homogeneous condensation in a vapour/gas mixture at high ... · homogeneous condensation in a...
TRANSCRIPT

Homogeneous condensation in a vapour/gas mixture at highpressures in an expansion cloud chamberCitation for published version (APA):Muitjens, M. J. E. H. (1996). Homogeneous condensation in a vapour/gas mixture at high pressures in anexpansion cloud chamber. Eindhoven: Technische Universiteit Eindhoven. https://doi.org/10.6100/IR471316
DOI:10.6100/IR471316
Document status and date:Published: 01/01/1996
Document Version:Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers)
Please check the document version of this publication:
• A submitted manuscript is the version of the article upon submission and before peer-review. There can beimportant differences between the submitted version and the official published version of record. Peopleinterested in the research are advised to contact the author for the final version of the publication, or visit theDOI to the publisher's website.• The final author version and the galley proof are versions of the publication after peer review.• The final published version features the final layout of the paper including the volume, issue and pagenumbers.Link to publication
General rightsCopyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright ownersand it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights.
• Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal.
If the publication is distributed under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license above, pleasefollow below link for the End User Agreement:www.tue.nl/taverne
Take down policyIf you believe that this document breaches copyright please contact us at:[email protected] details and we will investigate your claim.
Download date: 30. Mar. 2020


Homogeneaus condensation in a vapour /gas mixture at high pressures
in an expansion cloud chamber

Copyright @1996 M.J.E.H. Muitjens Omslagontwerp: Ben Mobach, TUE Druk: Universiteitsdrukkerij, TUE
Muitjens, Marcel Johannes Elisabeth Hubertus
Homogeneous condensation in a vapour I gas mixture at high pressures in an expansion cloud chamber I Marcel Muitjens. Eindhoven: Eindhoven University of Technology Proefschrift Eindhoven. - Met lit. opg. ISBN 90-386--0199-9

Homogeneons condensation in a vapour /gas mixture at high pressures
in an expansion cloud chamber
PROEFSCHRIFT
ter verkrijging van de graad van doctor aan de Technische Universiteit Eindhoven, op gezag van de Rector Magnificus, prof. dr. M. Rem, voor een commissie aangewezen door het College van Dekanen in het openbaar te verdedigen op
dinsdag 17 december 1996 om 16.00 uur
door
Marcel Johannes Elisabeth Hubertus Muitjens
geboren te Nuth

Dit proefschrift is goedgekeurd door de promotoren:
Prof.dr.ir. M.E.H. van Dongen en
Prof.dr.ir. G.J.F. van Heijst.
This work was supported by Grant No. ETN 00.2347 of the Netherlands Foundation for Fundemental Research on Matter (FOM).

Voor mijn ouders

CONTENTS
1 Introduetion 1 1 3
1.1 General introduetion 1.2 Homogeneons nucleation at high pressures .................... .
1.2.1 Unary nucleation in an ideal vapour/gas mixture and the effect of total pressure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.2.2 Nucleation in binary mixtures of real gases . . . . . . . . . . . . . . . . . 8 1.3 Homogeneons condensation due to a continuons adiabatic expansion . . . . . . . 10 1.4 Homogeneaus condensation of natura! gas due to a continuons adiabatic expansion 11 1.5 Thesis overview . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
2 Homogeneons Condensation 17 2.1 Unary nucleation theory . . . . . . . . . 18
2.1.1 Classical nucleation theory . . . . 19 2.1.2 The size of a newly born droplet . 23 2.1.3 A Semi-phenomenological theory 25
2.2 Binary nucleation theory . . . . . . . . . 27 2.2.1 Binary classical nucleation theory 28 2.2.2 Quasi-one-component theory . . . 31
2.3 Droplet growth in binary mixtures of real gases 33 2.3.1 The Droplet Growth Model . . . . . . . 35
3 Homogeneons condensation due to a continuons adiabatic expansion 43 3.1 Governing equations . . . . . . . . . . . . . . . . . . . . 44 3.2 Light scattering by small spherical particles: Mie-theory 46 3.3 Numerical method . 49 3.4 Asymptotic methad . 55
4 Experimental metbod 67 4.1 The expansion cloud chamber 67
4.1.1 The experimentalset-up 67 4.1.2 Opties . . . . . . . . . . 70
4.2 Experimental procedure .... 4.2.1 Initia! composition of the vapour/gas mixture
4.3 A typkal experimént . . . . . . . . . . . . . . . .
71 73
75

i i Contents
5 Nucleation rate data from homogeneons condensation experimentsin an ex-pausion cloud chamber 5.1 Homogeneaus condensation of water vapour
5.1.1 Results for water/helium .. 5.1.2 Results for water/nitrogen ..... . 5.1.3 Discussion and conclusions ..... .
5.2 Homogeneaus condensation of n-nonane/methane 5.2.1 Initial composition of the n-nonane/methane mixture 5.2.2 Results for n-nonane/methane . . . . 5.2.3 Discussion and condusion ..... .
5.3 A Gaussian Model for the Nucleation Pulse . 5.3.1 The Gaussian model ..... . 5.3.2 Results: Numerical Simulations 5.3.3 Results: Experiments . . . . 5.3.4 Discussion a.nd conclusions .
6 Discussion and Conclusions
A Thermadynamie and Physical properties
B Binary Classical Nucleation Theory
C Convergence of the numerical solution
D Experimental data
E Enhanced solubility of water vapour in air, oxygen and nitrogen
Symbols
Summary
Samenvatting
Nawoord
Curriculum Vitae
77 77 81 84 87 88 89 90 94 95 96 97 99
101
105
107
118
117
119
128
125
129
183
137
139

Chapter 1
lNTRODUCTION
1.1 General introduetion
Homogeneous condensa.tion is the non-equilibrium vapour-to-liquid phase transition in the a.bsence of foreign particles, e.g. ions or dust partides. It is a two stage process: during the nucleation process, stabie nuclei of only a sma.ll number of molecules are formed, which then grow to macroscopie droplets.
A vapour can be brought into a thermodyna.mic state of non-equilibrium, by subjecting it to a sudden change in temperature and pressure, e.g. an adiabatic expansion. At such a state the vapour pressure p., is higher than the saturation pressure Pvs at the sa.me temperature T, and in thermodynamic equilibrium a liquid phase would a.lready exist. Such a non-equilibrium state is referred to as supersaturated and corresponds toa saturation ratio S, defined as S = Pvfp.,.(T), which exceeds unity.
In tigure 1.1 a p T diagram is shown fora vapour component. The saturation pressure as well as the adiabatic expansion of an initia.lly undersaturated vapour ( S < 1) are shown. As the expansion continues, the saturation curve (S = 1) is passed and the vapour becomes supersaturated. With ongoing expansion homogeneons condensation airoost instantaneously ends the supersaturated state.
This state of non-equilibrium has to be attained before the phase transition starts, due to the fact that a droplet surface induces an energy harrier for the formation of the drop let. The height of this energy harrier decreà.ses with increasing saturation ratio S. Once a droplet is formed, energy is gained by increasing its size. Therefore, the droplet will grow until thermodyna.mic equilibrium is established.
The vapour may consist of more than one component and a gas1 component may be present, which in general affects the equilibrium state. Therefore, a more general definition of S is
S Yv/Yvs(P, T) (1.1)
Here Yv is the molar fraction of a vapour component and Yvs the molar fraction of the vapour component in the gaseous phase in thermodyna.mic equilibrium with a liquid phase at
component is referred to as a gas component when îts crîtîcal temperature is less than the actual temperature. Otherwise the component is referred to as a vapour component.
1

2
Uquid
8<1
8 =
1 ~---/rr;C state
1-----s_,.--,-1 ~--~-
Vapeur
T
Introduetion
Figure 1.1: p-T diagram of a vapour component. Solid line Pvs(T), the vapourliquid equilibrium curve or saturated vapour pressure. Dotted line, an adiabatic expansion of an initially undersaturated vapour.
total pressure p and temperature T. For a mixture of an i deal inert gas component and an ideal vapour component expression (1.1) is equivalent to the ratio of the partial vapour pressure and the saturation vapour pressure S p"fp".(T).
Starting with Wilsou (1897), homogeneous condensation has been stuclied as an important process in many fields of applications. Wilson's expansion cloud chamber developed into a valuable detection system in high energy physics (Ehrler 1988). Homogeneaus condensation of water and water-acid mixtures is stuclied as an important process in the formation of rain and acid rain (Mirabel and Clavelin 1978, Wyslouzil et al. 1991, Rudolf 1994). It plays a role in weather forecast as well as environmental technology. Smoke gas deaning by means of condensate separation is another application of dropwise condensation in the field of environmental technology.
The work presented in this thesis originates from the petrochemical industry: the production, transport and handling of natura! gas.
Natura! gas is found at pressures up to 200 bar and at temperatures around 80 oe (353 K). It consists of many components, e.g. methane, nitrogen, carbon dioxide, and heavy hydrocarbons in very low concentrations, such as n-nonane and n-deca.ne. We, as consumers, rec~ive the natura! gas at a pressure a.bout 200 mbar a.bove ambient pressure. So, by the necessary pressure reductions, homogeneons condensation of the heavy hydrocarbons is to be expected, and is used for separation purposes.
The formation of such condensate in natura! gas leads to a. decrease of the quality of the gas and to increased rnainterrance of compressors and pipelines. Knowledge a.nd understanding of the nucleation and droplet growth processes in natura.! gas, enables engineers to take them into account while designing pipeline-systems.
The thermodynamic beha.viour of natura! gas differs completely from that of a mixture of an ideal gas a.nd an ideal vapour. The interactions of the molecules of the various components in

1.2 Homogeneaus nucleation at high pressures 3
natural gas can no langer be neglected for pressures above a few bar. The real gas behaviour of the mixture dominates its thermodynamics. The vapour-liquid equilibrium behaviour changes drastically and the composition of bath phases depends strongly on temperature and total pressure.
To avoid the complexity of the composition of natura! gas, we model it by a binary mixture of methane and a heavy hydrocarbon, n-nonane. This is a reasonable description due to the similar vapour-liquid equilibrium behaviour of both mixtures, as will be shown insection 1.4.
Mixtures of real gases becarne the subject of investigations on homogeneaus condensation rather recently. Our preliminary study on the homogeneaus condensation behaviour of natura! gas ( Muitjens et al. 1994) and the work of Looijmans (1995) on homogeneaus nucleation in n-nonane/methane and n-octane/methane mixtures are the first experimental studies reported in the literature.
Almost all other studies on homogeneons condensation reported in the literature concern mixtures of one (Allen and Kassner Jr. 1969, Viisanen et al. 1993, Hung et al. 1989) or two vapour components (Flageollet-Daniel et al. 1983, Zahoransky and Peters 1985) and a gas component which does not condense, the inert gas component. The total pressure of these mixtures does not exceed 10 bar. Non-ideal behaviour was present only in vapour activities not satisfying Raoult's law.
The study presented in this thesis focusses on the homogeneons condensation behaviour of a binary mixture of n-nonane/methane as a function of pressure and temperature. Here, methane is definitely no inert gas and its presence is very important for the homogeneaus condensation behaviour of the mixture. As a reference, homogeneaus condensation of water is stuclied in mixtures of water/helium and water/nitrogen as a function of total pressure.
The pressure range of interest varies from 10 to 100 bar, where pressures above 10 bar are referred to as high pressures.
1.2 Homogeneons nucleation at high pressures
The first stage of homogeneons condensation is the formation of stabie nuclei, the homogeneaus nucleation process.
Although in unary nudeation only the vapour component condenses, the pressure of the inert gas component still has an influence on the nucleation beha.viour of the va.pour. This is due to the influence of the total pressure on the saturation pressure of the vapour, the so-called Poynting or Kelvin-Helmholtz effect (Kestin 1979). In section 1.2.1 we will in general discuss unary nucleation and the effect of the total gas pressure.
In section 1.2.2 we will discuss the thermadynamie beha.viour of a mixture of a. rea.l gas a.nd a vapour component and its effect on the homogeneaus condensation process.

4 Introduetion
1.2.1 Unary nucleation in an ideal vapourjgas mixture and the effect of total pressure
Consider a system of a mixture of an ideal gas and an ideal vapour with total pressure p, temperature T, and partial vapour pressure Pv·
If the saturation ratio is less than unity, the vapour prefers to be in the vapour phase. Statistica! density fluctuations will form clusters of vapour molecules. However, since the chemica! potential of the vapour phase is less than the chemical potential of the liquid phase, this will be energetically unfavourable.
In case the saturation ratio is larger than unity, the formation of a liquid phase is energetically favourable. In the absence of foreign particles, condensation will occur by the formation of a droplet out of vapour molecules. The formation of the surface of a droplet has a positive contri bution to the Gihhs free energy of formation of a cluster aG1, while the volume of a droplet has a negative contrihution. As aresult the Gibhs free energy of formation of a droplet will have a maximum, the energy harrier, shown in figure 1.2.
Figure 1.2: Gibbs /ree energy of formation of a cluster AG as a function of the number of molecules n in the cluster. The Gibbs free energy of formation has a maximum AG* at the critical cluster size n• forS> 1.
In caseS is smaller than unity the formation of a cluster always costs energy, while forS larger than unity aG first increases with n but attains a. maximum value aa• at a critica! cluster size, n*. These cri ti cal clusters are in unstahle equilibrium with the supersaturated vapour. Clusters which are able to overcome this energy harrier will grow to macroscopie droplets, while dropiets smaller than n* will tend to evaporate.
1 Phase transition generally takes place at constant pressure ~d temperature or in a system wbere p and T change very slow with respect to the characteristic time scale for formation of a droplet. In such a system thermadynamie equilibrium is characterized by an extremum in the Gibbs free energy. A minimum (maximum) refers toa stabie (unstable) equilibrium (Abraham 1974).

1.2 Homogeneous nucleation at high pressures 5
The rate of formation of an n-cluster (group of n molecules) is purely a kinetic process. By the collision of a vapour molecule with a cluster, the cluster will gain one molecule and increase in size. By evaporation of a molecule the cluster may lose one molecule and decrease in size. It is on account of the difference in the rates of these two processes that a net rate of cluster formation exists. Once the clusters conta.in much more molecules than n* they have a negligible chance of re-evaporation and a droplet is born.
The nudeation rate J is the net rate at which dropiets are born per unit of volume and per unit of time. In general the riucleation rate can be expressed as:
J = Joexp( -t::..G*/ kT), (1.2)
where Jo is a kinetic prefactor, t::..G• is the height of the energy harrier and k is Boltzmann's constant.
The nucleation rate appea.rs to be extremely dependent on the temperature and on the saturation ratio. lt is this extréme sensitivity of Jon S and T, that is used in various experimental methods to study homogeneons condensation. It also explains the almost instant collapse of the supersa.tura.ted state in a continuons adiabatic expansion.
The first model on nudeation of a vapour component, developed by Becker and Döring (1935) and Zeldovich (1943), is termed the classica! nucleation theory (CNT). It is basedon the capillarity approximation: properties of the clusters, such as surface tension and liquid density, are assumed to be equal to their macroscopie values. With a typical critical cluster size of 100 molecules, this is rather questionable. Nevertheless good agreement is found between the nucleation rates predicted by the CNT and nucleation rate data for some vapour components (Allen and Kassner Jr. 1969, Wagner and Strey 1981) in a limited temperature range.
Since then many different models were formulated with varying success. Differences of many orders of magnitude for the nucleation rates predicted by the various models are reported in the literature. A microscopie approach by Lothe and Pound (1962) differed by 12 to 17 orders of magnitude in J compared to the classical nucleation theory.
Recently semi-phenomenological models were developed, taking into account the inherent real gas behaviour ofthe condensing va.pour (Dillmann and Meier 1991, Delale and Meier 1993) and (Kalikmanov and Van Dongen 1995b ). These models are successful over a wide range of vapour components and temperatures.
We will discuss the classical nudeation theory in chapter 2 to gain a general understanding of the problem of nudeation. As an example of the semi-phenomenological models, the model proposed by Kalikmanov and van Dongen ( 1995b) will be briefly discussed. Their model has the advantage above the other semi-phenomenological models that it was extended to binary nucleation (Kalikmanov and Van Dongen 1995a), which is useful for us.
The nucleation theories discussed so far do not take into account the presence of the secoud inert gas component. However, with increasing total pressure the solubility of the vapour in the gas in general increases.
Consicier our gas mixture of ideal components to be in equilibrium withaflat plane of liquid consisting of only the vapour component. The chemical potential ft" of the vapour component in the gas phase is unaffected · by the presence of the gas-component since there is no mutual intera.ction. It. can therefore be expressed as
ft 11 = ftref + kTln(pv/Pref ), (1.3)
6 Introduetion
where Jlref is the chemica! potential of a reference state at vapour pressure Pre/·
The liquid is in mechanica! equilibrium with the gas mixture above it and therefore the total pressure is the same in both phases. The liquid formed is in general considered to be incompressible. Therefore, the chemica! potential of the liquid can be written as
J11 = Jlref + (p - Pref )v
1' (1.4)
where v1 is the molecular volume in the liquid, and Jlref is the same reference state as for the vapour component in the gas phase. It is chosen to be the value at vapour-liquid equilibrium for the pure vapour component: Pref = Pvso(T), which is a known function of temperature reported in the literature for many substances.
Fora pure vapour the second term on the right-hand side (RHS) of expression (1.4) is zero. However, fora mixture of a vapour component and an inert gas component it is not negligible in generaL The saturated vapour pressure as a function of p and T follows from the equality of the expressions for the chemica! potentials in both phases and yields
( T) (T) ( (p- Pvso)v1
) Pvs p, = Pv.o exp kT .
E J 1.6
~ 1.4
1.2
HzO
1 0 20 40 60 80 100
p [bar]
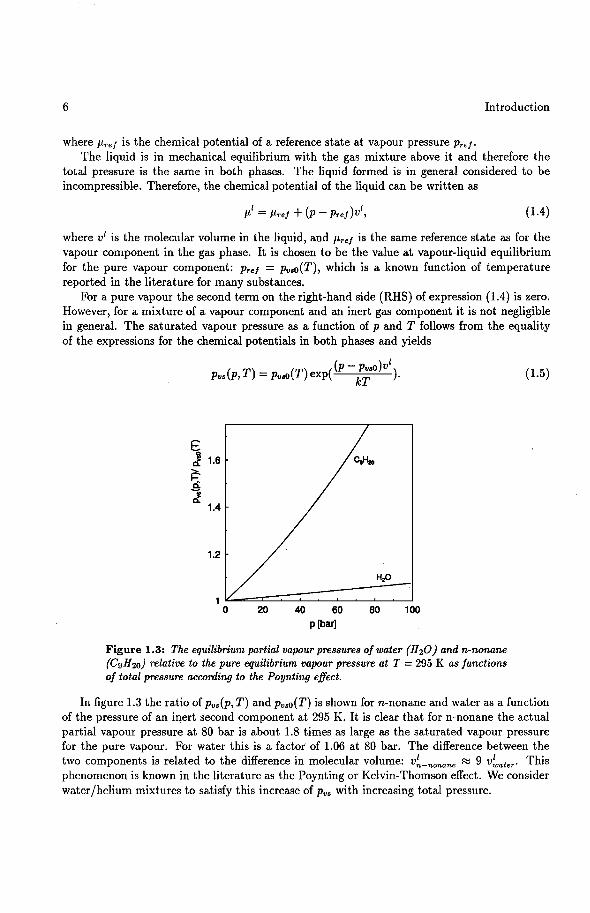
Figure 1.3: The equilibrium partial vapour pressures of water (H20) and n-nonane (C9H2o) relative to the pure equilibrium vapour pressure at T = 295 K as functions of total pressure according to the Poynting effect.
(1.5)
In figure 1.3 the ratio of Pvs(P, T) and Pvso(T) is shown for n-nonane and water as a function of the pressure of an inert second component at 295 K. It is clear that for n-nonane the actual partial vapour pressure at 80 bar is about 1.8 times as large as the saturated vapour pressure for the pure vapour. For water this is a factor of 1.06 at 80 bar. The difference between the two components is related to the difference in molecular volume: v~-nonane ~ 9 v~ater· This phenomenon is known in the literature as the Poynting or Kelvin-Thomson effect. We consider water/helium mixtures to satisfy this increase of Pvs with increasing total pressure.

1.2 Homogeneons nucleation at high pressures
1.3
1.2
1.1 Poyntirg effect: 0 "C
••••••• ••••••••••• 30't ...........
1 _,.::;_-•::.;·_·· .... ·_··_·_...__ _ _,__~_,_-_..J 0 20 40 60 80 100
p [bar]
Figure 1.4: The enhancement factor fw = Pvs(P, T)/Pvs(T) for water in air as a function of pressure for various temperatures . . Also the Poynting-effect is shown. Temperature changes 10 oe between two subsequent lines.
7
While the system water /helium behaves almast ideally, this is not so for the systems water/air or water/nitrogen. The molecular interactions of the componentsin the gaseous phase at, for example 100 bar, increase the saturated partial vapour pressure of water toabout 1.35 times its value for pure water at room temperature. Wylie and Fisher (1996) and Hyland (1975), determined this enhanced solubility of water vapour in air at vapour-liquid equilibrium as a function of total pressure for various temperatures. In figure 1.4 their results are shown (Wylie and Fisher 1996) tagether with the Poynting effect. It is clear that the Poynting effect accounts for only a small fraction of the increased solubility of water.
So, real gas effects are important for the equilibrium partlal vapour pressure. Nevertheless, the liquid formed consistsof almast pure water (Wylie and Fisher 1996) and the compressibility factor of the mixture is for pressures up to 100 bar close to unity. Both properties indicate that the gas can be described well by the ideal gas EOS and that the homogeneons condensation processis unary condensation. Therefore, from hereon mixtures of water/air (or nitrogen) are treated as mixtures of i deal components with a saturation pressure of water depending on total pressure and temperature as given in figure 1.4. In appendix E, the enhancement factor fw is given in a form similar to expression (1.5).
Due to the pressure dependenee of Pv., S decreases if p increases at constant Pv· It is clear from the strong functional dependenee of J on S that the total pressure of the inert component influences the nucleation process. Droplet growth is affected as well because the difference in the rnalar vapour fraction and the equilibrium molar vapour fraction, (y". y".), is the driving force for droplet growth.

8 Introduetion
1.2.2 Nucleation in binary mixtures of real gases
In the previous section we showed the indirect influence of the pressure of an inert gas component on the homogeneous condensation process. The liquid formed consisted of the vapour component only.
When the pressure increases, the intermolecular interactions can no longer he neglected. For mixtures such as natura! gas or n-nonane/methane this is already the case at pressures of a few bar. This behaviour is reflected in the equation of state (EOS). For mixtures of hydrocarbons a cubic equation of state such as the Redlich Kwong Soave (RKS)-EOS or the Peng Robinson (PR) EOS is very successful (Ahmed 1987, Martin 1979).
From such an equation of state, expressions for the chemica! potentials of the components in the vapour and liquid phase can he deduced. By equating the chemica! potentials of each component in both phases, the thermodynamic state at vapour-liquid equilibrium can he calculated.
Calculations of the equilibrium molar fraction of n-decane applying the RKS-EOS deviate not more than 10% from equilibrium data measured by Rijkers et al. (1992). Also the liquid molar volume predicted by the RKS equation of state in combination with the Peneloux correction (Reid et al. 1987) agreed within 1% with experimental data (Shipman and Kohn 1966). For these reasons, the RKS equation of state was used for all thermodynamic calculations throughout this thesis. It has the following form:
RT am P = V- bm- V(V + bm). (1.6)
Here V is the molar volume, and am and bm are the RKS properties for a mixture according to the mixing rules (Reid et al. 1987) as shown in appendix A.
In figure 1.5 the vapour-liquid equilibrium curve is shown in a p - T diagram for a binary mixture of n-nonane/methane at constant molar fraction n-nonane in the vapour phase. It exhibits the typical retrograde condensation behaviour. Starting above the equilibrium curve at high pressures, liquid will even he formed when the pressure is decreased isothermally such that the coexistence region is entered (arrow in figure 1.5). In a mixture of an inert gas and a vapour component this would never occur.
When the system enters the region enclosed by the retrograde condensation curve, liquid will form, thereby changing the composition of the vapour phase. Thermodynamic equilibrium will he reached between a liquid and vapour phase, where the latter phase contains less of the vapour component. Both phases consist of both components.
The liquid formed consists of both components present in the initia! mixture and this will also hold for a critica! cluster. The Gibbs free energy of formation of a cluster is a function of the number of molecules of both components (n1 , n2 ) present in the cluster. The energy harrier for unary nucleation in figure 1.2 will he replaced by a saddle point in the (nt, n2 ) plane as is shown in figure 1.6.
The thermodynamic behaviour of such a mixture strongly depends on the pressure, the temperature and the composition of the mixture. Theoretica! models for nucleation rates and droplet growth rates have to take these effects into account.

1.2 Homogeneous nucleation a.t high pressures
80
60
I Q. 40
Vapour + Liquid
20
0 200 220 240 260 280 300
T[K]
Fi.gure Ui: p-T diagram: the vapour-liquid equilibrium curve for a mixture of nonane and methane at constant vapour composition according to the RKS~EOS. Ynonane = 1.0 · 10-4
•
9
The classical binary nucleation theory (BCNT) a.s proposed by Reiss (1950), Ka.tz et al. (1966) a.nd Wilemski (1987), was applied by Looijmans et al. (1995) to binary mixtures of real gases. A quasi-one-component (QOC) model for nucleation was developed by Kalikmanov a.nd van Dongen (1995a) ba.sed on their semi-phenomenological unary nucleation model. Both roodels wîll be discussed in chapter 2.
For the second stage in the condensa.tion process we developed a droplet growth model for a droplet in a binary mixture of real ga.ses, presented inthelast section of cha.pter 2.
AG
0
Figure 1.6: The Gibbs free energy of formation of a cluster consisting of molecules of component 1, e.g. n-nonane, and component 2, e.g. methane.

10 Introduetion
1.3 Homogeneons condensation due to a continuons adiabatic expansîon
Many experimental techniques used to study homogeneous condensation make use of an adiabatic2
expansion to bring the vapour/gas mixture into a supersaturated state. Two of these experimental methods are used or frequently referred to in this work. For an overview of more experimental techniques reported in the literature, we refer to the workof Erhler (1988).
In the fust method, the mixture is expanded to a desired final pressure which is then kept constant for a short period of time. A small recompression follows to decrease the saturation ratio enough to stop the nuclea.tion process hut such that droplet growth continues. This technique is called the nudea.tion pulse technique and crea.tes a monodisperse cloud of dropiets of which radius and numher density can be monitored a.ccurately via light scattering and light attenuation (Wa.gner and Strey 1981 ). Nucleation and droplet growth can be stuclied very a.ccurately by this technique. A disadvantage of the method is the limited range of droplet concentrations (read 'nucleation rates') that can be handled experimentally and the related difficulty of "tuning" the initial conditions to be able to perform a successful experiment.
The second method is based on a continuons adiabatic expansion. The expansion cloud chamber is the setup we use and is discussed in detail in chapter 4. When pressure decreases adiabatically, temperature decreases as well and the mixture will become supersaturated as is illustrated in ligure 1. 7. Droplets will form, and with ongoing pressure decrease the saturation ratio will at first increase. Also the nudeation rate increases and more and more dropiets are formed which all start to grow. This simultaneons growth and production of dropiets will cause. the vapour component to start to deplete. At first this will give rise to a maximum in the nucleation rate, immediately foliowed by a maximum in the saturation ratio. With decreasing nucleation rate, droplet production will come to an end but droplet growth continnes trying to restore thermodynamic equilibrium. As an example of homogeneons condensation due to a continuons adiabatic expansion, a numerical simulation of an expansion of water and nitrogen is shown in figure 1.7. It is clear that the collapse of the supersaturated state by simultaneous formation and growth of dropiets occurs in a reiative small interval of time compared to the characteristic time of the pressure reduction. It results in a doud of dropiets which are distributed over their size. As a consequence optical signals based on light scattering and light attenuation are much more difficult to interpret. Still, the onset of the change in the light attenuation can be recognized very dearly. The thermodyna.mic state of the mixture at the instant of this detected onset represented in a p- T diagram are referred to as a Wilson-point.
The advantage of such an expansion cloud chamber is the large range of parameters it is suited for. The initia! vapour concentration, the initial total pressure, and the rate of expansion can be varied, leading to a large range of pressures, temperatures, and nucleation rates for the homogeneons condensation process.
Further, the set-up does not need "tuning". A continuous adiaba.tic expansion of a potentially condensable mixture will always lead to the a.ctual occurrence of the condensation process
4In this thesis, we wil! only use the term adiabatic. As long as no oondensation occurs the term isentropic is more accurate. However, the use ofboth terms and interchanging betweenthem 118 soon 118 dropiets are formed, may be confusing and so we only use the more general term adiabatic.

1.4 Homogeneons condensation of natura! gas due to a continuons adiaba.tic expa.nsion 11
1 -
0.8
0.6
0.4
0.2
time [ms]
Fignre 1.7: Results of a numerical simulation of the homogeneaus condensation process due toa continous adiabatic expansion of water/nitrogen. Using the classica[ nucleation theory (chapter 2}, and the droplet growth model of Gyarmathy (chapter 2). P/Po, J /Jma:r:, S/ Sma:r:' and the molar fraction of liquid formed relative to the initia! molar fraction of water vapour present YI/Yvo are shown as functions of time. Po= 50 bar, To = 295 K, PvO = 1000 Pa (Yvo = 2·10-4). Jmax = 6.1·1018 m-3s-I, Sma:z: = 22.62.
a.nd only a minimal amount of condensate is necessary to detect changes in the optical signals.
1.4 Homogeneons condensation of natura! gas due to a continuons adiabatic expansion
As an example of the homogeneons condensation process due to a continuons expansion, we will briefly discuss an experimental series with Groningen Natura! gas. This experimental series was done as part of a prelimina.ry study on the homogeneons condensation behaviour on na.tural gas in collaboration with the 'Nederlandse Gasunie N. V.' Aim of this investigation was to determine the onset of homogeneons condensation due to a continuons adiabatic expa.nsion of natural gas. The full description of this workis presented in a paper by Muitjens et. al. (1994).
A continuons adiabatic expa.nsion of na.tural gas was a.chieved by applying an expa.nsion _cloud chamber. The expansion doud chamber is a small bottle-shaped vessel (see also chapter 4) which is connected toa large low-pressure tank via an electromagnetic valve. Windows in the side walls of the expa.nsion cloud chamber make it optically accessible. One can detect the formation of dropiets by measuring the intensity of a light beam passing the gas/droplet

12
100
80
! 60
0.. 40
20
··' • 0 200 220 240 280
T[K]
o FII'Ststr"fl))e OFilst""""" • 5eoondSIIIfPie • 5eoond"""""
0
"' ~ "' • t 0 8
f 280 300
Introduetion
Figure 1.8: p-T diagram. The retrogmde condensation curve of Groningen natural gas is shown (provided by Gasunie). Also the initial states of the experimental series and the corresponding Wilson points are shown.
mixture or by light scattering techniques. Droplets scatter light in all directions, thereby lowering the intensity of the light beam
passing through the expansion cell. The Wilson point is defined as the thermodynamic state of the mixture at the moment the optica! signals change significantly (tJ.I / Io = 0.005). In figure 1.8 the initia! states of two series of experiments for two samples of natura! gas and the corresponding Wilson-points are shown. Also the retrograde condensation curve for the natural gas used is shown.
Natura! gas is a mixture of many components. The gas analysis technique applied by Gasunie (Kuijk et al. 1991) made a distinction of 44 components. However, even though natura! gas definitely consists of more than two components, the retrograde condensation curve shown in figure 1.8, resembles the one shown in figure 1.5 of the binary mixture of metharre and nnonane. Thermodynamically they are closely related and it is for this reason that we focus on a binary mixture of metharre and n-nonane as a model gas for actual natura! gas.
The first experimental series has an average temperature difference between the Wilsonpoints and the retrograde curve at the same pressure, called undercooling, of about 32 ± 3 K, whereas the second series has an undercooling of 60 ± 5 K. The experimental procedure for both series was identical. We concluded that the composition of the natural gas in both samples differed, even though both samples were taken successively at the same location. A possible explanation is tha.t small differences in the composition of the natura! gas samples existed, which may have a large influence on the onset of homogeneaus condensation.
Using the light scattering technique we found that liquid in the form of dropiets was detected in the expansion cloud chamber prior to the expansion. The total molar concentration of these dropiets was w-s. This is about one order less than the detection limit of the gas analysis technique. According to the retrograde condensation curve in figure 1.8 there was no condensate to he expected. These dropiets were very persistent and did not re-evaporate, so the dropletfnatural gas mixture apparently was in equilibrium. This illustrates that a retrograde

1.5 Thesis overview 13
condensation curve based on the results of a gas analysis technique is to a certain extent an artificial one, since its contour depends on the accuracy of the compositional analysis.
The main condusion that can be drawn from this preliminary study on the non-equilibrium condensation of natura! gas, is that the composition of the initia! gas mixture is crucial for the condensation behaviour, This leads to an essential question in studying homogeneons condensation: 'l:t'hat is the initia! composition of the mixture?'
1.5 Thesis overview
The homogeneons condensation process in binary mixtures, in partienlar mixtures of real gases, at high pressures is the subject of this investigation.
As mentioned before, homogeneons condensation is a two stage process of nucleation and droplet growth. Therefore, in chapter 2 we will first discuss some roodels for una.ry and bina.ry nucleation. Once a cluster has passed the critica! size it can increase unlimited as far as the energy of formation is concerned. However, condensing molecules have to be available and the latent heat releJ!Sed has to be removed. Therefore, mass and energy flows in the vapour phase control the growth of the droplets. To predict the growth process of a droplet in a binary mixture of real gases we developed a droplet growth model based on the work of Gyarmathy (1982). This model is the subject of the last section of the second chapter. We also discuss the influence of the differences between the roodels of Gyarmathy and Young (1993).
In chapter 3 homogeneons condensation due to a continuons adiabatic expansion of a vapourfgas mixture is considered. Taking the pressure to he a known function of time, the process is simulated numerically. Both, mixtures of an inert gas component and a vapour, and binary mixtures of real gases can be simulated. The numerical code uses the roodels for nucleation and droplet growth discussed in chapter 2.
The molar fraction of formed liquid, gl, is the key quantity for the homogeneons condensation process during the adiabatic expansion. From the expression for the molar fraction of formed liquid it follows that g1 is determined by an integral of the nucleation rate over time. The nucleation rate, as we see from figure 1.7, is a narrow pulse-shaped function of time. Therefore, an analytica! approach using asymptotic techniques is applied as was clone by Blythe and Shih (1976), Clarke and Delale (1986), and Delale et al. (1993a, 1993b) for homogeneons condensation in nozzle flows. We present this analytica! approach in the second part of chapter 3.
To study homogeneons condensation we made use of an expansion cl oud chamber. In chapter 4 the setup and the optica! detection system is discussed.
The initia! state of the mixture is characterised by the pressure, temperature, and the initial composition of the mixture. To determine this initia! composition for a mixture of n-nonane and methane we applied a metbod based on gas chromatography, section 4.2.1. Finally, we also discuss some typ i cal experiment al signals for a continuons adiabatic expansion of water /nitrogen in the expansion doud chamber.
In chapter 5 we will present the results. We will discuss the quality of the va.rious nucleation roodels by comparing measured optica! signals to the signals determined by the numerical

14 Introduetion
simulation based on such a nucleation model. This method of interpreting the experimental signals is related to the method applied by Wegener (1964, 1972) in nozzle flows. Also a new experimental method is proposed to determine nucleation rate data from the measured light transmission signals without the necessity of any knowledge of a nucleation model. The results of nucleation rates determined by this latter method are compared with the results obtained from the full numerical simulation.
Finally, chapter 6 contains a concluding discussion.
References
ABRAHAM, F. 1974. Homogeneaus nucleation theory. New York and London: Academie Press.
AHMED, T. 1987. Comparative study of eight equations of state for predicting hydracarbon volumetrie phase behavior. SPE 15673:1-15.
ALLEN, L., & J. KASSNER JR. 1969. The nucleation of water va por in the absence of partienlate matterand ions. J. Colloid and Interface Sci. 30(1):81-93.
BECKER, R., & W. DÖRING. 1935. Kinetische Behandlung der Keimbildung in übersättigten Dämpfen. Ann. Phys. 5(24):719-752.
BLYTHE, P ., & C. Sam. 1976. Condensation shocks in nozzleflows. J. Fluid Mech. 76(3):593-621.
CLARKE, J ., & C. DELALE. 1986. Nozzle flows with nonequilibrium condensation. Phys. Fluids 29(5):1398-1413.
DELALE, C., & G. MEIER. 1993. A semi-phenomenological droplet model of homogeneaus nucleation from the vapor phase. Journat of Chemica[ Physics 98:9850--9858.
·DELALE, C., G. SCHNERR, & J. ZIEREP. 1993a. Asymptotic salution of transonic nozzle flows with homogeneons condensation. I. subcritical flows. Phys. Fluids A 5(11 ):2969-2981.
DELALE, C., G. SCHNERR, & J. ZIEREP.l993b. Asymptoticsolutionoftransonicnozzleflows with homogeneaus condensation. II. supercritical fiows. Phys. Fluids A 5(11):2982-2992.
DILLMANN, A., & G. MEIER. 1991. A refined droplet approach to the problem of homogeneons nucleation from the vapor phase. J. Chem. Phys. 94(5):3872-3884.
EHRLER, F. 1988. Spontane kondensation. VDI- Wärmeatlas 5:Jel-Je20.
FLAGEOLLET-DANIEL, C., J. GARNIER, & P. MIRABEL. 1983. Microscopie surface tension and binary nucleation. J. Chem. Phys. 78(5):2600-2606.
GYARMATHY, G. 1982. The spherical droplet in gaseous carrier streams: review and synthesis. In Multiphase science and technology, 1. Washington: Hemisphere Publishing Corporation.
HUNG, C.-H., M. KRASNOPOLER, & J. KATZ. 1989. Condensation of a supersaturated vapor. VIII. The homogeneous nudeation of n-nonane . .J. Chem. Phys. 90(3):1856-1865.

