Bioresour Technol2011 102 1389
[12] D M Alonso S G Wettstein M A MellmerE I Gurbuz J A Dumesic Energy EnvironSci 2012 6 76
[13] J S Luterbacher J M Rand D M Alonso JHan J T Youngquist C T Maravelias B FPfleger J A Dumesic Science 2014 343 277
[14] J S Luterbacher D M Alonso J M Rand YM Questell-Santiago J H Yeap B F PflegerJ A Dumesic ChemSusChem 2015 8 1317
[15] H Tadesse R Luque Energy Environ Sci2011 4 3913
[16] J B Binder R T Raines J Am Chem Soc2009 131 1979
[17] J S Luterbacher J W Tester L P WalkerBiotechnol Bioeng 2010 107 451
[18] D B Hodge M N Karim D J Schell J DMcMillan Bioresour Technol 2008 99 8940
[19] J S Luterbacher Q Chew Y Li J W TesterL P Walker Energy Environ Sci 2012 56990
[20] T J Schwartz B J OrsquoNeill B H Shanks J ADumesic ACS Catal 2014 4 2060
[21] G W Huber J N Chheda C J Barrett J ADumesic Science 2005 308 1446
[22] G W Huber R D Cortright J A DumesicAngew Chem Int Ed 2004 43 1549
[23] J N Chheda Y Romaacuten-Leshkov J ADumesic Green Chem 2007 9 342
[24] D W Rackemann W O Doherty BiofuelsBioprod Biorefining 2011 5 198
[25] J N Chheda G W Huber J A DumesicAngew Chem Int Ed 2007 46 7164
[26] A V Bandura S N Lvov J Phys Chem RefData 2006 35 15
[27] H Xiong H N Pham A K Datye GreenChem 2014 16 4627
[28] K P De Jong lsquoSynthesis Of Solid CatalystsrsquoWiley-VCHVerlag GmbH amp Co KGaA 2009
[29] F Heacuteroguel G Siddiqi M D Detwiler DY Zemlyanov O V Safonova C Copeacuteret JCatal 2015 321 81
[30] T W Hansen A T DeLaRiva S R Challa AK Datye Acc Chem Res 2013 46 1720
[31] B J OrsquoNeill C Sener D H K Jackson T FKuech J A Dumesic ChemSusChem 2014 73247
[32] B J OrsquoNeill J T Miller P J Dietrich FG Sollberger F H Ribeiro J A DumesicChemCatChem 2014 6 2493
[33] B J OrsquoNeill D H K Jackson A J Crisci CA Farberow F Shi A C Alba-Rubio J Lu PJ Dietrich X Gu C L Marshall et al AngewChem 2013 125 14053
[34] H Zhang Y Lei A J Kropf G Zhang J WElam J T Miller F Sollberger F Ribeiro MC Akatay E A Stach J A Dumesic C LMarshall J Catal 2014 317 284
[35] H ZhangY Lei A Jeremy Kropf G Zhang JW Elam J T Miller F Sollberger F RibeiroM Cem Akatay E A Stach J A Dumesic CL Marshall J Catal 2015 323 165
[36] J Lee D H K Jackson T Li R E Winans JA Dumesic T F Kuech G W Huber EnergyEnviron Sci 2014 7 1657
[37] T van Haasterecht C C I Ludding K P deJong J H Bitter J Catal 2014 319 27
[38] P Sun G Gao Z Zhao C Xia F Li ACSCatal 2014 4 4136
[39] J Lu B Fu M C Kung G Xiao J W ElamH H Kung P C Stair Science 2012 3351205
[40] T J Schwartz R L Johnson J Cardenas AOkerlund N A Da Silva K Schmidt-Rohr JA Dumesic Angew Chem Int Ed 2014 5312718
[41] H N Pham A E Anderson R L Johnson KSchmidt-Rohr A K Datye Angew Chem IntEd 2012 51 13163
[42] H Xiong T J Schwartz N I Andersen J ADumesic A K Datye Angew Chem Int Ed2015 early view DOI 101002anie201502206
[43] P A Zapata J Faria M P Ruiz R E JentoftD E Resasco J Am Chem Soc 2012 1348570
[44] H N Pham Y J Pagan-Torres J C Serrano-Ruiz D Wang J A Dumesic A K DatyeAppl Catal Gen 2011 397 153
[45] Y J Pagaacuten-Torres J M R Gallo D WangH N Pham J A Libera C L Marshall J WElam A K Datye J A Dumesic ACS Catal2011 1 1234
[46] A G Gayubo B Valle A T Aguayo MOlazar J Bilbao Energy Fuels 2009 23 4129
[47] S R Challa A T Delariva T W Hansen SHelveg J Sehested P L Hansen F Garzon AK Datye J Am Chem Soc 2011 133 20672
[48] J A Moulijn A E van Diepen F KapteijnAppl Catal Gen 2001 212 3
[49] S L Wegener T J Marks P C Stair AccChem Res 2012 45 206
[50] B J OrsquoNeill D H K Jackson J Lee CCanlas P C Stair C L Marshall J W ElamT F Kuech J A Dumesic G W Huber ACSCatal 2015 5 1804
[51] D M King J A Spencer II X Liang L FHakim A W Weimer Surf Coat Technol2007 201 9163
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 591
[52] J A McCormick B L Cloutier AWWeimerS M George J Vac Sci Technol A 2007 2567
[53] D Longrie D Deduytsche C Detavernier JVac Sci Technol A 2014 32 010802
[54] J Lu J W Elam P C Stair Acc Chem Res2013 46 1806
[55] L De Rogatis M Cargnello V GombacB Lorenzut T Montini P FornasieroChemSusChem 2010 3 24
[56] A G Sergeev J D Webb J F Hartwig J AmChem Soc 2012 134 20226
[57] J He C Zhao J A Lercher J Am Chem Soc2012 134 20768
[58] Q Song F Wang J CaiY Wang J Zhang WYu J Xu Energy Environ Sci 2013 6 994
[59] R Dehghan-Niri J C Walmsley A HolmenP A Midgley E Rytter A H Dam A BHungria J C Hernandez-Garrido D ChenCatal Sci Technol 2012 2 2476
[60] M Morales P Y Dapsens I Giovinazzo JWitte C Mondelli S PapadokonstantakisK Hungerbuumlhler J Peacuterez-Ramiacuterez EnergyEnviron Sci 2015 8 558
[61] O A Abdelrahman H Y Luo A Heyden YRomaacuten-Leshkov J Q Bond J Catal 2015329 10
[62] D M Alonso S G Wettstein J A DumesicChem Soc Rev 2012 41 8075
[63] W Li L Ye P Long J Chen H Ariga KAsakura YYuan RSC Adv 2014 4 29072
[64] G Prieto J Zecevic H Friedrich K P deJong P E de Jongh Nat Mater 2013 12 34
[65] Z Zhang J E Jackson D J Miller BioresourTechnol 2008 99 5873
[66] H Luo T Prasomsri Y Romaacuten-Leshkov TopCatal 2015 394
[67] J Mazumder H I de Lasa Catal Today 2014237 100
[68] H Xiong H N Pham A K Datye J Catal2013 302 93
[69] H Xiong M Nolan B H ShanksA K DatyeAppl Catal Gen 2014 471 165
[70] P A Zapata Y Huang M A Gonzalez-BorjaD E Resasco J Catal 2013 308 82
[71] A Corma Chem RevWash DC U S 1995 95559
[72] P A Jacobs M Dusselier B F Sels AngewChem Int Ed 2014 53 8621
[73] M Milina S Mitchell J Peacuterez-Ramiacuterez CatalToday 2014 235 176
[74] R M Ravenelle F Schuumlssler A DrsquoAmico NDanilina J A van Bokhoven J A Lercher CW Jones C Sievers J Phys Chem C 2010114 19582
[75] D W Gardner J Huo T C Hoff R LJohnson B H Shanks J-P Tessonnier ACSCatal 2015 DOI 101021acscatal5b00888
[76] P M Dove N Han J J D Yoreo Proc NatlAcad Sci 2005 102 15357
[77] T Ennaert J Geboers E Gobechiya C MCourtin M Kurttepeli K Houthoofd C E AKirschhock P C M M Magusin S Bals P AJacobs B F Sels ACS Catal 2015 5 754
[78] B Beverskog I Puigdomenech Corros Sci1997 39 969
[79] D A Palmer P Beacuteneacutezeth C Xiao D JWesolowski L M Anovitz J Solut Chem2011 40 680
[80] J H Vleeming B F M Kuster G B Marin FOudet P Courtine J Catal 1997 166 148
[81] A G Gayubo A T Aguayo M Olazar RVivanco J Bilbao Chem Eng Sci 2003 585239
[82] A de Lucas P CanizaresA DuraacutenA CarreroAppl Catal Gen 1997 154 221
[83] CAellig D Scholz PY Dapsens C MondelliJ Peacuterez-Ramiacuterez Catal Sci Technol 2014 5142
[84] C P Canlas J Lu N A Ray N A Grosso-Giordano S Lee J W Elam R E Winans RP Van Duyne P C Stair J M Notestein NatChem 2012 4 1030

586 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
the nanoparticles from sintering whereassintering was observed during calcinationin the absence of overcoat
The methods above stabilized metalnanoparticles by adding a separate protec-tive layer over selected parts of the supportand metal particle surface Sintering andleaching of base metal catalysts could alsobe limited by embedding the metal into astructure[55] For instance Co nanoparti-cles were loaded into an HZSM-5 zeoliteproviding a bifunctional catalyst for theconversion of levulinic acid into valericbiofuel where cobalt performed hydroge-nation while the acidic support catalyzedγ-valerolactone (GVL) ring opening (Fig3c)[38] In contrast to the incipient wetnessimpregnation approach cobalt was em-bedded into HZSM-5 by nucleation andgrowth of HZSM-5 crystals onto Co
3O
4
SiO2whereas SiO
2was progressively dis-
solved thanks to the basicity of the systemH
2-TPR showed a lower temperature for
the reduction of cobalt oxide for the im-pregnated (CoHZSM-5) compared to theembedded (CoHZSM-5) catalyst dem-onstrating a higher metal-support interac-tion for the latter The strong interactionhad a limited influence on the Co acces-sibility as demonstrated by a similar H
2up-
take for both materials Structural charac-terization was performed on CoHZSM-5and CoHZSM-5 before and after cataly-sis to identify stability differences FirstTEM revealed a stable Co NPs size forCoHZSM-5 while for CoHZSM-5 theaverage Co NP size increased from 25 to32 nm Second coke formation was onlydetected for CoHZSM-5 by thermogravi-metric analysis (TGA) as well as N
2physi-
sorption (decrease of surface area and porevolume) Finally XRD patterns showedthat both materials maintained their crys-tal structure
Similar to Cu and Co supported Niparticles are relevant systems for the low-temperature conversion of biomass-de-rived compounds ndash notably for lignin hy-drogenolysis[56ndash58] However as for manybase metals the Ni particles suffer fromsintering Nanoconfinement of Ni par-ticles by self-assembly of Ni atoms insidethe nanoribbons of hydrotalcite-derivedmixed oxide provided increased catalyticstability for methane steam reformingcompared to Ni supported on commercialsupport surface[59] Metal embedment hasalso proven to be an efficient stabilizationmethod for lactic acid production fromglycerol Morales et al showed that MFIconstituted a stabilizing matrix for Sn andsignificantly decreased leaching of said Snwhile slowing down deactivation in com-parison to bifunctional carbon-silica com-posites and BEA zeolites[60]
Above we discussed various methodsdemonstrating that controlling local nano-
structures around the active metal catalystcould increase stability Abdelrahman etal recently showed that a simple judiciouschoice of the support could limit particlesintering during the hydrogenation oflevulinic acid[61] In fact fast deactivationwas observed for ruthenium particles sup-ported on SiO
2while increased stability
was achieved by using carbon and TiO2as
supports Al2O
3was the support that led to
the most stable catalyst which the authorsattributed to its lowest electronegativity
Another strategy is to tune the com-position of the metal itself Formation ofalloys for improved catalyst performanceshas often been used including in reactionsrelevant to biomass conversion[62] In thecase of SBA-15 the use of supported Ru-Fe catalysts led to stable conversion andselectivities over 300 h with no aggrega-tion of bimetallic particles no structurecollapse and improved selectivity for hy-drogenation of acetic acid to ethanol com-pared to monometallic catalysts[63]
Finally while many efforts have beendedicated to the control of individual char-acteristics of supported metal nanopar-ticles and their immediate environmentPrieto et al showed that controlling thecollective properties of supported metalnanoparticles led to increased stability[64]By achieving near-maximum interpar-ticle spacing for silica-supported coppernanoparticles deactivation during metha-nol synthesis was limited with deactivationrates which were an order of magnitudelower than those observed with a referencecatalyst that had a non-uniform spatial dis-tribution of particles
3 Poisoning of the Active Sites
In addition to the reduction of the num-ber of active sites by sintering and leach-ing competitive adsorption of undesirable
compounds such as coke or sulfur is a ma-jor catalyst deactivation pathway becauseit leads to the blocking of active sitesSome poisons including coke can oftenbe removed by regenerating the catalystunder oxidative or reductive media to formvolatile species However these methodsare sometimes incompatible with certainsupports such as carbon which has lowthermal stability and will degrade duringthese regeneration treatments In additionprocesses not requiring periodic catalystregeneration are generally favored foreconomic and practical reasons Henceseveral paths have been explored to limitcatalyst poisoning
Catalyst poisoning is often describedas especially problematic in the contextof fermentation-derived media which hasbeen proposed as an important source ofbiomass-derived platform molecules[65] Inparticular sulfur-containing amino acidsseem to be particularly detrimental to cat-alyst activity and are commonly found infermentation broths[65] To alleviate deacti-vation by coke deposition of alumina-sup-ported Pd catalysts during hydrogenationof fermentation-derived triacetic acid lac-tone (TAL) Schwartz et al formed equi-molar bimetallic PdAu particles Howeverthe alloy still suffered from sulfur poison-ing similar to the parent monometalliccatalyst when co-feeding model biogenicimpurities such as methionine The au-thors reasoned that a microenvironmentunfavorable to polar species would limitdeactivation by biogenic impurities whilethe reaction with the less polar substratewould still go forward[40] TAL is highlysoluble in alcohols whereas amino acidsare not hence a poly(vinyl alcohol) (PVA)overcoat was selected to form a polymericpseudo-solvent microenvironment (Fig5a) The PVA-overcoated PdAuAl
2O
3catalyst displayed a deactivation rateof 002 hndash1 vs 012 hndash1 for the parent Pd
CuAl2O3sintering and leaching
coke deposition
Al2O3CuAl2O3low sintering and leaching
coke deposition
MgOxCuAl2O3sintering and leachinglow coke deposition
Al2O3MgOxAl2O3CuAl2O3low sintering and leaching
low coke deposition
OH OH OH
OH OH OH OH
OH
OO
OH
S OH
O
NH2
substrate poison
a) b)
Fig 5 a) Creation of microenvironment unfavorable to poisons Adapted with permission fromWiley-VCH Verlag from ref [40] b) Stabilization of Cu(black)Al2O3 via ALD of Al2O3 (violet) andMgOx (blue)
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 587
Al2O
3for initial conversions between 50
and 70 Stabilization by PVA was alsohighly efficient when using TAL producedby fermentation without any feed purifi-cation Comparison of 13C NMR spectraof 13C-enriched methionine adsorbed onPVA-covered and non-covered PdAl
2O
3showed that the polymer acts as a lsquosolidsolventrsquo by limiting methionine adsorptionon Pd
The ALD overcoat strategy describedin the previous section for metal sinteringand leaching prevention has also provenuseful for limiting coke deposition Lu etal reported low reversible deactivation ofovercoated PdAl
2O
3for the dehydroge-
nation of ethane to ethylene at 650 degC[39]After using 45ALDcycles to form anAl
2O
3thin film the amount of coke deposited asdetermined by thermogravimetric analy-sis was more than 16 times lower than theamount measured for the parent PdAl
2O
3catalyst The reduction of the amount ofcoke formed was attributed to the blockingof the low-coordination Pd surface siteswhich were preferentially coated In factthe later sites favored CndashC bond scissionand hydrogen stripping resulting in the un-desirable formation of coke CH
4 CO and
CO2 Hence in addition to a lower catalyst
deactivation coating led to higher ethyleneyields
The Al2O
3overcoated CuAl
2O
3de-
scribed earlier suffered from reversiblecoke deposition In a separate study theauthors managed to limit this phenomenonby finely tuning the overcoat In addition totheAl
2O
3overcoat which provided particle
stabilization basic MgOxwas deposited to
reduce the overcoatrsquos acidity which wasassumed to catalyze the formation of coke(Fig 5b)[31] Interestingly pure MgO
xdid
not provide stabilization against sinteringThe local structure of the active site can
also reduce poisoning by affecting localmass transfer To suppress deactivation byfouling due to coke deposition during theconversion of propanal to hydrocarbons onAl-MFI Luo et al used an open mesopo-rous structure based on nanosheets to limitthe effects of fouling[66] This strategy re-sulted in a five-fold decrease of deactiva-tion rates In addition to an increase in thenumber of pore openings improved sta-bility was attributed to the short diffusionpath for reagents in the single unit-cell-thick nanosheet material which limitedcoke formation that was favored by longercontact times with the reaction products
4 Increasing Support Stability inHydrothermal Conditions
In addition to deactivation due to metalleaching and sintering loss of surface areaand pore collapsing in oxide supports is a
major source of irreversible deactivationespecially in aqueous phase conditionsFor biomass conversion processes whichoften require liquid phase andor aqueousmedia at low pH hydrothermal stabilityis a major concern Metal oxides are of-ten used as supports due to their thermalstability and high surface area Howeverthey tend to have low stability under theaforementioned conditions[27] Such ox-ides can sometimes be substituted by morestable support such as carbon but someprocesses require specific surface func-tionalities such as the acidic sites presenton alumina In addition contrary to car-bon most oxides have excellent thermalstability which is necessary during certaincatalyst regeneration processes Prominentsupports such as alumina and silica-basedmaterials undergo dissolution by hydroly-sis of aluminoxane and siloxane bonds Inaddition to loss of surface area hydrolysisof oxide network can lead to modificationof chemical composition For instance hy-drolysis of the Si-O-Al bond in zeolites fa-vors dealumination This section describesthe main techniques used to stabilize metaloxides supports in the area of biomass con-version
