hmgb-1 release and the cd8 t cell response elicited by
TRANSCRIPT

HMGB-1 Release and the CD8+ T Cell Response Elicited by Radiation Treatment in Malignant Pleural Mesothelioma
by
Matthew Wu
A thesis submitted in conformity with the requirements for the degree of Master of Science
Institute of Medical Science University of Toronto
© Copyright by Matthew Wu 2015

ii
HMGB-1 Release and the CD8+ T Cell Response Elicited by Radiation Treatment in Malignant Pleural Mesothelioma
Matthew Wu
Master of Science
Institute of Medical Science
University of Toronto
2015
Abstract
Malignant pleural mesothelioma (MPM) is a cancer of the pleura that is associated with the
inhalation of asbestos. Treatments, such as radiation therapy, have direct cytotoxic effects on
cancer cells in addition to immune activating effects through the release of pro-inflammatory
molecules associated with cancer cell death. The goal of this project was to investigate the role
of High Mobility Group Box 1 (HMGB-1), a typical Danger Associated Molecular Pattern
(DAMP) protein released after radiation treatment, in CD8+ T Cell mediated tumor killing and in
patient survival. Radiated tumor cells release HMGB-1 in vitro. Furthermore, radiation
treatment leads to HMGB-1 release and correlates with higher survival in vivo. This thesis
demonstrates that radiation leads to HMGB-1 release which is correlated to MPM cell death and
increased survival in vivo. The findings of this project could be used to develop new therapies in
combination with radiation.

iii
Acknowledgments
Graduate school is an experience. Dr. Heath McMillan, a PhD student at the time whom I was
working with, once told me: “research is like a drug; the lows are very low, but the highs are
incredible”. At this point I can absolutely say that he was right. Sometimes research can be
disheartening as one failure is met with the next. However, finding a truly amazing result and
knowing that I’m the first person to be uncovering this individual piece of a complex biological
puzzle, is something that has kept me coming back for more. I have numerous people to thank
for this tremendous opportunity and for their support.
Firstly I would like to thank my supervisor Dr. Marc de Perrot for his unparalleled support and
guidance. From my time as a summer student he instilled in me a passion for cancer research, a
subject in which I had little previous experience. Now as I finish my Master of Science degree,
that passion continues. Marc has been a great mentor for me and I hope that I can inspire others
as he has inspired me.
The members of the lab have been huge supporters of my work. Dr. Licun Wu, Ms. Hana Yun,
and Dr. Yidan Zhao have taught me everything from cell culture, to flow cytometry and I am
incredibly thankful for their guidance. I appreciate all the administrative help from Ms. Corrina
Cufaro, Ms. Susan Beaudoin, and Ms. Ivone Ornelas. A big thank you to Dr. Luis De La Maza
who has not only collaborated and shared in the graduate experience with me but has also
become a great friend.
I want to thank everyone in the Latner Thoracic Surgery Research Laboratories for their warmth
and inclusiveness over these years. A special thank you to Ms. Guan Zehong for being so patient
with me and always offering technical support with my experiments. I would also like to thank

iv
my committee members Drs. Shaf Keshavjee, Ming Tsao, and Li Zhang for all of their guidance
and advice over the course of my masters. I also thank Dr. Stephen Albelda and Dr. Daniel
Drucker for graciously providing me with cell lines. I would also like to thank Drs. Mansour
Haeryfar and Brent Sinclair at Western University, for teaching me the fundamentals of scientific
research and for giving me the tools to succeed.
My success in graduate school would not have been possible without the encouragement of my
support network. I would like to thank my mother, Hazel Nightingale, for keeping me well-fed
after countless late nights in the lab. I also want to thank my father, Edmond Wu, for helping me
keep things in perspective and for teaching me to keep a calm and collected mind even in
difficult times. Lastly, all of my accomplishments to date would not be possible without the
support of my girlfriend, Ms. (soon to be Dr.) Kathryn Boult, who has endured the rants about
my failures but always revels with me in my successes.

v
Contributions
This work was supported by the Swiss National Science Foundation (SNSF), Toronto General
Hospital Foundation, and Princess Margaret Hospital Foundation Grants. Dr. Luis de la Maza
performed the image-guided irradiation of mice using the X-Rad 225Cx irradiator. Dr. Licun
Wu performed some of the injections of anti-HMGB-1 into the mice.

vi
Table of Contents
Abstract………………………………………………………………………………………........ii
Acknowledgments .......................................................................................................................... iii
Contributions ................................................................................................................................... v Table of Contents ........................................................................................................................... vi List of Tables ................................................................................................................................. ix List of Abbreviations ...................................................................................................................... x List of Figures ............................................................................................................................... xii
Chapter 1 ......................................................................................................................................... 1 1 General Background: Malignant Pleural Mesothelioma; Epidemiology, Development,
Pathogenesis, and Treatment ...................................................................................................... 1
1.1 Malignant Pleural Mesothelioma Background ................................................................... 1
1.1.1 The Mesothelium .................................................................................................... 1 1.1.2 Presentation ............................................................................................................. 2
1.2 Asbestos and Other Risk Factors for MPM ........................................................................ 3
1.3 MPM Development ............................................................................................................. 5
1.3.1 Histological Subtypes ............................................................................................. 7
1.4 Current Treatment Options ................................................................................................. 9
1.4.1 Surgery .................................................................................................................. 10
1.4.2 Chemotherapy ....................................................................................................... 11 1.4.3 Radiation Therapy ................................................................................................. 12
1.4.4 SMART Treatment ............................................................................................... 13
1.5 Future Treatment Options: Immunotherapy ..................................................................... 15
1.5.1 Cytokines .............................................................................................................. 16
1.5.2 Monoclonal Antibodies ......................................................................................... 17 1.5.3 Immune Modulating Antibodies ........................................................................... 18
1.6 Cancer Treatment and Immunogenic Cell Death .............................................................. 20
1.6.1 DAMPs ................................................................................................................. 20 1.6.2 DAMPs in Cancer Therapy ................................................................................... 21
1.7 HMGB-1 ........................................................................................................................... 23
1.7.1 Background ........................................................................................................... 23 1.7.2 HMGB-1 Signaling Pathways............................................................................... 24
1.8 The Pro-Cancer Side of HMGB-1 .................................................................................... 27 1.9 The Anti-Cancer Side of HMGB-1 ................................................................................... 29

vii
1.9.1 HMGB-1 Release .................................................................................................. 29
1.9.2 Active Release of HMGB-1 .................................................................................. 29 1.9.3 Passive Release of HMGB-1 ................................................................................ 30
1.10 The Immune Response and HMGB-1 ............................................................................... 32
1.10.1 T Cells ................................................................................................................... 33 1.10.2 The T Cell Immune Response ............................................................................... 34 1.10.3 The Three Signal Hypothesis ................................................................................ 36
1.11 Summary ........................................................................................................................... 39
2 Hypothesis and Aims ............................................................................................................... 41
3 Materials and Methods ............................................................................................................. 43
3.1 Human and Murine Cell Lines .......................................................................................... 43 3.2 Cell Line Radiation ........................................................................................................... 45
3.3 Flow Cytometry Cell Death Quantification ...................................................................... 45 3.4 HMGB-1 concentration of Mouse Serum or Cell Culture Supernatant by ELISA .......... 47 3.5 Mice .................................................................................................................................. 47
3.6 Radiation Mouse Model .................................................................................................... 48 3.7 Hybridoma Preparation and anti-HMGB-1 Ab Purification ............................................. 49
3.8 Hybridoma Anti-HMGB-1 Antibody Blocking Test ........................................................ 50 3.9 Patient Sample HMGB-1 Immunohistochemistry ............................................................ 50 3.10 Immunofluorescence ......................................................................................................... 52
3.11 Statistical Analysis ............................................................................................................ 52
4 Results ...................................................................................................................................... 53
4.1 In vitro HMGB-1 characterization .................................................................................... 53
4.1.1 Epithelioid and sarcomatoid human MPM cell lines differ in intracellular HMGB-
1 expression 24 hours after radiation treatment ................................................... 53 4.1.2 Epithelioid MPM cell line H226 releases more HMGB-1 than sarcomatoid line
H28 in response to γ-radiation .............................................................................. 55
4.1.3 Epithelioid lines H226 and H2452 release more HMGB-1 into culture medium 24
and 48 hours after 25Gy γ-radiation than sarcomatoid lines H28 and H2052 ...... 56
4.1.4 Radiation induced cell death in human cell lines .................................................. 59
4.1.5 AE-17-OVA epithelioid mouse cell line behaves similarly to H226 in terms of
HMGB-1 release after 25Gy radiation .................................................................. 61 4.1.6 AE-17-OVA epithelioid mouse cell line behaves similarly to human epithelioid
cell line H226 in terms of cell death when treated with 25Gy radiation .............. 63
4.2 Patient Sample and in vivo HMGB-1 ................................................................................ 65
4.2.1 No significant difference in HMGB-1 positivity by IHC staining between
untreated and SMART treated epithelioid MPM paraffin embedded patient
samples .................................................................................................................. 65

viii
4.2.2 Serum HMGB-1 is significantly higher 2 days after treatment in mice receiving
local radiation therapy. .......................................................................................... 68 4.2.3 Tumor Growth Curve ............................................................................................ 69 4.2.4 Percent HMGB-1 positive nuclei in tissues of mice receiving no treatment or
15Gy radiation. ..................................................................................................... 71 4.2.5 Percent HMGB-1 high, medium, and low positivity in mouse tissues from
untreated and 15Gy radiated mice ........................................................................ 72 4.2.6 Quantification of tumor infiltrating CD8+ T Cells in untreated and radiated mouse
tumor tissue ........................................................................................................... 74
4.2.7 6E6 hybridoma anti-HMGB-1 antibody is able decrease the signal of recombinant
HMGB-1 in vitro by ELISA ................................................................................. 76 4.2.8 Anti-HMGB-1 Antibody combined with LRT increases tumor growth compared
to LRT alone ......................................................................................................... 77
5 Discussion ................................................................................................................................ 79
5.1 In vitro radiation-mediated HMGB-1 release ................................................................... 80
5.2 Paraffin-embedded Patient Samples. ................................................................................ 88 5.3 In vivo mouse model ......................................................................................................... 89
6 Conclusions ............................................................................................................................ 100 7 Limitations ............................................................................................................................. 102 8 Future Directions .................................................................................................................... 104
References ................................................................................................................................... 108 Appendices .................................................................................................................................. 132
Copyright Acknowledgements .................................................................................................... 133

ix
List of Tables
Table I. Positive staining patterns of epithelioid and sarcomatoid MPM………………………..8
Table II. Cell lines used for experiments………………………………………………………..44
Table III. Patient Demographics………………………………………………………………...66

x
List of Abbreviations
Ab Antibody
APC Antigen Presenting Cell
ATP Adenosine Triphosphate
BSA Bovine Serum Albumin
CRT Calreticulin
CTCF Corrected Total Cell Fluorescence
CTLA-4 Cytotoxic T-Lymphocyte-Associated Antigen 4
DAMP Danger Associated Molecular Pattern
DC Dendritic Cell
EGF Epidermal Growth Factor
ELISA Enzyme-Linked Immunosorbent Assay
EPD Extended Pleurectomy-Decortication
EPP Extrapleural Pneumonectomy
ERK Extracellular Signal-Related Kinase
FBS Fetal Bovine Serum
Fv Variable Fragment
H2SeO3 Selenious Acid
HMGB-1 High Mobility Group Box 1
ICAM Intracellular Adhesion Molecule
ICD Immunogenic Cell Death
IL Interleukin
LFA Lymphocyte Function Associated Antigen
LRT Local Radiation Therapy
mAb Monoclonal Antibody

xi
Mac-1 Macrophage Receptor 1
MHC Major Histocompatibility
MPM Malignant Pleural Mesothelioma
NF-κB Nuclear Factor Kappa Light Chain Enhancer of Activated B Cells
NK Natural Killer
NLRP3 NOD-like Receptor Family Pyrin Domain Containing 3
PD-1 Programmed Death-1
PD-L1 Programmed Death Ligand 1
PAMP Pathogen Associated Molecular Pattern
PRR Pathogen Recognition Receptor
RAGE Receptor for Advanced Glycation End Products
ROS Reactive Oxygen Species
SMART Surgery for Mesothelioma After Radiation Therapy
SV40 Simian Virus 40
TCR T Cell Receptor
TLR Toll-Like Receptor
TNF Tumor Necrosis Factor
VCAM Vascular Cell Adhesion Molecule
VLA Very Late Antigen
WT-1 Wilms Tumor Protein

xii
List of Figures
Figure 1. Asbestos Carcinogenesis……………………………………………………………….4
Figure 2. H & E staining of patient biopsies. ……………………………………………... ……..9
Figure 3. Overall survival of patients with epithelial and biphasic subtypes of disease….. ……12
Figure 4. Radiation treatment of cancers leads to DNA damage. DNA damage may be repaired
and result in tumor cell survival……………………………………………………...13
Figure 5. HMGB-1 protein domains. ……………………………………………………………24
Figure 6. HMGB-1 binds RAGE receptor or TLR 2 or TLR 4………………………………….26
Figure 7. HMGB-1 and its multitude of intracellular and extracellular roles…………………..32
Figure 8. DAMPs released by secondary apoptotic or necrotic are sensed by DCs……………35
Figure 9. The 3-signal hypothesis in immunology involves the activation of the adaptive
response by signals provided by APCs……………………………………………...37
Figure 10. CD8+ T cell licensing………………………………………………………….........38
Figure 11. Quantification of cell death by flow cytometry and gating strategy………………..46
Figure 12. Schematic of untreated, radiated, and radiation + anti-HMGB-1 treatment in tumor
bearing mice…………..…………………………………………………………….49
Figure 13. Quantification of staining using image analysis software………………………….51

xiii
Figure 14. Expression of HMGB-1 in human MPM cells lines 24 hours after
radiation treatment………………………………………………………………….54
Figure 15. HMGB-1 released into media at each treatment dose………………………………56
Figure 16. HMGB-1 release into culture medium by non-treated or radiated epithelioid lines
H226 and H2452 and sarcomatoid cell lines H28 and H25052……………………58
Figure 17. Percentage cell death after no treatement or 25Gy radiation………………………60
Figure 18: HMGB-1 release into culture medium by non-treated or radiated epithelioid lines
H226 and H2452 and murine epithelioid cell line AE-17-OVA………………. …62
Figure 19. Percentage cell death after no treatement or 25Gy radiation………………….......64
Figure 20. High, medium, and low intesnsity nuclear staining of HMGB-1 in paraffin embedded
patient samples…………………………………………………………………….67
Figure 21. Serum HMGB-1 and tumor growth curves of non-treated and mice treated with 15 Gy
LRT………………………………………………………………………………..69
Figure 22. Tumor volume inreases more in untreated mice compared to mice receiving
LRT………………………………………………………………………………..70
Figure 23. Percent positive nuclei in tumor tissue without treatment or with 15Gy
radiation…………………………………………………………………………...71
Figure 24. Percent nuclei with low, medium, and high staining in tissues 2, 7, and 12 days after
LRT………………………………………………………………………………73
Figure 25. Immunofluorescent staining of untreated or 15Gy radiated moues tissue……….75
Figure 26. Anti-HMGB-1 Ab abrogates recombinant HMGB-1 signal by ELISA……….....76

xiv
Figure 27. LRT in combination with LRT demonstrates greater tumor growth compared with
LRT alone……………………………………………………………………………………78

1
Chapter 1
1 General Background: Malignant Pleural
Mesothelioma; Epidemiology, Development,
Pathogenesis, and Treatment
1.1 Malignant Pleural Mesothelioma Background
Malignant pleural mesothelioma (MPM) is a cancer that affects the serous membrane of the
pleura (Jaurand, et al. 2009). Known for its lack of response to treatment, MPM has a median
survival from presentation of 9-12 months (Armstrong, et al. 1984, Robinson, et al. 2005). The
incidence of MPM has increased by 65 percent over the past two decades worldwide and is
projected to continue to increase. MPM is associated with asbestos exposure and while asbestos
use has been controlled in North America, the incidence of disease is predicted to continue to
increase for at least the next 5 to 10 years (Peto, et al. 1995, Spirtas, et al. 1986, Walker, et al.
1983). The continued use of asbestos in the developing world suggests that the incidence of
MPM will continue to increase worldwide for the foreseeable future.
1.1.1 The Mesothelium
MPM affects the cells of the mesothelium. The concept of the mesothelium was first proposed
in 1890 from observations of epithelial-like cells coating the organs of the body (Minot 1890).
The mesothelium, consisting of a mono-layer of mesothelial cells, displays a morphological
cobblestone-like appearance. It is a functionally complex tissue, providing a non-adherent

2
surface for organs through fluid secretion. In addition to preventing organ abrasion, the
mesothelium also mediates many inflammatory processes such as antigen presentation and
chemokine production (Mossman, et al. 2013, Mutsaers 2004, Yung and Chan 2007).
Furthermore, the mesothelium mediates other functions such as the transportation of fluids
across serous membranes, and is essential to tumor cell adhesion in MPM (Mutsaers 2004).
1.1.2 Presentation
Mesotheliomas can affect the serous surfaces of the pleura, peritoneum, and pericardium. While
pleural mesotheliomas have been reported to represent around 73% of all mesothelioma
diagnoses, it is important to note that with progression of disease, this neoplasm can expand to
the other cavities of the body (Suzuki 2001).
MPM is a disease that affects more males than females with males reported to represent 68% to
79% of all MPM cases (Adams, et al. 1986, Antman, et al. 1988, Brenner, et al. 1982, Ratzer, et
al. 1967, Ruffie, et al. 1989). Many clinical studies universally confirm the greater incidence of
disease in males with history of asbestos exposure being a common factor. The mean age of
disease diagnosis is around 55. However, a wide range of diagnoses in older individuals as well
as in children has been reported (Fraire, et al. 1988).
Symptoms of MPM occur very slowly. Patients typically present with a characteristic
unexplained shortness of breath related to excess pleural fluid, known as pleural effusion, in
addition to chest wall pain (Ismail-Khan, et al. 2006). Pleural effusion in the pleural space
reduces the lungs capacity to expand and leads to breathing difficulty and chest pain localized to
the affected side (Chahinian, et al. 1982). The right side is more commonly affected,

3
representing around 60% of cases due to asbestos concentrating more readily in the right lung
(Pass, et al. 2005).
MPM progression and growth results in the gradual thickening of the pleura, leading to a loss of
the pleural space. The lung on the affected side is unable to inflate, resulting in progressing
symptoms of dyspnea and chest pain. MPM may invade surrounding areas such as the lung,
chest wall, and diaphragm. Additionally, MPM has been observed to expand to the contralateral
side and the peritoneum (Chahinian, et al. 1982).
1.2 Asbestos and Other Risk Factors for MPM
The toxicity of asbestos has been known much before its association with mesothelioma. In
1935, asbestos was first implicated in the development of lung carcinomas (Lynch and Smith
1935). In the 1950s, asbestos was being consistently shown to induce lung cancers in asbestos
workers (Braun and Truan 1958, Doll 1993).
The first link between asbestos and mesotheliomas was established by a fundamental study that
noted that while pleural mesothelioma is an uncommon tumor, 45 of the 47 identified cases of
mesothelioma in the north west of Cape Province, South Africa had a history of crocidolite
asbestos fiber exposure (Wagner, et al. 1960). In 1964 this finding was supported by a study
that found 4 mesotheliomas in 255 deaths among insulation workers in the Asbestos Workers
Union in New York (Selikoff, et al. 1964). In both cases, the authors described the number of
MPM cases as exceedingly high for such a rare tumor.

4
Figure 1. Asbestos carcinogenesis. Inhaled asbestos penetrates through the lung and becomes
lodged in visceral pleura where it may cause cells to undergo transformation into cancer.
MPM’s known association with asbestos exposure is unique as it is one of the few cancers with a
known causative agent. Asbestos is a naturally occurring mineral with common uses including
insulation for buildings. Its malleability, tensile strength, and heat resistance render it useful in
industrial processes. There is some evidence to suggest that exposure to asbestos can occur due
to naturally occurring asbestos-containing land forms (Pan, et al. 2005). However, occupation
remains the primary risk factor for developing MPM (Agudo, et al. 2000, Godleski 2004,
Goldberg, et al. 2000, Howel, et al. 1999, Iwatsubo, et al. 1998, Kishimoto, et al. 2004, Zellos
and Christiani 2004).
There is a higher incidence of MPM in males compared to females as a result of asbestos-related
work being traditionally performed by men. In addition, the husband’s profession remains a risk
factor for women who present with MPM as asbestos fibers brought home on clothing often lead
to low-levels of domestic exposure (Huncharek, et al. 1989, Roggli, et al. 1997). The mean time
between exposure to asbestos and onset of disease is around 30 to 40 years. While the risk of

5
mesothelioma 10 years after exposure to asbestos is virtually non-existent, the probability of
developing mesothelioma increases over time (Bianchi, et al. 1997, Boffetta 1998, Hillerdal
1999, Mossman and Gee 1989).
Asbestos exposure is the most commonly known cause of MPM however, there remains a wide
range of other possible inducers of malignant mesotheliomas. Radiation from treatment of other
cancers or from radioactive imaging compounds has been implicated in the development of
malignant mesotheliomas (Babcock, et al. 1976, Dahlgren 1967, Hirsch, et al. 1982, Maurer and
Egloff 1975, Sanders and Jackson 1972, Stock, et al. 1979). Simian virus 40 (SV40) infection
has also been reported as having a potential role in the occurrence of malignant mesothelioma
(Barbanti-Brodano, et al. 2004, Carbone, et al. 1997, Chang, et al. 1997, Cicala, et al. 1993, De
Luca, et al. 1997, Engels, et al. 2003, Gazdar and Carbone 2003, Shivapurkar, et al. 1999, Testa,
et al. 1998). In addition, chronic inflammation from previous lung disease, infection, or injury
has been linked to subsequent MPM development (Brenner, et al. 1982, Brown, et al. 1968,
Chahinian, et al. 1982, Martinis and Radovic 1965, Riddell, et al. 1981, Roggli, et al. 1982).
1.3 MPM Development
It is thought that the development of MPM is mediated by the asbestos fibers which penetrate
deep into the lung and scratch the mesothelial cells lining the lung. The asbestos fibers that
lodge in the lung are able to repeatedly damage the mesothelium and result in chronic
inflammation; a hallmark cause of cancer development (Hanahan and Weinberg , Thompson, et
al. 2014).
While the immune response is able to target and destroy cancerous cells, it is often aberrant
immune activation that leads to cancer development (Hanahan and Weinberg). Inflammatory

6
infiltrates from persistent inflammation consist primarily of macrophages that release pro-
inflammatory cytokines such as IL (Interleukin)-1β and TNF (Tumor Necrosis Factor)-α in
addition to reactive oxygen species (ROS). Asbestos results in reactive oxygen species
generation and aberrant growth receptor signaling from macrophages attempting to digest the
fibers (Scott, et al. 2012).
NLRP3 (NOD-like receptor family pyrin domain containing) inflammasome is a DAMP sensing
mechanism that may be important aspect of mesotheliogenesis. The NLRP3 inflammasome is
triggered by extracellular DAMP (Danger Associated Molecular Pattern) molecules as well as
silicates such as asbestos. Macrophages are unable to breakdown asbestos or other silicate fibers
due to the high aspect ratio (the ratio of length to diameter); leading to “frustrated” phagocytosis
(Hornung, et al. 2008). The lysosome, which provides a wide range of degrading enzymes to aid
in phagocytic breakdown, swells and ruptures when trying to destroy asbestos fibers. This
damage triggers the cathepsin-B pathway and subsequent activation of the NLRP3
inflammasome. The importance of the NLRP3 inflammasome is demonstrated by its activation
of IL-1β secretion. In non-infectious or injury settings, IL-1β is not secreted by tissue
macrophages. However, the presence of asbestos fibers can lead to the secretion of the pro-
inflammatory IL-1β; providing a setting for chronic inflammation and transformation of
mesothelial cells.
Recently, the NLRP3 inflammasome has been suggested as being dispensable in MPM
development as NLRP3-/- mice and wildtype mice exposed to asbestos showed similar survival
times (Chow, et al. 2012). This indicates that the NLRP3 inflammasome is not essential for
tumor development in mice exposed to asbestos. However, this NLRP3 has not been precluded

7
from being a non-essential contributor to mesothelioma development. Furthermore, other
pathways of inflammation may function as redundancy when NLRP3 genes are knocked out.
The inflammation from damage and repair contributes to DNA damage and subsequent
transformation of cells into cancerous cells. Asbestos fibers have also been implicated in
disturbing mitosis by damaging the mitotic spindle apparatus that occurs during cell division,
resulting in chromosomal damage and the potential for mutation (Dinarello 2009).
1.3.1 Histological Subtypes
There are various histological subtypes of disease that present different prognoses (De Pangher
Manzini, et al. 1993, Elmes and Simpson 1976, Hartmann and Schutze 1992, Ruffie, et al. 1989,
Sugarbaker, et al. 1993). Malignant pleural mesothelioma is defined by three major subtypes of
MPM to be epithelioid, sarcomatoid, and biphasic (Tischoff, et al. 2011). Epithelioid disease
displays round or cuboidal cell cobblestone-like appearance. Sarcomatoid MPM demonstrates a
typical spindle, elongated, and fibroblast-like appearance while biphasic MPM is a mixture of
the epithelioid and sarcomatoid cells in the same tumor. Finally, desmoplastic mesothelioma
diagnoses describe highly aggressive sarcomatoid disease (Husain, et al. 2009).
Epithelioid MPM is typically identified by immunohistochemical staining which is able to
distinguish between epithelioid MPM, sarcomatoid MPM, Biphasic MPM, sarcoma,
adenocarcinoma, and carcinosarcoma (Inai 2008). Markers for epithelioid MPM include positive
staining for Calretinin (Doglioni, et al. 1996, Gotzos, et al. 1996, Ordonez 2005, Ordonez 2007),
WT-1(Ordonez 2007), Podoplanin (Kimura and Kimura 2005, Ordonez 2005, Ordonez 2007),
and Keratin 5/6 (Ordonez 2007). Sarcomatoid MPM is not as easily identified and may be

8
differentiated from epithelioid MPM using AE1/AE3 and CAM5.2 (Ordonez 2007), WT-1(Inai
2008, Kushitani, et al. 2008), and Calretinin (Lucas, et al. 2003).
Table I. Positive staining patterns of epithelioid and sarcomatoid MPM.
Epithelioid Sarcomatoid
Calretinin 92-100% (Cury, et al. 2000,
Miettinen, et al. 2001, Ordonez
2003, Padgett, et al. 2008)
18-100% (Abutaily, et al. 2002,
Doglioni, et al. 1996, Leers, et al.
1998, Lucas, et al. 2003, Miettinen,
et al. 2001, Ordonez 2004, Roberts,
et al. 2001)
WT-1 69-99% (Kushitani, et al. 2007,
Ordonez 2003, Padgett, et al. 2008) 10% (Padgett, et al. 2008)
Podoplanin 66-93% (Hinterberger, et al. 2007,
Ordonez 2006, Padgett, et al. 2008) 30-72% (Hinterberger, et al. 2007,
Padgett, et al. 2008)
Keratin 5/6 92-100% (Cury, et al. 2000,
Ordonez 2003, Padgett, et al. 2008) 0%-29% (Abutaily, et al. 2002,
Attanoos, et al. 2000, Chu and Weiss
0000, Clover, et al. 1997, Miettinen,
et al. 2001, Padgett, et al. 2008)
AE1/AE3 100% (Kushitani, et al. 2007) 77-100% (Attanoos, et al. 2000,
Lao, et al. 2014)
CAM5.2 97-100% (Kushitani, et al. 2007,
Roberts, et al. 2001) 90-94% (Al-Izzi, et al. 1989,
Dejmek and Hjerpe 1994, Garcia-
Prats, et al. 1998, Roberts, et al.
2001)
Approximately 60% of all MPM cases are of the epithelioid subtype (Husain, et al. 2009).
Biphasic and sarcomatoid subtypes are less common and comprise of about 30% and 10% of
cases respectively. Biphasic disease is diagnosed when at least 10% of the visible cells are
epithelioid cells in sarcomatoid disease or when 10% of cells in a predominantly epithelioid
tumor are sarcomatoid in nature (Pass, et al. 2005, Tischoff, et al. 2011).
The epithelioid subtype of disease typically provides the best prognosis while biphasic and
sarcomatoid subtype of disease present shorter survival times and are significantly negative
prognostic factors (Neumann, et al. 2013). While MPM is resistant to the typical cancer

9
treatments chemotherapy, radiation, and surgery, better outcomes from chemotherapeutic
treatment are observed in the epithelioid subtype. In contrast, sarcomatoid disease is ordinarily
unresponsive to chemotherapy (van Zandwijk, et al. 2013). Furthermore, multimodality therapy
presents better outcomes for epithelioid disease than for biphasic or sarcomatoid subtypes
(Ceresoli, et al. 2001, Richards, et al. 2006).
Figure 2. H & E staining of patient biopsies. Epithelioid disease, sarcomatoid MPM, and the
biphasic subtypes are displayed. Biphasic MPM possesses cells of both the epithelioid and
sarcomatoid morphology.
1.4 Current Treatment Options
MPM is challenging to treat and presents a median survival time after diagnosis of less than 12
months (Vogelzang, et al. 2003). Patient prognoses after standard therapeutic interventions such

10
as surgery, radiation, and chemotherapy have remained bleak. There is a substantial need to
investigate new avenues of treatment to bring about positive change for MPM patients.
1.4.1 Surgery
Surgical intervention can be palliative, in which pleural effusion is drained, or therapeutic, in
which the tumor is removed or reduced in size (Haas and Sterman 2013). Pleurectomy and
decortication involves the removal of the pleura and any tumors present. This procedure is able
to palliate symptoms such as shortness of breath though the procedure alone does not offer any
increase in patient survival (van Ruth, et al. 2003). Median survival of pleurectomy and
decortication is 8-22 months (Aziz, et al. 2002, Flores, et al. 2006, Flores, et al. 2008, Moskal, et
al. 1998, Pass, et al. 1997, Pass, et al. 1998, Schipper, et al. 2008).
Extrapleural pneumonectomy (EPP) has the greatest cytoreductive potential by removing the
pleura and the lung as well as parts of the diaphragm and pericardium. EPP presents a median
overall survival of 10-35 months (Aziz, et al. 2002, Baldini, et al. 1997, Batirel, et al. 2008,
Krug, et al. 2009, Rusch and Venkatraman 1999, Weder, et al. 2007, Yan, et al. 2009). EPP is
similarly unable to provide an increase in survival without the use of adjuvant therapy (Haas and
Sterman 2013).
Extended pleurectomy-decortication (EPD) is a radical surgery that involves the removal of the
parietal and visceral pleura, diaphragm, and sometimes the pericardium leaving the lung denuded
but remaining in the body (Rusch 2012). EPD may be advantageous for older individuals as the
removal of the lung in EPP may not be well tolerated for individuals over 65. While EPP offers
better potential clearance, EPD alone offers similar survival to EPP (Nakas and Waller 2014).