1.5 Thesis overview 15
HYLAND, R. 1975. A correlation for the second virial coefficients and enhancement factors for moist air. J. of Research of the National Bureau of Standards - A. Physics and Chemistry 79(4):551-560.
KALIKMANOV, V., & M. VAN DONGEN. 1995a. Quasi-one-component theoryofhomogeneous hinary nucleation. Phys. Rev. E 51:4391-4399.
KALIKMANOV, V., & M. VAN DONGEN.l995h. Semiphenomenologicaltheoryofhomogeneous vapour-liquid nucleation. J. Chem. Phys. 103:425o-4255.
KATZ, J., H. SALTSBURG, & H. REISS. 1966. Nucleation in associated vapors. J. Colloid and Interface Sci. 21:560-568.
KESTIN, J. 1979. A course in thermodynamics, Vol. IJ. Washington, London.
KUIJK, 1., R. BEKS, M. STRUIS, & A. SMIT. 1991. Detailed analysis of natural gas in order to predict condensa.tion behaviour. In - (ED.), G.R.I Symposium on Gas Quality Measurement, Chicago, USA. -.
LOOIJMANS, K. 1995. Homogeneaus nucleation and droplet growth in the coexistence region of n-alkane/methane mixtures at high pressures. Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.
LOOIJMANS, K., C. LUIJTEN, G. HoFMANS, & M. VAN DoNGEN. 1995. Classica! binary nuclea.tion theory applied to the real mixture n-nonane/methane at high pressures. J. Chem. Phys. 102 (11):4531-4537.
LOTHE, J., & G. POUND. 1962. Reconsiderations of nudeation theory. J. Chem. Phys. 36:2080-2085.
MARTIN, J. 1979. Cubic equation of state which? Ind. Eng. Chem. Fundam. 18(2):81-97.
MIRABEL, P., & J. CLAVELIN. 1978. Experimental study of nucleation in binary mixtures: The nitric acid-water and sulfurie-water systems. J. Chem. Phys. 68(11):5020-5027.
MUITJENS, M., V. KALIKMANOV, M. V. DONGEN, A. HIRSCHBERG, & P. DERKS. 1994. On mist formation in natura! gas. Revue de l'Institut Français du Pétrole 49(1):63-72.
REm, R., J. PRAUSNITZ, & B. POLING. 1987. The Properties of Gases and Liquids. New York: McGraw-Hill Book Company.
REISS, H. 1950. The kinetica of phase transitionsin binary systems. J. Chem. Phys. 18(6):840-848.
RIJKERS, M., M. MALAIS, C. PETERS, & J. DESWAAN ARONS.1992. Measurementson the phase behavior of binary hydrocarhon mixtures for modeHing the condensa.tion behavior of natural gas. Fl-uid Phase Equilibria 71:143-168.

16 Introduetion
RU DOLF, R. 1994. Experimental investigation on condensation of supersaturated acid-water vapor mixtures by means of laser light scattering in a newly developed expansion chamber. Ph.d. thesis, Universität Wien, Formal- und Naturwissenschaftlichen Fakultät.
SHIPMAN, L., & J. KOHN. 1966. Heterogeneaus phase and volumetrie equilibrium in the methane-n-nonane system. J. Chem. Eng. Data 11(2):176-180.
VIISANEN, Y., R. STREY, & H. REISS. 1993. Homogeneons nucleation rates for water. J. Chem. Phys. 99(6):4680-4692.
WAGNER, P., & R. STREY. 1981. Homogeneaus nucleation rates of water vapor measured in a two-piston expansion chamber. J. Phys. Chem. 85(18):2694-2700.
WEGENER, P., J. CLUMPNER, & B. Wu. 1972. Homogeneons nudeation and growth of ethanol drops in supersonic flow. Phys. Fluids 15(11):1869-1876.
WEGENER, P., & A. POURING. 1964. Experiments on condensation of water vapor by homogeneons nucleation in nozzles. Phys. Fluids 7(3):352-361.
WILEMSKI, G. 1987. Revised classical binary nucleation theory for aqueous alcohol and acetone vapors. J. Phys. Chem. 91(10):2492-2498.
WILS ON, C. 1897. Condensation of water vapour in the presence of dustfree air and other gases. Philos. Trans. R. Soc. London A 189:265-307.
WYLIE, R., & R. FIS HER. 1996. Molecular interaction of water vapor and air. J. Chem. Eng. Data 41:133-142.
WYSLOUZIL, B., J. SEINFELD, & R. FLAGAN. 1991. Bina.ry nucleation in acid-water systems. II. Sulfucic acid-water and a comparison with methanesulfonic acid-wat.er. J. Chem. Phys. 94(10):6842-6850.
YOUNG, J. 1993. The condensation and evaporation of liquid dropiets at arbitrary Knudsen number in the presence of an inert gas. Int. J. Heat Mass Transfer 36(11):2941-2956.
ZAIIORANSKY, R., & F. PETERS. 1985. Bina.ry nucleation at low temperatures. J. Chem. Phys. 83(12):6425-6431.
ZELDOVICH, Y. 1943. Acta Physicochim. (URSS) 18.

Chapter 2
HOMOGENEOUS CONDENSATION
The first stage of homogeneaus condensation is the nucleation process. Stabie nuclei of several tens to hundreds of molecules are formed.
The classical unary nucleation theory ( CNT) is the nucleation model most widely known. Even though the model is based on the rather questionable capillarity approximation, nucleation rates predicted for some supersaturated vapour components agree well with experimental data (Allen and Kassner Jr. 1969). However, this success of the classical theory is limited to only a few vapour components, e.g. water, and in a limited temperature range.
In section 2.1 two roodels for the nucleation process in supersaturated mixtures of an inert gas and a vapour will be discussed. The classical nucleation theory developed by Becker and Döring (1935) and Zeldovich (1943) serves as a basis for the general understanding of the problem of nucleation. The nucleation model of Kalikmanov and van Dongen (1995b) is one of the group of semi-phenomenological theories. This model is the most successful for a wide range of vapour components and temperatures. It was also extended to binary nucleation and can be applied for multi-component mixtures (Kalikmanov and Van Dongen 1995a, Kalikmanov and Van Dongen).
In section 2.2 we discuss two nucleation roodels for binary nucleation in mixtures of a real gas and a real vapour: the binary classical nucleation theory (BCNT) developed by Reiss (1950), Stauffer (1976} and Wilemski (1987), and a quasi-one-component theory (QOC) developed by Kalikmanov and van Dongen (1995a}. Both roodels are extensions of the roodels discussed in section 2.1.
Once a droplet is formed the balance between mass and heat transfer to and from the drop let, determine the further growth of the droplet. For droplet growth in a mixture of an inert gas and a single condensing vapour we refer to the workof Gyarmathy (1982). We extended his modeltoa droplet in a binary mixture of a real gas and a real vapour as presented insection 2.3.
17

18 Homogeneons Condensation
2.1 Unary nucleation theory
The aim of any nudeation theory, is to determine an expression for the rate of formation of stabie nuclei, the so-called nucleation rate J.
The nucleation process is a kinetic process. Density fluctuations form clusters of vapour molecules. Clusters consisting of n molecules are called n-clusters or n-mers, and molecules are called monomers. In the kinetic process cluster-cluster interactions are neglected, only clustermorromer reactions are considered. Schematically, this is shown in figure 2.1. So, it is by the callision of a morromer with an n-cluster that an (n + 1)-cluster is formed. Evaporation of a molecule decreases the cluster size from n to (n 1).
Figure 2.1: Kinetic processes: C,. condensation rate, En evaporation rate, Jn net rate of clusters transiting from n to n+ 1.
The rate of change in number density Pn of n-clusters is given by the difference of the rates of n- and ( n + 1 )-cluster formation:
where
dp,. dt
(2.1)
(2.2)
The condensation rate Cn is the collision frequency or impingement rate of a vapour molecule on the cross-sectien of the cluster multiplied by the probability that the vapour molecule will stick to the cluster, the mass accomodation coefficient On:
(2.3)
The mass accommodation coefficient is in general taken to be unity. In expression (2.3), the general assumption is made that the n-cluster can be considered spherical. The radius is proportional to the radius of a molecule r 0 multiplied by n113 , the cross-section to the surface of one molecule a0 multiplied by n213 , and the volume to the volume of one molecule v0 multiplied by n.
In equilibrium, the cluster concentration p':, is time independent and Jn 0. This leads to the so-called detailed balance relation
(2.4)

2.1 U nary nudeation theory 19
where c~ is the Collision frequency in the equilibrium state, and P! is the number density of n-clusters in the equilibrium state
p~ = p~ exp( -i:l.G"(n)/kT). (2.5)
Here i:l.G"(n) is the Gibbs free energy of formation of an n-cluster at the equilibrium state. The evaporation rate is independent on vapour pressure and only depends on the temperature and cluster size (Oxtoby 1992), E~ = E ...
The solutions of the equations (2.1) and (2.2) reach a steady state within a characteristic time of the order of 1 p,s or less (Abraham 1974). Most of the homogeneons condensation processes occur on much larger time scales. Therefore, only the steady state is of interest to us and it yields a constant J independent of n. From (2.2) and (2.4) it follows that
J C,.p,. Pn+l
C•p• = C•p• - -P. n n n n n+l
(2.6)
From this expression in combination with (2.5) it will he shown that two probieros remain to he solved: First, the equilibrium state has to be defined and second, the Gibbs free energy of formation of an n-cluster in the equilibrium state has to he determined.
The model for the Gibbs free energy of formation of an n-cluster is the crudal point in any nucleation theory, since i:l.G"{n) is in the exponent of expression (2.5). The different roodels for this property cause ditierences between the predictions of J of many orders of magnitude.
For the equilibrium state, two methods are reported in the literature. The first method, used in the classica! nucleation theory we will refer to as the "classica! approach". The equilibrium is assumed to be a constraint equilibrium which would exist at the same temperature and supersaturation. lt is an artificial unstable state which can exist only by introducing so-called Maxwell demons: large clusters are removed from the system, separated into single molecules and returned to the system.
The secoud method of Katzand coworkers {1977, 1979) is called the "kinetic approach". The equilibrium state is taken to be the actual vapour-liquid equilibrium at the same tempera.ture, S = 1. This can be clone since the eva.poration rate is a function of temperature and cluster size only.
2.1.1 Classical nucleation theory
To complete the expression for the nudeation rate following either the classica! or the kinetic approach, weneed a model for the Gibbs free energy of formation of an n-cluster.
In the classica! nucleation theory as well as in the original work of Katz and coworkers, the vapour-droplet system is described by a sphericalliquid volume, the vapour phase surrounding the droplet, and the surface dividing the two phases. The vapour is assumed to be an ideal gas and the liquid is taken incompressible, with a constant molecular volume v1• The total pressure of the vapour and the inert gas component is p, the temperature is T, and the partial vapour pressure is Pv.

20 Homogeneons Condensa.tion
The pressure in the droplet p1 is given by Lapla.ce's equa.tion:
p1 = p+ 2ujr, (2.7)
with u is the macroscopie surface tension, and r the radius of the droplet.
When an n-cluster, formed at constant pressure and tempera.ture, is in equilibrium with the gas/vapour mixture, the Gibbs free energy of formation of the n-cluster t:.G attains an extremum. In case the cluster was formed at constant temperature, it would be the Helmholtz free energy t:.F. Abraham (1974) shows, assuming tha.t nis much less than the total number of vapour molecules in the volume of the system considered, that t:.F is equal to /j.G. F is an additive quantity for the droplet and vapour subsystems leading to
(2.8)
where F 1 is the free energy of the cluster, F" of the vapour phase surrounding the cluster, and F0 of the gas/vapour mixture without a cluster present. Fis defined as -pV +pN. Using the fact that the droplet's volume is v0n = v1n, expression (2.8) leads to,
fj.p /j.G = -nkTln(Pv~(T)) + un213
ao + nv1(p Pvs(T)). (2.9)
With expressions (1.5) and (1.1) this becomes
(2.10)
The saturation ratio is based on the equilibrium vapour pressure as a function of temperature and total pressure (Poynting effect), which is a generalisation of the original model by Becker and Döring.
When the saturation ratio is smaller than or equal to unity, /j.G is always positive and stabie dropiets can not be formed. An equilibrium Boltzmann distribution will exist due to statistica! density fluctuations. But for saturation ratios larger than unity, expression (2.10) has a negative and a positive term leading to an energy ·harrier for droplet formation. The Gibbs free energy of formation of a cluster reaches a maximum value t:.G* at the critical cluster size n*, corresponding to the critîcal radius r*:
n* = 47r(r*)3
(2.11) 3v1
r* 2uv1
(2.12) kT InS'
fj.G* ~1747r(r*)2 • (2.13)
Clusters smaller than the critical radius tend to evaporate due to the positive slope of /j.Q. lnversely, clusters larger than the critica! clustertend to grow due to the negative slope of t:.G.
Taking the properties, e.g. surface tension, of the droplet as macroscopie ( the capillarity approximation) we found an expression for t:.G( n) for a given thermodynamic state. Combining

2.1 Unary nucleation theory 21
this with a chosen equilibrium state by either the classica! or kinetic approach, we obtain an expression for the nucleation rate.
We will now proceed according to the classica! approach. For the constraint equilibrium the partial vapour pressure is equal to the supersaturated vapour pressure, hence with (2.3) Cn C~. The cluster equilibrium number density is found by substituting expression (2.10) in expression (2.5),
( un2f3a0)
p~ = piexp nln(S)-~ . (2.14)
The formation of a cluster is assumed to have a negligible effect on the number density of vapour molecules. So, the concentration of single vapour molecules is taken to he very large and equal to the equilibrium state value, p; = p1 • Then, summing (2.6) from 1 to a certain G yields
J t (-1-) l - PG+l (2.15) n=l Cnp~ Pb+1 ,
From expression (2.14) it is clear that for increasing n the equilibrium number density will increase to infinity once nis larger than n•. In the actual condensing vapour the concentration of large clustes is limited and the last term on the RHS of expression (2.15) can be neglected for large G.
Replacing the summation in expression (2.15) by an integration yields,
J-1 = l>O [-cl e] dn. 1 nPn
(2.16)
The main contribution to this integral sterns from the neighbourhood of n• where the number density is minima!. The nucleation rate can be found by applying LapJace's method (Erdelyi 1956) to the integral (2.16), which results in
(2.17)
Here, p~. = PÎ exp( -!:.G*fkT) and Zn• = ( -(B2A~L~~n)Mn* f 12 is the Zeldovich factor which
accounts for Pn being less than p~ and for the probability of supercritical clusters to re-evaporate. From expressions (2.10) and (2.11) it follows that
Zn•:::; {ij} ao(n:)2/3'
The typical value of Zn• is in between 10-2 and 10-1•
(2.18)
Substituting the expressions for z".. (2.18) , Cn• (2.3), and Pn• (2.14) together with (2.11), and (2.13), in (2.17) yields
(2.19)
Here, p" is the partial vapour pressure, q the macroscopie surface tension, fh the liquid mass density, m the molecular mass of the vapour component, and we used mf'fil = v0 • As one can

22 Homogeneous Condensation
1018
1018
1014 .......
;.,11) 1012
·e :;;; 1010
108
108
4 5 6 10 20
s Figure 2.2: Nucleation rate of water vapour as function of saturation ratio for various temperatures according to the CNT.
see, only macroscopie quantities are needed to calculate the nucleation rate according to the CNT.
Now we return to the kinetic approach of Ka.tz et al.. The equilibrium state is the va.pourliquid equilibrium indicated by the superscript s. Since the saturation ratio is equa.l to unity, the Gibbs free energy of formation of an n-cluster only contains the surface term,
D.G aon2f3o-,
and the equilibrium number density of an n-cluster is given by
p~ = p~ exp( -aon213o-fkT).
(2.20)
(2.21)
The ratio of ptf p~ is S and therefore from (2.3) also C"JC~ is equal to S. Dividing both sides of expression (2.6) by sn+t and summing to an arbitrary large N, yields
J'E (-1-) = 1 - PN+l . n:::l GnP':,Sn SN+IPN+l
(2.22)
For large N the last term on the RHS beoomes negligible. We now write Gnp~Sn as exp(H(n)), which with (2.21) results in
( ) l S 2/3 o-ao ( s) H n = n n + n kT + ln Cnp1 . (2.23)
Without the last term on the RHS expression (2.23) is identical to the expression for D.G( n) for the classica! approach (2.10). Replacing the summation in expression (2.22) by an integra.tion and applying Laplace's method at the minimum of H(n) yields
P~· (2.24)

2.1 Unary nucleation theory 23
Heren* is the solution of H'(n) = 0, and 1 = fn· The contribution of ln(Cn) in (2.23) to H" is negligible (Katz and Donohue 1979). The
final expression for J according to the kinetic approach is a factor S smaller compared to the expression for the classica! approach (2.19). This is a relative sma.U difference in view of the many orders of magnitude difference when compared toother roodels (Lothe and Pound 1962).
As an example, in figure 2.2 the nucleation rate for water according to the classica! nucleation theory is shown as a fundion of the sa.turation ratio for various temperatures. lncreasing the satura.tion ratio from 5 to 7 at a. tempera.ture of 260 K changes the nuclea.tion rate about 8 orders of magnitude. Va.rying the temperature with only 2.5 degrees at constant saturation ratio changes the nuclea.tion rate about one order of magnitude. So, it is clear that J is indeed a very sensitive function of S and T.
2.1.2 The size of a newly born droplet
The definition of the nucleation ra.te is usually given by: "The number of critica! clusters formed per unit of volume and unit of time". Surprisingly, it is also common practice to assume that the size of a newly born droplet to which droplet growth laws are applicable is equal to the critical size. However, this can not be true, since a critica! cluster is in equilibrium with the vapour, although unsta.ble, and does not grow. The kinetic process of duster formation by random molecular collisions is the only mechanism of increasing the size of a critica! cluster. Therefore, a newly born droplet for which droplet growth laws are applicable must have a size larger than the critica! size, with a negligible probability of re-evaporating.
Basedon the steady state expressions (so in the definition of J any cluster size can be used) the size of a newly born droplet is derived in this section. This is doneon the basis of the CNT.
The change of cluster size by the addition or loss of a molecule is regarcled to be a. small, in:finitesimal change. So, equation (2.1) can be written as
op,. oJn ( ) 7it = - fJn • 2.25
Combining (2.4) a.nd (2.5) gives
En+l = C,.exp((-ÀG(n) + ÀG(n + 1))/kT),
or, writing the exponential term as a first order Taylor expansion,
1 oÀG E,.+l = C.,(l + kT a;:).
Substituted in expression (2.2) the result is
1 oÀG J., = C,.(pn - Pn+l - Pn+l kT a;:),
or, consiclering only terms up to the :first order,
J -c op,. n non
(2.26)
(2.27)
(2.28)
(2.29)

24 Homogeneons Condensation
The expressions (2.25) and (2.29) represent the Fokker-Planck equations in the cluster size space. The condensation coefficient C .. is the duster size dilfusion coefficient and the drift coef-
ficient is represented by A= ~;en f)!G· Substituting this in expression (2.29) and assuming
steady state (Jn J for all n) yields
(2.30)
The second part on the right hand side is the part of J that is zero at the critica! size but dominates as the cluster size increases, the droplet growth stage. The driving force of this drift part is the first derivative of f:j,Q,
The derivative of f:j,Q with respectton is related to the equilibrium nuinber density for the constraint equilibrium state by
Therefore A equals A= c .. ap~.
p;" on Substituting this in (2.30) yields an expression for the actual number density p,.,
1.5
:t 0.5
0
·0.5 0
J
\__
2
n/n·
J2
3 4
Figure 2.3: DijJusion J1 and drift term J2 contributions to the steady state nucleation rate J as functions of cluster size. CNT for water; n* = 75, J = 1 m-3s-1 ,
T = 240 K, Pv = 225 Pa.
(2.31)
(2.32)
(2.33)
Fora given thermodynamic state, J is known through (2.19). Combining this with expressions (2.5) and (2.33), Pn and its derivative to n is known for all n. This enables us todetermine

2.1 Unary nucleation theory 25
theseparate terms on the RHS of expression (2.30). In figure 2.3 these contributions.are shown as a function of cluster size.
Figure 2.3 shows that for a cluster with a size of about 2n*, the ditfusion term is negligible and only the drift term remains. For these dropiets the probability of re-evaporation is negligible and a droplet is born. We shall assume from hereon that in general the size of a newly born droplet for unary condensation is 2n*.
2.1.3 A Semi-pbenomenological tbeory
Nucleation models differ mainly by different models for b..G. They combine this witheither the classical approach or the kinetic approach in determining the evaporation rate.
Although the expression for J according to the CNT is fully determined by macroscopie quantities, this is also the main drawback of the CNT. Clusters are no macroscopie systems. This is refiected in e.g. the surface tension of a cluster.
In the classical nucleation theory the droplet is defined by a discontinuons jump of the bulk liquid density to the density of the vapour phase, the surface of the droplet. Actually, the density of the liquid gradually changes from the bulk density in the core of the cluster to the vapour/gas density in the vapour phase. Therefore, the radius of the interface is not uniquely defined. These curvature effects are not taken into account in the CNT. This might be corrected by taking the macroscopie surface tension to he radius dependent, the Tolman correction (Tolman 1949).
In the semi-phenomenological model of Kalikmanov and van Dongen (1995b ), the Gibbs free energy of formation of an n-clustel' is found by applying Fisher's droplet model (Fisher 1967) with a microscopie surface tension. This model for b..G is inserted into the kinetic approach of Katz.
Fisher's droplet model is a microscopie treatment of a substance in the vapour phase. The vapour, a real gas, is assumed to he anideal collection of clusters of different sizes. So Dalton's law can he applied to find arelation between cluster number densities and the vapour pressure,
p.,. kT
(2.34)
Here, we immediately applied the kinetic approach where the vapour pressure is the saturated vapour pressure at temperature T. The overall vapour density is given by
00
Pv• = Lnp~, n=l
where p':. can be written in the Boltzma.nn form
(2.35)
(2.36)
b..f!':. is the grand potential of the n-cluster which can he expressed as a function of n, the configuration integral of the n-cluster q,., the thermal de Broglie wavelength of a molecule

26 Homogeneons Condensation
A, and the chemica! potential of the vapour at saturation p.'. This .6.0 (system with constant V,T,p.) is equivalent to .ó.G (system of constant p,T) for the condition that V1 <{:::V, where V 1 is the volume of the droplet and V the total volume of the droplet/vapour system (Dillmann and Meier 1991 ).
The main result of Fisher's droplet model is an expression for CJn· Substituted into the expression for .6.0, this yields
(2.37)
The last two terms on the RHS of expression (2.37) account for the extra degrees of freedom an n-cluster has compared to the bulk liquid, r In n, for the rotational and vibrational degrees of freedom, and q0 for the translational degrees of freedom.
The surface tension Umicro has to be considered as a microscopie surface tension. In this microscopie surface tension effects of curvature are incorporated, so that we write in accordance with Tolman (1949)
U micro = u(l + O:'un-l/3
). (2.38)
From the expressions (2.36) to (2.38), p~ is given by
p~ qoexp(-Bo(l + a"n-113 )n213- rlnn), (2.39)
uao where Oo = kT . .
This expression contains three unknown parameters: r, q0 , and a". These parameterscan he determined using experimental data available in literature. By extrapolating the model to the critica! point and substituting the critica! properties of the vapour component, Tc, Pc, and p0 , in the expressions (2.34) and (2.35), two equations are found from which Tand q0 can be determined:
Pc kTc
Pe
qo(( T ),
qo((r 1),
(2.40)
(2.41)
(2.42)
where ( is the Riemann zeta function (Abramowitz and Stegun 1965). The parameter a". follows from the known value of the saturated vapour pressure at the temperature T and expressions (2.34) and (2.39):
Pvz~) = f: qo exp( -Oo(l + a"n -ll3)n213 - TIn n ). n=l
(2.43)
The final result for the nucleation rate J, applying the kinetie approach, is given by
(2.44)

2.2 Binary nucleation theory 27
where n* is the real root of
1 S 29 2/3 1(} 1/3 2 - n n + 3 on + 3 oaun + T - 3 = 0 (2.45)
In figure 2.4 we compare the results for the nucleation rate according to the CNT and this semi-phenomenological theory for water vapour. It is clear that the differences in J for both theories are rather small when applied for water. In their paper Kalikmanov and van Dongen compared the predicted nucleation rates of various theories to nucleation rate data reported in the literature for various vapour components. Overall, the best agreement was found applying the theoretica! model outlined above.
1018
1018
1014 .,::--;.,rn 1012
g ., 1010
108
106
4 5 6 10 20
s Figure 2.4: Nucleation rate of water as a function of saturation ratio for various temperatures. Solid lines, the CNT. Dashed lines, the semi-phenomenological theory of Kalikmanov and van Dongen (1995).
2.2 Binary nucleation theory
For a supersaturated mixture of two vapour components or a supersaturated mixture of a real gas a.nd a real vapour, the homogeneons condensation process is a binary process. The clusters formed will contain molecules of both components. As a consequence, the Gibbs free energy of formation of a droplet is composition dependent and thé nucleation rate J is a vector, characterized by its size and direction.
Reiss (1950) extended the unary classica! nucleation theory to a binary system, assuming that the rate of cluster formation follows the direction of steepest descent in !lG. Stauffer (1976) showed that also the kinetic part of cluster formation is important. Finally, Wilemski (1987) formulated a model applying the capillarity approximation in combination with a droplet model including a bulk and a surface phase, which made the model thermodynamically consistent.

28 Homogeneons Condensation
Looijmans (1995) applied the model of Stauffer and Wilemski, referred to as the Binary Classica! Nucleation Theory (BCNT), to binary mixtures of real gases in the coexistence region. In section 2.2.1 we will briefiy describe the BCNT as presented in Looijmans et al. (1995) together with some results for the mixture n-nonane/methane.
Kalikmanov and van Dongen (1995a) extended their semi-phenomenological model for nudea.tion to binary mixtures in the coexistence region. They reduced their binary system to a.n effective one-component system and applied their unary nuclea.tion theory. Therefore we refer to this model as the quasi-one-component (QOC) model. We will briefiy discuss this QOC model in section 2.2.2.
2.2.1 Binary classical nucleation theory
The formation of a binary cluster is a process in two dimensional size space. Every (nt, n2 )
duster ( n1 molecules of component 1 and n 2 molecules of component 2) is a point on the lattice in the (nt,n2)-space. The kinetic processes to forma {nt,n2)-cluster are the collisions of (n1- i, n 2 - j)- and ( i,j)-clusters or the evaporation of such clusterfrom an (n1 +i, n2 + j)cluster. It is assumed that the (i,j) clusters are either single molecules of component 1 or single molecules of component 2, as shown schema.tically in figure 2.5.
a) b)
(0,1) (n, .~-1 ) (n1 .~)
• + of!. - .2.~ oo• ......--.-,oe
J 11,,1\ J,
~""-of!.+ • eo o• .,__ eo oa (n~o11:!+1) (n1,f10 (0,1)
-- gfi. - olg". + o ttO o• - 4to oe
n, (0,+1,fl0 (n1 .~) (1,0)
Figure 2.5: a) The nb n2 lattice. b) Schematic of the kinetic processes. Upper: condensation or coalescence of a molecule of either of the components on an ( n1 -
1, n2) or ( nl> n2 - 1) cluster. Lower: Dissociation or evaporation of a molecule from an (n1>n2 + 1)- or (n1 + 1, n2)-cluster.
The Gibbs free energy of formation for an (n1 , n2)-duster is according to Wilemski (1987)
AG=(p-p1)V1(n!>n2)+uA(n1,n2)+ L nlAtt;, (2.46) i=1,2
where Àtt; ttl(p1, T)- pi(pv, T). The droplet system consists of three pha.ses, the bulk liquid with nî molecules of component i, the surface with n'[ molecules, and the surrounding vapour

2.2 Binary nudeation theory 29
phase. The chemical potential of the bulk liquid and the surface of the droplet are assumed to be equal, and n! ni + nf . The droplet volume a.nd droplet surface are determined by the composition of the cluster. V1 = Ei;l,2 n~Vj :::::: 4/31ir3 , A = 41ir2• No te, that the surface molecules do not contribute to the volume of the droplet.
The saddle point is de:fined as the point in the ( n1, n2)-space where the partial derivatives of D..G with respect to the number öf mólecules of either of the two components are equal to zero. In appendixBit is shown that this 1eads to the Kelvin equations,
D..p~ + 2o-v; = 0 ' r•
(i 1,2), (2.47)
from which the bulk composition of the critica! clusters ca.n be determined. Here D..pJ Jkl(p, T)- Jtf<p, T).
The composition of the cri~ical cluster ca.n he determined without know:ing the number of molecules on the surface of the cluster. The height of the saddle point is found to he
1 D..G,P = 3o-A,
which is a similar expression as was found for the CNT.
(2.48)
In general, many combinations of clusters create a.n (nt. n2)-cluster. However, we assume that only collisions of single molecules of either of the components or evaporation of these molecules contribute to the formation of a.n (nt. n2) cluster. This is reflected in a condensation rate tensor R with two diagonal terms corresponding to the impingement rates of the molecules of either of the components on a.n ( n1, n2) cluster:
R _ (Ru 0) -o R22' (2.49)
where Ru = .BtA(n1,n2), R22 = ,82A(nl!n2 ), a.nd ,8; the impingement rates of molecules of component i as de:fined in (2.3).
The direction of cluster growth by condensation of single molecules on a.n (n1 , n2)-cluster may not a.gree with the direction of steepest descent of D..G( n1 , n2) near the saddle point. Therefore, a combination of both influences, the steepest descent of D..G, as well as the kinetic "tra.nslation" over the lattice, determine the value a.nd direction of the nuclea.tion ra.te. Furthermore, the nucleation rate is an integral over the ridge near the saddle point as shown in figure 2.6.
The :final expression for the nuclea.tion rate has the form (Looijmans et al. 1995)
(2.50)
where Rav is the average growth rate of the clusters, Z is the Zeldovich factor, Psp is the number density of critical clusters. In appendix B these expressions are worked out in detaiL
Looijma.ns (1995) applied the BCNT to a mixture of n-nona.ne/metha.ne. The necessary expressions for the chemical potentials of the components in the liquid a.nd vapour phase were obtained using the RKS-EOS. We collected the expressions for all thermodynamic parameters

30 Homogeneons Condensation
Figure 2.6: The ridge over the saddle point (SP). The total nucleation rate is the integral of the cluster flux over the ridge.
related to the EOS in appendix A. Macroscopie properties such as the surface tension, are determined by a combination of results from the RKS-EOS and empirical relations from Reid et al. (1987).
In figure 2, 7 we present a p - T diagram with the retrograde condensation curve of nnonane/methane with Yv = 1 · 10-4 and various lines of constant nucleation rate. The lines of constant nucleation rate have a shape similar to the retrograde condensation curve. Changing the temperature from 240 K at 35 bar to 237 K, increases the nucleation rate by 4 orders of magnitude. This again indicates the extreme sensitivity of the nucleation rateon its parameters.
80
60
~ 40 c..
20
0 200 220 240 260 280 300
T (K)
Figure 2.7: p-T diagram. Coexistence curve according to the RKS-EOS for a mixture of n-nonane/methane, Yn-nonane = 10-4 • Lines of constant nucleation rate, Jin m-3 s-1}.
In figure 2.8 we compare the effect of total pressure on the nudeation rate for a system of n-nonanefmethane at constant partial vapour density and temperature, applying the BCNT

