how does an enzyme evolved in vitro compare to naturally occurring homologs possessing the targeted...
TRANSCRIPT

How Does an Enzyme Evolved In vitro Compare toNaturally Occurring Homologs Possessing theTargeted Function? Tyrosine Aminotransferase fromAspartate Aminotransferase
Steven C. Rothman and Jack F. Kirsch*
Department of Molecular andCell Biology, University ofCalifornia, Berkeley, 229Stanley Hall #3206, BerkeleyCA 94720-3206, USA
Aspartate aminotransferase (AATase) and tyrosine aminotransferase(TATase) are Escherichia coli paralogs that share 43% sequence identity.A plausible model posits that TATase arose from a duplication of anancestral AATase-like enzyme. Directed evolution of AATase to anenzyme having TATase activity was undertaken in order to compare theevolved AATase variants with homologous TATases. Eight rounds ofDNA shuffling and in vivo selection followed by a backcross with WTAATase produced enzymes that exhibited 100–270-fold increases in kcat/Km
Phe and had as much as 11% of the tyrosine aminotransferase activity ofWT E. coli TATase. Amino acid substitutions in 11 clones from rounds 7and 8 were compared with conserved residues in AATases and TATases.The findings are conveniently and compactly illustrated by the use ofVenn diagrams and set theory notation. A statistically significant ð0:001 #p # 0:008Þ concentration of mutations occurs in a subset of positions (setAAT – TAT) that is conserved ($75% identical) in AATases and variable(,75% identical) in TATases. Very few mutations occur in the intersection(set AAT > TAT) of amino acid residues that are conserved in bothenzyme types. Seven mutations from set AAT – TAT were combined bysite-directed mutagenesis to give a construct that is 60% as active as thebest round 8 enzyme, which has 13 amino acid replacements. The Venndiagrams may provide a generally useful tool to highlight the mostimportant specificity determinants for rational redesign. Amino acidreplacements were mapped onto the crystal structure of a hydro-cinnamate complex of a designed TATase. Five of the seven positionsmost frequently substituted in the evolved clones are within 15 A of thephenyl side-chain, but only six of the 48 positions that were mutatedonce or twice are within that radius. Context dependence, neutralmutations, different selective pressures, and stochastic componentsprovide explanations for the observation that many of the substitutionsfound in the directly evolved enzymes differ from the correspondingamino acids found in the modern natural TATases.
q 2003 Elsevier Science Ltd. All rights reserved
Keywords: enzyme evolution; DNA shuffling; aminotransferase; substratespecificity; Venn diagrams*Corresponding author
0022-2836/03/$ - see front matter q 2003 Elsevier Science Ltd. All rights reserved
E-mail address of the corresponding author: [email protected]
Abbreviations used: PLP, pyridoxal 50-phosphate; aKG, a-ketoglutarate; AATase, aspartate aminotransferase (EC2.6.1.1); TATase, tyrosine aminotransferase (EC 2.6.1.5); eAAT, E. coli aspartate aminotransferase; eTAT, E. coli tyrosineaminotransferase; pTAT, P. denitrificans tyrosine aminotransferase; HO-HxoDH, 2-hydroxyisocaproate (i.e. 2-hydroxy-4-methyl-pentanoate) dehydrogenase; MDH, malate dehydrogenase; LDH, lactate dehydrogenase; SRHEPT, a mutantof AATase with the following seven substitutions: A12T/P13T/N34D/T109S/G261A/S285S/N297S; HEX, a mutant ofAATase with the following six substitutions: V39L/K41Y/T47I/N69L/T109S/N297S; HPP, hydroxphenylpyruvate; PP,phenylpyruvate.
doi:10.1016/S0022-2836(03)00095-0 J. Mol. Biol. (2003) 327, 593–608

Introduction
An important historical goal of protein engineer-ing has been to discover a minimal set of residuechanges that can convert the substrate specificityof one enzyme to that of another. Successfulrational redesigns, typically based on homologymodeling, provide insights into mechanisms ofsubstrate recognition and discrimination and laythe ground work for generating enzymes withdesired specificities. Important examples includethe redesign of subtilisin BPN0 to recognize the pre-ferred substrates of homologous proteases Kex21
and furin,2 switching the coenzyme specificity ofisocitrate dehydrogenase from NADPH toNADH,3 and conversion of lactate dehydrogenaseinto a malate dehydrogenase.4 In these cases, asmall number of amino acid residue replacementssufficed to produce significant changes in substratespecificity. On the other hand, the seemingly equiv-alent task of converting trypsin into an enzymewith substantial chymotrypsin-like activity provedto be a much more formidable challenge, whichultimately required the exchange of surface loops.5
To date, no generally applicable procedure tochange substrate specificity or catalytic mechanismby site-directed mutagenesis exists.
Onuffer & Kirsch applied homology modeling tobroaden the substrate specificity of the PLP-depen-dent enzyme aspartate aminotransferase (AATase),to that of tyrosine aminotransferase (TATase).6
AATase reacts predominately with aspartate, gluta-mate and their corresponding keto acids.7 Tyrosineaminotransferase (TATase), a close homolog ofAATase (43% sequence identity in Escherichia coli8),recognizes both dicarboxylic and aromatica-amino and a-keto acids.9,10 Six positions inE. coli AATase were changed to the correspondingE. coli TATase residues to achieve a 1500-foldincrease in single turnover kf/KD for phenylalanine,while retaining aspartate activity. The kf/KD
Phe valuefor this “HEX” mutant is within sevenfold of thecorresponding TATase value. The reverse muta-genesis of the six positions in the E. coli TATaseproduced the opposite effect of lowering steadystate kcat/Km
Phe by 700-fold with only a small changein aspartate activity.51
The HEX redesign was based on a set of sixactive site residues that are generally conserved inthe known AATase sequences but are different inE. coli TATase. With the increasing availability ofother prokaryotic TATase sequences,11 – 14 it becameclear that residues other than the six from theHEX redesign plan determine specificity in homo-logous TATases. Oue et al.11 characterized aParaccocus denitrificans TATase, in which four ofthe six HEX positions are identical with thosefound in E. coli AATase, rather than the TATase.Five of the six positions in the TATase fromPseudomonas aeruginosa are identical with the corre-sponding amino acid residues in E. coli AATase.12,15
Jensen & Gu concluded that within the frameworkof such closely related aminotransferases, multiple
combinations of substitutions that broaden sub-strate specificity exist.16
Directed evolution provides a powerful methodto explore the sequence space accessible fromsingle base mutations for a given protein. Success-ful applications employing random mutagenesisor DNA shuffling have produced enzymes withnovel substrate specificities, enhanced thermo-stability, and improved heterologous expression.17
Particularly impressive examples include: (1) thework of Stemmer to generate variants of the TEM-1 b-lactamase that confer a 32,000-fold increase incefotaxime resistance;18 (2) the evolution of E. coliaspartate aminotransferase to recognizeb-branched chain amino acids19,20 (a 105-foldincrease in the catalytic efficiency with valine wasreported); (3) the alteration of b-glucuronidase tohydrolyze a b-galactoside substrate, with a 500-fold increase in kcat/Km;21 and (4) the inversion ofthe enantioselectivity of hydantoinase.22
Most reported examples of directed evolutionhave been focused on the introduction of a targetedproperty into a starting protein. No closely relatedhomologous protein having the desired activitywas available. The purpose of the present investi-gation was not to use directed evolution of AATaseto produce TATase, since the latter enzyme alreadyexists. Rather, the motivation was to explore mini-mal sets of mutations that suffice to convert anAATase to an enzyme that possesses catalyticproperties substantially like those of the extantand closely related TATases. Analysis of theevolved AATase variants reveals that severalfrequent amino acid replacements are derivedfrom a small subset of amino acids that are evolu-tionarily conserved in the initial protein, but arevariable in the paralog that has the targetedactivity.
Results
Selection scheme and directed evolution
An E. coli genetic selection was developed to iso-late aspartate aminotransferase mutants withenhanced tyrosine aminotransferase activity. Thenaturally occurring E. coli TATase, which isencoded by the tyrB gene, catalyzes the terminalstep in the biosyntheses of phenylalanine and tyro-sine (Figure 1). Coupling of growth rate to amino-transferase activity requires rescue of Phe and Tyrauxotrophies in TATase deficient strains. Selectionwas initially evaluated in E. coli strain MG204.This strain lacks functional tyrosine, aspartate(encoded by aspC), and branched chain (encodedby ilvE) aminotransferases. It was observed thatMG204 would grow on phenylalanine and tyrosine-free media if transformed with the gene for WTAATase on a plasmid. This means that WT AATasepossesses sufficient aromatic aminotransferaseactivity to convert the endogenous supply ofphenylpyruvate (PP) and hydroxyphenylpyruvate
594 Evolution of L-Asp to L-Tyr Aminotransferase

(HPP) to Phe and Tyr, respectively, thus sustaininggrowth. Gelfand & Steinberg inactivated the tyrAgene (see Figure 1), in order to eliminate the bio-synthesis of HPP, the endogenous direct precursorto Tyr.23 With this strain they observed a goodrelationship between tyrosine aminotransferaseactivity and growth on tyrosine-free plates sup-plemented with HPP. Similar manipulations wereperformed here to provide a stringent selection.Engineered strains SR224 and SR250 have fivegene deletions: tyrA, pheA, tyrB, aspC, and ilvE.recA is also inactivated in SR250. Growth in thesestrains depends absolutely on an exogenous sourceof HPP, and is nearly completely dependent onadded PP, as well as gained aromatic amino-transferase activity. Suitable concentrations of
added HPP and PP were found empirically. TheHPP and PP concentrations and levels of themutant gene expression were lowered in laterrounds of selection to modulate stringency.