11
1.4.2 Chemotherapy
Non-surgically resectable disease is treated with the first-line chemotherapy regimen of
Pemetrexed and Cisplatin. Cisplatin is a platinum-based drug that causes apoptosis through the
cross-linking of DNA (Tanida, et al. 2012). Pemetrexed acts by inhibiting the enzyme
thymidylate synthase, effectively preventing DNA synthesis (Vogelzang, et al. 2003). In a phase
III clinical trial, Vogelzang and colleagues found median survival time of patients receiving
Pemetrexed and Cisplatin of 12.1 months compared to 9.3 months in patients receiving only
Cisplatin. Currently, there is no standard second-line chemotherapy regimen (Ceresoli, et al.
2011, Pasello, et al. 2011, Stebbing, et al. 2009, Xanthopoulos, et al. 2008, Zauderer and Krug
2012, Zucali, et al. 2008). Moderate increase in survival times have been reported in the use of
second-line chemotherapeutics, however, low patient numbers in these studies warrants further
investigation.
A previous study involving the use of bevacizumab, an anti-VEGF antibody, in addition to the
Pemetrexed and Cisplatin combination failed to demonstrate any benefit in survival of
mesothelioma patients (Dowell, et al. 2012). However, a randomized phase 3 trial with a greater
number of patients presented at the 2015 American Society of Clinical Oncology (ASCO) annual
meeting found a significant increase in overall survival of patients receiving Pemetrexed,
Cisplatin and bevacizumab triplet therapy (median: 18.8 months) compared to Pemetrexed and
Cisplatin only (median: 16.1 months) (Zalcman, et al. 2015). These results indicate that triplet
therapy may become the new first line treatment for unresectable MPM.

12
1.4.3 Radiation Therapy
Radiation therapy is typically provided as part of multimodality treatment including surgery and
radiation. While there have been no randomized trials demonstrating its effectiveness, in a phase
I/II trial, Surgery for Mesothelioma After Radiation Therapy (SMART) has displayed promising
results with epithelial MPM patients experiencing a 3-year survival of 84% up from 30% with
previous radiotherapy regimens (Cho, et al. 2014, Haas and Sterman 2013). This is in sharp
contrast to a previous report on 140 asbestos workers with pleural mesothelioma in which only
one patient survived longer than 3 years after diagnosis without treatment (Ribak and Selikoff
1992). Furthermore, median survival of patients with biphasic disease without treatment is
estimated to be around 4-5 months (Moore, et al. 2008).
Figure 3. Overall survival of patients with epithelial and biphasic subtypes of disease. At 3
years, overall survival in epithelioid MPM patients was 84% (Figure reproduced from previous
article in our lab (Cho, et al. 2014)).

13
1.4.4 SMART Treatment
SMART is a new methodology of administering a short course of high-dose radiation before
surgery. This method differs from previous approaches in which surgery is used to remove
macroscopic disease followed by adjuvant radiation or chemotherapy given after surgical
resection in order to remove remaining microscopic disease.
The increase in survival is thought to occur as a result of the specific immune activation effects
(Lee, et al. 2009, Levy, et al.). A short course of high dose radiation can activate the immune
system by antigen release from dying tumor cells. A negative aspect of radiation treatment is
that radiation also kills CD8+ T Cells. However, hypofractionated radiation, as used in SMART,
does not deplete CD8+ T cells for as great a time as longer low dose regimens do, potentially
allowing for more CD8+ T cell mediated tumor cell killing (Siva, et al. 2015).
Figure 4. Radiation treatment of cancers leads to DNA damage. DNA damage may be
repaired and result in tumor cell survival. Greater number of DNA double-stranded breaks
leads to irreparable damage and either programmed cell death or necrosis.

14
1.4.4.1 Abscopal Effect and Immunogenic Radiation
A strong indicator of the immune system activating effects of radiation is the presence of the
abscopal effect; the shrinkage of a distant tumor outside the field of treatment after radiating a
tumor mass (Antoniades, et al. , Cotter, et al. 2011, Isobe, et al. 2009, Lakshmanagowda, et al.
2009, Takaya, et al. 2007). In clinical reports and animal studies, distant tumors can regress as a
result of irradiation of the primary tumor. The radiation induced abscopal effect is thought to
occur due to immune system activation towards antigens shared by both local and distant tumors.
This may allow for the killing of distant tumors and the prevention of recurrence. This effect has
been observed clinically in many cancers including lymphoid malignancies, hepatocellular
carcinoma, uterine cervical carcinoma, Merkel cell carcinoma, uterine cervical carcinoma, and
melanoma.
A study from our lab demonstrated the immunogenic aspect of radiation by reproducing the
abscopal effect in a concomitant immunity model where two MPM tumors were injected
concurrently on the leg and on the flank (Wu, et al. 2015). While the irradiated leg tumor shrank
in size due to radiation-induced DNA damage and cell death, the flank tumor outside of the field
of radiation also shrank presumably as a result of immune system involvement. This idea is
supported by the observation that the abscopal effect is not present in immunodeficient NOD-
SCID mice. Furthermore, this effect was enhanced by the immune activating anti-CTLA-4
checkpoint antibody (Wu, et al. 2014, Wu, et al. 2015).
It is thought that radiation is able to overcome the pro-growth tumor microenvironment by re-
initiating immune recognition of the tumor through release of danger signals also known as
DAMP molecules (Matzinger 1994, Matzinger 2002). This may allow immune cells to
recognize and destroy radiated as well as distant tumors in an in vivo vaccination-like effect.

15
Ionizing radiation is able to elicit the release of DAMPs such as HMGB-1, ATP, and
Calreticulin. These molecules are able to promote antigen processing and presentation by
Dendritic Cells (DC)s and thus the priming of anti-tumor T cells (Apetoh, et al. 2007,
Ghiringhelli, et al. 2009). In humans, radiation treatment is able lead to CD8+ T cell recruitment
and to the elimination of tumors (Lugade, et al. 2008).
1.5 Future Treatment Options: Immunotherapy
Immunotherapy is a growing field in which the patient’s immune system is used to destroy
cancer. Our lab has sought to improve upon MPM treatment by combining radiation and
immunotherapy.
There has been a paradigm shift in goals for treating cancer. While cancer cell death is the
ultimate objective, the activation of the immune system towards cancer may be more valuable
than the immediate killing effects of treatments such as surgery, radiation, or chemotherapy as
they may leave microscopic disease behind. While cancers display various other hallmark
behaviours including evasion of cell death and self-sufficient growth signals, evasion of the
immune system is a large part of tumor progression (Hanahan and Weinberg). Immune
surveillance is a well-known theory that posits that the immune system continually monitors and
eliminates cells that have undergone mutations towards a cancerous phenotype. This idea has
been supported by observation that various cancers are highly prevalent in immunocompromised
people (Vajdic and van Leeuwen 2009). Furthermore, genetically engineered mice that are
deficient in CD8+ T cells, CD4+ T helper cells, or NK cells, also demonstrate increased tumor
incidence compared to wildtype mice (Kim, et al. 2007, Teng, et al. 2008). In our lab, we have
also observed that tumor incidence is much higher in engineered mice whose entire CD8+ T cell

16
repertoire binds only to ovalbumin peptide; perhaps impairing its ability to recognize other tumor
antigens (Unpublished Data).
The goal of immunotherapy is to eradicate this immune evasion and promote tumor cell killing.
Immunotherapy can function through passive immunity, involving monoclonal antibodies
against specific tumor antigens, or active immunity in which the host immune system is educated
to recognize and destroy cancerous cells that have previously become invisible to immune
defenses.
1.5.1 Cytokines
Cytokines are secreted proteins that affect neighbouring cells with the appropriate receptors.
Cytokines comprise of a large part of the immune response and thus have been used in
treatments to sway the immune system against cancer (Murphy 2012).
IFN-α was the first immunotherapeutic agent used in cancer for the treatment of advanced
melanoma. After approval as adjuvant treatment, many groups have shown that IFN-α
administration is able to reduce the risk of recurrence and improve long-term survival rates. As a
result, this cytokine has also been used in other cancers including renal carcinoma and various
leukemias (Baxevanis, et al. 2009, Smyth, et al. 2004).
In MPM, IFN-α alone has little to no benefit (Ardizzoni, et al. 1994, Sterman, et al. 2011).
However, there have been promising results demonstrating increased disease free survival and
survival time when administering IFN-α intrapleurally before surgery (Sterman, et al. 2011).

17
1.5.2 Monoclonal Antibodies
Monoclonal antibodies directed against tumor-specific antigens have shaped oncologic
treatment. Antibodies against tumor associated antigens in melanoma and prostate cancer have
paved the way for use in other cancers (Baxevanis, et al. 2009). In addition, the HER2 binding
trastuzumab is a well-known mAb used in the treatment of HER2+ breast cancers. Furthermore,
antibodies are currently in pre-clinical and clinical phases for cancers such as lymphoma,
leukemia, and others (Perez, et al. 2007).
mAbs in cancer work through a wide range of methods. They may induce apoptosis or
neutralize growth receptors and arrest tumor cell growth (Murphy 2012). Abs may also be
conjugated to toxins that localize cytotoxic treatments to the tumor (Hughes 2010). The binding
of antibodies to tumor antigens may also elicit the immune response through recruitment of
immune cells such as monocytes and macrophages by Fc receptor binding the Fc region of the
Ab. Furthermore, tumor cell death may occur by Ab-mediated complement deposition and cell
death by membrane rupture (Scott, et al. 2012). There also exist mAbs that target the tumor
microenvironment through mechanisms such as targeting stromal cells or preventing
angiogenesis (Deckert 2009, Schliemann and Neri 2010, Welt, et al. 1994).
mAbs towards Mesothelin have been used in mesothelin expressing MPM cases (Kreitman, et al.
2009). A phase I clinical trial in which anti-mesothelin antibody variable fragments (Fv) were
administered to mesothelioma patients with known mesothelin expression found partial response
in 4 patients and disease stability in 19 patients out of 34 total patients. Further studies have
investigated combination therapy of anti-mesothelin mAb with Gemcitabine chemotherapy.
While mesothelin may be a potential therapeutic target, MPM currently lacks any reliable tumor
associated antigens. Furthermore mesothelin expression may be present in some patients with

18
the epithelial subtype of disease, biphasic disease is much more varied in expression and
sarcomatoid typically does not express mesothelin (Hassan, et al. 2010).
1.5.3 Immune Modulating Antibodies
1.5.3.1 Anti-CTLA-4
Antibodies may also act through immunomodulation by binding to checkpoint molecules and
swaying the immune system towards an anti-cancer response. One method includes blocking
surface receptors that normally function to inhibit the immune response. In the three signal
hypothesis, Cytotoxic T-Lymphocyte-Associated Antigen 4 (CTLA-4) is present on T Cells and
is able to bind to the co-stimulatory signal B7.1 and B7.2 on dendritic cells and inhibit the
adaptive immune response. The blocking of this interaction with an antibody prevents T-cell
inhibition and enables T-cell activating interactions to occur (Murphy 2012).
Anti-CTLA-4 antibody has demonstrated efficacy in the clinical setting. It was first approved in
2011 for use in treatment of metastatic melanoma and has since demonstrated an increase in
survival when compared to chemotherapy treatment (Robert, et al. 2011). A phase I trial of
ipilimumab, an anti-CTLA-4 antibody, on prostate, and ovarian cancer has showed some
efficacy through reduction of tumor size and lymphocyte infiltration (Hodi, et al. 2003, Ribas
2007, Small, et al. 2007). Furthermore, combination with chemotherapy has shown an
improvement in the rate of complete response (Goff, et al. 2009, Yuan, et al. 2011).
There are few studies investigating CTLA-4 in MPM treatment. A recent phase II clinical trial
studied the use of CTLA-4 for patients with un-resectable disease and disease progression after
chemotherapy. CTLA-4 in this study was administered alone once every 90 days and

19
demonstrated limited efficacy with no patients demonstrating complete response and 2 out of 29
patients displaying a partial response (Calabro, et al. 2013). Data from mouse studies in our lab
suggest that anti-CTLA-4 mAb alone does not offer prolonged survival. However, synergistic
effects were observed when combined with radiation therapy, leading to greater survival than
radiation alone (Wu, et al. 2015).
1.5.3.2 Anti-PD-1
The Programmed Death-1 (PD-1) Programmed Death Ligand 1 (PD-L1) pathway has garnered
interest as another potential therapeutic target. PD-1 is expressed on T cells and triggers T cells
to undergo apoptosis when binding and interacting with PD-L1. PD-L1 is expressed on various
cell types including epithelial cells and hematopoietic cells in response to the cytokine IFN-γ. It
is thought that this mechanism of T cell down-regulation prevents excess damage of surrounding
tissues during inflammation. Tumor cells are able to hijack this mechanism by expressing PD-L1
in order to evade T cell-mediated destruction by triggering T cell programmed cell death.
Binding of PD-1 blocks this interaction and counteracts this evasion mechanism (Carreno and
Collins 2002, Greenwald, et al. 2005, Keir, et al. 2008, Pentcheva-Hoang, et al. 2009).
Clinically, anti PD-1 mAb has been used in melanoma patients. In one phase I clinical study,
objective responses were achieved in 28% of the 94 total patients that had advanced melanoma
(Topalian, et al. 2012). PD-1 has been studied in MPM as well. PD-L1 expression is correlated
with decreased survival as patients with PD-L1 positive tumors show a median survival of 4.79
months compared to 16.3 months in patients with PD-L1 negative tumors. It has also been
reported that at least 20% of MPM tumors are PD-L1 positive (Cedres, et al. 2015). While this
molecule may be an important therapeutic pathway for some, there still remains a number of
MPM patients that do not have PD-L1 expressing tumors.

20
Studies in our lab have demonstrated limited therapeutic effect of anti PD-1 mAb alone on
subcutaneous MPM tumors (Unpublished Data). However, tumor shrinkage or slowed tumor
growth is observed when combined with local radiation therapy, similar to the combination of
radiation and CTLA-4 inhibition.
1.6 Cancer Treatment and Immunogenic Cell Death
Treatments such as radiation, chemotherapy, and immunotherapy are able to elicit cell death of
tumor cells. However, an important aspect in successful treatment involves immunogenic cell
death (ICD), in which tumor cell death triggers a lasting immune response through the release of
DAMP molecules.
1.6.1 DAMPs
DAMP molecules are a major concept in understanding how immune responses are elicited in
the absence of pathogens in conditions such as injury, autoimmunity, and cancer (Matzinger
1994). DAMPs are also important in rendering normally immunogenically silent forms of cell
death immunogenic. The DAMP theory builds upon the Pathogen Recognition Receptors
(PRRs) model. PRRs describe a diverse family of receptors that bind to Pathogen Associated
Molecular Pattern (PAMP) molecules. PAMPs are a broad term defining evolutionarily
conserved pathogen molecules that bind to PRRs to elicit a host immune response against the
invading pathogen (Murphy 2012). The PAMP-PRR theory is a fundamental theory in
immunology that explains how the inflammation in infection arises.
The DAMP theory, proposed by Matzinger, describes self-molecules that have normal
intracellular functions apart from their extracellular immunostimulatory effects. Upon release of

21
these molecules by dying or stressed cells, they are liberated and thus able to bind to PRR
receptors and begin the pathways to inflammation in the absence of infection (Matzinger 1994).
Among the numerous DAMPs that exist, the most well-studied include Calreticulin (CRT),
adenosine triphosphate (ATP), and High-Mobility Group Box 1 (HMGB-1) protein (Kroemer, et
al. 2013, Krysko, et al. 2012, Matzinger 2002).
1.6.2 DAMPs in Cancer Therapy
The potential importance of DAMPs has been observed in numerous mouse animal models
where radiation or chemotherapy are used to induce ICD.
1.6.2.1 Calreticulin
Apoptosis occurs constantly in normal tissue turnover and does not result in inflammation. CRT
is a DAMP that may be important in cancer therapy by rendering apoptotic cancer cells
immunogenic. UV radiation and various chemotherapy drugs, such as anthracyclines, are able to
cause immunogenic apoptosis through the shuttling of CRT to the surface of the dying cell
(Apetoh, et al. 2007, Ghiringhelli, et al. 2009, Obeid, et al. 2007). Surface CRT promotes DC
phagocytosis, tumor antigen processing, presentation, and the incitement of an immune response
towards the tumor cells. CRT’s importance may be evidenced by animal experiments in which
inhibiting CRT in anthracycline treated tumor cells weakens its immunogenicity in mice (Obeid,
et al. 2007).

22
1.6.2.2 Adenosine Triphosphate
ATP release has been demonstrated in separate experiments of tumor cells being treated with
chemotherapy or radiation (Martins, et al. 2009, Ohshima, et al. 2010). ATP is another DAMP
that, upon release into the extracellular environment, is recognized by P2Y2 receptors on
phagocytes that result in recruitment into sites of inflammation (Elliott, et al. 2009). In an
experiment demonstrating ATP released by tumor cells undergoing autophagy in response to
chemotherapy, RNA interference of pathways required for ATP release during autophagy
abolished the anti-tumor response (Michaud, et al. 2011). Thus ATP is a key player in ICD.
1.6.2.3 HMGB-1
HMGB-1 release is associated with multiple forms of cell death including, sustained autophagy,
late apoptosis, and necrosis (Tang, et al. 2010, Tang, et al. 2010). These forms of ICD occur as a
result of DNA-damaging treatments such as radiation or chemotherapy. HMGB-1 release is
crucial for antigen presenting cell (APC) activation and subsequent adaptive immune response
towards tumor cells (Lotze and Tracey 2005). Vaccination of mice with tumor cells undergoing
ICD is able to provide a protective effect upon re-challenging the mouse with live tumor cells.
Conversely, in this prophylactic vaccination model, live tumor cells are able to grow on re-
challenge if HMGB-1 in the ICD tumor vaccine is blocked (Apetoh, et al. 2007).
HMGB-1 is a key molecule in MPM that may contribute to the success of SMART radiation
treatment by mediating ICD. Thus the focus of this project is on HMGB-1 in radiation treatment
for MPM.

23
1.7 HMGB-1
1.7.1 Background
HMGB-1 is a DAMP protein with effects in both the development of various cancers as well as
in promoting the host anti-cancer immune response. The duality of HMGB-1 necessitates the
elucidation of its role in the context of mesothelioma.
The discovery of High Mobility Group Box 1 protein was first published by Goodwin and
colleagues in 1973 where they named the protein for its high mobility in gel electrophoresis
(Goodwin, et al. 1973). HMGB-1 is essential for survival as HMGB-1 knockout mice die shortly
after birth (Calogero, et al. 1999). Found in the nucleus of all nucleated cells, HMGB-1 plays a
role in DNA transcription replication, and recombination (Bianchi and Manfredi 2007). Nuclear
HMGB-1 expression levels differ by cell type in both normal and cancerous cells but are often
overexpressed in cancers (Choi, et al. 2003, He, et al. 2000, Ishiguro, et al. 2005, Poser, et al.
2003, Sharma, et al. 2008, Shen, et al. 2009, Volp, et al. 2006, Yamazaki, et al. 2014).
HMGB-1 is a highly conserved protein belonging to the HMGB family of proteins with a greater
than 99% sequence homology among all mammals studied (Ferrari, et al. 1996, Paonessa, et al.
1987, Rauvala, et al. 1988). This protein is composed of HMG boxes A-box (amino acids 1-79)
and B-box (amino acids 89-163) as well as a glutamate and aspartate rich negatively charged C-
terminal domain (Bustin, et al. 1990, Goodwin, et al. 1973). HMGB-1 uses these domains to
bind non-specifically to the minor grooves of DNA (Agresti and Bianchi 2003, Bustin 1999).
This binding is reported to bend DNA and promote transcription factor assembly on DNA targets
(Agresti and Bianchi 2003). In addition, HMGB-1 is able to associate with histones and unravel
chromatin coils to allow transcription factors to interact with uncoiled DNA (He, et al. 2000).

24
Interestingly, HMGB-1 is known to up-regulate transcription factors of genes involved in cancer
development such as p53, p73, and estrogen receptor (Banerjee and Kundu 2003, Das, et al.
2004, El Marzouk, et al. 2008, Jayaraman, et al. 1998, Livesey, et al. 2008, McKinney and Prives
2002, Stros, et al. 2002, Stros, et al. 2004, Uramoto, et al. 2003, Verrier, et al. 1997, Zhang, et al.
1999).
Figure 5. HMGB-1 protein domains. HMGB-1 consists of multiple domains that confer
specific functions including intracellular DNA binding and other nuclear functions as well as
extracellular RAGE receptor binding.
1.7.2 HMGB-1 Signaling Pathways
HMGB-1 is known to mediate effects through a variety of receptors: the Receptor for Advanced
Glycation End Products (RAGE), Toll-like receptor (TLR) 2, TLR 4, and TLR 9.
RAGE was the first receptor found to bind HMGB-1 (Neeper, et al. 1992). The HMGB-1-
RAGE interaction is a mediator of inflammation. Additionally, HMGB-1-RAGE mediates a
broad range of immune effects through chemotaxis, proliferation, and differentiation, of immune
cells (Ryckman, et al. 2003). HMGB-1-RAGE results in NF-κB activation through either the
prototypical MyD88 pathway or through extracellular signal-related kinase (ERK) 1, ERK2, and
p38. This leads to pro-inflammatory cytokine production such as TNF, IL-6, and IFN-γ. HMGB-

25
1 binding to TLR-2 and TLR-4 leads to NF-κB translocation into the nucleus and translation of
inflammatory genes through the MyD88 pathway (Neeper, et al. 1992).
TLR-9 is known to bind CpG DNA that is normally found in pathogen organisms such as
bacteria. Recent research has demonstrated that HMGB-1 is able to preferentially bind to CpG
DNA. This complex is able to enhance the immunostimulatory effect of CpG DNA when
detected by TLR-9 in macrophages, DCs, and B cells. Released HMGB-1 is able to enter
endosomes and accelerate the formation of the CpG DNA and TLR-9 complex. This then results
in the secretion of pro-inflammatory cytokines IL-6, IL-12, IFN-α, and TNF-α (Ivanov, et al.
2007, Tian, et al. 2007).

26
Figure 6. HMGB-1 binds RAGE receptor or TLR 2 or TLR 4. The signalling pathways
functions through the typical MyD88 pathway or through a pathway yet to be characterized;
involving ERK 1/2 and p38 (Lotze and Tracey 2005).

27
1.8 The Pro-Cancer Side of HMGB-1
1.8.1.1 The Role of HMGB-1 in Development
In development of disease, asbestos has been demonstrated to cause mesothelial cell necrosis and
the release of HMGB-1 from the nucleus into the extracellular milieu (Yang, et al. 2010). A
possible pathway of development involves macrophage TNF-α release in response to HMGB-1
in the extracellular environment. TNF-α is thought to mediate mesothelial cell survival by
causing NF-κB translocation into the nucleus. Additionally, mesothelial cells are normally
quiescent with only 0.16% to 0.5% of cells undergoing mitosis (Mutsaers 2004). However, in
the presence of inflammation from injury, 30% to 80% of mesothelial cells undergo cell division
and proliferation (Mutsaers, et al. 2002). Cells that are stimulated to survive and proliferate can
accumulate DNA damage, divide, and may eventually give rise to a cancerous MPM cell (Yang,
et al. 2006). The HMGB-1-TNF- α pathway may play a central role in MPM development as
TNF-α receptor knockout mice exposed to asbestos do not develop the fibroproliferative lesions
observed when wild-type mice are exposed to asbestos.
1.8.1.2 HMGB-1 in Disease Progression
After transformation, HGMB-1 has been described as having a role in the progression of disease.
MPM patients have been shown to have higher serum HMGB-1 concentrations than individuals
exposed to asbestos who have not developed MPM (Tabata, et al. 2013). Furthermore, a recent
study demonstrated that treatment of mice with anti-HMGB-1 antibody slows the growth of the
intraperitoneal injected human epithelioid malignant mesothelioma cell line, REN (Jube, et al.
2012). Thus HMGB-1 may promote the progression of disease.

28
Interestingly, in pancreatic tumor cell lines it has been shown that HMGB-1-RAGE signaling
enhances the activity of mitochondrial complex I in tumor cells, thus promoting greater ATP
production (Kang, et al. 2014). This ATP production is thought to allow for proliferative and
migratory abilities of tumor cells. This finding supports the observation of greater RAGE
expression in gastric and colorectal cancer patients being associated with greater invasiveness
and metastasis (Kuniyasu, et al. 2002, Sasahira, et al. 2005). HMGB-1 may play a similar role in
MPM and thus blocking this protein may lead to lower ATP production and slower growth and
prolonged survival as observed in MPM patients (Tabata, et al. 2013).
During treatment, HMGB-1 also has roles in rendering cancerous cells resistant to therapy.
Studies in other cancers such as leukemia and osteosarcoma have demonstrated that HMGB-1
suppression by RNAi increases the anti-cancer effectiveness of cytotoxic agents such as
chemotherapy and radiation (Huang, et al. 2012, Liu, et al. 2011, Livesey, et al. 2012). In
addition, HMGB-1 over expression has rendered certain cell types resistant to these treatments.
HMGB-1 in pancreatic cancer cells is able to counter cytotoxic agents by interfering with cell
death pathways (Tang, et al. 2010). This protein is able to confer survival to cancer cells
exposed to chemotherapy by inducing a Beclin-1 dependent autophagy response. Autophagy is
the degradation of organelles and is important in tumor cell survival as it allows for recycling of
ATP and biosynthetic molecules under conditions of nutrient limitation or metabolic stresses
such as chemotherapy and radiation (Degenhardt, et al. 2006). Autophagy allows for cancer
cells to enter a state of dormancy and resume growth after treatment has finished; leading to the
recurrence of disease. Intrinsic and extrinsic apoptosis is inhibited by HMGB-1; further
supporting the role of HMGB-1 in the survival of cancerous cells.

29
1.9 The Anti-Cancer Side of HMGB-1
1.9.1 HMGB-1 Release
HMGB-1 has a diverse range of functions on the immune system. Apart from its intranuclear
role, HMGB-1 can be released extracellularly and function as a DAMP to trigger the immune
response (Andersson, et al. 2000, Wang, et al. 1999). This protein can facilitate a multitude of
immunogenic effects through both active and passive release.
1.9.2 Active Release of HMGB-1
Active release of HMGB-1 occurs primarily through secretion by immune cells. HMGB-1 active
release has been best characterized in macrophages and monocytes stimulated by TNF which
were shown to release HMGB-1 in the absence of cell death. Actively secreted HMGB-1 has a
critical role in sepsis (Abraham, et al. 2000, Qin, et al. 2006, Suda, et al. 2006, Wang, et al. 1999,
Yang, et al. 2004). Sepsis, systemic inflammation against pathogen infection, can result in
considerable damage and hypoperfusion to the host’s organs through the increase in vascular
permeability required for immune cell recruitment. HMGB-1 has been shown to be released as a
late proinflammatory mediator in a variety of lipopolysaccharide sepsis models. In fact, the
blocking of HMGB-1 with neutralizing antibodies is able to rescue mice and rats dose-
dependently from lethal sepsis incurred by Cecal Ligation Puncture model.
Active HMGB-1 release has also been demonstrated in DCs (Lotze and Tracey 2005) pituicytes
(glial cells of the posterior pituitary) and human umbilical venous endothelial cells(Wang, et al.
1999). Additionally, HMGB-1 has been shown to be actively released by dendritic cells in order
to stimulate clonal expansion and polarization of naïve CD4+ T cells (Dumitriu, et al. 2005).

30
The mechanism of active release is best characterized in macrophages and monocytes. Active
release involves the endolysosome secretory pathway (Gardella, et al. 2002). While HMGB-1 is
normally able to travel in between the nucleus and cytoplasm through nuclear pores,
inflammatory signals such as LPS or TNF prevent the re-entry of HMGB-1 to the nucleus by the
acetylation of specific lysine amino acids. HMGB-1 is then taken up by endolysosomes through
an unknown mechanism. Endolysosomes then fuse with the membrane and release their
contents. It has been previously demonstrated that the released HMGB-1 is pre-synthesized and
that new HMGB-1, shown by the incorporation of radio-labelled peptides, is translated
approximately 16 hours after the original stimuli (Wang, et al. 1999).
1.9.3 Passive Release of HMGB-1
Passive HMGB-1 release occurs through diffusion out of the cell due to the loss of membrane
integrity most often related to cell death. HMGB-1 has been known to be released by cells that
are permeabilized, undergoing necrosis, or late apoptosis (Degryse, et al. 2001, Falciola, et al.
1997, Muller, et al. 2001). Regulated necrosis, referred to as necroptosis, also leads to HMGB-1
release (Kaczmarek, et al. 2013) and sustained autophagy is yet another mechanism of HMGB-1
to be released (Tang, et al. 2010).
The immunogenic potential of HMGB-1 was demonstrated in a study that measured HMGB-1
release in culture supernatant after triggering necrosis through repeatedly freeze-thawing
fibroblasts (Scaffidi, et al. 2002). Further experiments by this group displayed HMGB-1’s
ability to stimulate macrophage TNF release with necrotic fibroblasts but abrogating this effect
with HMGB-1 deficient fibroblasts (Scaffidi, et al. 2002).

31
Apoptosis is considered to be immunogenically silent (Bianchi and Manfredi 2007). Supporting
this idea, HMGB-1 remains bound to DNA and histones during due to underacetylation of
histones during apoptosis. During apoptosis, HMGB-1 remains in the nucleus and is unable to
stimulate an immune response. Thus, the passive release of HMGB-1 through cell death is a
crucial aspect of the immune response.
While both mechanisms of HMGB-1 release are pro-inflammatory in nature, actively released
HMGB-1 tends to be a late mediator of inflammation while passive HMGB-1 by cells
undergoing cell death is an early mediator. Furthermore, HMGB-1 is hyper-acetylated as part of
the active secretion pathway by immune cells and non-acetylated when liberated passively
(Bonaldi, et al. 2003). The functional significance of this difference remains unclear.