Formation of mixed oxides or supportdoping can be used to limit this phenome-non For instance Pham et al reported thatthe stability of Pdniobia during transfor-mation of γ-valerolactone (GVL) to pen-tanoic acid could be improved by additionof 5 SiO
2into niobia[44] Niobia could
not be entirely substituted by silica be-cause acid sites are required for GVL ringopening in addition to metal sites for thehydrogenation reaction However the ad-dition of a small portion of SiO
2improves
stability while retaining activity The com-parison of the N
2physisorption isotherms
of a Pdniobia HY commercial referencecatalyst before and after the GVL reactionshowed a dramatic surface area loss from150 to 8 m2g while the porous structurewas affected to a lesser extent for PdNb-Sioxide with a decrease from 81 to 31 m2gSimilarly HAADF-STEM and HRTEMrevealed a lower Pd particle growth uponreaction for the stabilized system than forthe PdHY reference catalyst SimilarlyMazumder et al reported the promotionof Niγ-Al
2O
3by 5 wt La
2O
3leading to
dramatic improvement of activity and sta-bility for the catalytic gasification of glu-cose compared to the parent catalyst whichsuffered from metal sintering[67]
The stabilizing oxide can also be in-troduced by ALD similar to the thin over-coats deposited for the stabilization ofsupported metal particles as described inSection 2 For instance hydrothermallystable Nb-Si oxide catalysts were preparedusing ALD of niobia within the pores ofmesoporous silica SBA-15 using between10 and 30 cycles which alternated expo-sure of the Nb(OCH
2CH
3)4precursor and
H2O to the SBA-15 surface (Fig 6a)[45]
The consumption of silanol group on thesurface was complete after 19 cycles as
a)
b)
c)
Fig 6 a) Strategyfor the synthesis ofhydrothermally stablehighly ordered niobiamesoporous materialsby ALD Reproducedwith permission fromref [45] Copyright2011 AmericanChemical Societyb) Strategy for theformation of niobiacarbon compositecatalysts Adaptedwith permission ofElsevier from ref [68]c) Deposition of gra-phitic carbon layer onalumina powder andpellets by chemicallayer deposition fromCH4 Reproducedwith permission fromWiley-VCH Verlagfrom ref [42]
588 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
demonstrated by the disappearance of theSiOH band at 3735 cmndash1 in the FTIR spec-tra Hydrated amorphous niobia was iden-tified as a major species by Raman spec-troscopy for samples presenting 10 19 and30 ALD layers N
2physisorption showed
a decrease of surface area pore volumeand pore diameter with increasing niobialoadings The presence of acid sites formedupon niobia deposition was verified byNH
3-TPD and hydrothermal stability was
tested by treating the materials in liquidwater for 12 h at 200 degC and 28 bar argonThe structure of the coated materials aftertreatment was confirmed by X-ray diffrac-tion with the persistence of the hexagonalmesoporous structure pattern while the lat-ter pattern was suppressed after hydrother-mal treatment of the parent SBA-15 TEManalysis confirmed the stability differencesobserved by XRD A bifunctional catalystfor the transformation of GVL to pentanoicacid was also formed by subsequent depo-sition of Pd nanoparticles As for the sup-port this catalyst showed higher stabilitythan the conventional PdHY-340
Improved hydrothermal stability ofa niobia-supported Pd catalyst was alsoachieved by deposition of the latter ona more stable surface such as carbonHighly dispersed spherical particles of aniobiacarbon composite were formed bythe reaction of d-glucose and ammoniumniobium oxalate in an autoclave at 200degC (pH was controlled by urea decom-position) followed by pyrolysis at 400degC under N
2(Fig 6b)[68] Hydrothermal
stability was investigated during liquid-phase butanol dehydration at 240 degC and51 bar While initial activity of the com-posite bifunctional catalyst was lower thanthe commercial HY-340 catalyst the lat-ter quickly deactivated during the first 20h of reaction while the modified catalystsremained very stable and surpassed HY-340 after 8 h Interestingly the analogousmaterial obtained by incipient wetness im-pregnation of niobia led to the formation oflarge niobia crystallites underscoring theimportance of strong interaction betweenniobia and carbon[69] Finally bifunctionalcatalysts were prepared by incipient wet-ness impregnation of Pd(NO
3)2and tested
during the conversion of GVL to pentanoicacid leading to improved hydrothermalstability for the niobiacarbon compositecatalysts just like the metal-free system
For silica and alumina improved hy-drothermal stability above 100 degC wasachieved by deposition of a thin film of car-bon formed from simple sugars Pham et aldeposited carbon by pyrolysis of adsorbedsucrose at 400 degC under N
2flow on SBA-
15 silica gel and fumed alumina leadingto improved stability after treatment in liq-uid water at 100 degC for 12 h[41] MoreoverPd was deposited on the carbon-coated
oxides to test their ability as supports forthe catalytic hydrogenation of acetylenein the presence of excess ethylene For acomparable Pd particle size (1ndash15 nm)the authors observed formation of ethaneat high acetylene conversion on both SiO
2andAl
2O
3 while carbon-coating prevented
over-hydrogenation to ethane The FTIRstudy showed a low hydration potential forthe coated materials compared to SiO
2and
Al2O
3 Therefore the higher selectivity to
ethylene for the coated materials was at-tributed to their increased hydrophobicity
More recently Xiong et al reported onthe deposition of a graphitic carbon layerover alumina by chemical vapor deposition(CVD) from CH
4(Fig 6c)[42] The graphit-
ic carbon layer provided higher stabilitythan the carbon deposited from pyrolysisof sugars as demonstrated by the higherstability achieved under flowing air (500vs 250 degC) As monitored by the growthof the characteristic graphitic peak at 24deg2θ on the XRD pattern the amount of de-posited carbon was controlled by the CH
4deposition time (from 05 to 6 h) whereasother peaks attributed to γ-Al
2O
3were un-
touched TEM and energy-filtered TEM at-tested that the carbon coating was uniformon the oxide support Raman spectros-copy mapping confirmed that depositionwas uniform over the entire surface of thespheres Furthermore the relatively highratio (071) between the peak intensitiesfor G and D bands (at 1590 and 1350 cmndash1respectively) in the Raman spectrum of gc-Al
2O
3revealed a high degree of graphitiza-
tion on the entire surface No such featureswere observed for blank alumina and pyro-lyzed carbon on alumina
The hydrothermal stability of the cata-lyst was investigated using a treatmentunder static conditions at 220 degC underautogenic pressure in an autoclave usingwater After treatment the XRD pattern ofγ-Al
2O
3displayed the well-defined diffrac-
tion peaks of the boehmite phase attestingto a reaction with water Simultaneouslyno significant change was observed forgraphitic-carbon covered γ-Al
2O
3(gc-
Al2O
3) Improved stability for gc-Al
2O
3was confirmed by N
2physisorption results
which showed that no significant decreaseof surface area pore volume and poresize occurred during hydrothermal treat-ment while γ-Al
2O
3saw its surface area
decrease by 80 during the same treat-ment Stability of gc-Al
2O
3as a support
was tested in the aqueous phase reforming(APR) of ethylene glycol at 250 degC and70 bar after deposition of platinum par-ticles Conversion using Ptgc-Al
2O
3was
twice as high as for Ptγ-Al2O
3 This result
was consistent with the leached Al and Ptdetected in the liquid phase after reactionby ICP-OES for the uncoated materialIn addition to full conversion of γ-Al
2O
3
to boehmite observed by XRD STEMshowed Pt particle size increase for Ptγ-Al
2O
3 Hence deactivation of Ptγ-Al
2O
3was also attributed to Pt particle sinteringin addition to leaching whereas the use ofgraphitic carbon overcoat prevented bothphenomena under APR conditions Thisapproach was also successful when usingRugc-Al
2O
3for lactic acid hydrogenation
Overall graphitic carbon layers depositedthrough vapor phase route provided higherhydrothermal stability than pyrolytic car-bon-coatings and seems to be a promisingstabilization method for a wide range ofapplications
In addition to the deposition of thinfilm precise surface functionalization canalso be used to improve catalyst stabilityFor instance the generation of hydropho-bic surfaces by grafting of octadecyltri-chlorosilane groups onHY zeolite prevent-ed the support structure from collapsingduring stability tests which mimicked theupgrading of biomass-derived liquid com-pounds in biphasic systems at 200 degC[43]Stabilization was attributed to a limitedcontact between the zeolitersquos hydrophobicsurface and the aqueous phase The sameauthors also reported a systematic studywith various alkylsilanes from C
2to C
18[70]
Hydrothermal stability of zeolites hasbeen studied in great detail as zeolites arethemost widely used solid acid catalysts[71]and are often proposed for biomass con-version[7273] As shown by detailed char-acterization using 27Al and 29Si solid-stateNMR and NH
3-TPD siloxane bridge hy-
drolysis was the main deactivation routein the presence of hot liquid water whilezeolites treated under steam suffered fromdealumination[74] After a treatment in hotwater at 200 degC mimicking APR condi-tions ZSM-5 was stable independent ofits SiAl ratio while degradation of zeoliteY increased with increasing SiAl ratioas opposed to the better stability of zeo-lites with higher SiAl in the presence ofsteam Finally Al-rich zeolite Y could bestabilized by incorporation of lanthanumcations[74]
Recently Gardner et al reported thattesting the hydrothermal stability of zeo-lites in distilled water was not completelyrelevant to biomass conversion reactionconditions[75] In fact dry biomass con-tains about 15 wt of inorganic saltssuch as NaCl and the dissolution rate ofmetal oxides could be dramatically en-hanced by cations in solution[76] Catalytictests showed that the presence of NaCl hadno influence on glucose conversion duringthe liquid-phase transformation of glucoseto HMF on ZSM-5 However zeolite hy-drothermal stability was significantly per-turbed by the presence of NaCl leading tothe hydrolytic attack of extra-frameworkspecies andor defects sites Interestingly
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 589
leached Al species were active in the con-version of glucose to fructose
Together with metal particles zeolitesconstitute an important class of bifunc-tional catalysts While studies mentionedabove aimed at adding a stabilizing ele-ment to an existing material Ennaert et alassessed the stability of Ruzeolite H-USYin hot liquid water for the hydrolytic hy-drogenation of cellulose using variousSiAl ratios in order to select the optimalzeolite composition[77] They identifiedthat the lattice was stabilized by frame-work aluminum atoms which preventedhydrolysis of framework bonds while theextra-framework aluminum atoms locatedat the external surface prevented zeolitesolubilization With a SiAl ratio greaterthan 3 a stable quantitative hexitol yieldwas obtained
5 Improving Catalyst Stability byProcess Design
Previous sections demonstrated howimportant smart catalyst design can be forprolonged catalyst lifetime In additionimproved stability for a given catalyst canbe achieved by tuning process parametersIn fact proper reactions conditions such asthe choice of gas atmosphere (eg hydro-gen versus an inert gas) or concentrationof reagents and solvents can have dra-matic effects on processes As mentionedpreviously leaching and sintering of metalnanoparticles tends to be more pronouncedat low pH Nickel at pH values below 7 canbe oxidized by water or acids[78] to formNi2+ species[79] To counterbalance this ef-fect van Haasterecht et al used additionof KOH in ethylene glycol conversion overNiCNF catalyst with KOH concentrationbetween 0ndash12 M to increase the pH andimprove catalyst stability (Fig 7)[37] Niparticles were less stable in hydrothermalconditions in comparison to ethylene gly-col In both cases increasing the pH hada significant impact though it was not aspronounced in ethylene glycol Moreoverat very high pH (gt13) sintering occurredagain which was probably due to a for-mation of highly mobile hydroxide nickelspecies
A reductive gas atmosphere can lead toincreased catalyst stability A typical ex-ample is the aqueous phase reforming ofethylene glycol conducted over Ni-basedcatalysts[37] Introduction of hydrogen inthe gas atmosphere suppressed the growthof nickel nanoparticles The average par-ticle size after reaction in a hydrogen andan inert atmosphere was 58 and 32 nmrespectively Moreover leaching of thenickel decreased leading to 026 catalystmass loss under inert atmosphere com-pared to 015 mass loss in hydrogen
However the use of hydrogen is notalways beneficial It was reported that forplatinum supported on graphite at highpH under a hydrogen-rich atmosphereand at an elevated temperature agglom-eration of platinum could occur[80] Thereason was that removal of the graphiteoxygen functional groups by hydrogenlead to lower adhesion and thereforehigher mobility of the nanoparticles onthe surface
Coke formation over the catalyst canalso be inhibited by co-feeding other sub-stances to the reactor For instance in cata-lytic conversion of bio-oil into hydrocar-bons a stream of bio-oil was diluted witha ratio 12 with water which decreased themass of carbon deposits on microporousZSM-5[46] Unfortunately this treatmentled to irreversible catalyst deactivation dueto dealumination of the zeolite caused bywater at high temperature[8182] Howeverthe same authors found that the use of a70 wt methanol with a bio-oil mixturecould decrease coke formation up to 70and prevent dealumination and thereforemaintain activity of the catalyst
Preventing deactivation can also beachieved by controlling the reaction me-dia During the process of producing fur-fural from xylans high deactivation ofthe GaUSY catalyst was observed whenusing water as a solvent XRD analysis ofthe spent catalyst confirmed the collapseof the zeolite structure during reactionMoreover gallium content dramaticallydecreased after 24 hours of reaction from59 wt to 027 wt To avoid the nega-tive effects of water on the catalyst thebiphasic system waterndashmethyl isobutyl
ketone (MIBK) was used with a ratio of2080 Gallium leaching decreased duringthe first few hours (36 wt Ga) and con-tinued but at a lower rate to reach a metalcontent of 19 wt at the end of the reac-tion Furthermore XRD confirmed that thezeolite retained its structure at the end ofthe experiment confirming the importanceof using a co-solvent[83]
6 Conclusions and Outlook
Deactivation of catalysts under theharsh conditions used for biomass conver-sion is a bottleneck in the developmentof competitive processes for bioresourcevalorization to renewable alternatives tofossil-based fuels and chemicals Biomass-derived molecules that must be further up-graded are usually produced in relativelydilute liquid (often aqueous) streamswhich contain biogenic and inorganic im-purities This introduces significant chal-lenges for heterogeneous catalysts whichhave often been developed in the context ofreforming fairly pure gas-phase hydrocar-bons An important category of catalystsfor catalytic biomass upgrading are oxide-supported metal particles due to their usein hydrogenation and hydrodeoxygenationreactions are essential for removing oxy-gen from bio-based molecules Several re-searchers have sought to reduce the maindeactivation pathways for these catalystsnotably their irreversible deactivation viametal leaching and sintering in the liquidphase as well as their reversible deactiva-tion by poisoning Leaching and sinteringoccur preferentially due to the instability
