identification of calcium binding sites on calsequestrin 1 and their implications for polymerization
TRANSCRIPT

This journal is c The Royal Society of Chemistry 2013 Mol. BioSyst.
Cite this: DOI: 10.1039/c3mb25588c
Identification of calcium binding sites on calsequestrin 1and their implications for polymerization†
Amit Kumar,za Harapriya Chakravarty,zb Naresh C. Bal,z*c Tuniki Balaraju,b
Nivedita Jena,d Gauri Misra,e Chandralata Bal,b Enrico Pieroni,a Muthu Periasamycf
and Ashoke Sharon*b
Biophysical studies have shown that each molecule of calsequestrin 1 (CASQ1) can bind about
70–80 Ca2+ ions. However, the nature of Ca2+-binding sites has not yet been fully characterized. In this
study, we employed in silico approaches to identify the Ca2+ binding sites and to understand the
molecular basis of CASQ1–Ca2+ recognition. We built the protein model by extracting the atomic
coordinates for the back-to-back dimeric unit from the recently solved hexameric CASQ1 structure
(PDB id: 3UOM) and adding the missing C-terminal residues (aa350–364). Using this model we
performed extensive 30 ns molecular dynamics simulations over a wide range of Ca2+ concentrations
([Ca2+]). Our results show that the Ca2+-binding sites on CASQ1 differ both in affinity and geometry. The
high affinity Ca2+-binding sites share a similar geometry and interestingly, the majority of them were
found to be induced by increased [Ca2+]. We also found that the system shows maximal Ca2+-binding to
the CAS (consecutive aspartate stretch at the C-terminus) before the rest of the CASQ1 surface becomes
saturated. Simulated data show that the CASQ1 back-to-back stacking is progressively stabilized by the
emergence of an increasing number of hydrophobic interactions with increasing [Ca2+]. Further, this
study shows that the CAS domain assumes a compact structure with an increase in Ca2+ binding, which
suggests that the CAS domain might function as a Ca2+-sensor that may be a novel structural motif to
sense metal. We propose the term ‘‘Dn-motif’’ for the CAS domain.
Introduction
CASQ1 is the major Ca2+ buffering protein present in thesarcoendoplasmic reticulum (SR) of the striated muscles1–5
where it also regulates Ca2+ release by its interaction with theRyanodine receptor (RyR) complex.1 Crystallographic studiesshowed that the CASQ1 monomer consists of three a–b globular
domains formed by five b-strands arranged in a sheet sand-wiched by four a-helices, which is structurally similar toubiquitous redox protein ‘‘thioredoxin’’.6,7 Beyond the struc-tural domain III, CASQ1 possesses a unique C-terminus that iscomposed of repeating aspartic acid residues. The length ofthis consecutive aspartate stretch (CAS) varies between speciesand typically among mammals it is of 9–14 residues. Biophysico-chemical studies suggest that CASQ1 binds a large amount ofCa2+ ions (B70–80 Ca2+ per molecule) with low affinity.3,8–11
However, CASQ1 lacks any known Ca2+ binding motif likeEF-hand6,12–14 or double-clamp15 and the nature of Ca2+-bindingsites is not fully characterized. It has been proposed that Ca2+
binding to CASQ1 is purely a surface phenomenon governed bythe highly negatively charged molecular surface.7,11
Based on biochemical and structural observations it hasbeen proposed that CASQ1 forms a polymeric structure in aCa2+ concentration ([Ca2+]) dependent manner.6,7,9 At first, twomonomers interact to form a very stable front-to-front dimer byinsertion of 14 N-terminal residues into the partner monomer.7,11,16
Then, two such front-to-front dimers stack by back-to-backinteractions leading to formation of a tetramer and subsequently
a CRS4, Bioengineering group, Science and Technology Park Polaris, Piscina Manna,
09010 Pula, CA, Italyb Department of Applied Chemistry, Birla Institute of Technology, Mesra, Ranchi,
Jharkhand 835215, India. E-mail: [email protected];
Fax: +91-651-2275401; Tel: +91-651-2276531c Department of Physiology and Cell Biology, The Ohio State University,
College of Medicine, Columbus, OH 43210, USA. E-mail: [email protected];
Fax: +1 614-292-4888; Tel: +1 614-688-4635d College of Pharmacy, The Ohio State University, Columbus, OH 43210, USAe Institute of Biotechnology, Amity University, Noida, Indiaf Davis Heart and Lung Research Institute, Ohio State University, Columbus, Ohio,
USA
† Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mb25588c‡ These authors contributed equally.
Received 19th December 2012,Accepted 9th April 2013
DOI: 10.1039/c3mb25588c
www.rsc.org/molecularbiosystems
MolecularBioSystems
PAPER
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 01
/05/
2013
07:
32:5
9.
Publ
ishe
d on
10
Apr
il 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3M
B25
588C
View Article OnlineView Journal

Mol. BioSyst. This journal is c The Royal Society of Chemistry 2013
resulting in the formation of a linear polymer in a Ca2+-dependent manner. However, detailed structural informationabout the interactions at the back-to-back interface in theCASQ1 polymer has been speculative and it is currently unclearif the back-to-back stacking is dependent on [Ca2+]. Further, theCAS residues are located at the back-to-back interface and cansignificantly influence the stacking interactions.17 It is alsounclear if the CAS domain plays a role in facilitating back-to-back stacking and Ca2+-binding, given that the structure of theCAS residues has not been resolved in any of the currentlyavailable CASQ1 structures.