2.2 Binary nucleation theory 31
and the CNT. When assuming that methane is inert, the nucleation rate according to the CNT decreases 6 orders of magnitude when pressure is increased from 1 bar to 80 bar according to the Poynting effect.
1012
1010
~ 108 11)
"? 106 E ,9..
104 ..., 102
0 20
Pnonane "' 0.25 moVm3
240K
····-·········· inert gas ... ··············· .....
40 60 80
p (bar)
Figure 2.8: Nucleation rate as a function of total pressure at constant partial density n-nonane: Pn-nonane = 0.25 mol m-3 . T = 240 K. CNT for n-nonane, regarding methane as inert. BCNT for n-nonanejmethane.
For a constant partial density of n-nonane the molar fraetion of n-nonane decreases with increasing total pressure. The equilibrium partial density of n-nonane at constant temperature is an increasing function of pressure, as is shown in figure 2.9. At a certain pressure the value of the satura.ted partial density n-nonane and the chosen value of Pn-nonn.ne in figure 2.8 will become equal a.nd thus S = L Increa.sing the pressure even more leads to an undersa.tura.ted state. Therefore, the nuclea.tion ra.te will eventually become zero according to the BCNT, a.s can be seen in figure 2.8. Starting at low pressures, the mixture is almost an i deal mixture and J is equal to. the unary nucleation rate. With increasing pressure, J initially increases due to the strong pressure dependenee of the composition of the critica! cluster. The molar fraction of methane in such a cluster increases with increasing total pressure, decreasing the surface tension of the cluster. From expression (2.48) it is clear that this will decrease the height of the saddle point and thus increase J. However, the decrease of S when further increasing p will eventually make J vanish.
2.2.2 Quasi-one-component theory
The classica! binary nucleation theory discussed in the previous section uses a model for b.G based on the capillarity approximation. Differences in composition of the surface and the bulk liquid, surface enrichment, and curvature effects are not taken into account. Due to the strong influence of the total pressure on the composition of the critical cluster it is most likely that the curvature effects will be very important for binary nucleation.
The main property for any nucleation theory is the Gibbs free energy of formation of a cluster D.G. Kalikmanov and van Dongen (1995a) proposed a model by which the binary mixture is

32
8
8 "b "':'
4 l :>;
2
I I
I I
I I I // .. y,..., // I /
\ ~,'~ \ -- _,---~ , __________ _
20 40
p [bar]
60
Homogeneons Condensation
0.5
0.4
"" 0.3 .§. .. 0.2 \
ei 0.1
0
Figure 2.9: Equilibrium partial density and molar fraction of n-nonane as a Junction of total pressure according to the RKS-EOS. T = 240 K.
reduced to a qua.si-one-component systern such that !::J.G* is not altered. Once the system is reduced to an effective one component system with effective one-component properties, they apply their semi-phenomenological model for unary nudeation, a.s described in section 2.1.3.
Comparing the expressions for !::J.G* for unary cla.ssical nucl('la.tion theory and binary dassical nudeation theory one can write them in the same form under the assumption that (Kalikmanov and Van Dongen 1995a)
s· = Sf'' s~·· (I + w*), (2.51)
where S* is the effective saturation ratio of the unary system, 81 , and 82 the metastability parameters of both componentsas defined by (1.1), Xts, x2• the molar fractions of both components in the liquid phase at vapour-liquid equilibrium, and I)!* is a correction which accounts for the difference in the composition of the critica! cluster compared to the bulk liquid in the equilibrium state. The equilibrium composition of the liquid at given p, T and 'Uv is calculated on the basis of the RKS-EOS.
In their paper Kalikmanov and van Dongen show that the correction term can be included in the microscopie surface tension as an extra curvature effect,
(2.52)
By oomparing this expression to (2.38), it is clear that a.n- 1113 is the extra correction term. In a modified version of their model (Kalikmanov and Van Dongen ), they show that it is plausible to take h = 2.
In order to apply the unary semi-phenomenological theory as described in section 2.1.3 the unknown parameters q0 , r, a.,., and the fourth parameter, a., have to be determined.
The effective q0 and T are found by introducing mixing rules which in the limit of unary nucleation (x1 or x2 1) yield the known results from section 2.1.3:
qo XtsqO,l + x2.qo,2,
T = X1s1"1 + X2sT2.
(2.53)
(2.54)

2.3 Droplet growth in binary mixtures of real gases 33
Here q0,;, T; are the pure component parameters, which can be found as shown insection 2.1.3. For the two unknowns, a.,., and a., we have the expressions for the effective satura.tion
pressure and the saturation density (2.34) a.nd (2.35) respectively. The value of the saturation pressure is determined with the equilibrium compositions of the vá.pour and liquid phase of the actual binary system by
Psat = [. IT (y?)"?] P· •=1,2
(2.55)
The saturation vapour density is given with the help of the virial expansion to the second order. The virial coefficients for the mixture can be found applying mixing rules for the second virial coefficients of the pure components as given in the text hooks (Reid et al. 1987). From these two equations, the two unknown parameters can be determined.
Once these parameters are found, J can be determined by applying similar expressions as given in section 2.1.3. However, O'micro should be replaced by expression (2.52).
In figure 2.10 the results for the nucleation rate in a mixture of n-nonanefmethane are shown as a function of the saturation ratio of n-nonane. As a reference also the curves according to the BCNT are shown. It is clear that at this temperature, the effective one component model prediets higher nucleation rates at smaller saturation ratios.
10 20 100
s Figure 2.10: Binary nucleation mte as a function of satumtion mtio for various pressures in a mixture of n-nonane/methane at T 240K. Solid line, BCNT, dotted line, QOC-model.
2.3 Droplet growth in binary mixtures of real gases
Once the cluster size becomes much larger than the critica! size, the probabîlity of re-evaporation has become negligible and the newly born droplet will continue to grow. The growth process

34 Homogeneous Condensation
is governed by mass- and heat-transfer towards and away from the droplet. The growth process of dropiets in a gaseous carrier consisting of a perfect inert gas and
a perfect vapour has been subject of many investigations (Maxwell 1877, Gyarmathy 1982, Wagner 1982, Davis 1983). Two explicit growth laws are valid in case the Knudsen number is either very smal! (Kn < 0.01) or very large (Kn > 10). These growth laws are known as the continuurn limit and the free molecular limit, respectively. The Knudsen number is the molecular mean free path length l relative to the radius of the droplet:
where l is given according to Gyarmathy as
l = 2rf'ffiT, p
(2.56)
(2.57)
and 'fJ is the dynamic viscosity of the gas mixture, Ël is the mass averaged specific gas constant of the gas mixture. 1
To determine droplet growth rates in the intermediate range of the Knudsen number, various transition models were suggested (Fuchs 1934, Gyarmathy 1982, Young 1993). Two of them are mentioned in this section: the model of Gyarmathy (1982) and the more recent model of Young (1993). The first model is a modification of an interpolation formula originally proposed by Fukuta and Walter (1956), whereas the latter model is basedon a more rigorous treatment applying a kinetic theory.
Experimental studies of Peters and Paikert (1989, 1994) show a good agreement between measured droplet growth rates in water/argon mixtures and predicted growth rates by the model of Gyarmathy. Looijmans (1995) compared the measured growth rates of water dropiets in a mixture of water and nitrogen with predictions of the model of Gyarmathy and found agreement within 10%. In section 2.3.1 we will show that the differences between the models of Gyarmathy and Young yield differences in growth rates of more than 20% under the conditions of the experiments mentioned. Young's model would yield a less satisfactory comparison with the experimental results. For this reason we adopt Gyarmathy's model as the basis of the droplet growth model for binary mixtures of real gases.
We consider dropiets in a mixture of a gas and a vapour component in the vapour-liquid coexistence region. We assume that
• the droplet is spherical with droplet radius rd.
• the droplet velocity is equal to the velocity of the gas-vapour mixture: no slip.
• the vapour molar fraction is small: Yv ~ 1.
• the total pressure in the gas mixture is less than 100 bar.
1 The mean free path as defined here is related to the gas molecules (y2 > y1) and as such applicable for the process of heat transfer. For dilfusion of the vapour only the mean free path between collisions of a vapour molecule and gas molecules are important, ld. The estimated difference is, e.g. water/nitrogen : ld/1 = 1.1, n-nonanefmethane 0.9, waterfhelium 1.5.

2.3 Droplet growth in binary mixtures of real gases 35
Insection 2.3.1 similar expressions for the mass flow rate M and the heat flow rate Q as given by Gyarmathy are deduced for a mixture of real gases.
The main problem for modelling droplet growth under high pressures in a mixture of real gases is the temperature, pressure, andfor composition dependenee of the diffusion coefficient and ma.ny properties, such as latent heat, and surface tension. In the literature limited data, if present at all, can be found on any of these properties. Therefore, we made use of several (semi-) empirica! relations reported in the work of Reid et al. (1987) in combination with thermodynamic expressions based on the RKS-EOS. The expressions related to the EOS are reported in appendix A.
2.3.1 The Droplet Growth Model
The dilfusion coefficient in liquids is many orders of magnitude less tha.n the diffusion coefficient in gases. We assume that the concentration gradients in combination with the dilfusion coefficient are small enough to neglect diffusion inside the droplet on the time scale of droplet growth, typically 10 ms.
For a binary mixture of real gases the liquid in the droplet consists of both components. In case the droplet would evaporate, diffusion of the most volatile component in the droplet towards the droplet surface would he the limiting process for evaporation (Hage et al. 1993). This may lead to a droplet which becomes overheated such that it actually explodes. These processes are not taken into account in the model presented here. In this section we only describe a growing droplet, i.e. condensation. Further, we assume that (Gyarmathy 1982)
• the temperature in the droplet is uniform and equal to Td.
• the droplet is in quasi-steady equilibrium with the gas mixture in its immediate surroundings.
• dropiets grow independently.
• coalescence, pressure diffusion, and thermal diffusion are negligible.
The system of growing dropiets is divided in equal spherical cells with radius r 00 containing a droplet with radius rd of bulk liquid, indicated by the superscript l, a.n intermediate surface layer of zero thickness, and a bulk vapour phase, indicated by the superscript v and subscript d for the vapour phase immediately near the droplet, and subscript oo far away from the droplet, as shown in figure 2.11.
Mass and energy conservation over the droplet yield
(2.58)
(2.59)

36 Homogeneaus Condensation
Figure 2.11: Schematic of the spherical cell, with radius r00 , containing a liquid droplet with radius rd indicated by superscript l, with droplet temperature Tr1, total pressure rl and mass fraction of the . vapour component Xt· The bulk vapour phase near the droplet is indicated by a superscript v and a subscript d, with temperature Tr1, mass fraction Ytá and pressure p~. The vapour phase far away from the droplet is indicated by superscript v and subscript oo with temperature T 00 , mass fraction ihoo, and pressure p~. The mass flow M and the heat flows Q and Q int are indicated.
where Mis the total ma.ss flow, Q;nt is the heat flow warming the droplet, and h.l the enthalpy of the liquid. The condensing ma.ss flow for each component M; is given by (Oswa.titsch 1990)
M;=x;M, (2.60)
where x; represents the mass fraction of component i in the liquid. From hereon, we a.ssume that component 1 is the vapour component and component 2 is the gas component. The ~ indicates that the quantity is to be considered per unit of ma.ss or related to a ma.ss fraction, wherea.s the symbol without- is per mole or a. molar fraction.
A growing droplet is a heat souree due to the latent heat relea.sed by condensation. The rate of heat relea.sed is the product of the mass flow and the latent heat L and is transferred to the gas mixture, Q, and the droplet, Qint· Energy conserva.tion at the droplet surface yields
ML= Q-Q;nt· (2.61)
The mass flow towards the droplet can be expressed as a. function of the ditfusion flow Jd of the vapour component as (Oswatitsch 1990)
(2.62)
Gyarma.thy shows that even for very fast nozzle flows, the environmental conditions of the droplet vary slowly enough to consider the ditfusion and the heat conduction processes around

2.3 Droplet growth in binary mixtures of real gases 37
the droplet as stationary. Although affected by the high pressures, the transfer processes around a droplet are still to he considered stationary in mixtures of real gases: for a typical radius of O.lj.tm at a total pressure of 100 bar, pressure changes may have a. charaderistic time of w-F s. Thè pressure changes considered in this thesis have a charaderistic time at least one order of magnitude larger.
The solutions to the stationary problem for ditfusion and heat conduction around a sphere in the continuurn limit are known analytically, yielding for Jd to he
(2.63)
Here the subscript m indicates that these quantities have to he evaluated at a temperature between the droplet temperature Td and the gas temperature T00 • Hubbard et al. (1975) show that the 1/3 ruleis appropriate:
1 Tm= 3(2Td + Too)
Expression (2.63) finally leads to an expression for M:
M _ 4 - D (Ytoo- Ytd) roo - 1rrpdm m (- - ) ( rd)' Xtd- Ytd roo
For the heat flow Q the expression at the continuurn limit is
Q 47rrdÀm(Td-Too)( roo ) roo- rd
(2.64)
(2.65)
(2.66)
It will prove convenient to introduce the Nusselt numbers for the transfer processes as
M 21f'rdfidmDm(Ytoo- Ytd)'
(2.67) Num
Nuq = Q
(2.68)
where Num is the Nusselt number for mass transfer and Nuq is the Nusselt number for heat transfer. From expressions (2.67) and (2.65), and (2.68) and (2.66) it follows tha.t for the continuurn limit (indicated by superscript ct)
2 1 roo
(xt- Yld)(roo rd)' = 2 roo
(roo - rd) ·
(2.69)
(2.70)
For a droplet in an infinite surrounding of a mixture of a vapour and an inert gas (x1 = 1) these expressions simplify to the well-known results Nu';= Nu~= 2.
For very large Knudsen numbers, e.g. low pressures, the mass flow depends on the impingement rate /3, (2.3), of va.pour molecules on the droplet. M and Q are given by
M 47rr~(.Btoo - f3ld), (2. 71)
Q (2. 72)
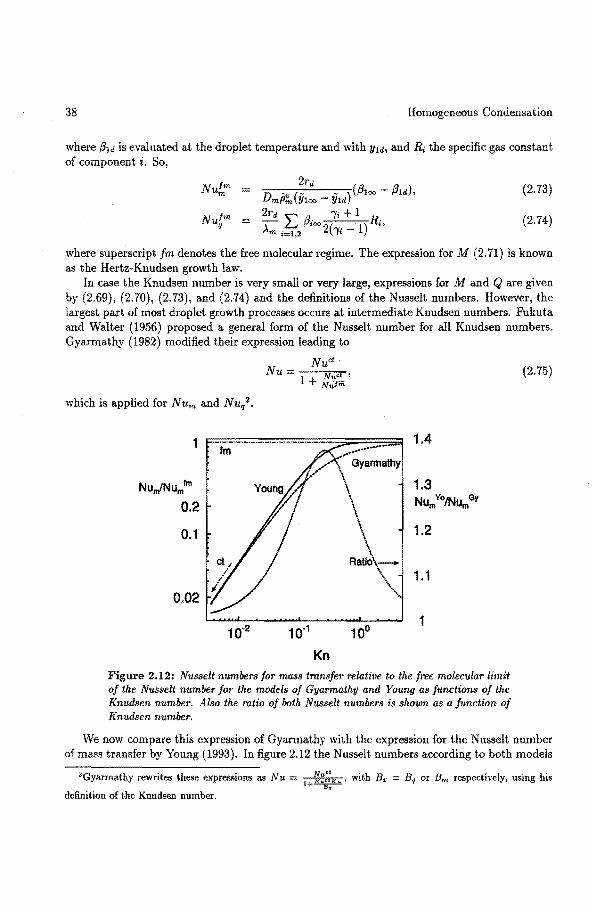
38 Homogeneaus Condensation
where /31d is evaluated at the droplet temperature and with Ytd, and Bi the specific gas constant of component i. So,
Nufm q
(2.73)
(2.74)
where superscript fm denotes the free molecular regime. The expression for M (2. 71) is known as the Hertz-Knudsen growth law.
In case the Knudsen number is very small or very large, expressions for M and Q are given by (2.69), (2.70), (2.73), and (2.74) and the definitions of the Nusselt numbers. However, the largest part of most droplet growth processes occurs at intermediate Knudsen numbers. Fukuta and Walter (1956) proposed a general form of the Nusselt number for all Knudsen numbers. Gyarma.thy (1982) modified their expression teading to
Nu
which is applied for Num andNu92•
1 1.4 " . ...----····
.,.....·\ Gyarmathy
'\ 1.3 NumYo/NUm Gy
1.2
R~ 1.1 .. ,
0.1
0.02
10"1 10° 1
Kn Figure 2.12: Nusselt numbers for mass transfer relative tothefree molecular limit of the Nusselt number for the models of Gyarmathy and Young as functions of the Knudsen number. A lso the ratio of both Nusselt numbers is shown as a function of Knudsen number.
(2.75)
We now oompare this expression of Gyarmathy with the expression for the Nusselt number of mass transfer by Young (1993). In figure 2.12 the Nusselt numbers according to both models
2Gyarmathy rewrites these expressions as Nu l+~~=:K., with B., B1 or Bm respectively, using his ---,;;;--
definition of the Knudsen number.

2.3 Droplet growth in binary mixtures of real gases 39
are shown for water in nitrogen as functions of the Knudsen number. They are presented relative to the Nu/m as given by expression (2.73). The behaviour at intermediate Knudsen numbers is different. The transition regime in Young's model is restricted to a small range of Knudsen numbers frorn Kn = 0.02 to Kn = 0.5. Gyarmathy's model approxirnates the Hertz-Knudsen expression at a. Knudsen number of about 5, and deviations frorn the continuurn expression appear at Kn 0.01.
For an a.rnbient pressure of 1 bar, the mean free path is a.bout 0.1 p,m. The radius of a newly born droplet has a typical value of 1 nm yielding a Knudsen number of 100. A droplet of 1 p,m corresponds to a Knudsen number of 0.1. A droplet growing from its critical size to lp,m spends most of its life-tirne at intermediate Knudsen numbers. At these Knudsen nurnbers, the average growth rate according to Young is more than 20% larger than the growth rate according to Gyarrnathy.
Peeters and Paikert (1989, 1994) rneasured the growth rate of water dropiets in a mixture of water/ argon at pressures below arnbient pressure. They found that the growth curves predicted by the model of Gyarmathy agreed within 10% with the measured growth curves, where they used the Chapman-Enskog relation for the di:ffusion coefficient.
Looijmans (1995) determined the growth curves of water dropiets in mixtures of water and nitrogen. Luijten () found an improved correlation for the di:ffusion coefficient of water in nitrogen with respect to the relation of Fuller (Reid et al. 1987). Applying this rela.tion for the di:ffusion coefficient, the experimentally deduced growth curves of Looijmans agree within 5% with the growth curves according to the model of Gyarmathy.
With increasing total pressure droplet growth will start at smaller Knudsen numbers (for 50 bar, {/2r* (see section 2.1.2) corresponds to Kn = 2). Their final size in the time interval of interest decreases to typically O.lp,m, due to the decreasing ditfusion coefficient with increasing pressure. This corresponds to Kn = 0.02. So, at higher pressures droplet growth will he in the range of Knudsen numbers where the deviation between the two models is largest. Since the predictions of Gyarmathy agree better with the experimental results reported in the literature we use expression (2. 75).
To determine the droplet growth rate for a droplet of given radius rd in a gas mixture at total pressure p~, ternperature T00 and with a rnass fraction of the vapour component Yv, the droplet temperature Td, the composition of the liquid at the droplet surface x1 and the composition of the vapour phase at the droplet surface Ytd have to be determined.
From the metastable equilibrium of the droplet with its immediate surrounding vapour phase, x1 and Ytd are found by the equality of the chernical potentials of both phases for both components:
llrd(p~,Td,fjld) = fl-id(p1,Td,xt), P,~,b~, Td, iJ·u) = fJ~iP1 , Td, x2),
combined with the mechanica! equilibrium of the droplet,
., + u(Td,p1,p~,Xt.Ytd) Poo ·
rd
(2.76)
(2. 77)
(2.78)
Here we assume that the surface tension u exhibits no curvature e:ffects. The surface tension is determined by applying the Macleod-Sugden relation (see appendix A).

40 Homogeneous Condensation
The droplet temperature is found by solving the energy equation over the droplet surface (2.61);
ML = Q -Q;.,.t.
In many cases Q;nt can be neglected (Gyarmathy 1982, Smolders 1992) and this equation reduces to the wet bulb approximation,
ML=Q. (2.79)
The equations (2.76), (2.77), (2.78), and (2.61) or (2.79) can be solved iteratively, using the expressions for the chemical potential and surface tension given in appendix A.
The expressions for the mass and heat flow can he applied for a binary mixture of real gases as well as a mixture of a vapour and an inert gas component. In case of an inert gas component the term (xt - Ytd) in the expression for Num (2.67) must be replaced by unity. The mass fraction of the vapour component near the droplet is found from the Kelvin equation for unary nucleation (2.12). The droplet temperature is found by solving the impHeit eqnations (2.61) or (2.79).
References
ABRAHAM, F. 1974. Homogeneous nucleation theory. New York and London: Academie Press.
ABRAMOWITZ, M., & A. STEGUN. 1965. Handhook of Mathematica[ Functions. New York: Dover.
ALLEN, 1., & J. KASSNER JR. 1969. The nudeation of water vapor in the absence of partienlate matterand ions. J. Colloid and Interface Sci. 30(1):81-93.
BECKER, R., & W. DÖRING. 1935. Kinetische Behandlung der Keimbildung in übersättigten Dämpfen. Ann. Phys. 5(24):719-752.
DAVIS, E. 1983. Transport phenomena with single aerosol particles. Aerosol Sci. Techno/. 2:121-144.
DILLMANN, A., & G. MEIER. 1991. A refined droplet approach to the problem of homogeneons nncleation from the vapor phase. J. Chem. Phys. 94(5):3872-3884.
ERDELYI, A. 1956. Asymptotic expansions. New York: Dover.
FISHER, M. 1967. The theory of condensation and the critica! point. Physics 3:255-283.
FUCHS, N. 1934. Ueber die verdampfungsgeschwindigkeit kleiner tröpfchen in einer gasatmo-sphäre. Phys. Z. Sowjet 6:224-243.
FUKUTA, N., & L. WALTER. 1956. Kinetics of hydrometeor growth from a vapor-spherical model. J. Atmos. Sci. 27(8):116Q-1172.
GYARMATHY, G. 1982. The spherical droplet in gaseous carrier streams: review and synthesis. In Multiphase science and technology, 1. Washington: Hemisphere Publishing Corporation.

2.3 Droplet growth in binary mixtures of real gases 41
HAGE, P., C. HACKENBERG, & R. RANGEL. 1993. Non-ideal vaporization of dilating binary dropiets with variabie properties. Int. J. Heat Mass Transfer 36(15):3731-3741.
HUBBARD, G., V. DENNY, & A. MILLS. 1975. Droplet evaporation: effects of transients and variabie properties. Int. J. Heat Mass Transfer 18:1003-1008.
KALIKMANOV, V., & M. VAN DoNGEN. n.d. Multicomponent homogeneons condensation: an effective medium approach. J. Chem. Phys.
KALIKMANOV, V., & M. VAN DONGEN.l995a. Quasi-one-componenttheoryofhomogeneous binary nudeation. Phys. Rev. E 51:4391-4399.
KALIKMANOV, V., & M. VAN DoNGEN.1995b. Semiphenomenologicaltheoryofhomogeneous vapour-liquid nucleation. J. Chem. Phys. 103:4250-4255.
KATZ, J., & M. DONOHUE. 1979. A kinetic approach to homogeneons nudeation theory. Adv. Chem. Phys. 40:137-155.
KATZ, J., & H. WIEDERSICH. 1977. Nucleation theory without Maxwell demons. J. Colloid Interface Sci. 61(2):351-355.
LooiJMANS, K. 1995. Homogeneous nucleation and droplet growth in the coexistence region of n-alkane/methane mixtures at high pressures. Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.
LOOIJMANS, K., C. LUIJTEN, G. HoFMANS, & M. VAN DoNGEN. 1995. Classica! binary nucleation theory applied to the real mixture n-nonane/methane at high pressures. J. Chem. Phys. 102 (11):4531-4537.
LOTHE, J., & G. PoUND. 1962. Reconsiderations of nudeation theory. J. Chem. Phys. 36:2080-2085.
LUIJTEN, C., K. BossCHAART, & M. VAN DONGEN. n.d. A new method todetermine binary diffusion coefficients in dilute condensable vapors. submitted for publication.
MAXWELL, J. 1877. Diffusion. Encyclopedia Brittanica. 2:82.
ÛSWATITSCH, K. 1990. Verdampfen und Kondensieren binärer Gemische als Strömungsproblem. Wärme und Stofübertragung 25:45-58.
ÛXTOBY, D. 1992. Homogeneons nucleation: theory and experiment. J. Phys.: Condens. Matter 4:7627-7650.
PETERS, F., & B. PAIKERT. 1989. Nucleation and growth rates of homogeneously condensing water vapor in argon from shock tube experiments. Exp. Fluids 7:521-530.
PETERS, F ., & B. PAIKERT. 1994. Measurement and interpretation of growth and evaporation of monodispersed dropiets in a shock tube. Int. J. Heat Mass Transfer 37(2):293-302.
REID, R., J. PRAUSNITZ, & B. POLING. 1987. The Properties of Gases and Liquids. New York: McGraw-Hill Book Company.

42 Homogeneous Condensation
REISS, H. 1950. The kinetics of phase transitionsin binary systems. J. Chem. Phys. 18(6):840-848.
SMOLDERS, H. 1992. Non-Linear wave phenomena in a gas-vapour mixture with phase tran· sition. Ph.d. thesis, Eindhoven University of Technology, Fa.culty of Applied Physics.
STAUFFER, D. 1976. Kinetic theory of two-component ("heteromolecula.r") nucleation and condensation. J. Aerosol Sci. 7:319-333.
TOLMAN, R. 1949. The effect of droplet size on surface tension. J. Ohem. Phys. 17(3):333-337.
WAGNER, P. 1982. Aerosol growth by condensation. InW. MARLOW (ED.), Aerosol Microphysics, Berlin. Springer.
WILEMSKI, G. 1987. Revised classica! binary nucleation theory for aqueous alcohol and a.cetone vapors. J. Phys. Chem. 91(10):2492-2498.
YOUNG, J. 1993. The condensation and evaporation of liquid dropiets at arbitrary Knudsen number in the preserree of an inert gas. Int. J. Heat Mass Transfer 36(11):2941-2956.
ZELDOVICH, Y. 1943. Acta Physicochim. (URSS) 18.

Chapter 3
HOMOGENEDUS CONDENSATION DUE TO A
CONTINUDUS ADIABATIC EXPANSION
In the previous chapter, models fordroplet formation and droplet growth in a supersaturated vapourjgas mixture were discussed. The mixture ca.n he brought in a supersaturated state by an adiabatic expansion in for example an expansion cloud charnber, a two piston doud chamber, or an expansion/shock tube. The pressure history of the expansion determines how the homogeneons condensation process occurs.
In this chapter, the homogeneons condensation process due to a continuons adiabatic expansion in a.n expansion cloud chamber will he treated. As mentioned in chapter 1, the homogeneons condensation process during such an expansion is characterized by the simultaneons occurrence of droplet formation and droplet growth. The strong dependenee of J on the saturation ratio and the temperature results in a pulse-like nucleation rate as a function of time. During this nucleation pulse new dropiets are formed and earlier formed dropiets grow, thereby cha.nging the composition of the vapour/gas mixture. The molar vapour fraction1 91 starts to decrease, while the molar liquid fractions of both components 9~ and g; increases. This diminishing 91 causes the nucleation rate to attain a maximum and to decrease rapidly. The saturation ratio is still rather large and droplet growth continues. During this period the molar liquid fractions increase swiftly until the saturation ratio approaches unity.
Insection 3.1 the governing equations for the homogeneaus condensation process in a binary mixture of real gases are discussed. They serve as the basis from which expressions for a mixture of a vapour and an inert gas are easily deduced. The set of equations is solved numerically and the numerical method we used is described in section 3.3.
As mentioned in the first chapter, to detect homogeneaus condensation experimentally, we use light scattering techniques and light attenuation. The Mie-theory (Mie 1908) is an exact solution of the Maxwell equations for a dielectric sphere in a plane electromagnetic wave. This theory yields light scattering efficiencies and effective cross sections for light extinction by spherical droplets, as described in section 3.2. In combination with the numerical solution of
1To prevent difficulties in case of binary condensation the vapour is indicated by the subscript 1 and the gas component by the subscript 2. The molar vapour fraction of either of the components in the vapour phase is related to the total amount of both components present in the system. 91 = ntf(nt + ni + n2 + n~), while Yl = n1 /(n1 + n2). However, in case ni, n~ < n2 than g = y.
43

44 Homogeneaus condensation due to a continuons adiabatic expansion
the condensation process due to the adiabatic expansion, optical properties can be calculated as functions of time.
Some results for various thermadynamie and optical properties during the homogeneons condensation process in mixtures of water and nitrogen and in mixtures of n-nonane and methane are discussed insection 3.3.
The key property for the homogeneaus condensation process due to a continuons adiabatic expansion of a vapour and an inert gas is the molar fraction of liquid formed. This property is mainly determined by art integral over time of the nucleation rate. Since this is a narrow pulse it will appear that asymptotic techniques can be applied to find an approximate solution for 9{(t).
Blythe and Shih (1976) were the first to solve the problem of homogeneons condensation in nozzle flows analytically based on asymptotics. Clarke and Delale (1986), and Delale et al. (1993a, 1993b) modified and improved their work. Delale et al. (1995) also solved the problem of condensation in shock tubes by the same asymptotic techniques.
In section 3.4 we show analytica! solutions of the homogeneons condensation problem due to a continuons expansion for a prescribed pressure signa! as a function of time. The asymptotic expressions found are very complex and rather lengthy, therefore a general description of the asymptotic technique is given and some main results are shown. The full description of this analytica! work is given in Delale et al. (1996).
3.1 Governing equations
Consider a binary mixture of a vapour and a gas at an initia! pressure of p0 , an initia! temperature of To and an initia! molar vapour fraction of 91,0 . The mixture is endosed in a volume that changes such that the pressure is a known function of time, p(t). At t 0 the expansion starts. Due to this expansion the mixture will become supersaturated and homogeneaus condensation will occur. By simultaneous nucleation and droplet growth, liquid is formed and the composition of the vapour/gas mixture changes. Conservation of mass yields
91,0 = 91 + 9L 92,0 92 + 9~,
(3.1)
(3.2)
where 91,o, 92,0 are the molar vapour and gas fraction at t = 0, and 9l is their molar liquid fraction at time t. In case of an inert gas component the second expression simplifies to
(3.3)
since only the vapour component condenses.
The molar liquid fractions are for small liquid fractions given by the condensation rate equations written as
l t -1 3 1 4/37rp;rd(r,t) -( )J(r,91 )dr,
t, p T (3.4)

3.1 Governing equations 45
with
rd(r, t) = ro(r) + l O(t',gi, r)dt', (3.5)
where r 0 is the size of the newly born droplet and Pl = x;/ is the pa.rtialliquid number density of component i.
The time interval of integration is from the moment the vapourfgas mixture becomes saturated t. until the actual moment in timet. The last part of the integral of expression (3.4), J( r )dr, is the number of dropiets formed per unit of volume in the time interval between r and r + dr, p(r) is the number density of the vapourfgas mixture at timer, and 1/p(r) accounts for the ongoing expansion by which the molar density of already formed dropiets decreases. The total number of molecules of component i present in the droplet is given by the volume of a droplet formed at moment r and the average partial liquid number density pj. For a changing thermodynamic state and droplet radius, the composition of the liquid condensing on the droplet changes, see section 2.3. Since we neglect ditfusion in the droplet, its partialliquid molar density will he radius dependent and the evaluation of the total number of molecules of either of the componentsis given by a volume integral over the partialliquid number density2•
The radius of a droplet is characterized by the size at birth of the droplet r 0 and the integral of the growth rate 0 over the time interval between birth and the actual moment in time. The growth rate in general depends on the thermodynamic state and the radius of the droplet.
For one-component condensation, expression {3.4) reduces to
I r 411" I 3 J(r,gD g1 = lt, 3P1 [ro(r,t)] --pr;:)dr. (3.6)
The expansion is considered to be adiabatic, but condensation is an internal heat souree by the release of latent heat. Therefore, applying the wet bulb equation for droplet growth (Gyarmathy 1982), conservation of energy yields,
dh dp lt p-d = -d + (L(t, r) t t t,
1 p(t) t:.h(t, r)) M (t, r) p(
7) J( r)dr, (3.7)
where p is the molar density of the mixture, h is the molar enthalpy of the mixture in the vapour phase, M' is the molar flow condensing on a droplet formed at t = r, (M' = M/(x1m1 +x2m2)),
and L is the latent heat defined by,
(3.8)
where L1 and L2 are the molar latent heats of component 1 and 2 given in detail in appendix A. The condensing mixture removes enthalpy from the vapour/gas mixture surrounding the droplet with a different molar average than for the mixture itself. The last term under the integral in expression (3. 7) takes that into account, where t:.h is
(3.9)

46 Homogeneons condensation due to a continuons a.diabatic expansion
Again for one-component condensation the conservation of energy can be expressed in a less complex form as
(3.10)
Here, L is the latent heat of the condensing vapour component, which can be found explicitly as a function of temperature for various substances in the literature, e.g. for water see appendix A. Writing the rate of enthalpy change of the mixture as a rate of temperature change reduces the term with !::..h as in (3.7) to ep1(Td- T00 ). The temperature di:lference between a growing droplet and the surrounding vapour /gas mixture is for water vapour typically 10 K for low tot al pressures and becomes negligible at high pressures. Therefore, when compared to the latent heat of the oondensing vapour it is neglected.
Fora frozen expansion, where M' = gl = 0 for all t, expression (3. 7) reduces to the isentropic relation, which yields,
(3.11)
For a mixture of an ideal gas and an ideal vapour a 7, where ')' is the ratio of the compositional average of the specific heats,"'=~· Fora mixture of real gases a is a function of temperature and pressure and includes the Joule-Thomson cooling. The exponent a is then given by (Looijmans 1995)
a= !!:_z (1 + ~ ( 8z) ] . Cpm Z lJTP
(3.12)
The compressibility factor Z is calculated from the RKS-EOS.
3.2 Light scattering by small spherical particles: Mie-theory
A droplet in a beam of light will scatter light in all directions. The droplet size is of the order of the wavelength of visible light. Light scattering by such dropiets is described by the Mie-theory, which gives an exact solution of the Maxwell equations fora plane electromagnetic wave incident on a dielectric spherical particle. Both the scattering partiele and the surrounding medium are assumed to be homogeneons and isotropic3 •
The intensity of the scattered light is a function of the scattering angles 0 and /i, the radius of the droplet rd relative to the wavelengthof the light a 21f-rd/ >.., and the refra.ctive index of the droplet m relative to the surrounding medium. The definition of the two scattering angles and a schematic of a scattering partiele are shown in figure 3.1.
In several text hooks (Bohren and Huifman 1983, Kerker 1969, Van de Hulst 1981) the Mietheory is explained in detaiL Expressions for the intensity of light scattered in a certain direction are given. In this study we only consider single sca.ttering, light scattered once by one droplet is not scattered by another droplet. In figure 3.2 we show a.n example of the intensity of the
3 Although dropiets formed by binary homogeneaus condensation are assumed to. have a radius dependent composition, the effects on the refractive index are negleeted. Optically the dropiets are taken uniform and isotropie with a refractive index determined by the vapour component.