Fig 7 Approximation based on experimental data between nickelnanoparticles size of the spent NiCNF catalysts and the pH of the finalreaction mixture in aqueous phase reforming of ethylene glycol ThepH of the feed solutions was adjusted by addition of KOH (0-12 M) toaqueous solutions of 1 wt ethylene glycol (green line) and to water(blue line) Reactions were performed at 230 degC for 6 h under inert gasAdapted with permission of Elsevier from ref [37]
590 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
of undercoordinated metal atoms Suchatoms can also favor byproduct formationdue to their high reactivity These byprod-ucts include coke in which case these re-active atoms contribute to poisoning theother active sites by initiating the forma-tion of carbon deposits For these reasonsseveral authors have reported methods topassivate these undercoordinated atomsusing overcoats deposited either by ALDsugar pyrolysis or CVD thereby stabiliz-ing the catalyst of interest Another issueis the leaching and structure modificationsof oxide supports due to the poor hydro-thermal stability of these solids whichinclude silica and zeolites In this caseseveral authors have turned to fine-tuningthe local environment of these catalystsagain by physically covering the materialof interest The addition of hydrophobicsurface functionalities or overcoats hasbeen shown to reduce support hydrolysisbywater molecules Similarly depositing apolymeric layer over metal hydrogenationsites was shown to act as an organic pseu-do-solvent which reduced the presence ofpolar biogenic catalyst poisons and thuscatalyst deactivation Finally increasedcatalyst lifetime was also improved byprocess engineering In fact tuning thereaction conditions including by loweringpH or co-feeding organic solvents and hy-drogen proved to be efficient for limitingcatalyst deactivation
Further research opportunities exist tocombine the principle strategies mentionedabove to further improve catalyst stabiliza-tion Tailored overcoats could be used bothto stabilize undercoordinated atoms and atthe same time to provide favorable micro-environments that could reduce hydrolysisandor concentrations of catalyst poisonsSuch an approach was used in part by add-ingMgO
xto reduce the acidity of an alumi-
na overcoat which was thought to catalyzecoke formation while the overcoat wasused to stabilize undercoordinated atomsHowever this approach could be furtherstrengthened by adding functionality to theovercoat and further tailoring themoleculardesign of the catalystrsquos local environmentFurthermore controlling pore size open-ings within these overcoats could intro-duce shape selectivity and be used to sieveout undesired reactants as was recentlydemonstrated for an alumina overcoat on atitania catalyst used for photocatalysis[84]Finally it will become increasingly impor-tant to use real biomass-derived streamsfor testing catalytic stability The negativeeffects of using liquid or aqueous condi-tions can be compounded by the presenceof numerous perhaps yet unidentified in-organic or biogenic impurities This wasnotably demonstrated by the acceleratedstructure collapse of zeolite structures inthe presence of inorganic salts
In summary current synthetic tech-niques can lead to unprecedented controlof the nanostructures surrounding a cata-lystrsquos active site but harsh conditions pres-ent for biomass conversion are equally un-precedented in industrial catalytic process-ing As society comes under increasingpressure to develop routes to sustainablecarbon-based molecules these syntheticmethods will likely prove essential in ad-dressing the challenges posed by catalyti-cally upgrading renewable resources suchas biomass
AcknowledgementsThe authors acknowledge financial support
from the Swiss National Science Foundationthrough grant number 407040_153866 as partof the National Research Program 70 and grantnumber PYAPP2_154281 and Benjamin LeMonnier for help with the design of figures
Received June 17 2015
[1] K Sugiura M Ishihara T Shimauchi SHarayama Environ Sci Technol 1997 31 45
[2] S Matar M J Mirbach HA Tayim lsquoCatalysisin Petrochemical Processesrsquo Springer Scienceamp Business Media 1989
[3] O S Abdul-Hamid Monthly Oil MarketReport - May 2015 OPEC 2015
[4] The European Parliament and the Council of theEuropean Union OJ 2009 L140 16
[5] R Rinaldi F Schuumlth Energy Environ Sci2009 2 610
[6] A A Peterson F Vogel R P Lachance MFroumlling J Michael JAntal JW Tester EnergyEnviron Sci 2008 1 32
[7] J S Luterbacher D M Alonso J A DumesicGreen Chem 2014 16 4816
[8] A Rahimi A Ulbrich J J Coon S S StahlNature 2014 515 249
[9] A Eyal R Jansen R Mali A Vitner lsquoSugarMixtures and Methods for Production and UseThereofrsquo 2012 WO 2011161685 A3
[10] D V Kazachkin M Colakyan F J MoeslerlsquoSupercritical Hydrolysis of Biomassrsquo 2015US 8999065 B2
[11] D L Sills J M Gossett Bioresour Technol2011 102 1389
[12] D M Alonso S G Wettstein M A MellmerE I Gurbuz J A Dumesic Energy EnvironSci 2012 6 76
[13] J S Luterbacher J M Rand D M Alonso JHan J T Youngquist C T Maravelias B FPfleger J A Dumesic Science 2014 343 277
[14] J S Luterbacher D M Alonso J M Rand YM Questell-Santiago J H Yeap B F PflegerJ A Dumesic ChemSusChem 2015 8 1317
[15] H Tadesse R Luque Energy Environ Sci2011 4 3913
[16] J B Binder R T Raines J Am Chem Soc2009 131 1979
[17] J S Luterbacher J W Tester L P WalkerBiotechnol Bioeng 2010 107 451
[18] D B Hodge M N Karim D J Schell J DMcMillan Bioresour Technol 2008 99 8940
[19] J S Luterbacher Q Chew Y Li J W TesterL P Walker Energy Environ Sci 2012 56990
[20] T J Schwartz B J OrsquoNeill B H Shanks J ADumesic ACS Catal 2014 4 2060
[21] G W Huber J N Chheda C J Barrett J ADumesic Science 2005 308 1446
[22] G W Huber R D Cortright J A DumesicAngew Chem Int Ed 2004 43 1549
[23] J N Chheda Y Romaacuten-Leshkov J ADumesic Green Chem 2007 9 342
[24] D W Rackemann W O Doherty BiofuelsBioprod Biorefining 2011 5 198
[25] J N Chheda G W Huber J A DumesicAngew Chem Int Ed 2007 46 7164
[26] A V Bandura S N Lvov J Phys Chem RefData 2006 35 15
[27] H Xiong H N Pham A K Datye GreenChem 2014 16 4627
[28] K P De Jong lsquoSynthesis Of Solid CatalystsrsquoWiley-VCHVerlag GmbH amp Co KGaA 2009
[29] F Heacuteroguel G Siddiqi M D Detwiler DY Zemlyanov O V Safonova C Copeacuteret JCatal 2015 321 81
[30] T W Hansen A T DeLaRiva S R Challa AK Datye Acc Chem Res 2013 46 1720
[31] B J OrsquoNeill C Sener D H K Jackson T FKuech J A Dumesic ChemSusChem 2014 73247
[32] B J OrsquoNeill J T Miller P J Dietrich FG Sollberger F H Ribeiro J A DumesicChemCatChem 2014 6 2493
[33] B J OrsquoNeill D H K Jackson A J Crisci CA Farberow F Shi A C Alba-Rubio J Lu PJ Dietrich X Gu C L Marshall et al AngewChem 2013 125 14053
[34] H Zhang Y Lei A J Kropf G Zhang J WElam J T Miller F Sollberger F Ribeiro MC Akatay E A Stach J A Dumesic C LMarshall J Catal 2014 317 284
[35] H ZhangY Lei A Jeremy Kropf G Zhang JW Elam J T Miller F Sollberger F RibeiroM Cem Akatay E A Stach J A Dumesic CL Marshall J Catal 2015 323 165
[36] J Lee D H K Jackson T Li R E Winans JA Dumesic T F Kuech G W Huber EnergyEnviron Sci 2014 7 1657
[37] T van Haasterecht C C I Ludding K P deJong J H Bitter J Catal 2014 319 27
[38] P Sun G Gao Z Zhao C Xia F Li ACSCatal 2014 4 4136
[39] J Lu B Fu M C Kung G Xiao J W ElamH H Kung P C Stair Science 2012 3351205
[40] T J Schwartz R L Johnson J Cardenas AOkerlund N A Da Silva K Schmidt-Rohr JA Dumesic Angew Chem Int Ed 2014 5312718
[41] H N Pham A E Anderson R L Johnson KSchmidt-Rohr A K Datye Angew Chem IntEd 2012 51 13163
[42] H Xiong T J Schwartz N I Andersen J ADumesic A K Datye Angew Chem Int Ed2015 early view DOI 101002anie201502206
[43] P A Zapata J Faria M P Ruiz R E JentoftD E Resasco J Am Chem Soc 2012 1348570
[44] H N Pham Y J Pagan-Torres J C Serrano-Ruiz D Wang J A Dumesic A K DatyeAppl Catal Gen 2011 397 153
[45] Y J Pagaacuten-Torres J M R Gallo D WangH N Pham J A Libera C L Marshall J WElam A K Datye J A Dumesic ACS Catal2011 1 1234
[46] A G Gayubo B Valle A T Aguayo MOlazar J Bilbao Energy Fuels 2009 23 4129
[47] S R Challa A T Delariva T W Hansen SHelveg J Sehested P L Hansen F Garzon AK Datye J Am Chem Soc 2011 133 20672
[48] J A Moulijn A E van Diepen F KapteijnAppl Catal Gen 2001 212 3
[49] S L Wegener T J Marks P C Stair AccChem Res 2012 45 206
[50] B J OrsquoNeill D H K Jackson J Lee CCanlas P C Stair C L Marshall J W ElamT F Kuech J A Dumesic G W Huber ACSCatal 2015 5 1804
[51] D M King J A Spencer II X Liang L FHakim A W Weimer Surf Coat Technol2007 201 9163
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 591
[52] J A McCormick B L Cloutier AWWeimerS M George J Vac Sci Technol A 2007 2567
[53] D Longrie D Deduytsche C Detavernier JVac Sci Technol A 2014 32 010802
[54] J Lu J W Elam P C Stair Acc Chem Res2013 46 1806
[55] L De Rogatis M Cargnello V GombacB Lorenzut T Montini P FornasieroChemSusChem 2010 3 24
[56] A G Sergeev J D Webb J F Hartwig J AmChem Soc 2012 134 20226
[57] J He C Zhao J A Lercher J Am Chem Soc2012 134 20768
[58] Q Song F Wang J CaiY Wang J Zhang WYu J Xu Energy Environ Sci 2013 6 994
[59] R Dehghan-Niri J C Walmsley A HolmenP A Midgley E Rytter A H Dam A BHungria J C Hernandez-Garrido D ChenCatal Sci Technol 2012 2 2476
[60] M Morales P Y Dapsens I Giovinazzo JWitte C Mondelli S PapadokonstantakisK Hungerbuumlhler J Peacuterez-Ramiacuterez EnergyEnviron Sci 2015 8 558
[61] O A Abdelrahman H Y Luo A Heyden YRomaacuten-Leshkov J Q Bond J Catal 2015329 10
[62] D M Alonso S G Wettstein J A DumesicChem Soc Rev 2012 41 8075
[63] W Li L Ye P Long J Chen H Ariga KAsakura YYuan RSC Adv 2014 4 29072
[64] G Prieto J Zecevic H Friedrich K P deJong P E de Jongh Nat Mater 2013 12 34
[65] Z Zhang J E Jackson D J Miller BioresourTechnol 2008 99 5873
[66] H Luo T Prasomsri Y Romaacuten-Leshkov TopCatal 2015 394
[67] J Mazumder H I de Lasa Catal Today 2014237 100
[68] H Xiong H N Pham A K Datye J Catal2013 302 93
[69] H Xiong M Nolan B H ShanksA K DatyeAppl Catal Gen 2014 471 165
[70] P A Zapata Y Huang M A Gonzalez-BorjaD E Resasco J Catal 2013 308 82
[71] A Corma Chem RevWash DC U S 1995 95559
[72] P A Jacobs M Dusselier B F Sels AngewChem Int Ed 2014 53 8621
[73] M Milina S Mitchell J Peacuterez-Ramiacuterez CatalToday 2014 235 176
[74] R M Ravenelle F Schuumlssler A DrsquoAmico NDanilina J A van Bokhoven J A Lercher CW Jones C Sievers J Phys Chem C 2010114 19582
[75] D W Gardner J Huo T C Hoff R LJohnson B H Shanks J-P Tessonnier ACSCatal 2015 DOI 101021acscatal5b00888
[76] P M Dove N Han J J D Yoreo Proc NatlAcad Sci 2005 102 15357
[77] T Ennaert J Geboers E Gobechiya C MCourtin M Kurttepeli K Houthoofd C E AKirschhock P C M M Magusin S Bals P AJacobs B F Sels ACS Catal 2015 5 754
[78] B Beverskog I Puigdomenech Corros Sci1997 39 969
[79] D A Palmer P Beacuteneacutezeth C Xiao D JWesolowski L M Anovitz J Solut Chem2011 40 680
[80] J H Vleeming B F M Kuster G B Marin FOudet P Courtine J Catal 1997 166 148
[81] A G Gayubo A T Aguayo M Olazar RVivanco J Bilbao Chem Eng Sci 2003 585239
[82] A de Lucas P CanizaresA DuraacutenA CarreroAppl Catal Gen 1997 154 221
[83] CAellig D Scholz PY Dapsens C MondelliJ Peacuterez-Ramiacuterez Catal Sci Technol 2014 5142
[84] C P Canlas J Lu N A Ray N A Grosso-Giordano S Lee J W Elam R E Winans RP Van Duyne P C Stair J M Notestein NatChem 2012 4 1030

SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 587
Al2O
3for initial conversions between 50
and 70 Stabilization by PVA was alsohighly efficient when using TAL producedby fermentation without any feed purifi-cation Comparison of 13C NMR spectraof 13C-enriched methionine adsorbed onPVA-covered and non-covered PdAl
2O
3showed that the polymer acts as a lsquosolidsolventrsquo by limiting methionine adsorptionon Pd
The ALD overcoat strategy describedin the previous section for metal sinteringand leaching prevention has also provenuseful for limiting coke deposition Lu etal reported low reversible deactivation ofovercoated PdAl
2O
3for the dehydroge-
nation of ethane to ethylene at 650 degC[39]After using 45ALDcycles to form anAl
2O
3thin film the amount of coke deposited asdetermined by thermogravimetric analy-sis was more than 16 times lower than theamount measured for the parent PdAl
2O
3catalyst The reduction of the amount ofcoke formed was attributed to the blockingof the low-coordination Pd surface siteswhich were preferentially coated In factthe later sites favored CndashC bond scissionand hydrogen stripping resulting in the un-desirable formation of coke CH
4 CO and
CO2 Hence in addition to a lower catalyst
deactivation coating led to higher ethyleneyields
The Al2O
3overcoated CuAl
2O
3de-
scribed earlier suffered from reversiblecoke deposition In a separate study theauthors managed to limit this phenomenonby finely tuning the overcoat In addition totheAl
2O
3overcoat which provided particle
stabilization basic MgOxwas deposited to
reduce the overcoatrsquos acidity which wasassumed to catalyze the formation of coke(Fig 5b)[31] Interestingly pure MgO
xdid
not provide stabilization against sinteringThe local structure of the active site can
also reduce poisoning by affecting localmass transfer To suppress deactivation byfouling due to coke deposition during theconversion of propanal to hydrocarbons onAl-MFI Luo et al used an open mesopo-rous structure based on nanosheets to limitthe effects of fouling[66] This strategy re-sulted in a five-fold decrease of deactiva-tion rates In addition to an increase in thenumber of pore openings improved sta-bility was attributed to the short diffusionpath for reagents in the single unit-cell-thick nanosheet material which limitedcoke formation that was favored by longercontact times with the reaction products
4 Increasing Support Stability inHydrothermal Conditions
In addition to deactivation due to metalleaching and sintering loss of surface areaand pore collapsing in oxide supports is a
major source of irreversible deactivationespecially in aqueous phase conditionsFor biomass conversion processes whichoften require liquid phase andor aqueousmedia at low pH hydrothermal stabilityis a major concern Metal oxides are of-ten used as supports due to their thermalstability and high surface area Howeverthey tend to have low stability under theaforementioned conditions[27] Such ox-ides can sometimes be substituted by morestable support such as carbon but someprocesses require specific surface func-tionalities such as the acidic sites presenton alumina In addition contrary to car-bon most oxides have excellent thermalstability which is necessary during certaincatalyst regeneration processes Prominentsupports such as alumina and silica-basedmaterials undergo dissolution by hydroly-sis of aluminoxane and siloxane bonds Inaddition to loss of surface area hydrolysisof oxide network can lead to modificationof chemical composition For instance hy-drolysis of the Si-O-Al bond in zeolites fa-vors dealumination This section describesthe main techniques used to stabilize metaloxides supports in the area of biomass con-version
Formation of mixed oxides or supportdoping can be used to limit this phenome-non For instance Pham et al reported thatthe stability of Pdniobia during transfor-mation of γ-valerolactone (GVL) to pen-tanoic acid could be improved by additionof 5 SiO
2into niobia[44] Niobia could
not be entirely substituted by silica be-cause acid sites are required for GVL ringopening in addition to metal sites for thehydrogenation reaction However the ad-dition of a small portion of SiO
2improves
stability while retaining activity The com-parison of the N
2physisorption isotherms
of a Pdniobia HY commercial referencecatalyst before and after the GVL reactionshowed a dramatic surface area loss from150 to 8 m2g while the porous structurewas affected to a lesser extent for PdNb-Sioxide with a decrease from 81 to 31 m2gSimilarly HAADF-STEM and HRTEMrevealed a lower Pd particle growth uponreaction for the stabilized system than forthe PdHY reference catalyst SimilarlyMazumder et al reported the promotionof Niγ-Al
2O
3by 5 wt La
2O
3leading to
dramatic improvement of activity and sta-bility for the catalytic gasification of glu-cose compared to the parent catalyst whichsuffered from metal sintering[67]