The recently solved CASQ1 structure (PDB Id: 3UOM12) inthe hexameric state provides the opportunity to apply computa-tional approaches to define the physico-chemical propertiesof the Ca2+-binding sites and the role of the CAS domain in[Ca2+]-dependent protein–protein interaction. The hexamerconsists of three repeating dimeric units formed by front-to-front dimerization, which on the other hand are held byback-to-back interaction. In the light of this scenario, the goalsof this study are to (1) identify and characterize the Ca2+-binding sites on CASQ1; (2) better understand how the surfaceof CASQ1 reacts to Ca2+-binding and (3) gain insight into thefunctional role of the CAS domain in CASQ1–Ca2+ interaction.To accomplish these goals, we built a model of the back-to-backdimeric unit from the hexameric CASQ1 structure, addedthe missing CAS residues (aa350–364) and performed exten-sive molecular dynamics (MD) simulations over a wide rangeof [Ca2+].
Results and discussions[Ca2+]-dependent charge distribution on the CASQ1 surface
Before starting a detailed structural investigation, we firstanalysed the stability of the CASQ1 dimeric unit during MDsimulation by calculating the backbone RMSD and entropy ofthe system. The stability of the dimeric system increases athigher [Ca2+] as reflected in the decrease of both RMSD and
entropy values (ESI,† Table S1). Then, we determined the sur-face charge of CASQ1 after MD simulation in all the systemsexposed to different [Ca2+]. As shown in Fig. 1, the surface ofCASQ1 in a Ca2+ free system is highly acidic, as also previouslyreported.11,18 Then, as expected, a gradual surface neutraliza-tion, mediated by Ca2+ binding, was observed with increasing[Ca2+]. However, up to the presence of 40 Ca2+ ions, the surfacewas significantly negative and only some regions were found tobe neutralized by Ca2+-binding. The CASQ1 surface for thesystem having 60 Ca2+ ions seems to be further neutralizedand becomes fully neutral in the presence of 80 Ca2+ ions.Interestingly, the electrostatic surface charge calculation showsa highly electropositive surface for the CASQ1 in the systemhaving 120 Ca2+ ions.
Differential Ca2+-binding to CAS and the rest of CASQ1
Next, we calculated the number of Ca2+ ions that interact withthe CASQ1 protein after the MD simulation. Interestingly, wefound that the CAS residues get saturated with Ca2+ before therest of the protein surface. As shown in Fig. 2A, the CASresidues (350–364) of chains B and C bind to an increasingnumber of Ca2+ ions as [Ca2+] increases. However, in both thechains, CAS gets saturated when [Ca2+] reached 80 ions in thesystem, and after this threshold no further increase inthe number of bound Ca2+ was observed. This indicates thatthe maximum Ca2+ binding capacity of the CAS of chains B andC is B6 and B8 Ca2+ ions, respectively, and the CAS cannotaccount for more than 10% of the Ca2+ binding of each CASQ1monomer (binds B70–80 Ca2+). On the other hand, the surfaceof the rest of the protein (residues 1–350) binds progressively amore number of Ca2+ ions as the [Ca2+] was increased. From theslope of the Ca2+-binding curve (Fig. 2B), it appears that theCASQ1 surface has not reached saturation even in the systemwith 120 Ca2+ ions, where each chain has bound B55 Ca2+ ions.These findings, and the observation that CAS may hinder back-to-back stacking due to charge–charge repulsion, indicate thatthe CAS domain undergoes complete charge neutralization
Fig. 1 Changes in the surface charge pattern of CASQ1 with increasing Ca2+ concentrations. Surface charge was calculated for the average structure for each systemindicated in the figure. Red, blue and white indicate negative, positive and neutral charge on the molecular surface. The color scale used to represent the Poisson–Boltzmann electrostatic potential isosurface range is from �4 to +4 KT/e (where K – Boltzmann constant, T – temperature, and e – electron charge). CASQ1 gainselectro-positivity on the surface in the 120 Ca2+-system, even if the surface negative charge is almost neutralized already in the 80 Ca2+-system.
Paper Molecular BioSystems
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 01
/05/
2013
07:
32:5
9.
Publ
ishe
d on
10
Apr
il 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3M
B25
588C
View Article Online

This journal is c The Royal Society of Chemistry 2013 Mol. BioSyst.
before the rest of the CASQ1 surface gets saturated with Ca2+.Furthermore, these observations suggest that the affinity ofdifferent Ca2+-binding sites on CASQ1 must be different, insuch a way to allow systematic neutralization of the electro-negative surface with increasing [Ca2+]. The negative potentialneutralization process would then lead to the formationof globular CASQ1 conformation that is ready to undergopolymerization.
[Ca2+] independent high affinity Ca2+-binding sites
The recently solved crystal structure (PDB Id: 3UOM) providedinformation about only 15 Ca2+-binding sites,12 while oursimulated data show that each monomer (chain) binds about55 Ca2+ in the system with 120 Ca2+ (Fig. 2), indicating theexistence of more Ca2+-binding sites. Therefore, we performeddetailed investigation of the CASQ1 structures in the systemsexposed to different [Ca2+], to identify and characterize otherCa2+ binding sites, in particular those not revealed by thecrystallographic structure. Interestingly, the Ca2+-binding sitesidentified by the crystallographic study were also confirmed byMD simulation. The Ca2+-binding strength was analysed fora set of Ca2+-binding sites by determining the non-bonded(van der Waals + electrostatic) energy of interaction betweenthe residues of the site and the Ca2+ ion. Based on the Ca2+-binding strength (summarized in Table 1) we further refined
the classification of both high and low affinity sites intoindependent or dependent of [Ca2+]. The high affinity sites(sites 1, 2, 3 and 4) are strictly independent of [Ca2+] and bindCa2+ with high affinity as indicated by the high interactionenergy (o�400 kcal mol�1) in all the protein systems. Wefurther found that the [Ca2+]-independent high affinity sitesbind Ca2+ in a very favorable geometry in most of the systems atdifferent [Ca2+] (Fig. 3). However, only 13 [Ca2+]-independenthigh affinity sites on CASQ1 were found.
