highly specific auto-antibodies against claudin-18 isoform 2
TRANSCRIPT

Therapeutics, Targets, and Chemical Biology
Highly Specific Auto-Antibodies against Claudin-18 Isoform2 Induced by a Chimeric HBcAg Virus-Like Particle VaccineKill Tumor Cells and Inhibit the Growth of Lung Metastases
Thorsten Klamp1, Jens Schumacher1, Georg Huber1,5, Christoph K€uhne2, Ulrich Meissner2, Abderraouf Selmi1,Thomas Hiller1, Sebastian Kreiter3, J€urgen Markl2, Özlem T€ureci4, and Ugur Sahin1,3
AbstractStrategies for antibody-mediated cancer immunotherapy, such as active immunization with virus-like particle
(VLP)-based vaccines, are gaining increasing attention. We developed chimeric hepatitis B virus core antigen(HBcAg)-VLPs that display a surface epitope of the highly selective tumor-associated cell lineagemarker claudin-18 isoform 2 (CLDN18.2) flanked by a mobility-increasing linker. Auto-antibodies elicited by immunization withthese chimeric HBcAg-VLPs in 2 relevant species (mouse and rabbit) bind with high precision to nativeCLDN18.2 at physiologic densities on the surface of living cells but not to the corresponding epitope of theCLDN18.1 splice variant that differs by merely one amino acid. The induced auto-antibodies are capable ofefficiently killing CLDN18.2 expressing cells in vitro by complement-dependent and antibody-dependent cell-mediated cytotoxicity. Moreover, they provide partial protective immunity against the challenge of mice withsyngeneic tumor cells stably expressing CLDN18.2. Our study provides a first proof-of-concept that immuni-zation combining VLPs as antigen carriers with specific conformational epitopes of a highly selectivedifferentiation antigen may elicit auto-antibodies with high cytocidal and tumoricidal potential. Cancer Res;
71(2); 516–27. �2011 AACR.
Introduction
Monoclonal antibodies (mAb) against tumor-associatedsurface antigens have emerged as promising therapeutics inoncology (1), which execute their antitumoral activity viaseveral independent modes of action. Among these, anti-body-dependent cell-mediated (ADCC) and complement-dependent cytotoxicity (CDC) are considered the most rele-vant ones (2, 3). IgG FcgR polymorphisms were revealed to beindependent predictors of clinical outcome in patients treatedwith the antibody drugs cetuximab, trastuzumab, and ritux-imab (4–6), and polymorphisms in the complement factorC1qA correlate with prolonged response in follicular lym-phoma patients following rituximab therapy (7), further sup-porting the clinical relevance of CDC and ADCC.
The success of recombinant tumor-targeting mAbs (8, 9)has boosted various innovative antibody-based approaches.Among these, the active induction of auto-antibodies to
cancer-associated self-antigens has several theoreticaladvantages over passively administered mAbs (10). AlthoughmAbs are eliminated from the blood with half-lives rangingfrom days to weeks, the titer of vaccine-induced antibodiesmay persist for years. This and the fact that the cost of goodssold of recombinant vaccines is much lower comparedwith the cost of mAbs (11) is of key importance, as, besidescost considerations, there is no rationale for stoppingthe clinically effective administration of a recombinantantibody.
A prerequisite for the induction of specific auto-antibodiesagainst self-proteins is to break B-cell tolerance. This can beaccomplished by immunization with chimeric virus-likeparticles (VLP; ref. 12), as the highly repetitive, dense displayand spacing of the inserted epitopes seems optimal for B-cellreceptor cross-linking (13). One of the best characterizedcapsid proteins is the hepatitis B virus core antigen (HBcAg),which assembles spontaneously to particulate icosahedralnucleocapsids (14). The cloning of foreign epitopes into themajor immunodominant region (MIR) at the tip of the VLPspike confers high immunogenicity to the inserted hetero-logous sequence (15). Originally, VLPs were equipped withpathogen epitopes and were assessed as vaccines againstinfections (14). Later, self-antigen–derived epitopes, includ-ing those from cancer-associated molecules, were engi-neered into the HBcAg backbone (12). CTL responses aswell as auto-antibodies have been elicited by VLP immuni-zation (12). The capability of such auto-antibodies to med-iate cell killing via immune effector mechanisms and to
Authors' Affiliations: 1Department of Internal Medicine III, Translationaland Experimental Oncology, and 2Institute of Zoology, Johannes Guten-berg-University; 3TRON, Center of Translational Oncology and Immunol-ogy; and 4Ganymed Pharmaceuticals AG, Mainz, Germany; and 5Sectionfor Virology, Medical University Innsbruck, Innsbruck, Austria
Corresponding Author: Ugur Sahin, Department of Internal Medicine III,Translational and Experimental Oncology, Johannes Gutenberg-Univer-sity, Obere Zahlbacherstr. 63, 55131 Mainz, Germany. Phone: 49-(0)6131-17-8054; Fax: 49-(0)6131-17-8055; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-10-2292
�2011 American Association for Cancer Research.
CancerResearch
Cancer Res; 71(2) January 15, 2011516
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292

confer protection against tumor challenge, however, has sofar not been shown.The objective of our study was to evaluate VLP-induced
auto-antibodies for their capability to mediate ADCC or CDCand to provide a first proof-of-concept that prophylacticvaccination with chimeric VLPs protects mice against chal-lenge with syngeneic tumor cells.As a model antigen, we chose isoform 2 of the tight junction
molecule claudin-18 (CLDN18.2), which is associated withgastroesophageal, pancreatic, and other cancers (16, 17).CLDN18.2 is a highly selective cell-lineage marker and con-fined to short-lived differentiated epithelia of the gastricmucosa; it is absent from the gastric stem cell zone andany other healthy tissue (17). A recombinant mAb againstthis target is currently in clinical Phase II trials, which makesthe evaluation of CLDN18.2 as a target for active immuniza-tion therapy particularly interesting.
Materials and Methods
AnimalsMice were obtained from the Laboratory Animal Facility,
Johannes Gutenberg-University (Mainz, Germany), and rabbitswere obtained from Charles River Laboratories. All animalswere maintained under specific pathogen-free conditions.
Cell linesThe human gastric adenocarcinoma cell line NUGC-4,
endogenously expressing CLDN18.2, was obtained from JCRB.HEK293 and CHO-K1 cells were obtained from ATCC andstably transfected with CLDN18-encoding plasmids. MurineCT26 colon carcinoma cells from ATCC were stably trans-duced with mouse CLDN18.2.