32
Figure 7. HMGB-1 and its multitude of intracellular and extracellular roles. HMGB-1 has
normal intracellular DNA-binding activity. It also has effects on tumor cell metastasis. Its
release is an important mediator of inflammation through RAGE or TLR signalling. (Figure
reproduced with permission from Nature Reviews Immunology).
1.10 The Immune Response and HMGB-1
HMGB-1 plays a broad role in generating immune responses. The signaling cascades described
in the previous section are important for immune system activation against pathogens and cancer.
In addition, aberrant HMGB-1-mediated inflammation contributes to the pathogenesis of various
immunological conditions including septic shock (Abraham, et al. 2000, Qin, et al. 2006, Suda,

33
et al. 2006, Wang, et al. 1999, Yang, et al. 2004), rheumatoid arthritis (Kokkola, et al. 2002,
Kokkola, et al. 2005, Taniguchi, et al. 2003), and systemic lupus erythematosus (Barkauskaite, et
al. 2007, Popovic, et al. 2005, Urbonaviciute, et al. 2007).
The immune system is divided into the innate and adaptive systems (Murphy 2012). The innate
defense system provides us with immediate and rapid immunity against a broad range of
pathogens by recognizing evolutionary conserved molecules. The adaptive immune system
requires time for an antigen-specific response to be generated against pathogen molecules not
previously encountered by the host. In the absence of pathogens, HMGB-1 and other DAMPs
released by dying cells provide a mechanism of sterile immune activation. This project is
focused on the adaptive system as it provides a specific response as well as immune memory,
which prevents further disease by the same pathogen. This idea can also be applied to cancer in
which a successful adaptive response can prevent the recurrence of a cancer that is removed or
killed with treatment.
1.10.1 T Cells
The immune system consists of a wide range of cells that derive from the pluripotent
hematopoietic stem cells of the bone marrow (Murphy 2012). Cells of the lymphoid and
myeloid lines undoubtedly play a diverse range of roles in cancer. T cells, deriving their name
from their maturation in the thymus, are broadly delineated into cytotoxic, helper, and regulatory
T cells.
In cancer, T cells remain important for the large role they play in the immune response. CD8+
cytotoxic T cells, named for the CD8 surface receptor they possess, are effector cells that are
able to directly kill virally infected cells to prevent viral spread to surrounding cells. They are

34
also paramount in destroying cancerous cells. Numerous studies have examined and found
positive correlations between the number of tumor-infiltrating CD8+ T cells and better patient
prognosis Greater levels of tumor-infiltrating CD8+ T cells has be associated with better
prognoses in colorectal cancer, head and neck cancers, ovarian, and breast cancers (Bachmayr-
Heyda, et al. 2013, Balermpas, et al. 2014, Gisterek, et al. , Nosho, et al. 2010). .
Our lab has similarly found that high levels of CD8+ tumor-infiltrating lymphocytes after
Pemetrexed chemotherapy is associated with longer progression-free survival as well as delayed
recurrence in malignant pleural mesothelioma patients (Anraku, et al. 2008). Many immune
checkpoint therapies in cancer attempt to achieve this CD8+ T cell infiltration and tumor cell
killing in order to bring greater survival rates to patients. Blocking PD1 and TIM3 with
neutralizing antibodies has been investigated as a way to re-activate exhausted CD8+ T cells in
melanoma and colon cancer (Fourcade, et al. 2010, Sakuishi, et al. 2010). Bringing better
prognosis and the chance to permanently eliminate tumors, the focus of this work is on CD8+ T
cells and their ability to directly kill MPM cells.
1.10.2 The T Cell Immune Response
In an adaptive T cell response, APCs such as Immature DCs in peripheral tissue constantly
uptake antigens from the extracellular environment through micropinocytosis (Murphy 2012).
Binding of PAMPs or DAMPs to PRRs on immature DCs act stimulates their maturation.
HMGB-1 released through both active and passive mechanisms are able to stimulate the immune
response through PRR signaling on DC. HMGB-1 secreted by Natural Killer (NK) cells lead to
DC maturation (Semino, et al. 2005). In HeLa cells, HMGB-1 was shown to act as an immune
adjuvant as stimulating DCs with wild-type necrotic cells induced maturation while HMGB-1

35
deficient necrotic cells did not (Rovere-Querini, et al. 2004). Furthermore, in vitro recombinant
HMGB-1 is able to induce DC maturation (Messmer, et al. 2004).
DC maturation involves cleaving and processing previously up taken antigens for loading onto
the Major Histocompatibility (MHC) molecule (Murphy 2012). This binding also stimulates up-
regulation of chemokine receptor CCR7 which recognizes CCL 19 and CCL 21; targeting the
DC to the draining lymph node. MHC I-Peptide complexes are shuttled to the surface of the DC,
allowing it to present this complex to the T Cell Receptor (TCR) on T-cells.
Figure 8. DAMPs released by secondary apoptotic or necrotic are sensed by DCs. DCs
migrate to the lymphnode and present their MHC-Ag complex to T cells and trigger the adaptive
immune response.

36
1.10.3 The Three Signal Hypothesis
The three signal hypothesis is a fundamental concept in immunology involving three stimulatory
events that must occur to generate a T cell immune response (Murphy 2012). The first signal
involves the MHC-TCR interaction which is stabilized by either CD4 or CD8 molecules on T
cells. The second signal involves the costimulatory interaction of B7.1 and B7.2 on DCs binding
to CD28 on T cells. The last aspect involves cytokines secreted by APCs that differentiate T-
cells into one of many T cell subsets that include but are not limited to CD8 cytotoxic, Th1, Th2,
Th17, and T regulatory cells. Crucially, DAMPs such as HMGB-1 play a role in every aspect of
the three signals. DAMP signaling leads to antigen processing and DC maturation that is
responsible for antigen presentation on MHC molecules. TLR signaling has a significant role in
upregulating the costimulatory signal. NFκB signaling described previously leads to cytokine
secretion that affects differentiation of T cells.
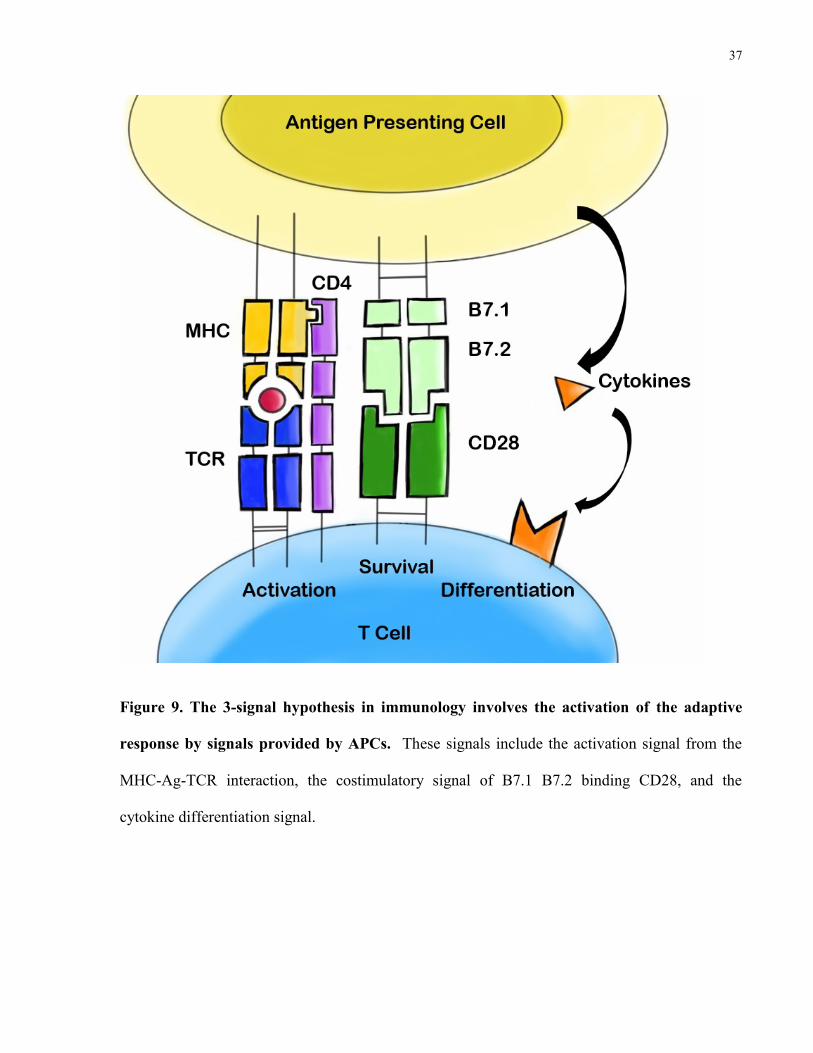
37
Figure 9. The 3-signal hypothesis in immunology involves the activation of the adaptive
response by signals provided by APCs. These signals include the activation signal from the
MHC-Ag-TCR interaction, the costimulatory signal of B7.1 B7.2 binding CD28, and the
cytokine differentiation signal.

38
HMGB-1 induces IL-2 up-regulation by DCs that allows for CD8 T cell licensing (Murphy 2012,
Zhu, et al. 2009). This process allows CD8+ T cells to exit into the periphery and mediate
specific cell killing. Once licensed CD8+ T cells exit into the bloodstream, HMGB-1-activated
endothelial cells are able to recruit circulating T cells by up-regulating ICAM-1, VCAM-1, and
E-selectin (Treutiger, et al. 2003). This allows circulating immune cells to bind and ultimately
extravasate into the inflamed tissue(Murphy 2012). ICAM-1 binds to LFA-1 or Mac1, VCAM-1
binds to VLA-4, and E-selectin binds to the ligand Sialyl-Lewis.
Figure 10. CD8+ T cell licensing. CD4+ T Cells and DCs function in concert to provide
specific activation signals to CD8+ T Cells, allowing them to travel to the periphery and perform
specific cell killing(Kurts, et al. 2010).

39
1.11 Summary
MPM is a cancer of the lung lining that is associated with the inhalation of asbestos fibers, a
fibrous compound still found in the insulation of older buildings. While asbestos use in North
America has been controlled, there still remains a risk of MPM development as older buildings
containing silicates are renovated or demolished. Due to a long incubation time between
exposure and onset of disease, the incidence of disease in North America has increased by 65
percent over the past two decades worldwide and is projected to continue to increase until at least
the year 2020. Moreover, the continued use in the developing world suggests that MPM will
continue to rise worldwide for the foreseeable future. Non-resectable MPM is treated with a
chemotherapy regiment that increases median survival from 9 to 12 months in epithelial disease.
Sarcomatoid and biphasic (a mix of epithelial and sarcomatoid disease) MPM subtypes offer
worse overall survival. Thus there is a substantial need to understand the different factors that
mediate treatment success in the MPM subtypes for novel treatments to be developed.
The de Perrot lab has developed a new methodology of administering short, but high-dose,
radiation fractions before surgery which differs from previous approaches in which radiation is
given after surgical resection. A feasibility study of Surgery for Mesothelioma After Radiation
Therapy (SMART) has demonstrated that this procedure results in a significant increase in 3-
year survival in patients with epithelial disease from 30% to 84% compared to previous
treatment modalities. However, biphasic overall survival was significantly lower at 13%. There
is therefore a substantial need to understand how different subtypes respond to radiation in the
context of host immune system stimulation.

40
New research suggests that high-dose hypofractionated radiation is able to stimulate the immune
system in addition to mediating direct tumor killing. Cytotoxic T lymphocytes circulate through
the body and are able to destroy tumors. However, an inhibitory tumor microenvironment
renders T cells unable to inhibit tumor growth. Furthermore, MPM has few known tumor
associated antigens; making it difficult to target treatment using engineered T-cell receptors or
neutralizing monoclonal antibodies. DAMP molecules offer a new avenue of treatment.
DAMPs, released from cells undergoing immunogenic cell death, are implicated in stimulating
the antigen uptake, maturation, and antigen presentation of dendritic cells; crucial activators of
cytotoxic T cells that mediate tumor killing. HMGB-1, a prototypical DAMP, has been
demonstrated as necessary to stimulate the immune system after chemotherapy in many animal
models of cancer and we are keen to observe its role in MPM.
In this chapter we have discussed the dual nature of the protein HMGB-1 in both development
and treatment of disease. We have studied HMGB-1 in tumor development and progression but
also in its role as a DAMP and as a mediator of the immune response. We have also examined,
in depth, the immunostimulatory effects of radiation through DAMP release and the benefit of
the CD8+ T cell immune response in eliminating MPM cells. The following chapter will outline
the hypothesis and aims of this study.

41
2 Hypothesis and Aims
Research in support of pro and anti-tumor roles of HMGB-1 has been increasing. Broadly,
HGMB-1 has been suggested to play a dual role in both cancer cell survival and death. HMGB-1
has been described as a negative prognostic marker in several cancers including MPM. However
after treatment, HMGB-1 is suggested to aid in the recruitment of the immune response. The
goal of this study was to elucidate the role of HGMB-1 in mediating an anti-tumor immune
response in radiation treatment of malignant pleural mesothelioma. The hope of this
translational research project was to use the findings of this study to understand and improve
treatment given to MPM patients.
The objectives of this study were threefold: 1) to determine if the different subtypes of MPM
differ in HMGB-1 release, 2) to correlate HMGB-1 release in paraffin-embedded patient samples
to survival, 3) to investigate the CD8+ T cell immune response elicited by HMGB-1 release. As
epithelial disease offers a greater survival than sarcomatoid disease, it is hypothesized that
epithelial disease releases more HMGB-1 by immunogenic cell death after radiation treatment
which leads to the generation of a specific CD8+ T cell response. This CD8+ T cell response
leads to specific tumor killing and results in greater overall and disease free survival in patients
after SMART.
To tackle the first objective, I postulated that HMGB-1 released as a result of radiation treatment
leads to an immune response that aids in improving patient survival. I suggest that epithelioid
subtypes which are commonly responsive to SMART treatment release more HMGB-1 by cell
death. Passively liberated HMGB-1 could then bind to RAGE and TLR molecules on DCs and
stimulate a T Cell immune response. This immune involvement would lead to specific tumor

42
cell killing and therefore longer survival. Conversely, I propose that sarcomatoid cells do not
release as much HMGB-1 in response to radiation induced cell death and therefore recruit the
immune system to a lesser extent, leading to a shorter patient survival time. For the second
objective, I postulated that higher staining intensity of HMGB-1 after treatment would correlate
with higher patient survival as HMGB-1 would recruit the immune system through binding to
APCs. In studying the last objective, I hypothesized that radiation in vivo would lead to tumor
cell death which would result in HMGB-1 release. This release would be responsible APC
maturation, tumor antigen presentation, and a CD8+ T Cell immune response measured by CD8+
T Cell infiltration.

43
3 Materials and Methods
3.1 Human and Murine Cell Lines
All cell lines used, with the exception of CRL-9444 (MeT-5A), were grown in RPMI 1640
culture media (Life Technologies Inc., Burlington ON, CAN) supplemented with 10% FBS.
CRL-9444 was grown in Medium 199 with 1.5g/L sodium bicarbonate, 3.3 nM EGF, 400 nM
hydrocortisone, 870 nM bovine insulin, 20 mM HEPES, 3.87 µg/L H2SeO3, and 10% FBS.
Additionally, cultures were grown in a 37oC and 5% CO2 environment.

44
Table I. Cell lines used for experiments.
Cell Line Cell Type Origin
NCI-H226 (CRL-5826) Human Epithelioid
Mesothelioma
Mesothelioma derived from
pleural effusion
NCI-H2052 (CRL-5915) Human Epithelioid
Mesothelioma
Stage 4 mesothelioma
derived from lymph node
metastasis
NCI-H28 (CRL-5820) Human Sarcomatoid
Mesothelioma
Stage 4 mesothelioma
derived from pleural
effusion
NCI-H2452 (CRL-5946) Human Sarcomatoid
Mesothelioma
Derived from lung tissue
MeT-5A (CRL-9444) Human Immortalized
Mesothelial Cell
Mesothelium cells derived
from pleural fluids of non-
cancerous individual
6E6 (PTA-5433) Murine BALB/c anti-HMGB-1
Hybridoma
Hybridoma line generated in
BALB/c mice in response to
vaccination with HMGB-1
protein
AB12 Murine BALB/c Epithelial
Mesothelioma
Malignant mesothelioma
from BALB/c mice exposed
to crocidolite asbestos
AE-17-OVA Murine C57BL/6 Ovalbumin-
transfected Epithelial
Mesothelioma
Malignant mesothelioma
cells from C57BL/6 mice
exposed to crocidolite
asbestos

45
3.2 Cell Line Radiation
2 million cells of the cell line of interest are plated in a 75 mm2 flask. Adherent cells are
irradiated for the time corresponding to the desired dose (Gy) in a Gammacell 40 Extactor
Caesium-137 irradiator (Best Theratronics Ltd., Ottawa ON, CAN). Radiation was given in 1
fraction.
3.3 Flow Cytometry Cell Death Quantification
2 million cells of the cell line of interest are plated in a 75 mm2 flask. Adherent cells are
irradiated for the time corresponding to the desired dose (Gy) in a Gammacell 40 Extactor
Caesium-137 irradiator (Best Theratronics Ltd., Ottawa ON, CAN)
After 1 fraction of 25Gy radiation, adherent cell lines were incubated for 1, 2, 4, 24, or 48 hours
before staining with 5µL/100µL of cells Annexin V Ab (Biolegend Inc., San Diego CA, USA)
and 1µL/1mL of cells eFluor450 viability dye (eBioscience Inc., San Diego CA, USA). Cells
were measured for fluorescence on a BD LSR II flow cytometer (BD Biosciences, Mississauga
ON, CAN). Analysis was performed using FlowJo V10 (FlowJo LLC, Ashland, USA) software.
Annexin V– eFluor450– cells were deemed to be viable cells and Annexin V+ eFluor450– cells
were considered apoptotic in nature. Annexin V+ eFluor450+ and Annexin V– eFluor 450+
cells were considered to be necrotic cells.

46
Figure 11. Quantification of cell death by flow cytometry and gating strategy. Annexin V–
eFluor450– cells were considered viable cells and Annexin V+ eFluor450– cells were considered
apoptotic. Annexin V+ eFluor450+ and Annexin V– eFluor 450+ cells were considered to be
necrotic cells.

47
3.4 HMGB-1 concentration of Mouse Serum or Cell Culture
Supernatant by ELISA
Cell culture supernatants were collected every 1, 2, 4, 24, or 48 hours after the single 25Gy
radiation fraction. Supernatant was collected from the corresponding time points in non-treated
cell lines. Mouse blood was collected in EDTA tubes to prevent coagulation and centrifuged at
3000 RPM for 15 minutes. Serum was collected to be used in HMGB-1 measurement. Media or
serum was placed in wells coated with anti-HMGB-1 antibody from a sandwich ELISA kit (IBL
International, Hamburg, GER). HMGB-1 in the samples were left overnight to bind to anti-
HMGB-1 antibodies. Secondary antibody conjugated to an enzyme was allowed to bind to the
captured HMGB-1 protein. Substrate was added and colour change reaction was observed.
Concentrations were determined by four-parameter logistic test using a standard curve. Samples
were measured in duplicate.
3.5 Mice
Eight to twelve week old female C57BL/6 and BALB/c wildtype mice were purchased from The
Jackson Laboratory (Maine, USA).
All mice were housed at the Toronto Medical Discovery Tower’s Animal Resource Centre under
pathogen-free conditions in accordance with institutional and national animal care and ethics
protocols.

48
3.6 Radiation Mouse Model
Eight to twelve week old C57BL/6 mice were injected subcutaneously in the right flank with 1 x
106 AE-17-OVA cells in 100µL of PBS. Mice were divided into two groups: 1) no treatment
and 2) local radiation therapy involving a total dose of 15 Gy over 3 days (3 fractions). At least
3 mice were used in each group. Radiation occurred on days 8, 9, and 10 after cell injection.
225 kV 13 mA radiation was given to mice under isofluorane anaesthesia using the X-Rad
225Cx (Precision X-Ray, Branford CT, USA) in which flank tumors were visually targeted using
X-ray images in x, y, and z dimensions. Imaging resolution was 0.2mm with an image
acquisition dose of 1-10 cGy. On day 12, 17, and 22, mice were sacrificed from each group and
blood and tissue samples were collected. Blood was taken by cardiac puncture method under
isofluorane for analysis of serum HMGB-1 level. Mice were then sacrificed by cervical
dislocation. Mouse tumors were measured and resected for use in immunohistochemical and
immunofluorescent staining. The longest two perpendicular measurements were taken and
tumor volume was calculated using the formula:
Length x Width2 x (π/6).
In a similar experiment, mice were measured for tumor growth and sacrificed when tumors
reached a volume of 250 mm3. In this experiment mice were either 1) untreated, 2) received
local radiation therapy of 15Gy over 3 days, or 3) received 15Gy local radiation therapy over 3
days and anti-HMGB-1 antibody (intraperitoneal 100µg injection per day for 5 days; days 8-12).
There were at least 3 mice per group.

49
Figure 12. Schematic of untreated, radiated, and radiation + anti-HMGB-1 treatment in
tumor bearing mice. In the treated group, days 12, 17, and 22 correspond to 2, 7, and 12 days
after LRT respectively.
3.7 Hybridoma Preparation and anti-HMGB-1 Ab Purification
BALB/c mice were given a 0.5mL Pristane (Sigma-Aldrich, Oakville ON, CAN) intraperitoneal
(IP) injection one week before IP injection of 5 x 106 cells in 0.5mL phosphate buffered saline
(PBS) of the 6e6 HMGB-1 hybridoma cell line (PTA-5433 from American Type Culture
Collection, Manassas VA, USA). One week after cell injection, mice are sacrificed and ascites
fluid with antibody, produced by the hybridoma tumor, is drained using a cannula into tubes with
EDTA to prevent coagulation.
Cell culture media or ascites fluid generated from the 6e6 hybridoma cell line was purified for
anti-HMGB-1 Abs through a protein A antibody purification kit (Abcam Inc., Toronto ON,

50
CAN). Desired sample is mixed with a protein A resin which binds to the Ab constant domain.
The protein A beads bound to the Ab are collected in a filter tube. Anti-HMGB-1 Abs are then
eluted and collected.
3.8 Hybridoma Anti-HMGB-1 Antibody Blocking Test
Anti-HMGB-1 isolated from ascites fluid generated by the anti-HMGB-1 6E6 hybridoma was
purified and tested for binding ability through ELISA assay. Recombinant HMGB-1 protein (80
ng/mL, HMGBiotech Srl., Milano, ITA) was allowed to bind to hybridoma-generated anti-
HMGB-1 antibody in a 1:1 or 1:2 ratio (80 ng/mL or 160 ng/mL Ab) for 30 minutes in PBS.
Ab-recombinant HMGB-1 mixture or recombinant HMGB-1 alone was measured for HMGB-1
concentration by ELISA kit (IBL International, Hamburg, GER) to determine the 6E6 antibody’s
ability to bind HMGB-1. Lack of signal in the mixture group is indicative of 6E6 antibody
binding.
3.9 Patient Sample HMGB-1 Immunohistochemistry
Mesothelioma tumor tissue samples were acquired and sent to pathology after patients underwent
biopsy, extrapleural pneumonectomy, decortication, or pleurectomy with decortication. Some
samples were snap frozen in liquid nitrogen and kept at –80oC on dry ice however most were
immediately fixed in paraformaldehyde for 48 hours before being embedded in paraffin blocks.
Frozen and paraffin embedded samples were sliced into 5µm thick sections using a microtome.
Frozen sections were fixed with 1% paraformaldehyde for 1 hour before staining. Paraffin
sections were deparaffinised with Xylene, 100% ethanol, 95% ethanol, and 70% ethanol
respectively. Paraffin sections underwent antigen retrieval by treating with 100oC citrate buffer
for 20 minutes. Sections were blocked with serum (5% BSA in Tris-buffered saline) for one

51
hour before the addition of primary anti-HMGB-1 antibody (Abcam Inc., Toronto ON, CAN) at
a 1:100 dilution. After incubating overnight at 4oC, sections were washed in TBS+0.2% Tween
20. Slides were subsequently incubated for 1 hour at room temperature with anti-rabbit-HRP
secondary Ab kit (Vector Laboratories Inc., Burlington ON, CAN). After washing, the HRP
substrate DAB (3, 3-diaminobenzidine) (Vector Laboratories Inc., Burlington ON, CAN) was
added to each slide at 100 µL per section. Sections were then counterstained with hematoxylin,
dehydrated, and mounted with mounting media (Fischer Scientific, Ottawa ON, CAN).
Immunostained slides were imaged at 200x magnification with the Aperio ImageScope digital
scanner and visualized with Aperio ImageScope Viewer version 12.1 (Vista, CA, USA).
Scanning was provided by the Advanced Optical Microscopy Facility (Toronto ON, CAN)
Semi-quantification of nuclear staining into high, medium and weak positive as well as negative
was performed using the Aperio nuclear v9 algorithm (Leica Microsystems Inc., Concord ON,
CAN). Distinction of nuclear staining intensity was based on observation and modelled using
the Aperio algorithm which applied observational parameters of all tissues studied.
Figure 13. Quantification of staining using image analysis software. Quantification of high,
medium, and low intensity staining is detected as well as negative cells. Image analysis can then
be applied to an entire tissue sample.

52
3.10 Immunofluorescence
Cell lines used for immunofluorescence were grown on glass slides and underwent the
designated treatment. Cell lines or frozen tissue samples on slides were fixed with cold acetone
for 10 minutes. Paraffin embedded samples were deparaffinised with Xylene, 100% ethanol,
95% ethanol, and 70% ethanol respectively and antigen retrieval was performed by immersing
samples in 100oC citrate buffer for 20 minutes. Samples were blocked with 5% BSA in Tris-
buffered saline for one hour before the addition of primary Ab. After incubating overnight at
4oC, sections were washed in TBS+0.2% Tween 20. Slides were subsequently incubated for 1
hour at room temperature with the appropriate fluorescently labelled secondary Ab. Slides were
further washed with TBS+0.2% Tween 20 before adding mounting media with DAPI nuclear
stain. Cover slips were placed on top and sealed with nail polish.
Fluorescently labelled cells or tissues were visualized with the WaveFX (Quorum Technologies
Inc, Guelph ON, CAN) confocal microscope system. Pictures were analyzed using ImageJ
V1.47 (National Institute of Health, USA). Corrected total cell fluorescence (CTCF) was
calculated by the formula CTCF = Integrated Density – (Area of cell x mean fluorescence of
background reading).
3.11 Statistical Analysis
Statistical analysis was performed with GraphPad Prism 5 (GraphPad Inc, La Jolla CA, USA).
More than 2 groups were compared using one-way ANOVA analysis. Student’s T-test was used
to analyze two groups. A P-value of less than 0.05 was considered statistically significant.
Results have been presented as Mean ± Standard Error of the Mean (SEM). * indicates p<0.05,
** indicates p<0.01, and *** indicates p<0.001 in all results figures.

53
4 Results
4.1 In vitro HMGB-1 characterization
4.1.1 Epithelioid and sarcomatoid human MPM cell lines differ in
intracellular HMGB-1 expression 24 hours after radiation
treatment
To determine the effect of γ-radiation on intracellular HMGB-1 expression, human epithelioid
MPM cell line H226 and human sarcomatoid MPM cell line H28 were irradiated over a dosing
rage of 5-75Gy in a single fraction. Staining of HMGB-1 by immunofluorescence was
performed and radiated cells were compared in fluorescent intensity to untreated cells.
Immunofluorescent staining of human epithelioid (H226) and sarcomatoid (H28) MPM cell lines
was performed 24 hours after the designated radiation doses were given (Figure 14A). In this
experiment it was found that fluorescently labelled intracellular HMGB-1 displayed decreasing
fluorescence intensity (488 nm) levels as radiation dose increased in the H226 subtype (Figure
14B) while fluorescent intensity increased across dose in H28 (Figure 14C). These findings
suggest that either HMGB-1 expression is affected by radiation or that intracellular HMGB-1 is
being lost or gained. HMGB-1 may be lost to the extracellular environment in epithelioid H226
while buildup or new synthesis of HMGB-1 may be occurring in sarcomatoid H28.

54
Figure 14. Expression of HMGB-1 in human MPM cell lines 24 hours after radiation
treatment. A) Immunofluorescent microscopy images (405 and 488 nm) of nuclei (DAPI 405
nm) and immunolabelled HMGB-1 (488 nm). Quantification of cell fluorescence at 488 nm for
B) H226 and C) H28.