Existence of [Ca2+]-induced Ca2+-binding sites
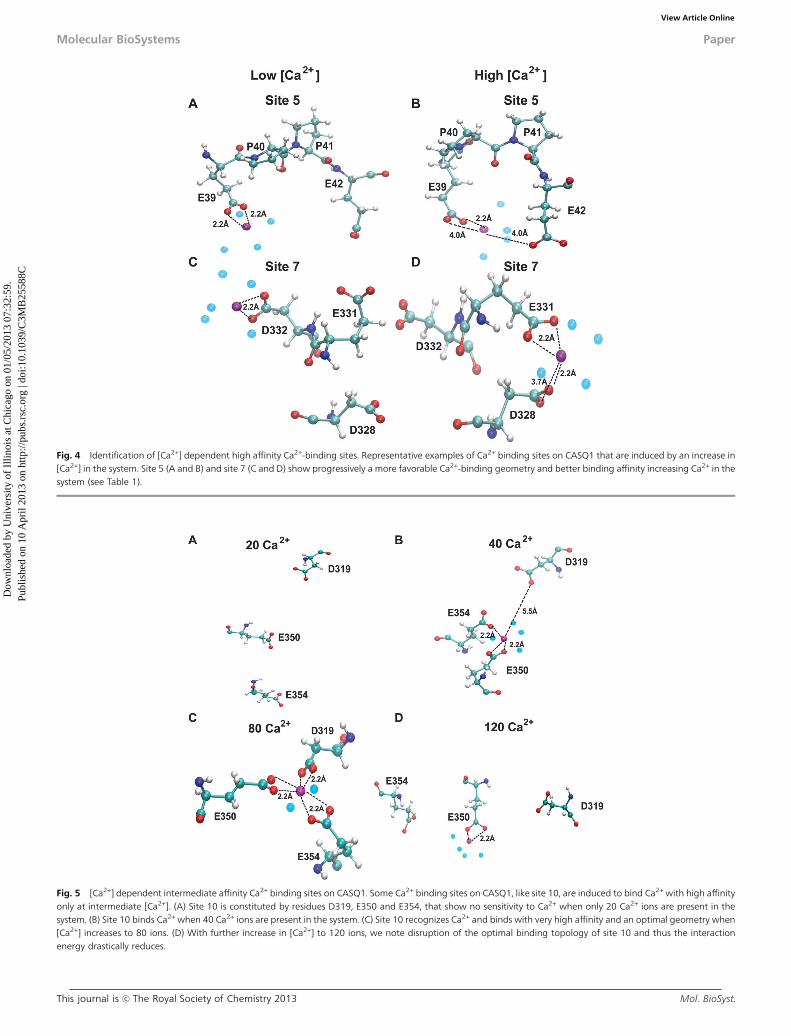
One of the most interesting findings of this study is that someCa2+-binding sites are solely dependent on [Ca2+]. We foundthat some sites, for instance sites 5, 6 and 7, bind Ca2+ withincreasingly high affinity, reaching the highest value (corre-sponding to energy of o�400 kcal mol�1) in the system with120 Ca2+ ions (Table 1). These sites are referred to as ‘‘[Ca2+]-dependent high affinity sites’’ and do not bind Ca2+ when thesystem has only 20 Ca2+ ions. Their interaction energy graduallyincreases as [Ca2+] increases, reaching a maximum value(o�400 kcal mol�1) for the system containing 120 Ca2+ ions.Moreover, sites 5, 6 and 7 show a more favorable geometry ofCa2+-binding when [Ca2+] increases, demonstrating that thesesites are induced upon Ca2+-binding (Fig. 4). Our data suggestthat the [Ca2+]-independent high affinity sites get occupied first
Fig. 2 Number of bound Ca2+ in the presence of increasing [Ca2+]. (A) Ca2+ bound to the Dn-motif (from amino acid 350 to 364 on the C-terminus) of each monomer.The Dn-motif at the dimeric interface binds less Ca2+ than the free Dn-motif. (B) Total number of Ca2+ ions bound to the rest of the protein (amino acids 1–350) surfaceof each monomer. Both the monomers bind almost a similar number of Ca2+ ions although at intermediate [Ca2+] the monomer ‘‘chain B’’ binds more Ca2+ ions than‘‘chain C’’. Observing the slope of the curves it seems that both the monomers still possess the ability to bind even a higher number of Ca2+ ions, if provided.
Table 1 Interaction energy of representative sites for the Ca2+ ion
Numberof Ca2+ inthe system
Concentration independenthigh affinity sites (kcal mol�1)
Concentration dependenthigh affinity sites (kcal mol�1)
Concentration dependentintermediate affinity sites(kcal mol�1)
Concentrationindependentlow affinity sites(kcal mol�1)
1(E39,E54 andD93)
2(E102and E169)
3(E194and D196)
4(D356and D357)
5(E39,P40 andE42)
6(D259and D261)
7(D328,E331 andD332)
8(D306and S308)
9(E326and E158)
10(D319,E350 andE354) 11(E66)
12(E199and T229)
120 �495 �458 �371 �506 �481 �389 �905 — �279 �313 — �25680 �560 �533 �386 �590 �355 �291 �785 �221 �274 �818 �198 —40 �471 �483 �375 �370 �158 �214 �312 �230 �545 �599 — �20720 �479 �551 �393 �530 — — — — �480 — �195 �276
Molecular BioSystems Paper
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 01
/05/
2013
07:
32:5
9.