Chimeric HBcAg-CLDN18.2-VLPsThe HBcAgD gene encodes a C-terminally truncated protein
(amino acid 1–150), which lacks the nucleotide bindingdomain and terminates with a short spacer sequence linkedto a hexahistidine-tag. This gene was codon-optimized forexpression in Escherichia coli modified to contain uniquecloning restriction sites, and cloned into plasmid pET21a(Merck). Two HBcAg-CLDN18.2 chimeras were derived fromthis backbone: CLDN-VLPs display the CLDN18.232–41 peptide(TQDLYNNPVT) in the MIR (18) replacing Pro-79 and Ala-80of HBcAgD, and CLDN-Link-VLPs feature this CLDN18.2epitope, flanked on both sides by glycine-rich linkers(G4SG4). All 3 HBcAg constructs contain a C-terminal hex-ahistidine-tag for purification (19) and for optimized assembly(20) and were expressed in the E. coli strain BL21 DE3 (Merck).For IPTG-induced large-scale expression, an inoculum wasdiluted in 2-L LB medium and incubated at 30�C for 8 to 12hours. After cell harvest, lysis, and debris removal, VLPs werepurified under mild denaturating conditions, reassembled invitro (19), and enriched by density gradient ultracentrifuga-tion. Pooled VLP-containing fractions were dialyzed againstPBS. The purity and integrity was analyzed by SDS-PAGE,nondenaturing agarose gels, and electron microscopy asdescribed previously (21, 22).
Electron microscopy and three-dimensional proteinreconstruction
Purified HBcAg-VLPs were adsorbed onto glow-dischargedcarbon-coated copper grids, negatively stained with 0.2% (w/v)uranyl acetate and analyzed with a Tecnai-12 electron micro-scope (FEI-Company) at a magnification of 68.000-fold. The 3D-reconstructions of chimeric HBcAg-CLDN18.2-VLPs with ico-sahedral triangulation numbers of T ¼ 4 were based on cryo-electron micrographs taken with a Tecnai-F20 microscope.Image analysis and 3D reconstruction was done as describedpreviously (23) with the Imagic5 software package (ImageScience); �1,400 single particles from each VLP species wereapplied. Homologymodelingwith the X-ray structure ofHBcAg-VLPs (24) as a template and modified HBcAg-sequences astargets was done in MODELLER-9v3 (http://salilab.org/model-ler/). Rigid-body and flexible fitting of the obtained molecularmodels into the cryo-EM structures were conducted with theUCSF-Chimera software (http://www.cgl.ucsf.edu/chimera/)and NMFF (http://mmtsb.org/software/nmff.html).
Immunization of animals, auto-antibody purification,and control antibodies
Immunizations were done with purified HBcAg-VLPs inPBS (Table 1); some of these were done with adjuvantsAbISCO-100 (Isconova), Montanide ISA720 (Seppic), orCFA/IFA (Sigma-Aldrich). Sera from immunized animals werecollected 1 week after each immunization and processed withthe Melon Gel IgG purification kit (Thermo Scientific).
Control mAbs ch-175D10 (IgG1, chimeric), its parentalmouse mAb mu-175D10 (IgG1, murine), and 163E12 (IgG3,murine) recognize conformational epitopes in the first extra-cellular domain of CLDN18.2 but not of CLDN18.1 and, exceptfor mu-175D10, exhibit strong target-specific CDC and ADCC.mAb 179 (IgG2a, murine) is cross-reactive with both CLDN18isoforms (17).
ELISA assaysMicroplates were coated with 500-ng antigen per well [BSA-
conjugated CLDN18.232–41 peptide (JPT Peptide Technologies)or HBcAgD-VLPs], blocked, washed, and incubated for 1 hourat room temperature with serially 10-fold diluted, nonpurifiedmouse or rabbit antisera in triplicates. Bound antibodies weredetected by species-specific, HRP-conjugated secondary anti-IgG antibodies (Jackson ImmunoResearch), followed by incu-bation with Turbo TMB (Thermo Fisher Scientific). Absor-bance at 450 nm was measured using a fluorescence platereader (Perkin Elmer). The endpoint ELISA antibody titer wascalculated as the highest dilution of antiserum, specifying anOD450 nm twice the mean, plus 3 SDs of preimmune sera at thesame dilution.
Flow cytometry–based assaysAntibody binding to native epitopes was analyzed by FACS
with CLDN18-transfected HEK293 cells or NUGC-4 cells. Cellswere washed and incubated with 1:50 diluted, Melon gel–purified, polyclonal mouse or rabbit IgGs or control mAbs(50 mg/mL) for 30 minutes at 4�C. Primary antibodies weredetected by staining with Alexa Fluor 647–conjugated goat
Auto-Antibody Induction against CLDN18.2 by VLP Immunization
www.aacrjournals.org Cancer Res; 71(2) January 15, 2011 517
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292
anti-mouse or anti-rabbit IgG secondary antibodies (Invitro-gen). Living cell counts were plotted against the fluorescenceintensity of cells in the Alexa647 channel using FACSCaliburequipment (BD Biosciences).
Immunofluorescence microscopyCHO-K1 cells were transiently transfected with plasmids
encoding CLDN18 orthologues from rabbits or mice, culturedfor 24 hours on cover slides, washed, and stained under nativeconditions with the respective antibody. The cells were fixatedin 100% methanol at �20�C, air-dried, and incubated withsecondary antibodies coupled to Cy3-fluorescent dyes (Jack-son ImmunoResearch) and with Hoechst 33342 (Invitrogen).Slides were washed, mounted in fluorescence mounting med-ium, and images were taken with a Leica DMR fluorescencemicroscope (Leica Microsystems).