3.2 Light scattering hy smal! spherical particles: Mie-theory
x
Figure 3.1: Geometry of scattering of light by a spherical particle. ti is the angle between the plane of scattering and the plane of polarization and () is the angle between the scattering direction and the direction of propagation of the incident wave.
47
scattered light as a function of the size parameter in a direction perpendicular to the beam of light illuminating the droplet. The refractive index for the dropiets is 1.405, which corresponds to n-nonane.
The intensity of light scattered hy particles with a size of the order of the wavelength of the light is characterized by the typical maxima and minima as shown in figure 3.2. It depends on the angles of detection at what sizes the extrema occur, while the refractive index influences the structure of the total pattern. These extrema in the scattered intensity have proven to he an accurate means to determine droplet growth (Wagner and Strey 1981 ).
For a monodispersed cloudof dropiets the intensity of the light scattered by a single sphere I. must he multiplied by the total number of scatterers N to yield the total intensity of scattered light. In case, the dropiets do not have the same size, the total scattered intensity 11 in a
0
88.85<9<91.15.
84 <li< 911"
~. w 5 10
a;- 21tr/À
w \fi J,_ ~
15 20
Figure 3.2: The intensity of scattered light as a function of the size parameter a 2ndf>.. m 1.405.

48 Homogeneons condensation dne to a continuons adiabatic expausion
certain direction is given by an integral over the size distribution function F(r) multiplied by the scattered intensity I.(O,ó,a) of a droplet of certain size rd:
(3.13)
Here F(rd)drd is the nnmber of dropiets with a droplet radius in between rd aud rd + drd. The energy of the scattered light is withdrawn from the incident light beam. The trausmitted
light beam will therefore he attenuated according to the law of Lambert-Beer:
I= Ioexp(-fJL), (3.14)
where I, I 0 are the intensities of the light beam with aud without auy scatterers respectively, L is the optical path length through the scattering medium, and (:J is the extinction coefficient. Absorption of light by the scatterers wilt be neglected since the imaginary part of the refractive index for the vapour components considered in this study is very small. The extinction coefficient is related to the total intensity of light scattered, aud for a monodispersed clond of dropiets it is given by (Van de Hulst 1981)
fJ = mrr3Qe:ct(a,m), (3.15)
where nis the droplet number density, aud Qem is the e:lfective cross section for light extinction, specified in the text hooks (Kerker 1969, Bohren and Hu:lfmau 1983). In figure 3.3 we show Qe:ct as a fundion of a for a refra.ctive index of 1.33 (water). For small a the extinction efficiency is varying with a 4 corresponding to the Rayleigh limit. With increasing size Q..,t increases at first until it reaches a maximum. Then Qezt shows a damped oscillation aud approaches a constant value of 2 as a -+ oo, the diifraction limit.
ot • 2n;rn..
Figure 3.3: Extinction efficiency Q..,1 as a function of a: for a refractive index of 1.33.
For a polydispersed cloud of dropiets the total extinction coe:lficient is given by an integral over size of the size distribution function and the extinction coefficient of a droplet with radius

3.3 Numerical method 49
(3.16)
Here f(rd)drd is the number density of dropiets with a radius in between rd and rd + drd.
To detect homogeneons condensation during a continuons adiabatic expansion in an expan· sion doud chamber we use the attenuation of light of two different wavelengtbs (section 4.1.1). Therefore, we give an expression for the extinction coefficient /3 as a function of time during the homogeneons condensation process in terms of nudeation rates and droplet sizes. With expression (3.16), this leads to
!3(t) 11 2 p~) 1frd(t,r) Q •• :t(a:(t,r),m)-( )J(r)dr.
I, p T (3.17)
Here ;[ ~ J ( r )dr represents the number density at time t of dropiets formed between r and
r + dr, which is f(rt:~)drd.
3.3 Numerical metbod
Expression (3.4) is a non-linear Volterra integral equation coupled to the conservation equations (3.1), {3.2), and (3.7). Due to the implicit expression for the droplet growth rate (2.61), it is only possible to find the solutions of these equations numerically. Especially, for a binary mixture of real gases the more complex thermodynamica make it even more difficult and numerical methods are inevitable.
To he able to calculate g) as functions of time, expressions for the nucleation rate J and the droplet growth rate !l are needed. These are taken from chapter 2, where we discussed various nudeation models for unary as well as binary nucleation and the droplet growth model of Gyarmathy which was extendedtoa binary mixture of a real gas and a real vapour. The expressions are completed with the thermodynamic expressions basedon the RKS-EOS, given in appendix A.
Numerical scheme
We applied an explicit first-order differential scheme to determine the time history of the homogeneons condensation process during the adiabatic expansion. The total outline of the numerical FORTRAN-code is shown in the flowchart of figure 3.4. Several routines from the NAG-library are used. ·
At time tn, the pressure, temperature, g1 and gj are known. These are kept constant during a timestep !::.t. Also the number densities and radii of earlier formed dropiets are known.
New dropiets are formed with a nudeation rate J(tn), and their number density is Pd,n = J(t")!::.t. Since they are formed during one time step, they canon the average grow for only

50 Homogeneous condensation due to a continuous adiabatic expansion
Figure 3.4: Flowchart of the numerical code for simulating the homogenrous condensation process in a binary mixture of a vapour and a gas. The pressure history is a known function of time p(t). To prevent extensive computing time, a small value for J is taken as a lower limit for droplet formation. The actual moment in time is indicated by n while the moments of birth of earlier formed dropiets is indicated by i.
half the time interval. The radius of a newly bom droplet at the end of the time step is given by
(3.18)
where ro(tn) ~r*(tn) for unary nucleation as shown insection 2.1.2. For binary nucleation a similar expression for r0 could not he obtained. We take r0 r* + Ll, where Ll has been determined by trial a.nd error from the saddle point region and the direction of steepest descent.
Existing dropiets grow during the time interval with a growth rate n( tn, 91 ( tn), r( T, tn) ). At the end of the timestep their radius has increased to,
(3.19)
By the increase of droplet volume multiplied by their number density and the partial liquid number density of the condensing liquid for all T, the change in 9! is known. At the end of the timestep this yields the corrected molar fraction of both components in the

3.3 Numerical method 51
vapour phase for the next moment in time according to (3.2). Also the extinction coefficient for light of a given wavelength is calculated according to expression (3.17).
Conservation of energy is split in two parts. First the amount of latent heat released is cakulated sinûlarto gJ. Then the temperature is correctedat constant pressure (p(t,.)) neglecting the temperature differences between dropiets and vapour/gas mixture and the pressure derivative of the enthalpy. This yields
,.,.. ( M' fl.tL )n,tota/ J.n+ .
C"mP (3.20)
After which the pressure is reduced instantaneously to p(t"+l) in an isentropic way, (3.11) yielding Tn+l·
The numerical convergence of this first-order scheme as to he expected is linear with the size of the timestep Ll.t. Knowing this, wedetermine the "exact" solution by extrapolating between solutions at the samemoment in time found from calculations using different time steps. This is done in the following way:
F _ F( Ll. ) _ Ll. (F(t,Ll.t2)- F(t,Ll.tt)) exact - t, t1 t1 (fl.t
2 fl.tl) , (3.21)
where Ll.t2 > ótt. and F can be g!, the nucleation rate J, the average droplet radius, the temperature T, saturation ratio S, or the extinction coefficient fJ. In appendix 0 we show that this "exact" solution is indeed independent of the the timesteps Ll.t1 and Ll.t2•
Results
The pressure history is assumed to be a known fundion of time. This can either be the experimental pressure signa! during an expansion of a vapour/gas mixture in an expansion cloud chamber or a presumed analytica! form. For the results presented in this chapter we assume that
p(t) = Po(0.05 + 0.95exp( -t/t2)),
where t2 is the cha.racteristic time of the expansion.
(3.22)
First we discuss the expansion of a mixture of water and nitrogen. Nitrogen is considered to be an inert gas component. The nucleation model is the CNT and the droplet growth model of Gyarmathy for one component is used under the wet-bulb approximation (2. 79) (Smolders 1992). The initia! conditions are: po = 5 bar, To = 295 K, and Pvo 1000 Pa. In figure 3.5 the nucleation rate, the molar liquid fraction, the supersaturation and the pressure are shown as functions of time. With decreasing pressure and temper at ure, the saturation ratio increases and dropiets are formed. Growth of these dropiets combined with simultaneons droplet production increases the molar liquid fraction and decreases the mola.r vapour fraction. Eventually, this leads to a maximum in the nucleation rate followed by a maximum in the saturation ratio. With decreasing temperature and sa.turation ratio, the nucleation rate rapidly diminishes and only droplet growth continues, hereby decreasing the saturation ratio.
With the formation of dropiets the light extinction coefficient as given in (3.17) increases. This is shown in figure 3.6 for light of two wavelengtbs in which also the nucleation rate is

52 Homogeneous condensation due to a continuous adiabatic expansion
t [ms]
Figure 3.5: Homogeneaus condensation process in water/nitrogen for an initial pressure of 5 bar, initial temperature 295 K, initial partial vapour pressure 1000 Pa. The characteristic time of the expansion t2 = 10 ms. Jmaz = 5.1 · 1017 m-3
Sma.:z: = 17.9-f.
shown. It is clear that the extinction coefficients increase most after the nucleation period. Very small dropiets are uneffeetive scatterers. The largest dropiets contribute most to light attenuation close to the Rayleigh limit, where {3 varies with r6 and one droplet of 2r is as
10
a 0.8
'i 6 o.s I cO. ~
4 0.4
2 0.2
0 0 5 10 15 20 25
t [msJ
Figure 3.6: Nucleation rate and extinction roefficients for light of two wavelengths À1 = 543.5 nm, Àz = 815 nm as functions of time during the homogeneaus rondensation of water due to a continuous adiabatic expansion of water and nitrogen: t 2 = JO ms, Po 5 bar, To = 295 K, Pv0 = 1000 Pa.

3.3 Numerical method 53
e:ffective for light extinction as 64 dropiets of size r. The extinction efficiency attains a maximum for au a of about 5. From then on it decreases
aud light with the smallest wavelength would he attenuated less strongly thau the light with the larger wavelength. From figure 3.6 it is clear that for this example the extinction coefficient /3s43.s is always larger thau /3815 aud so the average droplet radius is smaller than about 0.5 p,m (R! a= 5), as is shown in figure 3.7.
Figure 3.7: Size distribution function at t = 10 ms during the homogeneons con~ densation process of water. Expansion of water and nitrogen: t2 = 10 ms, Po = 5 bar, To = 295 K, Pv,o = 1000 Pa.
After maximum nucleation, the saturation ratio decreases and droplet growth rates become smalL The droplet radii attain a final value. However, the droplet number density decreases due to the ongoing expausion. Therefore, the extinction coefficient attains a maximum, where the growth aud expausion balauce, after which its va.lue decreases.
In figure 3.8 the influence of the total pressure is shown ·for au expausion with au initia! pressure of 50 bar. The nucleation rate attains higher values compared to the situation for an expausion starting at 5 bar. This is caused by the smaller droplet growth rates due to a smaller di:ffusion coefficient. The molar fraction of liquid formed needs more time to deplete the vapour enough to yield a maximum in the nucleation rate (about one order of magnitude di:fference). So, S increases to higher va.lues lea.ding to higher nucleation rates and more but smaller dropiets will be the final result of this expausion. This is also dear from the moment in time the nudeation rate attains its maximum value. For the results shown in figure 3.5 this is at about 8.5 ms while for the results shown in figure 3.8 this is at about 9 ms. The higher total pressure has an effect, similar to a fa.ster expansion (t2 smaller in (3.22)). In a faster expausion the charaderistic time for droplet growth increases with respect to the cooling rate of the mixture. Larger supersaturations are attained since droplet growth is not capable of depleting the vapour sufficiently. The result is a cloud of dropiets consisting of more but smaller dropiets than for slower expausions.

54 Homogeneous condensation due to a continuons adiabatic expansion
time [ms]
Figure 3.8: Pressure, nucleation rote, saturation ratio, and molar liquid fraction as functions of time. Homogeneaus condensation processin water/nitragen for an initial pressure of 50 bar, initial temperature 295 K, initial partial vapour pressure 1000 Pa. t2 = 10 ms; lmos = 6.2·1018 m-3 s-1, Smax = 22.64-
For the extinction coe:fficient we see in figure 3.9 that its maximum value decreases rather drastically to about 1.5 for light with a wavelength of 543.5 nm. Although more draplets are formed, their decreased size with respect to the situation shown in figure 3.6 has a larger effect
2
1.5 0.8
':' 0.6 lil .§.
E
cc. ~ 0.4
0.5 ~~----- 13u15 0.2 ," -... --......... _____
I
0 0 5 10 15 20
time [ms]
Figure 3.9: Nucleation rate and extinction coefficients for light of two wovelengths Àt 543.5 nm, À 2 = 815 nm as functions of time during the homogeneaus conden-Bation of water due to a continuous adiabatic expansion of water and nitrogen: t2 ·
10 ms. Po = 50 bar, To 295 K, Pv0 = 1000 Pa.

3.4 Asymptotic method 55
on the extinction coefficient and in total this decreases the extinction coefficient.
For a mixture of n-nonane and metha.ne we applied the BCNT in combination with the droplet growth model iil binary mixtures or real gases under the wet bulb approximation. In figure 3.10 a.n expa.nsion of such a mixture is shown at an initial pressure of 50 bar, initial temperature of 295 K, and initia! molar fraction of n-nonane of 1 · 10-4 •
0.8
0.6
0.4
0.2
5 10 t[ms]
15 20
Figure 3.10: Pressure, temperature, nucleation rate, saturation ratio, and malar liquid fraction as functions of time. Homogeneaus condensation process in nnonanejmethane for an initia/ pressure of 50 bar, initia/ temperature 295 K, initia/ molar fraction ofn-nonane 1·10-4
• Jmax = 5.9·1018 m-3 s-1, Sma~~: = 122.5.
The shape of all functions shown in figure 3.10 is idenÜcal to the ones shown previously. The saturation ratio attains highervalues while the nucleation rate has its maximum value at the same order of magnitude as for a.n expansion of water and nitrogen a.t an initial pressure of 50 bar.
3.4 Asymptotic method
In this section we consider . a mixture of a vapour and an inert gas, which is expanded in an expansion cloud chamber. The pressure signal is a. known function of time. The initia! conditions of the mixture are given by initia! pressure Po, initial tempera.ture To, a.nd initia! molar vapour fraction 9t,o·
With decreasing pressure and temperature, the saturation ratio increases, which leads to the simultaneons formation and growth of droplets. The molar fraction of liquid formed is the key qua.ntity governing the homogeneons condensation process. Taking the general form of the nuclea.tion rate as
(3.23)

56 Homogeneons condensation due to a continuons adiahatic expansion
the molar liquid fraction can, in combination with (3.1) be expressed as,
I = r 411' I [ ( t)]3 Jo(r,gD ( ilG*(gi,r))d Yt lt. 3 p r r, p(r) exp kT T.
(3.24)
The exponential term in this expression is responsihle for the extreme dependenee of J on S and T, which is the reason for the pulse shaped nucleation rate during the continuons adiabatic expansion.
The evaluation of equation (3.24) can be simplified by a.pplying asymptotic techniques. The asymptotic techniques provide us with an analytical structure of the condensation process in asymptotically distinct intervals. The actual models for droplet growth and nucleation and the components of the mixture can he left arbitrary until the end.
To he able to use the asymptotic techniques, we first normalize all variables such tha.t their va.riational part is of order unity. The Gibbs free energy of formation ilG* I kT is expressed as BIK, where B is the dimensionless activation function of order unity and K is the nucleation parameter. The nucleation parameter is a smal! quantity cortesponding toa high activation energy, K <: 1.
An analytical solution of ui during the homogeneons condensation process can only he given when the droplet growth rate n can he expressed as an explicit growth law. Therefore, the energy equations (2.61) or (2.79), are approximated by the assumption that Td = T for all droplets. Further, the Kelvin effect is neglected. The terms preceding the exponential term in (3.24) are norma.lized such that they CIIJl he written as >.3 h(t,r}, where À is related to the model for droplet growth.
aÎ i !lîne l
... ::~::::::! ' ' i IGZ! ~ + l
I FGZi ' :
ts At
B
~-G-~ .. J NZ ! DGZ
~~-~--f-·-·Bt
Figure 3.11: The activation function B as a function of time during the homogeneaus condensation process due toa continuous adiabatic ezpansion of a vapaurjgas mixture. The different asymptotic zones are indicated as well as the Taylor approximations of B: first-order in the initial growth zone (line) and second-order in the rnpid growth zone (parnbola).

3.4 Asymptotic metbod 57
In figure 3.11, the typkal shape of B(t) is shown for the nucleation pulse as presented in figure 3.5. Also various periods in time are indicated using the notation of Delale (1993a). At t. the saturation ratio is unity and B is infinitely large. With increasing saturation ratio, B decreases leading to an increasing J. Initially dropiets are formed in very low concentrations and the growth of these dropiets does not influence the thermodynamic state. The temperature is found from the pressure and the isentropic relation (3.11). This is .an interval in time which we will refer to as initial growth wne IGZ. The activation function is equal tothefrozen (gi = 0) activation function B,. With ongoing expansion, J increases further and Yi starts to have a small influence on B such that B differs numerically from B,. This interval in time where the infl.uence is present but rather small corresponds to the further growth zone FGZ. In the next period the influence of g{ on B increases and the shape of B starts to deviate significantly from the solution without condensation B 1. This period is referred to as the rapid growth zone, RGZ. This zone ends at t., where the nucleation rate attains its maximum value corresponding toa minimum for B. The following period is called the nucleation zone NZ. Here, the nucleation rate decreases rapidly to zero. Now, droplet formation has stopped and only droplet growth continues. This period is referred to as the droplet growth wne DGZ.
These zones differ also analytically. If K -+ 0 the main con tribution to the integral equation (3.24) arises from the neighbourhood of the moment in time where B attains its smallest value. This makes it attractive to apply tbe asymptotic metbod of Laplace (Erdelyi 1956).
Up to the moment t. where B attains its global minimum, the smallest value of the activation function in the interval of integration is the end point t. When applying LapJace's metbod for the evaluation of the condensation rate equation, the activation function B must be known well for an interval of time !::.t preceding t sucb that D.B over that interval is large compared to K. Then, the contributions to tbe integral (3.24) for tbe preceding time interval, t. < T < t D.t, can be neglected.
In the IGZ .and the FGZ, as is shown in figure 3.11, the linear approximation of B is sufficient. The tangent of the activation function at t is a good approximation for B(t) over a time interval from t - D.t to t. Tberefore, in these wnes B is described by a tirst-order Taylor expansion at T = t, which yields
dB B(r) = B(t) + dtfr=t(T- t). (3.25)
The tirst-order approximation is no longer sufficient in tbe RGZ, wbere, by definition, the second derivative of B with respect to time becomes important. So, B is approximated by
dB lJlB B(r) = B(t) + dtlr=t(T- t) + 2 dt2 lr:t(T t?. (3.26)
Higher order terros are not taken into account. The rapid growth zone is valid up to tbe moment of maximum nucleation t.. There, the
first order derivative of B is zero. For t > t. the minimum value of B will be witbin the interval of integration and tbe activation function is given by
{3.27)
This interior minimum approximation is again divided in two wnes. In the first, the nucleation zone, the variation of B is such tbat in tbe interval between t. and t nucleation is still

58 Homogeneons condensation due to a continuons adiabatic expansion
significant. This NZ is a rather short period of time in which it is assumed that the droplet growth rate remains constant. In the following period the nucleation has stopped and only growth of the earlier formed dropiets contributes to the integral (3.24). In this zone g~ rapidly increases by vapour depletion and the growth rate relaxes to zero.
Normalization and droplet growth rates
These approximations for B valid in the different zones willlead to analytica! expressions from which gi(t) can he determined.
To solve the problem of homogeneons condensation in a vapour/gas mixture analytically, all quantities will he normalized and a general form for the explicit droplet growth rate and the nucleation rate are given.
The expression for the droplet growth rate combined with the appropriate expressions for B in the various zones, determine whether the integral equation (3.24) is analytically solvable. Blythe and Shih (1976) showed that forthefree molecular limit this is possible, while Clarke and Delale ( 1988) generalized the growth equation to hold for any droplet growth law in between the Hertz-Knudsen limit and the continuurn limit:
drm
dt (3.28)
where mis a constant; m = 1 corresponds to the Hertz-Knudsen limit (HK), and m 2 to the continuurn limit (CT).
In general however, the droplet growth will take place at intermediate Knudsen numbers and will he radius dependent, as was discussed in section 2.3. A model such as Gyarmathy's growth model (even when excluding the Kelvin effect) does not yield analytica! solutions for g{ in the various asymptotic zones. Delale et al. (1996) formulated an explicit droplet growth rate U for arbitrary Knudsen numbers, yielding an !lm in between the growth rates according to the modelsof Young and Gyarmathy. This model resulted in analytica! expressions for the condensation rate equation for the asymptotic zones. The resulting analytica! expressions are very complex and lengthy. Although physically more sound, the final form of g{(t) and J(t) as functions of time are comparable to the results for the relatively more easy cases of HertzKnudsen and continuurn growth laws. Therefore, we will restriet ourselves in this section to the two limiting situations for droplet growth: m = 1 and m = 2.
In normalized form the growth rate yields
dr" = Àmfim(il!,T), (3.29)
where Àm is the growth parameter and the quantities, indicated by a bar, are normalized with respect to the moment the vapourjgas mixture is saturated t., subscript s:
OT
P - T p=-,T= !h - p p=-,
p. Yl,s Ps
- t r t = -,f= -,
tr rn
(3.30)
(3.31)

3.4 Asymptotic method 59
h t . h . al f h . t l dp I d .!\ Bl,Of/s J f 11 f w ere r IS t e t1me sc e o t e expans10n, r = --d t=O, an r;;, = 4 J -1' n o ows rom p t 3'/f ntrl'
the normalization of the nucleation rate, which can be expressed as
( t::.G*) J = E(gt,T)exp - kT .
In normalized form this leads to
So Jn is the normalization constant of the prefactor E.
Asymptotic solutions for various zones
Substituting the normalized quantities in (3.24) yields
with
-I Bt
t Ï:(f) ).3/m f M-exp(-K- 1B)df
m lt. p(r) '
(3.32)
(3.33)
(3.34)
(3.35)
Startingat the IGZ or FGZ, we substitute the fust-order Taylor approximation (3.25) in expression (3.34) and apply Laplace's metbod for the end point minimum. In good approximation ui hecomes
(3.36)
This can he worked out in more detail and the final analytica! solution of this integral is given in the appendix of the paper of Delale et al. (1996).
Expression (3.36) contains the first derivative of B with respect to time at r = t, which is given by
dB oB oBdüi = at+ oui df' (3.37)
where oB/ot is the separate effect of the expansion while the latter term on the RHS of expression (3.37) is the effect of condensation.
Delale et al. show that the energy equation coupled to the ideal gas equa.tion of state and the known pressure history yield explicit expressions for the temperature and number density of the gas mixture. For now it is suflident to know that T(l, gi) and p(t, gi) are known functions of their parameters. So, the partial derivatives of B with respect to either time or ffi are known and can he determined from the nucleation.model, p(l) and the relations for T and p. For the evaluation of (3.36) and (3.37) it is clear that also dgifdt must be known. It can be found from

60 Homogeneons condensation due to a continuons adiabatic expansion
the analytica! expression for Bi· The expressions for gi(f), dgifdt, and expression (3.37) form a transeendental system for Bi and dgUdt, which can be solved iteratively starting from the known frozen solution Bi = 0.
In the RGZ the activation function is approximated by asecond-order Taylor expansion. The molar liquid fraction is then,
-I Bt =
This expression can he solved analytically and the results are shown in Delale et al. (1996). This expression for ui combined with
(3.39)
and the expressions for duUdt and tPüUdP, form thesetof equations for the RGZ. For the nucleation zone, the integral is determined by an interior minimum for which the
first derivative of B with respect to r is zero. This leads to
-I Bt =
(3.40)
Once t. is found, the second derivative of B with respect to t at t. is known. The liquid fraction can he found directly as shown in Delale et al. (1996).
For the droplet growth zone, vapour depletion becomes important and the growth rate Öm varies with time. Here, it is assumed that up to l. the growth rate is constant and equal to Öm(t.) while for f > l., Öm varies. The solution for ui (t) takes the form
1 [-]3/m Bt = R , (3.41)
where R satisfies the relaxation rate equation
dR Öm - = ------=---' dx Om(t.)
(3.42)
and x is a rescaling of time, A(f- f.), where A is related to the second-order time derivative of Bat l
Matching between the different zones
IGZ and FGZ

3.4 Asymptotic metbod 61
From the known pressure history and the relations for temperature T( t, g{) and number density p(t,gi), the activation function forthefrozen situation, g{ = 0, can be found. From the frozen solutions, B1 and dB1/dl, we evaluate the expressions for 9i (3.36) and diA/dl. The solution for g{ is now re-entered in the relations for temperature and number density, yielding a new value for B(t) and dB/dt. These again determine g{ (3.36) and dg{/ dl and this processis repeated until it converges. Once the solution is found for one moment in time, Band dB/dl are the input parameters for the progressing moment in time.
The second-order Taylor approximation for B in the rapid growth zone is also applicable in the IGZ and FGZ. However, the influence of the second derivative is negligible in the first two zones. Still, the expressions for ui and its time derivatives in the RGZ are evaluated. The moment the solutions for g{ according to the expressions for the IGZ/FGZ and RGZ start to deviate, the rapid growth zone starts.
NZ and DGZ
With increasing g{, the nucleation rate will attain a maximum value at t •. This corresponds to the moment in time dB/ dl is zero. At that moment the NZ starts.
From the moment of maximum nucleation rate, g{ will increase rapidly. Vapour depletion becomes important and the droplet growth rate will decrease rapidly. This variation of the droplet growth rateis given by a relaxation rate equation ( Delale et al. 1996). The initia! value for the relaxation process is found as a solution to the smooth matching of ui in the NZ to the solution in the DGZ.
Results
We now compare the results obtained from the asymptotic metbod to the results obtained from the numerical simulation. For both calculations we use either the Hertz-Knudsen ot the continuurn growth law, without the Kelvin-effect. The nuclea.tion rate is prescribed by the CNT.
A mixture of water and nitrogen is expanded a.diabatically with a pressure history as prescribed in expression (3.22) for three different expansion rates. The initial state of the mixture is Po 5 bar, To 295 K, and P'!JO = 1000 Pa.
In figure 3.12 the nucleation rates according to the numerical and asymptotic metbod with the Hertz-Knudsen growth law are shown as functions of time. The fastest expansion reaches the highest nucleation rates at the latest moment in time relative to t 2 , so the lowest temperatures. As explained previously, the growth of dropiets in a fast expansion does not have the time to deplete the vapour sufticiently, so more dropiets are formed compared toa slow( er) expansion. For the IGZ and the FGZ the solutions are very close to the frozen solutions and the results for the asymptotic method and for the numerical simulation are identical. In the rapid growth zone the nucleation rate according to the asymptotic method is larger than for the numerical method. The maximum nucleation rate is attained later in time and reaches a value about one order of magnitude larger tha.n Jma:c for the numerical method.
This can be understood when looking at the second derivative of B with respect to time. In

62 Homogeneons condensation due to a continuons adiabatic expansion
0.5 0.75 1
Vl.:! 1.25 1.5
Figure 3.12: Nucleation rate according to the numerical method and the asymptotic method as functions of time for the Hertz-Knudsen growth law. A continuons expansion of a water/nitrogen mixture. a: t2 = 10 ms. b: t2 = 1 ms. a: t2 = 0.1 ms. Po = 5 bar, To = 295 K, Ut,o 2 · w-3
•
figure 3.13, the first and second derivative of B with respect to time are shown as functions of time during the homogeneons condensation process according to the asymptotic method. The first derivative of B with respect to time is an increasing nmction of time and becomes zero at t •. The second derivative decreases in the IGZ and the FGZ and becomes almost constant at the beginning of the RGZ. A pproaehing the moment of maximum nucleation d2 B / dt2 increases again strongly. Then, approximating the activation function by a Taylor series up to the second-
:i .!li.
~ :;
.!li. -,
~ 0 't:l
4.5 5 5.5 6 6.5 7 7.5
t[ms]
Figure 3.13: The first and second derivative of B with respect to time according to the asymptotic solution for the homogeneous condensation process of water in water/nitrogen due to a continuous adiabatic expansion. Po 5 bar, To = 295 K, Yt,o == 2 · I0-3 •

3.4 Asymptotic method 63
order near te underestimates the molar liquid fraction. The parabola as sketched in tigure 3.11 will not be a good approximation during the whole interval b.t which contributes most to the condensation rate equation. The actual activation function B( r) will be overestimated by the parabola and consequently the nucleation rate will be underestimated for a part of the integration interval. This leads to a value for gi which is too small. The vapour depletion will be underestimated and the nucleation rate will attain higher values than for the numerical simulation.
Figure 3.14: Molar liquid fraction as functions of time for the Hertz-Knudsen growth law. According to the numerical method and the asymptotic method. A continuous expansion of a water/nitragen mixture. t2 = 10 ms. Po = 5 bar, To = 295 K, 91,0 = 2 · 10-3 .
In tigure 3.14 the molar liquid fractions for the numerièal and asymptotic method are shown. As discussed, the asymptotic result for 9i attains smallervalues in the RGZ than the numerical result. However, due to the higher value of Jmax, the molar liquid fraction in the droplet growth zone increases more rapidly and even becomes larger for the asymptotic metbod compared to the numerical solution. Eventually, since both calculations were done with the same amount of water vapour, the relaxation of 9i will yield identical results, 9i --+ 1.
The continuurn results for the nucleation rate for a mixture at an initia! pressure of 50 bar are shown in figure 3.15. The differences between numerical and asymptotic results are smaller due to a smaller growth rate in the continuurn range for the dropiets which number density is underestimated by the second-order expansion of B.
The results for the nucleation rate show a rather large difference between the approximate asymptotic solution and the more rigorous numerical solution. The onset of the molar liquid fraction occurs near the end of the nucleation pulse. Globally the solutions for both methocis predict the same interval in time where the molar liquid fractions increases from zero to unity. During this period all of the latent heat is released. For nozzle flows this leads to a static pressure which shows a small spatial disturbance at some distance from the nozzle

64 Homogeneons condensation due to a continuons adiabatic expansion
10~.--------------------------. --· nurrerical solution - asyrllliOtic solutlon
Figure 3.15: Nucleation rate according to the numerical method and the asymptotic method as functions of time for the continuurn growth law. A continuous expansion of a water/nitragen mixture. t2 = 10 ms. Po= 5 bar, To = 295 K, 91,0 = 2 ·10-3 •
throat, see figure 3.16. For this phenomenon asymptotic techniques proved to yield satisfactory results (Blythe and Shih 1976, Clarke and Delale 1986, Delale et al. 1993a), in the sense that the position of the onset of g{ is predicted well.
Figure 3.16: Lower: Static pressure along the axis of a nozzle. The static pressure in case ( no) condensation occurs is indicated as a (b). U pper: Homogeneous condensation due to the nozzle :flow of a vapour/gas mixture.
However, when applying the asymptotic approximation for opticalproperties, the size distribution function is the most important quantity. Since the nucleation pulse is predicted poorly, the asymptotic method can not be applied to determine extinction coefficients accurately. Still,

3.4 Asymptotic metbod 65
in section 5.3 we wiJl propose a metbod to determine nucleation rate data from transmission measurements during a continuons adiabatic expansion of a vapour/gas mixture for which we were inspired by a result of the asymptotic approximation.
References
BLYTHE, P ., & C. SHIH. 1976. Condensation shocks in nozzleflows. J. Fluid Mech. 76{3):593-621.
BOHREN, C., & D. HUFFMAN. 1983. Absorption and scattering of light by smal! particles. New York: John Wiley & Sons.
CL ARKE, J., & C. DELALE. 1986. Nozzle fiows with nonequilibrium condensation. Phys. Fluids 29(5):1398-1413.
CL ARKE, J., & C. DELALE, 1988. Expansion flows on walls with nonequilibrium condensation. Quarterly of Appl. Math. 46(1):121-143.
DELALE, C., M. MUITJENS, & M. V. DONGEN. 1996. Asymptotic solution and numerical simulation of homogeneaus condensation in expansion cloud chambers. J. Chem. Phys. 105(18):accepted for publication.
DELALE, C., G. SCHNERR, & J. ZIEREP. 1993a. Asymptotic salution of transonic nozzle flows with homogeneons condensation. I. subcritical flows. Phys. Fluids A 5(11):2969-2981.
DELALE, C., G. SCHNERR, & J. ZIEREP.1993b. Asymptoticsolutionoftransonicnozzlefiows with homogeneons condensation. II. supercritical flows. Phys. Fluids A 5(11):2982-2992.
DELALE, C., G. SCHNERR, & J. ZIEREP. 1995. Asymptotic solution of shock tube fiows with homogeneaus condensation. J. Fluid Mech. 287:93-118.
ERDELYI, A. 1956. Asymptotic expa~sions. New York: Dover.
GYARMATHY, G. 1982. The spherical droplet in gaseaus carrier streams: review and synthesis. In Multiphase science and technology, 1. Washington: Hemisphere Publishing Corporation.
KERKER, M. 1969. The scattering of light and other electromagnetic radiation. Academie Press, New York.
LOOIJMANS, K. 1995. Homogeneaus nucleation and droplet growth in the coexistence region of n-alkanejmethane mixtures at high pressures. Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.
MIE, G. 1908. Beitrage zur Optik trüber Medien speziell kolleidaler Metallösungen. Ann. Phys. 25:377-445.
SMOLDERS, H. 1992. Non-Linear wave phenomena in a gas-vapour mixture with phase transition. Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.

66 Homogeneons condensation due to a continuons adiabatic expansion
VAN DE HULST, H. 1981. Light scattering by smal/ particles. New York: Dover.
WAGNER, P ., & R. STREY. 1981. Hornogeneous nucleation rates of water vapor rneasured in a two-piston expansion charnber. J. Phys. Ghem. 85(18):2694-2700.