Eight rounds of directed evolution by DNAshuffling were performed with E. coli AATase.Rapidly growing colonies were isolated on medialacking tyrosine and on media lacking phenyl-alanine. Several fast-growing positives fromrounds 7 and 8 were chosen for analysis. Positives7-2, 8-1, and 8-2 were specifically picked from asecondary screen on microtiter plates for highlyactive variants. Eight additional round 7 cloneswere taken without screening. DNA from positive8-2 was shuffled with WT AATase (3:1) in a back-cross to remove non-essential mutations. Theresultant 8-2B colony was isolated by subsequentselection and screening.
Kinetic parameters of evolved AATase mutants
Enzymes from multiple colonies from rounds 7and 8 were purified. The steady-state kinetic par-ameters with aromatic and dicarboxylic substratesare given in Table 1. The mutant enzymes exhibitincreases in kcat/Km of 100–270-fold for phenyl-alanine and 40–150-fold for tyrosine. That pre-pared from positive 8-2B displays the largestactivity improvements. kcat/Km
Phe and kcat/KmTyr are
within 30- and tenfold, respectively, of the WTE. coli TATase values. WT E. coli TATase andAATase exhibit similar kinetic parameters foraspartate, but this activity varies more significantlyin the enzymes realized from directed evolution.The kcat/Km value for positive 7-1 with aspartate isreduced by 30-fold from that of the WT enzyme,while that for positive 8-2 is within threefold of
Table 1. Steady-state kinetic parameters for clones from rounds 7 and 8 and wild-type aminotransferases
Phe Tyr Asp Leu Ala
Enzymevariant
kcat/Km
(M21 s21) £ 1022
kcat
(s21)kcat/Km
(M21 s21) £ 1022
kcat
(s21)kcat/Km
(M21 s21) £ 1022
kcat
(s21) kcat/Km (M21 s21) £ 1022
AATase 1.19a Ns 5.50 (0.03) Ns 910b 159b 0.055(0.003)
0.0068(0.0007)
7-1 200 (30) 11.4(0.5)
230 (10) 12.1(0.3)
35 (0.3) 10.7(0.3)
2.56(0.09)
0.253(0.007)
7-2 130 (2) 33 (2) 170 (1) 32 (2) 120 (2) 8.3(0.3)
1.18(0.09)
0.131(0.002)
8-1 220 (40) 15.5(1.3)
255 (30) 18.4(1.1)
64 (4) 12.0(0.2)
2.27(0.05)
0.211(0.003)
8-2 270 (30) 15.0(0.6)
410 (2) 17.8(0.3)
294 (8) 33 (6) 12.3 (0.2) 0.74 (0.02)
8-2B 320 (10) 45.1(0.7)
720 (20) 63 (1) 1070 (60) 41.0(0.5)
5.74(0.08)
1.18 (0.02)
TATase 9600c 250c 6500c 250c 370c 140c 2.56(0.03)
0.0273(0.0003)
Assay conditions for original data: 200 mM Taps (pH 8.0), 140 mM KCl, ,0.15–0.2 mM NADH, 2–20 mM aKG, 20 mM PLP, 25 8C[HO-HxoDH] ¼ 0.3–3.0 mM for Tyr, Phe, Leu assays; [MDH] ¼ 4–10 units/ml for Asp assays; [LDH] ¼ 10–20 units/ml for Alaassays. Standard errors are in parentheses; Ns, saturation was not observed for the substrate concentrations used.
a From Luong & Kirsch.39
b From Gloss & Kirsch.48
c From Hayashi et al.10
Figure 1. Terminal steps in the biosyntheses of tyrosineand phenylalanine. pheA encodes the dual functionchorismate mutase-prephenate dehydratase. tyrAencodes chorismate mutase-prephenate dehydrogenase.Tyrosine aminotransferase catalyzes the final step inboth pathways with glutamate as an amino donor.
Evolution of L-Asp to L-Tyr Aminotransferase 595

Table 2. Amino acid substitutions found in rounds 7 and 8 clones, HEX, and TATase homologs
(continued)
596 Evolution of L-Asp to L-Tyr Aminotransferase

that of WT AATase. kcat/KmAsp for positive 8-2B is
roughly the same as WT AATase, although kcat isreduced fourfold.
Aminotransferase activity towards alanine andleucine was measured to investigate the nature ofsubstrate specificity enhancements in the evolvedclones. Data from Table 1 indicate that improve-ments in Ala and Leu reactivity are similar tothose observed for Phe and Tyr. Relative kcat/Km
increases for alanine and leucine in positive 8-2are within twofold of that for phenylalanine.Activity improvements for positives 7-1, 7-2, and8-1 are only fivefold greater towards Phe than Leuor Ala. E. coli TATase is far more specific for aro-matic substrates than are the evolved variants.kcat/Km for Ala is only fourfold greater in TATasethan AATase. Leucine, which shares more struc-tural similarity with phenylalanine than alanine, istransaminated with a 100-fold greater efficiency inTATase than AATase. Yet, this improvement isonly 1/100th of the 10,000-fold activity increasefor Phe.
Sequences of evolved clones
The DNA sequences of nine clones from round 7were determined. Clones 8-1, 8-2, and 8-2B werealso sequenced. WT AATase amino acid residues8
and substitutions in evolved variants and in theHEX mutant6 are presented in Table 2, togetherwith the corresponding amino acid residues inWT TATases from E. coli8 and P. denitrificans.11
Clones resulting from directed evolution containnine to 17 replacements in the 396 amino acidresidue sequence of aspartate aminotransferase.A total of 158 amino acid substitutions at 67positions was found (clone 8-2B was omitted fromthe analysis). A total of 39 of the 67 positions weremutated only in a single clone, while only sevenpositions were mutated in a majority of clones.
Clone 8-2 displayed the largest activity improve-ments among the characterized round 7 and 8enzymes. The backcross with WT AATase shouldhelp to remove any deleterious or neutral
mutations.18 Mutations S139G, V26A, and R282Cwere reversed in the backcross clone 8-2B. S139Gis present in all round 7 and 8 clones. A possiblereason for its reversal is considered in Discussion.V26A is also found in a majority of sequencedclones. A novel substitution, R129G, aroseuniquely in the backcross. Along with S139G,P13T was observed in all round 7 and 8 clones. Allfive sequenced clones from rounds 2 and 4 havethe P13T replacement (data not shown).
Kinetic comparisons of site-directed mutants
A construct, SRHEPT, was created by site-directed mutagenesis of WT AATase. It containsthe substitutions A12T, P13T, N34D, T109S,G261A, S285G, and N297S. These amino acid repla-cements were present in at least three clones fromrounds 7 and 8, and are in the group of aminoacid residues that is conserved in AATases but is
Table 2 continued
Amino acids highlighted in shaded boxes indicate that these residues are also found in at least some evolved clones. Amino acidssegregated by unshaded boxes are identical with those found in the corresponding positions in E. coli AATase.
a Sequence numbering is based on the pig cytosolic aspartate aminotransferase.50
b The clone 8-2B contains the substitutions resulting from a backcross of clone 8-2 with WT AATase.c eTAT and pTAT are TATases from E. coli and P. denitrificans.d Total number of positions mutated in at least one of the 11 clones from rounds 7 and 8. The numbers that follow in this row are
the total number of non-silent mutations in the given clone.
Table 3. Steady-state kinetic parameters exhibited bysite-directed mutants and wild-type enzymes
kcat/Km (M21 s21) £ 1022
Enzyme variant Phe Asp Ala
AATase 1.19a 900b 0.0068 (0.0007)T109S AATase 3.00a 86a NdT109S/N297S AATase 3.30a 120a 0.0063 (0.0003)SRHEPTc 165 (7) 240 (2) 0.43 (0.01)HEXd 370 (20) 340 (50) 0.402 (0.005)TATase 9600e 370e 0.0273 (0.0003)
Assay conditions for original data: 200 mM Taps (pH 8.0 or8.4), 140 mM or 100 mM KCl, ,0.15–0.2 mM NADH, 2–20 mMaKG, 20 mM PLP, 25 8C. [HO-HxoDH] ¼ 0.3–3.0 mM for Pheassays; [MDH] ¼ 4–10 units/ml for Asp assays; [LDH] ¼ 10–20 units/ml for Ala assays; Nd, Not determined.
a Luong & Kirsch.39
b Gloss & Kirsch.48
c SRHEPT: A12T/P13T/N34D/T109S/G261A/S285G/N297S.Constructed by combining frequent round 7 and 8 substitutionsðn . 2Þ at positions conserved in AATases but variable inTATases.
d HEX: V39L/K41Y/T47I/T109S/N297S made in E. coliAATase6.
e Hayashi et al.10
Evolution of L-Asp to L-Tyr Aminotransferase 597
variable in TATases (see Discussion). The kineticparameters for SRHEPT and selected kineticallycharacterized site-directed mutants of AATase arecollected in Table 3. SRHEPT displays a 140-foldincrease in kcat/Km
Phe over WT AATase. HEX exhibitsa 310-fold improvement. Both SRHEPT and HEXretain aspartate activity. SRHEPT, which containsonly two (T109S, N297S) of the six designed HEXmutations, is nonetheless comparable in activity toHEX.