Complement-dependent cytotoxicity assayCHO-K1 target cells stably transfected with human
CLDN18 variants were transiently transfected with lucifer-ase encoding in vitro–transcribed RNA, seeded in 96-welltissue culture plates, and incubated overnight at 37�C. The1:10 diluted, polyclonal antisera from immunized animalswere added to the cells in triplicate wells per serum sampleand incubated for 30 minutes at 37�C. Heat-inactivated oractive human serum, as a complement source, and D-Luci-ferin (BD Biosciences) containing reaction buffer wereadded, and luminescence signals were measured with anInfinite 200 Reader (Tecan). Cytolytic activity was calculatedas follows:
%Cell lysis¼
100%� Signal antiserum�Signal 100% killing
Signal untreated cells
� ��100
� �
Table 1. Overview of performed immunization experiments
Recognition of
Species Group/Animal no.a Immunogen Adjuvantb Route Amount linear epitope native epitopeELISAc IF/FACSd
Mouse 1
CLDN-Link-VLPs
w/o s.c. 3 � 50 mg 4/4 1/42 w/o i.v. 2 � 50 mg 5/5 2/53 CFA/IFA s.c. 2 � 50 mg 4/5 1/54 CFA/IFA s.c. 3 � 50 mg 5/5 1/55 ISA720 s.c. 3 � 50 mg 3/5 0/56 Abisco s.c. 4 � 100 mg 5/5 2/57
CLDN-VLPsw/o s.c. 3 � 50 mg 0/5 0/5
8 CFA/IFA s.c. 3 � 50 mg 3/5 0/59 ISA720 s.c. 3 � 50 mg 2/4 0/410 KLH-conjugated
CLDN18.2peptide
w/o s.c. 3 � 50 mg 1/3 0/311 CFA/IFA s.c. 2 � 50 mg 5/5 0/512 CFA/IFA s.c. 3 � 50 mg 3/3 0/313 ISA720 s.c. 3 � 50 mg 2/3 0/3
14 Degraded CLDN-Link-VLPs
CFA/IFA s.c. 3 � 50 mg 0/5 0/5
Rabbit 1 HBcAgD-VLPs w/o i.d. 4 � 100 mg � �2 CFA/IFA s.c. 4 � 100 mg � �3 CLDN-Link-
VLPsw/o i.d. 4 � 100 mg þ þ
4 CFA/IFA s.c. 4 � 100 mg þ þ5 CFA/IFA s.c. 3 � 200 mg þ þ6 ISA720 s.c. 3 � 200 mg þ þ7 CLDN-VLPs CFA/IFA s.c. 3 � 200 mg þ �8 ISA720 s.c. 3 � 200 mg þ þ
aGroups of mice consisted of 5 animals, except for groups 1 and 9 (4 animals) and groups 10, 12, and 13 (3 animals).bImmunizations were performed without adjuvants (w/o) or with CFA for primary and IFA for booster immunizations. Alternatively,Montanide ISA720 (ISA720) or AbISCO-100 (Abisco) was used.cThenumber ofmicepergroupdisplaying reactivity against the linearBSA-conjugatedCLDN18.2 32–41 peptidewasdeterminedbyELISA.þ and� symbols indicate the reactivity of rabbit sera. Animals with endpoint titers lower than�log10=2.875 were classified as negative.dThe number of mice per group displaying reactivity against CLDN18.2 in its native conformation on the surface of living cells asdetermined by IF or FACS. þ and � symbols indicate the reactivity of rabbit sera.
Klamp et al.
Cancer Res; 71(2) January 15, 2011 Cancer Research518
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292

Antibody-dependent cellular cytotoxicity assayNUGC-4 target cells were transiently transfected with luci-
ferase encoding RNA, seeded in 96-well tissue culture plates,and incubated overnight at 37�C. Plates were incubated withpurified polyclonal IgG from the sera of immunized animals.Ficoll-Paque–purified human PBMCs were added as effectorcells (effector to target ratio¼ 10:1) and incubated for 5 hours,followed by the addition of D-Luciferin containing reactionbuffer. Cytolytic activity was determined as described above.Cells incubated only with antibodies, untreated, and comple-tely lysed cells served as a reference.
Assessment of prophylactic vaccination efficacy in vivoBALB/c mice were vaccinated 3 times (day 1, day 14, and
day 28) with 50 mg HBcAgD-, CLDN-Link-VLPs, or PBS ascontrol, all formulated in AbISCO-100 (Isconova). Two weeksafter the last immunization, 1 � 105 syngeneic CT26 coloncancer cells stably expressing murine CLDN18.2 were admi-nistered into the tail vein. Thirteen days later, the mice weresacrificed and their lungs were weighed and subjected tomicroscopic analysis to assess the load of pulmonary metas-tases. Statistical analysis of lung weights was done by ANOVAfollowed by the Tukey test. For histopathologic assessment, 3-mm thick sections of formalin-fixed and paraffin-embeddedlungs were deparaffinized and rehydrated, followed by heat-induced epitope retrieval in a citrate buffer at pH 6. After thequenching of endogenous peroxidases by H2O2, unspecificantibody binding sites were blocked with 10% goat serum,followed by overnight incubation with polyclonal rabbit anti-CLDN18 (Mid) (Invitrogen) at 4�C. For the detection of bind-ing, an HRP-conjugated secondary antibody (BrightVisionPoly-HRP–Anti-rabbit, Immunologic) and the Vector NovaR-EDTM kit (Vector Laboratories) were used. After hematoxylincounterstaining, dehydration, and mounting, sections weredocumented using a MIRAX SCAN (Zeiss). Ratios of tumorand normal tissue areas were found using ImageJ Softwarev.1.44. Statistical differences between groups were assessed byANOVA, followed by Dunn's test.
Results
Design of chimeric HBcAg-CLDN18.2-VLPs for theinduction of CLDN18.2 specific antibodies againstorthologues from 3 speciesOur objective was to design a VLP-based vaccine (i) for the
induction of auto-antibodies with specificity against thenative CLDN18.2 protein, (ii) with potent cytocidal effectorfunctions and (iii) that was suitable for valid proof-of-conceptstudies in animal models. These specifications imposed sev-eral requirements on the epitope to be selected. First, cross-reactivity of antibodies with the highly related splice variant 1of CLDN18, which is expressed in healthy lung tissue, had to beprevented to avoid harm to this toxicity-relevant organ (17,25). Both splice variants of the tetraspanin CLDN18 vary intheir first extracellular domain (8 amino acids of 51 differbetween both isoforms; Fig. 1A); however, the second extra-cellular loop is identical in both isoforms and, thus, is notsuitable for discrimination. Second, the localization of the
targeted epitope close to the cell membrane was regarded asbeneficial for the Fc-mediated cytolytic activity of inducedantibodies (26, 27). Third, biophysical and biochemical prop-erties had to facilitate VLP assembly (28). Fourth, since wewanted to evaluate these VLPs in mice and rabbits as proof-of-concept for subsequent clinical testing in humans, the epitopehad to be conserved across these species.
All criteria were satisfied by the CLDN18.232–41 peptide(TQDLYNNPVT) derived from the first extracellular loop ofCLDN18.2. Most importantly, this peptide differed from thecorresponding CLDN18.1 epitope by a single amino acid(Fig. 1A). The sequence was cloned into the MIR of theHBcAgD carrier molecule either directly (resulting inCLDN-VLPs) or flanked by flexible glycine-rich linkers (yield-ing CLDN-Link-VLPs; Fig. 1B).