55
4.1.2 Epithelioid MPM cell line H226 releases more HMGB-1 than
sarcomatoid line H28 in response to γ-radiation
While intracellular HMGB-1 is hidden from the immune system, extracellar HMGB-1 functions
as a pro-inflammatory DAMP. In order to confirm the differences observed in
immunofluorescent staining of H226 and H28 cell lines and their relation to extracellular release,
media HMGB-1 concentration was measured. HMGB-1 concentration in the media and
therefore release from cells was determined by HMGB-1 ELISA analysis of culture media 24
hours after each treatment. Radiation was given in a single fraction.
In the no treatment group, HMGB-1 release from H226 (7.113 ± 0.4929 ng/mL) and H28 (4.844
± 1.567 ng/mL) was not significantly different (P=0.2165). 24 hours after treatment with 5 Gy γ-
radiation, HMGB-1 in the media was significantly higher in H226 (8.847 ± 0.2496 ng/mL) than
H28 (7.155 ± 0.0925 ng/mL, P=0.0238). At 25Gy, H226 (24.99 ± 6.066 ng/mL) released
significantly more HMGB-1 into the media than H28 (8.090 ± 0.5087 ng/mL, P=0.0213). This
trend was also seen at 50Gy (H226, 60.99 ± 4.601 ng/mL, vs. H28, 6.001 ± 0.5240 ng/mL,
P=0.0070). Lastly, 24 hours after 75 Gy γ-radiation treatment, H226 media HMGB-1
concentration was significantly higher than H28 (37.95 ± 3.733 ng/mL vs. 8.061 ± 0.5600
ng/mL, P=0.0156).
Overall, this data displayed a trend that in response to radiation, epithelioid H226 cells release
more HMGB-1 into the media at all doses compared to sarcomatoid H28 cells. As extracellular
HMGB-1 release has been reported to occur as a result of necrotic cell death, this data suggests
that until 24 hours, epithelioid H226 is more susceptible to radiation than sarcomatoid H28.
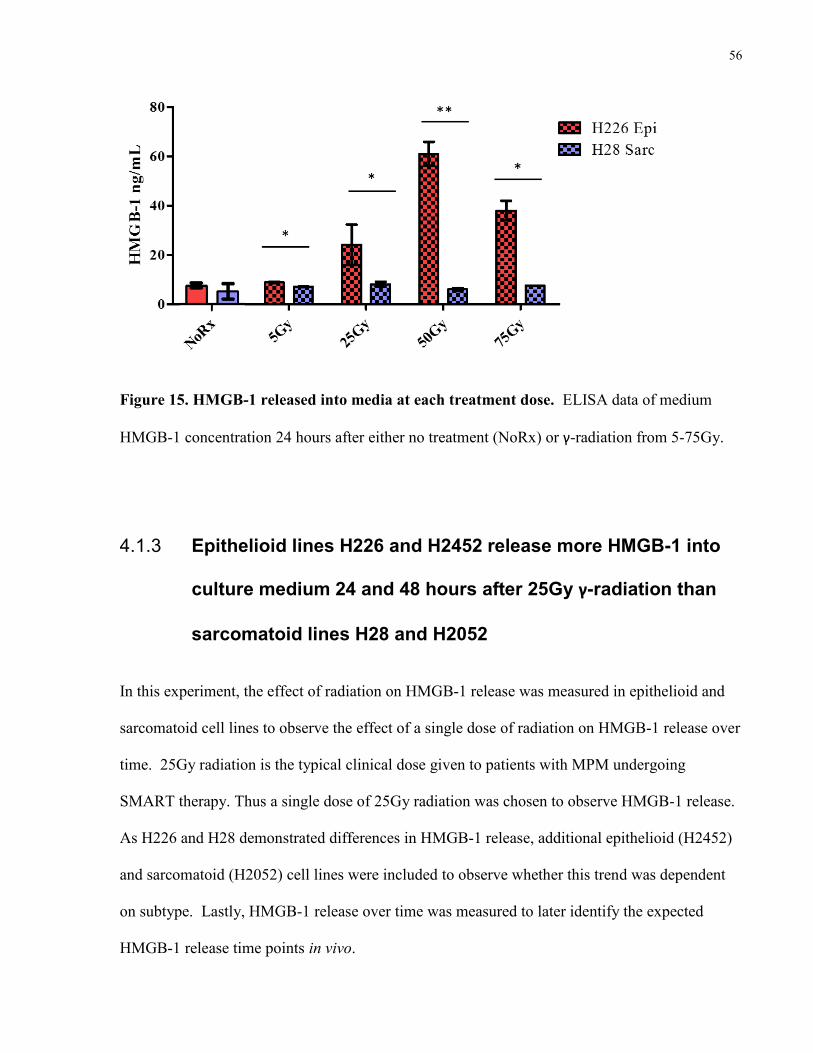
56
Figure 15. HMGB-1 released into media at each treatment dose. ELISA data of medium
HMGB-1 concentration 24 hours after either no treatment (NoRx) or γ-radiation from 5-75Gy.
4.1.3 Epithelioid lines H226 and H2452 release more HMGB-1 into
culture medium 24 and 48 hours after 25Gy γ-radiation than
sarcomatoid lines H28 and H2052
In this experiment, the effect of radiation on HMGB-1 release was measured in epithelioid and
sarcomatoid cell lines to observe the effect of a single dose of radiation on HMGB-1 release over
time. 25Gy radiation is the typical clinical dose given to patients with MPM undergoing
SMART therapy. Thus a single dose of 25Gy radiation was chosen to observe HMGB-1 release.
As H226 and H28 demonstrated differences in HMGB-1 release, additional epithelioid (H2452)
and sarcomatoid (H2052) cell lines were included to observe whether this trend was dependent
on subtype. Lastly, HMGB-1 release over time was measured to later identify the expected
HMGB-1 release time points in vivo.

57
Media release of HMGB-1 was measured by ELISA at each time point after 25Gy radiation
treatment or at the same point in untreated cells. We found no significant differences at time
points before 24 hours. Media HMGB-1 was significantly increased in treated H226 cells (18.61
± 0.9562 ng/mL) compared to untreated H226 cells (5.807 ± 0.4632 ng/mL) 24 hours after
radiation (P< 0.0001). Similarly, HMGB-1 concentration was significantly increased 24 hours
after radiation in radiated H2452 cells (29.50 ± 1.500 ng/mL) compared to untreated (4.917 ±
0.5290 ng/mL) H2452 cells (P=0.0042). No significant difference was observed between
untreated and radiated H28 cells at 24 hours (7.054 ± 0.5747 vs. 9.654 ± 2.693 ng/mL, P=
0.4008). No significant difference was observed between untreated and radiated H2052 cells at
the 24 hour time point (3.478 ± 0.3181 vs. 3.545 ± 0.8395 ng/mL, P=0.9446).
At the 48 hour time point, a significant increase was observed in treated H226 cells (42.88 ±
4.240 ng/mL) compared to untreated H226 cells (8.371 ± 0.6406, P<0.0001). Similarly, treated
H2452 (91.50 ± 11.50 ng/mL) cells showed greater media HMGB-1 concentrations than
untreated H2452 cells (8.447 ± 1.923 ng/mL, P=0.0191). Conversely, there was no significant
difference between untreated and treated H28 cells (9.524 ± 0.1926 vs. 11.57 ± 0.9960 ng/mL,
P=0.0538) or between untreated and treated H2052 cells (4.062 ± 0.3347 vs. 7.048 ± 1.617
ng/mL, P=0.1448).
These findings overall suggest that, at least until 48 hours after treatment, epithelioid MPM cell
lines are more susceptible to radiation in terms of HMGB-1 release while sarcomatoid MPM cell
lines are less susceptible; unchanging in HMGB-1 release after radiation treatment.

58
Figure 16. HMGB-1 release into culture medium by non-treated or radiated epithelioid
lines H226 and H2452 and sarcomatoid cell lines H28 and H25052. ELISA measurement of
medium HMGB-1 concentration at time points (1-48 hours) after either no treatment (NoRx) or
25 Gy.

59
4.1.4 Radiation induced cell death in human cell lines
To test the susceptibility of epithelioid MPM and sarcomatoid MPM to radiation, cell death was
measured in untreated and single fraction 25Gy radiated cells. After each time point, floating
and adherent cells were collected for analysis. Cell death was measured by flow cytometric
analysis of Annexin V and eFluor450 viability staining.
The percentage of apoptotic cells (Annexin V+ efluor 450–) did not change significantly over
time. At 48 hours after 25Gy radiation, radiated H226 cells displayed significantly higher
percentage of necrotic cells (Annexin V+ efluor450+ and Annexin V– efluor450+) than untreated
H226 cells out of total events measured (21.28 ± 0.7804 % vs. 48.41 ± 2.109 %, P=0.0003). 48
hours after treatment, 25Gy radiated H226 cell culture demonstrated a higher percentage of
necrotic cells than 25Gy radiated H28 (48.41 ± 2.109% vs. 12.52 ± 0.8741 %, P<0.0001). The
difference between treated and untreated H28 cells 48 hours after radiation was not significant
(9.047 ± 1.448 vs. 12.52 ± 0.8741, P=0.1094).
These findings suggest that in H226, necrotic cell death is the predominant form of cell death
elicited by 25Gy radiation treatment and occurs at least 48 hours after treatment. Conversely,
sarcomatoid H28 is resistant to 25Gy radiation by not undergoing necrotic cell death at least until
48 hours after treatment.

60
Figure 17: Percentage cell death after no treatement or 25Gy radiation. A) Apoptotic cell
death and B) necrotic cell death was measured for H226 and H28 untreated or 25Gy radiated cell
lines.

61
4.1.5 AE-17-OVA epithelioid mouse cell line behaves similarly to
H226 in terms of HMGB-1 release after 25Gy radiation
This experiment investigated HMGB-1 release after one dose of 25Gy radiation in AE-17-OVA
to determine if it behaved similarly to human epithelioid MPM cell lines. HMGB-1 release from
AE-17-OVA cells was determined by ELISA of culture media. Cells were either untreated or
subjected to 25Gy radiation by a Caesium-137 irradiator. Culture media was collected 1, 2, 4, 24
and 48 hours after treatment. Media was then analyzed by an HMGB-1 ELISA kit to determine
supernatant concentration.
At the 4 hour time point, HMGB-1 in radiated AE-17-OVA (12.08 ± 3.840 ng/mL) was
increased compared to no treatment (2.622 ± 0.4585 ng/mL, P=0.1283). At 24 hours, media
HMGB-1 concentrations were significantly higher in radiated AE-17-OVA than untreated cells
(84.630 ± 14.3500 ng/mL, P=0.0484). Lastly, 48 hours after treatment, media HMGB-1
concentration was higher in radiated cells (140.800 ± 14.9200 ng/mL) than in unirradiated cells
(34.021 ± 1.4110 ng/mL, P=0.0191).
The results demonstrate that murine epithelioid AE-17-OVA behaves similarly to human
epithelioid H226 as radiated groups release more HMGB-1 than untreated cells. However AE-
17-OVA differs in that HMGB-1 release appears to begin 4 hours after radiation while radiated
H226 begins releasing HMGB-1 around the 24 hour time point.

62
Figure 18. HMGB-1 release into culture medium by non-treated or radiated human
epithelioid line H226 and H2452 and murine epithelioid cell line AE-17-OVA. ELISA
measurement of medium HMGB-1 concentration at time points (1-48 hours) after either no
treatment (NoRx) or 25 Gy.

63
4.1.6 AE-17-OVA epithelioid mouse cell line behaves similarly to
human epithelioid cell line H226 in terms of cell death when
treated with 25Gy radiation
The purpose of this experiment was to investigate whether, similar to H226, murine epithelioid
MPM cell line AE-17-OVA cell death occurs at the same time as HMGB-1 release. Radiated
cells were given 25Gy in a single fraction.
After each time point, floating and adherent cells were collected for analysis. Cell death was
measured by flow cytometric analysis of Annexin V and eFluor450 viability staining. Apoptotic
cells were considered as Annexin V+ efluor 450–. Annexin V+ efluor450+ and Annexin V–
efluor450+ cells were considered necrotic.
We found that the percentage of apoptotic cells did not change significantly over time in AE-17-
OVA. Basal rate of apoptosis in AE-17-OVA of both untreated and radiated cells is increased
compared to H226. 4 hours after treatment, the percentage of AE-17-OVA cells undergoing
necrosis in the radiated group was increased compared to untreated (15.05 ± 3.126% vs. 23.82 ±
1.040%, P=0.0562). 24 hours after treatment, necrotic cell death in radiated AE-17-OVA (38.33
± 4.410%) was significantly higher than in untreated AE-17-OVA cells (10.18 ± 7.410%,
P=0.0309).
At 48 hours after 25Gy radiation, radiated H226 cells displayed significantly higher percentage
of necrotic cells than untreated H226 cells out of total events measured (21.28 ± 0.7804 % vs.
48.41 ± 2.109 %, P=0.0003). Similarly, in AE-17-OVA radiated cells necrotic cell death (69.78
± 8.851%) was significantly higher than in untreated murine cells (14.29 ± 3.873%, P=0.0046).

64
Overall, AE-17-OVA behaves similarly to H226 in that radiated cells undergo more necrotic cell
death compared to untreated cells. Differently however, AE-17-OVA demonstrates increased
necrotic cell death at 24 hours after treatment in radiated cell lines. These data suggest that AE-
17-OVA behaves similarly to H226 in terms of necrotic cell death but succumbs to radiation-
induced cell death at an earlier time point.
Figure 19: Percentage cell death after no treatement or 25Gy radiation. A) Apoptotic cell
death and B) necrotic cell death was measured for murine epithelioid AE-17-OVA and human
epithelioid H226 untreated or 25Gy radiated cell lines.

65
4.2 Patient Sample and in vivo HMGB-1
4.2.1 No significant difference in HMGB-1 positivity by IHC staining
between untreated and SMART treated epithelioid MPM
paraffin embedded patient samples
Epithelioid patient samples were stained for HMGB-1 to investigate whether HMGB-1 staining
in treated patients correlated with longer survival times of MPM patients.
Using Aperio Image Scope nuclear analysis software, the strength of HMGB-1 nuclear staining
in paraffin embedded patient samples was measured. Cells were designated by the software as
being either high, medium, or low strength in staining. Furthermore, percent of HMGB-1
positive nuclei was measured by counting positive nuclei regardless of strength of staining.
Epithelioid MPM samples were used for this experiment.
The percentage of highly positive nuclei in non-treated or SMART treated MPM patients did not
differ significantly (32.47 ± 5.59% vs 31.69 ± 5.44%, P=0.9372). No treatment (24.44 ± 2.48%)
and SMART treatment (19.95 ± 1.91%) patient samples did not differ significantly in the
percentage of medium intensity positive nuclei (P=0.2216). Similarly, percent low intensity
positive nuclei of no treatment (19.74 ± 3.30%) and SMART treated (17.76 ± 1.95%) samples
did not differ significantly (P=0.6107). Finally, the overall percentage of positive nuclei in the
samples of each group did not display any significant difference (76.64 ± 2.41% vs 69.40 ±
5.229%, P=0.4328). The no treatment group was N=5 while the SMART treated group was
N=14.

66
While MPM patients receiving SMART treatment have significantly higher survival than
untreated patients, the lack of difference in HMGB-1 staining intensities between the groups
suggest HMGB-1 immunohistochemistry alone is not an adequate predictor of survival in
untreated or SMART treated MPM patients.
Table III. Patient Demographics.
Age (yr) 66 ± 9.7
Sex
Male 14
Female 5
No Treatment N=5
SMART N=14

67
Figure 20. High, medium, and low intesnsity nuclear staining of HMGB-1 in paraffin
embedded patient samples. Percentage of total nuclei with A) high, B) medium, and C) low
intensity staining was quantified in MPM patient samples. D) Total positively stained nuclei was
also measured. Typical images of patient samples from epithelioid MPM patients who received
E) no treatment or F) SMART treatment are displayed.

68
4.2.2 Serum HMGB-1 is significantly higher 2 days after treatment
in mice receiving local radiation therapy.
Mice with subcutaneous epithelioid MPM from AE-17-OVA inoculation were either untreated or
radiated to observe whether HMGB-1 is released in vivo in response to radiation. Mice in the
15Gy treatment group had their tumors locally irradiated by image guidance. Mice received 3
fractions of 5Gy (1 per day) for a total dose of 15 Gy. HMGB-1 ELISA was performed on
mouse serum on days 12 (2 days after LRT), 17 (7 days after LRT), and 22 (12 days after LRT)
after subcutaneous MPM tumor inoculation.
At the time point 2 days after completing LRT, there was a significant increase in mean serum
HMGB-1 concentration of radiated mice (N=5, 52.77 ± 8.380 ng/mL) compared to mice who did
not receive treatment (N=9, 11.51 ± 3.381 ng/mL, P=0.0002). At 7 days after completion of
LRT, there was no significant difference in the mean serum HMGB-1 concentration of non-
treated (N=8, 84.61 ± 37.41) and radiation treated (N=8, 47.95 ± 19.59) mice (P=0.3999).
Similarly there was no significant difference in mean serum HMGB-1 concentration between
radiated mice (N=7, 133.9 ± 51.30) 12 days after completing LRT and no treatment mice (N=6,
85.38 ± 32.14) at the same time point (P=0.4579). These results suggest that radiating the tumor
leads to an acute release of HMGB-1 into the serum 2 days after treatment and that this effect
disappears at later time points.

69
Figure 21. Serum HMGB-1 and tumor growth curves of non-treated and mice treated with
15 Gy LRT. A) at days 12, B) 17, and C) 22, serum HMGB-1 was measured. 15Gy LRT mice
had significantly greater serum HMGB-1 than untreated mice at day 12 (2 days after LRT).
4.2.3 Tumor Growth Curve
Tumor size measurements were taken to observe the effect of radiation on tumor size. Tumor
growth was measured for untreated and 15 LRT treated mice. At day 12 there was no significant
difference between untreated (218.6 ± 55.30 mm3, N=6) and radiated (78.8 ± 15.43 mm3, N=3)
mice (P=0.2162). At day 17, untreated mice (425.3 ± 122.4 mm3, N=4) were significantly higher
in tumor volume than treated mice (64.9 ± 24.99 mm3, N=4, P=0.0279). Finally on day 22,
untreated mice (898.3 ± 473.70 mm3, N=3) displayed a significantly greater tumor volume than
LRT treated mice (66.85 ± 22.81 mm3, N=6, P=0.0106). Collectively, these findings
demonstrate that radiation is able to slow tumor growth while in untreated groups, tumors
continue to increase in size over time.
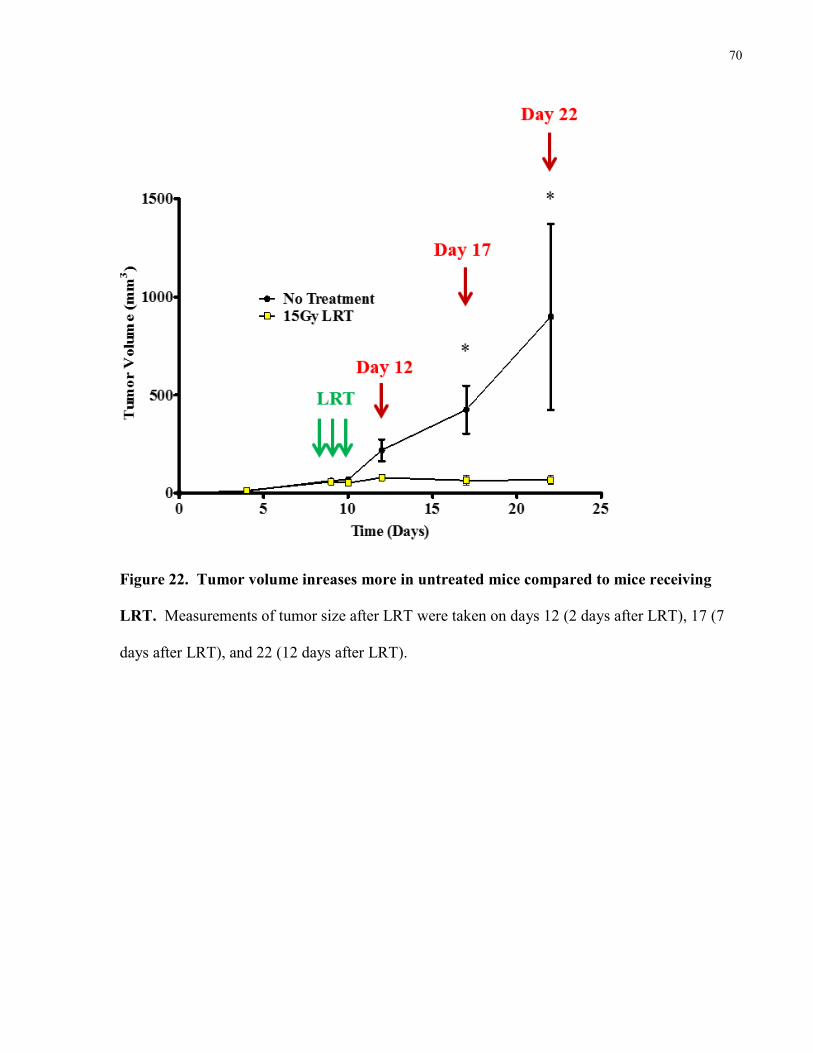
70
Figure 22. Tumor volume inreases more in untreated mice compared to mice receiving
LRT. Measurements of tumor size after LRT were taken on days 12 (2 days after LRT), 17 (7
days after LRT), and 22 (12 days after LRT).

71
4.2.4 Percent HMGB-1 positive nuclei in tissues of mice receiving
no treatment or 15Gy radiation.
The purpose of this experiment was to quantify HMGB-1 staining of untreated and radiated
murine epithelioid MPM tumors. The goal was to study whether radiation treatment has an
effect on HMGB-1 expression in tissue. The percentage of HMGB-1 positive cells of untreated
and radiated tumor tissue at each time point was measured. 2 days after the completion of LRT,
there was no significant difference in the mean percent positive cell staining of NoRx (88.97 ±
2.157%) and 15Gy (93.89 ± 1.347%) groups (P=0.1016). NoRx (81.84 ± 3.792%) and 15Gy
(95.20 ± 1.101%) groups showed a significant difference in percent HMGB-1 positive nuclei 7
days after completing LRT (P=0.0148). Lastly, 12 days after completion of LRT, NoRx (89.27 ±
2.758%) and 15Gy (95.71 ± 1.081%) did not significantly differ in mean percent positive nuclei
(P=0.0724). Despite the lack of significant difference at 2 days and 12 days after LRT, there was
a trend towards increased percent positive nuclei in 15Gy radiated tumors. These results propose
that radiation may increase tumor HMGB-1 expression.
Figure 23. Percent positive nuclei in tumor tissue without treatment or with 15Gy
radiation. Staining was performed on tissues A) 2 days, B) 7 days, or C) 12 days after
completing radiation treatment or on non-treated tissues of the same time point.

72
4.2.5 Percent HMGB-1 high, medium, and low positivity in mouse
tissues from untreated and 15Gy radiated mice
Percent positive cells were divided into high, medium and low intensity staining to model our
observations of tumor tissue in which there are varying degrees of positive staining intensity.
Staining of tissues for HMGB-1 was performed by immunohistochemistry and analyzed using
Aperio image analysis software.
Significant differences were observed in low intensity staining 2 days after LRT. Tissues treated
with LRT were significantly lower (7.323 ± 1.538 %) in percent low intensity stained cells than
untreated tissues (13.63 ± 1.958 %, P=0.0446). At 7 days after LRT, there was a significant
increase in high intensity stained cells in radiated tumors (65.28 ± 5.285%) compared with
untreated tumors (24.16 ± 8.434%, P=0.0061). High intensity staining is higher in radiated
tumors at 2 and 7 days after treatment (P=0.0750 and P= 0.0061 respectively) while low intensity
staining is lower in radiated tumors at the 2, 7, and 12 day time points (P=0.0446, P= 0.0534, and
P=0.0788 respectively).
Overall, low and medium intensity staining tends to be lower in radiated tumors while high
intensity staining tends to be higher in radiated tumors at 2 and 7 days after treatment. These
trends tend to be less clear at day 12 after treatment. This suggests radiation may have a role in
either increasing HMGB-1 expression of tumor cells, thus leading to higher levels of high
intensity staining. Alternatively, radiation may cause HMGB-1 release, leading to lower levels
of low and medium intensity staining.

73
Figure 24. Percent nuclei with low, medium, and high staining of tissues 2, 7, and 12 days
after LRT. 12 days after LRT, A) low, B) medium, and C) high positive nuclei are displayed. 7
days after the completion of LRT, D) low, E) medium, and F) high IHC staining is quantified.
Finally, 2 days after the completion of LRT, the mean percentage of G) low, H) medium, and I)
high positively stained nuclei is reported for untreated and 15Gy LRT treated mouse tissues.

74
4.2.6 Quantification of tumor infiltrating CD8+ T Cells in untreated
and radiated mouse tumor tissue
CD8+ T Cells are responsible for mediating antigen-specific tumor cell killing. Staining for
CD8+ T Cells was performed on untreated and radiated tumors to study the immunogenic effect
of radiation in terms of CD8+ T Cell recruitment. The recruitment of the CD8+ T Cell immune
response was measured using immunofluorescent staining of tumor tissue for CD3+CD8+ cells by
staining CD3 (488 nm) and CD8 markers (633nm). Untreated and 15Gy radiated tumor samples
from mice were taken 2, 7, and 12 days after LRT.
Quantification at each time point was performed by averaging the cell counts of 5 random fields.
The number of CD3+CD8+ cells counted 2 days after LRT was not found to be significantly
different between untreated and radiated tissues (18.40 ± 4.739 vs. 15.20 ± 2.818 cells, P=
0.5775). The number of CD3+CD8+ cells counted 7 days after LRT was significantly increased
in radiated tissue (32.20 ± 8.225 cells) compared to untreated tissue (10.60 ± 3.076 cells,
P=0.0398). 12 days after LRT, radiated tissue was similarly increased in the number of
CD3+CD8+ cells compared with untreated tissue (55.60 ± 13.07 vs. 1.600 ± 0.5099 cells,
P=0.0033). Overall, 2 days after LRT, untreated and radiated tumors have similar staining for
CD3 and CD8 markers However over time, CD3 and CD8 staining decreases in the untreated
group while CD3 and CD8 staining increases in the 15Gy radiated group. The peak staining for
CD3 and CD8 occurs 12 days after receiving radiation.
In summary, these findings indicate that local radiation treatment of tumors is able to stimulate
the recruitment of CD8+ T Cells to tumor tissue.

75
Figure 25. Immunofluorescent staining of untreated or 15Gy radiated moues tissue. A)
Immunofluorescent images of DAPI, CD3, and CD8 merged staining (405 blue, 488 green, and
633nm red respectively). B) Quantification of CD3+ CD8+ cells per x200 magnified field.

76
4.2.7 6E6 hybridoma anti-HMGB-1 antibody is able decrease the
signal of recombinant HMGB-1 in vitro by ELISA
The purpose of this experiment was to observe whether anti-HMGB-1 antibody is able to bind
recombinant HMGB-1 for later in vivo use. Anti-HMGB-1 Abs were generated from ascites
fluid after injection of the hybridoma cell line 6E6. Recombinant HMGB-1 (80 ng/mL) was
measured by ELISA. Recombinant HMGB-1 was also mixed with anti-HMGB-1 Ab generated
from the 6E6 hybridoma in a 1:1 ratio (80 ng/mL of recombinant HMGB-1 mixed with 80
ng/mL of anti-HMGB-1 Ab). The mixture was then plated on the ELISA kit to measure signal
abrogation and the ability of anti-HMGB-1 to bind recombinant HMGB-1.
The recombinant HMGB-1 and anti-HMGB-1 Ab mixture (6.835 ± 1.714 ng/mL) measured a
significantly lower concentration of HMGB-1 than recombinant HMGB-1 alone (74.83 ± 1.146
ng/mL, P=0.0009). The ability of anti-HMGB-1 to block the recombinant HMGB-1 signal in the
ELISA kit indicates that it is able to bind and neutralize HMGB-1.
Figure 26. Anti-HMGB-1 Ab abrogates recombinant HMGB-1 signal by ELISA. Wells
were measured in duplicate.

77
4.2.8 Anti-HMGB-1 Antibody combined with LRT increases tumor
growth compared to LRT alone
In this experiment, the goal was to determine the role of HMGB-1 in mediating an anti-tumoral
effect. Since HMGB-1 is hypothesized to mediate anti-tumoral immunity it was predicted that if
HMGB-1 was blocked during radiation the immune response would not be formed against the
tumor. AE-17-OVA mice either received no treatment (N=5), local radiation therapy (N=4), or
local radiation therapy in combination with anti-HMGB-1 antibody (N=5, 100µg per day on days
8-12 after tumor inoculation). Radiation was given in 3 5Gy fractions (1 fraction per day) for a
total of 15Gy. Tumor volume was measured using the longest length and width measurements
and calculated with the formula: Length x Width2 x (π/6).
NoRx showed the most rapid tumor growth and greatest tumor volumes. LRT+anti-HMGB-1
and LRT alone groups displayed lower tumor volumes than NoRx (Figure 29A). In observing
just LRT+anti-HMGB-1 and LRT groups, there was a trend towards greater tumor volume at
days 11 (P=0.0535) and a weaker trend at 14 (P=0.2669) in the LRT+HMGB-1 group. Tumor
growth is trending towards being greater in the LRT+anti-HMGB-1 over time. This trend is
greatest at days 38 (P=0.0769) and 40 (P=0.0441).
Overall, blockade of HMGB-1 during radiation treatment displayed a trend of greater tumor
volume over time compared to LRT alone. The results of this experiment suggest that HMGB-1
is an important beneficial aspect of radiation therapy as its neutralization during LRT increases
tumor growth.

78
Figure 27. LRT in combination with HMGB-1 demonstrates greater tumor growth
compared with LRT alone. A) Untreated mice (NoRx) demonstrate the greatest tumor growth
compared to LRT + anti-HMGB-1 and LRT groups. B) Mice receiving LRT + anti-HMGB-1
exhibit greater tumor volume than mice treated with LRT alone.

79
5 Discussion
The aims of this study were to study radiation treatment in MPM and factors that are crucial to
successful treatment. As radiation causes cell death, HMGB-1 is an important molecule released
from dying cells that is responsible for triggering inflammation. Thus 1) I investigated the
release of HMGB-1 as a result of radiation treatment in different MPM subtypes. 2) I examined
whether HMGB-1 release is related to the difference in survival observed in patients by staining
paraffin-embedded patient samples for HMGB-1. 3) I studied the CD8+ T cell immune response
generated by HMGB-1 release in radiation treatment.
The first set of experiments set out to characterize HMGB-1 release by radiation in vitro using
MPM cell lines. As there is a worse prognosis for MPM patients with biphasic and sarcomatoid
subtypes of disease compared with epithelioid disease, these experiments aimed to determine if
this difference relates to their response to radiation treatment. Based on the SMART protocol
and other groups using radiation therapy, epithelioid disease responds better than the other
subtypes. Indeed in this study the in vitro model mimicked what is observed in the clinic.
Radiation treatment resulted in more necrotic cell death and HMGB-1 release in epithelioid lines
than sarcomatoid lines.
In tackling the second goal, the HMGB-1 staining of patient samples was performed.
Chemotherapy has been shown to increase survival by a few months. However, the SMART
feasibility study demonstrated a significant increase in 3 year survival from 30% to 84% in
epithelioid patients. Thus, the HMGB-1 staining was compared between the two patient groups
and compared to survival. No significant difference was found in the intensity of staining of
either treatment group, suggesting a much more complex clinical picture

80
The last group of experiments focused on in vivo models to observe radiation treatment, HMGB-
1 release, and the potential for CD8+ T cell recruitment and tumor cell killing. Significant
differences were found in HMGB-1 release and CD8+ T cell recruitment in untreated and
radiated mice. This study indicates that HMGB-1 may be a major player in generating an anti-
cancer immune response after radiation therapy.
An in-depth view of each of the aims of this project is discussed below.
5.1 In vitro radiation-mediated HMGB-1 release
HMGB-1 Immunofluorescence Expression
The H226 epithelioid and H28 sarcomatoid cell lines were studied for differences in HMGB-1
immunofluorescent staining after no treatment or 5-75Gy radiation (Figure 15). Quantification
of fluorescent intensity revealed opposing patterns of each cell line. There was a trend towards
decreasing intensity across dose in the H226 line while the H28 cell line displayed an increase in
intensity. HMGB-1 immunofluorescence after radiation treatment has received limited study and
may identify behaviours that can be applied to clinical treatment options.
Epithelioid MPM is more susceptible to SMART treatment involving radiation as well as
chemotherapeutic treatment in the clinical setting. Furthermore, sarcomatoid disease is a
significant negative prognostic factor (Neumann, et al. 2013). Decreasing fluorescence may
indicate release of HMGB-1 into the extracellular environment. Liberated HMGB-1 could then
bind to APCs and trigger an anti-cancer immune response. The release of HMGB-1 in H226
may be responsible for the better outcomes seen in patients with epithelioid disease.