Publ
ishe
d on
10
Apr
il 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3M
B25
588C
View Article Online

Mol. BioSyst. This journal is c The Royal Society of Chemistry 2013
and then the binding events initiate structural alterations, bringingthe residues involved in the [Ca2+]-dependent high affinity sites in afavorable geometry for subsequent Ca2+-binding.
Interestingly, some inducible sites (sites 8, 9 and 10) wereobserved to bind Ca2+ with high affinity only at intermediate[Ca2+] (Table 1), thus we refer to them as ‘‘[Ca2+]-dependentintermediate affinity sites’’. The geometry of site 10 in systemswith different [Ca2+] is shown in Fig. 5. As can be seen in Fig. 5,the residues at site 10 are not in a proper orientation to supportCa2+ recognition in the system having 20 Ca2+ ions. In thesystem having 80 Ca2+ ions, these residues gradually reorient toa correct geometry for ion Ca2+-binding, with all three acidicresidues involved in bidentate interaction with the Ca2+ ion.However, further increase in [Ca2+] in the system disrupted theoptimal geometry of the site 10, thus leading to a sharp decline ofthe interaction energy (Fig. 5 and Table 1). Distance probabilitydistributions also showed that in the system having 80 Ca2+ ionsthe distance between the residues and Ca2+ is the least value(ESI,† Fig. S1). As a whole, we have found 25 inducible Ca2+-binding sites in CASQ1. Additionally, we also found that CASQ1has many low affinity sites (for instance sites 11 and 12) that areformed by either an acidic residue (Asp or Glu) alone or incombination with any other supporting residue (ESI,† Fig. S1D).These sites bind Ca2+ with low affinity, with an interaction energyhigher than�250 kcal mol�1 independent of [Ca2+] in the system.
[Ca2+]-mediated dimer stabilization by hydrophobicinteractions
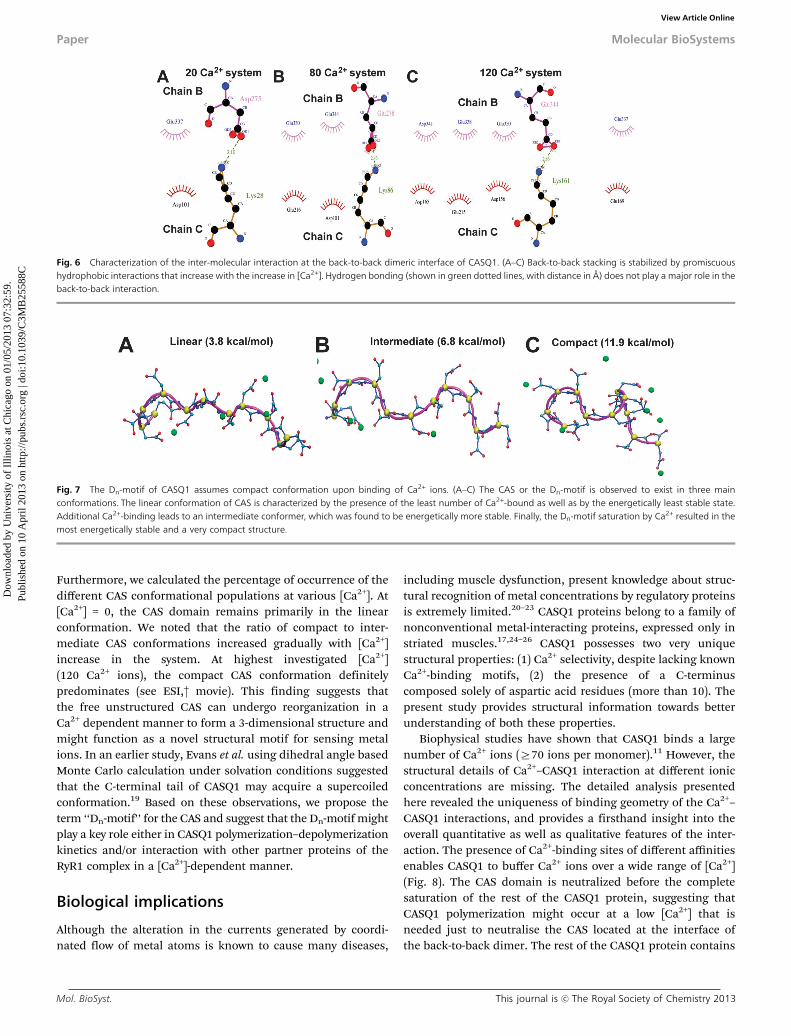
We investigated the structure of the back-to-back dimeric inter-face in all the protein systems. Importantly, we found only onehydrogen bond at the interface, while interestingly the number
of hydrophobic interactions increased with the rise in [Ca2+](Fig. 6). Then, upon increasing the number of Ca2+ ions, weobserved a gradual increase of hydrophobic interactions, and amaximum of four hydrophobic interactions were found in thesystem with 120 Ca2+ ions. However, we did not observe anycommitted residue-to-residue contact, rather a set of interfacialresidues that promiscuously established hydrophobic contacts as[Ca2+] increased. Nevertheless, all the hydrophobic interactionswere found between acidic (glutamic and aspartic) residues. Thissuggests that the Ca2+-binding mediated charge neutralization ofthese interfacial acidic residues provides them the ability toestablish hydrophobic interactions. We also observed that thenumber of bound water molecules in the interfacial region and tothe CAS of chain B is unchanged in all the protein systemsindicating that water plays only a minor role in back-to-backstacking. On the other hand, the free CAS of chain C binds morewater with increasing [Ca2+] in the system (ESI,† Fig. S2).