Discussion
Structural mapping of amino acidreplacements in evolved enzymes
The 11 sequenced AATase variants chosen fromrounds 7 and 8 contain a total of 158 amino acidsubstitutions at 67 positions. Figure 2 depicts the
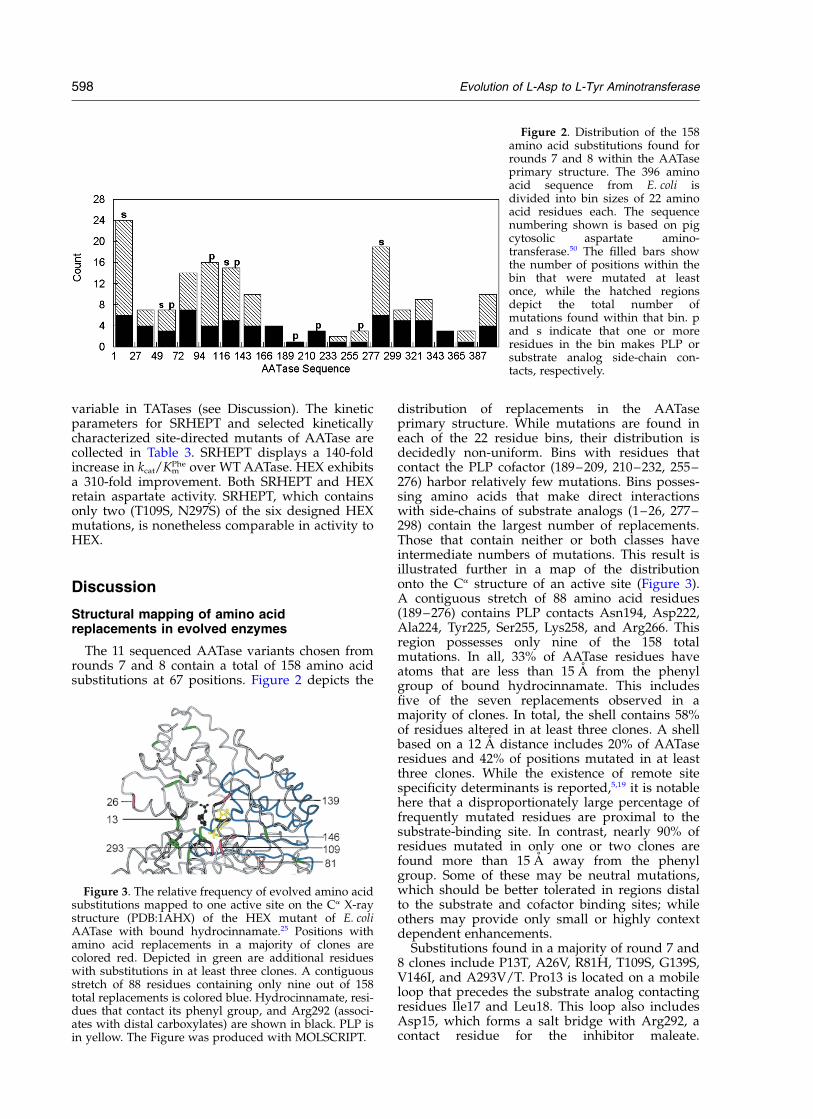
distribution of replacements in the AATaseprimary structure. While mutations are found ineach of the 22 residue bins, their distribution isdecidedly non-uniform. Bins with residues thatcontact the PLP cofactor (189–209, 210–232, 255–276) harbor relatively few mutations. Bins posses-sing amino acids that make direct interactionswith side-chains of substrate analogs (1–26, 277–298) contain the largest number of replacements.Those that contain neither or both classes haveintermediate numbers of mutations. This result isillustrated further in a map of the distributiononto the Ca structure of an active site (Figure 3).A contiguous stretch of 88 amino acid residues(189–276) contains PLP contacts Asn194, Asp222,Ala224, Tyr225, Ser255, Lys258, and Arg266. Thisregion possesses only nine of the 158 totalmutations. In all, 33% of AATase residues haveatoms that are less than 15 A from the phenylgroup of bound hydrocinnamate. This includesfive of the seven replacements observed in amajority of clones. In total, the shell contains 58%of residues altered in at least three clones. A shellbased on a 12 A distance includes 20% of AATaseresidues and 42% of positions mutated in at leastthree clones. While the existence of remote sitespecificity determinants is reported,5,19 it is notablehere that a disproportionately large percentage offrequently mutated residues are proximal to thesubstrate-binding site. In contrast, nearly 90% ofresidues mutated in only one or two clones arefound more than 15 A away from the phenylgroup. Some of these may be neutral mutations,which should be better tolerated in regions distalto the substrate and cofactor binding sites; whileothers may provide only small or highly contextdependent enhancements.
Substitutions found in a majority of round 7 and8 clones include P13T, A26V, R81H, T109S, G139S,V146I, and A293V/T. Pro13 is located on a mobileloop that precedes the substrate analog contactingresidues Ile17 and Leu18. This loop also includesAsp15, which forms a salt bridge with Arg292, acontact residue for the inhibitor maleate.
Figure 2. Distribution of the 158amino acid substitutions found forrounds 7 and 8 within the AATaseprimary structure. The 396 aminoacid sequence from E. coli isdivided into bin sizes of 22 aminoacid residues each. The sequencenumbering shown is based on pigcytosolic aspartate amino-transferase.50 The filled bars showthe number of positions within thebin that were mutated at leastonce, while the hatched regionsdepict the total number ofmutations found within that bin. pand s indicate that one or moreresidues in the bin makes PLP orsubstrate analog side-chain con-tacts, respectively.
Figure 3. The relative frequency of evolved amino acidsubstitutions mapped to one active site on the Ca X-raystructure (PDB:1AHX) of the HEX mutant of E. coliAATase with bound hydrocinnamate.25 Positions withamino acid replacements in a majority of clones arecolored red. Depicted in green are additional residueswith substitutions in at least three clones. A contiguousstretch of 88 residues containing only nine out of 158total replacements is colored blue. Hydrocinnamate, resi-dues that contact its phenyl group, and Arg292 (associ-ates with distal carboxylates) are shown in black. PLP isin yellow. The Figure was produced with MOLSCRIPT.
598 Evolution of L-Asp to L-Tyr Aminotransferase

Kawaguchi et al.24 postulated that the loop’s greaterflexibility in TATase, which has Ala13, than inAATase is an important specificity determinant.They generated a chimera by replacing this loopin E. coli TATase with the corresponding E. coliAATase residues. A significant loss in activity foraromatic substrates was reported.24 The presentresults support the contention that replacement ofPro13 in AATase does enhance recognition of aro-matic substrates. Malashkevich et al.25 solved thestructure of the HEX mutant, and reported thatT109S permits two additional water molecules toaccess the binding pocket. They were proposed toparticipate in a new hydrogen bonding network,which is more favorable for binding aromatic sub-strates. Yano et al.20 evolved E. coli AATase torecognize b-branched chain amino acids andfound S139G and A293V as frequent substitutions.S139G was proposed to increase flexibility in aloop that contains the substrate-contacting residueTrp140. Ile146 makes contacts with residues atpositions 293 and 109 in the P. denitrificans TATaseX-ray structure.26 Mutations A26V and R81H areboth surface residues several A from the activesite.
Adaptation via specificity relaxation
Aspartate aminotransferases react nearly exclu-sively with dicarboxylic a-amino acids and theircorresponding a-keto acids while tyrosine amino-transferases recognize both dicarboxylic andaromatic a-amino and a-keto acids. E. coli AATaseand TATase transaminate other amino acids withcatalytic efficiencies (kcat/Km values) that areseveral orders of magnitude less than those for thepreferred substrates. kcat/Km for alanine is close to1 M21 s21 in both enzymes. Similar specificity pre-ferences are reported for eukaryotic AATases27 – 30
and the P. denitrificans TATase.11 AATase variantsfrom rounds 7 and 8 exhibit relative activityincreases for alanine and leucine that are compar-able to those for phenylalanine and tyrosine. Thissuggests that adaptation in directed evolution wasachieved by a general broadening of specificity.Interestingly, both SRHEPT and HEX exhibit con-siderable enhancements in kcat/Km for alaninedespite the fact that four of the seven SRHEPTreplacements are changes to residues commonlyfound in TATases and all six substitutions in HEXare to E. coli TATase residues. The general activityincrease in the HEX mutant is likely attributed tothe mutations V39L, K41Y, T47I, and N69L. TheT109S/N297S mutant has a kcat/Km for alaninesimilar to the value for WT AATase. Previousstudies have also shown that V39L increases thekcat/Km values for many substrates.31,32 Based onthe solved HEX structure, Malashkevich et al.25
concluded that mutations V39L, K41Y, T47I, andN69L stabilize a closed active conformation of theenzyme.