81
The greater fluorescent intensity in the H28 sarcomatoid cell line may represent a mechanism of
survival. In other studies, RNAi suppression of HMGB-1 renders leukemia and osteosarcoma
cell lines more susceptible to chemotherapy and radiation (Huang, et al. 2012, Liu, et al. 2011,
Livesey, et al. 2012). Conversely, pancreatic tumor cell lines have been shown to be more
resistant to cytotoxic therapies when HMGB-1 is overexpressed (Tang, et al. 2010). In the H28
cell line, increased expression of HMGB-1 may be a survival mechanism used to counteract the
radiation treatment. With its roles in non-specific DNA binding and DNA transcription
promotion, there remains many possibilities in terms of up-regulation of other pro-survival
proteins such as DNA repair proteins. Based on these findings HMGB-1 release was studied.
Cell death will also be discussed later.
HMGB-1 media release
It was hypothesized that the decreasing intensity in H226 epithelioid line was due to release of
HMGB-1 into the extracellular milieu. Complementing this, the increase in staining of the H28
sarcomatoid cell line was predicted to correlate with a lack of release of HMGB-1 into the
media; instead remaining intracellular. The next experiment employed ELISA to measure media
HMGB-1 concentration of the same cell lines 24 hours after no treatment or 5-75Gy γ-radiation
(Figure 16). We found a significant trend across the dosing range in which the H226 epithelioid
line released more HMGB-1 into the media than the H28 sarcomatoid cell line at every radiation
dose. While higher doses tended to increase HMGB-1 release in H226, H28 remained similar in
HMGB-1 release over the entire dosing range.
These findings complement the previous experiment. In both cell lines, higher HMGB-1
fluorescence corresponded with lower media release and vice versa. Even at high doses, H28
displayed high fluorescent intensity and low release of HMGB-1.

82
As was previously covered, HMGB-1 release occurs during late apoptotic or necrotic cell death
when the membrane of the cell becomes permeable (Degryse, et al. 2001, Falciola, et al. 1997,
Muller, et al. 2001). Thus the decreasing fluorescent intensity and increasing HMGB-1 release
observed in the H226 cell line may relate to increasing levels of necrotic cell death. Therefore
high intensity H28 staining and low HMGB-1 release could indicate a lack of cell death and the
sequestration of HMGB-1.
Radiation results in DNA damage which can either be repaired, resulting in cell survival, or
remain irreparable leading to cell death by apoptosis. Higher doses lead to necrosis and
uncontrolled cell death (Tesniere, et al. 2008). As observed in H226, the decrease in fluorescent
intensity with increasing radiation dose and the concomitant increase in HMGB-1 observed in
the media indicates greater levels of necrotic cell death. Conversely in H28, the high fluorescent
intensity and the lack of HMGB-1 media increase 24 hours after treatment indicates cell death is
not occurring and HMGB-1 is remaining intracellular. In these initial experiments H226 and
H28 differ in their response to radiation in terms of HMGB-1 release. However, we wanted to
observe the response to radiation over time. Furthermore, as these are just 2 cell lines, we
wanted to confirm that the differences observed were as a result of differing subtypes. Thus the
next experiment focused on observing the radiation response of more cell lines as well as the
examining the release of HMGB-1 over time.
HMGB-1 release over time by multiple cell lines
Patients undergoing SMART treatment often receive 25Gy hypofractionated radiation treatment
before surgery and surgery is typically performed 2-7 days after radiation. Hence there is a short

83
window in which the immune response is formed. To study whether the differences in response
to radiation by H226 and H28 were based on their subtype differences rather than just chance,
multiple cell lines were added. To better model the clinical dose, H226 and H2452 epithelioid
MPM cell lines and H28 and H2052 sarcomatoid lines were observed at various time points until
48 hours after receiving 25Gy radiation.
At the early time points of 1-4 hours after treatment, radiated lines tended to release similar
levels of HMGB-1 as their un-irradiated counterparts. However at 24 and 48 hours, the greatest
differences are found. At 24 hours, both radiated the epithelioid lines differed significantly in
HMGB-1 release compared to untreated epithelioid cells as well as untreated and radiated
sarcomatoid cells. Furthermore, radiated sarcomatoid lines release more HGMB-1 than
untreated sarcomatoid cells. A similar trend occurs at 48 hours after treatment. There is a
difference in HMGB-1 concentration in media of untreated and treated sarcomatoid cells. While
treated sarcomatoid cells release more HMGB-1 than untreated, they release significantly less
than radiated epithelioid cell lines. Radiated epithelioid lines release 5 to 8 fold more HMGB-1
than untreated epithelioid cells. This is starkly contrasted with radiated sarcomatoid releasing at
most 1.5 fold more HGMB-1 than untreated sarcomatoid cells.
These findings may be important to what occurs in vivo. Rapid growth and the lack of
vasculature support often leads to cell death by necrosis in the center of tumors. As discussed
above, necrosis leads to HMGB-1 release which may facilitate tumor survival through neo-
angiogenesis mediated by recruited pro-angiogenic macrophages (Yang, et al. 2014). While
chronic release is thought to promote angiogenesis, the sudden acute release of HMGB-1 by
cytotoxic therapies is able to trigger the immune response (Venneri, et al. 2007). It is therefore
possible that while radiated sarcomatoid cell lines release more HMGB-1 than untreated, the

84
difference is quite small compared to the potential 8 fold increase seen in epithelioid cells. It is
conceivable that in patients with sarcomatoid MPM, radiation triggers a slight increase in
HMGB-1 by cell death, but not enough to overcome the chronic HMGB-1 release that promotes
tumor growth. On the contrary, epithelioid MPM may release a large acute burst of HMGB-1, as
seen in these in vitro experiments, thus overcoming the pro-tumor microenvironment and
promoting a host anti-tumor response. These in vitro differences may provide insight into
clinical disease.
Interestingly, the findings of this set of experiments disagree with an article studying H28,
H2452 and H2052 basal HMGB-1 release. The authors claim that HMGB-1 release is highest in
H28 and H2052 while lower in H2452. However the differences were marginal and differently
from this study, cell culture conditions in their work did not include serum (Tabata, et al. 2013).
Murine epithelioid line AE-17-OVA HMGB-1 Release
AE-17-OVA cells were observed for release of HMGB-1 after LRT. As AE-17-OVA was later
used in in vivo experiments, the purpose of this experiment was to determine whether the in vitro
activity of murine epithelioid and human epithelioid cell lines were similar. The ultimate goal
was to ensure consistency in vitro so that later in vivo results could potentially be extrapolated to
human interventions.
AE-17-OVA cells were treated with 25Gy radiation and observed over 48 hours. Similar to
human epithelioid cell lines H226 and H2452, AE-17-OVA treated with radiation at 24 and 48
hours after treatment showed the greatest media release of HMGB-1 compared to untreated cells.
Despite the lack of significant difference, there is a trend towards higher HMGB-1 release 4

85
hours after treatment. At 24 and 48 hours there is a significantly higher media HMGB-1
concentration in radiated AE-17-OVA compared to untreated cells.
Similar to human cell lines, the hypothesis for this finding was that radiation resulted in necrotic
cell death, leading to an acute release of HMGB-1 in radiated cell cultures. Discussed above was
the hypothesis that human epithelioid MPM cell acute release of HMGB-1 results in an immune
response that translates into longer survival. The goal of the findings with AE-17-OVA, given
that they behave similarly to human epithelioid MPM in vitro, was to later investigate the
HMGB-1-mediated immune response in mice.
Human cell line In vitro cell death
The purpose of this aspect of study was to determine whether HMGB-1 release is correlated to
necrotic cell death. Epithelioid cell line H226 was found to undergo a significant increase in
necrotic cell death 48 hours after treatment compared to untreated cells. Conversely,
sarcomatoid H28 did not demonstrate an increase in necrotic cell death. Furthermore, apoptotic
cell death was consistent at all time points. Thus HMGB-1 release appears to correlate with
necrotic cell death as the maximum HMGB-1 concentration and maximal necrotic cell death
levels occur 48 hours after treatment.
An interesting trend to note is that HMGB-1 release tends to appear in the epithelioid cell lines
around 24 hours after radiation while necrotic cell death differences are not apparent until 48
hours after treatment. This may be a result of HMGB-1 being released by necrosis before
staining for necrosis by eFluor viability dye is possible. Additionally, there may be other forms
of cell death that are resulting in HMGB-1 release. Sustained autophagy and necroptosis were

86
not measured in this experiment but have been reported to result in HMGB-1 release (Tang, et al.
2010).
These in vitro experiments may build the initial parts of a larger picture. MPM patients
receiving radiation differ in their prognoses depending on the subtype of disease present.
Epithelioid patients tend to experience longer survival after treatment. The SMART feasibility
study did not demonstrate any survival benefit for biphasic patients. Other studies have had
similar findings in which biphasic and sarcomatoid MPM disease does not respond as well to
chemotherapy or radiation. What has been demonstrated here is that, in vitro, both epithelioid
cell lines behave similarly by responding to radiation, in terms of HMGB-1 release, while the
sarcomatoid subtypes both do not respond as strongly to radiation. This may represent an
inherent subtype similarity. Furthermore this may offer insight into clinical cases. Epithelioid
disease may be more susceptible to radiation treatment. Epithelioid disease may be more likely
to undergo uncontrolled necrosis and release HMGB-1 into the extracellular environment and
possibly the blood (more on serum release discussed later). This would enable patient antigen
presenting cell activation and an anti-cancer response. While some cell death does occur in
sarcomatoid cells, there may not be enough HMGB-1 released to trigger an immune response.
The difference in survival in the clinic may be dependent on the immune response in addition to
the immediate cytotoxic effects of treatment.
In the sarcomatoid type, lower levels of necrosis suggests a natural resistance to radiation and
lower levels of cell death compared to epithelioid disease. Thus sarcomatoid disease may
survive, keeping HMGB-1 sequestered intracellularly and hidden from the immune system. This
dissimilarity from epithelioid cell lines may be due to the cell lines’ differing abilities to resist
radiation treatment or to repair DNA damage. In other studies, sarcomatoid H28 was shown to

87
be more resistant to chemotherapy as a result of higher basal mRNA levels of DNA synthesis
enzymes thymidylate synthase (TS). Epithelioid H2052 was shown to be more sensitive to
chemotherapeutic agents as a result of low TS mRNA level (Giovannetti, et al. 2011).
Furthermore, high expression of TS has been attributed to radiation resistance in human uterine
cervical cancer cell lines (Saga, et al. 2002). This is one option among many potential pathways
that may render sarcomatoid cells resistant to radiation.
Murine AE-17-OVA in vitro Cell Death
With the goal of moving to animal studies, the murine epithelioid cell line AE-17-OVA was used
to determine if, similar to human epithelioid cell lines, HMGB-1 release was correlated to cell
death. After 25Gy radiation, apoptotic cell death and necrotic cell death were observed over
time. Differently from H226, AE-17-OVA demonstrated a trend towards increasing apoptotic
cell death in radiated cells 48 hours after treatment. However, the difference was not significant.
Furthermore, at 24 hours after treatment, radiated AE-17-OVA cells displayed a trend towards
more necrotic cell death than untreated cells while H226 did not. Similar to the human
epithelioid cell line H226, necrotic cell death was increased in radiated AE-17-OVA cells 48
hours after treatment compared to untreated cells.
While the trends are similar, AE-17-OVA and human epithelioid cell lines there are subtle
differences to note. AE-17-OVA appears to have a higher basal level of apoptotic cell death
which may represent a faster natural turnover rate as AE-17-OVA was observed to have a much
more rapid division time than all human MPM cell lines. Furthermore, the high division rate
may explain the higher percentage of necrotic cell death starting 24 hours after radiation as the
effects of DNA damage potentially manifest more quickly in rapidly dividing cells.

88
5.2 Paraffin-embedded Patient Samples.
This study of HMGB-1 was geared towards translating findings into clinical use. Therefore, the
next area of interest was the use of MPM patient samples. Paraffin-embedded epithelioid MPM
samples were stained by immunohistochemistry for HMGB-1 expression (Figure 21). The
hypothesis was that high expression after treatment would give rise to the potential for greater
HMGB-1 release. Extracellular HMGB-1 would trigger APC maturation and the recruitment of
the immune response. In studying the effect of radiation on HMGB-1 release, samples from
patients who did not receive treatment were compared to those who received SMART treatment.
We did not observe any significant difference in percent positive nuclei between the treatment
groups. Furthermore, no trends were observed when splitting the overall percent positive nuclei
into high, medium, and low staining intensities. The means of untreated and SMART treated
patient samples were relatively similar with a high variability among the SMART treated
samples.
A significant challenge to this area of the study was the timeline between when patients receive
surgery and radiation. Tumor samples were either removed by biopsy or through surgical
intervention such as EPP. Patients receiving SMART underwent radiation before having the
tumor removed by EPP. Since surgery occurred between 2 and 7 days after radiation, it became
difficult to compare staining; especially since the in vitro experiments demonstrated that levels
of HMGB-1 measured depend on the time point observed after treatment.
The clinical picture is also more complex than in vitro analysis. While HMGB-1 was a potential
measure for response to radiation in cell culture, tumors are extremely heterogeneous and may
contain multiple cell types that may respond differently to radiation; they may have tumor stem

89
cells and multiple lineages of differentiated cells. Furthermore, stromal cells and immune cells
are a large part of tumors that HMGB-1 staining alone can not differentiate between. The
heterogeneity of the tumor cells and tumors between patients may make it difficult to discern
HMGB-1’s by patient staining alone.
The composition of the tumors was also very different. Tumors differed in cellularity from very
cellular to virtually acellular. Furthermore, within subtypes, such as epithelioid and sarcomatoid,
there are a variety of other classifications that may influence tumor behaviour (Arrossi, et al.
2008). Percent positive nuclei was used to standardize samples that varied in cellularity.
However, it is possible that relatively acellular tumors act differently than tumors with less
connective tissue and more cells. This may be one of many factors that lead to the lack of trends
observed.
In the next set of experiments mouse models were used to ensure that treatment timelines and
tumor sizes were standardized. Furthermore, tumor pathology was consistent as tumors were
generated in mice by injecting a syngeneic tumor cell lines.
5.3 In vivo mouse model
HMGB-1 has a dual role. High HMGB-1 in the blood has been demonstrated as a negative
prognostic factor in cancer. In mesothelioma, HMGB-1 was been shown to be elevated in the
blood of patients with tumor and not in non-tumor bearing patients exposed to asbestos (Tabata,
et al. 2013). However, HMGB-1 is also implicated in generating an immune response by being
released from cells undergoing necrotic cell death. In this set of experiments, the aim was to
investigate the role of HMGB-1 in generating an immune response against radiation-treated

90
tumors. Mice were irradiated with a maximum tolerable dose of 3 doses of 5Gy radiation which
is similar to the SMART study in which 5 doses of 5Gy was used in humans.
Serum HMGB-1 Measurement
The first part of this experiment involved observing whether HMGB-1 is released into the blood
after radiating subcutaneous mouse tumors. Subcutaneous injection of the epithelioid AE-17-
OVA tumor cell line allowed for the measurement of the tumor and easy targeting of the
radiation. Mice were then sacrificed 2 days, 7 days, and 12 days after completion of LRT.
We found no significant difference in serum HMGB-1 between untreated and radiated mice at 7
and 12 days post LRT. However, 2 days after LRT, there was a significant increase in serum
HMGB-1 concentration in mice receiving 15Gy LRT to the tumor. Similar to the in vitro studies
of radiation induced cell death, the acute HMGB-1 release 2 days after treatment may have been
due to the direct cytotoxic effects of radiation treatment. Tumor cells would have then released
HMGB-1 into the local extracellular environment and eventually into the bloodstream.
At later time points, there is a greater concentration of HMGB-1 in the bloodstream of both
untreated and radiated mice. Previous research has noted the dual role of HMGB-1 in both
facilitating tumor growth as well as recruiting the immune system. While there are high levels of
HMGB-1 in the blood of untreated and treated mice, their impact may differ vastly. In untreated
mice, it is possible that HMGB-1 is being actively released by the tumor in order to enable ATP
production in neighbouring tumor cells and ultimately cell division and tumor progression. It’s
also possible that while untreated tumors did not visually display any necrosis during surgery, at
even the latest time point of this experiment, it remains possible that the anoxic center of the

91
tumor was undergoing necrotic cell death and releasing HMGB-1. The high levels in treated
mice may relate more to tumor cell death and the recruitment of the immune system.
Tumor volumes in untreated and radiated tumors may support these ideas. Tumor volume was
measured over the course of treatment (Figure 23). Tumor volume and HMGB-1 concentration
in tended to increase together; supporting the idea that untreated tumor HMGB-1 may be
necessary for the tumor’s life cycle. Conversely, radiated mice have increased serum HGMB-1
in the absence of tumor volume increase. In the radiated mice, tumors are actively shrinking or
stable in size, decreasing the likelihood of active secretion from the tumor.
Radiation has a large impact on tumor growth as untreated mice had tumors that were
significantly greater in volume than radiated tumors, especially at later time points (Figure 23).
High levels of HMGB-1 in radiated tumors may have been released by direct cytotoxicity of
radiation as well as due to immune system recruitment and its release by cells such as monocytes
and macrophages (Gardella, et al. 2002). In addition, CD8+ T Cell-mediated tumor killing may
also result in HMGB-1 release by killed tumor cells. CD8+ T Cell killing of tumor cells may
explain the lack of increasing tumor volume in radiated mice. The increase in CD8+ T cells may
support this hypothesis (discussed below).
This study demonstrates that the setting of observation is important for determining the pro or
anti-tumor effects of HMGB-1. Previous studies in gastric cancer, non-small cell lung cancer,
hepatocellular carcinoma, leukemia and MPM have correlated serum HMGB-1 to increasing
tumor volume and disease progression (Cheng, et al. 2008, Chung, et al. 2009, Jube, et al. 2012,
Kang, et al. 2007, Shang, et al. 2009, Sheng, et al. 2009). This study establishes that serum
HMGB-1 can increase in the absence of tumor volume increase; as in the case of mice receiving
LRT that have stable tumor growth, longer survival, and high serum HMGB-1 concentration.

92
T Cell Tumor Infiltration
There is a striking difference between the tissues of mice having remained untreated or receiving
15Gy radiation. The staining for CD3 and CD8 markers demonstrate T Cell recruitment at later
time points in radiated mice. Untreated mice present some T Cells at 2 days and less at 7 days.
By 12 days, there are few to no T Cells in the entire tissue section of untreated tumor.
To tackle the observation of disappearing T Cells in untreated mice there are several hypotheses.
The initial injection of tumor cells may have triggered the immune response, leading to similar
levels of T Cells at the first time point (2 days after LRT). However, immunoediting and an
immunosuppressive microenvironment may have led to the downregulation and apoptosis of
tumor infiltrating T Cells in untreated mice. In one study on multiple myeloma, CD8+ T Cells
were expanded ex vivo and infused into tumor-bearing mice. In mice with recurrent disease, the
authors identified specific loss of MHC mutations and loss of tumor associated antigen
expression; allowing tumor cells to evade specific immune destruction (Klippel, et al. 2014).
Furthermore immunosuppressive tumor environments involving infiltrating T-regulatory Cells,
M2 macrophages, PD-L1 expression, IL-10 secretion, CTLA-4 expression, and TGFβ expression
are known immune downregulators in MPM (Stevenson, et al. 2013, Tey, et al. 2006, Wong, et
al. 2014). Radiation is thought to change the stromal environment and remove some of the
immune inhibiting factors (Wong, et al. 2014). Thus radiation treatment and the release of pro-
inflammatory molecules such as HMGB-1 may serve to stimulate an infiltrating CD8+ T Cell
response by removing the immunosuppressive tumor microenvironment.

93
It was observed that radiated mice have fewer T Cells initially compared to untreated mice. At
early time points even untreated mice are able to recruit TILs. Literature suggests that the initial
lower number of CD8+ T Cells in radiated tumors is most likely due to radiation treatment to the
tumor which inevitably kills tumor as well as the infiltrated T Cells (Siva, et al. 2015). At 7 days
untreated mice have fewer T Cells than treated mice. Treated mice are more numerous in
infiltrating T Cells likely due to radiation necrosis releasing antigens and DAMPs such as
HMGB-1 that triggered the immune response. As 7 to 9 days are required for the adaptive T
Cell immune response to be active in mice, this may provide reasons as to why T Cells appeared
the most in the day 7 and 12 time points (Busch, et al. 1998).
Radiated tumor mice demonstrated an acute HMGB-1 release into the serum 2 days after LRT
that is higher than in untreated mice. In radiated tumors, the serum HMGB-1 level 7 and 12 days
after LRT is not significantly different from serum HMGB-1 levels of untreated mice. However,
radiated mice have much smaller tumors at the same time points as untreated mice. Furthermore,
radiated mice display significant CD8+ T Cell tumor infiltration which likely aids in slowing
tumor growth. There are several hypotheses for this finding. 1) The acute and gradual release of
HMGB-1 have different effects. Gradual HMGB-1 release by tumor cells during development
(as seen in untreated mice) may act to down regulate or de-sensitize the immune response.
Furthermore, tumors often have necrotic centers that release HMGB-1 and do not recruit the
immune response, providing another potential mechanism of immune de-sensitization. Radiation
treatment result in an acute release that may be necessary to overcome inhibitory or de-
sensitization effects of high HMGB-1 levels. The literature suggests that while HMGB-1 has
pro-tumor effects in growth and macrophage inhibition, pulsatile release by chemotherapy or
radiation therapy promotes antigen processing and tumor-specific T Cell responses (Apetoh, et
al. 2007, Campana, et al. 2008, Kuniyasu, et al. 2005).

94
2) The redox state of HMGB-1 may also differ based on the setting of release. Radiation
treatment may promote a pro-inflammatory redox state of HMGB-1. The redox state has been
reported as being an important factor in determining immunogenic or tolerogenic aspects of
HMGB-1. It has been reported that reduced HMGB-1 promotes anti-tumor inflammation while
oxidized HMGB-1 promotes immune tolerance. While necrotic cell death can lead to reduced
HMGB-1 release, redox state variability in cancer is a factor due to tolerogenic and
immunogenic cell death occurring at the same time and due to the tumor’s unique
microenvironment (Tang, et al. 2010). These may explain how a poor prognostic marker in the
blood may lead to very different results.
While it has been previously reported by Tabata et al. 2013 that high serum HMGB-1 is an
indicator of poor prognosis in MPM patients, the findings of this project may suggest otherwise
when measured after receiving radiation treatment. Differently from the previous study,
radiation treatment provides longer survival and is correlated with high serum HMGB-1.
Without treatment serum HMGB-1 may be a poor prognostic marker, however, this study
demonstrates for the first time that HMGB-1 release after treatment may be beneficial for
survival in MPM as it is correlated with longer survival and T Cell infiltration. While HMGB-1
serum after treatment has not been extensively studied, a study on esophageal squamous cell
carcinoma has shown that serum HMGB-1 levels increase in patients who received preoperative
chemotherapy and radiation treatment compared to untreated patients. This increase in HMGB-1
was also correlated with tumor antigen-specific T cell responses (Suzuki, et al. 2012). As our
study also demonstrates a rise in serum HMGB-1 post radiation being correlated with TILs, there
may be a role in detecting serum HMGB-1 as a post-treatment prognostic marker.

95
HMGB-1 Immunohistochemical Staining
HMGB-1 immunohistochemical staining was observed in order to understand how radiation
affects HMGB-1 expression in tumor tissue. HMGB-1 positive nuclei were significantly
increased in 15Gy radiated mice compared to untreated mice 7 days after completing LRT
(Figure 25). While, the other time points were not significant, there is a trend towards increased
percent positive nuclei in the radiated group at 2 and 12 days after LRT as well. It’s possible that
the greater percent HMGB-1 positive nuclei is indicative of radiation-induced HMGB-1
overexpression. Initial overexpression may be important for later release during cell death when
HMGB-1 can then be released. Indeed in breast cancer cells, estrogen treatment is able to
increase HMGB-1 mRNA levels which has been demonstrated to sensitize these cells to
chemotherapy treatment (He, et al. 2000).
Percent positive nuclei was broken down into intensity of staining (Figure 26) for each time
point in order to quantify the different staining levels seen in tumor tissue. Significant
differences were found between groups in low staining 2 days after LRT (Figure 26 G). There
was also a significant difference in medium and high staining 7 days after LRT (Figure 26 E and
F). While other groups did not demonstrate significant differences, there trends were quite
consistent and most were approaching significance. Overall, at days 2 and 7 after LRT, 15Gy
radiated tumors tended to display a lower percentage of low intensity stained nuclei. This was
similarly observed for medium staining. However, in high intensity staining, radiated tumors
tended to display a higher percentage of highly stained nuclei at the same time points.
HMGB-1 staining overall increased in radiated tumors compared to untreated tumors. This
increased staining may relate to an increasing in expression of the tumor cells which may be
necessary for HMGB-1 release. Supporting this idea, a study using glioblastoma cell lines found

96
that radiation resulted in increased nuclear HMGB-1 staining which was correlated with HMGB-
1 media release in vitro.
In comparing human and murine HMGB-1 immunohistochemical tumor staining, it was
predicted that positive HMGB-1 staining be higher in radiated tumors. While this was the case
for murine samples, patient staining did not display any trend in relation to treatment or survival.
Murine tumors were generated from an epithelioid cell line while all clinical samples selected
were of the epithelioid subtype and either received no treatment or radiation. Patient staining
may have differed from the staining observed from mouse tissues due to mouse tissues being
subcutaneously injected, differing from the pleural tissue in which MPM is found. As many
patients had previous asbestos exposure, they presumably developed MPM from long-term
inflammation as a result of asbestos fibers causing HMGB-1 release by damaging mesothelial
cells. This is then able to lead to TNF-α release by macrophages and many other pro-
transformation events. The long-term pro-inflammatory loop experienced by patients may not be
experienced by mice that have tumor cells injected directly. Furthermore, injecting asbestos in
order to develop tumors in mice was not feasible for the timeline of this project. Murine human
differences may be explained by the standardized radiation timeline as well as the consistency of
tumor pathology. Moreover, as injection of cells occurred on the same day, tumor size was very
similar until treatment. This differs from the clinic in which different stages of disease and
different volumes of tumors were irradiated for SMART treatment. Nevertheless, this
experiment may allow us to understand what occurs in radiation treatment of MPM.
HMGB-1 Blockade
LRT was shown to result in serum release of HMGB-1. HMGB-1 is released by necrotic cells
and is an important factor in DC maturation, leading to Ag presentation and an adaptive immune

97
response. LRT was also correlated with CD3+ CD8+ T Cell infiltration into the tumor. In order
to investigate the role of HMGB-1 in the radiation-induced immune response, tumor growth was
measured in untreated mice, radiated mice, and mice that were radiated and given anti-HMGB-1
Ab. As HMGB-1 is an important factor in generating an immune response, it was expected that
blocking HMGB-1 would result in lower survival than mice receiving LRT alone. It was
predicted that if HMGB-1 was blocked, there would be less HMGB-1 available to bind APCs
and the immune response would be less able to be generated.
Untreated mice displayed the fastest tumor growth. Both LRT and LRT+HMGB-1 Ab groups
displayed slower growth compared to no treatment. While, LRT and LRT+HMGB-1 Ab groups
did not demonstrate a significant difference at most time points, there was nevertheless a trend
towards LRT treated mice displaying the slowest tumor growth. At the latest time point, this
trend reached statistical significance. Thus it appears that HMGB-1 release during radiation
induced cell death plays a major role in inhibiting tumor growth.
A similar theme is emerging in that the scenario of HMGB-1 release is crucial for determining its
prognostic value. In animal models of MPM, blocking HMGB-1 was shown to slow tumor
growth and correlate with longer survival, thus supporting HMGB-1’s role as a pro-tumor agent
(Jube, et al. 2012). However our work demonstrates that during LRT, HMGB-1 may have an
effect on tumor size as blocking HMGB-1 during LRT results in faster tumor growth. In the
setting of LRT-induced cell death, HMGB-1 plays a role in mediating anti-tumor effects.
Similar to our study, higher serum HMGB-1 after chemotherapy has been correlated with longer
overall survival in esophageal squamous cell carcinoma patients (Suzuki, et al. 2012).
Furthermore in a tumor vaccination model involving a mouse colon cancer and sarcoma cell line,
injecting cells exposed to a lethal dose of radiation led to a protective memory response that

98
rejected a subsequent injection of live tumor cells. However, adding blocking HMGB-1 Ab to
the radiation exposed cells led to tumor growth upon subsequent live tumor cell injection
(Apetoh, et al. 2007). Similar to our findings, this study supports the idea that HMGB-1 is
necessary in forming an immune response against dying tumor cells.
Despite the HMGB-1 blockade, there was still slower tumor growth in LRT+HMGB-1 Ab
treated mice than in untreated mice. There are several possibilities for the tumor growth to still
be impaired.
1) The immediate effects of radiation may have resulted in cell death. Thus tumor growth was
slowed compared to untreated mice. The tumor would then take time to divide and become as
large as the tumors in the untreated group.
2) HMGB-1 may have been incompletely blocked in terms of concentration as well as over
time. HMGB-1 may have still been able to trigger an immune response if the Ab concentration
was not high enough to completely negate its effects. Incomplete blocking may have occurred
over time as there was a large spike in growth during and shortly after HMGB-1 Ab was
administered however, the tumors shrank extensively after HMGB-1 Ab was no longer being
given. The immune response may have been inhibited temporarily during the spike in tumor
growth and returned once the Ab was no longer present. As in vitro necrosis was high 48 hours
after radiation, anti-HMGB-1 Ab was given during LRT and until 48 hours after completion of
LRT. However, necrotic cell death may have been occurring extensively past 48 hours after
LRT in vivo; resulting in the observed decrease in tumor growth following the cessation of
HMGB-1 Ab.