Ca2+-induced CAS folding
Next, we investigated the alteration in the structure of CASupon Ca2+-binding throughout the MD trajectory in all thesystems. One of the most exciting findings of this study is thatthe CAS acquires a compact 3-dimensional structure upon Ca2+-binding. Analysis of the MD simulation trajectory suggests thatthe CAS domain can exist in three distinct conformations:linear, intermediate and compact, as shown in Fig. 7. Thelinear conformation of CAS forms 2–3 hydrogen bonds, theintermediate conformation 4–5 hydrogen bonds, and the com-pact conformation is stabilized by 6–8 hydrogen bonds. Then,we calculated the energy of the different CAS conformationalpopulations, which is pertinent to the number of hydrogen bonds.
Fig. 3 Identification of high affinity Ca2+-binding sites independent of [Ca2+]. Sites 1 and 3 are representative examples of high affinity Ca2+ binding sites on CASQ1that are independent of [Ca2+]. Site 1 (A and B) and site 3 (C and D) bind Ca2+ with very similar geometry and affinity (see Table 1). System with 20 Ca2+ ions is referredto as ‘‘Low [Ca2+]’’, whereas System with 80 Ca2+ ions is referred to as ‘‘High [Ca2+]’’. The protein residues are represented in ball–stick representation, Ca2+ ions inmagenta and water molecules that are present within 3.0 Å of Ca2+ ions in light blue.
Paper Molecular BioSystems
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 01
/05/
2013
07:
32:5
9.
Publ
ishe
d on
10
Apr
il 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3M
B25
588C
View Article Online
This journal is c The Royal Society of Chemistry 2013 Mol. BioSyst.
Fig. 4 Identification of [Ca2+] dependent high affinity Ca2+-binding sites. Representative examples of Ca2+ binding sites on CASQ1 that are induced by an increase in[Ca2+] in the system. Site 5 (A and B) and site 7 (C and D) show progressively a more favorable Ca2+-binding geometry and better binding affinity increasing Ca2+ in thesystem (see Table 1).
Fig. 5 [Ca2+] dependent intermediate affinity Ca2+ binding sites on CASQ1. Some Ca2+ binding sites on CASQ1, like site 10, are induced to bind Ca2+ with high affinityonly at intermediate [Ca2+]. (A) Site 10 is constituted by residues D319, E350 and E354, that show no sensitivity to Ca2+ when only 20 Ca2+ ions are present in thesystem. (B) Site 10 binds Ca2+ when 40 Ca2+ ions are present in the system. (C) Site 10 recognizes Ca2+ and binds with very high affinity and an optimal geometry when[Ca2+] increases to 80 ions. (D) With further increase in [Ca2+] to 120 ions, we note disruption of the optimal binding topology of site 10 and thus the interactionenergy drastically reduces.
Molecular BioSystems Paper
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 01
/05/
2013
07:
32:5
9.
Publ
ishe
d on
10
Apr
il 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3M
B25
588C
View Article Online
Mol. BioSyst. This journal is c The Royal Society of Chemistry 2013
Furthermore, we calculated the percentage of occurrence of thedifferent CAS conformational populations at various [Ca2+]. At[Ca2+] = 0, the CAS domain remains primarily in the linearconformation. We noted that the ratio of compact to inter-mediate CAS conformations increased gradually with [Ca2+]increase in the system. At highest investigated [Ca2+](120 Ca2+ ions), the compact CAS conformation definitelypredominates (see ESI,† movie). This finding suggests thatthe free unstructured CAS can undergo reorganization in aCa2+ dependent manner to form a 3-dimensional structure andmight function as a novel structural motif for sensing metalions. In an earlier study, Evans et al. using dihedral angle basedMonte Carlo calculation under solvation conditions suggestedthat the C-terminal tail of CASQ1 may acquire a supercoiledconformation.19 Based on these observations, we propose theterm ‘‘Dn-motif’’ for the CAS and suggest that the Dn-motif mightplay a key role either in CASQ1 polymerization–depolymerizationkinetics and/or interaction with other partner proteins of theRyR1 complex in a [Ca2+]-dependent manner.
Biological implications
Although the alteration in the currents generated by coordi-nated flow of metal atoms is known to cause many diseases,
including muscle dysfunction, present knowledge about struc-tural recognition of metal concentrations by regulatory proteinsis extremely limited.20–23 CASQ1 proteins belong to a family ofnonconventional metal-interacting proteins, expressed only instriated muscles.17,24–26 CASQ1 possesses two very uniquestructural properties: (1) Ca2+ selectivity, despite lacking knownCa2+-binding motifs, (2) the presence of a C-terminuscomposed solely of aspartic acid residues (more than 10). Thepresent study provides structural information towards betterunderstanding of both these properties.
Biophysical studies have shown that CASQ1 binds a largenumber of Ca2+ ions (Z70 ions per monomer).11 However, thestructural details of Ca2+–CASQ1 interaction at different ionicconcentrations are missing. The detailed analysis presentedhere revealed the uniqueness of binding geometry of the Ca2+–CASQ1 interactions, and provides a firsthand insight into theoverall quantitative as well as qualitative features of the inter-action. The presence of Ca2+-binding sites of different affinitiesenables CASQ1 to buffer Ca2+ ions over a wide range of [Ca2+](Fig. 8). The CAS domain is neutralized before the completesaturation of the rest of the CASQ1 protein, suggesting thatCASQ1 polymerization might occur at a low [Ca2+] that isneeded just to neutralise the CAS located at the interface ofthe back-to-back dimer. The rest of the CASQ1 protein contains
Fig. 6 Characterization of the inter-molecular interaction at the back-to-back dimeric interface of CASQ1. (A–C) Back-to-back stacking is stabilized by promiscuoushydrophobic interactions that increase with the increase in [Ca2+]. Hydrogen bonding (shown in green dotted lines, with distance in Å) does not play a major role in theback-to-back interaction.