Chapter 4
EXPERIMENTAL METHOD
In chapter 3, the homogeneaus condensation process due toa continuons adiabatic expa.nsion of a vapour/gas mixture was discussed. Solutions for the nucleation rate, the molar liquid fraction a.nd the extinction coefficients were found numerically. For a mixture of a vapour and a.n inert gas also an a.na.lytical approxima.tion for the liquid mola.r fra.ction as a function of time was discussed.
To study homogeneaus condensation due to a continuous a.dia.batic expa.nsion experimentally, we developed a.n expa.nsion cloud cha.mber suited for pressures up to 100 ba.r. Insection 4.1 the expa.nsion cloud cha.mber is described in more detail as wellas the procedure preceding a.n experiment. To detect homogeneons condensation a. method basedon light attenuation was used. The optical set-up is described in section 4.1.2. A gas-analysis method is applied to determine the initial composition of a mixture of n-nona.ne/methane prior to an experiment, which is described insection 4.2.1. Some typical signals measured during an experiment with the expansion cloud chamber are discussed in section 4.3.
4.1 The expansion cloud chamber
In essence, the expansion cloud chamber consists of a.n expansion cell connected to a lowpressure tank via an electroma.gnetic va.lve. When the valve is opened, the mixture of vapour and gas will flow to the low-pressure tank. The pressure and temperature in the expa.nsion cell will decrease a.nd homogenrous condensa.tion will occur. The homogeneons condensa.tion process during the expansion in an expa.nsion cloud cha.mber is infiuenced by the initial state of the vapourfga.s mixture a.nd the expansion ra.te tr (3.4).
4.1.1 The experimentalset-up
The expa.nsion cloud chamber is shown schematica.lly in figure 4.1. The expansion cell Eis a sta.inless steel cell with a tota.l volume of about 4 · 10-5 m3 •
From the bottorn plate up to the first three centimeters in height the cross-section of the test
67

68 Experimental method
Figure 4.1: Schematic of the expansion cloud chamber.
cellis rhombic (see section 4.1.2). Then it gradually changes to become axially symmetrie with a fin al diameter of 8 mm at the orifice 0. The minimum surface area of the orifice determines the rate of expansion (tr ), slnce there the gas flow will be lirnited by the choking condition. The orifice is formed by two parallel rods perpendicular to the gas flow. As shown in figure 4.2, each of the rods has three notches of different size, which in various orientations determine the surface area of the orifice in between 8 mm2 and 50 mm2• It is possible to change this surface area of the orifice easily, even at a pressure of 100 bar inside the expansion cell.
In the bottorn plate of the expansion cell two pressure transducers are mounted. The transient pressure in the expansion cell is measured by a piezo-electric pressure transducer (Kistler, type 603B) and the initia! pressure is measured by a piezo-resistive transducer (Druck type PDCR 200), which is suited for total pressures in between 10 and 100 bar. During an expansion, the veloeities of the vapour/gas mixture in the expansion cell are small compared to the speed of sound except near the orifice. So, the pressure measured at the bottorn plate is equal to the pressure at the core of the expansion cell. During the expansion the pressure is known with a relative error of 5 · 10-3 .
In each of the side-walls of the rhombic part of the test cell a window (MK7) is placed to make the expansion cell optically accessible. A fifth window is positioned in the centre of the bottorn plate.
A stainless steel plate of 5 mm thickness is inserted just above the observation windows. The plate is perforated with about 100 small cylindrical holes (diameter 1 mm ). Any instahilities moving upstream in the direction of the windows are assumed to break up at the plate1•
1It was observed that in the absence of this plate a laser beam passing through the expansion cellis deflected in a stochastic manner during an expansion of only a gas component. It was also observed that this disturbance

4.1 The expansion cloud chamber
cfcp-·-1 ~ jau~
c1~-·- ~ j Gullow
~P-'- 9 Figure 4.2: The orifice of the expansion cell given for a side and upper view. The gas flow is indicated by the arrows for the side view, and is perpendicular to the plane of the figure for the upper view. Some of the orientations of the rods are shown including the minimal A .and maximal surface area C.
69
The expansion cellis connected toa large low-pressure tank V with a volume of 3 · 10-3
m3 via an electromagnetic valve M (Asco SCX B223A003). The low-pressure tank is suited for pressures up to 100 bar. So, the relative expansion depth can he varled in between zero and the volume ratio of the low-pressure tank and the expansion cell ( ~ 1/75). The initia! pressure inside the low-pressure tank is measured by the sametype of piezo-resistive pressure transducer as mounted in the expansion doud chamber (see figure 4.1).
The low-pressure tank is connected to a vacuum pump (Balzers DUO 1.5A) which can evacuate the whole system to a pressure less than 100 Pa. At about a pressure of 300 Pa, a turbo pump (Balzers TPH 055) can be activated, which reduces the pressure to less than 1 Pa (measuring uncertainty of a low-pressure detector (Edwards, barocel, type 600 AB) situated in the mixing circuit LP).
The expansion doud chamber is connected to a mixing circuit via a piston valve in the bottorn plate of the expansion cell and a piston valve above the orifice. The main part of this mixing circuit is a mixing pump developed by Looijmans (1995). The direction of the gas flow induced by the mixing pump is indicated in figure 4.1. The performance of the mixing pump can be monitored by a transient pressure transducer (Kistler, type 603B) near one end of the pump.
lnitially, when the set-up is evacuated, the vapour component is injected into the set-up in liquid form through a septurn (Chrompac, chromsep blue) with a syringe (Hamilton 10 pl- 2.5 ml) at the injection point lP. The gas component can belet in by opening a pneumatic ball-valve GI. For experiments with water, the relative humidity is measured by a Humicap (HMP 124 B).
moved away from the orifice in the direction of the bottorn plate, independent of the orientation of the expansion cell with respect to gravity. We condurled that the special shape of the expansion cell may cause flow-instahilities moving upstream in the gas flow. This leadstostrong density gradients ~n the gas which deflect the laser beam. By inserting the plate these effects were suppressed.

70 Experimental method
The whole set-up is placed in a thermally controlled room, the temperatnre of which ca.n be regulated in between 10 oe and 35 oe. The initial temperature is measnred with a PtlOO with an uncertainty of 0.1 K.
4.1.2 Opties
To detect the process of homogeneons condensation during a continuons expansion in an expansion cloud chamber we measure the at tennation of two laser beams of different wavelengths. The lasers are a He-Ne laser (Melles Griot, À = 543.5 nm, 1.5 mW) and asolid state laser (Philips, type QL70, À = 815 nm).
lens
~--~~~~' Diode
À=815nm
Îl.=543.5nm
Figure 4.3: Schematic of the optical set-up for the expansion cloud chamber.
In figure 4.3 the optical set-up is shown. A laser beam passes through the expansion cell via the windows in the side walls of the cell. The laser beam is focussed (J = 10 cm) on a photodiode through a diaphragm with a diameter of 1 mm. The aperture of this set-up is kept as smal! as possible to avoid detecting light scattered in forward directions. The signal-to-noise ratiosof the laser-diode combinations are 2000 for the infrared laser and 1000 for the "green" laser.
The four windows are placed in the rhombic part of the expansion cell in the same horizontal plane as shown in figure 4.4. Two windows are facing each other, while the third window is shifted with respect to the axis of the fourth window. A laser beam passing through the expansion cellis reflected at the glass/gas interfaces. The reileetion originating from the glassjgas interface entering the inner part of the expansion cell ( 1) is indicated in figure 4.4. This beam while being reflected inside the first window at interface (2) and exiting at the same interface

4.2 Experimental procedure
Figure 4.4: Cross-section of the rhombic part of the expansion cell. The Jour windows are shown. Two windows are facing each other. The other two are slightly shifted. The laser beam passes through the first window. The reflection of the laser beam, starting at the windowjgas interface at the inside of the expansion cell, is also shown. To prevent this reftection from interfering with the original beam, the test cell is rhombic and the windows are shifted.
71
( 1) as it was formed, transverses through the expansion cell parallel to the initia.l beam. If the laser beam has a diameter la.rger than the geometrica.l difference between the reflected and initia.l beam, they pa.rtia.lly overlap. This can lead to interference at the optica! detection system when a relative phase change occurs (by inhomogeneons changes in windows or vapour /gas mixture). To prevent this, the beam has to be tilted with respect to the plane of the surface of the window, thus increasing the spatia.l difference between the two beams such that they are geometrically separated. The rhomb shape ensures that a laser beam transversing horizontally is a.lready tilted with respect to the interface of the two windows. The windows are shifted with respect to each other because the beam is displaced by a sma.ll distance as shown in figure 4.4. The angle of 84° is chosen to create a geometrie& difference between the reflected and initia.l beam of 2 mm which is about twice the diameter of a He-Ne laser beam. The second laser passes through the other two windows and is tilted itself with respect. to the glass/gas interfaces of those windows.
4.2 Experimental procedure
To perform an experiment, first the system of expansion cell, mixing circuit, and low-pressure tank is evacuated. The vacuum pump is active forabout 1/2 hour fora series of experiments invalving the same vapour/gas mixture. When an experimental series with a different vapour component starts, the turbo pump is activated for at least two days, Collowed by an experiment& run of severa.l "dry" experiments with only the gas component of interest to ensure tha.t all of the former vapour component is gone. Finally theset-upis evacuated again fora. 1/2 hour.
Then the magnetic va.lve is closed, sepa.rating the low-pressure tank from the rest. The desired amount of vapour component is injected by a syringe at the injection point (n-nonane: Fluka chemika :> 99%, distilled water). The vapour has to spread through the set-up first

72 Experimental metbod
and after about 10 minutes, the gas component is let in (Hoekloos: nitrogen > 99.9%, helium > 99.996%, methane > 99.5%) until the desired initial pressure is reached. Meanwhile the mixing pump is activated. Once the mixture has reached the initia! total pressure the pneumatic hall valve GI is dosed and the miocture is circulated foranother 5 minutes. While activated, the temperature of the moving parts of the mixing pump increases rapidly and this influences the temperature of the vapour f gas mixture. Temperature differences of a few degrees between parts of the mixing pump, mixing circuit and the expansion cell are observed. Therefore, the pump is cooled by air (metal parts) and water (coils) and is activated for periods of notmore than 10 minutes. This is controlled by monitoring the temperature at various positions in the mixing circuit, which are kept within 0.1 K. Every 1/2 hour the pump is reactivated for 10 minutes. This is repeated for at least 2.5 hours.
In case the vapour component is water, the relative humidity can he monitored as a fundion of time through the output of the Humicap. In figure 4.5 a typical output signa! is shown schematically. At the moment of injection the humidity increases almost stepwise. Adsorption and a.bsorption of the water vapour at the walls of the system decrease the vapour pressure exponentially in time. By filling the set-up with the gas component, the concentration of water decreases locally due to the dry gas, but the mixing pump restores a homogeneaus mixture within 5 minutes. The magnetic valve does not seal off the cloud chamber instantly, but only from the moment the pressure difference over the valve is more than 1 bar. So at the moment the pressure increa.ses from almost vacuum to about 1 bar some of the water vapour is flusbed out of the set-up into the vacuum tank. This is the reason for the lower level of rela.tive humidity obtained after the mixing pump restored a homogeneaus mixture. The temperature of the mixing circuit and the vapour/gas mixture is affected by the heat genera.ted in the mixing pump. The remaining effects are due to temperature distributions in the mixing circuit. After 3 hours the variations of the relative humidity when activating the mixing pump are within 1 % of the relative humidity.
Adlabsorption
1
Gasinlet
' / Teflll"'lllureeflect
0 0.1 0.4 0.75 1.5
t (hours)
2.25
Figure 4.5: Schematic of a typical Humicap output as a function of time.

4.2 Experimental procedure 73
4.2.1 Initial composition of the vapour/gas mixture
Once the mixture of a vapour and a gas component is homogeneously mixed, an experiment can he 'done. However, the initia! state characterized by the initial pressure, temperature and composition of the gasfvapour mixture, must he determined. The initial pressure is determined from the piezo-resistive pressure transducer, while the initial temperature is determined with a PtlOO. -
At the beginning of the experimental procedure the expansion celland mixing circuit are evacuated and the vapour component is injected in liquid form. The liquid will (partially) evaporate încreasing the vapour pressure in the system of mixing circuit and expansion cell. The vapour will spread through theset-upand partially adsorb on the roetal surfaces and absorb in the 0-rings present in the set-up. The amount of vapour component already present on the walls or in 0-rings also affects the fina.l vapour pressure. By injecting the gas component the total pressure increases and the equilibrium between ad/absorption and desorption of the vapour component may change. Especially when consirlering a mixture of n-nonane and methane, the solubility of n-nonane in methane is astrong function of pressure, see figure 2.9. So, the initial composition of the vapour/gas mixture is determined by the amount of liquid injected in the set-up, the history of the set-up and the desired total pressure. A direct relation between the amount of liquid injected and the initial molar vapour fraction can not be deduced and the initial molar vapour fraction must be measured.
The metbod to determine the initial composition of the mixture depends on the mixture of interest. In this study the mixtures are water in nitrogen or helium, and n-nonane in methane.
In case of water-vapour the Humicap is used todetermine the relative humidity. However, it appeared that the gas component and its total pressure affect the capacitance measurement of the Humicap. For a mixture of water and helium the calibration at low-pressures proved to be valid for high pressures also, when including the Poynting effect. The uncertainty in the calibration is 5%.
For a mixture of water and nitrogen the Humkap showed a strong pressure dependenee and could not he calibrated accurately enough (inaccuracy > 10%). Therefore, only saturated water/nitrogen mixtures were prepared. The composition was determined by the saturated partial vapour pressure given by the enhancement factor (appendix E) and the saturated vapour pressure of pure water.
Fora mixture of n-nonane and methane, on-line measurement of the n-nonane concentration as the Humicap does for water, could not be realised. Therefore, a gas analysis method based on gas chromatography was applied.
In figure 4.6 the sampling set-up is shown schematically. A tube segment, containing an adsorption material (J.T. Baker, Bakerbond Octadecyl, 40 p,m), is mounted toa valve in the mixing circuit. At the other side of the adsorption column, it is connected to two valves. Through one valve an inert gas component can be let in to pressurize the adsorption tube up to the same pressure as in the mixing circuit. The second valve is a needie valve. The outlet of the needie valve is connected to a plastic tube, which leads to a measuring jug placed upside

74 Experimental metbod
Mixing cirt:u~
Gasvolume
Figure 4.6: Schematic of the sampling set-up for the compositional analysis of n-nonanejgas mixtures.
down under water. To take a sample, the first valve is opened to pressurize the adsorption tube. Once this is
clone the valve is closed a.nd the valveto the mixing circuit is opened together with the needie valve. The small outlet of the needie valve ensures that the gas sample can be taken in a controlled way without decreasing the pressure in the mixing circuit too much. The adsorption tube takes out all n-nonane from the mixture passing it. The remairring metha.ne is collected in the measuring jug a.nd when the volume of the gas sample is about 0.25 l the valve at the mixing circuit is closed. The remaining pressure in the adsorption tube decreases nuther until it dropped to ambient pressure. Then, the needie valve is closed a.nd the total gas volume is measured. This sample volume is corrected for the volume of the inert gas to pressurize the column. From the final volume of the gas out of the mixing circuit, Vg (typically 250 ml ± 2.5 ml), the ambient pressure p0 , the water temperature Ta a.nd the ideal gas law, the total moles n2 of metha.ne ca.n he determined:
( 4.1)
The n-nona.ne is desorbed from the adsorption material by flushing it with n-hexa.ne (MerckSchuchardt: > 96% ). The liquid mixture of n-nona.ne and n-hexa.ne with a total weight of Ms is analysed with a gas chromatograph (GC-a.nalysis). This analysis yields the mass concentration Ct of n-nonane in n-hexa.ne (uncertainty of 10%). From this the total number of moles n1 of n-nona.ne adsorbed in the tube ca.n he determined:
(4.2)
where m1 is the molar weight of n-nona.ne. The ratio of the number of moles of n-nonane in the sample a.nd the number of moles of the
gas component yields the initial molar concentration of n-nona.ne: y1,0 = nt/(n1 + n 2 ) .
After the mixing procedure, three samples are taken as described above. Then the piston valves a.re closed, the initial pressure and temperature are determined a.nd an experiment is

4.3 A typical experiment 75
done by opening the magnetic valve. The transient pressure and the intensities of the two laser beams are recorded hy a Lecroy 6810 wave form recorder.
4.3 A typical experiment
In this section we will describe some typical experimental signals. A mixture of water and nitrogen was prepared. The initial pressure is 58.5 bar, the initial temperature is 21.9 ac, and the initia! saturation ratio is l.O.
In :figure 4. 7 the pressure and the extinction coeflicients for the light of the two laser beams are shown as functions of time. The arrow indicates the moment in time the magnetic valve is opened and the pressure starts to decrease. The time scale originates at the moment the pressure transducers triggers the Lecroy wave form recorder. Momentsin time before the trigger pulse are labeled with a negative value.
The extinction coeflicients show a small increase as soon as the pressure starts to decrease. This small variation is caused by the change in the reileetion coeflicient at the glassfgas interfaces at the inside of the expansion cell. The density of the vapour f gas mixture determines its refractive index. With decreasing density the refractive index of the vapour/gas mixture decreases giving rise to an increasing reileetion coeflicient. So the intensity of the light beam arriving at the photodiode deercases hy twice the change in reflection coeflicient. For an isentropic expansion of nitrogen from 50 bar to 10 bar the decrease of the transmitted intensity was 0.002. 50
40 0.8
30 0 0.6 .e. i E
0. i 20 as:: 0.4 I /,, ____
11 - 10
0.2 /f •I
0 0 -10 0 10 20 30 40 50
time [ms]
Figure 4.7: Typical experiment: Pressure, f3>.=Sl5nm 1 and JÎ>.=543.5nm as functions of time. The mixture of water and nitrogen has initial pressure 50 bar, initia/ temperature 293.2 K, and initia! saturation ratio 1.0.
The very drastic changes in both extinction coeflicients at t 12 ms correspond to the moment where homogeneons condensation hecomes detectable. The light with the shortest wavelength yields the largest size parameter and therefore the extinction coeflicient for the

76 Experimental metbod
light of 543.5 nm increases more rapid than for the extinction coefficient for the light of 815 nm. We define the Wilson point as that state of the expanded gas at which the extinction coefficient of the 543.5 nm laser beam has a value of 0.07. In this definition we corrected for the effect of the changing reileetion coefficient at the glass/gas interfaces. The temperature is determined from the pressure history, the initia! temperature and expression (3.11).
As described in chapter 3, the extinction coefficients at first increases very rapidly due to the simultaneous formation and growth of droplets. Even when nudeation bas stopped, droplet growth leads to a strong change in {3. However, as the saturation ratio decreases to unity, the droplet growth rate diminishes and the effect of the continuons expansion beoomes apparant. Due to the decreasing pressure, the number density of the dropiets decreases, resulting in a decreasing extinction coefficient.
Camparing both extinction coefficients for the light of the two different wavelengths, it is dear that the {3 for the 815 nm beam is always smaller than for the 543.5 nm beam. The extinction efficiency bas a maximumfora size parameter of about 5 (rd ~ 0.5p,m ). If the average droplet size for the experiment shown here would he larger than this 0.5p,m, the extinction coefficient for the 815 nm beam would attain values close to or even larger than the extinction coefficient of the green light, À = 543.5nm. As a condusion we can say that for the experiment considered in figure 4. 7 the average droplet size is much less than 0.5 p,m. The behaviour of the extinction coefficients for both wavelengtbs differ in the sense that the relative decrease of f3s43.s following the moment it attained its maximum value is more rapid than for {3815• So, the average size parameter for the "green" light will be in a less steep area of Qe:ct than for the infrared light.
References
LOOIJMANS, K. 1995. Homogeneaus nucleation and droplet growth in the coexistence region of n-alkane/methane mixtures at high pressures. Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.

Chapter 5
NUCLEATION RATE DATA FROM HOMOGENEOUS
CONDENSATION EXPERIMENTSIN AN EXPANSION
CLOUD CHAMBER
As shown inthelast chapter, the intensity of light transmitted through a continuously expanding vapour/gas mixture exhibits a rapid change at the moment homogeneons condensation occurs. Wilson-pointscan he determined very easily from such signals, but information on nudeation rates or droplet growth rates can not he ohtained directly. In chapter 3 we showed that the extinction coefficient can be calculated numerically for the homogeneons condensation process due toa continuons expansion of a vapour/gas mixture. The similarity between the results from the numerical simulation and the deduced extinction coefficients from the experiments is obvious.
In the next section we present a method to determine nudeation rate data from the extinction coefficient of laser light measured during experimentsin an expansion doud chamber. It is based on a comparison of the experimental extinction coefficients and the numerically simulated extinction coefficients using the measured pressure history of the experiment. Results for water/helium and water/nitrogen mixtures are presented insection 5.1, and for n-nonane/methane mixtures in section 5.2.
In the last section, 5.3, of this chapter, we will propose a different method for which the beginning of the nucleation pulse is approximated by a Gaussian shape. Then, it is possible to deduce nudeation rate data from the experimental extinction coefficient in a small period of time during the expansion without any a priori knowledge of a nucleation modeL
5.1 Homogeneons condensation of water vapour
In chapter 3 we discussed the governing equations of the homogeneons condensation process due to a continuons adiabatic expansion. As stated there, it is possible to calculate optica! signals, iri partienlar extinction coefficients. This enables us to compare these numerically generated signals with the actual measured transmission coefficient.
The main parameter determining the onset of the extinction coefficient is the nucleation rate. The moment of maximum nucleation rate can he regarcled as the origin in time for the
77

Nuclea.tion ra.te data. from homogeneaus condensation experimentsin a.n expansion cloud 78 cha.mber
changes in all optical properties of the vapour/gas mixture.
1~r-------------------~
" " .. ..
220
<>-<l Vtisanen et al . .. looijmans
240
T[K)
260
Figure 5.1: Nucleation rate data of water relative to the nucleation rate according to the CNT as a function of temperature. Low pressure data. Circles: Viisanen et al. {1993), Inverse triangles: Looijmans {1995,1996).
Wegener (1964) measured the static pressure deviations in nozzle flows due to the homogeneous condensation process of water in a carrier gas, see figure 3.16. A numerical simula.tion ba.sed on the classical nucleation theory, the Hertz-Knudsen growth la.w, and the geoii1etry of the nozzle yielded numerical results for the static pressure along the nozzle axis. By oomparing the two, he determined the best fit value for the reduced nucleation rate r, defined as
J = fJoNT· (5.1)
This is useful when the deviation hetween the CNT and the a.ctua.l nucleation rate is indeed a constant factor during the nucleation pulse. From different studies on homogeneous nucleation in water vapour (Hung et al. 1989, Wagner a.nd Strey 1981) it was concluded that the CNT descrihes the saturation dependenee of J a.ccurately, hut shows a different tempera.ture dependenee compared to the nucleation rate data. In :figure 5.1 we see some of the nucleation rate data of water reported in the literature (Viisanen et al. 1993, Looijma.ns 1995) relative to nucleation rates according to the classica! nucleation theory as a. fundion of tempera.ture. It is clear that this ratio is a relative weak function of temperature. It only varies two orders of magnitude over a temperature range of 60 K, while the value of the nucleation rate varies orders of magnitude in only a few degrees (see section 2.1.1). So, the reduced nudeation rate for water vapour is in good approximation constant for the limited tempera.ture ra.nge of a few degrees for the nuclea.tion pulse1•
Nucle<~ticm rate data is to be regarcled on a logaritbmic scale, because even the slightest variation in one of the parameters of interest, e.g. S or T, yields changes in the nudeation rate of an order of magnitude. Therefore, we regard the methods proposed in this sectien as accurate if the results found for r are correct within one order of magnitude.

5.1 Homogeneons condensation of water vapeur 79
At increasing total pressure the effect of condensation on the static pressure will become negligible because of the diminishing infl.uence of the latent heat released. Therefore, we propose a method, in which the reduced nucleation factor r is found by matching the measured and numerically calculated extinction coefficients. The :final result of this procedure is the nucleation rate during the time interval of the pulse. The reduced nucleation rate relates this result to the nucleation modelused in the numerical simulation.
The combined processes of droplet formation and droplet growth determine the shape of the extinction coefficient for a given expansion. For the numerical evaluation of the growth rate and the nucleation rate, the relations given in appendix A are used. The effect of a variatien in the main parameters for nucleation and droplet growth, the surface tension and the diffusion coefficient, on the numerical result for the extinction coefficient is shown in :figure 5.2.
8
6
i 4
ct:l.
2
0 6 8 10 12 14
t [ms]
Figure 5.2: The extinction coefficient for light with a wavelength of 5..{3.5 nm as a function of time during an adiabatic expansion of water in nitrogen. Using the CNT, the droplet growth model of Gyarmathy, and the Mie-theory. p(t)/Po = 0.05 + 0.95 exp( -t/t2), Po = 50 bar, To :::: 295 K, Pv,o 1000 Pa, 'Yl,O :::: 1 · 1tr3, t2 = 10 ms. a: "Exact", reference values for dijjusion coefficient D and surface tension 0'. b D = 0.8 Dref· c: D = 1.2 Dref• d: 0' = 1.05 O'ref
The diffusion coefficient was taken from fits reported in literature (Smolders 1992), e.g. water/nitrogen, or from the general semi-empirical relation of Reid (1987), e.g. water/helium (see appendix A). The uncertainty in the diffusion coefficient according to these relations is 5 to 10 % and infiuences the growth ra.te of the droplets. When applying the growth model of Young instead of that of Gya.rmathy, the differences in growth rates would be more than 20 %. To illustrate the effect of the diffusion coefficient and the different growth roodels we show in figure 5.2 the effect that a. variatien of 20% in the diffusion coefficient would have on the f3 signal. It is obvious that such a variatien does not have a large infiuence on the onset of the optical signal but mainly affects the behaviour in the droplet growth zone.

Nucleation rate data from homogeneons condensation experimentsin an expansion cloud 80 chamher
A variation of the surface tension ( and other thermophysical parameters of the components of the mixture, e.g. ,o;) affects the results of the simulation hy changing the valnes of the nucleation rate as predicted hy the model, JeNT· lncreasing the surface tension increases the energy harrier for droplet formation and suhsequently J decreases. A larger saturation ratio is needed to attain the same nucleation rate as fora smaller value of u. The nucleation pulse as well as the change in f3 appear later in time.
A higher nucleation rate results in a shift of the onset zone backward in time, while the shape of f3(t) is affected most hy a change in the droplet growth rate but ma.inly in the droplet growth zone. It is for this reason that for romparing the results of the numerical simulation with the measured extinction coefficients we focus on the onset zone of the extinction coefficient.
1.5•1018 ....-------------....,
t --------------, I
I ' I
I
8
6
---- D = 0.8 0., 2 ·- 0=1.201,1
- a=1.05a,. - •exact•
8 9 10 11 12
t [ms)
Figure 5.8: The nucleation rate and the extinction coefficient at 543.5 nm as functions of time after matching to the exact signal. The matched {3 for a variation in u coincides with the {3for D = 1.2 Djit·
Given the variations mentioned above, let us assume that the reference f3 signal is the experimental one and that in our analysis we use the "wrong" value of the diffusion coefficient or surface tension. What would he the error in the experimental nucleation rate? We foliowed the normal "experimental" matching procedure and determined the factor r hased on a comparison of the reference f3 and the signals basedon the "wrong" values of D and u.
The results for the reduced nuclea.tion rate are represented in table 5.1, while the fina.l nucleation pulses and the matebed signals are shown in figure 5.3. It is clear that for a change · of 5 % in the surface tension, a reduced nudea.tion rate of 100 is needed for the onset of f3 to occur at the same period in time as the exact solution. But, even though r has the value of 100 the final nucleation pulse has almost the same form and height as the "exact" pulse.
Variations in the diffusion coefficient affect the growth process and lead to a change of r such that the final nucleation pulse differs from the exact solution. However, the uncertainty in the nucleation rate introduced by this uncertainty in the growth rate is only a factor 1.5.
So, matching the numerical and experimental extinction coefficient results in a nudeation

5.1 Homogeneons condensation of water vapour
! Situation Jmax [m-3 s-1] T[K]I Ut r "exact" 1.04 · 1016 235. 1 9.6o . w-4 1.0 D = 0.8 Dfit 1.38 . 1018 235.07 ! 9.55 . w-4 1.7 D = 1.2 DJit 8.29 . 1017 234.71
1
9.57 . w-4 . 0.66 0" = 1.05 O'HzO i 1.37. 1018 235.07 9.63 . 10-4 100
Table 5.1: The nucleation rate, temperature and molar vapour fraction at the moment of maximum nucleation. A lso the reduced nucleation rate is given after matching the extinction coefficients of the changed situations to the exact solution.
81
pulse which is virtually independent of the actual values of the properties used to evaluate the condensation process numerically.
5.1.1 Results for waterfhelium
40
30
-i co.
10
0
2 3 4 5 6
t [ms]
Figure 6.4: The extinction coefficients for the 543.5 nm and 815 nm beams as functions of time during an adiabatic expansion of water in helium. Experiment nr. m30: Po = 42.1 bar, To 294.7 K, Pv,o = 2650 Pa, Dotted linea: Numerical simulation. Full Jines: Experimental signals.
In figure 5.4 the final result of the fit procedure is shown for an experiment with a water/helium mixture. It is clear that the onset zone is described very well for the extinction coefficients of the light of both lasers. Later in time there is a. slight discrepa.ncy between the

Nuclea.tion rate data from homogeneons condensation experimentsin an expansion cloud 82 chamber
numerical results and experimentally deduced extinction coeffidents. This is related to influences of uncertainties in the initia! state of the mixture, uncertainties in properties used during the numerical simulation, and by influences of the experimental set-up;
First of all, the expansion of the vapourfgas mixture is regarcled to he adiabatic. Once the temperature of the mixture decreases, the walls of the expansion cloud chamber will transfer heat towards the core of the cell. Droplets within the boundary layer might evaporate or at least grow with a smaller growth rate. Therefore, the extinction coefficient deduced from the experiment signals is an underlimit for the extinction coefficient according to the numerical simulation. The penetration depth of the thermal boundary layer develops into the expansion cel! roughly as 4v'at, where a )..j pCp. With increasing total pressure the effect of the thermal boundary layers decreases. For initia! pressures above 10 bar the thermal boundary layer will he less than 10% of the width of the expansion cell for the experimental recorded time interval (Morgenstern 1990). So, the experimentally deduced extinction coefficients will he a.ffected also less than 10% .
Secondly, the result for the theoretical extinction coefficient depends strongly on the initial partial vapour pressure, which directly influences the saturation ratio and therefore also JeNT· If the initia! partial vapour pressure is overestimated, the saturation is larger than according to the actual experimental situation. The calculated nucleation rateis therefore overestimated and r will he too low. Is the initia! vapour pressure taken too small, than the reduced nucleation rate is overestimated. The initial vapour pressure is determined with the Humicap with an uncertainty of 5 %. According to the classica! nucleation theory, this results in an uncertainty for r of a factor 5.
Finally, the temperature is determined from expression (3.11), taking"' as 1.667 (helium)2,
and the measured pressure history. With a relative error of 0.005 for the total pressure of the mixture, this yields an error in the temperature at a typical pressure of 0.6 Po of about 0.5 K. The corresponding error in r is within a factor 3.
In figure 5.5 the results for r of an experimental series with water vapour in helium are shown as a function of temperature for various total pressures of helium. Also low pressure data of Looijmans (1995) and Viisanen et al. (1993) are shown. The temperature and pressure for our experiments correspond to the thermodynamic state at the moment of maximum nucleation. The range of maximum nucleation rates attained during the nucleation pulses for the experiments shown in figure 5.5 varies in between f017 m-3 s-1 and 1019 m-3 s-1 for total pressures around 5 bar and 60 bar, respectively.
It can he concluded that the reduced nucleation rate for water in helium is independent of the total pressure. The variation of r for the experiments with different total pressures but at the same temperature are well within the uncertainty of a factor 15. The values of r show a good agreement with the data of Looijmans, while the comparison with data of Viisanen et al. is less satisfactory.
The experimental technique of Viisanen et al. as of Looijmans is based on the nucleation pulse method. Viisanen et al. prepared the mixture of water and a carrier gas outside their setup in a large storage tank. By flushing the set-up with this mixture for some time, the mixture
total pressures above 10 bar the mol ar fraction of water is less than 2.5 ·10-3 . So, 1 has a value in between 1.665 and 1.667. This yields a difference in the temper at ure at pfpo = 0.5 of less than 0.1 K taking 1 or 'Yhelium.
Therefore, we neglect the influence of the water vapour on the 1·

5.1 Homogeneons condensation of water vapour
10S o obar 0 10-15bar
1cfl .. 0 20-25bar .. <> 45-55bar ,. ,. .. " Pv,o • p,.(p,1)
~ ,. ()-() Viisanen et al
I" Looijmans !!>
()" !.!>1' '[I ,. ,.
·~" ... .. ~~ , __ 10-3
200 220 240
T[K]
260
Figure 5.5: The reduced nucleation mte r (J_,/lcNT) for water as a function of tempemture. Experiments of water/helium for various total pressures, data of Looijmans (1995}, experiments of water/helium with initially satumted mixtures, and data of ViiBanen et al. (1993).
83
intheset-up was assumed to have the same composition as prepared in the storage tank. No measurement of the initia.l composition of the mixture in the set-up prior to an experiment was done.
Looijmans determined the nucleation rate of water in mixtures of water in nitrogen at low pressures in a nucleation pulse tube (Looijmans et al. 1993). The initia.l partia.l vapour pressure was determined with the help of a Humicap similar to the one we used. He showed that a systematic deviation of 10 % between the initia} partial vapour pressure for his and Viisanen's series of experimentscan explain the difference in r.
The expansion doud chamber and the experimental/numerical methad todetermine r differ completely from the nucleation pulse tube and its optical detection method. The only similarity between the two set-ups is that a Humicap was used to determine the initia} partiai vapour pressure of water. To investigate whether this device may have led to a systematic error in the reduced nucleation rate, we also present an experimental series with saturated mixtures of water in helium in figure 5.5, see also section 5.1.2. For these experiments, the initia! partial vapour pressure is derived from the initial temperature, the initial pressure, and expression (1.5)3 The results for the reduced nucleation rate of this experimental series agree perfectly with the results obtained by usîng the Humicap. This indicates that determining the initia! composition of the water/helium mixture with the Humicap is accurate and does not result in systematic errors.
In figure 5.6 the results for the series with saturated mixtures and the results for the red u eed
is a noble ga.s which only affeets the saturated vapour pressure of water by the Poynting effect.
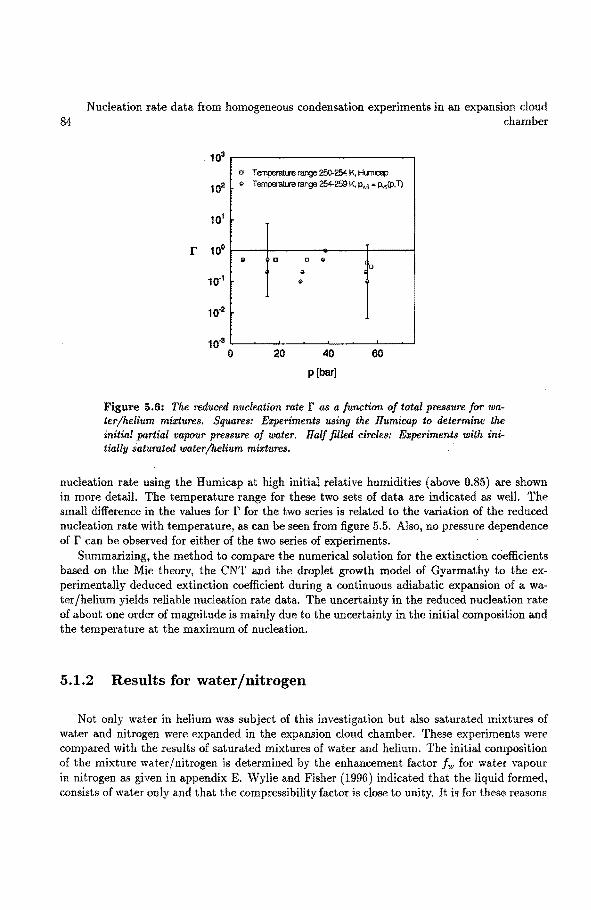
Nucleation rate data from homogeneons condensation experimentsin an expansion cloud 84 chamber
10" 0
0
"'
"
T empemture range 250·254 K Humicap T empemture mnge 254-259 K. Pv.o = p",(p,T}
D .. ..
20
0 ~
40
p [bar]
0
60
Figure 5.6: The reduced nucleation rate r as a function of total pressure for water/helium mixtures. Squares: Ezperiments using the Humicap to determine the initial partial vapour pressure of water. Half fl.lled circles: Ezperiments with initially saturated water /helium miztures.
nucleation rate using the Humicap at high initia! relative humidities (above 0.85) are shown in more detail. The temperature range for these two sets of data are indicated as well. The small difference in the valnes for r for the two series is related to the variation of the reduced nucleation rate with temperature, as can beseen from figure 5.5. Also, no pressure dependenee of r can be observed for either of the two series of experiments.
Summarizing, the method to compare the numerical solution for the extinction coefficients based on the Mie theory, the CNT and the droplet growth model of Gyarmathy to the experimentally deduced extinction coefficient during a continuons adiabatic expansion of a water /helium yields reliable nucleation rate data. The uncertainty in the reduced nucleation rate of about one order of magnitude is mainly due to the uncertainty in the initial composition and the temperature at the maximum of nucleation.
5.1.2 Results for waterfnitrogen
Not only water in helium was subject of this investigation but also saturated mixtures of water and nitrogen were expanded in the expansion cloud chamber. These experiments were compared with the re1mlts of saturated mixtures of water and helium. The initia! composition of the mixture waterfnitrogen is determined by the enhancement factor fw for water vapour in nitrogen as given in appendix E. Wylie and Fisher (1996) indicated that the liquid formed, consists of water only and that the compressibility factor is close to unity. It is forthese reasons