99
3) There are likely multiple other DAMPs released during cell death that are important in
immune response generation. It’s possible that other DAMPs could function similarly or as
redundancy for HMGB-1, and thus blocking HMGB-1 may not completely abolish the
immunogenic effect of radiation. Indeed there are studies that support other molecules as being
crucial for the immunogenicity of dying cancer cells. One study found that exposure of
calreticulin, an “eat me” signal for DCs, was crucial for immunogenicity in a colon cancer cell
line. Dying cancer cells exposed to CRT-inducing anthracycline drugs were able to elicit
immune memory and the rejection of live tumor cells upon re-challenge. However, blocking
CRT eliminated this effect and live tumor cell rechallenge was able to grow (Obeid, et al. 2007).
It is likely that all of the above reasons contributed to lack of anti-HMGB-1 Ab removing the
anti-tumoral effects of radiation. Nevertheless, the increased tumor growth observed in mice
receiving anti-HMGB-1 Ab in combination with radiation compared to mice receiving radiation
alone suggests that HMGB-1 in the context of cytotoxic therapy is important in generating an
anti-tumoral effect.

100
6 Conclusions
The objective of this project was to understand how radiation in SMART treatment recruits the
immune system against malignant pleural mesothelioma and how this can be used to improve
treatments given to patients. Based on the findings of SMART treatment, we postulated that the
benefits seen in MPM treatment are largely due to the activation of the immune system. We
hypothesized that the differences in survival observed in each subtype of disease was the result
of different degrees immune avoidance by releasing more or less HMGB-1; lower patient
survival relating to low HMGB-1 release and low immune recruitment and longer patient
survival correlating with high HMGB-1 release and increase immune involvement.
First, in vitro radiation studies were used to understand HMGB-1 release. In vitro cell cultures
were radiated and the greatest release of HMGB-1 tended to be exhibited by epithelioid cell lines
around 24 to 48 hours after receiving radiation. Furthermore, the release of HMGB-1 tended to
correlate with necrotic cell death which similarly increased 48 hours after radiation treatment.
HMGB-1 release is likely related to the loss of membrane integrity in uncontrolled cell death.
The mouse cell line AE-17-OVA behaved similarly, allowing this effect to be modelled in the in
vivo mouse model. We therefore confirmed that γ-radiation is able to induce cell death and
subsequent HMGB-1 release in vitro in human and mouse cell lines.
MPM patient samples were used to correlate HMGB-1 expression with patient survival. While
untreated and SMART treated patients experienced a significant difference in survival, their
HMGB-1 staining remained similar. HMGB-1 immunohistochemical staining alone may not be
representative of the immunological complexity of clinical cases and therefore alone is unlikely
to be a predictor of patient outcome.

101
In vivo modelling of disease and treatment yielded some noteworthy data. Radiated tumors grew
more slowly than untreated tumors. Interestingly, radiation resulted in a release of HMGB-1
into the blood stream 2 days after the completion of local radiation therapy. Radiated tumors
also stained more intensely for HMGB-1 than untreated tumors by immunohistochemistry.
Furthermore, radiation treatment correlated with T-cell recruitment, suggesting an immunogenic
role for radiation by HMGB-1 release.
From a basic science standpoint, this project sheds light on the intricacy of the immune system
during radiation treatment. HMGB-1, a typically pro-inflammatory molecule, is shown to be
concentrated in the serum of tumor bearing untreated and treated mice. However, in untreated
mice, there is a distinct lack of tumor-infiltrating T Cells. Conversely T Cell numbers increase in
radiated mice. Despite the presence of HMGB-1 in the serum, there is a substantially different
effect between treated and untreated mice. HMGB-1 prominently plays a dual role in each
system and the setting in which it is released is likely very important.
The goal of this project was to translate findings into clinical practice. In vivo data suggests that
radiated tumors release HMGB-1 by necrosis and trigger an anti-tumor T Cell response. We
support the idea that radiation is effective outside of direct cytotoxic effects and that patients can
benefit from immune involvement. Radiation-induced immunogenicity warrants further
investigation and supports the potential benefit of immunomodulating therapy in combination
with high dose radiation therapy.

102
7 Limitations
A challenge in the in vitro cell death experiments of the study is the lack of observation after 48
hours. Further time points were observed however in a cell culture model, the cells quickly
overgrew the flask. Furthermore, measuring media ELISA meant that media could not be
replaced as it would change the HMGB-1 concentration. After 48 hours, cell death occurred in
all cell lines of both untreated and radiated samples due to overgrowth, build-up of metabolites,
and lack of nutrients. Furthermore, cell death staining was performed using Annexin V and
eFluor viability stain for assessing apoptosis and necrosis. However, there are many other forms
of cell death including senescence and necroptosis that may contribute to HMGB-1 release.
Nevertheless, the trends seen up to 48 hours were important foundations for the in vivo aspects of
this project.
A weakness of this study was that each mouse was sacrificed at the point of measurement of
HMGB-1, thus only one measurement exists per mouse. It may be beneficial to be able to draw
serum samples over time to measure how serum HMGB-1 changes over time in each mouse.
Limited mouse blood volume and the high volume required for the ELISA assay necessitated
only one blood draw per mouse. Thus larger animals may be necessary to draw blood over time
to correlate individual serum HMGB-1 and tumor size.
In an effort to translate basic science findings into the clinic, we believe that a more clinically
relevant mouse model could be used. Subcutaneously injected MPM cells was used in order to
easily measure tumor growth and target radiation. However, in observing AE-17-OVA in vivo,
the tumor remains encapsulated and does not spread to other areas of the body. Furthermore, in
other studies from our lab (unpublished data), we have found that aggressive surgery is often

103
curative. These features differ from clinical MPM which may spread into the chest wall
contralaterally. In addition surgery alone in humans is not usually curative and does not offer
any increase in patient survival. While posing significant challenges, an intrapleural tumor
model may better display the pertinent aspects of disease in patients.
An inherent weakness in this study is the use of cell lines to simulate MPM. Re-implantation of
the cell line AE-17-OVA into syngeneic mice may not yield the same microenvironment as
spontaneously developed MPM. Where asbestos requires much time and inflammation to
develop a tumor, tumor implantation is immediate and may lead to a less relevant
microenvironment. Asbestos leads to uncontrolled inflammation which slowly selects for non-
immunogenic cancer cell clones. Conversely, injection of one million tumor cells may
overwhelm the immune system by sheer cell number rather than immune evasion. Supporting
this idea is the information that AE-17-OVA is transfected with the chicken ovalbumin peptide
which has been reported as being an immunogenic antigen that can be rejected by some hosts.
Our lab and others have found that injecting high cell numbers supports tumor formation in
almost 100% of hosts while the rejection rate tends to increase when injecting lower cell
numbers.

104
8 Future Directions
An important future experiment involves the analysis of cytoplasmic HMGB-1 in tissue stains.
While nuclear staining may be important for understanding expression levels, HMGB-1 passes
through the cytoplasm before being released to the extracellular milieu during necrotic cell death.
While the Aperio algorithm did not offer an accurate means of quantifying cytoplasmic staining,
it would be beneficial to work with pathologists in order to assess the role of cytoplasmic
HMGB-1 staining in radiation-induced immunogenicity in MPM.
One significant experiment in this project was the generation of hybridoma anti-HMGB-1 Ab.
Sufficient Ab was generated for the initial blocking experiment with three groups: 1) No
treatment, 2) Radiation and anti-HMGB-1, and 3) Radiation only. However, insufficient Ab was
generated for the inclusion of a necessary control. Thus, we would like to explore future
experiments in which an additional group is included: 4) anti-HMGB-1 Ab alone. This would
ensure that the observed trend is not due to just anti-HMGB-1 but rather a combined effect of
LRT and anti-HMGB-1.
To compare epithelioid and sarcomatoid subtypes further, in vivo comparisons could be made
between AE-17-OVA and a mouse sarcomatoid cell line. If radiating sarcomatoid tumors results
in less HMGB-1 release than radiating epithelioid AE-17-OVA tumors, this could explain the
lower survival observed in patients with sarcomatoid MPM. This would be able to confirm the
hypothesis that the sarcomatoid MPM patients release less HMGB-1 in response to radiation and
therefore do not recruit the immune response as intensely as epithelioid disease.
In vivo HMGB-1 release was predicted to occur as a result of necrosis of tumor tissue. To
confirm the mode of cell death that relates to HMGB-1 release, tumor tissue could be stained for

105
apoptosis and necrosis markers as well as HMGB-1 by immunofluorescence. Co-localized
staining of necrosis and HMGB-1 markers would indicate that HMGB-1 release by radiation is
indeed mediated by necrosis. Furthering this, an important area of future study is the
measurement of multiple other forms of cell death that may contribute to HMGB-1 release.
Starting with in vitro studies, this could be extended to animal tissue staining.
The cell line AE-17-OVA was used as the subcutaneous MPM model. The transfection of this
cell line with OVA chicken peptide gives rise to the potential for tracking tumor specific
responses against OVA. As OVA tetramers have been used in our lab, a future experiment could
involve staining tumor infiltrating lymphocytes with OVA tetramer, thus identifying a whether
the infiltrating T Cells are indeed tumor specific.
To achieve a more clinically relevant model, it would be beneficial to establish an intrapleural
model of MPM. As discussed above, there are some weaknesses with using a subcutaneous
model of MPM. Mapping growth may be addressed using MRI, or fluorescently labelled tumor
cells that could be imaged in vivo. Similar technologies could also be used to target treatment to
the tumor.
In further modelling human MPM, future studies could involve mouse MPM developed from
asbestos exposure. Spontaneously developed tumors may be less immunogenic than cell lines
due to slow selection of cancer cell clones that escape the immune system, potentially better
modelling clinical cases of MPM. Spontaneous development is possible as asbestos tumor
development in mice has been extensively demonstrated. However, long incubation period and
low number of mice that develop tumors after asbestos exposure still poses a potential challenge.

106
Studying HMGB-1 levels in patient blood is a promising area of future study. While it has been
reported that MPM patients have higher serum HMGB-1 than individuals exposed to asbestos
without a tumor, we have demonstrated that HMGB-1 serum levels can increase as a result of
radiation. Studying HMGB-1 in serum samples of patients before and after radiation could
provide more elucidation of HMGB-1’s role in development and treatment in the context of
MPM. HMGB-1 levels after treatment could be an indicator of successful treatment.
New DAMPs are constantly being discovered and it is very likely that many DAMPs play a role
in radiation-induced immunogenicity. Our earlier studies (unpublished data) found differences
in calreticulin expression between radiated and untreated cells. Calreticulin and ATP have been
highly studied as danger signals and further study in MPM may yield novel interventions.
DAMPs have been suggested to work as redundancy as well as in concert with other DAMPs.
Therefore, studying other DAMPs in addition to HMGB-1 may allow for interventions that
amplify the immunogenic effect of radiation treatment for MPM.
SMART treatment in patients involves radiation of the tumor mass before surgical resection.
Previous methods involved performing surgery followed by radiation. The hypothesis behind
SMART’s success in lengthening patient survival was that the tumor was irradiated first,
allowing it to release danger signals an antigens before removal. Conversely, previous methods
would remove the tumor and subsequent radiation would not serve to activate the immune
system as there would be little danger signals or tumor antigen release. Our lab has previously
established a SMART mouse model which finds greater survival in mice treated with SMART in
comparison to surgery followed by radiation. To further understand if the increase in survival in
patients is immune-mediated, HMGB-1 serum levels and tumor infiltrating lymphocytes could
be measured after the completion of each treatment. Furthermore, paraffin-embedded patient

107
samples could be stained by immunohistochemistry or immunofluorescence for CD8+ T Cells
whose presence would suggest specific immune-mediated tumor cell killing. SMART treated
and surgery then radiation treated patients could be compared.

108
References
1 Abraham, E., Arcaroli, J., Carmody, A., Wang, H.and Tracey, K. J. HMG-1 as a mediator
of acute lung inflammation. J Immunol 165, 2950-2954 (2000).
2 Abutaily, A. S., Addis, B. J.and Roche, W. R. Immunohistochemistry in the distinction
between malignant mesothelioma and pulmonary adenocarcinoma: a critical evaluation
of new antibodies. J Clin Pathol 55, 662-668 (2002).
3 Adams, V. I., Unni, K. K., Muhm, J. R., Jett, J. R., Ilstrup, D. M.and Bernatz, P. E.
Diffuse malignant mesothelioma of pleura. Diagnosis and survival in 92 cases. Cancer
58, 1540-1551 (1986).
4 Agresti, A. and Bianchi, M. E. HMGB proteins and gene expression. Curr Opin Genet
Dev 13, 170-178 (2003).
5 Agudo, A., Gonzalez, C. A., Bleda, M. J., Ramirez, J., Hernandez, S., Lopez, F., Calleja,
A., Panades, R., Turuguet, D., Escolar, A., Beltran, M.and Gonzalez-Moya, J. E.
Occupation and risk of malignant pleural mesothelioma: A case-control study in Spain.
Am J Ind Med 37, 159-168 (2000).
6 Al-Izzi, M., Thurlow, N. P.and Corrin, B. Pleural mesothelioma of connective tissue
type, localized fibrous tumour of the pleura, and reactive submesothelial hyperplasia. An
immunohistochemical comparison. J Pathol 158, 41-44 (1989).
7 Andersson, U., Wang, H., Palmblad, K., Aveberger, A. C., Bloom, O., Erlandsson-Harris,
H., Janson, A., Kokkola, R., Zhang, M., Yang, H.and Tracey, K. J. High mobility group 1
protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J
Exp Med 192, 565-570 (2000).
8 Anraku, M., Cunningham, K. S., Yun, Z., Tsao, M. S., Zhang, L., Keshavjee, S.,
Johnston, M. R.and de Perrot, M. Impact of tumor-infiltrating T cells on survival in
patients with malignant pleural mesothelioma. J Thorac Cardiovasc Surg 135, 823-829
(2008).
9 Antman, K., Shemin, R., Ryan, L., Klegar, K., Osteen, R., Herman, T., Lederman, G.and
Corson, J. Malignant mesothelioma: prognostic variables in a registry of 180 patients, the
Dana-Farber Cancer Institute and Brigham and Women's Hospital experience over two
decades, 1965-1985. J Clin Oncol 6, 147-153 (1988).
10 Antoniades, J., Brady, L. W.and Lightfoot, D. A. Lymphangiographic demonstration of
the abscopal effect in patients with malignant lymphomas. International Journal of
Radiation Oncology • Biology • Physics 2, 141-147.
11 Apetoh, L., Ghiringhelli, F., Tesniere, A., Obeid, M., Ortiz, C., Criollo, A., Mignot, G.,
Maiuri, M. C., Ullrich, E., Saulnier, P., Yang, H., Amigorena, S., Ryffel, B., Barrat, F. J.,
Saftig, P., Levi, F., Lidereau, R., Nogues, C., Mira, J. P., Chompret, A., Joulin, V.,
Clavel-Chapelon, F., Bourhis, J., Andre, F., Delaloge, S., Tursz, T., Kroemer, G.and

109
Zitvogel, L. Toll-like receptor 4-dependent contribution of the immune system to
anticancer chemotherapy and radiotherapy. Nat Med 13, 1050-1059 (2007).
12 Apetoh, L., Ghiringhelli, F., Tesniere, A., Criollo, A., Ortiz, C., Lidereau, R., Mariette,
C., Chaput, N., Mira, J. P., Delaloge, S., Andre, F., Tursz, T., Kroemer, G.and Zitvogel,
L. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer
chemotherapy and radiotherapy. Immunol Rev 220, 47-59 (2007).
13 Ardizzoni, A., Pennucci, M. C., Castagneto, B., Mariani, G. L., Cinquegrana, A., Magri,
D., Verna, A., Salvati, F.and Rosso, R. Recombinant interferon alpha-2b in the treatment
of diffuse malignant pleural mesothelioma. Am J Clin Oncol 17, 80-82 (1994).
14 Armstrong, B. K., Musk, A. W., Baker, J. E., Hunt, J. M., Newall, C. C., Henzell, H. R.,
Blunsdon, B. S., Clarke-Hundley, M. D., Woodward, S. D.and Hobbs, M. S.
Epidemiology of malignant mesothelioma in Western Australia. Med J Aust 141, 86-88
(1984).
15 Arrossi, A. V., Lin, E., Rice, D.and Moran, C. A. Histologic assessment and prognostic
factors of malignant pleural mesothelioma treated with extrapleural pneumonectomy. Am
J Clin Pathol 130, 754-764 (2008).
16 Attanoos, R. L., Dojcinov, S. D., Webb, R.and Gibbs, A. R. Anti-mesothelial markers in
sarcomatoid mesothelioma and other spindle cell neoplasms. Histopathology 37, 224-231
(2000).
17 Aziz, T., Jilaihawi, A.and Prakash, D. The management of malignant pleural
mesothelioma; single centre experience in 10 years. Eur J Cardiothorac Surg 22, 298-
305 (2002).
18 Babcock, T. L., Powell, D. H.and Bothwell, R. S. Radiation-induced peritoneal
mesothelioma. J Surg Oncol 8, 369-372 (1976).
19 Bachmayr-Heyda, A., Aust, S., Heinze, G., Polterauer, S., Grimm, C., Braicu, E. I.,
Sehouli, J., Lambrechts, S., Vergote, I., Mahner, S., Pils, D., Schuster, E., Thalhammer,
T., Horvat, R., Denkert, C., Zeillinger, R.and Castillo-Tong, D. C. Prognostic impact of
tumor infiltrating CD8+ T cells in association with cell proliferation in ovarian cancer
patients--a study of the OVCAD consortium. BMC Cancer 13, 422 (2013).
20 Baldini, E. H., Recht, A., Strauss, G. M., DeCamp, M. M., Jr., Swanson, S. J., Liptay, M.
J., Mentzer, S. J.and Sugarbaker, D. J. Patterns of failure after trimodality therapy for
malignant pleural mesothelioma. Ann Thorac Surg 63, 334-338 (1997).
21 Balermpas, P., Rödel, F., Weiss, C., Rödel, C.and Fokas, E. Tumor-infiltrating
lymphocytes favor the response to chemoradiotherapy of head and neck cancer.
Oncoimmunology 3 (2014).
22 Banerjee, S. and Kundu, T. K. The acidic C-terminal domain and A-box of HMGB-1
regulates p53-mediated transcription. Nucleic Acids Res 31, 3236-3247 (2003).

110
23 Barbanti-Brodano, G., Sabbioni, S., Martini, F., Negrini, M., Corallini, A.and Tognon,
M. Simian virus 40 infection in humans and association with human diseases: results and
hypotheses. Virology 318, 1-9 (2004).
24 Barkauskaite, V., Ek, M., Popovic, K., Harris, H. E., Wahren-Herlenius, M.and Nyberg,
F. Translocation of the novel cytokine HMGB1 to the cytoplasm and extracellular space
coincides with the peak of clinical activity in experimentally UV-induced lesions of
cutaneous lupus erythematosus. Lupus 16, 794-802 (2007).
25 Batirel, H. F., Metintas, M., Caglar, H. B., Yildizeli, B., Lacin, T., Bostanci, K., Akgul,
A. G., Evman, S.and Yuksel, M. Trimodality treatment of malignant pleural
mesothelioma. J Thorac Oncol 3, 499-504 (2008).
26 Baxevanis, C. N., Perez, S. A.and Papamichail, M. Cancer immunotherapy. Critical
Reviews in Clinical Laboratory Sciences 46, 167-189 (2009).
27 Bianchi, C., Giarelli, L., Grandi, G., Brollo, A., Ramani, L.and Zuch, C. Latency periods
in asbestos-related mesothelioma of the pleura. Eur J Cancer Prev 6, 162-166 (1997).
28 Bianchi, M. E. and Manfredi, A. A. High-mobility group box 1 (HMGB1) protein at the
crossroads between innate and adaptive immunity. Immunol Rev 220, 35-46 (2007).
29 Boffetta, P. Health effects of asbestos exposure in humans: a quantitative assessment.
Med Lav 89, 471-480 (1998).
30 Bonaldi, T., Talamo, F., Scaffidi, P., Ferrera, D., Porto, A., Bachi, A., Rubartelli, A.,
Agresti, A.and Bianchi, M. E. Monocytic cells hyperacetylate chromatin protein HMGB1
to redirect it towards secretion. EMBO J 22, 5551-5560 (2003).
31 Braun, D. C. and Truan, T. D. An epidemiological study of lung cancer in asbestos
miners. AMA Arch Ind Health 17, 634-653 (1958).
32 Brenner, J., Sordillo, P. P., Magill, G. B.and Golbey, R. B. Malignant mesothelioma of
the pleura: review of 123 patients. Cancer 49, 2431-2435 (1982).
33 Brown, J. W., Kristensen, K. A.and Monroe, L. S. Peritoneal mesothelioma following
pneumoperitoneum maintained for twelve years. Report of a case. Am J Dig Dis 13, 830-
835 (1968).
34 Busch, D. H., Pilip, I. M., Vijh, S.and Pamer, E. G. Coordinate regulation of complex T
cell populations responding to bacterial infection. Immunity 8, 353-362 (1998).
35 Bustin, M., Lehn, D. A.and Landsman, D. Structural features of the HMG chromosomal
proteins and their genes. Biochim Biophys Acta 1049, 231-243 (1990).
36 Bustin, M. Regulation of DNA-Dependent Activities by the Functional Motifs of the
High-Mobility-Group Chromosomal Proteins. Molecular and Cellular Biology 19, 5237-
5246 (1999).

111
37 Calabro, L., Morra, A., Fonsatti, E., Cutaia, O., Amato, G., Giannarelli, D., Di Giacomo,
A. M., Danielli, R., Altomonte, M., Mutti, L.and Maio, M. Tremelimumab for patients
with chemotherapy-resistant advanced malignant mesothelioma: an open-label, single-
arm, phase 2 trial. Lancet Oncol 14, 1104-1111 (2013).
38 Calogero, S., Grassi, F., Aguzzi, A., Voigtlander, T., Ferrier, P., Ferrari, S.and Bianchi,
M. E. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes
lethal hypoglycaemia in newborn mice. Nat Genet 22, 276-280 (1999).
39 Campana, L., Bosurgi, L.and Rovere-Querini, P. HMGB1: a two-headed signal
regulating tumor progression and immunity. Curr Opin Immunol 20, 518-523 (2008).
40 Carbone, M., Rizzo, P., Grimley, P. M., Procopio, A., Mew, D. J., Shridhar, V., de
Bartolomeis, A., Esposito, V., Giuliano, M. T., Steinberg, S. M., Levine, A. S., Giordano,
A.and Pass, H. I. Simian virus-40 large-T antigen binds p53 in human mesotheliomas.
Nat Med 3, 908-912 (1997).
41 Carreno, B. M. and Collins, M. The B7 family of ligands and its receptors: new
pathways for costimulation and inhibition of immune responses. Annu Rev Immunol 20,
29-53 (2002).
42 Cedres, S., Ponce-Aix, S., Zugazagoitia, J., Sansano, I., Enguita, A., Navarro-Mendivil,
A., Martinez-Marti, A., Martinez, P.and Felip, E. Analysis of Expression of Programmed
Cell Death 1 Ligand 1 (PD-L1) in Malignant Pleural Mesothelioma (MPM). PLoS One
10, e0121071 (2015).
43 Ceresoli, G. L., Locati, L. D., Ferreri, A. J., Cozzarini, C., Passoni, P., Melloni, G.,
Zannini, P., Bolognesi, A.and Villa, E. Therapeutic outcome according to histologic
subtype in 121 patients with malignant pleural mesothelioma. Lung Cancer 34, 279-287
(2001).
44 Ceresoli, G. L., Zucali, P. A., De Vincenzo, F., Gianoncelli, L., Simonelli, M., Lorenzi,
E., Ripa, C., Giordano, L.and Santoro, A. Retreatment with pemetrexed-based
chemotherapy in patients with malignant pleural mesothelioma. Lung Cancer 72, 73-77
(2011).
45 Chahinian, A. P., Pajak, T. F., Holland, J. F., Norton, L., Ambinder, R. M.and Mandel, E.
M. Diffuse malignant mesothelioma. Prospective evaluation of 69 patients. Ann Intern
Med 96, 746-755 (1982).
46 Chang, T. H., Ray, F. A., Thompson, D. A.and Schlegel, R. Disregulation of mitotic
checkpoints and regulatory proteins following acute expression of SV40 large T antigen
in diploid human cells. Oncogene 14, 2383-2393 (1997).
47 Cheng, B. Q., Jia, C. Q., Liu, C. T., Lu, X. F., Zhong, N., Zhang, Z. L., Fan, W.and Li, Y.
Q. Serum high mobility group box chromosomal protein 1 is associated with
clinicopathologic features in patients with hepatocellular carcinoma. Dig Liver Dis 40,
446-452 (2008).

112
48 Cho, B. C., Feld, R., Leighl, N., Opitz, I., Anraku, M., Tsao, M. S., Hwang, D. M., Hope,
A.and de Perrot, M. A feasibility study evaluating Surgery for Mesothelioma After
Radiation Therapy: the "SMART" approach for resectable malignant pleural
mesothelioma. J Thorac Oncol 9, 397-402 (2014).
49 Choi, Y. R., Kim, H., Kang, H. J., Kim, N. G., Kim, J. J., Park, K. S., Paik, Y. K., Kim,
H. O.and Kim, H. Overexpression of high mobility group box 1 in gastrointestinal
stromal tumors with KIT mutation. Cancer Res 63, 2188-2193 (2003).
50 Chow, M. T., Tschopp, J., Moller, A.and Smyth, M. J. NLRP3 promotes inflammation-
induced skin cancer but is dispensable for asbestos-induced mesothelioma. Immunol Cell
Biol 90, 983-986 (2012).
51 Chu, P. G. and Weiss, L. M. Expression of Cytokeratin 5//6 in Epithelial Neoplasms: An
Immunohistochemical Study of 509 Cases. Mod Pathol 15, 6-10 (0000).
52 Chung, H. W., Lee, S. G., Kim, H., Hong, D. J., Chung, J. B., Stroncek, D.and Lim, J. B.
Serum high mobility group box-1 (HMGB1) is closely associated with the clinical and
pathologic features of gastric cancer. J Transl Med 7, 38 (2009).
53 Cicala, C., Pompetti, F.and Carbone, M. SV40 induces mesotheliomas in hamsters. Am J
Pathol 142, 1524-1533 (1993).
54 Clover, J., Oates, J.and Edwards, C. Anti-cytokeratin 5/6: a positive marker for
epithelioid mesothelioma. Histopathology 31, 140-143 (1997).
55 Cotter, S. E., Dunn, G. P., Collins, K. M., Sahni, D., Zukotynski, K. A., Hansen, J. L.,
O'Farrell, D. A., Ng, A. K., Devlin, P. M.and Wang, L. C. Abscopal effect in a patient
with metastatic Merkel cell carcinoma following radiation therapy: potential role of
induced antitumor immunity. Arch Dermatol 147, 870-872 (2011).
56 Cury, P. M., Butcher, D. N., Fisher, C., Corrin, B.and Nicholson, A. G. Value of the
mesothelium-associated antibodies thrombomodulin, cytokeratin 5/6, calretinin, and
CD44H in distinguishing epithelioid pleural mesothelioma from adenocarcinoma
metastatic to the pleura. Mod Pathol 13, 107-112 (2000).
57 Dahlgren, S. Effects of locally deposited colloidal thorium dioxide. Ann N Y Acad Sci
145, 786-790 (1967).
58 Das, D., Peterson, R. C.and Scovell, W. M. High mobility group B proteins facilitate
strong estrogen receptor binding to classical and half-site estrogen response elements and
relax binding selectivity. Mol Endocrinol 18, 2616-2632 (2004).
59 De Luca, A., Baldi, A., Esposito, V., Howard, C. M., Bagella, L., Rizzo, P., Caputi, M.,
Pass, H. I., Giordano, G. G., Baldi, F., Carbone, M.and Giordano, A. The retinoblastoma
gene family pRb/p105, p107, pRb2/p130 and simian virus-40 large T-antigen in human
mesotheliomas. Nat Med 3, 913-916 (1997).

113
60 De Pangher Manzini, V., Brollo, A., Franceschi, S., De Matthaeis, M., Talamini, R.and
Bianchi, C. Prognostic factors of malignant mesothelioma of the pleura. Cancer 72, 410-
417 (1993).
61 Deckert, P. M. Current constructs and targets in clinical development for antibody-based
cancer therapy. Curr Drug Targets 10, 158-175 (2009).
62 Degenhardt, K., Mathew, R., Beaudoin, B., Bray, K., Anderson, D., Chen, G.,
Mukherjee, C., Shi, Y., Gelinas, C., Fan, Y., Nelson, D. A., Jin, S.and White, E.
Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and
tumorigenesis. Cancer Cell 10, 51-64 (2006).
63 Degryse, B., Bonaldi, T., Scaffidi, P., Muller, S., Resnati, M., Sanvito, F., Arrigoni,
G.and Bianchi, M. E. The high mobility group (HMG) boxes of the nuclear protein
HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J
Cell Biol 152, 1197-1206 (2001).
64 Dejmek, A. and Hjerpe, A. Immunohistochemical reactivity in mesothelioma and
adenocarcinoma: a stepwise logistic regression analysis. Apmis 102, 255-264 (1994).
65 Dinarello, C. A. Immunological and inflammatory functions of the interleukin-1 family.
Annu Rev Immunol 27, 519-550 (2009).
66 Doglioni, C., Dei Tos, A. P., Laurino, L., Iuzzolino, P., Chiarelli, C., Celio, M. R.and
Viale, G. Calretinin: a novel immunocytochemical marker for mesothelioma. Am J Surg
Pathol 20, 1037-1046 (1996).
67 Doll, R. Mortality from lung cancer in asbestos workers 1955. Br J Ind Med 50, 485-490
(1993).
68 Dowell, J. E., Dunphy, F. R., Taub, R. N., Gerber, D. E., Ngov, L., Yan, J., Xie, Y.and
Kindler, H. L. A multicenter phase II study of cisplatin, pemetrexed, and bevacizumab in
patients with advanced malignant mesothelioma. Lung Cancer 77, 567-571 (2012).
69 Dumitriu, I. E., Baruah, P., Valentinis, B., Voll, R. E., Herrmann, M., Nawroth, P. P.,
Arnold, B., Bianchi, M. E., Manfredi, A. A.and Rovere-Querini, P. Release of high
mobility group box 1 by dendritic cells controls T cell activation via the receptor for
advanced glycation end products. J Immunol 174, 7506-7515 (2005).
70 El Marzouk, S., Gahattamaneni, R., Joshi, S. R.and Scovell, W. M. The plasticity of
estrogen receptor-DNA complexes: binding affinity and specificity of estrogen receptors
to estrogen response element half-sites separated by variant spacers. J Steroid Biochem
Mol Biol 110, 186-195 (2008).
71 Elliott, M. R., Chekeni, F. B., Trampont, P. C., Lazarowski, E. R., Kadl, A., Walk, S. F.,
Park, D., Woodson, R. I., Ostankovich, M., Sharma, P., Lysiak, J. J., Harden, T. K.,
Leitinger, N.and Ravichandran, K. S. Nucleotides released by apoptotic cells act as a
find-me signal to promote phagocytic clearance. Nature 461, 282-286 (2009).