DNA shuffling combined with a genetic selec-tion generated an AATase variant that exhibited a100,000-fold improvement in activity for valine,measured as kf/Kd under pre-steady-stateconditions.20 A significant portion of the enhancedactivity is manifested in a general improvement inrates of transamination of all other tested aminoacid substrates (with the exceptions of the nativeAATase substrates Asp and Glu). Adaptationthrough specificity relaxation, in other enzymesresulting from directed evolution experiments, is acommon observation.21 Two explanations havebeen offered: (1) the in vitro experiments do notselect for specificity, while natural evolution clearlymust. (2) Modern enzymes likely evolved fromancestors possessing broad specificities,33 whiledirected evolution typically initiates with enzymes
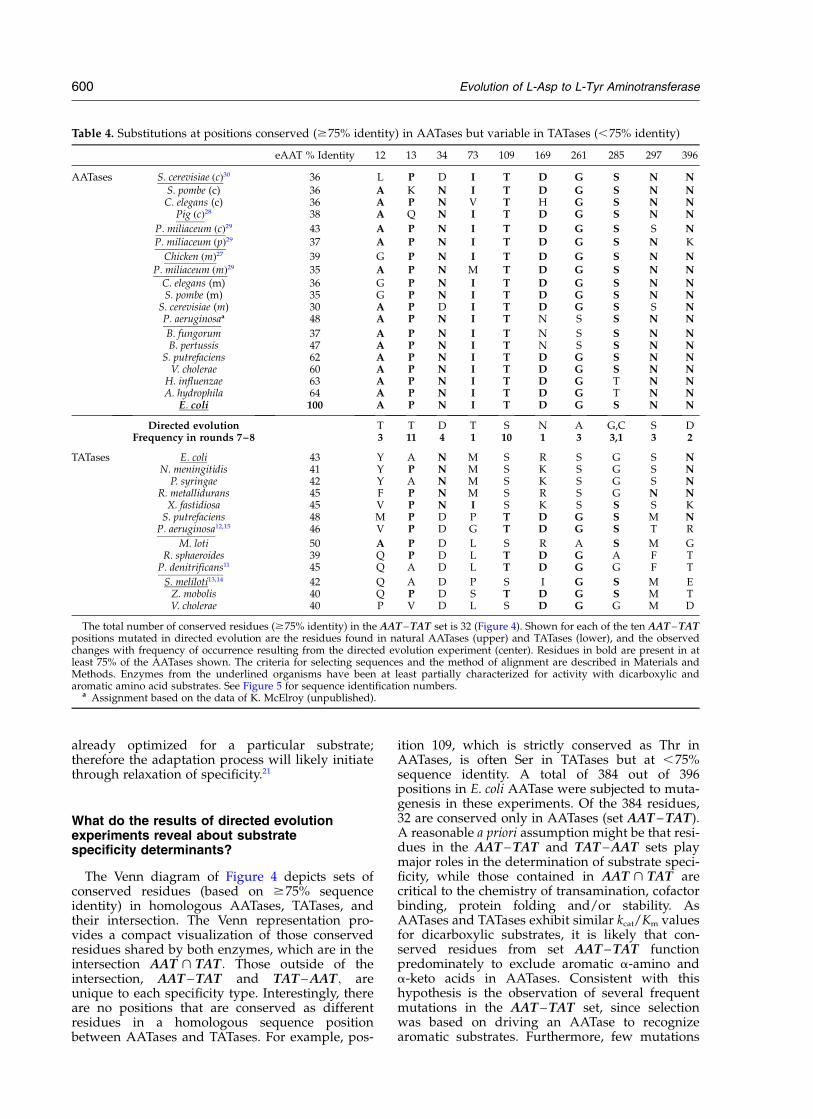
Figure 4. Venn diagram of theintersection of conserved AATase(left circle) and TATase (right circle)residues for subfamily Ia amino-transferases. The assignments ofconserved residues are based on75% sequence identity. Underlinedplain style residues were found tobe mutated only once in the 11clones sequenced in rounds 7 and8. Underlined bold-face residueswere mutated more than once. Theconsensus residues shown in plainstyle in the set TAT – AAT; whileless than 75% conserved inAATases, happen to be identicalwith the corresponding E. coliAATase residue. Those shown initalics are conserved residues thatare not identical with the corre-sponding E. coli AATase aminoacid residues. Consensus residuesin italics in sets AAT – TAT andAAT > TAT are also different inthe E. coli AATase.
Evolution of L-Asp to L-Tyr Aminotransferase 599
already optimized for a particular substrate;therefore the adaptation process will likely initiatethrough relaxation of specificity.21
What do the results of directed evolutionexperiments reveal about substratespecificity determinants?
The Venn diagram of Figure 4 depicts sets ofconserved residues (based on $75% sequenceidentity) in homologous AATases, TATases, andtheir intersection. The Venn representation pro-vides a compact visualization of those conservedresidues shared by both enzymes, which are in theintersection AAT > TAT: Those outside of theintersection, AAT – TAT and TAT – AAT; areunique to each specificity type. Interestingly, thereare no positions that are conserved as differentresidues in a homologous sequence positionbetween AATases and TATases. For example, pos-
ition 109, which is strictly conserved as Thr inAATases, is often Ser in TATases but at ,75%sequence identity. A total of 384 out of 396positions in E. coli AATase were subjected to muta-genesis in these experiments. Of the 384 residues,32 are conserved only in AATases (set AAT – TAT).A reasonable a priori assumption might be that resi-dues in the AAT – TAT and TAT – AAT sets playmajor roles in the determination of substrate speci-ficity, while those contained in AAT > TAT arecritical to the chemistry of transamination, cofactorbinding, protein folding and/or stability. AsAATases and TATases exhibit similar kcat/Km valuesfor dicarboxylic substrates, it is likely that con-served residues from set AAT – TAT functionpredominately to exclude aromatic a-amino anda-keto acids in AATases. Consistent with thishypothesis is the observation of several frequentmutations in the AAT – TAT set, since selectionwas based on driving an AATase to recognizearomatic substrates. Furthermore, few mutations
Table 4. Substitutions at positions conserved ($75% identity) in AATases but variable in TATases (,75% identity)
eAAT % Identity 12 13 34 73 109 169 261 285 297 396
AATases S: cerevisiae ðcÞ30 36 L P D I T D G S N N
S. pombe (c) 36 A K N I T D G S N NC. elegans (c) 36 A P N V T H G S N N
Pig ðcÞ28 38 A Q N I T D G S N N
P: miliaceum ðcÞ29 43 A P N I T D G S S N
P: miliaceum ðpÞ29 37 A P N I T D G S N K
Chicken ðmÞ27 39 G P N I T D G S N N
P: miliaceum ðmÞ29 35 A P N M T D G S N N
C. elegans (m) 36 G P N I T D G S N NS. pombe (m) 35 G P N I T D G S N N
S. cerevisiae (m) 30 A P D I T D G S S NP: aeruginosaa 48 A P N I T N S S N N
B. fungorum 37 A P N I T N S S N NB. pertussis 47 A P N I T N S S N N
S. putrefaciens 62 A P N I T D G S N NV. cholerae 60 A P N I T D G S N N
H. influenzae 63 A P N I T D G T N NA. hydrophila 64 A P N I T D G T N N
E: coli 100 A P N I T D G S N N
Directed evolution T T D T S N A G,C S DFrequency in rounds 7–8 3 11 4 1 10 1 3 3,1 3 2
TATases E: coli 43 Y A N M S R S G S NN. meningitidis 41 Y P N M S K S G S N
P. syringae 42 Y A N M S K S G S NR. metallidurans 45 F P N M S R S G N N
X. fastidiosa 45 V P N I S K S S S KS. putrefaciens 48 M P D P T D G S M N
P: aeruginosa12,15 46 V P D G T D G S T R
M. loti 50 A P D L S R A S M GR. sphaeroides 39 Q P D L T D G A F T
P: denitrificans11 45 Q A D L T D G G F T
S: meliloti13,14 42 Q A D P S I G S M EZ. mobolis 40 Q P D S T D G S M TV. cholerae 40 P V D L S D G G M D
The total number of conserved residues ($75% identity) in the AAT – TAT set is 32 (Figure 4). Shown for each of the ten AAT – TATpositions mutated in directed evolution are the residues found in natural AATases (upper) and TATases (lower), and the observedchanges with frequency of occurrence resulting from the directed evolution experiment (center). Residues in bold are present in atleast 75% of the AATases shown. The criteria for selecting sequences and the method of alignment are described in Materials andMethods. Enzymes from the underlined organisms have been at least partially characterized for activity with dicarboxylic andaromatic amino acid substrates. See Figure 5 for sequence identification numbers.
a Assignment based on the data of K. McElroy (unpublished).
600 Evolution of L-Asp to L-Tyr Aminotransferase

were acquired at residues in the AAT > TAT set.Ten of the 67 positions mutated in the 11 evolvedclones that were sequenced belong to setAAT – TAT: Amino acids resident at these tenpositions in homologous aminotransferases areshown in Table 4. The 32 positions in AAT – TATrepresent 8.3% of the 384 potentially mutatableresidues. Yet, the total number of replacements inTable 4 (42) accounts for 26.5% of the observed158 substitutions from rounds 7 and 8. On theother hand, only 2.5% of the substitutions (4) arein AAT > TAT; while this set of 82 amino acid resi-dues encompasses 21% of the sequence subjectedto mutagenesis. None of the 32 residues in the setTAT – AAT was found to be mutated in the 11clones from the directed evolution experiment.However, for 26 of the 32 residues, the amino acidpresent in E. coli AATase is identical with theTATase consensus. These 26 would not be expectedto change from a TATase conserved amino acidgiven the selection for enhanced activity withTATase substrates.
The probability that the observed clustering of 42of the 158 mutations would, by chance, be in asubset of 32 of the 384 possible positions wascalculated. This hypothesis was initially testedwith a model of sampling with replacement. The158 substitutions were treated as independentsampling events from a 384-member pool. Theprobability of $42 being found in a randomlychosen subset of 32 members was determined bysummation of the individual probabilitiescalculated from the binomial distribution equation(1):34
Px ¼n
x
!pxqn2x; PTotal
x ¼X158
x¼42
Px ð1Þ
where x, n, p, q are defined as the number clusteredð42 # x # 158Þ in the desired set of 32, the totalnumber of independent samples (158), the prob-ability of a single sampling being included in thechosen subset (32/384), and 1 2 p; respectively. Px
is the probability of sampling exactly x substi-tutions from the 32 member subset and PTotal
x isthe sum of probabilities for x ¼ 42 to x ¼ 158: Theresult was that there is less than a 1/1010 prob-ability of the observed clustering occurring byrandom chance. However, this method certainlyoverestimates the significance, as multiple occur-rences of the same substitution (i.e. P13T is presentin 11 clones) are not independent, because eachsuccessive round of directed evolution initiateswith beneficial realized mutations from the pre-vious rounds. Therefore, the opposite extreme wasevaluated by sampling without replacement,where a residue mutated in a single clone is con-sidered fixed for all clones and thus removedfrom the pool of 384 initial members. Thishypothesis was tested for the probability of $10(Table 4) of 67 total positions mutated (Table 2)occurring in a random cluster of 32. The hyper-geometric distribution34 and summation of
individual probabilities are given by equation (2):
Px ¼
M
x
!N 2 M
n 2 x
!