Generation and characterization of HBcAg-CLDN18.2-VLPs fulfilling integrity and quality criteria
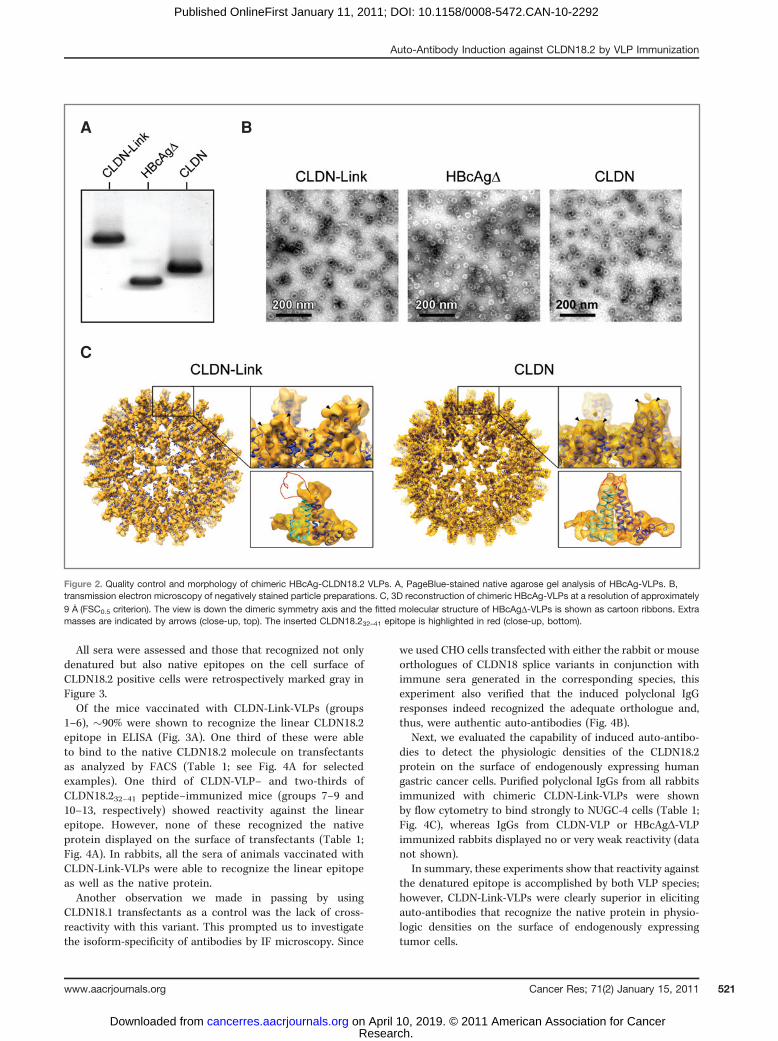
Purified chimeric and HBcAgD-VLPs were found to migrateas distinct sharp bands in native agarose gel electrophoresis(Fig. 2A), indicating the particulate nature of the proteins.Ethidium bromide staining of native agarose gels did notreveal the incorporation of nucleic acids into the VLPs (datanot shown).
Negative-staining TEM confirmed VLP formation and theintegrity of all constructs. Moreover, it showed an almostuniform size distribution of chimeric HBcAg-CLDN18.2-VLPs,which had a capsid diameter of �35 nm and were morpho-logically indistinguishable from HBcAgD-VLPs (Fig. 2B). The3D-reconstructions were processed from cryo-electron micro-graphs of chimeric VLPs. Into these 3D volumes, we con-ducted rigid-body fitting of the HBcAgD-VLP X-ray structureand revealed additional masses at the tips of their capsidspikes (Fig. 2C). Homology modeling and flexible fittingshowed that these extra masses on the tip of CLDN-VLPscorresponded to the theoretical volume of the insertedCLDN18.232–41 peptide. The calculated loop structure of theinserted epitope was further supported by a Ramachandranplot (data not shown). Extra masses on the tip of the CLDN-Link-VLP spikes, in contrast, displayed a volume too small forthe CLDN18.232–41 epitope plus flanking linkers. Modeling ofthe entire insert (close-up in Fig. 2C) indicated that the apicalloop region of CLDN-Link-VLPs exhibits a high morphologicflexibility. The failure of the applied computational recon-struction method to resolve the structure by picture averagingresulted in a blurring of this region.
In summary, these data imply that the intrinsic assemblycapacity of the HBcAg backbone is not hampered by theinserted CLDN18.2 epitope and that flanking linkers inCLDN-Link-VLPs provide a high conformational flexibility.
Immunization of mice and rabbits with HBcAg-CLDN18.2-VLPs results in anti-CLDN18.2 antibodiesrecognizing a linear epitope of this self-protein
The potency of chimeric VLPs to induce antibody responsesagainst the inserted CLDN18.2 epitope was analyzed throughthe immunization of mice and rabbits. Importantly, theselected epitope and the tissue distribution of the orthologous
Auto-Antibody Induction against CLDN18.2 by VLP Immunization
www.aacrjournals.org Cancer Res; 71(2) January 15, 2011 519
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292

proteins with strict restriction to short-lived gastric cells areconserved in all 3 species (manuscript in preparation).
BALB/c mice were immunized with CLDN-, CLDN-Link-VLPs, or with KLH-conjugated linear CLDN18.232–41 peptideas a control, and antibody reactivity against the linearCLDN18.232–41 peptide or the HBcAg backbone was foundby measuring the ELISA endpoint titer (Fig. 3A). Differentimmunization protocols were applied—varying the adju-vant, administration route, and immunogen amount—eachto groups of 3 to 5 mice (Table 1). We observed that serafrom mice immunized with CLDN-Link-VLPs displayed ahigher specific reactivity against the linear BSA-conjugatedCLDN18.232–41 peptide as compared with mice in othergroups (Fig. 3A top panel). Interestingly, mice immunizeds.c. with CLDN-Link-VLPs without the addition of adjuvant(group 1 in Fig. 3A) revealed the highest mean endpoint titeragainst the peptide. Immunization with CLDN-VLP or pep-tide vaccine formulations, in contrast, resulted less fre-quently in high endpoint titers (nearly all vs. one third;Table 1). All VLPs induced antibodies against the HBcAg
backbone with similar endpoint titers (Fig. 3A bottompanel). Heat denaturation of chimeric VLPs abrogated theircapability to induce peptide-binding antibodies withoutcompromising the development of antibodies against thebackbone.
Also, in rabbits, we accomplished the induction of high-titerIgG responses against the inserted peptide epitope. CLDN-Link-VLPs gave moderately higher endpoint titers as com-pared with CLDN-VLPs, again without clear benefit from aco-application of adjuvant (Fig. 3B).
Immunization with CLDN-Link-VLPs results in splice-variant–specific auto-antibodies recognizing CLDN18.2on the surface of endogenously expressing cancer cells
Since antibodies binding to a denatured epitope in a pep-tide ELISA are not necessarily capable of recognizing theCLDN18.2 protein in its native conformation on living cells,we tested polyclonal IgG purified from the sera of all immu-nized animals on unfixated target-positive cells by FACS andIF microscopy.