5.1 Homogeneons condensation of water va.pour
0 Tideal 0 TS)<:!Ev
101
r " " " D
"
20
" "
" " 0
"
40
p [bar)
l 0 " ()CCI ()
<>
"
60
Figure 5. 7: The reduced nucleation rate r for water as a function of total pressure during an adiabatic e:xpansion of water vapour in· nitrogen. No pressure dependenee of the surface tension of water is used. The results for a temperature to the ideal behaviour of nitrogen : 7 1.4, and the EOS of Sychev. Temperature range: 254.6 < T;t~.eat < 257.8 K, 253.9 < Tsychw < 256.6 K
85
that the ideal gas behaviour of both components is assumed and tha.t the CNT is used as a reference for the nucleation process of water in nitrogen.
To prepare a sa.turated mixture of water and either nitrogen or helium the set-up was evacuated for 30 minutes. Then a. large amount of liquid water was injected at the injection point, see figure 4.1. Based on the volume of the set-up including mixing circuit, 2 pl should be su:fficient to obta.in a sa.turated water vapour in the set-up. Ad- and absorption processes cause this volume to increase drastically. We varied the a.mount of liquid water injected in between 100 ttl and 1 ml. After a. few experiments, in which the injected volume ranged from 100 pl to 1 ml, no varia.tion in the results for r was observed. From then on 400 pl was used as the injection volume for water to obtain a saturated mixture.
In figure 5.7 the results for r for the satura.ted water/nitrogen mixture are shown as a function of the total pressure. The differences between the experimental and numerical extinction signals a.fter the matching procedure is similar to the differences shown in figure 5.4. Two different equations of state are used to evaluate the temperature: the ideal gas EOS with a constant ratio of specific heats of 1.4 and the EOS of Sychev et al. (1987). This latter EOS is determined by a multi-parameter fit of all available p-V-T data found in the literature. For an expa.nsion of 80 bar to 50 bar the "ideal" temperature is about 1.2 K higher than the "Sychev" temperature. This leadstoa difference in JeNT of a factor 8 (J(Tidea.t) < J(Tsychev)). In figure 5. 7 the difference in r due to the extra cooling of the gas is shown. At high pressures the effect is largest and for the expansion leading to a nucleation pulse at 60 bar the difference between the two reduced nucleation rates is a factor 11.
It is clear that the reduced nucleation rate increases with increasing total pressure. At a

Nuclea.tion ra.te data. frorn homogeneons condensa.tion experirnents in an expa.nsion cloud 86 cha.rnber
0.9 tf ti
0.8
0.7 l.-...o--'-~-'-~-'--........_,.___.__,_ _ _,_,
0 20 40 60 80 100 120
p [bar)
Figure 5.8: The effect Qf total pressure on the surface tension of water for various gases, T = 25 °C. Slowinski et al. {1957).
pressure of 60 bar the nuclea.tion ra.te increa.ses a.bout 2 orders of magnitude compared to low pressures. The ternperature at the maximurn nudeation ra.te increa.ses slightly (see appendix D). So, even though the diffusion process is slowed down by the higher total pressure4 and the nucleation pulse is expected to occur at a later moment in time (lower ternperatures), it shifts a.head in time towards a higher ternperature. As a conclusion, the energy harrier for droplet formation of water molecules must decrea.se with increa.sing total pressure of nitrogen.
Most probably, the energy harrier decrea.ses due to the decrea.sing surface tension of water with increasing total pressure of nitrogen. Slowinski et al. (1957) discussed the effect of the total pressure on the surface tension of water for various ga.ses. Their results are represented in figure 5.8. The surface tension of water increa.ses very slightly for heliurn while for nitrogen it decrea.ses by 7.5% when the total pressure of the water/nitrogen mixture changes frorn 1 to 80 bar. This is caused by the adsorption of nitrogen molecules on the liquid surface. It is a.ssurned that no nitrogen is present in the bulk liquid ( Slowinski et al. 1957, Wylie and Fisher 1996).
The effect of decrea.sing surface tension is incorporated in the numerical sirnulation in the approxirnate way as suggested by Slowinski et al. They a.ssurned that the surface tension at a given ternpera.ture T decrea.ses linearly as the product of the total pressure pand ternperature, starting at the surface tension as given in appendix A for pure water vapour, u(T):
u,(T,p) = u(T)- CpT, (5.2)
where C is a constant deterrnined forrn figure 5.8. In figure 5.9 the results for the reduced nucleation rate are shown when corrected for the
effect of the total pressure on the surface tension and when applying the EOS of Sychev to deterrnine the ternperature during the expansion. Also the results for r for saturated mixtures of water and heliurn are shown. As it is clear, the pressure dependenee of the red u eed nucleation rate for the water/nitrogen experirnents ha.s disa.ppeared a.lrnost cornpletely. At 60 bar the effect
enhancement factor fw increases toabout 1.4 at 100 bar. The growth rate would increase by this larger Pv but the dilfusion coefficient (~ 1/p) deereases much faster.

5.1 Homogeneous condensation of water vapour
t. ~
" i-ipiN.z: T-arxi CJCIJ!l!CÜOO
10-3 0
t.
" y
" "
20
t.
" "
A
" ..
40
p [bar]
.. " ..
60
Figure 5.9: The reduced nucleation mte r (Je:xrp/JcNT) for water as a function of total pressure of either helium or nitrogen. For nitrogen: The tempemture is calculated based on the EOS of Sychev and the surface tension of water is corrected for the inftuence of the total pressure of nitrogen. 25-t.6 < Thelium < 257.8 K, 253.9 < Tsychev < 256.6 K.
87
of the pressure dependenee of u on the reduced nudeation rateisabout 3 orders of magnitude. This temperature effect seems to decrease the reduced nucleation rate, as is clear from figure 5.1. The results for r in case of helium as carrier gas are identical to the corrected results for nitrogen.
In expression (5.2) we have assumed C to be temperature independent. This assumption is questionable. The temperature dependenee of C is related to the adsorption of carrier gas molecules on the liquid surface (Langmuir 1918). A more fundamental study on the behaviour of the nucleation rate of water in nitrogen as a function of total pressure and temperature is needed.
5.1.3 Discussion and conclusions
The metbod outlined in this section is based on the assumption tha.t the reduced nuclea.tion rate r is almost constant for the temperature range of the nucleation pulse (typical variation 5 K).
A slight variation of the actua.l r with temperature as shown in figure 5.1, changes the form of the nucleation pulse. This may affect the size distribution of the dropiets a.nd hence the extinction coefficient. This can lead to a systematic error, the value of which can be estimated from figure 5.1. It is expected to be within a factor 3 for a temperature range of 5 K.

Nucleation rate data from homogeneous condensation experimentsin an expa.nsion cloud 88 chamber
The values for the reduced nucleation rate found from the experimental series with water/helium agree well with the low pressure data of water in nitrogen reported hy Looijmans (1995). The value of the actual nucleation rate of water is not affected hy the total pressure of helium.
Our results for r differ from the data of Viisa.nen et al. (1993). This may he related to a difference in the way of determining the initial partial vapour pressure of water. We found a very good agreement hetween our results for r for initially undersaturated mixtures using the Humicap a.nd initially saturated mixtures.
The red u eed nucleation rate for the experimental series with water /nitrogen shows a pressure dependence. When calculating the temperature hased on a constant"/ of 1.4, the temperature is overestimated for expa.nsions at higher initial pressures. The use of the EOS of Sychev et al. (1987) is recommended, which yields temperatures which are less, e.g. ahout 1.2 K less for a.n expansion of 80 har to 50 har. Since the nucleation rateis strongly dependent on temperature, this difference is very relevant.
Taking the temperature according to the EOS of Sychev et al., still a pressure dependenee of r is ohserved. At a total pressure of 60 har the nucleation rate increased hy ahout 2 orders of magnitude compared to low pressures.
We have found strong indieations that the apparent pressure dependenee of the redueed nucleation rate is related to the pressure dependenee of the surface tension of water. This pressure dependenee is rather strong for water/nitrogen systems, a.nd approximately absent for waterfhelium mixtures. Incorporating the pressure dependenee of the surface tension in the theoretica} model for nucleation yields values for the reduced nucleation rate that are almost independent of pressure. A more fundamental study on nucleation hehaviour of water in nitrogen as a fundion of temperature and. pressure is neeessary.
The results for r for water/nitrogen after these modifications agree with the results found for saturated waterfhelium mixtures.
5.2 Homogeneons condensation of n-nonanefmethane
The enha.ncement factor of water in nitrogen is mueh larger tha.n due to the Poynting effect alone, the temper at ure is best determined with the Syehev EOS, and the surface tension of water is affected by the adsorption of nitrogen molecules. This all indicates that water/nitrogen mixtures form the intermediate hetween ideal mixtures with one component condensation and systems in which hoth eomponents are present in the hulk liquid. The effect of real gas behaviour of a vapour /gas mixture on the homogeneons condensation process due to a continuons adiabatic expansion is much more dominant for the mixture of n-nonane/methane. We performed three experimental series with n-nonane/methane each with a different initial temperature, 11 oe, 22 oe, and 29 oe respectively.

5.2 Homogeneous condensation ofn-nonane/methane 89
5.2.1 Initial composition of the n-nonane/methane mixture
The initial composition of the n-nonane/methane mixtures corresponded to the saturated state. The injected volume of liquid n-nonane was 1 ml. Reproducing an experiment a few times yielded no variation of the state at the Wilson point and the inner side of the observation windows was wetted with n-rionane. This indicated that the initial composition of the nnonane/methane mixtures was indeed saturated.
As described in the experimental procedure (section 4.2.1) three gas samples were taken for each experiment. The average initial molar fractions of n-nonane for the three samples is presented in figure 5.10 for the three experimental series.
0.0015
0.001
Ourdata: o T·29"C A T-22'C 0 T-11"C
Rl\8: - T- 29'C --- T·22'C - T-11"C
0'--~--'----'-~-'-~--'---.L...l
0 20 40 60 80 100
P {bar]
Figure 5.10: The saturated molar vapour fraction of n-nonane as a function of pressure for n-nonanejmethane mixtures. Markers: Our experimental series with saturated mixtures of n-nonanejmethane for three different tempemtures. Lines: the satumted molar vapour fmction of n-nonane according to the RKS-EOS for the three tempemtures.
The initial molar vapour fractions of n-nonane for the three gas samples of the same experiment exhibit variations which can be much more than the inaccuracy of 10% from the GC-analysis. Variations up to 50 %are observed, see also appendix D.
The method of sampling involves many separate steps: connecting the adsorption column gas tight to the set-up, pressurizing the sample loop and determining the pressure, measuring the gas volume, flushing the adsorption column with n-hexane, collecting and weighing the liquid sample, and analyzing this liquid with the help of a gas chromatograph. Each of these steps introduces possible errors. Furthermore, the processes of ad- and absorption of n-nonane at "dry spots" in the sample duet between the set-up and the adsorption tube influence the result for the molar vapour fraction of n-nonane in an irreproducable way.

Nucleation rate data from homogeneons condensation experimentsin an expansion cloud 90 chamber
The mixing procedure when preparing a mixture of n-nonane and methane was identical to the procedure for water vapour in helium or nitrogen. For some experiments the vapourfgas mixture was prepared as described insection 4.2 after which we waited an extra 16 hours (in two cases even 65 hours), see appendix D, and proceeded the mixing procedure foranother one or two mixing cycles. The results for y1,o for those samples did not differ systematically from the results for mixtures prepared within 3 hours. So, the mixtures for all experiments were homogeneons and within the error bounds also saturated.
The experimentally determined initial molar fraction of n-nonane in methane as a function of pressure behaves qualitatively as predicted by the RKS-EOS. However, its value seems to be lower than the result for y1 ,sat according to the RKS-EOS by a constant difference for each initia! temperature. It is in between 10 % and 25 % for a total pressure varying from 20 to 100 bar. The initia! molar fraction of n-nonane determined by the gas analysis metbod is known with an average uncertainty of 15 %.
5.2.2 Results for n-nonanefmethane
---0.8
l ! ! !
0.6 I
0.4 1/lo, 543.5 nm J
I I
0.2 I 1/lo. B15nm
I 0
·10 0 10 20 t[ms]
30 40 4
,, ,. lllo. 543.5 nm :\
~ \ •' \\ l/lo.615nm \1 ,, \\
6 8 10 t[ms]
12
Figure 5.11: Pressure and transmission of the 543.5 nm and 815 nm lasers as functions of time. n-nonanefmethane mixture, exp. nr. m92., Po = 74.75 bar. Full signals (left) and stretched (right) in the time interval near the onset.
The experimental signals for the pressure and transmission coefficients as shown in tigure 5.11 have similar shapes as the signals for experiments with water, see tigure 4.7. The droplet growth rates for n-nonane/methane are larger than for water due to the smaller latent heat of the mixture, and the large molar fraction of n-nonane. Combined with a larger refractive index this leads to an extinction coefficient which increases rapidly to values which can be much larger than 300. So, the doud of dropiets formed during the expansion of saturated n-nonanefmethane suddenly becomes optically very thick such that the laser light eaunotpass through the expansion chamber.
In tigure 5.12 the Wilsou points are shown for the experiments with n-nonane/methane in

5.2 Homogeneons condensation of n-nonane/methane
80
60
p [bar)
40
•
: ~i --"-. ·_...______.__·~ 220 230 240 250 260 270 280
T[K]
Figure 5.12: P-T diagram: The Wilson points for the experiments with nnonane/methane for three different initial temperatures.
the expansion cloud chamber.
91
We detected homogeneons condensation in the expansion cloud chamber in a pressure range from 5 bar to 75 bar and a temper at ure range from 225 K to 278 K. So fa.r, this is the largest range of pressures and temperatures for which homogeneons condensation of n-nonane/methane mixtures has been stuclied experimentally.
For an initial state corresponding to a saturated mixture at the same initia! temperature but for different initia.l pressures, the Wilson points form one smooth curve in a p- T diagram. For our experiments, slight variations from such a curve can be observed: largest for the series with the highest initia.l temperature and smallest for the series with the lowest initia! temperature. This ma.y be caused by an initia.l saturation ratio of n-nonane which does not have the same va.lue for all experiments corresponding to the same initial temperature. At a lower temperature less n-nonane dissolves in methane and saturation may have been achieved earlier than for higher tempera.tures. Variations of only a few percent in S0 can cause the Wilson point to vary a few degrees in temper at ure. F'l1rther, the pressure transducers were affected by the preserree of n-nonane in liquid form. Severa.l calibra.tions of the transducers showed a slight change in the sensitivity of the transducers during the course of the experiments. This affected the experiments done later in time most, the series of 22 oe and 29 oe. Forthese experiments the relative uncertainty in p(t) increased toabout 0.01.
Similar to the experiments with water, we now ca.lculate the extinction coefficients numerica.lly by using the measured pressure history, the RKS-EOS, the BCNT and the droplet growth model of Gyarmathy as discussed in chapter 2. However, matching the measured extinction coefficient and the numerica.lly ca.lculated extinction coefficient will be done differently.
In figure 5.13 the nudeation rate as a function of the saturation ratio is shown for a mixture

Nudeation rate data from homogeneons condensation experimentsin an expansion cloud 92 chamber
1018 p (bar)~
1017
::-' 1016 : . ., . g - BCNT .... 1015 0 10bar
• 20bar 1014 0 30bar . 40bar 1013
2 10 20 100
s
Figure 5.13: The nucleation rate as a function of saturation ratio for nnonanejmethane at 240 K. Solid lines: BCNT. Markers: Data of Looijmans (1995).
of n-nonane/methane for various pressures at a temperature of 240 K. The results for the BCNT and the data of Looijmans (1995) are shown. It is clear that, especially at high pressures, discrepancies of many orders of magnitude appear between the nucleation rate data and the predictions of the BCNT. The reduced nucleation rate r can attain large values, of the order of 103 or more, so a different parameter is used.
The nucleation rate data of Looijmans is located parallel to the J- S curve according to the BCNT in the J-S diagram. Therefore, we introduce an effective molar vapour fraction Yl,eff defined by
Yt,ef 1 = K Y1> (5.3)
which is used to evaluate the nucleation rate of the BCNT at a saturation ratio Self which is J( times larger than the actual saturation ratio. This means that we rescale the S-axis in figure 5.13 such that the experimental points shift horizontally to the curve of the BCNT at the same pressure and temperature. The parameter K will be referred to as the saturation sealing factor.
Rescaling the saturation ratio influences the Kelvin equations and as such the critica! radius for which the droplet growth rate is zero. So, the same correction for S must he incorporated in the model for the droplet growth rate. However, the actual molar fraction does not vary ancl therefore the growth rateis adjusted by replacingD(yi- y1,d) by D/K(y1,eff- Y~,d)· Here, we mark Yl,d with a' to emphasize that the value of y1 ,á clepends on S because of the Kelvin effect. In this way the growth ra te for a high supersaturation is proportional to D /I< (YI,ef f) = Dy1, so its value does not change.
The pressure dependenee of the composition of the saturated mixture is described qualitatively by the RKS-EOS. In figure 5.10 we observe that for a given temperature T the experimental valnes of Yl,sal are on the average a constant value 6.y1(T) lower than the RKS values, with some scatter. In order to rednee the effect of the inaccuracy in the indivicliual molar fractions we acloptecl the following procedure. For a given initial state we determined first YI,RKS,
the saturatecl value for the RKS-EOS. Then we subtracted 6.y1 (T) for that temperature to find

5.2 Homogeneons condensation of n-nonane/methane
2.5
2
K 1.5
0.5
Loai;nans: <> T.227.5K o T·237.5K CJ T ·249K
Present : + 229 < T < 235 K • 23S<T <252K A. 252<T <2S2K • 2ll2<T<2i'8K
ç: ~cab ; I~ . =- d
• •I.ni ~ • - l • 1 •• I· · r· I
20 40
p [bar]
60 80
Figure 5.14: The saturation sealing factor K related to the BCNT (K ::::: 1} as a function of pressure. Our results for various temperat-ure ranges, the data of Looijmans for J in between HJ14 and liP m-3 s-1 and the QOC-model for J around HP m-3 s-1 are shown: a: 290 K, b: 240 K, c: 250 K, and d: 260 K. As an example the error bounds for one series of experiments are indicated.
93
the actual molar fraction. The initia! molar liquid fraction found in this way is à.ssumed to be accurate within 10% and the results are given in appendix D.
With this initia! composition, the final results for the matching procedure are shown in figure 5.14. The uncertainty in the sealing factorKof 15 %is determined by the large uncertainty in y1,o and T.
The maximum nucleation rates attained during the expansions shown in figure 5.14 range from 1017 m-3 s-1 to 1019 m-3 s-1 fortotal pressures in between 10 bar and 75 bar, respectively.
Figure 5.14 shows that the saturation sealing factor K varies from a value of 1.2 ± 0.2 at 10 bartoa value of 0.8 ± 0.1 at 75 bar. At 30 barK equals 1.0 ± 0.15, which means that the BCNT prediets nucleation rates quite welL We reeall that a saturation scaler K > 1 implies that a certain nucleation rate is found experimentally at a value of the saturation ratio which is lower than the saturation ratio that corresponds to this rate according to the BCNT. Not only the pressure varies in figure 5.14 but also the nucleation temperature varies. The different symbols correspond to temperature ranges indicated in figure 5.14. We see at pressures between 50 and 75 bar that the results for K for different temperature ranges almost coincide. Similar results are found for other pressures and temperature ranges. This suggests that in the parameter range investigated there is no strong temperature dependenee of K.
Some results for the QOC-theory at nucleation rates of 1017-1018 m-3 s-1 arealso shown in figure 5.14 for four different temperatures. The QOC-theory prediets the sa.me nucleation rates as the BCNT at a saturation ratio of about 1.5 to 1.8 times smaller.

Nucleation rate data from homogeneons condensation experimentsin an expansion cloud 94 chamber
In figure 5.14 also some experimental results are shown obtained by Looijmans (1995), with the nucleation pulse tube. We recalculated the nucleation temperatures at the pressure dip for the data of Looijmans using the RKS-EOS insteadof the Sychev-EOS (Looijmans et al. 1995, Looijmans 1995). His nucleation rates varied in between 1014 and 1017 m-3 s-1 fora temperature range of 227.5 to 250 K and a pressure range of 10 to 40 bar.
Some experimental points agree well with our data, while others do not. Agreement is found for the 10 bar experiments and for the experiments with the highest temperatures, 250 K. For low temperatures and pressures of 30 and 40 bar Looijmans found Kvalues of 1.5 and even higher, up to 3.9. Until now, there is no clear explanation of the apparent discrepancy.
Looijmans determined the initia! molar fraction of n-nonane from the experimental droplet growth curves and the continuurn limit of the growth model. The accuracy of the molar fraction determined in this way strongly depends on the accuracy of the relation used to determine the diffusion coefficient, the Fuller relation (Reid et al. 1987) combined with the high pressure correction of Takahashi (1974). We evaluated these relations for the diffusion coefficient of nnonane in methane by repeating some nucleation experiments in the nucleation pulse tube by Looijmans while applying our method of gas analysis. This did not lead to a reinterpretation of Looijmans' data and to a better understanding of the differences between his results and the present ones.
To he more conclusive in comparing the results of both experimental methods the overlap in the range of pressures and temperatures of the experiments must be increased. This will give a means to verify the increase of the saturation scaler for lower temperatures and higher pressures as the data of Looijmans indicate, 2.8 for 240 K and 40 bar and 3.9 for 230 K and 30 bar.
5.2.3 Discussion and condusion
Three series of homogeneons condensation experiments with initially saturated mixtures of n-nonane/methane were done in the expansion cloud chamber at different initial temperatures.
The initia! molar fraction of n-nonane was determined with the help of the gas analysis method as described in section 4.2.1. We found that the results for y1,0 from the three gas samples taken prior to one experiment can have mutual variations of about a factor 2. On average the inaccuracy of the initial molar fraction of n-nonane for one experiment is 15 %. To deduce nucleation rates from an experiment with the expansion cloud chamber or the nucleation pulse tube, an uncertainty of 15 %in the initial molar vapour fraction is rather large. To improve the quality of the nucleation rate data and to he more conclusive about the comparison of both methods the accuracy for y1,0 must he improved. An on-line measurement of the concentration of n-nonane, avoiding all the separate steps of the present method, will be more appropriate.
The results for the initial molar fraction of n-nonane as a function of total pressure, are described qualitively well by the results for YI,sat according to the RKS-EOS. However, a constant difference between the two results exists.
The pressure and temperature range of the Wilson-points of the experimentsin the expan-sion cloud chamber is in between 5 and 75 bar and 225 and 278 K, respectively. ·

5.3 A Gaussian Model for the Nucleation Pulse 95
The experimentally determined extinction coefficients were matebed to the theoretica! extinction coefficients, introducing a saturation sealing factor K. The results for the saturation scaler of our experiments show that the BCNT describes the nucleation hehaviour of the mixture of n-nonane/methane more accurate than the QOC-theory. Still, K exhibits a slight pressure dependence, while there is no clear temperature dependenee within the experimental parameter range. Around 30 bar and in the temperature range of 240 to 260 K the experimental nucleation rates are well described hy the BCNT.
The results for the saturation scaler were compared to the values for K from the nucleation rate data of Looijmans for mixtures of n-nonane/methane. The pressure and temperature ranges of our experiments and the data of Looijmans only overlap in a. very small parameter range. The results for K for those experiments agree within the (large) error bounds. The strong variation of the scale parameter with pressure for temperatures helow 240 K, as Looijmans found, must be verified by extending the tempera.ture range of the expansion cloud chamber to lower temperatures. This can he realized by experiments with initially undersaturated mixtures.
Thus far we did not discuss the influence of the EOS on the results for K. We found that the composition of the saturated n-nonane/methane mixtures was not described well quantita.tively. This affects the value of the satura.tion ratio directly. Systematic errors in Sof the order of 15 to 25 % are possible, which is quite relevant when comparing nucleation data with theoretica! ones. What would happen when we use a different EOS, that describes the initia! state of saturation accurately? It is not to he expected that this would lead to an essentially different value for the experimental nucleation rate. But it would certainly affect the corresponding value of the saturation ratio and therefore the saturation sealing factor. Of course, the comparison of nucleation data obtained with different methods is possible, irrespective of the EOS.
5.3 A Gaussian Model for the Nucleation Pulse
From the full numerical simulation it is possible, by introducing an adjustment parameter, to match the extinction coefficient calculated numerically to the extinction coefficient deduced from a condensation experiment with the expansion cloud chamber. The results for the oudeation ra te as a function of time are very accurate. However, roodels for the droplet growtb ra te and the nucleation rate are necessary.
For mixtures like water/helium or even n-nonane/methane appropriate nudeation and droplet growth roodels are a.vaila.hle. But for mixtures like natura} gas no nucleation roodels are available and tbe metbod described in the previous sections can not be used.
In this section we will use some of the results of the asymptotic tecbnique to propose a new experimental metbod by which nucleation rate data can be obtained from experiments with the expansion doud chamber. Fortbis metbod it is only necessary to know an approximate droplet growth rate in the rapid growth zone (RGZ), e.g. the continuurn limit at high total pressures or the Hertz-Koudsen growth law for low pressures. This growth rateis taken radius independent and constant during the time interval of interest.

N ucleation ra te data from homogeneons condensation experiments in an expansion cl oud 96 chamber
6 7 8
t [ms]
9
Figure 5.Hi: The nucleation rate as a function of time. The numerical salution of the nucleation pulse and a Gaussian-shaped approximation. Numerical solution: Po = 5 bar, To = 295 K, Pv,o = 1000 Pa, tz = 10 ms. The CNT applied for water/nitragen (without correction of the surface tension and 1 1.../).
5.3.1 The Gaussian model
From figure 3.13 we know that the first and second derivative of the activation function with respect to time change relatively slowly and are almost constant in a period just before the maximum nucleation rate. This results in a time dependenee of J which can he approximated by a Gaussian shape,
(5.4)
In tigure 5.15 it is shown that the nucleation rate in the period from t; to t1 prior to the moment of maximum nucleation for an expansion of water/nitrogen, see also figure 3.5, can indeed bedescribed well by a Gaussian shape. This period corresponds to the RGZ in terms of asymptotic zones.
For this period in time, when we neglect the initial radius of the newly born droplet and substitute (5.4) in expression (3.17), the result is
/3( t) = r 1frd( t, T )2Qe:xt( a(t, T ), m) p(( t))Jo exp( -( T- te)2 j(.CJ.t)2)dr. (5.5) lt, p T
The integral (5.5) will be evaluated in a limited time interval and therefore the effect of the ongoing expansion will he neglected.
For a constant growth rate in the continuurn limit (2.65), a relation between the square of a droplet radius and the time interval (t- r) is found
(5.6)

5.3 A Gaussian Model for the Nucleation Pulse 97
Taking the growth rate constant is appropriate because the liquid molar fraction in the RGZ is very small, so g1 is almost g1,0 • Also the temperature and pressure changes during this period in time are small, yielding a constant dilfusion coefficient.
When fl2 is known, integral (5.5) depends on three parameters, the nucleation rate J0 , the time of maximum nudeation t. and the width of the nucleation pulse t:.t. Dividing the integral by a reference value of (3 at a moment in time tI> yields the shape of the extinction coefficient versus time curve which is independent of Jo:
(J{t)/ fli K(t)/IC(ti) (5.7)
where K(t) is
(5.8)
So in the time interval where the Gaussian shape for J is adequate, the shape of f3(t) depends only on t. and .!lt. These two parameters are found by minimizing the difference of the f3(t)/ fli curve given by (5.7) and the experimental valnes in the (ilt, t.) space. Once these two parameters are known the absolute value of {3( ti) yields the value of J0 •
The only problem we are facing is to determine the time interval for which (5. 7) is valid.
5.3.2 Results: Numerical Simulations
The value of f3 at a given moment in time does not reveal whether or not the moment of maximum nucleation still has to come. Only a procedure of trial and error will yield the correct interval.
The Gaussian approximation of the nucleation behaviour before the maximum of nucleation is regarcled as adequate if the nucleation rate is described well during a time interval .!lt preceding ti such that contributions to (3 of dropiets formed earlier in time are negligible throughout the time interval between t; and ti. Slight varia ti ons in t 1 wîll not require a completely different Gaussian approximation.
To determine the appropriate time interval we propose the following: from the sudden increase of the extinction coefficient, an interval in time is estimated for which expression (5.7) is valid. It starts at the moment t; corresponding with {3; = 0.001. This value sterns from experimental signal-to-noise ratios5• The interval ends at th where f3 is about L Then the two parameters t. and Llt are determined numerically for this time intervaL We now vary t 1 such that the solution fort. satisfies t. = t,. This solution indicates that the moment of maximum nucleation is attained at the moment the interval ends. At this point, the assumptions made to deduce (5.7) areabout to be violated strongly. Fora solution to be valid, the value of ti must be reduced. If the Gaussian shape is appropriate for the onset of nucleation, all of the solutions of J(t) found for different ti (smaller than t1 t.) will give values, which in their mutual period of time almost coincide. The nucleation rates in that time interval are "independent" of ti and form the solution of the method. We refer to "independent" if the solutions for different t 1 yield nucleation rates at the same moment in time within a factor of 1.5.
t; and thus (3(t;) doesnotaffect the solution found forte and At

Nucleation rate data from homogeneons condensation experiments in an expansion cloud 98 chamber
1019
1018 0.8
.;:;;;- 1017 0.6 ':§:
"' §. (Q. .., 1018
0.4
1015 0.2
1014 0 7 8 9 10 11
t[ms)
Figure 5.16: The nucleation rate and extinction coefficient at 543.5 nm as functions of time. The numerical salution for J and f3 (solid curve}, and various solutions for the approximate Gaussian shape for the nucleation rate for different ti. t; = 8.5 ms, f3(t;) = 10-3 • Waterjnitrogen: CNT, na u correction and 1 = 1.4. t2 = 10 ms, Po = 50 bar, To = 295 K, Pv,o = 1000 Pa.
To verify this procedure, we genera.ted two f3 signals numerically. In figures 5.16 and 5.17 we show the results for the minirnizing procedure for various time intervals for mixtures of water/nitrogen and n-nonane/methane. These calculations correspond to the situation shown in figures 3.8 and 3.10.
First of all we see in figure 5.16 that for water, the values of f3 are very small during the nucleation pulse. At a value of ti of 9.1 ms the solution forte is equal tot i· For the approximate Gaussian shaped solutions for J(t) a.t t1 less than 9.1 ms, we see a large interval in which the solutions for J coincide but in the period just before iJ they differ. Droplets formed at the end of the interval do not contribute significantly to the shape and value of f3 due to the r6
dependenee of {3 in the Ra.yleigh limit. Subsequently, the nucleation ra.tes found from the minimizing procedure are not reliable for the last part of the time interval.
As can he seen in figure 5.16, in the time interval during which the salution for J(t) is independent of t" the val u es for J differ less than a factor of 2 with the numerical solution. The approximate Gaussian solutions are shifted in time with respect to the numerical solution. This can be understood when realizing that the Kelvin effect, taken into account in the numerical solution, decreases the droplet growth rate for smal! droplets. With increasing size their growth rate increa.ses and for large dropiets the growth rate becomes radius independent and ( approximately) equal to the continuurn limit. The Gaussian model takes this final growth ra te already at the moment of birth of a droplet. So these dropiets increa.se much more quickly in size. This is compensated by the approximate model by forming dropiets a little later in time such that the increase of f3 is described accura.tely.
For a mixture of n-nonane/methane the values for f3 during the nucleation pulse are much larger than for water. This is caused by larger droplet growth rates due to a lower droplet

5.3 A Gaussian Model for the Nucleation Pulse
1019 3
~-2.5 1018
···<~'\ 2
;::- 1017 \., -... \, 1.5 ~ .,
.5. \ .... cc. .., 1018
.....
1015 0.5
1014 0 10 11 12 13
t[msJ
Figure 5.17: The nucleation ra te and extinction coefficient at 543.5 nm as functions of time. The numerical solution for J and (3 (solid curve), and various solutions for the approximate Gaussian shape for the nucleation rate for different t 1. t; 10.67 ms, (3(t;) = 10-3 • n-nonanejmethane. BCNT and RKS-EOS. t2 = 10 ms, Po 50 bar, To = 295 K, Y1,0 = 10-4
•
99
temperature (latent heat is less) and a larger refractive index. Therefore, the interval for which the approximate model can he applied is larger. In figure 5.17 we see four Gaussian solutions for four different values of t1. The solution d must he discarded since t. is less than t1. Again, for the time interval during which the solution for J(t) is independent of t" the nucleation
-rates are accurate within a factor 2 compared to the numerical solution. In conclusion, applying the a.pproxima.te model as outlined in section 5.3.1 and following
the procedure described above, we find a certa.in range of tempera.tures and supersaturations in which the nucleation ra.te is found within a factor 2 a.ccuracy.
5.3.3 Results: Experiments
The size parameter a for the light with the shortest wavelength is largest. This makes this light most sensitive for light attenuation by droplets. Thus, to detect the changes in the intensity of a laser as early as possible, the laser with the shortest wavelength is used. A change in (3 of 0.07 corresponds to the signa! to noise ratio of the "green" laser. Therefore, ti is located just before the fJ signal exceeds 0.07. To apply the Gaussian model successfully the change in {3 must he much la.rger than 0.07 hefore the maximum nucleation rate is a.ttained.
From the last section we know that for water this may he prohlematic. Nevertheless we applied the model for experimental signals for mixtures of n-nonanefmethane and for water/ nitrogen.
In figures 5.18 and 5.19 the results are shown for fits at (3543•5 for two experiments with

N ucleation rate data from homogeneons condensation experiments in an expansion cl oud 100 chamber
60 1018
40
1016 ':' 30:-
U) g "' .5. 1014 co. ., 20
1012 10
0 6 6 7 8 9
t[ms]
Figure 5.18: The nucleation rate and extinction ooefficient at 543.5 nm as functions of time. The experimental {3 (dashed curve), the numerical solutions after matching as described in the second section of this chapter (solid curves), and various solutions for the approximate Gaussian curve for the nucleation rate for different fit-intervals, all starting at t; and ending at different t f indicated by a, b, c, d. Exp. nr. m92: n-nonanejmethane, Po = 74.8 bar.
n-nonane/methane. Indicated are the different intervals for which te and !:J.t were determined. Also the final results for the matching procedure described in the previous section are shown. We see that the extinction coefficient at 543.5 nm for these experiments obtains very large values even before the maximum of nucleation is attained. The matching of the experimental and numerically calculated extinction coefficient was done at small values of {J. For larger values, later in time, a small discrepancy occurs between the two. This is caused by the uncertainties in the initial molar fraction of n-nonane, the temperature, and diffusion coeffident. It does indicate the possible error in the location of the nudeation pulse due to these uncertainties. In figure 5.18 the numerical solution for {:J after matching to the experimental extinction coefficient is larger than the experimental signal for momentsin time beyond the matching zone, while in figure 5.19 it is vice versa.
On average, the results for the nucleation rates obtained with the Gaussian model correspond to the valnes for J for the numerical solution within a factor 4.
In figure 5.20 the matched numerical solution and the solutions for the Gaussian curves are shown for an experiment with an initially saturated mixture of water /nitrogen. Even though the valnes for t 1 are clearly beyond the moment of maximum nucleation according to the numerical solution, the valnes of the approximate Gaussian shapes coincide at valnes of J which agree within a factor of 3 with the numerical solution. This supports the applicability of the method.
For this Gaussian method an uncertainty of the dilfusion coefficient has similar effects as described in section 5.1. So, a factor of 2 must be regarcled the inaccuracy of the nucleation rate data obtained for an uncertainty of 20 % in the diffusion coefficient.