The stabilizing oxide can also be in-troduced by ALD similar to the thin over-coats deposited for the stabilization ofsupported metal particles as described inSection 2 For instance hydrothermallystable Nb-Si oxide catalysts were preparedusing ALD of niobia within the pores ofmesoporous silica SBA-15 using between10 and 30 cycles which alternated expo-sure of the Nb(OCH
2CH
3)4precursor and
H2O to the SBA-15 surface (Fig 6a)[45]
The consumption of silanol group on thesurface was complete after 19 cycles as
a)
b)
c)
Fig 6 a) Strategyfor the synthesis ofhydrothermally stablehighly ordered niobiamesoporous materialsby ALD Reproducedwith permission fromref [45] Copyright2011 AmericanChemical Societyb) Strategy for theformation of niobiacarbon compositecatalysts Adaptedwith permission ofElsevier from ref [68]c) Deposition of gra-phitic carbon layer onalumina powder andpellets by chemicallayer deposition fromCH4 Reproducedwith permission fromWiley-VCH Verlagfrom ref [42]
588 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
demonstrated by the disappearance of theSiOH band at 3735 cmndash1 in the FTIR spec-tra Hydrated amorphous niobia was iden-tified as a major species by Raman spec-troscopy for samples presenting 10 19 and30 ALD layers N
2physisorption showed
a decrease of surface area pore volumeand pore diameter with increasing niobialoadings The presence of acid sites formedupon niobia deposition was verified byNH
3-TPD and hydrothermal stability was
tested by treating the materials in liquidwater for 12 h at 200 degC and 28 bar argonThe structure of the coated materials aftertreatment was confirmed by X-ray diffrac-tion with the persistence of the hexagonalmesoporous structure pattern while the lat-ter pattern was suppressed after hydrother-mal treatment of the parent SBA-15 TEManalysis confirmed the stability differencesobserved by XRD A bifunctional catalystfor the transformation of GVL to pentanoicacid was also formed by subsequent depo-sition of Pd nanoparticles As for the sup-port this catalyst showed higher stabilitythan the conventional PdHY-340
Improved hydrothermal stability ofa niobia-supported Pd catalyst was alsoachieved by deposition of the latter ona more stable surface such as carbonHighly dispersed spherical particles of aniobiacarbon composite were formed bythe reaction of d-glucose and ammoniumniobium oxalate in an autoclave at 200degC (pH was controlled by urea decom-position) followed by pyrolysis at 400degC under N
2(Fig 6b)[68] Hydrothermal
stability was investigated during liquid-phase butanol dehydration at 240 degC and51 bar While initial activity of the com-posite bifunctional catalyst was lower thanthe commercial HY-340 catalyst the lat-ter quickly deactivated during the first 20h of reaction while the modified catalystsremained very stable and surpassed HY-340 after 8 h Interestingly the analogousmaterial obtained by incipient wetness im-pregnation of niobia led to the formation oflarge niobia crystallites underscoring theimportance of strong interaction betweenniobia and carbon[69] Finally bifunctionalcatalysts were prepared by incipient wet-ness impregnation of Pd(NO
3)2and tested
during the conversion of GVL to pentanoicacid leading to improved hydrothermalstability for the niobiacarbon compositecatalysts just like the metal-free system
For silica and alumina improved hy-drothermal stability above 100 degC wasachieved by deposition of a thin film of car-bon formed from simple sugars Pham et aldeposited carbon by pyrolysis of adsorbedsucrose at 400 degC under N
2flow on SBA-
15 silica gel and fumed alumina leadingto improved stability after treatment in liq-uid water at 100 degC for 12 h[41] MoreoverPd was deposited on the carbon-coated
oxides to test their ability as supports forthe catalytic hydrogenation of acetylenein the presence of excess ethylene For acomparable Pd particle size (1ndash15 nm)the authors observed formation of ethaneat high acetylene conversion on both SiO
2andAl
2O
3 while carbon-coating prevented
over-hydrogenation to ethane The FTIRstudy showed a low hydration potential forthe coated materials compared to SiO
2and
Al2O
3 Therefore the higher selectivity to
ethylene for the coated materials was at-tributed to their increased hydrophobicity
More recently Xiong et al reported onthe deposition of a graphitic carbon layerover alumina by chemical vapor deposition(CVD) from CH
4(Fig 6c)[42] The graphit-
ic carbon layer provided higher stabilitythan the carbon deposited from pyrolysisof sugars as demonstrated by the higherstability achieved under flowing air (500vs 250 degC) As monitored by the growthof the characteristic graphitic peak at 24deg2θ on the XRD pattern the amount of de-posited carbon was controlled by the CH
4deposition time (from 05 to 6 h) whereasother peaks attributed to γ-Al
2O
3were un-
touched TEM and energy-filtered TEM at-tested that the carbon coating was uniformon the oxide support Raman spectros-copy mapping confirmed that depositionwas uniform over the entire surface of thespheres Furthermore the relatively highratio (071) between the peak intensitiesfor G and D bands (at 1590 and 1350 cmndash1respectively) in the Raman spectrum of gc-Al
2O
3revealed a high degree of graphitiza-
tion on the entire surface No such featureswere observed for blank alumina and pyro-lyzed carbon on alumina
The hydrothermal stability of the cata-lyst was investigated using a treatmentunder static conditions at 220 degC underautogenic pressure in an autoclave usingwater After treatment the XRD pattern ofγ-Al
2O
3displayed the well-defined diffrac-
tion peaks of the boehmite phase attestingto a reaction with water Simultaneouslyno significant change was observed forgraphitic-carbon covered γ-Al
2O
3(gc-
Al2O
3) Improved stability for gc-Al
2O
3was confirmed by N
2physisorption results
which showed that no significant decreaseof surface area pore volume and poresize occurred during hydrothermal treat-ment while γ-Al
2O
3saw its surface area
decrease by 80 during the same treat-ment Stability of gc-Al
2O
3as a support
was tested in the aqueous phase reforming(APR) of ethylene glycol at 250 degC and70 bar after deposition of platinum par-ticles Conversion using Ptgc-Al
2O
3was
twice as high as for Ptγ-Al2O
3 This result
was consistent with the leached Al and Ptdetected in the liquid phase after reactionby ICP-OES for the uncoated materialIn addition to full conversion of γ-Al
2O
3
to boehmite observed by XRD STEMshowed Pt particle size increase for Ptγ-Al
2O
3 Hence deactivation of Ptγ-Al
2O
3was also attributed to Pt particle sinteringin addition to leaching whereas the use ofgraphitic carbon overcoat prevented bothphenomena under APR conditions Thisapproach was also successful when usingRugc-Al
2O
3for lactic acid hydrogenation
Overall graphitic carbon layers depositedthrough vapor phase route provided higherhydrothermal stability than pyrolytic car-bon-coatings and seems to be a promisingstabilization method for a wide range ofapplications
In addition to the deposition of thinfilm precise surface functionalization canalso be used to improve catalyst stabilityFor instance the generation of hydropho-bic surfaces by grafting of octadecyltri-chlorosilane groups onHY zeolite prevent-ed the support structure from collapsingduring stability tests which mimicked theupgrading of biomass-derived liquid com-pounds in biphasic systems at 200 degC[43]Stabilization was attributed to a limitedcontact between the zeolitersquos hydrophobicsurface and the aqueous phase The sameauthors also reported a systematic studywith various alkylsilanes from C
2to C
18[70]
Hydrothermal stability of zeolites hasbeen studied in great detail as zeolites arethemost widely used solid acid catalysts[71]and are often proposed for biomass con-version[7273] As shown by detailed char-acterization using 27Al and 29Si solid-stateNMR and NH
3-TPD siloxane bridge hy-
drolysis was the main deactivation routein the presence of hot liquid water whilezeolites treated under steam suffered fromdealumination[74] After a treatment in hotwater at 200 degC mimicking APR condi-tions ZSM-5 was stable independent ofits SiAl ratio while degradation of zeoliteY increased with increasing SiAl ratioas opposed to the better stability of zeo-lites with higher SiAl in the presence ofsteam Finally Al-rich zeolite Y could bestabilized by incorporation of lanthanumcations[74]
Recently Gardner et al reported thattesting the hydrothermal stability of zeo-lites in distilled water was not completelyrelevant to biomass conversion reactionconditions[75] In fact dry biomass con-tains about 15 wt of inorganic saltssuch as NaCl and the dissolution rate ofmetal oxides could be dramatically en-hanced by cations in solution[76] Catalytictests showed that the presence of NaCl hadno influence on glucose conversion duringthe liquid-phase transformation of glucoseto HMF on ZSM-5 However zeolite hy-drothermal stability was significantly per-turbed by the presence of NaCl leading tothe hydrolytic attack of extra-frameworkspecies andor defects sites Interestingly
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 589
leached Al species were active in the con-version of glucose to fructose
Together with metal particles zeolitesconstitute an important class of bifunc-tional catalysts While studies mentionedabove aimed at adding a stabilizing ele-ment to an existing material Ennaert et alassessed the stability of Ruzeolite H-USYin hot liquid water for the hydrolytic hy-drogenation of cellulose using variousSiAl ratios in order to select the optimalzeolite composition[77] They identifiedthat the lattice was stabilized by frame-work aluminum atoms which preventedhydrolysis of framework bonds while theextra-framework aluminum atoms locatedat the external surface prevented zeolitesolubilization With a SiAl ratio greaterthan 3 a stable quantitative hexitol yieldwas obtained
5 Improving Catalyst Stability byProcess Design
Previous sections demonstrated howimportant smart catalyst design can be forprolonged catalyst lifetime In additionimproved stability for a given catalyst canbe achieved by tuning process parametersIn fact proper reactions conditions such asthe choice of gas atmosphere (eg hydro-gen versus an inert gas) or concentrationof reagents and solvents can have dra-matic effects on processes As mentionedpreviously leaching and sintering of metalnanoparticles tends to be more pronouncedat low pH Nickel at pH values below 7 canbe oxidized by water or acids[78] to formNi2+ species[79] To counterbalance this ef-fect van Haasterecht et al used additionof KOH in ethylene glycol conversion overNiCNF catalyst with KOH concentrationbetween 0ndash12 M to increase the pH andimprove catalyst stability (Fig 7)[37] Niparticles were less stable in hydrothermalconditions in comparison to ethylene gly-col In both cases increasing the pH hada significant impact though it was not aspronounced in ethylene glycol Moreoverat very high pH (gt13) sintering occurredagain which was probably due to a for-mation of highly mobile hydroxide nickelspecies
A reductive gas atmosphere can lead toincreased catalyst stability A typical ex-ample is the aqueous phase reforming ofethylene glycol conducted over Ni-basedcatalysts[37] Introduction of hydrogen inthe gas atmosphere suppressed the growthof nickel nanoparticles The average par-ticle size after reaction in a hydrogen andan inert atmosphere was 58 and 32 nmrespectively Moreover leaching of thenickel decreased leading to 026 catalystmass loss under inert atmosphere com-pared to 015 mass loss in hydrogen
However the use of hydrogen is notalways beneficial It was reported that forplatinum supported on graphite at highpH under a hydrogen-rich atmosphereand at an elevated temperature agglom-eration of platinum could occur[80] Thereason was that removal of the graphiteoxygen functional groups by hydrogenlead to lower adhesion and thereforehigher mobility of the nanoparticles onthe surface
Coke formation over the catalyst canalso be inhibited by co-feeding other sub-stances to the reactor For instance in cata-lytic conversion of bio-oil into hydrocar-bons a stream of bio-oil was diluted witha ratio 12 with water which decreased themass of carbon deposits on microporousZSM-5[46] Unfortunately this treatmentled to irreversible catalyst deactivation dueto dealumination of the zeolite caused bywater at high temperature[8182] Howeverthe same authors found that the use of a70 wt methanol with a bio-oil mixturecould decrease coke formation up to 70and prevent dealumination and thereforemaintain activity of the catalyst
Preventing deactivation can also beachieved by controlling the reaction me-dia During the process of producing fur-fural from xylans high deactivation ofthe GaUSY catalyst was observed whenusing water as a solvent XRD analysis ofthe spent catalyst confirmed the collapseof the zeolite structure during reactionMoreover gallium content dramaticallydecreased after 24 hours of reaction from59 wt to 027 wt To avoid the nega-tive effects of water on the catalyst thebiphasic system waterndashmethyl isobutyl
ketone (MIBK) was used with a ratio of2080 Gallium leaching decreased duringthe first few hours (36 wt Ga) and con-tinued but at a lower rate to reach a metalcontent of 19 wt at the end of the reac-tion Furthermore XRD confirmed that thezeolite retained its structure at the end ofthe experiment confirming the importanceof using a co-solvent[83]
6 Conclusions and Outlook
Deactivation of catalysts under theharsh conditions used for biomass conver-sion is a bottleneck in the developmentof competitive processes for bioresourcevalorization to renewable alternatives tofossil-based fuels and chemicals Biomass-derived molecules that must be further up-graded are usually produced in relativelydilute liquid (often aqueous) streamswhich contain biogenic and inorganic im-purities This introduces significant chal-lenges for heterogeneous catalysts whichhave often been developed in the context ofreforming fairly pure gas-phase hydrocar-bons An important category of catalystsfor catalytic biomass upgrading are oxide-supported metal particles due to their usein hydrogenation and hydrodeoxygenationreactions are essential for removing oxy-gen from bio-based molecules Several re-searchers have sought to reduce the maindeactivation pathways for these catalystsnotably their irreversible deactivation viametal leaching and sintering in the liquidphase as well as their reversible deactiva-tion by poisoning Leaching and sinteringoccur preferentially due to the instability
Fig 7 Approximation based on experimental data between nickelnanoparticles size of the spent NiCNF catalysts and the pH of the finalreaction mixture in aqueous phase reforming of ethylene glycol ThepH of the feed solutions was adjusted by addition of KOH (0-12 M) toaqueous solutions of 1 wt ethylene glycol (green line) and to water(blue line) Reactions were performed at 230 degC for 6 h under inert gasAdapted with permission of Elsevier from ref [37]
590 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
of undercoordinated metal atoms Suchatoms can also favor byproduct formationdue to their high reactivity These byprod-ucts include coke in which case these re-active atoms contribute to poisoning theother active sites by initiating the forma-tion of carbon deposits For these reasonsseveral authors have reported methods topassivate these undercoordinated atomsusing overcoats deposited either by ALDsugar pyrolysis or CVD thereby stabiliz-ing the catalyst of interest Another issueis the leaching and structure modificationsof oxide supports due to the poor hydro-thermal stability of these solids whichinclude silica and zeolites In this caseseveral authors have turned to fine-tuningthe local environment of these catalystsagain by physically covering the materialof interest The addition of hydrophobicsurface functionalities or overcoats hasbeen shown to reduce support hydrolysisbywater molecules Similarly depositing apolymeric layer over metal hydrogenationsites was shown to act as an organic pseu-do-solvent which reduced the presence ofpolar biogenic catalyst poisons and thuscatalyst deactivation Finally increasedcatalyst lifetime was also improved byprocess engineering In fact tuning thereaction conditions including by loweringpH or co-feeding organic solvents and hy-drogen proved to be efficient for limitingcatalyst deactivation