A
B
Figure 1. CLDN18.2 epitopeselection and design of chimericHBcAg-CLDN18.2 constructs.A, cross-species sequencealignment of the first extracellulardomain (black rectangle) ofCLDN18 isoform orthologues andCLDN18.232–41 epitope (gray box)selected for insertion into HBcAg.B, schematic representations ofVLP constructs. The nucleotide-binding domain of wild-typeHBcAg was replaced by a glycine-glycine-serine linker and ahexahistidine-tag (black box). Inchimeric HBcAg-CLDN18.2constructs, aa 79 and 80(P, proline; A, alanine) werereplaced by the selectedCLDN18.232–41 epitope(TQDLYNNPVT; CLDN-VLP) or bythe epitope flanked with glycine-rich linkers (amino acid sequenceG4SG4; CLDN-Link-VLP).
Klamp et al.
Cancer Res; 71(2) January 15, 2011 Cancer Research520
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292
All sera were assessed and those that recognized not onlydenatured but also native epitopes on the cell surface ofCLDN18.2 positive cells were retrospectively marked gray inFigure 3.Of the mice vaccinated with CLDN-Link-VLPs (groups
1–6), �90% were shown to recognize the linear CLDN18.2epitope in ELISA (Fig. 3A). One third of these were ableto bind to the native CLDN18.2 molecule on transfectantsas analyzed by FACS (Table 1; see Fig. 4A for selectedexamples). One third of CLDN-VLP– and two-thirds ofCLDN18.232–41 peptide–immunized mice (groups 7–9 and10–13, respectively) showed reactivity against the linearepitope. However, none of these recognized the nativeprotein displayed on the surface of transfectants (Table 1;Fig. 4A). In rabbits, all the sera of animals vaccinated withCLDN-Link-VLPs were able to recognize the linear epitopeas well as the native protein.Another observation we made in passing by using
CLDN18.1 transfectants as a control was the lack of cross-reactivity with this variant. This prompted us to investigatethe isoform-specificity of antibodies by IF microscopy. Since
we used CHO cells transfected with either the rabbit or mouseorthologues of CLDN18 splice variants in conjunction withimmune sera generated in the corresponding species, thisexperiment also verified that the induced polyclonal IgGresponses indeed recognized the adequate orthologue and,thus, were authentic auto-antibodies (Fig. 4B).
Next, we evaluated the capability of induced auto-antibo-dies to detect the physiologic densities of the CLDN18.2protein on the surface of endogenously expressing humangastric cancer cells. Purified polyclonal IgGs from all rabbitsimmunized with chimeric CLDN-Link-VLPs were shownby flow cytometry to bind strongly to NUGC-4 cells (Table 1;Fig. 4C), whereas IgGs from CLDN-VLP or HBcAgD-VLPimmunized rabbits displayed no or very weak reactivity (datanot shown).
In summary, these experiments show that reactivity againstthe denatured epitope is accomplished by both VLP species;however, CLDN-Link-VLPs were clearly superior in elicitingauto-antibodies that recognize the native protein in physio-logic densities on the surface of endogenously expressingtumor cells.
BA
C
Figure 2. Quality control and morphology of chimeric HBcAg-CLDN18.2 VLPs. A, PageBlue-stained native agarose gel analysis of HBcAg-VLPs. B,transmission electron microscopy of negatively stained particle preparations. C, 3D reconstruction of chimeric HBcAg-VLPs at a resolution of approximately9 A
�(FSC0.5 criterion). The view is down the dimeric symmetry axis and the fitted molecular structure of HBcAgD-VLPs is shown as cartoon ribbons. Extra
masses are indicated by arrows (close-up, top). The inserted CLDN18.232–41 epitope is highlighted in red (close-up, bottom).
Auto-Antibody Induction against CLDN18.2 by VLP Immunization
www.aacrjournals.org Cancer Res; 71(2) January 15, 2011 521
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292

Auto-antibodies against CLDN18.2 induced byimmunization with CLDN-Link-VLPs kill target positivecells by CDC and ADCC
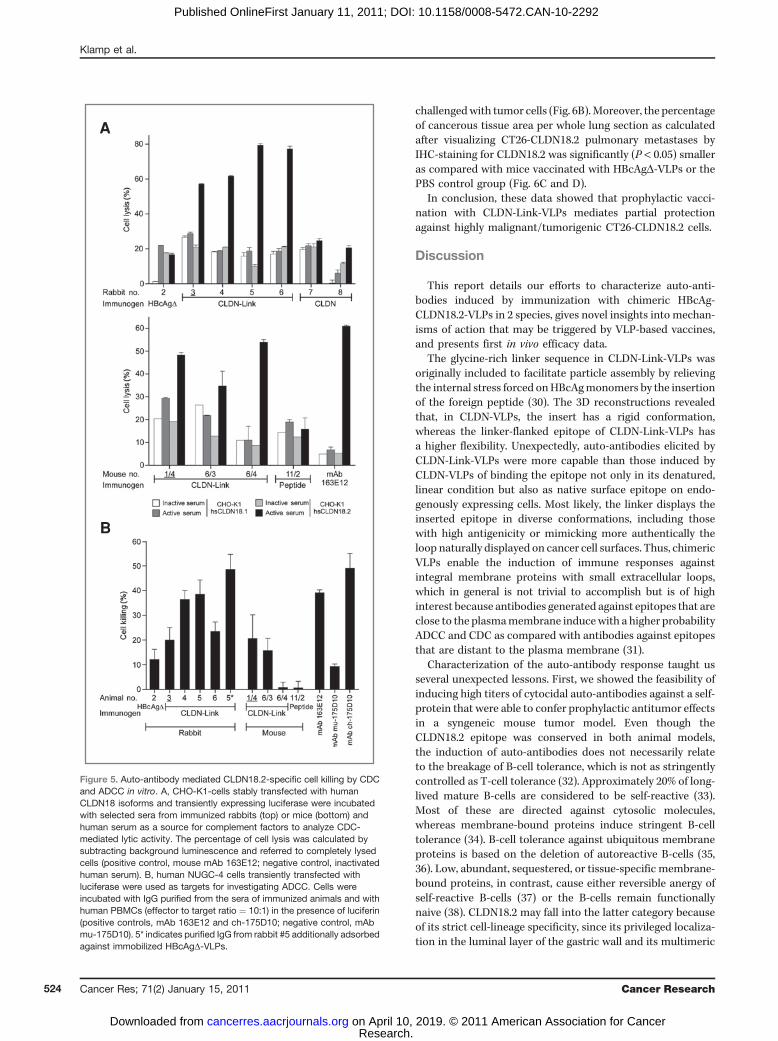
A key objective of this study was to induce specific auto-antibodies, capable of lysing cells by Fc-mediated immuneeffectormechanisms such as ADCC and CDC. Purified IgG fromsera, for which we had shown target-specific binding to cellsurface–displayed epitopes, were subjected to luciferase-basedCDC and ADCC in vitro assays and compared with recombinantmAbs 163E12 and ch-175D10 as positive controls.