Fig. 7 The Dn-motif of CASQ1 assumes compact conformation upon binding of Ca2+ ions. (A–C) The CAS or the Dn-motif is observed to exist in three mainconformations. The linear conformation of CAS is characterized by the presence of the least number of Ca2+-bound as well as by the energetically least stable state.Additional Ca2+-binding leads to an intermediate conformer, which was found to be energetically more stable. Finally, the Dn-motif saturation by Ca2+ resulted in themost energetically stable and a very compact structure.
Paper Molecular BioSystems
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 01
/05/
2013
07:
32:5
9.
Publ
ishe
d on
10
Apr
il 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3M
B25
588C
View Article Online

This journal is c The Royal Society of Chemistry 2013 Mol. BioSyst.
many Ca2+-binding sites of different affinities, suggesting thatthe CASQ1 polymer can sequester and deliver Ca2+ ions withoutthe need to depolymerize, in the normal physiological range of[Ca2+] oscillation in the skeletal muscles. Further, our datashowed that promiscuous interfacial hydrophobic interactionsstabilize the back-to-back dimer in a [Ca2+] dependent manner,which suggests that the back-to-back stacking is not extremelystable. Such a labile, but energetically stable back-to-back stackingcould support the dynamic depolymerization of CASQ1 if requiredduring extreme Ca2+ demand (Fig. 8). Dynamic depolymerizationof the CASQ2 isoform has been suggested recently,27,28 and suchan interpretation is also experimentally supported by the recentdynamic measurement of the calcium buffering properties of thesarcoplasmic reticulum in the mouse skeletal muscle by Mannoet al.29 Our current study shows that MD simulations of aqueoussolution of metals (at various concentrations) can be successfullyapplied to gain a structural and functional insight into protein–metal interaction of metalloproteins.
We would like to emphasize that the presence of more than10 consecutive aspartic acid residues (Dn-motif) in a proteinsequence is a very rare feature that is found only in about20 proteins in the human genome, out of more than 30 000predicted proteins. An evident common property shared amongthese proteins is metal binding capability, which indicates thatthe Dn-motif might be important as a metal concentrationsensor or for the metal sensing and binding, and to the bestof our knowledge, has not been investigated so far. Forinstance, these 20 proteins include; zinc finger protein castor
homolog 1 (genebank accession id: NP_001073312), histidine-rich calcium-binding protein (AAI12356), ring finger protein 34(EAW98263), E3 ubiquitin-protein ligase RNF34 isoform 3(NP_001243787), and asporin (CAI16698). Searches in the PDBindicate that the structure of the Dn-motif has also not beendetermined through experimental methods for any protein andthe functional role of the Dn-motif in metalloproteins iscurrently unknown. Our results showed that the Dn-motif ofCASQ1 undergoes folding at increasing [Ca2+], which suggeststhat this motif might have a metal-sensing function.
Materials and methodCASQ1 model building with the CAS motif
The recent crystal structure of the hexameric model of skeletalcalsequestrin (PDB Id: 3UOM30) was adopted as a base modelfor our MD studies. The protein preparation wizard of theSchrodinger suite was used to optimize the model structurefor its chemical correctness.31 In general, before simulations,the crystal structures require fixing the common issues relatedto the experimentally adopted methodology, that is, missinghydrogen atoms, missing side chains, bond order assignments,charge states, and conformational orientations of symmetricalgroups. During the structure refinement the orientation ofhydroxyl (or thiol) groups, the terminal amide groups inasparagine (Asn) and glutamine (Gln), and the ring of histidine(His), those not already determined in the X-ray structure, werecorrected. Flipping the terminal amide groups and the histidine
Fig. 8 Proposed model for Ca2+–CASQ1 interaction and polymerization. The centre of each domain of the CASQ1 molecule is hydrophobic and must undergohydrophobic collapse to form the domains. The core region of CASQ1 at the interface, between the three domains is acidic, which is a characteristic feature of theCASQ-protein family. The inter-domain charge neutralization by Ca2+ is necessary for the three domains to come to close proximity to form a compact monomer. TheDn-motif of CASQ1 is composed of 13 aspartic acids that cannot bind more than 8 Ca2+ ions, whereas the CASQ1 monomer has capacity to bind 70–80 Ca2+ ions.Therefore, the CASQ1 structural domains can bind more than 50 Ca2+. Existence of sites that can switch to high affinity sites at varying [Ca2+] allows CASQ1 to releaseCa2+ without the necessity of undergoing depolymerization. This observation holds in most part of the ‘‘physiological range of Ca2+ variations’’. Dotted lines indicatethe extreme Ca2+ concentrations that might be important only during higher physiological calcium demand.
Molecular BioSystems Paper
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 01
/05/
2013
07:
32:5
9.