N
n
! ; PTotalx ¼
X32
x¼10
Px ð2Þ
Here x has values ð10 # x # 32Þ; n ¼ 67; M ¼ 32and N ¼ 384: The result is that the observed clus-tering would only be observed in 3% of suchsamplings. This number, representing the oppositeextreme, is an underestimate of significance,because all mutations were assigned the sameweight independently of their frequency of occur-rence. Therefore, a more realistic scenario wasinvestigated by numerical simulation, in whichthe 67 positions were weighted according to thefrequencies of substitution in the 11 sequencedclones. The details are described in Materials andMethods. The result is that there is a 1 in 1000chance that at least 42 of the 158 observed substi-tutions would belong to a randomly chosen set of32 residues. The simulation was also applied toestimate the significance of only four substitutionsbeing found in the AAT > TAT set. Less than oneout of 10,000 random sets of 82 residues wouldcontain four or fewer changes. The probability ofobtaining no substitutions in the 32 residues of setTAT – AAT is less than 1/500. Numerical simu-lations were repeated with only the sequencesfrom round 7 clones, and yielded similar prob-abilities. An alternate hypothesis, suggested by areferee, is that the apparent enrichment formutation in set AAT – TAT mainly derives from avery strong selection against substitutions in setsAAT > TAT and TAT – AAT: The simulations werethus repeated to compare the mutation enrichmentin set AAT – TAT only with mutations observed inpositions outside the conserved AATase andTATase sets. This set is designated as “Global-AAT < TAT”, where the global set consists of 384mutatable positions and AAT < TAT has 146 resi-dues. The enrichment in set AAT – TAT remainssignificant ðp ¼ 0:008Þ: This finding supports thecontention that the directed evolution experimentenriched for substitutions in the set AAT – TAT overany other group of residues. Simulations performedwith sets based on conservation criteria rangingfrom 65% to 85% sequence identity also gave resultsconsistent with a highly significant observed cluster-ing. These numbers strongly support the contentionthat the observed concentration of mutations inAAT – TAT and paucity of substitutions in AAT >TAT and TAT – AAT are highly significant.
The contribution of substitutions from setAAT – TAT to the enhanced activity towardsTATase substrates is further demonstrated by thekinetic parameters of SRHEPT. This constructedE. coli AATase variant contains the seven most fre-quently observed amino acid replacements of theten shown in Table 4. kcat/Km
Phe is 60% of that of themost highly active evolved clone 8-2 (Table 1),
Evolution of L-Asp to L-Tyr Aminotransferase 601

which has 13 mutations. kcat/KmAsp values for
SRHEPT, round 8-2, and WT TATase are within afactor of 1.6. While the AATase specificity determi-nants are not exclusively localized in setAAT – TAT; nonetheless it should be considered asa forcing set, in which a mutation is more likely toeffect specificity than one made outside of the set.
Comparison of substitutions in evolved cloneswith residues in E. coli TATase and E. coliAATase variants
Examination of the data of Table 2 appears to indi-cate that few of the observed substitutions arechanges to the corresponding endogenous E. coliTATase residue. The 13 amino acid replacements tothe native residues found in the E. coli TATase rep-resent just 18% of the 74 different substitutions. How-ever, a single base mutation of a given codon onaverage results in 5.7 amino acid changes.35 Thus,there are roughly 384 £ 5.7 < 2200 accessible aminoacid replacements. Only ,6% of these are to E. coliTATase residues†, suggesting that the observed 18%represents some bias towards E. coli TATase residuesin the directed evolution experiment.
Of the six designed HEX mutations, T109S ispresent in ten clones while N297S occurs in three.Of the other four positions characterizing the HEXconstruct, only Asn69 was found to be mutated inthe directed evolution experiment. This is leucinein HEX, but is serine in the evolved enzymes.HEX substitutions N69L and K41Y were notexpected as both require two base mutations inthe WT AATase DNA sequence. K126S was theonly replacement in the directed evolution experi-ment that necessitated two base changes. However,this residue was mutated to arginine in two otherclones. Both K126R and R126S require only a singlebase change; thus providing a functional route toSer126.36
AATase variants evolved to recognizeb-branched substrates have substitutions at onlytwo (N34D, N297S) of the subset of ten conservedpositions listed in Table 4.20 N34D and N297S areof particular significance because they are two ofthe five substitutions found in all of the sequencedclones from that study. The remaining three areN142T/I, I37M, and S139G. Asn142 belongs to setAAT > TAT: I37M is proposed to relieve stericinteractions for b-branched substrates in the activesite.20 The implied role of position 139 resultingfrom both studies is curious. The resident aminoacid is Thr or Ser in all AATases and TATases, yetit was invariably changed to Gly in the presentexperiments, as well as in those of Yano et al.20
This mutation was lost in the backcross clone 8-2Bwith WT AATase, accompanying the restoration ofaspartate activity without a decrease in kcat=Km foreither Phe or Tyr. Oue et al.37 reversed the substi-tutions at positions 139 and 142 from the evolvedvariants by site-directed mutagenesis with a result-ing 60-fold gain in kf/KD
Glu to WT AATase levelswith only small changes in kf/KD values for Phe,Tyr, Val, and Ala. It thus appears that these indepen-dent studies to evolve transamination activity todifferent new substrates both elicited a mutationthat mainly reduces activity towards the originalsubstrates. This could be advantageous if the in vivoconcentrations of the dicarboxylic a-keto and a-amino acids are great enough in toto so as to saturatethe enzyme partially. These alterations should be lessbeneficial in later rounds or in a natural TATase.
Evolution of TATase specificity in subfamilyIa aminotransferases
Aminotransferase subfamily Ia (also designatedas aminotransferase subfamily AT I, group a) com-prises a set of closely related AATases and TATasesincluding the two E. coli enzymes under presentconsideration.16,38 Figure 5 is a phylogram con-structed from amino acid sequence alignments ofsubfamily members. Eukaroytic AATases, with theexception of an outlier from Saccharomyces cerevisiae,cluster together. The rest of the tree representsenzymes found in proteobacteria. The phylogramindicates that the AATase/TATase divergenceoccurred twice, as indicated by the fences. AATasesare distributed throughout the major branches ofthe tree, suggesting that the AATase conserved resi-dues (sets AAT > TAT and AAT – TAT) were fixedrelatively early in evolutionary time. An evolution-ary model embodying an AATase ancestor is dis-cussed in the next paragraph. As AATases andTATases further evolved from the common ancestor,residues in set AAT > TAT remained constant forboth substrate specificities, and can be consideredessential for the reasons given above. The necessityto transaminate Asp and Glu with high catalytic effi-ciencies and concomitantly to restrict binding ofcompeting a-amino and a-keto acids, served to fixthe members of set AAT – TAT in AATases, whereasreplacements in TATases led to broadened specificity.Substitutions at other positions not conserved inAATases also contributed to the enhanced TATaseactivity. Jensen & Gu16 pointed out that differentcombinations of amino acid substitutions generateTATase specificity in the subfamily Ia framework.This is clearly illustrated in TATases for the tenAAT – TAT positions shown in Table 4. The pheno-type of a given substitution will generally be con-text-dependent. It is notable that replacementsN34D, T109S, G285S, and N297S occur in multipleevolved clones as well as in homologous TATases. Itis likely that these substitutions have high impactfor Phe reactivity and relatively low context depen-dence as has been established for T109S and N297S.39
† Of the 384 mutable positions, 164 are identical inE. coli AATase and TATase. A TATase residue can beaccessed by a single base change in AATase at 129 of the220 remaining positions. Thus, 129 equals the number ofamino acid changes out of 2200 possible (,6%) that areto residues in the E. coli TATase.
602 Evolution of L-Asp to L-Tyr Aminotransferase

Figure 6 depicts a landscape that accommodatesthe results from both natural and directedevolution based on a hypothetical model for evol-ution from an AATase-like ancestor. This modelwas chosen because AATase, unlike TATase, is con-stitutive and because aspartate is directly linked tothe TCA cycle and is utilized in the biosyntheticpathways for purines, pyrimidines, several aminoacids, and cofactors in E. coli†. Such an enzyme
may have possessed some TATase activity suffi-cient for low-level production of Tyr and Phe asneeded. It is also possible that the requisite TATaseactivity was provided by the ancestral histidinolphosphate aminotransferase (subfamily Ib) orbranched chain aminotransferase. Gene dupli-cation of the ancestral AATase followed by sub-strate specificity broadening produced the modernsubfamily Ia TATases, which are regulatedenzymes specialized for generation of aromaticamino acids. The directed evolution experimentinitiated with the modern E. coli AATase. Thefitness landscape shows multiple pathways lead-ing to different peaks with high TATase activity(Figure 6). It is evident that there are at least asmany sequences compatible with a given functionas there are organisms having this function, as allorthologs differ in sequence to some degree in thedifferent organisms.
Materials and Methods
Strains and plasmids
E. coli strain KB224 was a gift from Rog Yocum (Omni-Gene Bioproducts). Its genotype is DðpheA-tyrA-aroFÞ;DðargF-lacZYAÞU169; thi1, endA1, hsdR17, supE44. Selec-tion strains SR224 (additional modifications: hppþ;DtyrB < spcr; DaspC < tetr; DilvE < kanr), and SR250(additional modifications: hppþ; DtyrB < spcr; DaspC <tetr; DilvE < genr; recA < kanr) were derived from KB224as described below. Aminotransferase-deficient E. colistrain MG204 (gift from Ian Fotheringham, Nutrisweet
Figure 6. Contour representation of the results ofdirected and natural evolution of TATase activity. Themodern E. coli TATase gene (T), whose product has highactivity for both aromatic and dicarboxylic substrates,arose by gene duplication of an ancestor possessingmainly AATase activity ( p ). The AATase-like enzymecontinued to evolve, constrained in part by the impera-tive that it not acquire TATase activity, to point A. Thedirected evolution experiment began with the modernAATase (A), and the resulting evolved TATase is at thenearby peak D. There is no monotonically increasingpathway for acquisition of TATase activity leading frompoint A to T, but there is at least one from A to D; there-fore many of the beneficial amino acid substitutions innaturally and directly evolved TATases are different.The remaining peaks represent schematically some ofthe other naturally occurring TATase sequences (see thetext).