5.3 A Ga.ussian Model for the Nucleation Pulse
10
1017 8
1016 ';'
","'
.§. 1015 ..,
2
0 19 20 21 22
t [ms]
Figure 5.19: The nucleation rate and e::tinction coefficient at 5./.3.5 nm as functions of time. The ea:perimental (3 (dashed curve), the numerical solution after matching as described in the second section of this chapter (solid curve}, and various solutions for the approrimate Gaussian shape for the nucleation rate for different fit-intervals, all startingat t; and ending at different t1 indicated by a, b, c, d. Ea:p. nr. m80: n-nonanejmethane, Po = 29.3 bar.
5.3.4 Discussion and conclusions
101
In this section we propose an approximate method to determine nucleation rate data from expansion cloud chamber experiments. It is based on a Gaussian approximation of the nucle-
1018 3
1018 2.5
1017 2 -':" ';' "?liJ
1016 1.5 .5.. .5.. <1!1. ..,
1015
1014 0.5
1013
9 10 11 12
t[ms]
Figure 5.20: The nucleation rate as a function of time. The numerical salution for the matching procedure described in the first section of this chapter, and various solutions for the approximate Gaussian curve for the nucleation rate for different fit-intervals, all starting at t; and ending at different t 1 indicated by a, b, c. Exp. nr. m54: Po = 94.7 bar. Water/nitrogen.

Nucleation rate data from homogeneons condensation experimentsin an expansion cloud 102 chamber
ation rate in the onset zone of nucleation. The growth rate is taken constant and is for high pressures rela.ted to the continuurn limit. With these approximations the extinction coefficient is calculated.
The shape of the {3(t) in the interval of the Gaussia.n approximation is determined by two parameters, the moment of maximum nucleation t. and the width of the Gaussian pulse t:J.t. These parameters are found by a two parameter fit procedure of the experimenta.l extinction coefficient to the theoretica.l one. From the absolute value of the extinction coefficient at a moment in time the absolute value of the nucleation rate is deduced.
The problem of the proposed metbod is to determine the time interval for which the Gaussian approximation of J is appropriate. This can not be determined directly from the experimenta.lly deduced extinction coefficient.
We propose a procedure which was verified by generating "experimental" {3 signa.ls using the numerical simulation of the condensation process. Applying the procedure to these signa.ls yields nucleation rates in a sma.ll time interval prior to the moment of maximum nucleation which are accurate within a factor 2. So, if the experimenta.l signa.l can be measured accurately enough (smal} {3 values), the approximate metbod will yield accurate nucleation rate data.
The approximate model has been applied tosome experimentsof mixtures of n-nonane/methane and water/nitrogen. The nucleation rates found from the matching procedure described in the first two sections of this cha.pter agree with the nudeation rates found with the approximate metbod within a factor of 4. This is within the error bounds of both methods.
References
HUNG, C.-H., M. KRASNOPOLER, & J. KATZ. 1989. Condensation of a supersaturated vapor. VIII. The homogeneons nucleation of n-nonane. J. Chem. Phys. 90(3):1856-1865.
LANGMUIR, I. 1918. J. Am. Chem. Soc, 40:1361.
LOOIJMANS, K. 1995. Homogeneous nucleation and droplet growth in the coexistence region of n-alkane/methane mixtures at high pressures. Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.
LOOIJMANS, K., P. KRIESELS, & M. VAN DoNGEN.1993. Gasdynamicaspectsofamodified expansion-shock tube for nucleation and condensation studies. Exp. Fluids 15:61-64.
LOOIJMANS, K., C. LUIJTEN, & M. VAN DONGEN. 1995. Binary nucleation rate measurementsof n-nonane/methane mixtures at high pressures. J. Chem. Phys. 103(11 ):1714-1719.
MORGENSTERN, M. 1990. Homogeneous and heterogeneous nucleation in a Wilson cloud chamber. Thesis, Institute for Continuing Education, Eindhoven University of Technology, Eindhoven.
REID, R., J. PRAUSNITZ, & B. PoLING. 1987. The Properlies of Gases and Liquids. New York: McGraw-Hill Book Company.
SLOWINSKI, E., E.E. GATES JR., & C. WARING. 1957. The effect of pressure on the surface tension of liquids. J. Phys. Chem. 61:808-810.

References 103
SMOLDERS, H. 1992. Non-Linear wave phenomena in a gas-vapour mixture with phase transition. Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.
SYCHEV, V., A. VASSERMAN, A. KOZLOV, G. SPIRIDONOV, & V. TSYMARNY.1987. Thermodynamic properties of nitrogen. Berlin: Springer.
TAKAHASHI, S. 1974. Preparation of a generalized chart for the diffusion coefficient of gases at high pressures. J. Chem. Eng. Japan 6:417-420.
VIISANEN, Y., R. STREY, & H. REISS. 1993. Homogeneous nucleation rates for water. J. Chem. Phys. 99(6):468G-4692.
WAGNER, P., & R. STREY. 1981. Homogeneous nucleation rates of water vapor measured in a two-piston expansion chamber. J. Phys. Chem. 85(18):2694-2700.
WEGENER, P., & A. POURING. 1964. Experiments on condensation of water vapor by homogeneons nucleation in nozzles. Phys. Fluids 7(3):352-361.
WYLIE, R., & R. FISHER. 1996. Molecular interaction of water vapor and air. J. Chem. Eng. Data 41:133-142.

Nucleation rate data from homogeneous condensation experimentsin an expansion cloud 104 chamber

Chapter 6
DISCUSSION AND CONCLUSIONS
The expansion cloud chamber that has beendevelopedis suited for pressures up to 100 bar. It was placed in a room in which the temperature could be regulated in between 10 and 35 °C. These ranges of the initia) temperature and the initial pressure of a vapour/gas mixture result in a very large range of temperatures and pressures where homogeneons condensation has been studied. The expansion doud chamber is an easy-to-use experimental technique. Once the expansion of a vapour/gas mixture has started, homogeneons condensation will occur and can easily be detected by measuring the transmission of a laser beam during the expansion. This experimental technique combined with the matching procedure of the experimental extinction coefficient with the theoretica) one, based on a nucleation model and a droplet growth model, yields an accurate means to determine nucleation rate data. This data is obtained for a small interval in time, the nucleation pulse, which corresponds to a range in pressures, temperatures and saturation ratios. The nucleation rate data is as accurate as the data obtained with the nucleation pulse tube. The expansion cloud chamber lacks the experimental difficulty of the nucleation pulse tube in performing a successful experiment due to the limited range of nucleation rates it is suited for. However, it does not offer the possibility to study droplet formation and droplet growth separately. We performed experimental series for water vapour, in water /helium mixtures and water/ ni trogen mixtures, foliowed by three experimental series with n-nonane/metha.ne mixtures. It appeared that this order in vapour/gas mixtures formed the transition from ideal (helium) to real (methane) gas behaviour of the gas component via the semi-real gas behaviour of nitrogen. The homogeneons condensation process in water /helium systems is accurately described by the ideal gas behaviour of helium and unary condensation of water. For mixtures of water/nitrogen, the solubility of water vapour is much larger than according to the Poynting effect. But the gas phase can still be described as "ideal", although the EOS of Sychev et al. (1987) is more accurate, especially for temperature calculations. The bulk liquid formed by the condensation process consists of water but some nitrogen is adsorbed on the surface of the droplet. Finally, for n-nonane/methane mixtures the liquid phase consists of both components and the RKS-EOS is needed to describe this binary condensation behaviour. From the matching procedure, the reduced nucleation rate r was determined for initially saturated and undersaturated mixtures of water/helium and initially saturated mixtures of water/nitrogen. For water/helium mixtures r was found to be independent of the total pressure, while the temperature dependenee agrees well with the low pressure data of Looijmans (1995).
105

106 Discussion and Conclusions
For water/nitrogen mixtures the sa.turated vapour pressure was ca.lculated using the enhancement factor fw as discussed by Wylie and Fisher (1996). The ma.in effect of the enhanced solubility of water vapour in nitrogen is the increase of the growth rates of the formed droplets. The temperature of the water/nitrogen mixtures was determined by the EOS of Sychev et al .. The reduced nucleation rate of water in nitrogen increases about 2 orders of magnitude in between 5 and 60 bar. This can be expla.ined by a decreasing surface tension of water with increasing tota.l pressure of nitrogen. Slowinsky et al. (1957) showed that the surface tension of liquid water is affected by the presence of an adsorbing gas component. This pressure dependenee of the surface tension was incorporated in the numerical simulation of the experiments. The reduced nuclea.tion ra.tes when eva.luated with this pressure dependenee of u were almost independent of the tota.l pressure of nitrogen. The condensation process in mixtures of n-nonane/methane involves both components. The binary classica.l nucleation theory describes the homogeneons oondensation behaviour well for the parameter range of this investigation. Experiments at lower temperatures and higher pressures need to he done to be able to make a better comparison with those results of Looijma.ns (1995). To increase the accuracy of the saturation sealing parameter and the qua.lity of the nudeation rate data, a.lso an on-line gas ana.lysis method should he implemented.
References
LOOIJMANS, K. 1995. Homogeneaus nucleation and droplet growth in the coexistence region of n-alkanejmethane mixtures at high pressures: Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.
LoOIJMANS, K., C. LUIJTEN, & M. VAN DONGEN. 1995. Binary nucleation rate measurementsof n-nonane/methane mixtures at high pressures. J. Chem. Phys. 103(11):1714-1719.
SYCHEV, V., A. VASSERMAN, A. KOZLOV, G. SPIRIDONOV, & V. TSYMARNY.l987. Thermadynamie properties of nitrogen. Berlin: Springer.
WYLIE, R., & R. FISHER. 1996. Molecular interaction of water vapor and air. J. Chem. Eng. Data 41:133-142.

Appendix A
THERMODYNAMIC AND PHYSICAL PROPERTIES
Material properties of gases a.nd liquids: critical molar volume V.,, critical pressure Pc, critical temperature Tc, Pitzer's acentric factor w, Rackett compressibility factor ZRA, molar mass M, parachor P, refractive index m, liquid density ji, surface tension u, saturation vapour pressure Peq, second virial coefficient B. For temperature dependent properties the absolute temperature T in K must he inserted:
water: unit literature Pc= 221.2 bar (a) Tc= 647.3 K (a) V., = 57.1 cm3 (a) M = 18.015 kg kmol-1 (a) m = 1.334 (c) [i = 999.84 + 0.086(T- 273.15)-
0.0108(T- 273.15? kg m-3 (d) u= 0.111773(1- T/T0 )
0•712012 N m-1 (c) Peq 610.8exp( -5.1421ln(T /273.15)-
6828.77(1/T- 1/273.15)) Nm-2 (f) B = 17.1 - 102.9(To/T)2
-
33.6 X 10-3T/T0 exp(5.255T0 /T) cm3 moi-1 (e)
nitrogen: Pc= 33.9 bar (a) Tc 126.2 K (a) V., 89.8 cm3 (a) w = 0.039 (a) ZRA = 0.290 (a) M = 28.013 kg kmol-1 (a) p = 60.4 gl/4cm3s-l/2moi-l (a)
helium: Pc= 2.27 bar (a) Tc= 5.19 K (a) V., = 57.4 cm3 (a)
107

108
w = -0.365 ZRA = M = 4.003 p = 50.0
methane: Pc= 46.0 Tc= 190.4 Vc = 99.2 w = 0.011 ZRA 0.2892 M 16.043 p = 81.0
n-nonane: Pc= 22.9 Tc= 594.6 Vc = 548 w = 0.445 ZRA 0.2543 M = 128.259 p = 387.6 m = 1.405 B = 369.2 705.3Tc/T + 17.9(Tc/T)2
-
427(Tc/T)3 - 8.9(Tc/T)8
Thermodynamic and Physical properties
kg kmol-1
gl/4cm3s-t/2mol-l
bar K cm3
kg kmoi-1
gl/4cm3s-1/2mol-I
bar K cm3
kg kmol-1
gl/4cm3s-l/2mol-1
cm3 moi-1
(a) (a) (a) (a)
(a) (a) (a) (a) (a) (a) (a)
(a) (a) (a) (a) (a) (a) (a) (c)
(g)
Ditfusion coefficients of waterfhelium and n-nonane/methane are calculated according to the method proposed by Fuller et al. (1987), and for n-nonane/methane combined with the Takahashi correlation T (1974) for the effect of pressure on the ditfusion coefficient. Temperatures are inserted in K, pressure in bar:
Dt2 o.oot43Tt.n . T (Tr, Pr) pM:/2 [(E,):/3 +(1:")~13]
T(Tr,Pr) = 1 +Pr ÜTr -0.583) for helium T 1.0 M12 = 2 [(1/Mt) + (1/M2)r1
T _T r- Te
Tc YtTc,I + Y2Tc,2 Pr = .11..
Pc
Pc = YtPc,t + Y2Pc,2 methane: I:" = 25.4 helium: 'Ev 2.67
(a)
(a) (a)

Thermadynamie and Physical properties
water: E" 13.1 n-nonane: E" = 189.3
(a) (a)
109
The diffusion coefficient for water in nitrogen is given by the relation of Smolders (1992) or (Luijten et al. ). Insert pressure in bar and T in K:
Literature: (a) Reid et al. (1987) (b) Knappet al. {1982) (c) Landolt-Börnstein (1960, 1962) (d) Pruppacher and Klett (1978) (e) Dillmann and Meier (1991) (f) Vargaftik (1975) (g) Adamset al. (1984)
(A.1)
The Redlich-K wong-Soave equation of statefora mixture of real gases is given by (Reid et al. 1987)
RT am (A.2) p- -
- VRKs- b,.. VRKs(VRKs +bm)'
where
bm L:v;b; i
b; 0.08664RT.,;
= p.,;
am = L:LYïYi(a;ai)112(1- k;j) i i
a; = 0.42:R2T~ [ 1 + f(w;) ( 1 - {f)] 2
f(w;) = 0.48 + 1.574w;- 0.176wf
Here am, and bm arethe RKS properties for amixtureaccordingto themixingrules (Reid et al. 1987), T.,;, Pci is the critical temperature and critica! pressure of the i-th component respectively, R is the univeraal gas constant, VRKs the molar volume, w; Pitzers acentric factor for the i-th component (Reid et al. 1987).
Binary interaction parameters k;j used in the RKS equation of state:
n-nonane/methane k;j = 0.0448 (b)

110 Thermodynamic and Physical properties
The compressibility factor is given by:
z = ___..!!____, pR:l'
(A.3)
Radulovic (1991) deduced expressions for the chemical potential of both components in the liquid and vapour phases fora bina.ry mixture of real ga.ses described by the RKS-EOS. The vapour-liquid equilibrium for component i is determined from
[-R:fln (V"- b~) b;RT _ ( 2A1 _ a~b;) ln (yv + b~) _ a:;,b; ] _
Y·R:J.' + Vv - b" ' b" V" bv (V" + bv ~ M m m m
[-RTl (V1 b~) b;RT _ (2A1 _ a:._b,) l (V'+ b:,_). _ a:._b, ] = O
n y·RT + VI M • b1 n V 1 . bi. (V1 + bi. ' ' M m m m
where Ai is y;a; + Yi(l - A;J)(a;aj)112 , for ~ the mola.r fraction y; must he replaced by the mola.r liquid fraction x;.
The surface tension t:r of a vapour/liquid surface of a bina.ry mixture is determined with the Macleod-Sugden relation (Reid et al. 1987):
n
t:rl/4 = L [P;] (p'x;- p"y;), (A.4) i=l
where pis in molfm3 and P; is the Pa.rachor for component i. The density of both pha.ses is calculated according to the RKS-EOS and corrected with the Peneloux correction (Reid et al. 1987).
The latent heatfora mixture of a supercriticaland subcritical component wa.s deduced by Van Beeck (1992) and yields
(all{ all'î ) (a"~ a"2 ) L = XtT (IT lp,xl,x2- (IT jp,yl,y2 + X2T (IT lp,xl,x2- (IT lp,yl,y2 • (A.5)
References
ADAMS, G., J. SCHMITT, & R. ZALABSKY. 1984. The homogeneous nucleation of nonane. J. Chem. Phys. 81(11):5074-5078.
DILLMANN, A., & G. MEIER. 1991. A refined droplet approach to the problem of homogeneous nucleation from the vapor pha.se. J. Chem. Phys. 94(5):3872-3884.
KNAPP, H., R. DÖRING, L. ÛELLRICH, U. PLÖCKER, & J. PRAUSNITZ. 1982. Vapor-Liquid Equilibria for Mixtures of Low Boiling Substances. Frankfurt am Main: Deutsche Gesellachaft für Chemisehes Appa.ratewesen.

Thermodynamic and Physical properties 111
LANDOLT-BÖRNSTEIN. 1960. Zahlenwerte und Funktionen. Vol. II Band, 2a. Teil. Berlin: Springer Verlag.
LANDOLT-BÖRNSTEIN. 1962. Zahlenwert und Funktionen. VoL li Band, 8. Teil. Berlin: Springer Verlag.
LUIJTEN, C., K. BOSSCHAART, & M. VAN DONGEN. n.d. A new method todetermine binary diffusion coefficients in dilute condensable vapors. submitted for publication.
PRUPPACHER, H., & J. KLETT. 1978. Microphysics of clouds and precipitation. Dordrecht, Holland: ReideL
RADULOVIC, S. 1991. Nucleation and Condensation in Mixtures of Hydrocarbons. Master thesis, R-1101-A, Eindhoven University of Technology, Faculty of Applied Physics.
REm, R., J. PRAUSNITZ, & B. PoLING. 1987. The Properties of Gases and Liquids. New York: McGraw-Hill Book Company.
SMOLDERS, H. 1992. Non-Linear wave phenomena in a gas-vapour mixture with phase transition. Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.
TAKAHASHI, S. 1974. Prepara.tion of a generalized chart for the diffusion coefficient of gases at high pressures. J. Chem. Eng. Japan 6:417-420.
VAN BEECK, J. 1992. Een reëel druppelgroeimodel, belicht in een binair mengsel. Master thesis , R-1169-A, Eindhoven University of Technology, Faculty of Applied Physics.
VARGAFTIK, N. 1975. Tables on the thermophysical properties of liquids and gases 2nd edition. New York: Wiley.

112 Thermodynamic and Physical properties

Appendix B
BINARY CLASSICAL NUCLEATION THEORY
Here we discuss the binary clas!!ical nucleation theory in more detail. We follow the paper of Looijma.ns (Looijma.ns et al. 1995), who applied the BONT model of Reiss (1950), Stauffer (1976), a.nd Wilemski (1984) for a mixture of n-nona.ne/metha.ne. The Gibbs free energy of formation of a n1, n2 cluster is given by (Wilemski 1984)
~G = (p" -l)V1 +erA+ 2: (~tl- ~ti}nl + i=l,2
2: (Jtî - pi)ni, (B.l) i=1,2
where p1 is the pressure inside the cluster, V1 the volume of the duster, n~ the number of molecules inside the duster, nt the number of molecules at the clusterfvapour interface, also referred to as excess molecules, a.nd the chemical potentials p~, 1'': are evaluated at the temperature T, and the total pressure in the duster or vapour phase respectively. It is assumed that the surface of the duster is in equilibrium with the interior of the duster such that p~ = p~. Then ~G ca.n he written as
~G = (p"- p1)V1 +erA+ 2: ~~t;n~. i=1,2
Here ~/'i = P!(p1, T) - ~t'!(p", T) a.nd n~ = ni + nt.
The saddle point of this ~G pla.ne is determined by
8
8~? = o, i= 1,2. ni
(B.2)
(B.3)
This, in combination with expression (B.2), the Gibbs-Duhem relation for the bulk liquid
S1dT- V1dp1 + 2: nldJ.tl(p1,T) = 0, (B.4) i:l,2
and the Gibbs-adsorption equation for the surface,
S 8 dT +Ader+ L nidpi = 0 (B.5) i=:;l,2
yield Àp; = 0. (B.6)
113

114 Binary Classica! Nucleation Theory
The liquid is assumed to be incompressible, p.[(p, T) = p.l(p", T) + v;(p1 - p"). In combination with LapJace's equation, the equilibrium of the cluster and the surrounding vapour phase can be expressed by the Kelvin equations,
A • 2uv; 0 P-i+ -r- = (i=l,2), (B.7)
where Apj = PHP", T) - p'f(p", T). All variables intheKelvin equations are functions of the bulk composition of the duster. So, from the saddle point only the bulk composition can he determined and not the excess and thus total composition.
The Gibbs free energy of formation of a duster combined with the kinetics of cluster formation will yield an expression for the nucleation rate. For the kinetic process only monomer contributions to cluster growth are taken into account. As a result the matrix R is diagonal,
R = (Ru 0), 0 Rzz
(B.S)
where Ru = C1A(nt. n 2),R22 = C2A(nt. n 2), and C; the impingement rates of molecules of component i. The final expression for the nucleation rateis (Looijmans 1995)
where Rav is the average growth of the clusters, and Z is the Zeldovich factor. Here,
Psp PN exp( -AG."/ kT), Ra" = det(R)/(Rusin2 </> + Rz2cos2tjJ- 2R12sintjJcostjJ),
(B.9)
(B.lO) (B.ll)
PN is the number density of the single molecules, D is the matrix containing the secoud deriva· tives of t!:..G at the saddle point with respect to the total number of molecules of the components, and tjJ is the angle of the nucleation rate J and the n1 axis, see figure 2.6. This tjJ is used for a coordinate transformation such that the cluster :flux over the saddle point can he written as
(B.12)
The x-axis is paralel to the direction of the nucleation rate over the saddle point and the y-axis is perpendicular to this direction. The width of the saddle point region W is unknown and has to he determined along with !/;. Stauffer (1976) showed that these two parameters are found as the solutions of
W/ D12 = (sin,Pcosf/Jt1(tan</>- R22D22/ RuDt2)/tan,P + R22/(Rutan,P)
tan!/; = s + Vs2 + r,
where s = 1/2( -Du/ D12 + R22D22/ RnDt2), and D;j the secoud derivatives of t!:..G.
(B.l3)
(B.14)

Binary Classical Nudeation Theory 115
The Zeldovich factor Z is determined by
Z = -~ (iPAG(x,y)jàx2).'{1/(-det(D))112• (B.15)
The Gibbs free energy of formation AG and its derivatives with respect to the total number of molecules are determined by contributions of n1 and n•. From the solution of the Kelvin equa.tions, n~ is known at the saddle point. The number of surface molecules is not known, but as Wilemski (1984) pointed out, the influence of surface molecules is limited to the rela.tively unimportant kinetic prefactor Z and therefore does not play an important role.
References
LOOIJMANS, K. 1995. Homogeneous nucleation and droplet growth in the coexistence region of n-alkane/methane mixtures at high pressures. Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.
LOOIJMANS, K., C. LUIJTEN, G. HOFMANS, & M. VAN DONGEN. 1995. Classical binary nucleation theory applied to the rea.l mixture n-nonane/methane at high pressures. J. Chem. Phys. 102 (11):4531-4537.
REISS, H. 1950. The kinetics of phase transitionsin hinary systems. J. Chem. Phys. 18(6):840-848.
STAUFFER, D. 1976. Kinetic theory of two-component ("heteromolecular") nuclea.tion and condensa.tion. J. Aerosol Sci. 7:319-333.
WILEMSKI, G. 1984. Composition of the critica.! nucleus in multicomponent va.por nuclea.tion. J. Ghem. Phys. 80(3):1370-1372.

116 Binary Classical Nucleation Theory

Appendix C
CONVERGENGE OF THE NUMERICAL SOLUTION
In tigure C.l the nucleation rate is shown as a function of time for an expansion of water/nitrogen. The solution is determined using the procedure described in chapter 3 and applying expression (3.21) for different sets of time steps ((50, 25 p. s), (20, 10 p. s), and (10, 5 p. s)).
t [ms]
Figure C.l: Nucleation mte as a function of time for three sets of time steps: (50, 25} indicated by t.J.t = 50 p. s, (20, 10} indicated by t.J.t 20 p. s, and (10, 5) 11 s indicated by t.J.t = 10 p. s. Homogeneom condensation processin waterfnitrogen for an initial pressure of 5 bar, initial temperature 295 K, initial partial vapour pressure 1000 Pa. The chamcteristic time of the ezpansion tz = 10 ms. Jmax = 5.1 · 1017
m-3 s-1, Smax = 17.94.
It is obvious that the solution for the nucleation pulse is independent of the size of the time steps as long as these are not chosen extremely large with respect to the width of the nudeation pulse. The largest time step used to find the results shown in figure C.l is 50 p s. This timestep yields less than 50 steps during the main part of the nucleation pulse, where the nudeation rate varles from 1013 to 5.1 · 1017• However, the final solution for J according to (3.21) for Llt1
50 and Àt2 ::::: 25 p. s is still very accurate compared to the solutions for even smaller time steps. The solutions for J for time steps of (10, 5 p. s) and (20, 10 p. s) are identical.
117
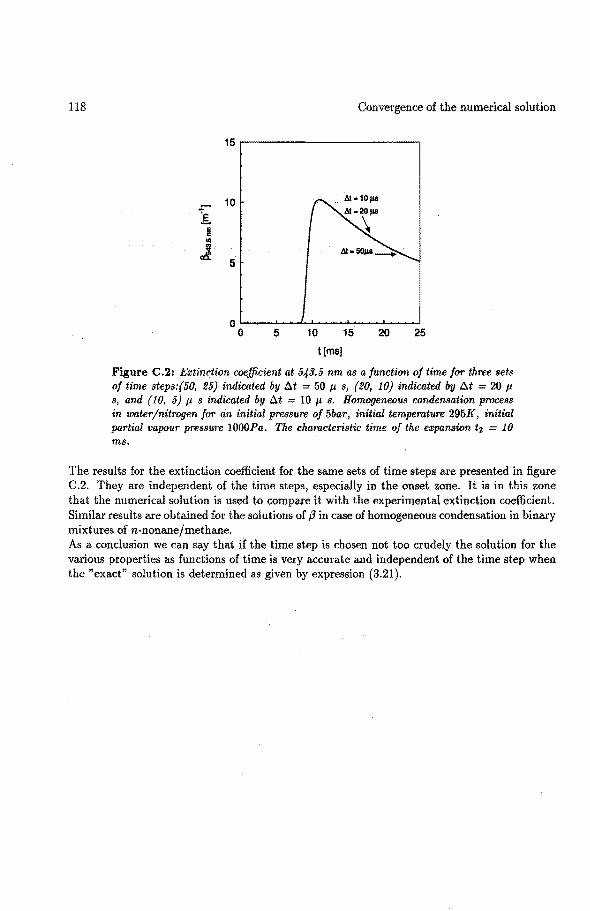
118 Convergenre of the numerical solution
t[ms]
Figure C.2: Extinction coefficient at 549.5 nm as a function of time for three sets of time steps:(50, 25} indicated by !lt = 50 p s, {20, 10} indicated by !lt = 20 p. s, and (10, 5} p s indicated by !lt = 10 p. s. Homogeneaus condensation process in water/nitragen for an initia[ pressure of 5bar, initial temperature 295K, initial partial vapour pressure lOOOPa. The characteristic time of the expansion tz = 10 ms.
The results for the extinction coefficient for the same sets of time steps are presented in figure C.2. They are independent of the time steps, especially in the onset zone. It is in this zone that the numerical solution is used to compare it with the experimental extinction coefficient. Similar results are obtained for the solutions of fJ in case of homogeneons condensation in binary mixtures of n-nonane/methane. As a condusion we can say that if the time step is chosen not too crudely the solution for the various properties as functions of time is very accurate and independent of the time step when the "exact" solution is determined as given by expression (3.21).