114
72 Elmes, P. C. and Simpson, J. C. The clinical aspects of mesothelioma. Q J Med 45, 427-
449 (1976).
73 Engels, E. A., Katki, H. A., Nielsen, N. M., Winther, J. F., Hjalgrim, H., Gjerris, F.,
Rosenberg, P. S.and Frisch, M. Cancer incidence in Denmark following exposure to
poliovirus vaccine contaminated with simian virus 40. J Natl Cancer Inst 95, 532-539
(2003).
74 Falciola, L., Spada, F., Calogero, S., Langst, G., Voit, R., Grummt, I.and Bianchi, M. E.
High mobility group 1 protein is not stably associated with the chromosomes of somatic
cells. J Cell Biol 137, 19-26 (1997).
75 Ferrari, S., Finelli, P., Rocchi, M.and Bianchi, M. E. The active gene that encodes human
high mobility group 1 protein (HMG1) contains introns and maps to chromosome 13.
Genomics 35, 367-371 (1996).
76 Flores, R. M., Krug, L. M., Rosenzweig, K. E., Venkatraman, E., Vincent, A., Heelan, R.,
Akhurst, T.and Rusch, V. W. Induction chemotherapy, extrapleural pneumonectomy, and
postoperative high-dose radiotherapy for locally advanced malignant pleural
mesothelioma: a phase II trial. J Thorac Oncol 1, 289-295 (2006).
77 Flores, R. M., Pass, H. I., Seshan, V. E., Dycoco, J., Zakowski, M., Carbone, M., Bains,
M. S.and Rusch, V. W. Extrapleural pneumonectomy versus pleurectomy/decortication in
the surgical management of malignant pleural mesothelioma: results in 663 patients. J
Thorac Cardiovasc Surg 135, 620-626, 626.e621-623 (2008).
78 Fourcade, J., Sun, Z., Benallaoua, M., Guillaume, P., Luescher, I. F., Sander, C.,
Kirkwood, J. M., Kuchroo, V.and Zarour, H. M. Upregulation of Tim-3 and PD-1
expression is associated with tumor antigen-specific CD8+ T cell dysfunction in
melanoma patients. J Exp Med 207, 2175-2186 (2010).
79 Fraire, A. E., Cooper, S., Greenberg, S. D., Buffler, P.and Langston, C. Mesothelioma of
childhood. Cancer 62, 838-847 (1988).
80 Garcia-Prats, M. D., Ballestin, C., Sotelo, T., Lopez-Encuentra, A.and Mayordomo, J. I.
A comparative evaluation of immunohistochemical markers for the differential diagnosis
of malignant pleural tumours. Histopathology 32, 462-472 (1998).
81 Gardella, S., Andrei, C., Ferrera, D., Lotti, L. V., Torrisi, M. R., Bianchi, M. E.and
Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical,
vesicle-mediated secretory pathway. EMBO Rep 3, 995-1001 (2002).
82 Gazdar, A. F. and Carbone, M. Molecular pathogenesis of malignant mesothelioma and
its relationship to simian virus 40. Clin Lung Cancer 5, 177-181 (2003).
83 Ghiringhelli, F., Apetoh, L., Tesniere, A., Aymeric, L., Ma, Y., Ortiz, C., Vermaelen, K.,
Panaretakis, T., Mignot, G., Ullrich, E., Perfettini, J. L., Schlemmer, F., Tasdemir, E.,
Uhl, M., Genin, P., Civas, A., Ryffel, B., Kanellopoulos, J., Tschopp, J., Andre, F.,
Lidereau, R., McLaughlin, N. M., Haynes, N. M., Smyth, M. J., Kroemer, G.and

115
Zitvogel, L. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-
dependent adaptive immunity against tumors. Nat Med 15, 1170-1178 (2009).
84 Giovannetti, E., Zucali, P. A., Assaraf, Y. G., Leon, L. G., Smid, K., Alecci, C.,
Giancola, F., Destro, A., Gianoncelli, L., Lorenzi, E., Roncalli, M., Santoro, A.and
Peters, G. J. Preclinical emergence of vandetanib as a potent antitumour agent in
mesothelioma: molecular mechanisms underlying its synergistic interaction with
pemetrexed and carboplatin. Br J Cancer 105, 1542-1553 (2011).
85 Gisterek, I., Frydecka, I., ŚWiĄToniowski, G., Fidler, S.and Kornafel, J. Tumour-
infiltrating CD4 and CD8 T lymphocytes in breast cancer. Reports of Practical Oncology
& Radiotherapy 13, 206-209.
86 Godleski, J. J. Role of asbestos in etiology of malignant pleural mesothelioma. Thorac
Surg Clin 14, 479-487 (2004).
87 Goff, S. L., Robbins, P. F., El-Gamil, M.and Rosenberg, S. A. No correlation between
clinical response to CTLA-4 blockade and presence of NY-ESO-1 antibody in patients
with metastatic melanoma. J Immunother 32, 884-885 (2009).
88 Goldberg, M., Banaei, A., Goldberg, S., Auvert, B., Luce, D.and Gueguen, A. Past
occupational exposure to asbestos among men in France. Scand J Work Environ Health
26, 52-61 (2000).
89 Goodwin, G. H., Sanders, C.and Johns, E. W. A new group of chromatin-associated
proteins with a high content of acidic and basic amino acids. Eur J Biochem 38, 14-19
(1973).
90 Gotzos, V., Vogt, P.and Celio, M. R. The calcium binding protein calretinin is a selective
marker for malignant pleural mesotheliomas of the epithelial type. Pathol Res Pract 192,
137-147 (1996).
91 Greenwald, R. J., Freeman, G. J.and Sharpe, A. H. The B7 family revisited. Annu Rev
Immunol 23, 515-548 (2005).
92 Haas, A. R. and Sterman, D. H. Malignant Pleural Mesothelioma: Update on Treatment
Options with a Focus on Novel Therapies. Clin Chest Med 34, 99-111 (2013).
93 Hanahan, D. and Weinberg, Robert A. Hallmarks of Cancer: The Next Generation. Cell
144, 646-674.
94 Hartmann, C. A. and Schutze, H. [Frequency of metastases and survival in histologic
subtypes of pleural mesothelioma. Autopsy study of 106 cases]. Pathologe 13, 259-268
(1992).
95 Hassan, R., Schweizer, C., Lu, K. F., Schuler, B., Remaley, A. T., Weil, S. C.and Pastan,
I. Inhibition of mesothelin-CA-125 interaction in patients with mesothelioma by the anti-
mesothelin monoclonal antibody MORAb-009: Implications for cancer therapy. Lung
Cancer 68, 455-459 (2010).

116
96 He, Q., Liang, C. H.and Lippard, S. J. Steroid hormones induce HMG1 overexpression
and sensitize breast cancer cells to cisplatin and carboplatin. Proc Natl Acad Sci U S A
97, 5768-5772 (2000).
97 Hillerdal, G. Mesothelioma: cases associated with non-occupational and low dose
exposures. Occup Environ Med 56, 505-513 (1999).
98 Hinterberger, M., Reineke, T., Storz, M., Weder, W., Vogt, P.and Moch, H. D2-40 and
calretinin - a tissue microarray analysis of 341 malignant mesotheliomas with emphasis
on sarcomatoid differentiation. Mod Pathol 20, 248-255 (2007).
99 Hirsch, A., Brochard, P., De Cremoux, H., Erkan, L., Sebastien, P., Di Menza, L.and
Bignon, J. Features of asbestos-exposed and unexposed mesothelioma. Am J Ind Med 3,
413-422 (1982).
100 Hodi, F. S., Mihm, M. C., Soiffer, R. J., Haluska, F. G., Butler, M., Seiden, M. V., Davis,
T., Henry-Spires, R., MacRae, S., Willman, A., Padera, R., Jaklitsch, M. T., Shankar, S.,
Chen, T. C., Korman, A., Allison, J. P.and Dranoff, G. Biologic activity of cytotoxic T
lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic
melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U S A 100, 4712-4717
(2003).
101 Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L.,
Fitzgerald, K. A.and Latz, E. Silica crystals and aluminum salts activate the NALP3
inflammasome through phagosomal destabilization. Nat Immunol 9, 847-856 (2008).
102 Howel, D., Gibbs, A., Arblaster, L., Swinburne, L., Schweiger, M., Renvoize, E., Hatton,
P.and Pooley, F. Mineral fibre analysis and routes of exposure to asbestos in the
development of mesothelioma in an English region. Occup Environ Med 56, 51-58
(1999).
103 Huang, J., Ni, J., Liu, K., Yu, Y., Xie, M., Kang, R., Vernon, P., Cao, L.and Tang, D.
HMGB1 promotes drug resistance in osteosarcoma. Cancer Res 72, 230-238 (2012).
104 Hughes, B. Antibody-drug conjugates for cancer: poised to deliver? Nat Rev Drug Discov
9, 665-667 (2010).
105 Huncharek, M., Capotorto, J. V.and Muscat, J. Domestic asbestos exposure, lung fibre
burden, and pleural mesothelioma in a housewife. Br J Ind Med 46, 354-355 (1989).
106 Husain, A. N., Colby, T. V., Ordonez, N. G., Krausz, T., Borczuk, A., Cagle, P. T.,
Chirieac, L. R., Churg, A., Galateau-Salle, F., Gibbs, A. R., Gown, A. M., Hammar, S.
P., Litzky, L. A., Roggli, V. L., Travis, W. D.and Wick, M. R. Guidelines for pathologic
diagnosis of malignant mesothelioma: a consensus statement from the International
Mesothelioma Interest Group. Arch Pathol Lab Med 133, 1317-1331 (2009).
107 Inai, K. Pathology of mesothelioma. Environ Health Prev Med 13, 60-64 (2008).

117
108 Ishiguro, H., Nakaigawa, N., Miyoshi, Y., Fujinami, K., Kubota, Y.and Uemura, H.
Receptor for advanced glycation end products (RAGE) and its ligand, amphoterin are
overexpressed and associated with prostate cancer development. Prostate 64, 92-100
(2005).
109 Ismail-Khan, R., Robinson, L. A., Williams, C. C., Jr., Garrett, C. R., Bepler, G.and
Simon, G. R. Malignant pleural mesothelioma: a comprehensive review. Cancer Control
13, 255-263 (2006).
110 Isobe, Y., Aritaka, N., Sasaki, M., Oshimi, K.and Sugimoto, K. Spontaneous regression
of natural killer cell lymphoma. J Clin Pathol 62, 647-650 (2009).
111 Ivanov, S., Dragoi, A. M., Wang, X., Dallacosta, C., Louten, J., Musco, G., Sitia, G.,
Yap, G. S., Wan, Y., Biron, C. A., Bianchi, M. E., Wang, H.and Chu, W. M. A novel role
for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 110, 1970-
1981 (2007).
112 Iwatsubo, Y., Pairon, J. C., Boutin, C., Menard, O., Massin, N., Caillaud, D., Orlowski,
E., Galateau-Salle, F., Bignon, J.and Brochard, P. Pleural mesothelioma: dose-response
relation at low levels of asbestos exposure in a French population-based case-control
study. Am J Epidemiol 148, 133-142 (1998).
113 Jaurand, M.-C., Renier, A.and Daubriac, J. Mesothelioma: Do asbestos and carbon
nanotubes pose the same health risk? Particle and Fibre Toxicology 6, 16 (2009).
114 Jayaraman, L., Moorthy, N. C., Murthy, K. G., Manley, J. L., Bustin, M.and Prives, C.
High mobility group protein-1 (HMG-1) is a unique activator of p53. Genes Dev 12, 462-
472 (1998).
115 Jube, S., Rivera, Z. S., Bianchi, M. E., Powers, A., Wang, E., Pagano, I., Pass, H. I.,
Gaudino, G., Carbone, M.and Yang, H. Cancer cell secretion of the DAMP protein
HMGB1 supports progression in malignant mesothelioma. Cancer Res 72, 3290-3301
(2012).
116 Kaczmarek, A., Vandenabeele, P.and Krysko, D. V. Necroptosis: the release of damage-
associated molecular patterns and its physiological relevance. Immunity 38, 209-223
(2013).
117 Kang, R., Tang, D. L., Cao, L. Z., Yu, Y., Zhang, G. Y.and Xiao, X. Z. [High mobility
group box 1 is increased in children with acute lymphocytic leukemia and stimulates the
release of tumor necrosis factor-alpha in leukemic cell]. Zhonghua Er Ke Za Zhi 45, 329-
333 (2007).
118 Kang, R., Tang, D., Schapiro, N. E., Loux, T., Livesey, K. M., Billiar, T. R., Wang, H.,
Van Houten, B., Lotze, M. T.and Zeh, H. J. The HMGB1/RAGE inflammatory pathway
promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene
33, 567-577 (2014).

118
119 Keir, M. E., Butte, M. J., Freeman, G. J.and Sharpe, A. H. PD-1 and its ligands in
tolerance and immunity. Annu Rev Immunol 26, 677-704 (2008).
120 Kim, R., Emi, M.and Tanabe, K. Cancer immunoediting from immune surveillance to
immune escape. Immunology 121, 1-14 (2007).
121 Kimura, N. and Kimura, I. Podoplanin as a marker for mesothelioma. Pathol Int 55, 83-
86 (2005).
122 Kishimoto, T., Ozaki, S., Kato, K., Nishi, H.and Genba, K. Malignant pleural
mesothelioma in parts of Japan in relationship to asbestos exposure. Ind Health 42, 435-
439 (2004).
123 Klippel, Z. K., Chou, J., Towlerton, A. M., Voong, L. N., Robbins, P., Bensinger, W.
I.and Warren, E. H. Immune escape from NY-ESO-1-specific T cell therapy via loss of
heterozygosity in the MHC. Gene therapy 21, 337-342 (2014).
124 Kokkola, R., Sundberg, E., Ulfgren, A. K., Palmblad, K., Li, J., Wang, H., Ulloa, L.,
Yang, H., Yan, X. J., Furie, R., Chiorazzi, N., Tracey, K. J., Andersson, U.and Harris, H.
E. High mobility group box chromosomal protein 1: a novel proinflammatory mediator in
synovitis. Arthritis Rheum 46, 2598-2603 (2002).
125 Kokkola, R., Andersson, A., Mullins, G., Ostberg, T., Treutiger, C. J., Arnold, B.,
Nawroth, P., Andersson, U., Harris, R. A.and Harris, H. E. RAGE is the major receptor
for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol
61, 1-9 (2005).
126 Kreitman, R. J., Hassan, R., Fitzgerald, D. J.and Pastan, I. Phase I trial of continuous
infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res 15, 5274-
5279 (2009).
127 Kroemer, G., Galluzzi, L., Kepp, O.and Zitvogel, L. Immunogenic cell death in cancer
therapy. Annu Rev Immunol 31, 51-72 (2013).
128 Krug, L. M., Pass, H. I., Rusch, V. W., Kindler, H. L., Sugarbaker, D. J., Rosenzweig, K.
E., Flores, R., Friedberg, J. S., Pisters, K., Monberg, M., Obasaju, C. K.and Vogelzang,
N. J. Multicenter phase II trial of neoadjuvant pemetrexed plus cisplatin followed by
extrapleural pneumonectomy and radiation for malignant pleural mesothelioma. J Clin
Oncol 27, 3007-3013 (2009).
129 Krysko, D. V., Garg, A. D., Kaczmarek, A., Krysko, O., Agostinis, P.and Vandenabeele,
P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 12, 860-875
(2012).
130 Kuniyasu, H., Oue, N., Wakikawa, A., Shigeishi, H., Matsutani, N., Kuraoka, K., Ito, R.,
Yokozaki, H.and Yasui, W. Expression of receptors for advanced glycation end-products
(RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J
Pathol 196, 163-170 (2002).

119
131 Kuniyasu, H., Yano, S., Sasaki, T., Sasahira, T., Sone, S.and Ohmori, H. Colon cancer
cell-derived high mobility group 1/amphoterin induces growth inhibition and apoptosis in
macrophages. Am J Pathol 166, 751-760 (2005).
132 Kurts, C., Robinson, B. W. S.and Knolle, P. A. Cross-priming in health and disease. Nat
Rev Immunol 10, 403-414 (2010).
133 Kushitani, K., Takeshima, Y., Amatya, V. J., Furonaka, O., Sakatani, A.and Inai, K.
Immunohistochemical marker panels for distinguishing between epithelioid
mesothelioma and lung adenocarcinoma. Pathol Int 57, 190-199 (2007).
134 Kushitani, K., Takeshima, Y., Amatya, V. J., Furonaka, O., Sakatani, A.and Inai, K.
Differential diagnosis of sarcomatoid mesothelioma from true sarcoma and sarcomatoid
carcinoma using immunohistochemistry. Pathol Int 58, 75-83 (2008).
135 Lakshmanagowda, P. B., Viswanath, L., Thimmaiah, N., Dasappa, L., Supe, S. S.and
Kallur, P. Abscopal effect in a patient with chronic lymphocytic leukemia during
radiation therapy: a case report. Cases J 2, 204 (2009).
136 Lao, I., Chen, Q., Yu, L.and Wang, J. [Sarcomatoid malignant mesothelioma: a
clinicopathologic and immunohistochemical analysis of 22 cases]. Zhonghua Bing Li Xue
Za Zhi 43, 364-369 (2014).
137 Lee, Y., Auh, S. L., Wang, Y., Burnette, B., Wang, Y., Meng, Y., Beckett, M., Sharma,
R., Chin, R., Tu, T., Weichselbaum, R. R.and Fu, Y. X. Therapeutic effects of ablative
radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment.
Blood 114, 589-595 (2009).
138 Leers, M. P., Aarts, M. M.and Theunissen, P. H. E-cadherin and calretinin: a useful
combination of immunochemical markers for differentiation between mesothelioma and
metastatic adenocarcinoma. Histopathology 32, 209-216 (1998).
139 Levy, A., Chargari, C., Cheminant, M., Simon, N., Bourgier, C.and Deutsch, E. Radiation
therapy and immunotherapy: Implications for a combined cancer treatment. Critical
Reviews in Oncology / Hematology 85, 278-287.
140 Liu, L., Yang, M., Kang, R., Wang, Z., Zhao, Y., Yu, Y., Xie, M., Yin, X., Livesey, K.
M., Lotze, M. T., Tang, D.and Cao, L. HMGB1-induced autophagy promotes
chemotherapy resistance in leukemia cells. Leukemia 25, 23-31 (2011).
141 Livesey, K. M., Tang, D., Zeh, H. J.and Lotze, M. T. Not just nuclear proteins: 'novel'
autophagy cancer treatment targets - p53 and HMGB1. Curr Opin Investig Drugs 9,
1259-1263 (2008).
142 Livesey, K. M., Kang, R., Vernon, P., Buchser, W., Loughran, P., Watkins, S. C., Zhang,
L., Manfredi, J. J., Zeh, H. J., 3rd, Li, L., Lotze, M. T.and Tang, D. p53/HMGB1
complexes regulate autophagy and apoptosis. Cancer Res 72, 1996-2005 (2012).

120
143 Lotze, M. T. and Tracey, K. J. High-mobility group box 1 protein (HMGB1): nuclear
weapon in the immune arsenal. Nat Rev Immunol 5, 331-342 (2005).
144 Lucas, D. R., Pass, H. I., Madan, S. K., Adsay, N. V., Wali, A., Tabaczka, P.and
Lonardo, F. Sarcomatoid mesothelioma and its histological mimics: a comparative
immunohistochemical study. Histopathology 42, 270-279 (2003).
145 Lucas, D. R., Pass, H. I., Madan, S. K., Adsay, N. V., Wali, A., Tabaczka, P.and
Lonardo, F. Sarcomatoid mesothelioma and its histological mimics: a comparative
immunohistochemical study. Histopathology 42, 270-279 (2003).
146 Lugade, A. A., Sorensen, E. W., Gerber, S. A., Moran, J. P., Frelinger, J. G.and Lord, E.
M. Radiation-induced IFN-gamma production within the tumor microenvironment
influences antitumor immunity. J Immunol 180, 3132-3139 (2008).
147 Lynch, K. M. and Smith, W. A. Pulmonary Asbestosis III: Carcinoma of Lung in
Asbesto-Silicosis. The American Journal of Cancer 24, 56-64 (1935).
148 Martinis, U. J. and Radovic, V. R. PLEURAL MESOTHELIOMA IN PATIENT WITH
PULMONARY TUBERCULOSIS: REPORT OF A CASE. Dis Chest 47, 568-570
(1965).
149 Martins, I., Tesniere, A., Kepp, O., Michaud, M., Schlemmer, F., Senovilla, L., Seror, C.,
Metivier, D., Perfettini, J. L., Zitvogel, L.and Kroemer, G. Chemotherapy induces ATP
release from tumor cells. Cell Cycle 8, 3723-3728 (2009).
150 Matzinger, P. Tolerance, danger, and the extended family. Annu Rev Immunol 12, 991-
1045 (1994).
151 Matzinger, P. The danger model: a renewed sense of self. Science 296, 301-305 (2002).
152 Maurer, R. and Egloff, B. Malignant peritoneal mesothelioma after cholangiography
with thorotrast. Cancer 36, 1381-1385 (1975).
153 McKinney, K. and Prives, C. Efficient specific DNA binding by p53 requires both its
central and C-terminal domains as revealed by studies with high-mobility group 1
protein. Mol Cell Biol 22, 6797-6808 (2002).
154 Messmer, D., Yang, H., Telusma, G., Knoll, F., Li, J., Messmer, B., Tracey, K. J.and
Chiorazzi, N. High mobility group box protein 1: an endogenous signal for dendritic cell
maturation and Th1 polarization. J Immunol 173, 307-313 (2004).
155 Michaud, M., Martins, I., Sukkurwala, A. Q., Adjemian, S., Ma, Y., Pellegatti, P., Shen,
S., Kepp, O., Scoazec, M., Mignot, G., Rello-Varona, S., Tailler, M., Menger, L.,
Vacchelli, E., Galluzzi, L., Ghiringhelli, F., di Virgilio, F., Zitvogel, L.and Kroemer, G.
Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents
in mice. Science 334, 1573-1577 (2011).

121
156 Miettinen, M., Limon, J., Niezabitowski, A.and Lasota, J. Calretinin and other
mesothelioma markers in synovial sarcoma: analysis of antigenic similarities and
differences with malignant mesothelioma. Am J Surg Pathol 25, 610-617 (2001).
157 Minot, C.-S. The Mesoderm and the Coelom of Vertebrates. The American Naturalist 24,
877-898 (1890).
158 Moore, A. J., Parker, R. J.and Wiggins, J. Malignant mesothelioma. Orphanet Journal of
Rare Diseases 3, 34-34 (2008).
159 Moskal, T. L., Urschel, J. D., Anderson, T. M., Antkowiak, J. G.and Takita, H. Malignant
pleural mesothelioma: a problematic review. Surg Oncol 7, 5-12 (1998).
160 Mossman, B. T. and Gee, J. B. Asbestos-related diseases. N Engl J Med 320, 1721-1730
(1989).
161 Mossman, B. T., Shukla, A., Heintz, N. H., Verschraegen, C. F., Thomas, A.and Hassan,
R. New insights into understanding the mechanisms, pathogenesis, and management of
malignant mesotheliomas. Am J Pathol 182, 1065-1077 (2013).
162 Muller, S., Scaffidi, P., Degryse, B., Bonaldi, T., Ronfani, L., Agresti, A., Beltrame,
M.and Bianchi, M. E. New EMBO members' review: the double life of HMGB1
chromatin protein: architectural factor and extracellular signal. Embo j 20, 4337-4340
(2001).
163 Murphy, K. Janeway's Immunobiology. 8 edn, (Garland Science, Taylor & Francis
Group, 2012).
164 Mutsaers, S. E., Whitaker, D.and Papadimitriou, J. M. Stimulation of mesothelial cell
proliferation by exudate macrophages enhances serosal wound healing in a murine
model. Am J Pathol 160, 681-692 (2002).
165 Mutsaers, S. E. The mesothelial cell. Int J Biochem Cell Biol 36, 9-16 (2004).
166 Nakas, A. and Waller, D. Predictors of long-term survival following radical surgery for
malignant pleural mesothelioma. European Journal of Cardio-Thoracic Surgery 46, 380-
385 (2014).
167 Neeper, M., Schmidt, A. M., Brett, J., Yan, S. D., Wang, F., Pan, Y. C., Elliston, K.,
Stern, D.and Shaw, A. Cloning and expression of a cell surface receptor for advanced
glycosylation end products of proteins. J Biol Chem 267, 14998-15004 (1992).
168 Neumann, V., Löseke, S., Nowak, D., Herth, F. J. F.and Tannapfel, A. Malignant Pleural
Mesothelioma: Incidence, Etiology, Diagnosis, Treatment, and Occupational Health.
Dtsch Arztebl Int 110, 319-326 (2013).
169 Nosho, K., Baba, Y., Tanaka, N., Shima, K., Hayashi, M., Meyerhardt, J. A.,
Giovannucci, E., Dranoff, G., Fuchs, C. S.and Ogino, S. Tumour-infiltrating T-cell

122
subsets, molecular changes in colorectal cancer and prognosis: cohort study and literature
review. J Pathol 222, 350-366 (2010).
170 Obeid, M., Tesniere, A., Ghiringhelli, F., Fimia, G. M., Apetoh, L., Perfettini, J. L.,
Castedo, M., Mignot, G., Panaretakis, T., Casares, N., Metivier, D., Larochette, N., van
Endert, P., Ciccosanti, F., Piacentini, M., Zitvogel, L.and Kroemer, G. Calreticulin
exposure dictates the immunogenicity of cancer cell death. Nat Med 13, 54-61 (2007).
171 Ohshima, Y., Tsukimoto, M., Takenouchi, T., Harada, H., Suzuki, A., Sato, M., Kitani,
H.and Kojima, S. gamma-Irradiation induces P2X(7) receptor-dependent ATP release
from B16 melanoma cells. Biochim Biophys Acta 1800, 40-46 (2010).
172 Ordonez, N. G. The immunohistochemical diagnosis of mesothelioma: a comparative
study of epithelioid mesothelioma and lung adenocarcinoma. Am J Surg Pathol 27, 1031-
1051 (2003).
173 Ordonez, N. G. The diagnostic utility of immunohistochemistry in distinguishing
between mesothelioma and renal cell carcinoma: a comparative study. Hum Pathol 35,
697-710 (2004).
174 Ordonez, N. G. D2-40 and podoplanin are highly specific and sensitive
immunohistochemical markers of epithelioid malignant mesothelioma. Hum Pathol 36,
372-380 (2005).
175 Ordonez, N. G. Podoplanin: a novel diagnostic immunohistochemical marker. Adv Anat
Pathol 13, 83-88 (2006).
176 Ordonez, N. G. What are the current best immunohistochemical markers for the diagnosis
of epithelioid mesothelioma? A review and update. Hum Pathol 38, 1-16 (2007).
177 Padgett, D. M., Cathro, H. P., Wick, M. R.and Mills, S. E. Podoplanin is a better
immunohistochemical marker for sarcomatoid mesothelioma than calretinin. Am J Surg
Pathol 32, 123-127 (2008).
178 Pan, X. L., Day, H. W., Wang, W., Beckett, L. A.and Schenker, M. B. Residential
proximity to naturally occurring asbestos and mesothelioma risk in California. Am J
Respir Crit Care Med 172, 1019-1025 (2005).
179 Paonessa, G., Frank, R.and Cortese, R. Nucleotide sequence of rat liver HMG1 cDNA.
Nucleic Acids Res 15, 9077 (1987).
180 Pasello, G., Nicotra, S., Marulli, G., Rea, F., Bonanno, L., Carli, P., Magro, C., Jirillo,
A.and Favaretto, A. Platinum-based doublet chemotherapy in pre-treated malignant
pleural mesothelioma (MPM) patients: a mono-institutional experience. Lung Cancer 73,
351-355 (2011).
181 Pass, H. I., Temeck, B. K., Kranda, K., Thomas, G., Russo, A., Smith, P., Friauf, W.and
Steinberg, S. M. Phase III randomized trial of surgery with or without intraoperative

123
photodynamic therapy and postoperative immunochemotherapy for malignant pleural
mesothelioma. Ann Surg Oncol 4, 628-633 (1997).
182 Pass, H. I., Temeck, B. K., Kranda, K., Steinberg, S. M.and Feuerstein, I. R. Preoperative
tumor volume is associated with outcome in malignant pleural mesothelioma. J Thorac
Cardiovasc Surg 115, 310-317; discussion 317-318 (1998).
183 Pass, H. I., Carbone, M.and Vogelzang, N. Malignant Mesothelioma : Advances in
Pathogenesis, Diagnosis, and Translational Therapies. (Springer, 2005).
184 Pentcheva-Hoang, T., Corse, E.and Allison, J. P. Negative regulators of T-cell activation:
potential targets for therapeutic intervention in cancer, autoimmune disease, and
persistent infections. Immunol Rev 229, 67-87 (2009).
185 Perez, S. A., Karamouzis, M. V., Skarlos, D. V., Ardavanis, A., Sotiriadou, N. N.,
Iliopoulou, E. G., Salagianni, M. L., Orphanos, G., Baxevanis, C. N., Rigatos, G.and
Papamichail, M. CD4+CD25+ regulatory T-cell frequency in HER-2/neu (HER)-positive
and HER-negative advanced-stage breast cancer patients. Clin Cancer Res 13, 2714-2721
(2007).
186 Peto, J., Hodgson, J. T., Matthews, F. E.and Jones, J. R. Continuing increase in
mesothelioma mortality in Britain. Lancet 345, 535-539 (1995).
187 Popovic, K., Ek, M., Espinosa, A., Padyukov, L., Harris, H. E., Wahren-Herlenius, M.and
Nyberg, F. Increased expression of the novel proinflammatory cytokine high mobility
group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus.
Arthritis Rheum 52, 3639-3645 (2005).
188 Poser, I., Golob, M., Buettner, R.and Bosserhoff, A. K. Upregulation of HMG1 leads to
melanoma inhibitory activity expression in malignant melanoma cells and contributes to
their malignancy phenotype. Mol Cell Biol 23, 2991-2998 (2003).
189 Qin, S., Wang, H., Yuan, R., Li, H., Ochani, M., Ochani, K., Rosas-Ballina, M., Czura,
C. J., Huston, J. M., Miller, E., Lin, X., Sherry, B., Kumar, A., Larosa, G., Newman, W.,
Tracey, K. J.and Yang, H. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp
Med 203, 1637-1642 (2006).
190 Ratzer, E. R., Pool, J. L.and Melamed, M. R. Pleural mesotheliomas. Clinical experiences
with thirty-seven patients. Am J Roentgenol Radium Ther Nucl Med 99, 863-880 (1967).
191 Rauvala, H., Merenmies, J., Pihlaskari, R., Korkolainen, M., Huhtala, M. L.and Panula,
P. The adhesive and neurite-promoting molecule p30: analysis of the amino-terminal
sequence and production of antipeptide antibodies that detect p30 at the surface of
neuroblastoma cells and of brain neurons. J Cell Biol 107, 2293-2305 (1988).
192 Ribak, J. and Selikoff, I. J. Survival of asbestos insulation workers with mesothelioma.
British Journal of Industrial Medicine 49, 732-735 (1992).