Publ
ishe
d on
10
Apr
il 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3M
B25
588C
View Article Online

Mol. BioSyst. This journal is c The Royal Society of Chemistry 2013
ring can improve charge–charge interactions with neighboringgroups as well as hydrogen bonding. The 1801 flips preserve theheavy-atom placement deduced from the X-ray electron density.In addition, the protonation state of histidine is varied tooptimize hydrogen bonding and charge interactions. The opti-mization was done using exhaustive sampling, which allowedincreasing the number of iterations to an adequate level. Theprotein was further minimized using the protein preparationwizard using the OPLS2005 force field with the maximumconvergence of 0.3 Å of heavy atoms. As shown in ESI†(Fig. S3A), the CASQ1 monomers (labeled A, B, C, D, E and F)are arranged in a repetitive manner. AB, CD and EF are thefront-to-front dimers and back-to-back interactions occurbetween B and C chains. In this study we are interested inunderstanding the dynamics of back-to-back stacking andC-terminus-to-Ca2+ interaction, thus we chose to considerchains B and C only. The missing C-terminal residues wereadded using Prime and Builder modules of Schrodinger tothe chains B and C, without disturbing the structure of theremaining regions of the chains.32 After C-terminus addition,the residues were corrected by an extended serial loop samplingmethod, using the loop refinement module of Prime (ESI,†Fig. S3B).33 The dimeric structure with the CAS region wassubjected to global minimization through the Macromodel34
module of the Schrodinger suite using the OPLS 2005 forcefield with a GB/SA continuum water solvation model. Thiswas followed by a Polak–Ribiere Conjugate Gradient energyminimization stopped after 5000 steps or when the energydifference between two subsequent structures was inferior to0.05 kJ mol�1. Finally, a varying number of Ca2+ ions wereadded randomly to the system to build up the final CASQ1–Ca2+
complexes using the System Building Module of the Desmondsuite to perform the following MD simulations.35
Molecular dynamics simulations
The parameters for the protein and ions were assigned usingCHARMM27 force-field (C27-FF) parameters.36 The protein–protein complexes with different concentrations of Ca2+ ionswere alternately inserted in the water box, and additionalcounter-ions were added to neutralize the system, using themodules in VMD software.37 We used the TIP3P parameters forwater molecules.38 Standard protonation states were assignedto all residues using the propKa software.39 Each of the result-ing molecular system was energy minimized and slowlyheated to 300 K in steps of 30 K, with positional restraints of50 kcal mol�1 Å�2 on C-alpha atoms for a simulation time of0.2 ns. The positional restraints on the C-alpha atoms werethen slowly released in steps of 10 kcal mol�1 Å�2 and after0.3 ns of simulation all positional restraints on the C-alphaatoms were completely released. Then, equilibration of themolecular system was done for a simulation time of 3 ns.Subsequently, all production runs of 30 ns simulations wereperformed at 300 K and 1 atm pressure (NPT ensemble), usingperiodic boundary conditions. The initial dimensions of thesimulation box edges were [138, 110, 95] Å, for a total system ofB130 000 atoms. All bonds involving hydrogen atoms were
constrained using SHAKE,40 which allowed using an integra-tion time step of 2 fs. The long-range electrostatic interactionswere evaluated using particle mesh Ewald with a [148, 128, 96] Ågrid dimension.41 We used a 12 Å cut-off radius for bothvan der Waals and electrostatic interactions along with smoothparticle mesh Ewald.41 We used the NAMD software package42
to perform all-atom molecular dynamics (MD) simulations on a96 processor cluster.
Structure analysis and energy calculations
To monitor the stability of protein structure during MD simula-tions, we calculated the root mean square deviation (RMSD) onheavy protein atoms using VMD software. The configurationalentropy for the CAS domain during MD simulations wascalculated from covariance matrices of the heavy atom fluctuationsat 200 ps time step, using CARMA software.43–45 The interactionenergy for the Ca2+ ion in the many sites we identified was calcu-lated by evaluating non-bonded interactions (that is, van der Waalsand electrostatic) between the Ca2+ and the protein residues in theidentified sites, using a cutoff of 12.0 Å. For the electrostaticinteractions, we adopted the same scheme adopted for the wholeMD simulations. The total number of Ca2+ ions around the CASresidues (residue id 350–364) and the rest of the protein (residue id1–350) for the chains B and C was monitored using a distancecutoff of 5.0 Å, at every 20 ps time step of each MD trajectory.Similarly, the number of water molecules around the CAS residuesand the rest of the protein for the chains B and C was calculatedusing a distance cutoff of 3.0 Å. The H-bond and hydrophobicinteractions between the interface residues belonging to chains Band C were calculated using appropriate VMD scripts on theaverage structure of the protein calculated at intervals of 2 ns.The two dimensional schematic representation of the interactionnetwork was finally generated using the software LIGPLOT.46
Acknowledgements
AS acknowledges for modelling software support from theDepartment of Biotechnology (DBT), Delhi, through grantno. BT/PR14237/MED/29/196/2010. AK thanks the computationalfacility at CRS4 (Polaris), Pula, Italy. HC was supported by instituteresearch fellowship from Birla Institute of Technology, Mesra. TBthanks CSIR, Delhi, for providing Senior Research Fellowship.N.C.B. was supported by postdoctoral fellowship from theAmerican Heart Association. This work was supported in partby National Institutes of Health Grant R01 HL64014 to M.P.
References
1 N. A. Beard, D. R. Laver and A. F. Dulhunty, Prog. Biophys.Mol. Biol., 2004, 85, 33–69.
2 B. Caudwell, J. F. Antoniw and P. Cohen, Eur. J. Biochem.,1978, 86, 511–518.
3 A. Maurer, M. Tanaka, T. Ozawa and S. Fleischer, Proc. Natl.Acad. Sci. U. S. A., 1985, 82, 4036–4040.
4 S. E. Cala and L. R. Jones, J. Biol. Chem., 1983, 258,11932–11936.
Paper Molecular BioSystems
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 01
/05/
2013
07:
32:5
9.
Publ
ishe
d on
10
Apr
il 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3M
B25
588C
View Article Online

This journal is c The Royal Society of Chemistry 2013 Mol. BioSyst.