Further research opportunities exist tocombine the principle strategies mentionedabove to further improve catalyst stabiliza-tion Tailored overcoats could be used bothto stabilize undercoordinated atoms and atthe same time to provide favorable micro-environments that could reduce hydrolysisandor concentrations of catalyst poisonsSuch an approach was used in part by add-ingMgO
xto reduce the acidity of an alumi-
na overcoat which was thought to catalyzecoke formation while the overcoat wasused to stabilize undercoordinated atomsHowever this approach could be furtherstrengthened by adding functionality to theovercoat and further tailoring themoleculardesign of the catalystrsquos local environmentFurthermore controlling pore size open-ings within these overcoats could intro-duce shape selectivity and be used to sieveout undesired reactants as was recentlydemonstrated for an alumina overcoat on atitania catalyst used for photocatalysis[84]Finally it will become increasingly impor-tant to use real biomass-derived streamsfor testing catalytic stability The negativeeffects of using liquid or aqueous condi-tions can be compounded by the presenceof numerous perhaps yet unidentified in-organic or biogenic impurities This wasnotably demonstrated by the acceleratedstructure collapse of zeolite structures inthe presence of inorganic salts
In summary current synthetic tech-niques can lead to unprecedented controlof the nanostructures surrounding a cata-lystrsquos active site but harsh conditions pres-ent for biomass conversion are equally un-precedented in industrial catalytic process-ing As society comes under increasingpressure to develop routes to sustainablecarbon-based molecules these syntheticmethods will likely prove essential in ad-dressing the challenges posed by catalyti-cally upgrading renewable resources suchas biomass
AcknowledgementsThe authors acknowledge financial support
from the Swiss National Science Foundationthrough grant number 407040_153866 as partof the National Research Program 70 and grantnumber PYAPP2_154281 and Benjamin LeMonnier for help with the design of figures
Received June 17 2015
[1] K Sugiura M Ishihara T Shimauchi SHarayama Environ Sci Technol 1997 31 45
[2] S Matar M J Mirbach HA Tayim lsquoCatalysisin Petrochemical Processesrsquo Springer Scienceamp Business Media 1989
[3] O S Abdul-Hamid Monthly Oil MarketReport - May 2015 OPEC 2015
[4] The European Parliament and the Council of theEuropean Union OJ 2009 L140 16
[5] R Rinaldi F Schuumlth Energy Environ Sci2009 2 610
[6] A A Peterson F Vogel R P Lachance MFroumlling J Michael JAntal JW Tester EnergyEnviron Sci 2008 1 32
[7] J S Luterbacher D M Alonso J A DumesicGreen Chem 2014 16 4816
[8] A Rahimi A Ulbrich J J Coon S S StahlNature 2014 515 249
[9] A Eyal R Jansen R Mali A Vitner lsquoSugarMixtures and Methods for Production and UseThereofrsquo 2012 WO 2011161685 A3
[10] D V Kazachkin M Colakyan F J MoeslerlsquoSupercritical Hydrolysis of Biomassrsquo 2015US 8999065 B2
[11] D L Sills J M Gossett Bioresour Technol2011 102 1389
[12] D M Alonso S G Wettstein M A MellmerE I Gurbuz J A Dumesic Energy EnvironSci 2012 6 76
[13] J S Luterbacher J M Rand D M Alonso JHan J T Youngquist C T Maravelias B FPfleger J A Dumesic Science 2014 343 277
[14] J S Luterbacher D M Alonso J M Rand YM Questell-Santiago J H Yeap B F PflegerJ A Dumesic ChemSusChem 2015 8 1317
[15] H Tadesse R Luque Energy Environ Sci2011 4 3913
[16] J B Binder R T Raines J Am Chem Soc2009 131 1979
[17] J S Luterbacher J W Tester L P WalkerBiotechnol Bioeng 2010 107 451
[18] D B Hodge M N Karim D J Schell J DMcMillan Bioresour Technol 2008 99 8940
[19] J S Luterbacher Q Chew Y Li J W TesterL P Walker Energy Environ Sci 2012 56990
[20] T J Schwartz B J OrsquoNeill B H Shanks J ADumesic ACS Catal 2014 4 2060
[21] G W Huber J N Chheda C J Barrett J ADumesic Science 2005 308 1446
[22] G W Huber R D Cortright J A DumesicAngew Chem Int Ed 2004 43 1549
[23] J N Chheda Y Romaacuten-Leshkov J ADumesic Green Chem 2007 9 342
[24] D W Rackemann W O Doherty BiofuelsBioprod Biorefining 2011 5 198
[25] J N Chheda G W Huber J A DumesicAngew Chem Int Ed 2007 46 7164
[26] A V Bandura S N Lvov J Phys Chem RefData 2006 35 15
[27] H Xiong H N Pham A K Datye GreenChem 2014 16 4627
[28] K P De Jong lsquoSynthesis Of Solid CatalystsrsquoWiley-VCHVerlag GmbH amp Co KGaA 2009
[29] F Heacuteroguel G Siddiqi M D Detwiler DY Zemlyanov O V Safonova C Copeacuteret JCatal 2015 321 81
[30] T W Hansen A T DeLaRiva S R Challa AK Datye Acc Chem Res 2013 46 1720
[31] B J OrsquoNeill C Sener D H K Jackson T FKuech J A Dumesic ChemSusChem 2014 73247
[32] B J OrsquoNeill J T Miller P J Dietrich FG Sollberger F H Ribeiro J A DumesicChemCatChem 2014 6 2493
[33] B J OrsquoNeill D H K Jackson A J Crisci CA Farberow F Shi A C Alba-Rubio J Lu PJ Dietrich X Gu C L Marshall et al AngewChem 2013 125 14053
[34] H Zhang Y Lei A J Kropf G Zhang J WElam J T Miller F Sollberger F Ribeiro MC Akatay E A Stach J A Dumesic C LMarshall J Catal 2014 317 284
[35] H ZhangY Lei A Jeremy Kropf G Zhang JW Elam J T Miller F Sollberger F RibeiroM Cem Akatay E A Stach J A Dumesic CL Marshall J Catal 2015 323 165
[36] J Lee D H K Jackson T Li R E Winans JA Dumesic T F Kuech G W Huber EnergyEnviron Sci 2014 7 1657
[37] T van Haasterecht C C I Ludding K P deJong J H Bitter J Catal 2014 319 27
[38] P Sun G Gao Z Zhao C Xia F Li ACSCatal 2014 4 4136
[39] J Lu B Fu M C Kung G Xiao J W ElamH H Kung P C Stair Science 2012 3351205
[40] T J Schwartz R L Johnson J Cardenas AOkerlund N A Da Silva K Schmidt-Rohr JA Dumesic Angew Chem Int Ed 2014 5312718
[41] H N Pham A E Anderson R L Johnson KSchmidt-Rohr A K Datye Angew Chem IntEd 2012 51 13163
[42] H Xiong T J Schwartz N I Andersen J ADumesic A K Datye Angew Chem Int Ed2015 early view DOI 101002anie201502206
[43] P A Zapata J Faria M P Ruiz R E JentoftD E Resasco J Am Chem Soc 2012 1348570
[44] H N Pham Y J Pagan-Torres J C Serrano-Ruiz D Wang J A Dumesic A K DatyeAppl Catal Gen 2011 397 153
[45] Y J Pagaacuten-Torres J M R Gallo D WangH N Pham J A Libera C L Marshall J WElam A K Datye J A Dumesic ACS Catal2011 1 1234
[46] A G Gayubo B Valle A T Aguayo MOlazar J Bilbao Energy Fuels 2009 23 4129
[47] S R Challa A T Delariva T W Hansen SHelveg J Sehested P L Hansen F Garzon AK Datye J Am Chem Soc 2011 133 20672
[48] J A Moulijn A E van Diepen F KapteijnAppl Catal Gen 2001 212 3
[49] S L Wegener T J Marks P C Stair AccChem Res 2012 45 206
[50] B J OrsquoNeill D H K Jackson J Lee CCanlas P C Stair C L Marshall J W ElamT F Kuech J A Dumesic G W Huber ACSCatal 2015 5 1804
[51] D M King J A Spencer II X Liang L FHakim A W Weimer Surf Coat Technol2007 201 9163
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 591
[52] J A McCormick B L Cloutier AWWeimerS M George J Vac Sci Technol A 2007 2567
[53] D Longrie D Deduytsche C Detavernier JVac Sci Technol A 2014 32 010802
[54] J Lu J W Elam P C Stair Acc Chem Res2013 46 1806
[55] L De Rogatis M Cargnello V GombacB Lorenzut T Montini P FornasieroChemSusChem 2010 3 24
[56] A G Sergeev J D Webb J F Hartwig J AmChem Soc 2012 134 20226
[57] J He C Zhao J A Lercher J Am Chem Soc2012 134 20768
[58] Q Song F Wang J CaiY Wang J Zhang WYu J Xu Energy Environ Sci 2013 6 994
[59] R Dehghan-Niri J C Walmsley A HolmenP A Midgley E Rytter A H Dam A BHungria J C Hernandez-Garrido D ChenCatal Sci Technol 2012 2 2476
[60] M Morales P Y Dapsens I Giovinazzo JWitte C Mondelli S PapadokonstantakisK Hungerbuumlhler J Peacuterez-Ramiacuterez EnergyEnviron Sci 2015 8 558
[61] O A Abdelrahman H Y Luo A Heyden YRomaacuten-Leshkov J Q Bond J Catal 2015329 10
[62] D M Alonso S G Wettstein J A DumesicChem Soc Rev 2012 41 8075
[63] W Li L Ye P Long J Chen H Ariga KAsakura YYuan RSC Adv 2014 4 29072
[64] G Prieto J Zecevic H Friedrich K P deJong P E de Jongh Nat Mater 2013 12 34
[65] Z Zhang J E Jackson D J Miller BioresourTechnol 2008 99 5873
[66] H Luo T Prasomsri Y Romaacuten-Leshkov TopCatal 2015 394
[67] J Mazumder H I de Lasa Catal Today 2014237 100
[68] H Xiong H N Pham A K Datye J Catal2013 302 93
[69] H Xiong M Nolan B H ShanksA K DatyeAppl Catal Gen 2014 471 165
[70] P A Zapata Y Huang M A Gonzalez-BorjaD E Resasco J Catal 2013 308 82
[71] A Corma Chem RevWash DC U S 1995 95559
[72] P A Jacobs M Dusselier B F Sels AngewChem Int Ed 2014 53 8621
[73] M Milina S Mitchell J Peacuterez-Ramiacuterez CatalToday 2014 235 176
[74] R M Ravenelle F Schuumlssler A DrsquoAmico NDanilina J A van Bokhoven J A Lercher CW Jones C Sievers J Phys Chem C 2010114 19582
[75] D W Gardner J Huo T C Hoff R LJohnson B H Shanks J-P Tessonnier ACSCatal 2015 DOI 101021acscatal5b00888
[76] P M Dove N Han J J D Yoreo Proc NatlAcad Sci 2005 102 15357
[77] T Ennaert J Geboers E Gobechiya C MCourtin M Kurttepeli K Houthoofd C E AKirschhock P C M M Magusin S Bals P AJacobs B F Sels ACS Catal 2015 5 754
[78] B Beverskog I Puigdomenech Corros Sci1997 39 969
[79] D A Palmer P Beacuteneacutezeth C Xiao D JWesolowski L M Anovitz J Solut Chem2011 40 680
[80] J H Vleeming B F M Kuster G B Marin FOudet P Courtine J Catal 1997 166 148
[81] A G Gayubo A T Aguayo M Olazar RVivanco J Bilbao Chem Eng Sci 2003 585239
[82] A de Lucas P CanizaresA DuraacutenA CarreroAppl Catal Gen 1997 154 221
[83] CAellig D Scholz PY Dapsens C MondelliJ Peacuterez-Ramiacuterez Catal Sci Technol 2014 5142
[84] C P Canlas J Lu N A Ray N A Grosso-Giordano S Lee J W Elam R E Winans RP Van Duyne P C Stair J M Notestein NatChem 2012 4 1030

588 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
demonstrated by the disappearance of theSiOH band at 3735 cmndash1 in the FTIR spec-tra Hydrated amorphous niobia was iden-tified as a major species by Raman spec-troscopy for samples presenting 10 19 and30 ALD layers N
2physisorption showed
a decrease of surface area pore volumeand pore diameter with increasing niobialoadings The presence of acid sites formedupon niobia deposition was verified byNH
3-TPD and hydrothermal stability was
tested by treating the materials in liquidwater for 12 h at 200 degC and 28 bar argonThe structure of the coated materials aftertreatment was confirmed by X-ray diffrac-tion with the persistence of the hexagonalmesoporous structure pattern while the lat-ter pattern was suppressed after hydrother-mal treatment of the parent SBA-15 TEManalysis confirmed the stability differencesobserved by XRD A bifunctional catalystfor the transformation of GVL to pentanoicacid was also formed by subsequent depo-sition of Pd nanoparticles As for the sup-port this catalyst showed higher stabilitythan the conventional PdHY-340
Improved hydrothermal stability ofa niobia-supported Pd catalyst was alsoachieved by deposition of the latter ona more stable surface such as carbonHighly dispersed spherical particles of aniobiacarbon composite were formed bythe reaction of d-glucose and ammoniumniobium oxalate in an autoclave at 200degC (pH was controlled by urea decom-position) followed by pyrolysis at 400degC under N
2(Fig 6b)[68] Hydrothermal
stability was investigated during liquid-phase butanol dehydration at 240 degC and51 bar While initial activity of the com-posite bifunctional catalyst was lower thanthe commercial HY-340 catalyst the lat-ter quickly deactivated during the first 20h of reaction while the modified catalystsremained very stable and surpassed HY-340 after 8 h Interestingly the analogousmaterial obtained by incipient wetness im-pregnation of niobia led to the formation oflarge niobia crystallites underscoring theimportance of strong interaction betweenniobia and carbon[69] Finally bifunctionalcatalysts were prepared by incipient wet-ness impregnation of Pd(NO
3)2and tested
during the conversion of GVL to pentanoicacid leading to improved hydrothermalstability for the niobiacarbon compositecatalysts just like the metal-free system
For silica and alumina improved hy-drothermal stability above 100 degC wasachieved by deposition of a thin film of car-bon formed from simple sugars Pham et aldeposited carbon by pyrolysis of adsorbedsucrose at 400 degC under N
2flow on SBA-
15 silica gel and fumed alumina leadingto improved stability after treatment in liq-uid water at 100 degC for 12 h[41] MoreoverPd was deposited on the carbon-coated
oxides to test their ability as supports forthe catalytic hydrogenation of acetylenein the presence of excess ethylene For acomparable Pd particle size (1ndash15 nm)the authors observed formation of ethaneat high acetylene conversion on both SiO
2andAl
2O
3 while carbon-coating prevented
over-hydrogenation to ethane The FTIRstudy showed a low hydration potential forthe coated materials compared to SiO
2and
Al2O
3 Therefore the higher selectivity to
ethylene for the coated materials was at-tributed to their increased hydrophobicity
More recently Xiong et al reported onthe deposition of a graphitic carbon layerover alumina by chemical vapor deposition(CVD) from CH
4(Fig 6c)[42] The graphit-
ic carbon layer provided higher stabilitythan the carbon deposited from pyrolysisof sugars as demonstrated by the higherstability achieved under flowing air (500vs 250 degC) As monitored by the growthof the characteristic graphitic peak at 24deg2θ on the XRD pattern the amount of de-posited carbon was controlled by the CH
4deposition time (from 05 to 6 h) whereasother peaks attributed to γ-Al
2O
3were un-
touched TEM and energy-filtered TEM at-tested that the carbon coating was uniformon the oxide support Raman spectros-copy mapping confirmed that depositionwas uniform over the entire surface of thespheres Furthermore the relatively highratio (071) between the peak intensitiesfor G and D bands (at 1590 and 1350 cmndash1respectively) in the Raman spectrum of gc-Al
2O
3revealed a high degree of graphitiza-
tion on the entire surface No such featureswere observed for blank alumina and pyro-lyzed carbon on alumina
The hydrothermal stability of the cata-lyst was investigated using a treatmentunder static conditions at 220 degC underautogenic pressure in an autoclave usingwater After treatment the XRD pattern ofγ-Al
2O
3displayed the well-defined diffrac-
tion peaks of the boehmite phase attestingto a reaction with water Simultaneouslyno significant change was observed forgraphitic-carbon covered γ-Al
2O
3(gc-
Al2O
3) Improved stability for gc-Al
2O
3was confirmed by N
2physisorption results
which showed that no significant decreaseof surface area pore volume and poresize occurred during hydrothermal treat-ment while γ-Al
2O
3saw its surface area
decrease by 80 during the same treat-ment Stability of gc-Al
2O
3as a support
was tested in the aqueous phase reforming(APR) of ethylene glycol at 250 degC and70 bar after deposition of platinum par-ticles Conversion using Ptgc-Al
2O
3was
twice as high as for Ptγ-Al2O
3 This result
was consistent with the leached Al and Ptdetected in the liquid phase after reactionby ICP-OES for the uncoated materialIn addition to full conversion of γ-Al
2O
3
to boehmite observed by XRD STEMshowed Pt particle size increase for Ptγ-Al
2O
3 Hence deactivation of Ptγ-Al
2O
3was also attributed to Pt particle sinteringin addition to leaching whereas the use ofgraphitic carbon overcoat prevented bothphenomena under APR conditions Thisapproach was also successful when usingRugc-Al
2O
3for lactic acid hydrogenation
Overall graphitic carbon layers depositedthrough vapor phase route provided higherhydrothermal stability than pyrolytic car-bon-coatings and seems to be a promisingstabilization method for a wide range ofapplications
In addition to the deposition of thinfilm precise surface functionalization canalso be used to improve catalyst stabilityFor instance the generation of hydropho-bic surfaces by grafting of octadecyltri-chlorosilane groups onHY zeolite prevent-ed the support structure from collapsingduring stability tests which mimicked theupgrading of biomass-derived liquid com-pounds in biphasic systems at 200 degC[43]Stabilization was attributed to a limitedcontact between the zeolitersquos hydrophobicsurface and the aqueous phase The sameauthors also reported a systematic studywith various alkylsilanes from C
2to C
18[70]