124
193 Ribas, A. Anti-CTLA4 Antibody Clinical Trials in Melanoma. Update Cancer Ther 2,
133-139 (2007).
194 Richards, W. G., Zellos, L., Bueno, R., Jaklitsch, M. T., Janne, P. A., Chirieac, L. R.,
Yeap, B. Y., Dekkers, R. J., Hartigan, P. M., Capalbo, L.and Sugarbaker, D. J. Phase I to
II study of pleurectomy/decortication and intraoperative intracavitary hyperthermic
cisplatin lavage for mesothelioma. J Clin Oncol 24, 1561-1567 (2006).
195 Riddell, R. H., Goodman, M. J.and Moossa, A. R. Peritoneal malignant mesothelioma in
a patient with recurrent peritonitis. Cancer 48, 134-139 (1981).
196 Robert, C., Thomas, L., Bondarenko, I., O'Day, S., Weber, J., Garbe, C., Lebbe, C.,
Baurain, J.-F., Testori, A., Grob, J.-J., Davidson, N., Richards, J., Maio, M., Hauschild,
A., Miller, W. H., Gascon, P., Lotem, M., Harmankaya, K., Ibrahim, R., Francis, S.,
Chen, T.-T., Humphrey, R., Hoos, A.and Wolchok, J. D. Ipilimumab plus Dacarbazine
for Previously Untreated Metastatic Melanoma. New England Journal of Medicine 364,
2517-2526 (2011).
197 Roberts, F., Harper, C. M., Downie, I.and Burnett, R. A. Immunohistochemical analysis
still has a limited role in the diagnosis of malignant mesothelioma. A study of thirteen
antibodies. Am J Clin Pathol 116, 253-262 (2001).
198 Robinson, B. W. S., Musk, A. W.and Lake, R. A. Malignant mesothelioma. The Lancet
366, 397-408 (2005).
199 Roggli, V. L., McGavran, M. H., Subach, J., Sybers, H. D.and Greenberg, S. D.
Pulmonary asbestos body counts and electron probe analysis of asbestos body cores in
patients with mesothelioma: a study of 25 cases. Cancer 50, 2423-2432 (1982).
200 Roggli, V. L., Oury, T. D.and Moffatt, E. J. Malignant mesothelioma in women. Anat
Pathol 2, 147-163 (1997).
201 Rovere-Querini, P., Capobianco, A., Scaffidi, P., Valentinis, B., Catalanotti, F., Giazzon,
M., Dumitriu, I. E., Müller, S., Iannacone, M., Traversari, C., Bianchi, M. E.and
Manfredi, A. A. HMGB1 is an endogenous immune adjuvant released by necrotic cells.
EMBO Rep 5, 825-830 (2004).
202 Ruffie, P., Feld, R., Minkin, S., Cormier, Y., Boutan-Laroze, A., Ginsberg, R., Ayoub, J.,
Shepherd, F. A., Evans, W. K., Figueredo, A.and et al. Diffuse malignant mesothelioma
of the pleura in Ontario and Quebec: a retrospective study of 332 patients. J Clin Oncol 7,
1157-1168 (1989).
203 Rusch, V. W. and Venkatraman, E. S. Important prognostic factors in patients with
malignant pleural mesothelioma, managed surgically. Ann Thorac Surg 68, 1799-1804
(1999).
204 Rusch, V. W. Extrapleural pneumonectomy and extended pleurectomy/decortication for
malignant pleural mesothelioma: the Memorial Sloan-Kettering Cancer Center approach.
Annals of Cardiothoracic Surgery 1, 523-531 (2012).

125
205 Ryckman, C., Vandal, K., Rouleau, P., Talbot, M.and Tessier, P. A. Proinflammatory
activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil
chemotaxis and adhesion. J Immunol 170, 3233-3242 (2003).
206 Saga, Y., Suzuki, M., Mizukami, H., Urabe, M., Fukushima, M., Ozawa, K.and Sato, I.
Enhanced expression of thymidylate synthase mediates resistance of uterine cervical
cancer cells to radiation. Oncology 63, 185-191 (2002).
207 Sakuishi, K., Apetoh, L., Sullivan, J. M., Blazar, B. R., Kuchroo, V. K.and Anderson, A.
C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-
tumor immunity. J Exp Med 207, 2187-2194 (2010).
208 Sanders, C. L. and Jackson, T. A. Induction of mesotheliomas and sarcomas from "hot
spots" of 239 PuO 2 activity. Health Phys 22, 755-759 (1972).
209 Sasahira, T., Akama, Y., Fujii, K.and Kuniyasu, H. Expression of receptor for advanced
glycation end products and HMGB1/amphoterin in colorectal adenomas. Virchows Arch
446, 411-415 (2005).
210 Scaffidi, P., Misteli, T.and Bianchi, M. E. Release of chromatin protein HMGB1 by
necrotic cells triggers inflammation. Nature 418, 191-195 (2002).
211 Schipper, P. H., Nichols, F. C., Thomse, K. M., Deschamps, C., Cassivi, S. D., Allen, M.
S.and Pairolero, P. C. Malignant pleural mesothelioma: surgical management in 285
patients. Ann Thorac Surg 85, 257-264; discussion 264 (2008).
212 Schliemann, C. and Neri, D. Antibody-based vascular tumor targeting. Recent Results
Cancer Res 180, 201-216 (2010).
213 Scott, A. M., Wolchok, J. D.and Old, L. J. Antibody therapy of cancer. Nat Rev Cancer
12, 278-287 (2012).
214 Selikoff, I. J., Churg, J.and Hammond, E. C. ASBESTOS EXPOSURE AND
NEOPLASIA. Jama 188, 22-26 (1964).
215 Semino, C., Angelini, G., Poggi, A.and Rubartelli, A. NK/iDC interaction results in IL-18
secretion by DCs at the synaptic cleft followed by NK cell activation and release of the
DC maturation factor HMGB1. Blood 106, 609-616 (2005).
216 Shang, G. H., Jia, C. Q., Tian, H., Xiao, W., Li, Y., Wang, A. H., Dong, L.and Lin, D. J.
Serum high mobility group box protein 1 as a clinical marker for non-small cell lung
cancer. Respir Med 103, 1949-1953 (2009).
217 Sharma, A., Ray, R.and Rajeswari, M. R. Overexpression of high mobility group (HMG)
B1 and B2 proteins directly correlates with the progression of squamous cell carcinoma
in skin. Cancer Invest 26, 843-851 (2008).

126
218 Shen, X., Hong, L., Sun, H., Shi, M.and Song, Y. The expression of high-mobility group
protein box 1 correlates with the progression of non-small cell lung cancer. Oncol Rep
22, 535-539 (2009).
219 Sheng, X., Du, X., Zhang, X., Li, D., Lu, C., Li, Q., Ma, Z., Song, Q.and Wang, C.
Clinical value of serum HMGB1 levels in early detection of recurrent squamous cell
carcinoma of uterine cervix: comparison with serum SCCA, CYFRA21-1, and CEA
levels. Croat Med J 50, 455-464 (2009).
220 Shivapurkar, N., Wiethege, T., Wistuba, II, Salomon, E., Milchgrub, S., Muller, K. M.,
Churg, A., Pass, H.and Gazdar, A. F. Presence of simian virus 40 sequences in malignant
mesotheliomas and mesothelial cell proliferations. J Cell Biochem 76, 181-188 (1999).
221 Siva, S., MacManus, M. P., Martin, R. F.and Martin, O. A. Abscopal effects of radiation
therapy: a clinical review for the radiobiologist. Cancer Lett 356, 82-90 (2015).
222 Small, E. J., Tchekmedyian, N. S., Rini, B. I., Fong, L., Lowy, I.and Allison, J. P. A pilot
trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory
prostate cancer. Clin Cancer Res 13, 1810-1815 (2007).
223 Smyth, M. J., Cretney, E., Kershaw, M. H.and Hayakawa, Y. Cytokines in cancer
immunity and immunotherapy. Immunological Reviews 202, 275-293 (2004).
224 Spirtas, R., Beebe, G. W., Connelly, R. R., Wright, W. E., Peters, J. M., Sherwin, R. P.,
Henderson, B. E., Stark, A., Kovasznay, B. M., Davies, J. N.and et al. Recent trends in
mesothelioma incidence in the United States. Am J Ind Med 9, 397-407 (1986).
225 Stebbing, J., Powles, T., McPherson, K., Shamash, J., Wells, P., Sheaff, M. T., Slater, S.,
Rudd, R. M., Fennell, D.and Steele, J. P. The efficacy and safety of weekly vinorelbine in
relapsed malignant pleural mesothelioma. Lung Cancer 63, 94-97 (2009).
226 Sterman, D. H., Haas, A., Moon, E., Recio, A., Schwed, D., Vachani, A., Katz, S. I.,
Gillespie, C. T., Cheng, G., Sun, J., Papasavvas, E., Montaner, L. J., Heitjan, D. F.,
Litzky, L., Friedberg, J., Culligan, M., June, C. H., Carroll, R. G.and Albelda, S. M. A
trial of intrapleural adenoviral-mediated Interferon-alpha2b gene transfer for malignant
pleural mesothelioma. Am J Respir Crit Care Med 184, 1395-1399 (2011).
227 Stevenson, J. P., Kindler, H. L., Papasavvas, E., Sun, J., Jacobs-Small, M., Hull, J.,
Schwed, D., Ranganathan, A., Newick, K., Heitjan, D. F., Langer, C. J., McPherson, J.
M., Montaner, L. J.and Albelda, S. M. Immunological effects of the TGFβ-blocking
antibody GC1008 in malignant pleural mesothelioma patients. Oncoimmunology 2,
e26218 (2013).
228 Stock, R. J., Fu, Y. S.and Carter, J. R. Malignant peritoneal mesothelioma following
radiotherapy for seminoma of the testis. Cancer 44, 914-919 (1979).
229 Stros, M., Ozaki, T., Bacikova, A., Kageyama, H.and Nakagawara, A. HMGB1 and
HMGB2 cell-specifically down-regulate the p53- and p73-dependent sequence-specific
transactivation from the human Bax gene promoter. J Biol Chem 277, 7157-7164 (2002).

127
230 Stros, M., Muselikova-Polanska, E., Pospisilova, S.and Strauss, F. High-affinity binding
of tumor-suppressor protein p53 and HMGB1 to hemicatenated DNA loops.
Biochemistry 43, 7215-7225 (2004).
231 Suda, K., Kitagawa, Y., Ozawa, S., Saikawa, Y., Ueda, M., Ebina, M., Yamada, S.,
Hashimoto, S., Fukata, S., Abraham, E., Maruyama, I., Kitajima, M.and Ishizaka, A.
Anti-high-mobility group box chromosomal protein 1 antibodies improve survival of rats
with sepsis. World J Surg 30, 1755-1762 (2006).
232 Sugarbaker, D. J., Strauss, G. M., Lynch, T. J., Richards, W., Mentzer, S. J., Lee, T. H.,
Corson, J. M.and Antman, K. H. Node status has prognostic significance in the
multimodality therapy of diffuse, malignant mesothelioma. J Clin Oncol 11, 1172-1178
(1993).
233 Suzuki, Y. Pathology of human malignant mesothelioma--preliminary analysis of 1,517
mesothelioma cases. Ind Health 39, 183-185 (2001).
234 Suzuki, Y., Mimura, K., Yoshimoto, Y., Watanabe, M., Ohkubo, Y., Izawa, S., Murata,
K., Fujii, H., Nakano, T.and Kono, K. Immunogenic tumor cell death induced by
chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res 72,
3967-3976 (2012).
235 Tabata, C., Shibata, E., Tabata, R., Kanemura, S., Mikami, K., Nogi, Y., Masachika, E.,
Nishizaki, T.and Nakano, T. Serum HMGB1 as a prognostic marker for malignant pleural
mesothelioma. BMC Cancer 13, 205 (2013).
236 Takaya, M., Niibe, Y., Tsunoda, S., Jobo, T., Imai, M., Kotani, S., Unno, N.and
Hayakawa, K. Abscopal effect of radiation on toruliform para-aortic lymph node
metastases of advanced uterine cervical carcinoma--a case report. Anticancer Res 27,
499-503 (2007).
237 Tang, D., Loze, M. T., Zeh, H. J.and Kang, R. The redox protein HMGB1 regulates cell
death and survival in cancer treatment. Autophagy 6, 1181-1183 (2010).
238 Tang, D., Kang, R., Cheh, C. W., Livesey, K. M., Liang, X., Schapiro, N. E., Benschop,
R., Sparvero, L. J., Amoscato, A. A., Tracey, K. J., Zeh, H. J.and Lotze, M. T. HMGB1
release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299-
5310 (2010).
239 Tang, D., Kang, R., Livesey, K. M., Cheh, C. W., Farkas, A., Loughran, P., Hoppe, G.,
Bianchi, M. E., Tracey, K. J., Zeh, H. J., 3rdand Lotze, M. T. Endogenous HMGB1
regulates autophagy. J Cell Biol 190, 881-892 (2010).
240 Tanida, S., Mizoshita, T., Ozeki, K., Tsukamoto, H., Kamiya, T., Kataoka, H., Sakamuro,
D.and Joh, T. Mechanisms of Cisplatin-Induced Apoptosis and of Cisplatin Sensitivity:
Potential of BIN1 to Act as a Potent Predictor of Cisplatin Sensitivity in Gastric Cancer
Treatment. International Journal of Surgical Oncology 2012, 8 (2012).

128
241 Taniguchi, N., Kawahara, K., Yone, K., Hashiguchi, T., Yamakuchi, M., Goto, M.,
Inoue, K., Yamada, S., Ijiri, K., Matsunaga, S., Nakajima, T., Komiya, S.and Maruyama,
I. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of
rheumatoid arthritis as a novel cytokine. Arthritis Rheum 48, 971-981 (2003).
242 Teng, M. W., Swann, J. B., Koebel, C. M., Schreiber, R. D.and Smyth, M. J. Immune-
mediated dormancy: an equilibrium with cancer. J Leukoc Biol 84, 988-993 (2008).
243 Tesniere, A., Panaretakis, T., Kepp, O., Apetoh, L., Ghiringhelli, F., Zitvogel, L.and
Kroemer, G. Molecular characteristics of immunogenic cancer cell death. Cell Death
Differ 15, 3-12 (2008).
244 Testa, J. R., Carbone, M., Hirvonen, A., Khalili, K., Krynska, B., Linnainmaa, K.,
Pooley, F. D., Rizzo, P., Rusch, V.and Xiao, G. H. A multi-institutional study confirms
the presence and expression of simian virus 40 in human malignant mesotheliomas.
Cancer Res 58, 4505-4509 (1998).
245 Tey, S.-K., Bollard, C. M.and Heslop, H. E. Adoptive T-cell transfer in cancer
immunotherapy. Immunol Cell Biol 84, 281-289 (2006).
246 Thompson, J. K., Westbom, C. M.and Shukla, A. Malignant mesothelioma: development
to therapy. J Cell Biochem 115, 1-7 (2014).
247 Tian, J., Avalos, A. M., Mao, S. Y., Chen, B., Senthil, K., Wu, H., Parroche, P., Drabic,
S., Golenbock, D., Sirois, C., Hua, J., An, L. L., Audoly, L., La Rosa, G., Bierhaus, A.,
Naworth, P., Marshak-Rothstein, A., Crow, M. K., Fitzgerald, K. A., Latz, E., Kiener, P.
A.and Coyle, A. J. Toll-like receptor 9-dependent activation by DNA-containing immune
complexes is mediated by HMGB1 and RAGE. Nat Immunol 8, 487-496 (2007).
248 Tischoff, I., Neid, M., Neumann, V.and Tannapfel, A. Pathohistological diagnosis and
differential diagnosis. Recent Results Cancer Res 189, 57-78 (2011).
249 Topalian, S. L., Hodi, F. S., Brahmer, J. R., Gettinger, S. N., Smith, D. C., McDermott,
D. F., Powderly, J. D., Carvajal, R. D., Sosman, J. A., Atkins, M. B., Leming, P. D.,
Spigel, D. R., Antonia, S. J., Horn, L., Drake, C. G., Pardoll, D. M., Chen, L., Sharfman,
W. H., Anders, R. A., Taube, J. M., McMiller, T. L., Xu, H., Korman, A. J., Jure-Kunkel,
M., Agrawal, S., McDonald, D., Kollia, G. D., Gupta, A., Wigginton, J. M.and Sznol, M.
Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med
366, 2443-2454 (2012).
250 Treutiger, C. J., Mullins, G. E., Johansson, A. S., Rouhiainen, A., Rauvala, H. M.,
Erlandsson-Harris, H., Andersson, U., Yang, H., Tracey, K. J., Andersson, J.and
Palmblad, J. E. High mobility group 1 B-box mediates activation of human endothelium.
J Intern Med 254, 375-385 (2003).
251 Uramoto, H., Izumi, H., Nagatani, G., Ohmori, H., Nagasue, N., Ise, T., Yoshida, T.,
Yasumoto, K.and Kohno, K. Physical interaction of tumour suppressor p53/p73 with
CCAAT-binding transcription factor 2 (CTF2) and differential regulation of human high-
mobility group 1 (HMG1) gene expression. Biochem J 371, 301-310 (2003).

129
252 Urbonaviciute, V., Furnrohr, B. G., Weber, C., Haslbeck, M., Wilhelm, S., Herrmann,
M.and Voll, R. E. Factors masking HMGB1 in human serum and plasma. J Leukoc Biol
81, 67-74 (2007).
253 Vajdic, C. M. and van Leeuwen, M. T. Cancer incidence and risk factors after solid
organ transplantation. Int J Cancer 125, 1747-1754 (2009).
254 van Ruth, S., Baas, P.and Zoetmulder, F. A. Surgical treatment of malignant pleural
mesothelioma: a review. Chest 123, 551-561 (2003).
255 van Zandwijk, N., Clarke, C., Henderson, D., Musk, A. W., Fong, K., Nowak, A.,
Loneragan, R., McCaughan, B., Boyer, M., Feigen, M., Currow, D., Schofield, P., Nick
Pavlakis, B. I., McLean, J., Marshall, H., Leong, S., Keena, V.and Penman, A. Guidelines
for the diagnosis and treatment of malignant pleural mesothelioma. J Thorac Dis 5,
E254-307 (2013).
256 Venneri, M. A., De Palma, M., Ponzoni, M., Pucci, F., Scielzo, C., Zonari, E., Mazzieri,
R., Doglioni, C.and Naldini, L. Identification of proangiogenic TIE2-expressing
monocytes (TEMs) in human peripheral blood and cancer. Blood 109, 5276-5285 (2007).
257 Verrier, C. S., Roodi, N., Yee, C. J., Bailey, L. R., Jensen, R. A., Bustin, M.and Parl, F.
F. High-mobility group (HMG) protein HMG-1 and TATA-binding protein-associated
factor TAF(II)30 affect estrogen receptor-mediated transcriptional activation. Mol
Endocrinol 11, 1009-1019 (1997).
258 Vogelzang, N. J., Rusthoven, J. J., Symanowski, J., Denham, C., Kaukel, E., Ruffie, P.,
Gatzemeier, U., Boyer, M., Emri, S., Manegold, C., Niyikiza, C.and Paoletti, P. Phase III
study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with
malignant pleural mesothelioma. J Clin Oncol 21, 2636-2644 (2003).
259 Volp, K., Brezniceanu, M. L., Bosser, S., Brabletz, T., Kirchner, T., Gottel, D., Joos,
S.and Zornig, M. Increased expression of high mobility group box 1 (HMGB1) is
associated with an elevated level of the antiapoptotic c-IAP2 protein in human colon
carcinomas. Gut 55, 234-242 (2006).
260 Wagner, J. C., Sleggs, C. A.and Marchand, P. Diffuse Pleural Mesothelioma and
Asbestos Exposure in the North Western Cape Province. Br J Ind Med 17, 260-271
(1960).
261 Walker, A. M., Loughlin, J. E., Friedlander, E. R., Rothman, K. J.and Dreyer, N. A.
Projections of asbestos-related disease 1980-2009. J Occup Med 25, 409-425 (1983).
262 Wang, H., Vishnubhakat, J. M., Bloom, O., Zhang, M., Ombrellino, M., Sama, A.and
Tracey, K. J. Proinflammatory cytokines (tumor necrosis factor and interleukin 1)
stimulate release of high mobility group protein-1 by pituicytes. Surgery 126, 389-392
(1999).
263 Wang, H., Bloom, O., Zhang, M., Vishnubhakat, J. M., Ombrellino, M., Che, J., Frazier,
A., Yang, H., Ivanova, S., Borovikova, L., Manogue, K. R., Faist, E., Abraham, E.,

130
Andersson, J., Andersson, U., Molina, P. E., Abumrad, N. N., Sama, A.and Tracey, K. J.
HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248-251 (1999).
264 Weder, W., Stahel, R. A., Bernhard, J., Bodis, S., Vogt, P., Ballabeni, P., Lardinois, D.,
Betticher, D., Schmid, R., Stupp, R., Ris, H. B., Jermann, M., Mingrone, W., Roth, A.
D.and Spiliopoulos, A. Multicenter trial of neo-adjuvant chemotherapy followed by
extrapleural pneumonectomy in malignant pleural mesothelioma. Ann Oncol 18, 1196-
1202 (2007).
265 Welt, S., Divgi, C. R., Scott, A. M., Garin-Chesa, P., Finn, R. D., Graham, M., Carswell,
E. A., Cohen, A., Larson, S. M., Old, L. J.and et al. Antibody targeting in metastatic
colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein
of reactive tumor stromal fibroblasts. J Clin Oncol 12, 1193-1203 (1994).
266 Wong, R. M., Ianculescu, I., Sharma, S., Gage, D. L., Olevsky, O. M., Kotova, S., Kostic,
M. N., Grundfest, W. S., Hou, D.and Cameron, R. B. Immunotherapy for malignant
pleural mesothelioma. Current status and future prospects. Am J Respir Cell Mol Biol 50,
870-875 (2014).
267 Wu, L., Yun, Z., Tagawa, T., De la Maza, L., Wu, M. O., Yu, J., Zhao, Y.and de Perrot,
M. Activation of CD1d-restricted natural killer T cells can inhibit cancer cell
proliferation during chemotherapy by promoting the immune responses in murine
mesothelioma. Cancer Immunol Immunother 63, 1285-1296 (2014).
268 Wu, L., Onn Wu, M., Maza, L. D. l., Yun, Z., Yu, J., Zhao, Y., Cho, J.and Perrot, M. d.
Targeting the inhibitory receptor CTLA-4 on T cells increased abscopal effects in murine
mesothelioma model. Oncotarget; Vol 6, No 14 (2015).
269 Xanthopoulos, A., Bauer, T. T., Blum, T. G., Kollmeier, J., Schonfeld, N.and Serke, M.
Gemcitabine combined with oxaliplatin in pretreated patients with malignant pleural
mesothelioma: an observational study. J Occup Med Toxicol 3, 34 (2008).
270 Yamazaki, T., Hannani, D., Poirier-Colame, V., Ladoire, S., Locher, C., Sistigu, A.,
Prada, N., Adjemian, S., Catani, J. P., Freudenberg, M., Galanos, C., Andre, F., Kroemer,
G.and Zitvogel, L. Defective immunogenic cell death of HMGB1-deficient tumors:
compensatory therapy with TLR4 agonists. Cell Death Differ 21, 69-78 (2014).
271 Yan, T. D., Boyer, M., Tin, M. M., Wong, D., Kennedy, C., McLean, J., Bannon, P.
G.and McCaughan, B. C. Extrapleural pneumonectomy for malignant pleural
mesothelioma: outcomes of treatment and prognostic factors. J Thorac Cardiovasc Surg
138, 619-624 (2009).
272 Yang, H., Ochani, M., Li, J., Qiang, X., Tanovic, M., Harris, H. E., Susarla, S. M., Ulloa,
L., Wang, H., DiRaimo, R., Czura, C. J., Wang, H., Roth, J., Warren, H. S., Fink, M. P.,
Fenton, M. J., Andersson, U.and Tracey, K. J. Reversing established sepsis with
antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A 101,
296-301 (2004).

131
273 Yang, H., Bocchetta, M., Kroczynska, B., Elmishad, A. G., Chen, Y., Liu, Z., Bubici, C.,
Mossman, B. T., Pass, H. I., Testa, J. R., Franzoso, G.and Carbone, M. TNF-alpha
inhibits asbestos-induced cytotoxicity via a NF-kappaB-dependent pathway, a possible
mechanism for asbestos-induced oncogenesis. Proc Natl Acad Sci U S A 103, 10397-
10402 (2006).
274 Yang, H., Rivera, Z., Jube, S., Nasu, M., Bertino, P., Goparaju, C., Franzoso, G., Lotze,
M. T., Krausz, T., Pass, H. I., Bianchi, M. E.and Carbone, M. Programmed necrosis
induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein
release and resultant inflammation. Proc Natl Acad Sci U S A 107, 12611-12616 (2010).
275 Yang, S., Xu, L., Yang, T.and Wang, F. High-mobility group box-1 and its role in
angiogenesis. Journal of Leukocyte Biology 95, 563-574 (2014).
276 Yuan, J., Adamow, M., Ginsberg, B. A., Rasalan, T. S., Ritter, E., Gallardo, H. F., Xu,
Y., Pogoriler, E., Terzulli, S. L., Kuk, D., Panageas, K. S., Ritter, G., Sznol, M., Halaban,
R., Jungbluth, A. A., Allison, J. P., Old, L. J., Wolchok, J. D.and Gnjatic, S. Integrated
NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in
advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci U S A 108,
16723-16728 (2011).
277 Yung, S. and Chan, T. M. Mesothelial cells. Perit Dial Int 27 Suppl 2, S110-115 (2007).
278 Zalcman, G., Mazières, J., Margery, J., Audigier-Valette, C., Moro-Sibilot, D., Molinier,
O., Corre, R., Monnet, I., Gounant, V., Janicot, H., Gervais, R., Locher, C., Milleron, B.,
Tran, Q., Lebitasy, M., Morin, F., Creveuil, C., Parienti, J. & Scherpereel, A.
Bevacizumab 15mg/kg plus cisplatin-pemetrexed (CP) triplet versus CP doublet in
Malignant Pleural Mesothelioma (MPM): Results of the IFCT-GFPC-0701 MAPS
randomized phase 3 trial. in American Society of Clinical Oncology Annual Meeting
(ASCO, 2015). at <http://meetinglibrary.asco.org/content/150191-156>
279 Zauderer, M. G. and Krug, L. M. Novel therapies in phase II and III trials for malignant
pleural mesothelioma. J Natl Compr Canc Netw 10, 42-47 (2012).
280 Zellos, L. and Christiani, D. C. Epidemiology, biologic behavior, and natural history of
mesothelioma. Thorac Surg Clin 14, 469-477, viii (2004).
281 Zhang, C. C., Krieg, S.and Shapiro, D. J. HMG-1 stimulates estrogen response element
binding by estrogen receptor from stably transfected HeLa cells. Mol Endocrinol 13, 632-
643 (1999).
282 Zhu, X. M., Yao, Y. M., Liang, H. P., Xu, S., Dong, N., Yu, Y.and Sheng, Z. Y. The
effect of high mobility group box-1 protein on splenic dendritic cell maturation in rats. J
Interferon Cytokine Res 29, 677-686 (2009).
283 Zucali, P. A., Ceresoli, G. L., Garassino, I., De Vincenzo, F., Cavina, R., Campagnoli, E.,
Cappuzzo, F., Salamina, S., Soto Parra, H. J.and Santoro, A. Gemcitabine and
vinorelbine in pemetrexed-pretreated patients with malignant pleural mesothelioma.
Cancer 112, 1555-1561 (2008).

132
Appendices

133
Copyright Acknowledgements
Figure 3 used with permission from: A Feasibility Study Evaluating Surgery for Mesothelioma
After Radiation Therapy: The “SMART” Approach for Resectable Malignant Pleural
Mesothelioma. Jan 1 2014 – Volume 9 – Issue 3 – p 397-402. Wolters Kluwer Health Lippincott
Williams & Silkins©.
Figure 7 reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Immunology:
Lotze, M. T. and Tracey, K. J. High-mobility group box 1 protein (HMGB1): nuclear weapon in
the immune arsenal. Nat Rev Immunol 5, 331-342 (2005).