Figure 5. Phylogram of subfamily Ia amino-transferases. This tree is based on the multiple sequenceamino acid alignment generated with PILEUP. The treewas built with PAUP by applying the minimumevolutionary distance criterion and performing 1000replicate random heuristic searches. AATases annotated(c), (p), or (m) represent eukaroytic enzymes localized inthe cytosol, chloroplast, or mitochondria, respectively.The remaining enzymes are prokaryotic AATases andTATases found in proteobacteria. Entrez identificationnumbers are indicated. (a) Sources for these sequencesare given in Materials and Methods. (b) The assignmentof the X. fastidiosa gene product as a TATase is uncertain.It resembles a TATase at key positions such as 12, 109,297, and 396 but shares greater overall sequence identitywith AATases than with TATases. The coded geneproduct has not been characterized.
† An alternate model emanates from the “patchwork”hypothesis that postulates that modern enzymes evolvedfrom ancestors with broader specificities.33 According tothis view, the ancestral subfamily Ia aminotransferasewould have possessed both aromatic and dicarboxylicaminotransferase activity. This model is readilyaccommodated by small modifications to Figure 6.
Evolution of L-Asp to L-Tyr Aminotransferase 603

corporation) has the genotype his23; proB; trpA-605, lacI3;lacZ118; gyrA; rpsL; DaspC < kanr; tyrB; ilvE; recA : Tn10.Gene replacement vector pKO3 was a gift from GeorgeChurch. A pKO3/recA construct was prepared by KeithKoch. Plasmid pBSL141, containing the gentamicinresistance gene ðgentrÞ; and plasmids pBAD18 andpBAD33 were obtained from American Type CultureCollections. pNN602S,40 containing the spectinomycinresistance gene ðspcrÞ derived from pHP45V,41 was a giftfrom Hiroshi Nikaido. pUC4K, containing a kanamycinresistance gene ðkanrÞ; was purchased from AmershamPharmacia. pBR322, containing a tetracycline resistancegene ðtetrÞ; was purchased from New England Biolabs.Plasmids for E. coli AATase mutant T109S/N297S andTATase mutant retroHex were prepared by Tinh Luong.pMIASPC, consisting of the WT AATase gene clonedinto pBAD18, was constructed by Meghan Imrie.
Construction of KB224 derivative KBHPP2
The method described by Gelfand and Steinberg23 wasapplied to generate a KB224 derivative capable of utiliz-ing exogenous hydroxyphenylpyruvate (HPP) for con-version to Tyr within the cell. KB224 overnight cultureswere plated on tyrosine-free selection media (see belowfor a description of selection media) freshly spread with100 ml of 10 mg/ml HPP. The fastest growing colonywas isolated and grown in liquid culture. Cells werethen plated on selective media spread with 100 ml ofHPP at 3 mg/ml. HPP competent strain KBHPP2 wasisolated and used for subsequent modifications.
Deletion of tyrB (TATase gene) in KBHPP2 togenerate KBDTAT
Knockouts of tyrB and other genes were prepared bymodification of the procedure described by Link et al.42
in which deletions are integrated into the E. coli chromo-some with the gene replacement vector pKO3. Initially,120 bp of a tyrB derivative, retroHex, in vector pUC118was replaced with a spectinomycin resistance ðspcrÞgene. The spcr marker was amplified from plasmidpNN602S with primers 50-AAAGGTACCTTATTATTATTTGCCGACTACCTTGTTG-30 and 50-AAACCAT GGCGCTCGTTCGCCAGCC-30 and cloned into the KpnI andNcoI sites of retroHex. These restriction sites had beenengineered into the tyrB gene near DNA encodingamino acid positions 69 and 109. This construct servedas the starting material for a complete tyrB deletion.Chromosomal DNA fragments upstream and down-stream of tyrB were amplified from KB224 genomicDNA with the primer sets 50-CCGGAATTCACGCAATATCTATATCGATGACTCCTC-30/50-AAACCATGGGAGTGAGACTCATGACAAACGTACATC-30 and 50-AAAGGTACCCCCAATTATCTGGAATTCCTTATCCTG-30/50-AACCCAAGCTTGCGATTTTGTATTTTGCCCTGG-3 0
respectively. tyrB DNA in the retroHex/spcr constructwas replaced by the upstream chromosomal and down-stream fragments with restriction enzymes EcoRI/NcoIand KpnI/HindIII, respectively. The resulting constructcontains the spcr marker flanked by chromosomalfragments corresponding to regions upstream anddownstream of tyrB. This was amplified with primers50-AACGCGGATCCCCGTGCATAACGAAGAACAGC-30 and 50-AACGCCCCGGGCACTG ATTGCCTGTGTGATCTT-30 and cloned into the XmaI and BamHI sites ofpKO3 to generate TATase replacement plasmidpDtyrB=KO3:
Gene replacement with pDtyrB/KO3 in strainKBHPP2 was performed following the protocoldescribed by Link et al.42 The deletion of tyrB in spectino-mycin-resistant derivatives was confirmed by PCRanalysis of genomic DNA and comparisons of growthrates on tyrosine-free media supplemented with HPP(selection media described below). The resultant strainKBDTAT was employed for deletion of the aspC gene.
Deletion of aspC (AATase gene) in KBDTAT togenerate KBDAAT
The deletion was prepared by crossover PCR asdescribed by Link et al.42 with modifications. The follow-ing primers were used: (1) 50-AACGC GGATC CGGAAAAAG AAGAT GAGTT CTGG CTG-30, (2) 50-TTGCCCCTGCAG TAAAC TTAATGGC TCCG CTGTGCGAAG-30, (3) 50-TTAAGTTTACTGCAG GGGCAAGAC GTGAG ATTGC TCTGG-30, (4) 50-AACGCGGAT CCTGAA CTTCGC TGTTC AGTAC CTG-30.
Primers (1) and (2) were employed to clone the,700 bp N-terminal flanking region of aspC. Primers (3)and (4) were used to clone a ,700 bp C-terminal region.A Pst I site, located between the two fused regions inthe final 1.4 kb product, is underlined in primers (2) and(3). The 1.4 kb fragment was cloned into the BamHI siteof pKO3. A tetracycline resistance marker was amplifiedfrom pBR322 with primers 50-AAA ACTG CAGG GAATAAGG GCGA CAC GG-30 and 50-AAAA CTG CAGCGTATC GGTGA TTCATT CTGCT AACC-30 and clonedinto the Pst I site. The resulting plasmid, pDaspC/KO3,was used for gene replacement in strain KBDTAT. PCRanalysis of tetracycline-resistant colonies was performed.The resulting strain KBDAAT was auxotrophic for aspar-tate, consistent with previous reports for E. coli strainslacking both AATase and TATase.23
Deletion of ilvE (branched chain aminotransferasegene) in KBDAAT to generate SR224 and SR230
The ilvE deletion was prepared in a manner similar tothe aspC knockout. Primers were: (1) 50-CGCGGATCCGCC GTTGT TGTTAA AACAAC TGTC-30, (2) 50-CCCATC CTGCAG ACTAG TCGAC CGAC CGTGGGC TG-30, (3) 50-TCG ACTAGT CTGCAG GATGGGTC TTCA CTGG CGAA ACCGA-30,(4) 50-CGCGGAT CCGA GTTA GCGG TAAA CATC CCG GAG-30.
The underlined sequence is a PstI site that is flankedby the upstream and downstream fragments in the final1.5 kb product, which was cloned into the BamHI site ofpKO3. Two ilvE knockouts were derived from the result-ing construct. A kanamycin resistance gene was excisedfrom pUK4 and cloned into the PstI site to generatepDilvE/KO3(kan r). pDilvE/KO3(gentr), was prepared ina similar manner employing a gentamicin resistancemarker from plasmid pBSL141. Gene replacement inKBDAAT was performed with pDilvE/KO3(kanr) to gen-erate SR224. The pDilvE/KO3(gentr) plasmid was usedto generate SR230. Both strains are leucine auxotrophs.
Inactivation of recA in SR230 to generate SR250
The kanr marker from pUC4K was cloned into the PstIsite of pKO3/recA to generate a recA inactivation plas-mid. Inactivation of recA in SR230 was confirmed byPCR analysis and UV sensitivity. The resultant strainSR250 was employed in directed evolution.
604 Evolution of L-Asp to L-Tyr Aminotransferase

Library construction
DNA shuffling was performed following the methoddescribed by Stemmer43 as modified by Lorimer &Pastan.44 The AATase gene was amplified frompMIASPC or evolved variants with primers 50-GCGTAACAA AAGT GTCT ATATC ACGG CAG-30 and 50-CATCG GCGC TACG GCGT TTC-30. DNA was sub-sequently digested with DNase I in 0.1 mM MnCl2.Fragments of fewer than 250 bp were gel-purified. Re-assembly was performed with Taq polymerase in rounds1–6 and Pfu polymerase in rounds 7, 8, and in the 8-2Bbackcross. The AATase coding sequence was amplifiedfrom the re-assembly with 50-AAAG GTACC GGAGTGC CTC GTCATG TTTGA GAACA TTA CCG-30 andeither 50-AAA AGCA TGCT TATT AGTG ATGG TGATGGTG ATGC AGCA CTG CCA CAAT CGC-30 or50AAAG CATG CTTA TTAT TACA GCAC TGCCACAA TCGC-30. The former of the final two primersencodes a 6£ histidine tag. That primer was used onlyin rounds 1–4. The latter primer without the tag wasused for the remaining rounds. The AATase gene wascloned into the KpnI and SphI sites of pBAD18. pBAD33was used in rounds 8 and the 8-2/WT AATase backcrossin order to reduce expression levels. Ligated DNA wastransformed into selection strain SR250. SR224 was orig-inally employed, but smeared DNA from positivesselected with this RecAþ strain prompted the switch toRecA2 strain SR250. Library sizes were typically of theorder of 100,000 clones.