Hydrothermal stability of zeolites hasbeen studied in great detail as zeolites arethemost widely used solid acid catalysts[71]and are often proposed for biomass con-version[7273] As shown by detailed char-acterization using 27Al and 29Si solid-stateNMR and NH
3-TPD siloxane bridge hy-
drolysis was the main deactivation routein the presence of hot liquid water whilezeolites treated under steam suffered fromdealumination[74] After a treatment in hotwater at 200 degC mimicking APR condi-tions ZSM-5 was stable independent ofits SiAl ratio while degradation of zeoliteY increased with increasing SiAl ratioas opposed to the better stability of zeo-lites with higher SiAl in the presence ofsteam Finally Al-rich zeolite Y could bestabilized by incorporation of lanthanumcations[74]
Recently Gardner et al reported thattesting the hydrothermal stability of zeo-lites in distilled water was not completelyrelevant to biomass conversion reactionconditions[75] In fact dry biomass con-tains about 15 wt of inorganic saltssuch as NaCl and the dissolution rate ofmetal oxides could be dramatically en-hanced by cations in solution[76] Catalytictests showed that the presence of NaCl hadno influence on glucose conversion duringthe liquid-phase transformation of glucoseto HMF on ZSM-5 However zeolite hy-drothermal stability was significantly per-turbed by the presence of NaCl leading tothe hydrolytic attack of extra-frameworkspecies andor defects sites Interestingly
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 589
leached Al species were active in the con-version of glucose to fructose
Together with metal particles zeolitesconstitute an important class of bifunc-tional catalysts While studies mentionedabove aimed at adding a stabilizing ele-ment to an existing material Ennaert et alassessed the stability of Ruzeolite H-USYin hot liquid water for the hydrolytic hy-drogenation of cellulose using variousSiAl ratios in order to select the optimalzeolite composition[77] They identifiedthat the lattice was stabilized by frame-work aluminum atoms which preventedhydrolysis of framework bonds while theextra-framework aluminum atoms locatedat the external surface prevented zeolitesolubilization With a SiAl ratio greaterthan 3 a stable quantitative hexitol yieldwas obtained
5 Improving Catalyst Stability byProcess Design
Previous sections demonstrated howimportant smart catalyst design can be forprolonged catalyst lifetime In additionimproved stability for a given catalyst canbe achieved by tuning process parametersIn fact proper reactions conditions such asthe choice of gas atmosphere (eg hydro-gen versus an inert gas) or concentrationof reagents and solvents can have dra-matic effects on processes As mentionedpreviously leaching and sintering of metalnanoparticles tends to be more pronouncedat low pH Nickel at pH values below 7 canbe oxidized by water or acids[78] to formNi2+ species[79] To counterbalance this ef-fect van Haasterecht et al used additionof KOH in ethylene glycol conversion overNiCNF catalyst with KOH concentrationbetween 0ndash12 M to increase the pH andimprove catalyst stability (Fig 7)[37] Niparticles were less stable in hydrothermalconditions in comparison to ethylene gly-col In both cases increasing the pH hada significant impact though it was not aspronounced in ethylene glycol Moreoverat very high pH (gt13) sintering occurredagain which was probably due to a for-mation of highly mobile hydroxide nickelspecies
A reductive gas atmosphere can lead toincreased catalyst stability A typical ex-ample is the aqueous phase reforming ofethylene glycol conducted over Ni-basedcatalysts[37] Introduction of hydrogen inthe gas atmosphere suppressed the growthof nickel nanoparticles The average par-ticle size after reaction in a hydrogen andan inert atmosphere was 58 and 32 nmrespectively Moreover leaching of thenickel decreased leading to 026 catalystmass loss under inert atmosphere com-pared to 015 mass loss in hydrogen
However the use of hydrogen is notalways beneficial It was reported that forplatinum supported on graphite at highpH under a hydrogen-rich atmosphereand at an elevated temperature agglom-eration of platinum could occur[80] Thereason was that removal of the graphiteoxygen functional groups by hydrogenlead to lower adhesion and thereforehigher mobility of the nanoparticles onthe surface
Coke formation over the catalyst canalso be inhibited by co-feeding other sub-stances to the reactor For instance in cata-lytic conversion of bio-oil into hydrocar-bons a stream of bio-oil was diluted witha ratio 12 with water which decreased themass of carbon deposits on microporousZSM-5[46] Unfortunately this treatmentled to irreversible catalyst deactivation dueto dealumination of the zeolite caused bywater at high temperature[8182] Howeverthe same authors found that the use of a70 wt methanol with a bio-oil mixturecould decrease coke formation up to 70and prevent dealumination and thereforemaintain activity of the catalyst
Preventing deactivation can also beachieved by controlling the reaction me-dia During the process of producing fur-fural from xylans high deactivation ofthe GaUSY catalyst was observed whenusing water as a solvent XRD analysis ofthe spent catalyst confirmed the collapseof the zeolite structure during reactionMoreover gallium content dramaticallydecreased after 24 hours of reaction from59 wt to 027 wt To avoid the nega-tive effects of water on the catalyst thebiphasic system waterndashmethyl isobutyl
ketone (MIBK) was used with a ratio of2080 Gallium leaching decreased duringthe first few hours (36 wt Ga) and con-tinued but at a lower rate to reach a metalcontent of 19 wt at the end of the reac-tion Furthermore XRD confirmed that thezeolite retained its structure at the end ofthe experiment confirming the importanceof using a co-solvent[83]
6 Conclusions and Outlook
Deactivation of catalysts under theharsh conditions used for biomass conver-sion is a bottleneck in the developmentof competitive processes for bioresourcevalorization to renewable alternatives tofossil-based fuels and chemicals Biomass-derived molecules that must be further up-graded are usually produced in relativelydilute liquid (often aqueous) streamswhich contain biogenic and inorganic im-purities This introduces significant chal-lenges for heterogeneous catalysts whichhave often been developed in the context ofreforming fairly pure gas-phase hydrocar-bons An important category of catalystsfor catalytic biomass upgrading are oxide-supported metal particles due to their usein hydrogenation and hydrodeoxygenationreactions are essential for removing oxy-gen from bio-based molecules Several re-searchers have sought to reduce the maindeactivation pathways for these catalystsnotably their irreversible deactivation viametal leaching and sintering in the liquidphase as well as their reversible deactiva-tion by poisoning Leaching and sinteringoccur preferentially due to the instability
Fig 7 Approximation based on experimental data between nickelnanoparticles size of the spent NiCNF catalysts and the pH of the finalreaction mixture in aqueous phase reforming of ethylene glycol ThepH of the feed solutions was adjusted by addition of KOH (0-12 M) toaqueous solutions of 1 wt ethylene glycol (green line) and to water(blue line) Reactions were performed at 230 degC for 6 h under inert gasAdapted with permission of Elsevier from ref [37]
590 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
of undercoordinated metal atoms Suchatoms can also favor byproduct formationdue to their high reactivity These byprod-ucts include coke in which case these re-active atoms contribute to poisoning theother active sites by initiating the forma-tion of carbon deposits For these reasonsseveral authors have reported methods topassivate these undercoordinated atomsusing overcoats deposited either by ALDsugar pyrolysis or CVD thereby stabiliz-ing the catalyst of interest Another issueis the leaching and structure modificationsof oxide supports due to the poor hydro-thermal stability of these solids whichinclude silica and zeolites In this caseseveral authors have turned to fine-tuningthe local environment of these catalystsagain by physically covering the materialof interest The addition of hydrophobicsurface functionalities or overcoats hasbeen shown to reduce support hydrolysisbywater molecules Similarly depositing apolymeric layer over metal hydrogenationsites was shown to act as an organic pseu-do-solvent which reduced the presence ofpolar biogenic catalyst poisons and thuscatalyst deactivation Finally increasedcatalyst lifetime was also improved byprocess engineering In fact tuning thereaction conditions including by loweringpH or co-feeding organic solvents and hy-drogen proved to be efficient for limitingcatalyst deactivation
Further research opportunities exist tocombine the principle strategies mentionedabove to further improve catalyst stabiliza-tion Tailored overcoats could be used bothto stabilize undercoordinated atoms and atthe same time to provide favorable micro-environments that could reduce hydrolysisandor concentrations of catalyst poisonsSuch an approach was used in part by add-ingMgO
xto reduce the acidity of an alumi-
na overcoat which was thought to catalyzecoke formation while the overcoat wasused to stabilize undercoordinated atomsHowever this approach could be furtherstrengthened by adding functionality to theovercoat and further tailoring themoleculardesign of the catalystrsquos local environmentFurthermore controlling pore size open-ings within these overcoats could intro-duce shape selectivity and be used to sieveout undesired reactants as was recentlydemonstrated for an alumina overcoat on atitania catalyst used for photocatalysis[84]Finally it will become increasingly impor-tant to use real biomass-derived streamsfor testing catalytic stability The negativeeffects of using liquid or aqueous condi-tions can be compounded by the presenceof numerous perhaps yet unidentified in-organic or biogenic impurities This wasnotably demonstrated by the acceleratedstructure collapse of zeolite structures inthe presence of inorganic salts
In summary current synthetic tech-niques can lead to unprecedented controlof the nanostructures surrounding a cata-lystrsquos active site but harsh conditions pres-ent for biomass conversion are equally un-precedented in industrial catalytic process-ing As society comes under increasingpressure to develop routes to sustainablecarbon-based molecules these syntheticmethods will likely prove essential in ad-dressing the challenges posed by catalyti-cally upgrading renewable resources suchas biomass
AcknowledgementsThe authors acknowledge financial support
from the Swiss National Science Foundationthrough grant number 407040_153866 as partof the National Research Program 70 and grantnumber PYAPP2_154281 and Benjamin LeMonnier for help with the design of figures
Received June 17 2015
[1] K Sugiura M Ishihara T Shimauchi SHarayama Environ Sci Technol 1997 31 45
[2] S Matar M J Mirbach HA Tayim lsquoCatalysisin Petrochemical Processesrsquo Springer Scienceamp Business Media 1989
[3] O S Abdul-Hamid Monthly Oil MarketReport - May 2015 OPEC 2015
[4] The European Parliament and the Council of theEuropean Union OJ 2009 L140 16
[5] R Rinaldi F Schuumlth Energy Environ Sci2009 2 610
[6] A A Peterson F Vogel R P Lachance MFroumlling J Michael JAntal JW Tester EnergyEnviron Sci 2008 1 32
[7] J S Luterbacher D M Alonso J A DumesicGreen Chem 2014 16 4816
[8] A Rahimi A Ulbrich J J Coon S S StahlNature 2014 515 249
[9] A Eyal R Jansen R Mali A Vitner lsquoSugarMixtures and Methods for Production and UseThereofrsquo 2012 WO 2011161685 A3
[10] D V Kazachkin M Colakyan F J MoeslerlsquoSupercritical Hydrolysis of Biomassrsquo 2015US 8999065 B2
[11] D L Sills J M Gossett Bioresour Technol2011 102 1389
[12] D M Alonso S G Wettstein M A MellmerE I Gurbuz J A Dumesic Energy EnvironSci 2012 6 76
[13] J S Luterbacher J M Rand D M Alonso JHan J T Youngquist C T Maravelias B FPfleger J A Dumesic Science 2014 343 277
[14] J S Luterbacher D M Alonso J M Rand YM Questell-Santiago J H Yeap B F PflegerJ A Dumesic ChemSusChem 2015 8 1317
[15] H Tadesse R Luque Energy Environ Sci2011 4 3913
[16] J B Binder R T Raines J Am Chem Soc2009 131 1979
[17] J S Luterbacher J W Tester L P WalkerBiotechnol Bioeng 2010 107 451
[18] D B Hodge M N Karim D J Schell J DMcMillan Bioresour Technol 2008 99 8940
[19] J S Luterbacher Q Chew Y Li J W TesterL P Walker Energy Environ Sci 2012 56990
[20] T J Schwartz B J OrsquoNeill B H Shanks J ADumesic ACS Catal 2014 4 2060
[21] G W Huber J N Chheda C J Barrett J ADumesic Science 2005 308 1446
[22] G W Huber R D Cortright J A DumesicAngew Chem Int Ed 2004 43 1549
[23] J N Chheda Y Romaacuten-Leshkov J ADumesic Green Chem 2007 9 342
[24] D W Rackemann W O Doherty BiofuelsBioprod Biorefining 2011 5 198
[25] J N Chheda G W Huber J A DumesicAngew Chem Int Ed 2007 46 7164
[26] A V Bandura S N Lvov J Phys Chem RefData 2006 35 15
[27] H Xiong H N Pham A K Datye GreenChem 2014 16 4627
[28] K P De Jong lsquoSynthesis Of Solid CatalystsrsquoWiley-VCHVerlag GmbH amp Co KGaA 2009
[29] F Heacuteroguel G Siddiqi M D Detwiler DY Zemlyanov O V Safonova C Copeacuteret JCatal 2015 321 81
[30] T W Hansen A T DeLaRiva S R Challa AK Datye Acc Chem Res 2013 46 1720
[31] B J OrsquoNeill C Sener D H K Jackson T FKuech J A Dumesic ChemSusChem 2014 73247
[32] B J OrsquoNeill J T Miller P J Dietrich FG Sollberger F H Ribeiro J A DumesicChemCatChem 2014 6 2493
[33] B J OrsquoNeill D H K Jackson A J Crisci CA Farberow F Shi A C Alba-Rubio J Lu PJ Dietrich X Gu C L Marshall et al AngewChem 2013 125 14053
[34] H Zhang Y Lei A J Kropf G Zhang J WElam J T Miller F Sollberger F Ribeiro MC Akatay E A Stach J A Dumesic C LMarshall J Catal 2014 317 284
[35] H ZhangY Lei A Jeremy Kropf G Zhang JW Elam J T Miller F Sollberger F RibeiroM Cem Akatay E A Stach J A Dumesic CL Marshall J Catal 2015 323 165
[36] J Lee D H K Jackson T Li R E Winans JA Dumesic T F Kuech G W Huber EnergyEnviron Sci 2014 7 1657
[37] T van Haasterecht C C I Ludding K P deJong J H Bitter J Catal 2014 319 27
[38] P Sun G Gao Z Zhao C Xia F Li ACSCatal 2014 4 4136
[39] J Lu B Fu M C Kung G Xiao J W ElamH H Kung P C Stair Science 2012 3351205
[40] T J Schwartz R L Johnson J Cardenas AOkerlund N A Da Silva K Schmidt-Rohr JA Dumesic Angew Chem Int Ed 2014 5312718
[41] H N Pham A E Anderson R L Johnson KSchmidt-Rohr A K Datye Angew Chem IntEd 2012 51 13163
[42] H Xiong T J Schwartz N I Andersen J ADumesic A K Datye Angew Chem Int Ed2015 early view DOI 101002anie201502206
[43] P A Zapata J Faria M P Ruiz R E JentoftD E Resasco J Am Chem Soc 2012 1348570
[44] H N Pham Y J Pagan-Torres J C Serrano-Ruiz D Wang J A Dumesic A K DatyeAppl Catal Gen 2011 397 153
[45] Y J Pagaacuten-Torres J M R Gallo D WangH N Pham J A Libera C L Marshall J WElam A K Datye J A Dumesic ACS Catal2011 1 1234
[46] A G Gayubo B Valle A T Aguayo MOlazar J Bilbao Energy Fuels 2009 23 4129
[47] S R Challa A T Delariva T W Hansen SHelveg J Sehested P L Hansen F Garzon AK Datye J Am Chem Soc 2011 133 20672
[48] J A Moulijn A E van Diepen F KapteijnAppl Catal Gen 2001 212 3
[49] S L Wegener T J Marks P C Stair AccChem Res 2012 45 206
[50] B J OrsquoNeill D H K Jackson J Lee CCanlas P C Stair C L Marshall J W ElamT F Kuech J A Dumesic G W Huber ACSCatal 2015 5 1804
[51] D M King J A Spencer II X Liang L FHakim A W Weimer Surf Coat Technol2007 201 9163
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 591
[52] J A McCormick B L Cloutier AWWeimerS M George J Vac Sci Technol A 2007 2567
[53] D Longrie D Deduytsche C Detavernier JVac Sci Technol A 2014 32 010802
[54] J Lu J W Elam P C Stair Acc Chem Res2013 46 1806
[55] L De Rogatis M Cargnello V GombacB Lorenzut T Montini P FornasieroChemSusChem 2010 3 24
[56] A G Sergeev J D Webb J F Hartwig J AmChem Soc 2012 134 20226
[57] J He C Zhao J A Lercher J Am Chem Soc2012 134 20768
[58] Q Song F Wang J CaiY Wang J Zhang WYu J Xu Energy Environ Sci 2013 6 994
[59] R Dehghan-Niri J C Walmsley A HolmenP A Midgley E Rytter A H Dam A BHungria J C Hernandez-Garrido D ChenCatal Sci Technol 2012 2 2476
[60] M Morales P Y Dapsens I Giovinazzo JWitte C Mondelli S PapadokonstantakisK Hungerbuumlhler J Peacuterez-Ramiacuterez EnergyEnviron Sci 2015 8 558