5 C. Franzini-Armstrong, L. J. Kenney and E. Varriano-Marston,J. Cell Biol., 1987, 105, 49–56.
6 S. Wang, W. R. Trumble, H. Liao, C. R. Wesson,A. K. Dunker and C. H. Kang, Nat. Struct. Biol., 1998, 5,476–483.
7 H. Park, S. Wu, A. K. Dunker and C. Kang, J. Biol. Chem.,2003, 278, 16176–16182.
8 B. Cozens and R. A. Reithmeier, J. Biol. Chem., 1984, 259,6248–6252.
9 B. M. Aaron, K. Oikawa, R. A. Reithmeier and B. D. Sykes,J. Biol. Chem., 1984, 259, 11876–11881.
10 N. Ikemoto, M. Ronjat, L. G. Meszaros and M. Koshita,Biochemistry, 1989, 28, 6764–6771.
11 H. Park, I. Y. Park, E. Kim, B. Youn, K. Fields, A. K. Dunkerand C. Kang, J. Biol. Chem., 2004, 279, 18026–18033.
12 E. J. Sanchez, K. M. Lewis, B. R. Danna and C. Kang, J. Biol.Chem., 2012, 287, 11592–11601.
13 L. Fliegel, M. Ohnishi, M. R. Carpenter, V. K. Khanna,R. A. Reithmeier and D. H. MacLennan, Proc. Natl. Acad.Sci. U. S. A., 1987, 84, 1167–1171.
14 A. Zarain-Herzberg, L. Fliegel and D. H. MacLennan, J. Biol.Chem., 1988, 263, 4807–4812.
15 A. Mishra, S. K. Suman, S. S. Srivastava, R. Sankaranarayananand Y. Sharma, J. Mol. Biol., 2012, 415, 75–91.
16 E. Kim, B. Youn, L. Kemper, C. Campbell, H. Milting,M. Varsanyi and C. Kang, J. Mol. Biol., 2007, 373, 1047–1057.
17 M. Gaburjakova, N. C. Bal, J. Gaburjakova and M. Periasamy,Cell. Mol. Life Sci., 2012, DOI: 10.1007/s00018-012-1199-7.
18 N. C. Bal, A. Kumar, A. Chakravarty, T. Balaraju, C. Bal, N. Jena,A. Sharon and M. Periasamy, Biophys. J., 2013, 104, 173a.
19 J. S. Evans, S. I. Chan and W. A. Goddard, 3rd, Protein Sci.,1995, 4, 2019–2031.
20 S. G. Priori and S. R. Chen, Circ. Res., 2011, 108, 871–883.21 J. M. Berg, Cold Spring Harbor Symp. Quant. Biol., 1987, 52,
579–585.22 T. Ozawa, Mol. Med. Rep., 2010, 3, 199–204.23 A. F. Dulhunty, N. A. Beard and A. D. Hanna, J. Gen. Physiol.,
2012, 140, 87–92.24 N. Ikemoto, B. Nagy, G. M. Bhatnagar and J. Gergely, J. Biol.
Chem., 1974, 249, 2357–2365.25 N. C. Bal, N. Jena, D. Sopariwala, T. Balaraju, S. Shaikh,
C. Bal, A. Sharon, S. Gyorke and M. Periasamy, Biochem. J.,2011, 435, 391–399.
26 D. R. Scriven, P. Asghari and E. D. Moore, Cardiovasc. Res.,2013, 98, 169–176.
27 N. C. Bal, A. Sharon, S. C. Gupta, N. Jena, S. Shaikh,S. Gyorke and M. Periasamy, J. Biol. Chem., 2010, 285,17188–17196.
28 K. W. Lee, J. S. Maeng, J. Y. Choi, Y. R. Lee, C. Y. Hwang,S. S. Park, H. K. Park, B. H. Chung, S. G. Lee, Y. S. Kim,H. Jeon, S. H. Eom, C. Kang, H. Kim do and K. S. Kwon,J. Biol. Chem., 2012, 287, 1679–1687.
29 C. Manno, M. Sztretye, L. Figueroa, P. D. Allen and E. Rios,J. Physiol., 2013, 591, 423–442.
30 E. J. Sanchez, K. M. Lewis, B. R. Danna and C. Kang, J. Biol.Chem., 2012, 287, 11592–11601.
31 K. W. Park, J. H. Goo, H. S. Chung, H. Kim, D. H. Kim andW. J. Park, Gene, 1998, 217, 25–30.
32 M. Arai, N. R. Alpert and M. Periasamy, Gene, 1991, 109,275–279.
33 L. Fliegel, E. Leberer, N. M. Green and D. H. MacLennan,FEBS Lett., 1989, 242, 297–300.
34 E. Damiani, P. Volpe and A. Margreth, J. Muscle Res. CellMotil., 1990, 11, 522–530.
35 D. Biral, P. Volpe, E. Damiani and A. Margreth, FEBS Lett.,1992, 299, 175–178.
36 A. D. MacKerell, Jr., N. Banavali and N. Foloppe, Biopolymers,2000, 56, 257–265.
37 W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics,1996, 14, 33–38, 27–38.
38 W. L. Jorgensen, J. Chandrasekhar, J. D. Madura,R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79,926–935.
39 H. Li, A. D. Robertson and J. H. Jensen, Proteins, 2005, 61,704–721.
40 J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput.Phys., 1977, 23, 327–341.
41 U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Leeand L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593.
42 J. C. Phillips, R. Braun, W. Wang, J. Gumbart,E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kaleand K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802.
43 I. Andricioaei and M. Karplus, J. Chem. Phys., 2001, 115,6289–6292.
44 N. M. Glykos, J. Comput. Chem., 2006, 27, 1765–1768.45 T. Balaraju, A. Kumar, C. Bal, D. Chattopadhyay, N. Jena,
N. C. Bal and A. Sharon, Struct. Chem., 2012, DOI: 10.1007/s11224-012-0181-1.
46 A. C. Wallace, R. A. Laskowski and J. M. Thornton, ProteinEng., 1995, 8, 127–134.
Molecular BioSystems Paper
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 01
/05/
2013
07:
32:5
9.
Publ
ishe
d on
10
Apr
il 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3M
B25
588C
View Article Online