Sera from rabbits immunized with CLDN-Link-VLPs lysedCLDN18.2 transfected cells in a complement factor–depen-dent manner with killing rates of up to �80%. Most impor-tantly, CLDN18.1 transfected cells were not lysed, and serafrom rabbits immunized with CLDN-VLPs or HBcAgD-VLPshad no effect (Fig. 5A top panel). Analogously, exclusivelyantisera from mice immunized with CLDN-Link-VLPs andpreviously shown to recognize native CLDN18.2 protein, dis-played strong target-specific CDC with lytic activity of up to55% and was comparable to cell lysis obtained by mAb 163E12(Fig. 5A bottom panel).
ADCC was analyzed by incubating purified serum IgGs withluciferase expressing NUGC-4 cells as targets and humanPBMCs as effectors. IgG derived from rabbits immunized withCLDN-Link-VLPs resulted in cell killing of up to 45% (Fig. 5B),reaching levels observed with mAbs 163E12 and ch-175D10and known to bind CLDN18.2 with nanomolar affinities.Antibodies from rabbits immunized with HBcAgD-VLPs, incontrast, were only marginally active in ADCC (Fig. 5B). Theremoval of VLP-backbone reactive antibodies with column–immobilized HBcAgD-VLPs further augmented the ADCC ofthe respective serum (asterisk in Fig. 5B). Similarly, exclusivelyIgGs derived from CLDN-Link-VLP–immunized but not frompeptide-immunized mice mediated ADCC. However, the over-all activity was less profound, which is in line with the notionthat several mouse antibody isoforms (particularly the IgG1isotype antibodies) are known to be very inefficient in theinduction of ADCC with human effector cells (29). Accord-ingly, the murine IgG1 mAb mu-175D10 did only marginallyinduce ADCC, whereas the murine mAb 163E12 (IgG3 isotype)and a chimerized version of mAb 175D10 were found to bevery effective.
In summary, these results proved that CLDN-Link-VLPvaccination elicits auto-antibodies efficiently killing CLDN18.2expressing cells by CDC and ADCC.
Prophylactic vaccination with CLDN-Link-VLPs conferspartial protection in an immunocompetent syngeneicmouse tumor model
To evaluate the prophylactic in vivo efficacy of CLDN-Link-VLPs, we used a syngeneic tumormodel in immunocompetentBALB/c mice in which pulmonary metastasis formation wasinduced by the i.v. application of CT26 colon cancer cells thatwere stably transduced with murine CLDN18.2. Macroscopicanalysis of the lungs derived frommice vaccinated with CLDN-Link-VLPs revealed a smaller number of metastatic nodules ascompared with HBcAgD-VLP or PBS control groups (Fig. 6A)and significantly lower lung weights close to those of mice not
A
B
Figure 3. Serum antibody reactivity against the linear CLDN18.232–41epitope and the HBcAg carrier molecule. A, BALB/c mice (3–5 per group)were immunized with chimeric HBcAg-VLPs and KLH-conjugatedCLDN18.232–41 peptide as positive or heat-degraded CLDN-Link-VLPs (*)as negative control using different immunization protocols (see Table 1).Antibody endpoint titers against CLDN18.232–41 peptide (top) and againstthe HBcAg carrier (bottom) were determined by ELISA. Endpoint titers areindicated as reciprocals of the highest dilutions of sera required to yield anOD450 nm twice as high plus 3 SDs of preimmune sera at the same dilution.The mean value of each group is indicated by a bar. Endpoint titers lowerthan �log10 ¼ 2.9 (indicated by a dashed line) were classified as negative.B, NZW rabbits were subjected to a similar immunization procedure (seeTable 1) and the reactivity of sera was determined. Error bars represent theSD of triplicate experiments. Light gray–filled circles/bars indicate serarecognizing the protein expressed on cells in its native (versus onlydenatured¼ black circles/bars) conformation (see Fig. 4). Groups or singleanimals treated without adjuvant are underlined.
Klamp et al.
Cancer Res; 71(2) January 15, 2011 Cancer Research522
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292

A
C
B
HEK293 hsCLDN18.1
HEK293 hsCLDN18.2
Figure 4. Specificities of auto-antibodies to native CLDN18.2 expressed on the surface of living cells. All sera were investigated by FACS or IF forreactivity against the cell surface–displayed native epitope (see Table 1, last column). Selected examples are shown in this figure. A, HEK293 cells stablyexpressing human CLDN18 isoforms were stained under native conditions with polyclonal IgG purified from the sera of immunized rabbits or mice.The immunogens used for vaccination and identifiers of serum-donating animals (e.g., mouse 1/4 means mouse 4 in group 1) are indicated (see Table 1).B, CHO-K1 cells transiently expressing CLDN18 isoform orthologues from rabbits (oc) or mice (mm) were stained under native conditions with polyclonalIgG purified from the sera of CLDN-Link-VLP–immunized animals and analyzed by IF microscopy (controls, isoform cross-reactive mAb179 to show thefunctionality of constructs and CLDN18.2 specific mAb ch-175D10). C, binding of sera from immunized rabbits to NUGC-4 cells was found under nativeconditions by flow cytometry. Controls, prebleed sera and mAb ch-175D10.
Auto-Antibody Induction against CLDN18.2 by VLP Immunization
www.aacrjournals.org Cancer Res; 71(2) January 15, 2011 523
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292
challengedwith tumor cells (Fig. 6B).Moreover, the percentageof cancerous tissue area per whole lung section as calculatedafter visualizing CT26-CLDN18.2 pulmonary metastases byIHC-staining for CLDN18.2 was significantly (P < 0.05) smalleras compared with mice vaccinated with HBcAgD-VLPs or thePBS control group (Fig. 6C and D).
In conclusion, these data showed that prophylactic vacci-nation with CLDN-Link-VLPs mediates partial protectionagainst highly malignant/tumorigenic CT26-CLDN18.2 cells.
Discussion
This report details our efforts to characterize auto-anti-bodies induced by immunization with chimeric HBcAg-CLDN18.2-VLPs in 2 species, gives novel insights into mechan-isms of action that may be triggered by VLP-based vaccines,and presents first in vivo efficacy data.
The glycine-rich linker sequence in CLDN-Link-VLPs wasoriginally included to facilitate particle assembly by relievingthe internal stress forced onHBcAgmonomers by the insertionof the foreign peptide (30). The 3D reconstructions revealedthat, in CLDN-VLPs, the insert has a rigid conformation,whereas the linker-flanked epitope of CLDN-Link-VLPs hasa higher flexibility. Unexpectedly, auto-antibodies elicited byCLDN-Link-VLPs were more capable than those induced byCLDN-VLPs of binding the epitope not only in its denatured,linear condition but also as native surface epitope on endo-genously expressing cells. Most likely, the linker displays theinserted epitope in diverse conformations, including thosewith high antigenicity or mimicking more authentically theloop naturally displayed on cancer cell surfaces. Thus, chimericVLPs enable the induction of immune responses againstintegral membrane proteins with small extracellular loops,which in general is not trivial to accomplish but is of highinterest because antibodies generated against epitopes that areclose to the plasmamembrane inducewith a higher probabilityADCC and CDC as compared with antibodies against epitopesthat are distant to the plasma membrane (31).