Appendix D
EXPERIMENTAL DATA
Waterfhelium experiments: the experiment number, the initia[ pressure, the initial temperature, the initia/ saturation ratio, the pressure, temperafure and molar fraction at the moment of maximum nucleation, the value of the maximum nucleation rate, and the reduced nucleation rate.
ex:p.nr Po [bar] To [KJ So Pm (bar] Tm [KJ I Yl,m · 104 Jm (m-;ss 1] r mOl 44.0 294.5 0.18 22.4 224.8 1.01 1 . 101
" 10 m02 43.9 294.5 0.34 24.9 234.9 1.98 6. 1018 1.5 m03 44.2 294.9 0.31 24.6 233.4 1.81 6 . 1018 I m04 43.2 294.9 0.34 43.0 234.0 1.16 1 . 1019 1 m05 78.2 294.8 0.31 43.1 231.2 1.05 1 . 1019 0.5 m06 21.9 294.7 0.32 12.4 234.6 3.64 5 . 1018 1.5 m07 26.2 294.7 0.31 13.5 234.4 3.24 4 . 1018 2 mOS 26.4 294.8 0.14 12.9 221.1 1.33 6 . 1018 10 m09 27.8 294.7 0.16 13.9 223.1 1.43 6 . 1018 10
mlO 45.9 294.8 0.87 31.2 252.7 4.93 5. 1018 0.5 mll 46.0 294.7 0.88 31.3 252.5 4.92 5. 1018 0.5 m12 26.9 294.6 0.83 18.2 251.9 7.85
i 3 . 1018 0.5
m13 27.1 294.6 i 0.87 18.5 252.9 8.13 2 . 1018 0.5 m14 82.6 294.7 0.88 55.7 251.8 2.79 9 . 1018 0.5 ml5 85.0 294.8 0.88 57.4 251.8 2.75 9 . 1018 0.3
· m19 20.8 294.8 0.18 10.6 225.0 2.13 5 . 1018 8 i m20 40.4 294.8 0.24 21.1 227.1 1.46 6 . 1018 2
m23 10.5 294.9 1.00 7.6 262 30 4. 101' 4
m24 21.3 294.9 1.00 14.9 255.6 12.0 2. 1018 0.2 m25 41.3 294.9 1.00 28.5 254.4 6.28 3 . 1Ql8 0.1 m26 81.3 294.9 1.00 55.8 253.6 2.83 9 . 1018 0.1 m29 21.5 294.8 1.00 15.0 255.3 11.8 1 . 1018 0.5 m30 42.1 294.7 1.00 29.3 254.7 5.94 4 . 1Ql8 0.2 m31 7.3 294.9 1.00 5.2 258 35 3 . 1017 0.5 m32 79.7
i 295.0 1.00 55.1 254.4 3.32 8 . 1018 0.2
m57 54.3 294.6 1.00 37.8 255.0 4.73 i
6 . 1018 0.5 m58 55.2 i 294.7 1.00 38.7 255.9 4.78 6 . 1018 I 1
119

120 Experimental data
Water/nitrogen experiments: the experiment numbér, the initial pressure, the initial temperature, the initial saturation ratio, the pressure, molar vapour fraction, the ideal temperafure {I= 1.4) and the temperafure according to the Sychev-EOS at the moment of maximum nucleation, and the value of the maximum nucleation rate.
exp. nr Po [bar] To [K] So Pm [bar] YI,m ·104 Tm· m,S!!cheu [ K] Jm [m ·3s IJ m43 74.1 294.7 1.00 45.7 4.22 256.7 255.5 1 . 101!!
m45 21.7 294.8 1.00 13.2 . 12.2 255.7 255.2 2. 1017
m46 42.2 294.7 1.00 25.6 6.68 255.4 254.9 5. 1017
m47 75.9 294.5 1.00 46.4 4.10 255.8 254.6 1 . 1018
m48 12.4 294.7 1.00 7.6 21 255.8 255.4 8 . 1016
m49 32.1 294.8 1.00 19.3 8.63 254.8 254.1 3. 1017
m50 62.3 295.0 1.00 38.8 4.96 257.7 256.5 1 . 1018
m51 49.4 295.0 1.00 30.3 5.91 256.6 255.6 7 . 1017
m52 58.5 295.1 1.00 36.7 5.24 257.5 256.5 1 . 1018
m53 31.9 294.8 1.00 19.6 8.62 255.8 255.2 4. 1017
m54 94.7 294.8 1.00 60.3 3.53 257.8 256.5 2 . 1018
m55 83.2 294.8 1.00 52.4 3.94 257.8 256.6
I
2 . 1018
m56 69.0 294.6 1.00 43;5 4.42 257.3 256.2 1 . 1018
The experiment number, the reduced nucleation rates r1 determined without correction for the pressure dependenee of the surface tension and the temperafure found with 'Y 1.4, r 2 : same as r 1 but with Tsychev, and r with Tavchev and with the pressure effect of nitrogen on the surface tension of water (Slowinski et al.).
1 exp. nr l r 1 I r 2 1 r m43 10 1.6 0.07 m45 1.5 0.75 0.2 m46 1.8 0.63 0.069 m47 6.0 0.78 0.022 m48 0.5 0.26 0.13 m49 0.4 0.11 0.038 m50 45 5.9 0.26 m51 10 1.8 0.090 m52 25 4 0.11 m53 2.5 1 0.16 m54 350 32 0.045 m55 55 6.6 0.060 m56 50 7.0 0.056

Experimental data 121
n-Nonanefmethane experiments: the experiment number, the initial pressure and temperature, the saturated molar fraction of n-nonane according to the RKS-EOS, the initial molar fraction of n-nonane according to the GC method, the relative inaccuracy, the corrected initia! molar fraction, and the extra waiting time during the preparation of the mixture.
exp. nr. Result RKS Gas Ana. rel. error RKS+cor extra Po [bar] To [KJ Yl,sat ' 10
4 Yl,sat • 104 D..ytfyl% Yl,sat • 104 [h) m77 73.0 284.7 4.3
I 4.1 35 3.8 0
m78 82.3 284.3 5.2 4.4 10 4.7 0 m79 92.9 283.9 6.6 5.2 10 6.1 0 m80 23.3 284.3 2.2 1.7 10 1.7 i 0 m81 59.5 283.7 3.1 2.4 I 10 2.6 65 m82 100.6 i 284.2 8.1 12 40 7.6 0 m83 41.7 283.8 2.3 2.1 50 1.8 0 m84 93.6 301.6 12 12 20 11 16 m85 47.3 302.0 6.3 5.5 10 5.1 0 m86 64.7 302.4 7.8 6.5 15 6.5 0 m88 21.0 302.8 6.7 5.4 15 5.4 0 m89 90.4 302.3 12 10.5 10 10 65 m91 35.5 302.3 5.9 4.8 10 4.6 16 m92 74.8 302.9 9.2 7.8 10 7.9 0 m93 56.7 295.3 5.1 4.5 10 4.3 16 m94 49.0 295.2 4.6 3.8 10 3.8 0 m95 31.4 295.7 4.2 5 35 3.4 0 m96 93.9 295.3 9.8 9.3 10 9.1 16 m97 66.5 295.3 5.9 5.0 10 5.2 0 m98 83.9 295.3 8.1 7.3 i 10 7.4 16 m99 22.4 295.3 4.3 2.7 i 10 3.5 20

122 Experimental data
n-Nonane/methane: the experiment number, the pressure and temperafure at the Wilson point, the pressure and temperafure at the moment of maximum nucleation, the temperafure according to the EOS of Sychev, the value of the maximum nucleation rate, and the saturation sealing factor.
I exp. nr I Pw [bar] I Tw [KJ I Pm [bar] I Y1 m · 104 I Tm I Ts11cheu I Jm [m-3s 1] I I< I
m77 . 47.5 253.0 46.0 3.7 250.7 251.9 3. 101" 0.88 m78 56.6 256.6 54.1 4.5 253.4 254.4 5 . 1018 0.81 m79 67.0 259.8 64.8 6.0 257.3 258.1 9. 1018 0.78 m80 10.2 227.9 9.3 1.55 222.2 225.9 2. 1017 1.13 m81 35.9 246.9 33.6 2.5 242.4 244.2 2 . 1018 0.93 m82 75.3 262.8 73.2 7.5 260.7 261.4 1 . 1019 0.78 m83 21.5 237.1 20.5 1.75 233.8 236.4 6. 1017 1.08 m84 68.8 278.0 65.0 10.8 273.8 274.4 4 . 10'8 0.86 m85 26.6 259.6 24.8 5.0 254.8 256.5 5 . 1017 1.02 m86 40.7 267.7 38.3 6.3 263.3 264.3 1 . 1Q18 0.97 m88 9.7 248.2 8.8 5.1 242.0 244.5 9 . 1016 1.20 m89 64.4 276.5 60.8 9.5 272.1 272.7 4 . 1018 0.86 m91 19.1 263.3 17.9 4.5 253.2 254.8 2. 1017 1.34 m92 50.2 272.7 47.5 7.7 268.7 269.4 2 . 1018 0.95 m93 34.1 257.8 32.5 3.6 254.5 255.9 1 . 10u; 1.02 m94 29.0 255.3 26.3 3.7 250.0 251.7 7 . 1017 1.05 m95 14.8 242.4 13.7 3.2 237.6 240.3 3 . 1017 0.99 m96 68.0 270.9 65.2 9.0 267.9 268.6 7 . 1018 0.82 m97 41.5 260.2 39.7 5.0 257.1 258.2 2 . 1018 0.92 m98 59.8 269.6 56.2 7.3 265.2 265.9 4 . 1018 0.86 m99 9.0 232.3 8.8 3.3 230.6 233.9 2. 1017
I o.96,

Appendix E
ENHANCED SOLUBILITY OF WATER VAPOUR IN AIR,
OXYGEN AND NITROGEN
The enhancement factor of water in a gaseous carrier at temperature T and total pressure p is defined as
fw(p,T) (E.l)
The definition of Scan be reformulated using fw as
S = p" ~ Y~qP . p~s(p, T) f w(p, T)Pv.o(T)
(E.2)
Wylie and Fisher (1996) determined the enhancement factor of water vapour in mixtures of water/airand water/oxygen for various temperatures and pressures. Hyland and Wexler (1973, 1975) determined this enhancement factor also for ice/air. The departure of fw from unity is related to the imposed pressure exceeding the satl,liated pressure of pure water: &p = p- PvsO· Following Hyland and Wexler (1973), ln(fw) is expanded in terros of &p. It is found (Luijten and Van Dongen ) that a linear term is sufficient to descri he the pressure dependenee of fw:
ln(fw) = b(T)&p. (E.3)
They also showed that a third-order polonomial for b(T) is sufficient to perform an excellent match to the available data on fw in air and in oxygen. The liquid (or ice) formed consists only of water, which means that its chemica! potential depends on the total pressure as described insection 1.2.1. This chemical potential depends on the total pressure, temperature and the density of the liquid (or ice). The extra increase of the partial vapour pressure of water compared to the Poynting effect is therefore only related to the effect of the inter molecular interaction of the molecules in the gas phase. Luijten and van Dongen express fw as two separate contributions, one from the liquid phase fp011nting and one from the vapour phase ft, such that fw ft* !Poynting·
For temperatures below the freezing point only data on fw in air above ice are available. However, during condensation experiments undercooled liquid water is formed and so for theoretica! purposes fw above liquid water must be known. The difference between ice and undercooled water is the density of the condensed phase. This affects only the contribution of the Poynting effect to fw· Correcting the data on fw for this difference in density yields the appropriate values for fw for undercooled water. Luijten and Van Dongen showed that the relationship for b(T)
123

124 Enhanced solubility of water vapour in air, oxygen and nitrogen
found for this extrapolated data describes the data on fw well for the complete temperature range. From the data of fw in mixtures of water/airand water/oxygen, the results for fw in a mixture of water/nitrogen are deduced by a linear mixing rule (Luijten and Van Dongen). The results are shown in table E.I.
I Gas component Air (78.084 % nitrogen 21.916 % oxygen (lumped) Oxygen
en
4.186 . w-2 • 2.87 . w-4 T + 6.91 . w-" T 2 - 5.63 . w-10 T3
3.354 . w-2 • 2.30 . w-4 T + 5.50 . w-7 T 2 - 4.40 . w-10 T3
4.42 . w-2 - 3.03 . w-4 T + 7.31 . 10 7 T 2 • 5.98 . w-to T3
References
HYLAND, R. 1975. A correlation for the second virial coefficients and enhancement factors for moist air. J. of Research of the National Bureau of Standards- A. Physics and Chemistry 79(4):551-560.
HYLAND, R., & A. WEXLER. 1973. The enhancement of water vapor in carbon dioxide-free air at 30, 40 and 50 oe. J. of Research of the National Bureau of Standards - A. Physics and Chemistry 77(1 ):115-131.
LUIJTEN, C., & M. VAN DoNGEN. n.d. The enhancement factor of water in nitrogen, air, and oxygen. Private communication.
WYLIE, R., & R. FISHER. 1996. Molecular interaction of water vapor and air. J. Chem. Eng. Data 41:133-142.

SYMBOLS
This list of symbols contains the main symbols used in this thesis. Occasionally, the symbols can have a different meaning; this will he explicitly mentioned in the section concerned.
Roman symbols:
a ma a m2 A m2 A s-1 b J mol-1
B c m3 c m Cn s-1
c" J kg-1 K-t
c" J kg-I K-1 D m2 s-1
D J En s-1
F J g; G J D.G J H h J mol-1
J m-3 s-1
Jd kg s-1
k J K-t k;j
K K Kn l m L J kg-1 m m kg M kg s-1
coefficient RKS equation surface area surface area drift coefficient Fokker-Planck equation coefficient RKS equation activation fundion Peneloux correction speed of sound condensation rate isobaric specific heat isochoric specific heat dilfusion coefficient tensor with second derivatives of free energy evaporation rate Helmholtz free energy molar fraction Gibbs free energy Gibbs free energy of droplet formation Activation function for nucleation in Katz's approach molar enthalpy nucleation rate ditfusion flow Boltzmann's constant interaction parameter nucleation parameter fit parameter Knudsen number molecular mean free path latent heat of evaporation refractive index ( molecular) mass rate of mass transfer to a droplet
125

126
M n n! ' NA mol-1
NuM Nuq p p
qo
Pa kg1/4 m3 s-t/2 mol-1
Q J s-t
Qint J s-1
Q..,t -Td m R J mol-1 K-1
R s-1
Rav s-1
s t s ll.t s ll.t s T K V m3
V m 3
V m3
VRKS m 3
W m-2
Yi x m z z ZRA -
Greek symbols:
a a an /3 m-2 s-1
/3 m-1
1' Î
m-2 s-t
r
dimensionless mass of a droplet number of molecules number of excess molecules in critical cluster A vogadro's number Nusselt number for mass transfertoa droplet Nusselt number for heat transfer toa droplet pressure parachor parameter Fisher's droplet model rate of heat transfer to a droplet rate of heat transfer into a droplet extinction efficiency droplet radius universal gas constant growth rate tensor average growth rate saturation ratio time nucleation pulse duration timestep temper at ure molecular volume molar volume total volume molar volume in RKS equation width factor molar liquid fraction of component i molar vapour fraction of component i space coordinate Zeldovich factor compressibility factor Rackett compressibility factor
"cooling coefficient" size parameter mass accomodation coefficient impingement rate extinction coefficient ratio of specific heats evaporation coefficient Fit parameter: Jexvl JeNT
Symbols

Symbols
8 () 0
À m ,\ J m-1 s-1 K-1
A mK-1
"' J
(J m-3 (f N m-1
:E m-38-l T
'f'
\[1
w n ms-1
n J
Subscripts:
c eq
ij m 0
V
vs n 1 2 00
Superscripts:
e fm cl l s sp t
scattering angle scattering angle wavelength of light thermal conductivity constant related to surface tension chemica! potential number density surface tension kinetic prefactor parameter Fisher's droplet model angle determining direction of saddle point flux correction in e:lfective unary saturation ratio Pitzer's acentric factor growth rate Grand potential
pertains to critica! point pertains to thermadynamie equilibrium pertains to component i pertains to an (i,j) cluster . pertains to mixture (RKS equa.tion) pertains to molecule pertains to vapour perta.ins to saturated vapour pertains to cluster size perta.ins to component 1 pertains to component 2 pertains to fax field
perta.ins to equilibrium state perta.ins to free-molecular flow regime pertains to continuurn flow regime pertains to the liquid state pertains to the surface la.yer perta.ins to the saddle point pertains to the total cluster
127

128
V
* pertains to the vapour state pertains to the critical nucleus pertains to a dimensionless parameter pertains to a parameter per unit of mass
Symhols

SUMMARY
Inthls thesis we describe homogeneons condensation of a vapour/gas mixture due toa continuons adiabatic expansion in an expansion doud chamber at high pressures. When the interaction between the vapour and gas molecules is negligible the gas component can he regarcled as inert because it does not condense. With increasing pressure the interaction between vapour and gas molecules can no longer he neglected. At pressures over 10 bar these real gas effects are important for mixtures of alkanes, e.g. n-nonane and methane. The solubility of the vapour component increases with increasing pressure and both components are present in the liquid phase even at temperatures above the critica! temper at ure of the gas component. To study the homogeneons condensation processof vapour/gas mixtures at high pressures an expansion cloud chamber was developed, suited for pressures up to 100 bar. It was placed in a room where the temperature could be regulated between 10 oe and 35 oe. The processof homogeneons condensation is monitored by measuring the transmission of laser beams. In the expansion doud chamber, a vapourjgas mixture experiences a continuons adiabatic expansion due to which homogeneons condensation will occur. This processis characterized
' by the simultaneons formation and growth of droplets, which try to restore thermodynamic èquilibrium. During a small period of time, the nucleation pulse, dropiets are formed and existing dropiets grow, then foliowed by a period of droplet growth only. The final result of the condensation processes is a cloud of dropiets distributed over their sizes. The theoretical problem of homogeneons condensation in a prescribed continuons expansion was solved numerically. For mixtures of an ideal vapour component and an idea1 and inert gas component, e.g. waterfhelium or water/nitrogen, we applied the classical nucleation theory (CNT) and the model of Gyarmathy fordroplet growth. The saturated partial vapour pressure of water was determined by the Poynting effect for water/helium mixtures and by the empirical enhancement factor fw for mixtures of water/nitrogen. For a binary mixture of real gases, e.g. n-nonane/methane, we used the Redlich-Kwong-Soave equation of state (RKS-EOS) to describe the thermodynamic behaviour of the mixture, the binary classical nucleation theory (BONT) and the droplet growth model of Gyarmathy, which we extended to binary mixtures of real gases. Also, an approximate analytical solution of a mixture of an i deal vapour and an i deal inert gas was given based on asymptotic techniques. The asymptotic solution yields a nucleation pulse which reaches its maximum later in time than the numerical solution and at a higher value. However, the change in the molar liquid fraction occurs in the same period of time for both solutions. In this period all of the latent heat is released. For low total pressures, this explains the success of the asymptotic solution when comparing the predictions of the static pressure to measurements in nozzle flowsof a vapour/gas mixture. At high total pressures the effect of latent heat on the gasdynamics of the mixture betomes negligible and optical techniques are applied to detect homogeneons condensation. The optical properties are determined by the size distribution fundion of the cloud of droplets, which is not predicted well by the approximate
129

130 Summary
asymptotic solution. To calculate optica! properties of the expanding vapour/gas mixture the numerical solution of the condensation process is combined with tbe Mie theory.
The first series of experimentsin the expansion cl oud chamber was performed with water /helium mixtures. The initial pressure and initia! partial vapour pressure were varied, yielding the nucleation pulse to occur in a pressure range of 5 to 60 bar and a temperature range of 220 to 260 K We introduced a reduced nucleation rate r, i.e. the ratio of the experimental and tbe theoretica! nucleation rate, to match the experimentally deterrnined extinction . coefficients to the numerically calculated extinction coefficients. The metbod of matching the extinction coefficients yields reliable nucleation rate data wbich are only weakly dependent on the relations used for the various thermopbysical quantities and properties, e.g. surface tension or diffusion coefficient. Tbe reduced nucleation rate for the water/helium experiments showed no pressure dependenee and was independent of the way in which the initia! composition was determined (Humicap device or saturated mixtures). We compared the results of our series of water/helium experiments to nucleation rate data of Looijmans et al. (1995) and Viisanen et al. (1993). Tbe reduced nucleation rate agreed very well with the data of Looijmans, but showed a systematic difference with the experiments of Viisanen et al. Tbis may be related to a difference in the way the initia! composition of the mixtures was determined. Experiments with initially saturated mixtures of water/nitrogen yielded a reduced nucleation rate wbich depended on tbe total pressure. For a pressure of 60 bar the reduced nucleation rate increased by about a factor 100 compared to low pressures. So, nudeation is enhanced in such mixtures by adding nitrogen. There is strong evidence that this can he explained as an effect of the total pressure of nitrogen on tbe surface tension of water. We incorporated this effect in the numerical simulation and this resulted in a reduced nucleation rate which was almost independent of total pressure. The temperature dependenee of the pressure effect on the surface tensionwas taken linear, but from adsorption theory a much stronger temperature dependenee is to be expected. So, some questions are still open. The results for the reduced nucleation rate for water/nitragen with the effect of the total pressure on the surface tension agreed with the values for r found for experiments with initially saturated mixtures of water and helium.
We performed three series of experiments with saturated mixtures of n-nonane/methane in the expansion doud chamber each with a different initia! temperature. To determine the initia! composition of a mixture a metbod for gas analysis based on gas chromatography was implemented. The average uncertainty for the molar fraction of n-nonane for all experiments was 15 %. The RKS-EOS systematically predicted higher values for the initia! molar fraction of n-nonane, Yt,sat, for a saturated mixture of n-nonane/methane. For each initia! temperature there exists a constant difference for Yt.sat between the experimental results and the results for the RKS-EOS. Tbe range of pressures and temperatures at the detected onset of homogeneons condensation for the three experimental series range from 5 to 75 bar, and from 225 to 278 K, respectively. To match the experimentally deduced extinction coefficients and the numerically calculated ex-

References 131
tinction coefficients due to the homogeneons condensation processof a mixture of n-nonane/methane, we introduced a saturation sealing factor K. It is defined as the ratio of the actual value of S and the value of S for which a nucleation theory would adequately describe the experimental observation. The results for K for the three series of experiments were compared to the results for the saturation scaler from the data of Looijma.ns (1995) and the quasy one component ( QOC) theory. The saturation scaler for our experiments show that the BCNT describes the nuclea.tion behaviour of the n-nonane/metha.ne mixture better than the QOC-theory. A small extra pressure dependenee exists for the nucleation behaviour of the mixture relative to the BCNT. For the small number of experiments which have a range of pressures and temperatures which overlap with the data of Looijmans, the results for the saturation scaler agree rather well with results for the data of Looijmans. To increase the overlap, the temperature range for the experiments in the expansion cloud chamber must be extended to lower tempera.tures. This can be realized for a.n initia! mixture of n-nonane/methane which is undersa.turated. Also, the error bounds must be reduced for both methods to increase the quality of the nucleation ra.te data and the capability to compare the two experimental methods. This can be realized by an on-line measurement of the initia] composition of the n-nonane/methane mixture, avoiding all of the separate steps of the present metbod of gas analysis.
Finally we proposed a new model to determine nucleation rate data from experiments with the expansion cloud chamber. A priori knowledge of the nudeation behaviour of the mixture is not needed at all and only an approximate droplet growth rate is sufficient. The model is based on the assumption that the nudeation rate J(t) has a known functional form with three free parameters to he determined experimentally.
References
LOOIJMANS, K. 1995. Homogeneous nucleation and droplet growth in the coexistence region of n-alkanefmethane mixtures at high pressures. Ph.d. thesis, Eindhoven University of Technology, Faculty of Applied Physics.
LOOIJMANS, K., C. LUIJTEN, & M. VAN DONGEN. 1995. Binary nucleation rate measurementsof n-nonane/methane mixtures at high pressures. J. Chem. Phys. 103(11):1714-1719.
VIISANEN, Y., R. STREY, & H. REISS. 1993. Homogeneons nucleation rates for water. J. Chem. Phys. 99(6):468(}--4692.

132 Summary

SAMENVATTING
Dit proefschrift beschrijft het homogene condensatieproces van een damp/gas mengsel door het adiabatisch te expanderen in een expansie-nevelkamer. Bij een te verwaarlozen interactie tussen de damp- en gasmoleculen, condenseert het gas niet en kan het als inert worden beschouwd. Naarmate de druk toeneemt wordt de interactie tussen moleculen van de verschillende componenten steeds belangrijker. Deze reële gaseffecten zijn voor mengsels van alkanen, b.v. n-nonaan en methaan belangrijk bij een druk van meer dan 10 bar. De oplosbaarheid van de dampcomponent neemt toe als de druk stijgt en beide componenten zijn aanwezig in de vloeistof, zelfs bij temperaturen boven de kritische temperatuur van de gascomponent. We hebben het homogene condensatie proces van damp/gas mengsels bij hoge druk experimenteel onderzocht in een zelf ontworpen expansie-nevelkamer. Deze is geschikt tot een druk van 100 bar en stond in een kamer waarin de temperatuur gevarieerd kon worden tussen 10 en 35 oe. Door het meten van de transmissie coëfficiënt van laserbundels werd het condensatieproces gevolgd. In de nevelkamer ondergaat het damp/gas mengsel een continue adiabatische expansie waardoor homogene condensatie zal optreden. Dit proces wordt gekarakteriseerd door het simultaan onstaan en groeien van druppels, die proberen het thermodynamisch evenwicht te herstellen. Gedurende een korte periode, de nucleatie puls, worden nieuwe druppels gevormd, terwijl de reeds bestaande druppels groeien. Na deze periode van druppelvorming is er uitsluitend nog druppelgroei. Het uiteindelijke resultaat is een nevel van druppels die variëren in grootte. Het theoretische probleem van homogene condensatie in een voorgeschreven continue expansie werd numeriek opgelost. Voor mengsels van een ideale dampcomponent en een ideale inerte gascomponent, b.v. water/helium of water/stikstof, gebruikten we de klassieke nucleatie-theorie (CNT) in combinatie met het druppel-groei-model van Gyarmathy. De verzadigde partiële dampdruk van water is bepaald aan de hand van het Poynting effect voor mengsels van water/helium en aan de hand van de empirische "enhancement" faktor fw voor mengsels van water/ stikstof. De thermodynamische toestand van een mengsel van reële gassen, b.v. n-nonaan/methaan, beschrijven we aan de hand van de Redlich-Kwong-Soave toestandsvergelijking (RKS-EOS). Het condensatie gedrag beschrijven we aan de hand van de binaire klassieke nucleatie-theorie (BCNT) en het druppel-groei-model van Gya.rmathy, hetgeen we hebben uitgebreid tot binaire mengsels van reële gassen. Een benaderende analytische oplossing voor het condensatieproces van een ideale dampcomponent en een ideale inerte gascomponent wordt ook gepresenteerd. Deze oplossing is gebaseerd op asymptotische technieken. De asymptotische oplossing resulteert in een nuclea.tiepuls waarvan het maximum later optreedt en hoger is dan het maximum van de numerieke oplossing. Echter voor beide oplossingen vindt de toename van de molaire vloeistof fractie plaats in dezelfde periode. Gedurende deze periode komt alle latente warmte vrij. Dit verklaart het succes van deze asymptotische oplossing wanneer zij gebruik wordt om statische
133

134 Samenvatting
drukken te voorspellen in nozzle stromingen van een damp/gas mengsel bij lage druk. Bij hogere drukken wordt het effect van de vrij komende latente warmte verwaarloosbaar en worden optische technieken gebruikt om homogene condensatie te detecteren. De optische eigenschappen worden bepaald door de druppel-grootte-verdelingen. Deze worden niet goed voorspeld door de benaderende asymptotische oplossing. De optische eigenschappen van het expanderend damp/gas mengsel worden numeriek berekend door de oplossing van het condensatieproces te combineren met de Mie-theorie.
De eerste serie experimenten met de expansie-nevelkamer zijn uitgevoerd met mengsels van water/helium. Daarbij werd de initiële druk en initiële partiële dampdruk gevarieerd zodat de nucleatiepuls binnen een drukbereik van 5 tot 60 bar én een temperatutirbereik van 220 tot 260 K lag. We introduceerden een gereduceerdenucleatiesnelheid r, i.e. de verhouding van de experimentele en theoretische nucleatiesnelheid, waarmee de experimenteel bepaalde extinctiecoëfficiënt met de numeriek berekende extinctiecoëfficiënt in overeenstemming kan worden gebracht. Deze methodiek levert betrouwbare resultaten voor de nucleatiesnelheid die vrijwel onafhankelijk zijn van de relaties voor de verschillende thermische en fysische grootheden, zoals oppervlaktespanning en diffusiecoëfficiënt. De gereduceerde nucleatiesnelheid bepaald uit de water/helium experimenten vertoonde geen drukafhankelijkheid en was onatbankelijk van de manier waarop de initiële partiële dampdruk van water werd bepaald (Humkap of verzadigde toestand). De resultaten van onze meetserie met water/helium mengsels hebben we vergeleken met nudeatiesnelheidsdata van Looijmans et al. (1995) en Viisanen et al. (1993). The gereduceerde nudeatiesnelheid kwam goed overeen met de data van Looijmans, maar verschilde systematisch met de data van Viisanen. Dit kan veroorzaakt zijn door in verschil in de manier waarop de beginsamenstelling van het gas is bepaald. Experimenten met initiëel verzadigde waterfstikstof mengsels resulteerden in een gereduceerde nucleatiesnelheid die afhankelijk was van de totaal druk. Voor een druk van 60 bar was de gereduceerde nucleatiesnelheid toegenomen met een faktor 100 ten opzichte van de waarde bij lage drukken. Dus het nucleatieproces neemt in dergelijke mengsels toe door het verder toevoegen van stikstof. Er zijn sterke aanduidingen dat dit veroorzaakt wordt door de invloed van de stikstofdruk op de oppervlaktespanning van water. We hebben deze invloed in de numerieke simulatie meegenomen en dit resulteerde in een r die bijna onatbankelijk werd van de druk. De temperatuuratbankelijkheid van het druk-effekt op de oppervlaktespanning werd lineair verondersteld, maar op basis van adsorptietheorieën verwachtten we een veel sterkere temperatuuratbankelijkheid. Dit geeft aan dat er nog wat vragen open staan. De resultaten voor de gereduceerde nudeatiesnelheid voor mengsels van water/stikstof inclusief het druk-effekt op de oppervlaktespanning komen overeen met de waarden van r bepaald uit experimenten met initiëel verzadigde mengsels van water/helium.
We hebben drie series experimenten uitgevoerd met mengsels van n-nonaan en methaan, ieder bij een andere begintemperatuur. Om de initiële samenstelling van het gasmengsel te kunnen bepalen is een gas-analyse methode opgenomen in de meetprocedure. Deze gas-analyse methode maakt gebruik van een gas-chromatograaf. De gemiddelde nauwkeurigheid van de molaire fractie n-nonaan bedroeg

Referenties 135
15 %. De RKS toestandsvergelijking voorspelt systematisch een hogere waarde voor de molfractie van n-nonane, Yt,sat• in een verzadigd mengsel van n-nonaan en methaan. Voor iedere begintemperatuur blijkt een constant verschil te bestaan tussen de waarde van y1, sat bepaald uit het experiment en bepaald met de RKS toestandsvergelijking. Het bereik in drukken en temperaturen op het moment waarop het begin van homogene condensatie werd gedetecteerd voor de drie series experimenten bedraagt 5 to 75 bar en 225 tot 278 K. Om de experimentele bepaalde en numeriek berekende extinctiecoëfficiënten tijdens het homogene condensatieproces van een n-nonaanfmethaan mengsel op elkaar te leggen, introduceerden we een schalingsfaktor K voor de verzadigingsverhouding. Deze is gedefinieerd als de verhouding van de werkelijke verzadigingaverhouding S tot de waarde van S waarvoor een nucleatietheorie de experimentele waarneming goed beschrijft. De resultaten voor K voor de drie series van experimenten zijn vergeleken met met de resultaten voor de schalingsfaktor bepaald uit experimenten van Looijmans en met de quasi-een-component (QOC) theorie. De schalingsfaktor K bepaald uit onze experimenten liet zien dat de BCNT het nucleatiegedrag van n-nonaan/methaan mengsels beter beschrijft dan de QOC theorie. Het nucleatiegedrag vertoont een lichte extra drukafhankelijk in vergelijking met de BCNT. De resultaten voor K uit het beperkt aantal experimenten waarvoor het druk- en temperatuurbereik overlappen met het druk- en temperatuurbereik voor data van Looijmans komen goed overeen met de waarden van K voor de data van Looijmans. Deze overlap kan worden vergroot door het temperatuurbereik van de experimenten met de expansie nevelkamer naar lagere temperaturen uit te breiden. Daartoe moeten experimenten verricht worden met initiëel onderverzadigde mengsels n-nonaan/methaan. Verder moeten de foutenmarges verkleind worden voor beide experimentele technieken zodat de kwaliteit van de experimenteel bepaalde nucleatiesnelheden en de mogelijkheid om de twee experimentele technieken (nucleatie puls buis (Looijmans) en expansie nevelkamer) met elkaar te vergelijken vergroot wordt. Dit kan worden bereikt door een on-line meting van de samenstelling van het mengsel aan het begin van het experiment. Hiermee worden alle individuele stappen van de huidige gas-analyse methode omzeild.
Tot slot stelden we een nieuw model voor waarmee nucleatiesnelheden bepaald kunnen worden uit experimenten met de expansie-nevelkamer. Vooraf is geen enkele kennis van het nucleatiegedrag van het mengsel nodig, en een benadering voor de druppelgroeisnelheid is voldoende. Het model is gebaseerd op de veronderstelling dat de nucleatiesnelheid J(t) een bekend functioneel verband heeft met drie vrije parameters die experimenteel bepaald worden.
References
LOOIJMANS, K. 1995. Homogeneous nucleation and droplet growth in the coexistence region of n-alkane/methane mixtures at high pressures. Ph.d. thesis, Eindhoven U niversity of Technology, Faculty of Applied Physics.
VIISANEN, Y., R. STREY, & H. REISS. 1993. Homogeneous nucleation rates for water. J. Chem. Phys. 99(6):4680-4692.

136 Samenvatting

NAWOORD
Na een periode van iets meer dan vier jaar zit mijn promotiewerk erop. Ik heb die tijd in de zeer prettige omgeving van de vakgroep Transportfysica van de faculteit Technische Natuurkunde aan de Technische Universiteit Eindhoven gewerkt. Maar ook buiten de vakgroep ben ik door velen goed geholpen. In het bijzonder wil ik de groep instrumentele analyse van de faculteit scheikunde bedanken, met name Hans Gert Janssen en Marc van Lieshout. Beiden hebben mij en enkele van mijn afstudeerders geholpen bij het opstellen van de gas-analyse methode en ons met veel geduld begeleid bij het verrichten van vele analyses aan enkele van hun gaschromatografen. De medewerkers van de werkplaats van de faculteit Natuurkunde wil ik bedanken voor hun nauwkeurige werk waardoor ik met de expansie nevelkamer succesvolle metingen heb kunnen uitvoeren. Ik heb mijn werk verricht in de groep van Rini van Dongen, die ik voor zijn steun en vele waardevolle suggesties en discussies hartelijk wil danken. Verder heb ik heel veel steun en hulp gekregen van de "stille" krachten van de groep: Jan Willems, Louis Wasser, Eep van Voorthuisen, Harm Jager en Bram Wijnands. Zonder hen was ik nooit aan experimenteren toe gekomen. Speciaal wil ik Louis Wasser noemen door wie de gerealiseerde experimentele opstelling zeer mooi en uiterst functioneel is vorm gegeven. Karel Looijmans, Carlo Luijten, Geert Hofmans, Vitaly Kalikmanov, en Mico Hirschberg, ben ik zeer erkentelijk voor de vele leerzame en/ of onderhoudende discussies. De afstudeerders Jeroen van Beeck, Geert Hofmans, John van de Broek, Maykel Verschueren, Marc Roelands en Ruud van Eeghem hebben belangrijke bijdragen geleverd aan het onderzoek. Het verkennende werk van de stagiairs Harold Kloosterhof, Gino Thielens, Gino Lambert, en Rene Cortenraed hebben tot nuttige resultaten geleid. De afstudeerder en latere AI0-2 Kris Snoeijs is eigenlijk een hoofdstuk apart, maar zijn enthousiasme en vele ideëen zijn voor dit onderzoek belangrijk geweest. Carlo Luijten en Ed Niessen wil ik bedanken voor het correctiewerk van de grove versie(s) van dit proefschrift. Tot slot wil ik ook mijn familie, mijn vrienden en natuurlijk mijn vrouw, Jacqueline, bedanken voor hun steun en de interesse die zij voor mijn werk getoond hebben.
137

138 Nawoord

3 mei 1967
mei 1985
augustus 1990
oktober 1990
november 1991-februari 1996
1 juni 1996
CURRICULUM VITAE
geboren te Nuth
diploma Gymnasium B Bisschoppelijk College Sittard
diploma Technische Natuurkunde Technische Universiteit Eindhoven
vervullen van dienstplicht
onderzoeker in opleiding (OIO) Technische Universiteit Eindhoven Faculteit der Technische Natuurkunde vakgroep Transportfysica
medewerker afd. mechanische analyse ASM Litography Veldhoven
139

Stellingen behorende bij het proefschrift "Homogeneous condensation in a vapour /gas mixture at high pressures in an expansion
cloud chamber". door M.J.E.H. Muitjens
1. De kwaliteit van experimenteel bepaalde nucleatiesnelheden in een damp/ gas mengsel wordt voornamelijk bepaald door de nauwkeurigheid waarmee de samenstelling van dat mengsel bekend is.
Dit proefschrift.
2. Met de expansie nevelkamer zijn nucleatiesnelheden te bepalen met een gelijke nauwkeurigheid als met de nucleatiepuls-buis.
3. De benaderende asymptotische oplossing voor het condensatie probleem in een adiabatisch expanderend damp/gas mengsel, is voor het berekenen van optische eigenschappen van dit mengsel tijdens de expansie een te eenvoudige weergave van de werkelijkheid.
Delale C.F., Muitjens M.J.E.H., Van Dongen M.E.H., {1996), accepted for publication, J. Chem. Phys., Dit proefschrift.
4. Door het toevoegen van stikstof wordt homogene kernvorming bij druppelgewijze condensatie van water makkelijker gemaakt.
5. Voor het bepalen van de oppervlaktespanning van onderkoelde vloeistoffen biedt de 'density functional theory' uitkomst.
Lee D.J., Telo da Gama M., Gubbins K.E., {1984), Mol. Phys., 53{5):1113-1130

6. Bij de beschrijving van diffusie heeft het didactische voordelen om uit te gaan van de beschrijving van Maxwell-Stefan in plaats van de beschrijving volgens Fick.
Kuiken G.D.C., "Thermodynamics of Irreversible Processes", Wiley, (1994}
7. Voor de analyse van diffusie-nevelkàmer experimenten moet altijd rekening gehouden worden met stromingseffecten.
Eertelmans A., Heist R.H., "How does the walt ofthe dijjusion cloud chamber affect performance?", in: Nucleation and Atmospheric Aerosols, Kulmala M., Wagner P., {ed.}, Pergamom, (1996}
8. Gezien de gevoeligheid van ons gehoor, is de kwaliteit van muzikale klanken mede bepaald door kleine effecten. Men heeft reeds aandacht besteed aan de invloed van wandtrillingen bij blaasinstrumenten. Het is aanbevelenswaardig om meer aandacht te besteden aan de mogelijke invloed van vochttransport en condensatie op geluidsproduktie.
Hirschberg A., Kergomard J., Weinreich G., "Mechanics of Musical Instruments", Int. Centre for Mechanica[ Science, Springer, (1995)
9. Electronische post is, vanuit het oogpunt van de ontvanger, superieur boven mobiele telefonie.
10. Met de toename van de rekenkracht van computers is de aandacht voor elegante snelle rekenprocedures gedaald.
Eindhoven, 17 december 1996