Genetic selection
Transformants were washed in M9 salts and plated onselective media based on M9c as described by Kast et al.45
Media consisted of M9 minimal salts (Sigma), thiamine(10 mg/ml), 4-aminobenzoic acid (5 mg/ml), 4-hydroxy-benzoic acid (5 mg/ml), 2,3-dihydroxybenzoate (1.6 mg/ml), 0.4% (v/v) glycerol or 0.2% (w/v) glucose, 0.1 mMCaCl2, 2 mM MgSO4, Bactoagar (15 g/l), and either ampi-cillin (100 mg/ml) or chloramphenicol (20 mg/ml).Amino acids other than Phe or Tyr were typically sup-plemented at a concentration of 50 mg/ml. Arabinosewas added at 0.1% (w/v) for AATase expression. Fucose,an antagonist of arabinose, was added at concentrationsup to 0.3% (w/v) to increase stringency. Selection platescontained either HPP (5–120 mg/ml)/Phe (50 mg/ml) orPP(5–120 mg/ml)/Tyr (50 mg/ml). A total of 50–100colonies were picked from selection plates and trans-ferred to one or two LB plates containing either ampi-cillin (100 mg/ml) or chloramphenicol (20 mg/ml). Cellswere grown overnight, scraped off the plates with 4 mlof sterile water, and centrifuged. Plasmid DNA preparedfrom the cell pellets was used for the next round of DNAshuffling.
Microtiter plate assays
Round 7 and 8 positives were inoculated into 96-wellblocks (Qiagen) containing 1 ml of selection media (asabove, but supplemented with 0.2% arabinose and bothPhe and Tyr at 50 mg/ml instead of PP or HPP). Cellsfrom overnight cultures were pelleted and lysed in Cell-Lytice B II (Sigma) containing DNase I. Aromatic amino-transferase activity in crude extracts was determinedwith the HO-HxoDH coupled assay.46 Assays monitoreda decrease in absorbance at 340 nm in a SPECTRAmax250 microplate spectrophotometer (Molecular Devices).Reactions (200 ml) contained 200 mM Taps (pH 8.0),
150 mM NADH, 140 mM KCl, 20 mM PLP, 200 mM Tyr,and 20 mM aKG. Variants with the highest rates werere-assayed at additional Tyr concentrations to evaluateVmax and Km.
Site-directed mutagenesis and DNA sequencing
SRHEPT was derived from AATase mutant T109S/N297S through PCR-based site-directed mutagenesis.Mutagenic fragments were ligated into pUC119.Sequences of site-directed mutants and evolved cloneswere determined by automated DNA sequencing (UCBerkeley DNA Sequencing Facility or Elim Biopharma-ceuticals, Hayward, CA).
Enzyme purification
WT TATase, WT AATase, and AATase variants wereoverexpressed in aminotransferase-deficient strainMG204. Cells transformed with WT enzymes or site-directed mutants were grown ,30 hours in 2YT contain-ing 0.1% (w/v) pyridoxine and ampicillin (100 mg/ml).Cells transformed with evolved clones were grown,18 hours in M9 minimal salts (Sigma) supplementedwith thiamine (10 mg/ml), 0.1% pyridoxine, 0.4% gly-cerol, 0.4% arabinose, a complete set of amino acids(50 mg/ml), 0.1 mM CaCl2, 1 mM MgSO4, and eitherampicillin (100 mg/ml) or chloramphenicol (20 mg/ml).Harvested cells were lyzed in a French press and centri-fuged. Protein was purified from the supernatant follow-ing the protocol described by Herold & Kirschner,47 withmodifications by Onuffer & Kirsch.9
Steady-state kinetics
Transamination of (L-Phe, L-Tyr, and L-Leu), L-Asp,and L-Ala was monitored in HO-HxoDH,46 MDH,48 orLDH coupled assays, respectively, with an HP 8453spectrophotometer. Saturating levels of aKG weremaintained. See Tables 1 and 3 for details.
Sequence alignments and phylogenetic analyses
The sequences that are included in Table 4 and Figure5 were selected to generate a diverse set that was notheavily weighted to a particular group of closely relatedhomologs. All clones sharing 70% or higher amino acidsequence identity with a previously chosen orthologwere excluded. Additional available eukarotyic AATases(55–70% sequence identity with clones already included)were excluded in order to present a balanced distri-bution of eukaroytic and prokaryotic AATases. Thesequence data for B. pertussis AATase were produced bythe Bordetella pertussis Sequencing Group at the SangerInstitute†. Preliminary sequence data for enzymes fromR. metallidurans, R. sphaeroides, and B. fungorum wereobtained from The DOE Joint Genome Institute (JGI)‡.Preliminary sequence data for enzymes from S. putrefa-ciens and P. syringae were obtained from The Institutefor Genomic Research website§. The amino acidsequence for the TATase from Z. mobilis is from Jensen& Gu.16
† http://www.sanger.ac.uk/Projects/B_pertussis/‡ http://www.jgi.doe.gov/tempweb/JGI_microbial/
html/index.html§ http://www.tigr.org
Evolution of L-Asp to L-Tyr Aminotransferase 605

A multiple sequence alignment was constructed withPILEUP from the GCG suite of programs. Conservedpositions in sets AAT – TAT; AAT > TAT; andTAT – AAT were identified with the program venn.out.The phylogram was generated with PAUP in the GCGsuite and displayed in Treeview.
Numerical simulation
Subsets of the 384 AATase positions available formutation were assembled with the random number gen-erator Mersenne Twister.49 The number of members ineach was equal to the number of elements in AAT – TATor in AAT > TAT: These in turn were defined by the cho-sen conservation criteria. A typical simulation isdescribed: at 75% sequence identity 32 positions belongto set AAT – TAT; while 82 are in set AAT > TAT (Figure4). For rounds 7 and 8, 42 out of the 158 substitutions (67positions mutated in 11 sequenced clones) were found inset AAT – TAT: The 67 positions in which mutationswere found were weighted in proportion to the numberof mutations occurring at each site in the 11 sequencedclones. Residues in random sets were compared withthe 67 mutated positions. Random sets were scored andcompared to set AAT – TAT to determine the percentagethat would contain 42 or more substitutions. They werealso compared with set AAT > TAT to determine thepercentage that contains four or fewer and with setTAT – AAT to find the percentage containing 0substitutions.
Acknowledgements
We thank Dan Malashock for numerical simu-lations and for writing the venn.out program foridentifying conserved residues. He, Andrew Eliot,and Ichiro Matsumura provided critical reviews.We are grateful to Kathryn McElroy for assistancewith analysis software. We thank Francis Arnoldfor helpful advice and Keith Koch for assistancewith DNA shuffling. The unpublished sequencesused in alignments and in phylogenetic analysiswere from The DOE Joint Genome Institute (JGI)†.The Institute for Genomic Research website§, andthe Sanger Institute‡. This work was supported byNIH grant GM-35393. S.C.R. was supported, inpart, by the Applied Biology Bioprocess Engineer-ing Research Training Grant and was a HowardHughes Medical Institute Predoctoral Fellow.
References
1. Ballinger, M. D., Tom, J. & Wells, J. A. (1995). Design-ing subtilisin BPN0 to cleave substrates containingdibasic residues. Biochemistry, 34, 13312–13319.
2. Ballinger, M. D., Tom, J. & Wells, J. A. (1996).Furilisin: a variant of subtilisin BPN0 engineered for
cleaving tribasic substrates. Biochemistry, 35,13579–13585.
3. Chen, R., Greer, A. & Dean, A. M. (1995). A highlyactive decarboxylating dehydrogenase with ration-ally inverted coenzyme specificity. Proc. Natl Acad.Sci. USA, 92, 11666–11670.
4. Wilks, H. M., Hart, K. W., Feeney, R., Dunn, C. R.,Muirhead, H., Chia, W. N. et al. (1988). A specific,highly active malate dehydrogenase by redesign ofa lactate dehydrogenase framework. Science, 242,1541–1544.
5. Hedstrom, L., Szilagyi, L. & Rutter, W. J. (1992). Con-verting trypsin to chymotrypsin: the role of surfaceloops. Science, 255, 1249–1253.
6. Onuffer, J. J. & Kirsch, J. F. (1995). Redesign of thesubstrate specificity of Escherichia coli aspartateaminotransferase to that of Escherichia coli tyrosineaminotransferase by homology modeling and site-directed mutagenesis. Protein Sci. 4, 1750–1757.
7. Mavrides, C. & Orr, W. (1975). Multispecificaspartate and aromatic amino acid amino-transferases in Escherichia coli. J. Biol. Chem. 250,4128–4133.
8. Fotheringham, I. G., Dacey, S. A., Taylor, P. P., Smith,T. J., Hunter, M. G., Finlay, M. E. et al. (1986). Thecloning and sequence analysis of the aspC and tyrBgenes from Escherichia coli K12. Comparison of theprimary structures of the aspartate aminotransferaseand aromatic aminotransferase of E. coli with thoseof the pig aspartate aminotransferase isoenzymes.Biochem. J. 234, 593–604.
9. Onuffer, J. J., Ton, B. T., Klement, I. & Kirsch, J. F.(1995). The use of natural and unnatural amino acidsubstrates to define the substrate specificitydifferences of Escherichia coli aspartate and tyrosineaminotransferases. Protein Sci. 4, 1743–1749.
10. Hayashi, H., Inoue, K., Nagata, T., Kuramitsu, S. &Kagamiyama, H. (1993). Escherichia coli aromaticamino acid aminotransferase: characterization andcomparison with aspartate aminotransferase.Biochemistry, 32, 12229–12239.
11. Oue, S., Okamoto, A., Nakai, Y., Nakahira, M.,Shibatani, T., Hayashi, H. & Kagamiyama, H. (1997).Paracoccus denitrificans aromatic amino acid amino-transferase: a model enzyme for the study of dualsubstrate recognition mechanism. J. Biochem. 121,161–171.