[61] O A Abdelrahman H Y Luo A Heyden YRomaacuten-Leshkov J Q Bond J Catal 2015329 10
[62] D M Alonso S G Wettstein J A DumesicChem Soc Rev 2012 41 8075
[63] W Li L Ye P Long J Chen H Ariga KAsakura YYuan RSC Adv 2014 4 29072
[64] G Prieto J Zecevic H Friedrich K P deJong P E de Jongh Nat Mater 2013 12 34
[65] Z Zhang J E Jackson D J Miller BioresourTechnol 2008 99 5873
[66] H Luo T Prasomsri Y Romaacuten-Leshkov TopCatal 2015 394
[67] J Mazumder H I de Lasa Catal Today 2014237 100
[68] H Xiong H N Pham A K Datye J Catal2013 302 93
[69] H Xiong M Nolan B H ShanksA K DatyeAppl Catal Gen 2014 471 165
[70] P A Zapata Y Huang M A Gonzalez-BorjaD E Resasco J Catal 2013 308 82
[71] A Corma Chem RevWash DC U S 1995 95559
[72] P A Jacobs M Dusselier B F Sels AngewChem Int Ed 2014 53 8621
[73] M Milina S Mitchell J Peacuterez-Ramiacuterez CatalToday 2014 235 176
[74] R M Ravenelle F Schuumlssler A DrsquoAmico NDanilina J A van Bokhoven J A Lercher CW Jones C Sievers J Phys Chem C 2010114 19582
[75] D W Gardner J Huo T C Hoff R LJohnson B H Shanks J-P Tessonnier ACSCatal 2015 DOI 101021acscatal5b00888
[76] P M Dove N Han J J D Yoreo Proc NatlAcad Sci 2005 102 15357
[77] T Ennaert J Geboers E Gobechiya C MCourtin M Kurttepeli K Houthoofd C E AKirschhock P C M M Magusin S Bals P AJacobs B F Sels ACS Catal 2015 5 754
[78] B Beverskog I Puigdomenech Corros Sci1997 39 969
[79] D A Palmer P Beacuteneacutezeth C Xiao D JWesolowski L M Anovitz J Solut Chem2011 40 680
[80] J H Vleeming B F M Kuster G B Marin FOudet P Courtine J Catal 1997 166 148
[81] A G Gayubo A T Aguayo M Olazar RVivanco J Bilbao Chem Eng Sci 2003 585239
[82] A de Lucas P CanizaresA DuraacutenA CarreroAppl Catal Gen 1997 154 221
[83] CAellig D Scholz PY Dapsens C MondelliJ Peacuterez-Ramiacuterez Catal Sci Technol 2014 5142
[84] C P Canlas J Lu N A Ray N A Grosso-Giordano S Lee J W Elam R E Winans RP Van Duyne P C Stair J M Notestein NatChem 2012 4 1030
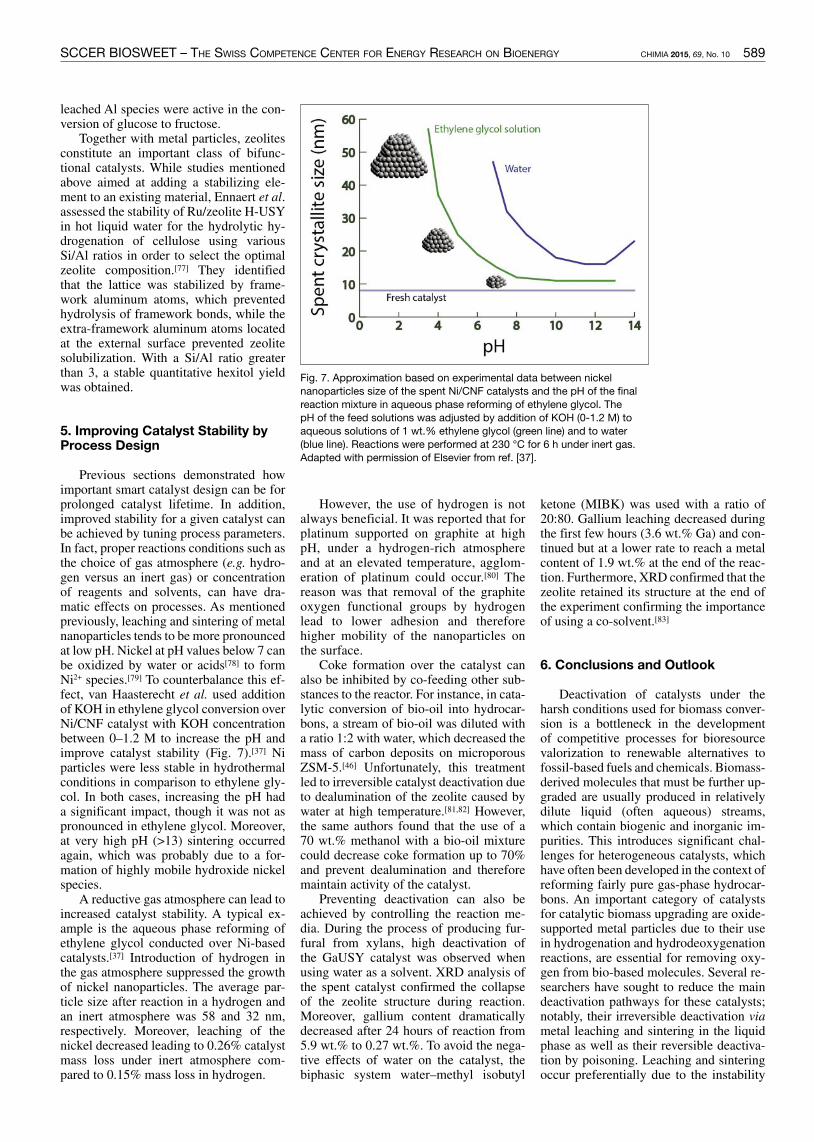
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 589
leached Al species were active in the con-version of glucose to fructose
Together with metal particles zeolitesconstitute an important class of bifunc-tional catalysts While studies mentionedabove aimed at adding a stabilizing ele-ment to an existing material Ennaert et alassessed the stability of Ruzeolite H-USYin hot liquid water for the hydrolytic hy-drogenation of cellulose using variousSiAl ratios in order to select the optimalzeolite composition[77] They identifiedthat the lattice was stabilized by frame-work aluminum atoms which preventedhydrolysis of framework bonds while theextra-framework aluminum atoms locatedat the external surface prevented zeolitesolubilization With a SiAl ratio greaterthan 3 a stable quantitative hexitol yieldwas obtained
5 Improving Catalyst Stability byProcess Design
Previous sections demonstrated howimportant smart catalyst design can be forprolonged catalyst lifetime In additionimproved stability for a given catalyst canbe achieved by tuning process parametersIn fact proper reactions conditions such asthe choice of gas atmosphere (eg hydro-gen versus an inert gas) or concentrationof reagents and solvents can have dra-matic effects on processes As mentionedpreviously leaching and sintering of metalnanoparticles tends to be more pronouncedat low pH Nickel at pH values below 7 canbe oxidized by water or acids[78] to formNi2+ species[79] To counterbalance this ef-fect van Haasterecht et al used additionof KOH in ethylene glycol conversion overNiCNF catalyst with KOH concentrationbetween 0ndash12 M to increase the pH andimprove catalyst stability (Fig 7)[37] Niparticles were less stable in hydrothermalconditions in comparison to ethylene gly-col In both cases increasing the pH hada significant impact though it was not aspronounced in ethylene glycol Moreoverat very high pH (gt13) sintering occurredagain which was probably due to a for-mation of highly mobile hydroxide nickelspecies
A reductive gas atmosphere can lead toincreased catalyst stability A typical ex-ample is the aqueous phase reforming ofethylene glycol conducted over Ni-basedcatalysts[37] Introduction of hydrogen inthe gas atmosphere suppressed the growthof nickel nanoparticles The average par-ticle size after reaction in a hydrogen andan inert atmosphere was 58 and 32 nmrespectively Moreover leaching of thenickel decreased leading to 026 catalystmass loss under inert atmosphere com-pared to 015 mass loss in hydrogen
However the use of hydrogen is notalways beneficial It was reported that forplatinum supported on graphite at highpH under a hydrogen-rich atmosphereand at an elevated temperature agglom-eration of platinum could occur[80] Thereason was that removal of the graphiteoxygen functional groups by hydrogenlead to lower adhesion and thereforehigher mobility of the nanoparticles onthe surface
Coke formation over the catalyst canalso be inhibited by co-feeding other sub-stances to the reactor For instance in cata-lytic conversion of bio-oil into hydrocar-bons a stream of bio-oil was diluted witha ratio 12 with water which decreased themass of carbon deposits on microporousZSM-5[46] Unfortunately this treatmentled to irreversible catalyst deactivation dueto dealumination of the zeolite caused bywater at high temperature[8182] Howeverthe same authors found that the use of a70 wt methanol with a bio-oil mixturecould decrease coke formation up to 70and prevent dealumination and thereforemaintain activity of the catalyst
Preventing deactivation can also beachieved by controlling the reaction me-dia During the process of producing fur-fural from xylans high deactivation ofthe GaUSY catalyst was observed whenusing water as a solvent XRD analysis ofthe spent catalyst confirmed the collapseof the zeolite structure during reactionMoreover gallium content dramaticallydecreased after 24 hours of reaction from59 wt to 027 wt To avoid the nega-tive effects of water on the catalyst thebiphasic system waterndashmethyl isobutyl
ketone (MIBK) was used with a ratio of2080 Gallium leaching decreased duringthe first few hours (36 wt Ga) and con-tinued but at a lower rate to reach a metalcontent of 19 wt at the end of the reac-tion Furthermore XRD confirmed that thezeolite retained its structure at the end ofthe experiment confirming the importanceof using a co-solvent[83]
6 Conclusions and Outlook
Deactivation of catalysts under theharsh conditions used for biomass conver-sion is a bottleneck in the developmentof competitive processes for bioresourcevalorization to renewable alternatives tofossil-based fuels and chemicals Biomass-derived molecules that must be further up-graded are usually produced in relativelydilute liquid (often aqueous) streamswhich contain biogenic and inorganic im-purities This introduces significant chal-lenges for heterogeneous catalysts whichhave often been developed in the context ofreforming fairly pure gas-phase hydrocar-bons An important category of catalystsfor catalytic biomass upgrading are oxide-supported metal particles due to their usein hydrogenation and hydrodeoxygenationreactions are essential for removing oxy-gen from bio-based molecules Several re-searchers have sought to reduce the maindeactivation pathways for these catalystsnotably their irreversible deactivation viametal leaching and sintering in the liquidphase as well as their reversible deactiva-tion by poisoning Leaching and sinteringoccur preferentially due to the instability
Fig 7 Approximation based on experimental data between nickelnanoparticles size of the spent NiCNF catalysts and the pH of the finalreaction mixture in aqueous phase reforming of ethylene glycol ThepH of the feed solutions was adjusted by addition of KOH (0-12 M) toaqueous solutions of 1 wt ethylene glycol (green line) and to water(blue line) Reactions were performed at 230 degC for 6 h under inert gasAdapted with permission of Elsevier from ref [37]
590 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
of undercoordinated metal atoms Suchatoms can also favor byproduct formationdue to their high reactivity These byprod-ucts include coke in which case these re-active atoms contribute to poisoning theother active sites by initiating the forma-tion of carbon deposits For these reasonsseveral authors have reported methods topassivate these undercoordinated atomsusing overcoats deposited either by ALDsugar pyrolysis or CVD thereby stabiliz-ing the catalyst of interest Another issueis the leaching and structure modificationsof oxide supports due to the poor hydro-thermal stability of these solids whichinclude silica and zeolites In this caseseveral authors have turned to fine-tuningthe local environment of these catalystsagain by physically covering the materialof interest The addition of hydrophobicsurface functionalities or overcoats hasbeen shown to reduce support hydrolysisbywater molecules Similarly depositing apolymeric layer over metal hydrogenationsites was shown to act as an organic pseu-do-solvent which reduced the presence ofpolar biogenic catalyst poisons and thuscatalyst deactivation Finally increasedcatalyst lifetime was also improved byprocess engineering In fact tuning thereaction conditions including by loweringpH or co-feeding organic solvents and hy-drogen proved to be efficient for limitingcatalyst deactivation
Further research opportunities exist tocombine the principle strategies mentionedabove to further improve catalyst stabiliza-tion Tailored overcoats could be used bothto stabilize undercoordinated atoms and atthe same time to provide favorable micro-environments that could reduce hydrolysisandor concentrations of catalyst poisonsSuch an approach was used in part by add-ingMgO
xto reduce the acidity of an alumi-
na overcoat which was thought to catalyzecoke formation while the overcoat wasused to stabilize undercoordinated atomsHowever this approach could be furtherstrengthened by adding functionality to theovercoat and further tailoring themoleculardesign of the catalystrsquos local environmentFurthermore controlling pore size open-ings within these overcoats could intro-duce shape selectivity and be used to sieveout undesired reactants as was recentlydemonstrated for an alumina overcoat on atitania catalyst used for photocatalysis[84]Finally it will become increasingly impor-tant to use real biomass-derived streamsfor testing catalytic stability The negativeeffects of using liquid or aqueous condi-tions can be compounded by the presenceof numerous perhaps yet unidentified in-organic or biogenic impurities This wasnotably demonstrated by the acceleratedstructure collapse of zeolite structures inthe presence of inorganic salts
In summary current synthetic tech-niques can lead to unprecedented controlof the nanostructures surrounding a cata-lystrsquos active site but harsh conditions pres-ent for biomass conversion are equally un-precedented in industrial catalytic process-ing As society comes under increasingpressure to develop routes to sustainablecarbon-based molecules these syntheticmethods will likely prove essential in ad-dressing the challenges posed by catalyti-cally upgrading renewable resources suchas biomass
AcknowledgementsThe authors acknowledge financial support
from the Swiss National Science Foundationthrough grant number 407040_153866 as partof the National Research Program 70 and grantnumber PYAPP2_154281 and Benjamin LeMonnier for help with the design of figures
Received June 17 2015
[1] K Sugiura M Ishihara T Shimauchi SHarayama Environ Sci Technol 1997 31 45
[2] S Matar M J Mirbach HA Tayim lsquoCatalysisin Petrochemical Processesrsquo Springer Scienceamp Business Media 1989
[3] O S Abdul-Hamid Monthly Oil MarketReport - May 2015 OPEC 2015
[4] The European Parliament and the Council of theEuropean Union OJ 2009 L140 16
[5] R Rinaldi F Schuumlth Energy Environ Sci2009 2 610
[6] A A Peterson F Vogel R P Lachance MFroumlling J Michael JAntal JW Tester EnergyEnviron Sci 2008 1 32
[7] J S Luterbacher D M Alonso J A DumesicGreen Chem 2014 16 4816
[8] A Rahimi A Ulbrich J J Coon S S StahlNature 2014 515 249
[9] A Eyal R Jansen R Mali A Vitner lsquoSugarMixtures and Methods for Production and UseThereofrsquo 2012 WO 2011161685 A3
[10] D V Kazachkin M Colakyan F J MoeslerlsquoSupercritical Hydrolysis of Biomassrsquo 2015US 8999065 B2
[11] D L Sills J M Gossett Bioresour Technol2011 102 1389
[12] D M Alonso S G Wettstein M A MellmerE I Gurbuz J A Dumesic Energy EnvironSci 2012 6 76
[13] J S Luterbacher J M Rand D M Alonso JHan J T Youngquist C T Maravelias B FPfleger J A Dumesic Science 2014 343 277
[14] J S Luterbacher D M Alonso J M Rand YM Questell-Santiago J H Yeap B F PflegerJ A Dumesic ChemSusChem 2015 8 1317
[15] H Tadesse R Luque Energy Environ Sci2011 4 3913
[16] J B Binder R T Raines J Am Chem Soc2009 131 1979
[17] J S Luterbacher J W Tester L P WalkerBiotechnol Bioeng 2010 107 451
[18] D B Hodge M N Karim D J Schell J DMcMillan Bioresour Technol 2008 99 8940
[19] J S Luterbacher Q Chew Y Li J W TesterL P Walker Energy Environ Sci 2012 56990
[20] T J Schwartz B J OrsquoNeill B H Shanks J ADumesic ACS Catal 2014 4 2060
[21] G W Huber J N Chheda C J Barrett J ADumesic Science 2005 308 1446
[22] G W Huber R D Cortright J A DumesicAngew Chem Int Ed 2004 43 1549
[23] J N Chheda Y Romaacuten-Leshkov J ADumesic Green Chem 2007 9 342
[24] D W Rackemann W O Doherty BiofuelsBioprod Biorefining 2011 5 198
[25] J N Chheda G W Huber J A DumesicAngew Chem Int Ed 2007 46 7164
[26] A V Bandura S N Lvov J Phys Chem RefData 2006 35 15
[27] H Xiong H N Pham A K Datye GreenChem 2014 16 4627
[28] K P De Jong lsquoSynthesis Of Solid CatalystsrsquoWiley-VCHVerlag GmbH amp Co KGaA 2009
[29] F Heacuteroguel G Siddiqi M D Detwiler DY Zemlyanov O V Safonova C Copeacuteret JCatal 2015 321 81
[30] T W Hansen A T DeLaRiva S R Challa AK Datye Acc Chem Res 2013 46 1720
[31] B J OrsquoNeill C Sener D H K Jackson T FKuech J A Dumesic ChemSusChem 2014 73247
[32] B J OrsquoNeill J T Miller P J Dietrich FG Sollberger F H Ribeiro J A DumesicChemCatChem 2014 6 2493
[33] B J OrsquoNeill D H K Jackson A J Crisci CA Farberow F Shi A C Alba-Rubio J Lu PJ Dietrich X Gu C L Marshall et al AngewChem 2013 125 14053
[34] H Zhang Y Lei A J Kropf G Zhang J WElam J T Miller F Sollberger F Ribeiro MC Akatay E A Stach J A Dumesic C LMarshall J Catal 2014 317 284
[35] H ZhangY Lei A Jeremy Kropf G Zhang JW Elam J T Miller F Sollberger F RibeiroM Cem Akatay E A Stach J A Dumesic CL Marshall J Catal 2015 323 165
[36] J Lee D H K Jackson T Li R E Winans JA Dumesic T F Kuech G W Huber EnergyEnviron Sci 2014 7 1657
[37] T van Haasterecht C C I Ludding K P deJong J H Bitter J Catal 2014 319 27
[38] P Sun G Gao Z Zhao C Xia F Li ACSCatal 2014 4 4136
[39] J Lu B Fu M C Kung G Xiao J W ElamH H Kung P C Stair Science 2012 3351205
[40] T J Schwartz R L Johnson J Cardenas AOkerlund N A Da Silva K Schmidt-Rohr JA Dumesic Angew Chem Int Ed 2014 5312718
[41] H N Pham A E Anderson R L Johnson KSchmidt-Rohr A K Datye Angew Chem IntEd 2012 51 13163
[42] H Xiong T J Schwartz N I Andersen J ADumesic A K Datye Angew Chem Int Ed2015 early view DOI 101002anie201502206
[43] P A Zapata J Faria M P Ruiz R E JentoftD E Resasco J Am Chem Soc 2012 1348570
[44] H N Pham Y J Pagan-Torres J C Serrano-Ruiz D Wang J A Dumesic A K DatyeAppl Catal Gen 2011 397 153
[45] Y J Pagaacuten-Torres J M R Gallo D WangH N Pham J A Libera C L Marshall J WElam A K Datye J A Dumesic ACS Catal2011 1 1234
[46] A G Gayubo B Valle A T Aguayo MOlazar J Bilbao Energy Fuels 2009 23 4129
[47] S R Challa A T Delariva T W Hansen SHelveg J Sehested P L Hansen F Garzon AK Datye J Am Chem Soc 2011 133 20672
[48] J A Moulijn A E van Diepen F KapteijnAppl Catal Gen 2001 212 3
[49] S L Wegener T J Marks P C Stair AccChem Res 2012 45 206
[50] B J OrsquoNeill D H K Jackson J Lee CCanlas P C Stair C L Marshall J W ElamT F Kuech J A Dumesic G W Huber ACSCatal 2015 5 1804
[51] D M King J A Spencer II X Liang L FHakim A W Weimer Surf Coat Technol2007 201 9163
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 591
[52] J A McCormick B L Cloutier AWWeimerS M George J Vac Sci Technol A 2007 2567
[53] D Longrie D Deduytsche C Detavernier JVac Sci Technol A 2014 32 010802
[54] J Lu J W Elam P C Stair Acc Chem Res2013 46 1806
[55] L De Rogatis M Cargnello V GombacB Lorenzut T Montini P FornasieroChemSusChem 2010 3 24
[56] A G Sergeev J D Webb J F Hartwig J AmChem Soc 2012 134 20226
[57] J He C Zhao J A Lercher J Am Chem Soc2012 134 20768
[58] Q Song F Wang J CaiY Wang J Zhang WYu J Xu Energy Environ Sci 2013 6 994
[59] R Dehghan-Niri J C Walmsley A HolmenP A Midgley E Rytter A H Dam A BHungria J C Hernandez-Garrido D ChenCatal Sci Technol 2012 2 2476
[60] M Morales P Y Dapsens I Giovinazzo JWitte C Mondelli S PapadokonstantakisK Hungerbuumlhler J Peacuterez-Ramiacuterez EnergyEnviron Sci 2015 8 558
[61] O A Abdelrahman H Y Luo A Heyden YRomaacuten-Leshkov J Q Bond J Catal 2015329 10
[62] D M Alonso S G Wettstein J A DumesicChem Soc Rev 2012 41 8075
[63] W Li L Ye P Long J Chen H Ariga KAsakura YYuan RSC Adv 2014 4 29072
[64] G Prieto J Zecevic H Friedrich K P deJong P E de Jongh Nat Mater 2013 12 34
[65] Z Zhang J E Jackson D J Miller BioresourTechnol 2008 99 5873
[66] H Luo T Prasomsri Y Romaacuten-Leshkov TopCatal 2015 394
[67] J Mazumder H I de Lasa Catal Today 2014237 100
[68] H Xiong H N Pham A K Datye J Catal2013 302 93
[69] H Xiong M Nolan B H ShanksA K DatyeAppl Catal Gen 2014 471 165
[70] P A Zapata Y Huang M A Gonzalez-BorjaD E Resasco J Catal 2013 308 82
[71] A Corma Chem RevWash DC U S 1995 95559
[72] P A Jacobs M Dusselier B F Sels AngewChem Int Ed 2014 53 8621
[73] M Milina S Mitchell J Peacuterez-Ramiacuterez CatalToday 2014 235 176
[74] R M Ravenelle F Schuumlssler A DrsquoAmico NDanilina J A van Bokhoven J A Lercher CW Jones C Sievers J Phys Chem C 2010114 19582
[75] D W Gardner J Huo T C Hoff R LJohnson B H Shanks J-P Tessonnier ACSCatal 2015 DOI 101021acscatal5b00888
[76] P M Dove N Han J J D Yoreo Proc NatlAcad Sci 2005 102 15357
[77] T Ennaert J Geboers E Gobechiya C MCourtin M Kurttepeli K Houthoofd C E AKirschhock P C M M Magusin S Bals P AJacobs B F Sels ACS Catal 2015 5 754
[78] B Beverskog I Puigdomenech Corros Sci1997 39 969
[79] D A Palmer P Beacuteneacutezeth C Xiao D JWesolowski L M Anovitz J Solut Chem2011 40 680
[80] J H Vleeming B F M Kuster G B Marin FOudet P Courtine J Catal 1997 166 148
[81] A G Gayubo A T Aguayo M Olazar RVivanco J Bilbao Chem Eng Sci 2003 585239
[82] A de Lucas P CanizaresA DuraacutenA CarreroAppl Catal Gen 1997 154 221
[83] CAellig D Scholz PY Dapsens C MondelliJ Peacuterez-Ramiacuterez Catal Sci Technol 2014 5142
[84] C P Canlas J Lu N A Ray N A Grosso-Giordano S Lee J W Elam R E Winans RP Van Duyne P C Stair J M Notestein NatChem 2012 4 1030

590 CHIMIA 2015 69 No 10 SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy
of undercoordinated metal atoms Suchatoms can also favor byproduct formationdue to their high reactivity These byprod-ucts include coke in which case these re-active atoms contribute to poisoning theother active sites by initiating the forma-tion of carbon deposits For these reasonsseveral authors have reported methods topassivate these undercoordinated atomsusing overcoats deposited either by ALDsugar pyrolysis or CVD thereby stabiliz-ing the catalyst of interest Another issueis the leaching and structure modificationsof oxide supports due to the poor hydro-thermal stability of these solids whichinclude silica and zeolites In this caseseveral authors have turned to fine-tuningthe local environment of these catalystsagain by physically covering the materialof interest The addition of hydrophobicsurface functionalities or overcoats hasbeen shown to reduce support hydrolysisbywater molecules Similarly depositing apolymeric layer over metal hydrogenationsites was shown to act as an organic pseu-do-solvent which reduced the presence ofpolar biogenic catalyst poisons and thuscatalyst deactivation Finally increasedcatalyst lifetime was also improved byprocess engineering In fact tuning thereaction conditions including by loweringpH or co-feeding organic solvents and hy-drogen proved to be efficient for limitingcatalyst deactivation
Further research opportunities exist tocombine the principle strategies mentionedabove to further improve catalyst stabiliza-tion Tailored overcoats could be used bothto stabilize undercoordinated atoms and atthe same time to provide favorable micro-environments that could reduce hydrolysisandor concentrations of catalyst poisonsSuch an approach was used in part by add-ingMgO
xto reduce the acidity of an alumi-
na overcoat which was thought to catalyzecoke formation while the overcoat wasused to stabilize undercoordinated atomsHowever this approach could be furtherstrengthened by adding functionality to theovercoat and further tailoring themoleculardesign of the catalystrsquos local environmentFurthermore controlling pore size open-ings within these overcoats could intro-duce shape selectivity and be used to sieveout undesired reactants as was recentlydemonstrated for an alumina overcoat on atitania catalyst used for photocatalysis[84]Finally it will become increasingly impor-tant to use real biomass-derived streamsfor testing catalytic stability The negativeeffects of using liquid or aqueous condi-tions can be compounded by the presenceof numerous perhaps yet unidentified in-organic or biogenic impurities This wasnotably demonstrated by the acceleratedstructure collapse of zeolite structures inthe presence of inorganic salts
In summary current synthetic tech-niques can lead to unprecedented controlof the nanostructures surrounding a cata-lystrsquos active site but harsh conditions pres-ent for biomass conversion are equally un-precedented in industrial catalytic process-ing As society comes under increasingpressure to develop routes to sustainablecarbon-based molecules these syntheticmethods will likely prove essential in ad-dressing the challenges posed by catalyti-cally upgrading renewable resources suchas biomass
AcknowledgementsThe authors acknowledge financial support
from the Swiss National Science Foundationthrough grant number 407040_153866 as partof the National Research Program 70 and grantnumber PYAPP2_154281 and Benjamin LeMonnier for help with the design of figures
Received June 17 2015
[1] K Sugiura M Ishihara T Shimauchi SHarayama Environ Sci Technol 1997 31 45
[2] S Matar M J Mirbach HA Tayim lsquoCatalysisin Petrochemical Processesrsquo Springer Scienceamp Business Media 1989
[3] O S Abdul-Hamid Monthly Oil MarketReport - May 2015 OPEC 2015
[4] The European Parliament and the Council of theEuropean Union OJ 2009 L140 16
[5] R Rinaldi F Schuumlth Energy Environ Sci2009 2 610
[6] A A Peterson F Vogel R P Lachance MFroumlling J Michael JAntal JW Tester EnergyEnviron Sci 2008 1 32
[7] J S Luterbacher D M Alonso J A DumesicGreen Chem 2014 16 4816
[8] A Rahimi A Ulbrich J J Coon S S StahlNature 2014 515 249
[9] A Eyal R Jansen R Mali A Vitner lsquoSugarMixtures and Methods for Production and UseThereofrsquo 2012 WO 2011161685 A3
[10] D V Kazachkin M Colakyan F J MoeslerlsquoSupercritical Hydrolysis of Biomassrsquo 2015US 8999065 B2
[11] D L Sills J M Gossett Bioresour Technol2011 102 1389
[12] D M Alonso S G Wettstein M A MellmerE I Gurbuz J A Dumesic Energy EnvironSci 2012 6 76
[13] J S Luterbacher J M Rand D M Alonso JHan J T Youngquist C T Maravelias B FPfleger J A Dumesic Science 2014 343 277
[14] J S Luterbacher D M Alonso J M Rand YM Questell-Santiago J H Yeap B F PflegerJ A Dumesic ChemSusChem 2015 8 1317
[15] H Tadesse R Luque Energy Environ Sci2011 4 3913
[16] J B Binder R T Raines J Am Chem Soc2009 131 1979
[17] J S Luterbacher J W Tester L P WalkerBiotechnol Bioeng 2010 107 451
[18] D B Hodge M N Karim D J Schell J DMcMillan Bioresour Technol 2008 99 8940
[19] J S Luterbacher Q Chew Y Li J W TesterL P Walker Energy Environ Sci 2012 56990
[20] T J Schwartz B J OrsquoNeill B H Shanks J ADumesic ACS Catal 2014 4 2060
[21] G W Huber J N Chheda C J Barrett J ADumesic Science 2005 308 1446
[22] G W Huber R D Cortright J A DumesicAngew Chem Int Ed 2004 43 1549
[23] J N Chheda Y Romaacuten-Leshkov J ADumesic Green Chem 2007 9 342
[24] D W Rackemann W O Doherty BiofuelsBioprod Biorefining 2011 5 198
[25] J N Chheda G W Huber J A DumesicAngew Chem Int Ed 2007 46 7164
[26] A V Bandura S N Lvov J Phys Chem RefData 2006 35 15
[27] H Xiong H N Pham A K Datye GreenChem 2014 16 4627
[28] K P De Jong lsquoSynthesis Of Solid CatalystsrsquoWiley-VCHVerlag GmbH amp Co KGaA 2009
[29] F Heacuteroguel G Siddiqi M D Detwiler DY Zemlyanov O V Safonova C Copeacuteret JCatal 2015 321 81
[30] T W Hansen A T DeLaRiva S R Challa AK Datye Acc Chem Res 2013 46 1720
[31] B J OrsquoNeill C Sener D H K Jackson T FKuech J A Dumesic ChemSusChem 2014 73247
[32] B J OrsquoNeill J T Miller P J Dietrich FG Sollberger F H Ribeiro J A DumesicChemCatChem 2014 6 2493
[33] B J OrsquoNeill D H K Jackson A J Crisci CA Farberow F Shi A C Alba-Rubio J Lu PJ Dietrich X Gu C L Marshall et al AngewChem 2013 125 14053
[34] H Zhang Y Lei A J Kropf G Zhang J WElam J T Miller F Sollberger F Ribeiro MC Akatay E A Stach J A Dumesic C LMarshall J Catal 2014 317 284
[35] H ZhangY Lei A Jeremy Kropf G Zhang JW Elam J T Miller F Sollberger F RibeiroM Cem Akatay E A Stach J A Dumesic CL Marshall J Catal 2015 323 165
[36] J Lee D H K Jackson T Li R E Winans JA Dumesic T F Kuech G W Huber EnergyEnviron Sci 2014 7 1657
[37] T van Haasterecht C C I Ludding K P deJong J H Bitter J Catal 2014 319 27
[38] P Sun G Gao Z Zhao C Xia F Li ACSCatal 2014 4 4136
[39] J Lu B Fu M C Kung G Xiao J W ElamH H Kung P C Stair Science 2012 3351205
[40] T J Schwartz R L Johnson J Cardenas AOkerlund N A Da Silva K Schmidt-Rohr JA Dumesic Angew Chem Int Ed 2014 5312718
[41] H N Pham A E Anderson R L Johnson KSchmidt-Rohr A K Datye Angew Chem IntEd 2012 51 13163
[42] H Xiong T J Schwartz N I Andersen J ADumesic A K Datye Angew Chem Int Ed2015 early view DOI 101002anie201502206
[43] P A Zapata J Faria M P Ruiz R E JentoftD E Resasco J Am Chem Soc 2012 1348570
[44] H N Pham Y J Pagan-Torres J C Serrano-Ruiz D Wang J A Dumesic A K DatyeAppl Catal Gen 2011 397 153
[45] Y J Pagaacuten-Torres J M R Gallo D WangH N Pham J A Libera C L Marshall J WElam A K Datye J A Dumesic ACS Catal2011 1 1234
[46] A G Gayubo B Valle A T Aguayo MOlazar J Bilbao Energy Fuels 2009 23 4129
[47] S R Challa A T Delariva T W Hansen SHelveg J Sehested P L Hansen F Garzon AK Datye J Am Chem Soc 2011 133 20672
[48] J A Moulijn A E van Diepen F KapteijnAppl Catal Gen 2001 212 3
[49] S L Wegener T J Marks P C Stair AccChem Res 2012 45 206
[50] B J OrsquoNeill D H K Jackson J Lee CCanlas P C Stair C L Marshall J W ElamT F Kuech J A Dumesic G W Huber ACSCatal 2015 5 1804
[51] D M King J A Spencer II X Liang L FHakim A W Weimer Surf Coat Technol2007 201 9163
SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 591
[52] J A McCormick B L Cloutier AWWeimerS M George J Vac Sci Technol A 2007 2567
[53] D Longrie D Deduytsche C Detavernier JVac Sci Technol A 2014 32 010802
[54] J Lu J W Elam P C Stair Acc Chem Res2013 46 1806
[55] L De Rogatis M Cargnello V GombacB Lorenzut T Montini P FornasieroChemSusChem 2010 3 24
[56] A G Sergeev J D Webb J F Hartwig J AmChem Soc 2012 134 20226
[57] J He C Zhao J A Lercher J Am Chem Soc2012 134 20768
[58] Q Song F Wang J CaiY Wang J Zhang WYu J Xu Energy Environ Sci 2013 6 994
[59] R Dehghan-Niri J C Walmsley A HolmenP A Midgley E Rytter A H Dam A BHungria J C Hernandez-Garrido D ChenCatal Sci Technol 2012 2 2476
[60] M Morales P Y Dapsens I Giovinazzo JWitte C Mondelli S PapadokonstantakisK Hungerbuumlhler J Peacuterez-Ramiacuterez EnergyEnviron Sci 2015 8 558
[61] O A Abdelrahman H Y Luo A Heyden YRomaacuten-Leshkov J Q Bond J Catal 2015329 10
[62] D M Alonso S G Wettstein J A DumesicChem Soc Rev 2012 41 8075
[63] W Li L Ye P Long J Chen H Ariga KAsakura YYuan RSC Adv 2014 4 29072
[64] G Prieto J Zecevic H Friedrich K P deJong P E de Jongh Nat Mater 2013 12 34
[65] Z Zhang J E Jackson D J Miller BioresourTechnol 2008 99 5873
[66] H Luo T Prasomsri Y Romaacuten-Leshkov TopCatal 2015 394
[67] J Mazumder H I de Lasa Catal Today 2014237 100
[68] H Xiong H N Pham A K Datye J Catal2013 302 93
[69] H Xiong M Nolan B H ShanksA K DatyeAppl Catal Gen 2014 471 165
[70] P A Zapata Y Huang M A Gonzalez-BorjaD E Resasco J Catal 2013 308 82
[71] A Corma Chem RevWash DC U S 1995 95559
[72] P A Jacobs M Dusselier B F Sels AngewChem Int Ed 2014 53 8621
[73] M Milina S Mitchell J Peacuterez-Ramiacuterez CatalToday 2014 235 176
[74] R M Ravenelle F Schuumlssler A DrsquoAmico NDanilina J A van Bokhoven J A Lercher CW Jones C Sievers J Phys Chem C 2010114 19582
[75] D W Gardner J Huo T C Hoff R LJohnson B H Shanks J-P Tessonnier ACSCatal 2015 DOI 101021acscatal5b00888
[76] P M Dove N Han J J D Yoreo Proc NatlAcad Sci 2005 102 15357
[77] T Ennaert J Geboers E Gobechiya C MCourtin M Kurttepeli K Houthoofd C E AKirschhock P C M M Magusin S Bals P AJacobs B F Sels ACS Catal 2015 5 754
[78] B Beverskog I Puigdomenech Corros Sci1997 39 969
[79] D A Palmer P Beacuteneacutezeth C Xiao D JWesolowski L M Anovitz J Solut Chem2011 40 680
[80] J H Vleeming B F M Kuster G B Marin FOudet P Courtine J Catal 1997 166 148
[81] A G Gayubo A T Aguayo M Olazar RVivanco J Bilbao Chem Eng Sci 2003 585239
[82] A de Lucas P CanizaresA DuraacutenA CarreroAppl Catal Gen 1997 154 221
[83] CAellig D Scholz PY Dapsens C MondelliJ Peacuterez-Ramiacuterez Catal Sci Technol 2014 5142
[84] C P Canlas J Lu N A Ray N A Grosso-Giordano S Lee J W Elam R E Winans RP Van Duyne P C Stair J M Notestein NatChem 2012 4 1030

SCCER BIOSWEET ndash ThE SWISS COmpETEnCE CEnTER fOR EnERgy RESEaRCh On BIOEnERgy CHIMIA 2015 69 No 10 591
[52] J A McCormick B L Cloutier AWWeimerS M George J Vac Sci Technol A 2007 2567
[53] D Longrie D Deduytsche C Detavernier JVac Sci Technol A 2014 32 010802
[54] J Lu J W Elam P C Stair Acc Chem Res2013 46 1806
[55] L De Rogatis M Cargnello V GombacB Lorenzut T Montini P FornasieroChemSusChem 2010 3 24
[56] A G Sergeev J D Webb J F Hartwig J AmChem Soc 2012 134 20226
[57] J He C Zhao J A Lercher J Am Chem Soc2012 134 20768
[58] Q Song F Wang J CaiY Wang J Zhang WYu J Xu Energy Environ Sci 2013 6 994
[59] R Dehghan-Niri J C Walmsley A HolmenP A Midgley E Rytter A H Dam A BHungria J C Hernandez-Garrido D ChenCatal Sci Technol 2012 2 2476
[60] M Morales P Y Dapsens I Giovinazzo JWitte C Mondelli S PapadokonstantakisK Hungerbuumlhler J Peacuterez-Ramiacuterez EnergyEnviron Sci 2015 8 558
[61] O A Abdelrahman H Y Luo A Heyden YRomaacuten-Leshkov J Q Bond J Catal 2015329 10
[62] D M Alonso S G Wettstein J A DumesicChem Soc Rev 2012 41 8075
[63] W Li L Ye P Long J Chen H Ariga KAsakura YYuan RSC Adv 2014 4 29072
[64] G Prieto J Zecevic H Friedrich K P deJong P E de Jongh Nat Mater 2013 12 34
[65] Z Zhang J E Jackson D J Miller BioresourTechnol 2008 99 5873
[66] H Luo T Prasomsri Y Romaacuten-Leshkov TopCatal 2015 394
[67] J Mazumder H I de Lasa Catal Today 2014237 100
[68] H Xiong H N Pham A K Datye J Catal2013 302 93
[69] H Xiong M Nolan B H ShanksA K DatyeAppl Catal Gen 2014 471 165
[70] P A Zapata Y Huang M A Gonzalez-BorjaD E Resasco J Catal 2013 308 82
[71] A Corma Chem RevWash DC U S 1995 95559
[72] P A Jacobs M Dusselier B F Sels AngewChem Int Ed 2014 53 8621
[73] M Milina S Mitchell J Peacuterez-Ramiacuterez CatalToday 2014 235 176
[74] R M Ravenelle F Schuumlssler A DrsquoAmico NDanilina J A van Bokhoven J A Lercher CW Jones C Sievers J Phys Chem C 2010114 19582
[75] D W Gardner J Huo T C Hoff R LJohnson B H Shanks J-P Tessonnier ACSCatal 2015 DOI 101021acscatal5b00888
[76] P M Dove N Han J J D Yoreo Proc NatlAcad Sci 2005 102 15357
[77] T Ennaert J Geboers E Gobechiya C MCourtin M Kurttepeli K Houthoofd C E AKirschhock P C M M Magusin S Bals P AJacobs B F Sels ACS Catal 2015 5 754
[78] B Beverskog I Puigdomenech Corros Sci1997 39 969
[79] D A Palmer P Beacuteneacutezeth C Xiao D JWesolowski L M Anovitz J Solut Chem2011 40 680
[80] J H Vleeming B F M Kuster G B Marin FOudet P Courtine J Catal 1997 166 148
[81] A G Gayubo A T Aguayo M Olazar RVivanco J Bilbao Chem Eng Sci 2003 585239
[82] A de Lucas P CanizaresA DuraacutenA CarreroAppl Catal Gen 1997 154 221
[83] CAellig D Scholz PY Dapsens C MondelliJ Peacuterez-Ramiacuterez Catal Sci Technol 2014 5142
[84] C P Canlas J Lu N A Ray N A Grosso-Giordano S Lee J W Elam R E Winans RP Van Duyne P C Stair J M Notestein NatChem 2012 4 1030