12. Zhao, G., Xia, T., Song, J. & Jensen, R. A. (1994).Pseudomonas aeruginosa possesses homologues ofmammalian phenylalanine hydroxylase and 4alpha-carbinolamine dehydratase/DCoH as part ofa three-component gene cluster. Proc. Natl Acad. Sci.USA, 91, 1366–1370.
13. Alfano, J. R. & Kahn, M. L. (1993). Isolation andcharacterization of a gene coding for a novel aspar-tate aminotransferase from Rhizobium meliloti.J. Bacteriol. 175, 4186–4196.
14. Watson, R. J. & Rastogi, V. K. (1993). Cloning andnucleotide sequencing of Rhizobium meliloti amino-transferase genes: an aspartate aminotransferaserequired for symbiotic nitrogen fixation is atypical.J. Bacteriol. 175, 1919–1928.
15. Gu, W., Song, J., Bonner, C. A., Xie, G. & Jensen, R. A.(1998). PhhC is an essential aminotransferase foraromatic amino acid catabolism in Pseudomonasaeruginosa. Microbiology, 144, 3127–3134.
16. Jensen, R. A. & Gu, W. (1996). Evolutionary recruit-ment of biochemically specialized subdivisions of
† http://www.jgi.doe.gov/tempweb/JGI_microbial/html/index.html
‡ http://www.sanger.ac.uk/
606 Evolution of L-Asp to L-Tyr Aminotransferase

Family I within the protein superfamily of amino-transferases. J. Bacteriol. 178, 2161–2171.
17. Petrounia, I. P. & Arnold, F. H. (2000). Designedevolution of enzymatic properties. Curr. Opin.Biotechnol. 11, 325–330.
18. Stemmer, W. P. (1994). Rapid evolution of a protein invitro by DNA shuffling. Nature, 370, 389–391.
19. Oue, S., Okamoto, A., Yano, T. & Kagamiyama, H.(1999). Redesigning the substrate specificity of anenzyme by cumulative effects of the mutations ofnon-active site residues. J. Biol. Chem. 274,2344–2349.
20. Yano, T., Oue, S. & Kagamiyama, H. (1998). Directedevolution of an aspartate aminotransferase withnew substrate specificities. Proc. Natl Acad. Sci. USA,95, 5511–5515.
21. Matsumura, I. & Ellington, A. D. (2001). In vitro evol-ution of beta-glucuronidase into a beta-galactosidaseproceeds through non-specific intermediates. J. Mol.Biol. 305, 331–339.
22. May, O., Nguyen, P. T. & Arnold, F. H. (2000).Inverting enantioselectivity by directed evolutionof hydantoinase for improved production ofL-methionine. Nature Biotechnol. 18, 317–320.
23. Gelfand, D. H. & Steinberg, R. A. (1977). Escherichiacoli mutants deficient in the aspartate and aromaticamino acid aminotransferases. J. Bacteriol. 130,429–440.
24. Kawaguchi, S., Nobe, Y., Yasuoka, J., Wakamiya, T.,Kusumoto, S. & Kuramitsu, S. (1997). Enzyme flexi-bility: a new concept in recognition of hydrophobicsubstrates. J. Biochem. 122, 55–63.
25. Malashkevich, V. N., Onuffer, J. J., Kirsch, J. F. &Jansonius, J. N. (1995). Alternating arginine-modu-lated substrate specificity in an engineered tyrosineaminotransferase. Nature Struct. Biol. 2, 548–553.
26. Okamoto, A., Nakai, Y., Hayashi, H., Hirotsu, K. &Kagamiyama, H. (1998). Crystal structures ofParacoccus denitrificans aromatic amino acid amino-transferase: a substrate recognition site constructedby rearrangement of hydrogen bond network. J. Mol.Biol. 280, 443–461.
27. Mavrides, C. & Christen, P. (1978). Mitochondrialand cytosolic aspartate aminotransferase fromchicken: activity toward aromatic amino acids.Biochem. Biophys. Res. Commun. 85, 769–773.
28. Pan, Q. W., Tanase, S., Fukumoto, Y., Nagashima, F.,Rhee, S., Rogers, P. H. et al. (1993). Functional rolesof valine 37 and glycine 38 in the mobile loop ofporcine cytosolic aspartate aminotransferase. J. Biol.Chem. 268, 24758–24765.
29. Taniguchi, M., Kobe, A., Kato, M. & Sugiyama, T.(1995). Aspartate aminotransferase isozymes inPanicum miliaceum L., an NAD-malic enzyme-typeC4 plant: comparison of enzymatic propertiesprimary structures, and expression patterns. Arch.Biochem. Biophys. 318, 295–306.
30. Yagi, T., Kagamiyama, H. & Nozaki, M. (1982).Aspartate: 2-oxoglutarate aminotransferase frombakers’ yeast: crystallization and characterization.J. Biochem. 92, 35–43.
31. Hayashi, H., Kuramitsu, S. & Kagamiyama, H.(1991). Replacement of an interdomain residueVal39 of Escherichia coli aspartate aminotransferaseaffects the catalytic competence without altering thesubstrate specificity of the enzyme. J. Biochem. 109,699–704.
32. Kohler, E., Seville, M., Jager, J., Fotheringham, I.,Hunter, M., Edwards, M. et al. (1994). Significant
improvement to the catalytic properties of aspartateaminotransferase: role of hydrophobic andcharged residues in the substrate binding pocket.Biochemistry, 33, 90–97.
33. Jensen, R. A. (1976). Enzyme recruitment inevolution of new function. Annu. Rev. Microbiol. 30,409–425.
34. Bain, L. J. & Engelhardt, M. (1987). Introduction toProbability and Mathematical Statistics, PWS-KENTPublishing Company, Boston.
35. Kuchner, O. & Arnold, F. H. (1997). Directedevolution of enzyme catalysts. Trends Biotechnol. 15,523–530.
36. Gloss, L. M., Spencer, D. E. & Kirsch, J. F. (1996).Cysteine-191 in aspartate aminotransferases appearsto be conserved due to the lack of a neutral mutationpathway to the functional equivalent, alanine-191.Proteins: Struct. Funct. Genet. 24, 195–208.
37. Oue, S., Okamoto, A., Yano, T. & Kagamiyama, H.(2000). Cocrystallization of a mutant aspartateaminotransferase with a C5-dicarboxylic substrateanalog: structural comparison with the enzyme-C4–dicarboxylic analog complex. J. Biochem. 127,337–343.
38. Mehta, P. K. & Christen, P. (2000). The molecularevolution of pyridoxal-50-phosphate-dependentenzymes. Advan. Enzymol. Relat. Areas Mol. Biol. 74,129–184.
39. Luong, T. N. & Kirsch, J. F. (2001). A general methodfor the quantitative analysis of functional chimeras:applications from site-directed mutagenesis andmacromolecular association. Protein Sci. 10, 581–591.
40. Ma, D., Alberti, M., Lynch, C., Nikaido, H. & Hearst,J. E. (1996). The local repressor AcrR plays amodulating role in the regulation of acrAB genes ofEscherichia coli by global stress signals. Mol. Microbiol.19, 101–112.
41. Prentki, P. & Krisch, H. M. (1984). In vitro insertionalmutagenesis with a selectable DNA fragment. Gene,29, 303–313.
42. Link, A. J., Phillips, D. & Church, G. M. (1997).Methods for generating precise deletions and inser-tions in the genome of wild-type Escherichia coli:application to open reading frame characterization.J. Bacteriol. 179, 6228–6237.
43. Stemmer, W. P. (1994). DNA shuffling by randomfragmentation and reassembly: in vitro recombina-tion for molecular evolution. Proc. Natl Acad. Sci.USA, 91, 10747–10751.
44. Lorimer, I. A. & Pastan, I. (1995). Random recombi-nation of antibody single chain Fv sequences afterfragmentation with DNaseI in the presence of Mn2þ.Nucl. Acids Res. 23, 3067–3068.
45. Kast, P., Asif-Ullah, M., Jiang, N. & Hilvert, D. (1996).Exploring the active site of chorismate mutase bycombinatorial mutagenesis and selection: the import-ance of electrostatic catalysis. Proc. Natl Acad. Sci.USA, 93, 5043–5048.
46. Luong, T. N. & Kirsch, J. F. (1997). A continuouscoupled spectrophotometric assay for tyrosineaminotransferase activity with aromatic and othernonpolar amino acids. Anal. Biochem. 253, 46–49.
47. Herold, M. & Kirschner, K. (1990). Reversibledissociation and unfolding of aspartate amino-transferase from Escherichia coli: characterization of amonomeric intermediate. Biochemistry, 29,1907–1913.
48. Gloss, L. M. & Kirsch, J. F. (1995). Decreasing thebasicity of the active site base, Lys-258, of Escherichia
Evolution of L-Asp to L-Tyr Aminotransferase 607

coli aspartate aminotransferase by replacement withgamma-thialysine. Biochemistry, 34, 3990–3998.
49. Matsumoto, M. & Nishimusa, T. (1998). MersenneTwister: a 623-dimensionally equidistributeduniform pseudorandom number generator. ACMTrans. Modeling Comput. Simul. 8, 3–30.
50. Mehta, P. K., Hale, T. I. & Christen, P. (1989). Evol-utionary relationships among aminotransferases.Tyrosine aminotransferase, histidinol-phosphate
aminotransferase, and aspartate aminotransferaseare homologous proteins. Eur. J. Biochem. 186,249–253.
51. Shaffer, W. A., Luong, T. N., Rothman, S. C. & Kirsch,J. F. (2002). Quantitative chimeric analysis of sixspecificity determinants that differentiate Escherichiacoli aspartate from tyrosine aminotransferase. ProteinSci. 11, 2848–2859.
Edited by P. Wright
(Received 2 August 2002; received in revised form 6 December 2002; accepted 13 January 2003)
608 Evolution of L-Asp to L-Tyr Aminotransferase