Characterization of the auto-antibody response taught usseveral unexpected lessons. First, we showed the feasibility ofinducing high titers of cytocidal auto-antibodies against a self-protein that were able to confer prophylactic antitumor effectsin a syngeneic mouse tumor model. Even though theCLDN18.2 epitope was conserved in both animal models,the induction of auto-antibodies does not necessarily relateto the breakage of B-cell tolerance, which is not as stringentlycontrolled as T-cell tolerance (32). Approximately 20% of long-lived mature B-cells are considered to be self-reactive (33).Most of these are directed against cytosolic molecules,whereas membrane-bound proteins induce stringent B-celltolerance (34). B-cell tolerance against ubiquitous membraneproteins is based on the deletion of autoreactive B-cells (35,36). Low, abundant, sequestered, or tissue-specific membrane-bound proteins, in contrast, cause either reversible anergy ofself-reactive B-cells (37) or the B-cells remain functionallynaive (38). CLDN18.2 may fall into the latter category becauseof its strict cell-lineage specificity, since its privileged localiza-tion in the luminal layer of the gastric wall and its multimeric
A
B
Figure 5. Auto-antibody mediated CLDN18.2-specific cell killing by CDCand ADCC in vitro. A, CHO-K1-cells stably transfected with humanCLDN18 isoforms and transiently expressing luciferase were incubatedwith selected sera from immunized rabbits (top) or mice (bottom) andhuman serum as a source for complement factors to analyze CDC-mediated lytic activity. The percentage of cell lysis was calculated bysubtracting background luminescence and referred to completely lysedcells (positive control, mouse mAb 163E12; negative control, inactivatedhuman serum). B, human NUGC-4 cells transiently transfected withluciferase were used as targets for investigating ADCC. Cells wereincubated with IgG purified from the sera of immunized animals and withhuman PBMCs (effector to target ratio ¼ 10:1) in the presence of luciferin(positive controls, mAb 163E12 and ch-175D10; negative control, mAbmu-175D10). 5* indicates purified IgG from rabbit #5 additionally adsorbedagainst immobilized HBcAgD-VLPs.
Klamp et al.
Cancer Res; 71(2) January 15, 2011 Cancer Research524
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292

complexing with other tight junction components causesmany epitopes to be shielded under physiologic conditions.Second, a single amino acid difference in the selected
CLDN18.2 epitope was sufficient to prevent cross-reactivity
with CLDN18 splice variant 1 with potentially harmful effectson lung tissue.
Third, we showed for the first time that VLPs may induceauto-antibodies, which were capable of efficiently killing
Figure 6. Prophylactic vaccinationof CLDN-Link-VLPs againstchallenge with syngeneicCLDN18.2 expressing tumor cells.A, selected lungs derived frommice vaccinated with therespective compounds incombination with AbISCO-100 asadjuvant. Arrowheads indicatepulmonary CT26-CLDN18.2metastases. NT, nonchallengedand nontreated control. B, scatterplot analysis of lung weightdistribution. The mean value ofeach group is indicated.Significant differences betweenexperimental groups wereassessed by ANOVA followed byTukey's test (P < 0.05). C, IHCanalysis of CLDN18.2 positivepulmonary metastases. Thesections correspond to the lungsshown in A. D, statistical analysisof the metastatic area. Thepercentage of metastatic tissuewas calculated in relation to thewhole area of the lung section. Themean, 95% confidence interval,and P-values (ANOVA followed byDunn's test) for each group areindicated. The mean of the PBScontrol group was set to 100%.
A
B
C
D
Met
asta
tic a
rea
(%)
Auto-Antibody Induction against CLDN18.2 by VLP Immunization
www.aacrjournals.org Cancer Res; 71(2) January 15, 2011 525
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292

target-positive cells by ADCC and CDC. VLP-based vaccineshave been primarily used to elicit immunity against infectionsor to develop treatments against chronic autoimmune dis-eases (12, 14, 39). In both indications, the objective is to induceauto-antibodies capable of blocking or neutralizing pathogensor autoreactivity-pertaining self-molecules (39–41). Modesof action required for immunotherapy for nonviral cancer,however, are different. Few studies have used VLPs to induceimmunity against cancer-associated self-antigens (42–50). Innone of these studies were auto-antibodies with antitumoralmode-of-action reported. Since we used cross-speciesreagents for conducting killing assays (human serum orPBMCs combined with mouse or rabbit antisera), cytocidalactivities might even be underestimated (29). Both modes ofaction are likely to also be responsible for the observedprotective immunity against syngeneic tumor challenge afterprophylactic vaccination with CLDN-Link-VLPs.
In summary, this is the first report demonstrating a chimericVLP-based vaccine that elicits high titers of polyclonal auto-antibodies that are capable of recognizing, with high precision,native epitopes of membrane-bound tumor-associated anti-gens on cancer cells and eliciting cytotoxic activity. This opensnew paths for the development of VLPs for cancer therapy.
Disclosure of Potential Conflicts of InterestSeveral authors (U. Sahin, Ö. T€ureci, T. Klamp, J. Schumacher, T. Hiller) are
inventors on a patent application describing HBcAg-CLDN18.2-VLPs for cancerimmunotherapy. Ö. T€ureci is the CEO/CSO of Ganymed Pharmaceuticals, acompany holding patent applications on CLDN18.2 as a therapeutic target forrecombinant antibody therapy of cancer. U. Sahin is the CEO of BioNTech, acompany developing VLP-based cancer vaccines. All other authors declare nopotential conflict of interest.
Acknowledgments
We are grateful for the immunochemistry analysis provided by Dr. ChristophRohde (Ganymed).
Grant Support
This work was supported by the Combined Project Grant SFB 432 (Ö. T€ureci,U. Sahin) and the Research Fund of the Johannes Gutenberg-University, Mainz(T. Klamp, U. Meissner).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received June 23, 2010; revised October 14, 2010; accepted November 1, 2010;published OnlineFirst January 11, 2011.
References1. Oldham RK, Dillman RO. Monoclonal antibodies in cancer therapy: 25
years of progress. J Clin Oncol 2008;26:1774–7.2. Griggs J, Zinkewich-Peotti K. The state of the art: immune-mediated
mechanisms of monoclonal antibodies in cancer therapy. Br J Cancer2009;101:1807–12.
3. Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. CurrOpin Immunol 2007;19:239–45.
4. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, ColombatP, et al. Therapeutic activity of humanized anti-CD20 monoclonalantibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene.Blood 2002;99:754–8.
5. Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G,et al. Immunoglobulin G fragment C receptor polymorphisms andclinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol 2008;26:1789–96.
6. Bibeau F, Lopez-Crapez E, Di FF, Thezenas S, Ychou M, Blanchard F,et al. Impact of Fc(gamma)RIIa-Fc(gamma)RIIIa polymorphisms andKRAS mutations on the clinical outcome of patients with metastaticcolorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol2009;27:1122–9.
7. Racila E, Link BK, Weng WK, Witzig TE, Ansell S, Maurer MJ, et al. Apolymorphism in the complement component C1qA correlates withprolonged response following rituximab therapy of follicular lym-phoma. Clin Cancer Res 2008;14:6697–703.
8. Dalle S, Thieblemont C, Thomas L, Dumontet C. Monoclonal anti-bodies in clinical oncology. Anticancer Agents Med Chem 2008;8:523–32.
9. Scolnik PA. mAbs: a business perspective. MAbs 2009;1:179–84.10. Xiang SD, Scalzo-Inguanti K, Minigo G, Park A, Hardy CL, Plebanski
M. Promising particle-based vaccines in cancer therapy. Expert RevVaccines 2008;7:1103–19.
11. Spohn G, Bachmann MF. Exploiting viral properties for the rationaldesign of modern vaccines. Expert Rev Vaccines 2008;7:43–54.
12. Chackerian B. Virus-like particles: flexible platforms for vaccine devel-opment. Expert Rev Vaccines 2007;6:381–90.
13. Milich DR, Chen M, Schodel F, Peterson DL, Jones JE, Hughes JL.Role of B cells in antigen presentation of the hepatitis B core. Proc NatlAcad Sci U S A 1997;94:14648–53.
14. Pumpens P, Grens E. HBV core particles as a carrier for B cell/T cellepitopes. Intervirology 2001;44:98–114.
15. Schodel F, Moriarty AM, Peterson DL, Zheng JA, Hughes JL, Will H,et al. The position of heterologous epitopes inserted in hepatitis Bvirus core particles determines their immunogenicity. J Virol1992;66:106–14.
16. Karanjawala ZE, Illei PB, Ashfaq R, Infante JR, Murphy K, Pandey A,et al. New markers of pancreatic cancer identified through differentialgene expression analyses: claudin 18 and annexin A8. Am J SurgPathol 2008;32:188–96.
17. Sahin U, Koslowski M, Dhaene K, Usener D, Brandenburg G, Seitz G,et al. Claudin-18 splice variant 2 is a pan-cancer target suitable fortherapeutic antibody development. Clin Cancer Res 2008;14:7624–34.
18. Salfeld J, Pfaff E, Noah M, Schaller H. Antigenic determinants andfunctional domains in core antigen and e antigen from hepatitis Bvirus. J Virol 1989;63:798–808.
19. Wizemann H, von BA. Purification of E. coli-expressed HIS-taggedhepatitis B core antigen by Ni2þ-chelate affinity chromatography. JVirol Methods 1999;77:189–97.
20. Vogel M, Vorreiter J, Nassal M. Quaternary structure is critical forprotein display on capsid-like particles (CLPs): efficient generation ofhepatitis B virus CLPs presenting monomeric but not dimeric andtetrameric fluorescent proteins. Proteins 2005;58:478–88.
21. Serwer P, Khan SA, Griess GA. Non-denaturing gel electrophore-sis of biological nanoparticles: viruses. J Chromatogr A 1995;698:251–61.
22. Harris JR. Negative staining of thinly spread biological samples.Methods Mol Biol 2007;369:107–42.
23. Meissner U, Gatsogiannis C, Moeller A, Depoix F, Harris JR, Markl J.Comparative 11A
�structure of two molluscan hemocyanins from 3D
cryo-electron microscopy. Micron 2007;38:754–65.24. Wynne SA, Crowther RA, Leslie AG. The crystal structure of the
human hepatitis B virus capsid. Mol Cell 1999;3:771–80.25. Niimi T, Nagashima K, Ward JM, Minoo P, Zimonjic DB, Popescu NC,
et al. Claudin-18, a novel downstream target gene for the T/EBP/NKX2.1 homeodomain transcription factor, encodes lung- and sto-mach-specific isoforms through alternative splicing. Mol Cell Biol2001;21:7380–90.
Klamp et al.
Cancer Res; 71(2) January 15, 2011 Cancer Research526
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292

26. Cragg MS, Walshe CA, Ivanov AO, Glennie MJ. The biology of CD20and its potential as a target for mAb therapy. Curr Dir Autoimmun2005;8:140–74.
27. Ruuls SR, Lammerts van Bueren JJ, van de Winkel JG, Parren PW.Novel human antibody therapeutics: the age of the UmAbs. Biotech-nol J 2008;3:1157–71.
28. Karpenko LI, Ivanisenko VA, Pika IA, Chikaev NA, Eroshkin AM,Veremeiko TA, et al. Insertion of foreign epitopes in HBcAg: how tomake the chimeric particle assemble. Amino Acids 2000;18:329–37.
29. Ortaldo JR, Woodhouse C, Morgan AC, Herberman RB, Cheresh DA,Reisfeld R. Analysis of effector cells in human antibody-dependentcellular cytotoxicity with murine monoclonal antibodies. J Immunol1987;138:3566–72.
30. Kratz PA, Bottcher B, Nassal M. Native display of complete foreignprotein domains on the surface of hepatitis B virus capsids. Proc NatlAcad Sci USA 1999;96:1915–20.
31. Beers SA, Chan CH, French RR, Cragg MS, Glennie MJ. CD20 as atarget for therapeutic type I and II monoclonal antibodies. SeminHematol 2010;47:107–14.
32. Zinkernagel RM, Pircher HP, Ohashi P, Oehen S, Odermatt B, Mak T,et al. T and B cell tolerance and responses to viral antigens intransgenic mice: implications for the pathogenesis of autoimmuneversus immunopathological disease. Immunol Rev 1991;122:133–71.
33. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nus-senzweig MC. Predominant autoantibody production by early humanB cell precursors. Science 2003;301:1374–7.
34. Dyer MR, Renner WA, Bachmann MF. A second vaccine revolution forthe new epidemics of the 21st century. Drug Discov Today2006;11:1028–33.
35. Goodnow CC. B-cell tolerance. Curr Opin Immunol 1992;4:703–10.36. Ferry H, Jones M, Vaux DJ, Roberts IS, Cornall RJ. The cellular
location of self-antigen determines the positive and negative selectionof autoreactive B cells. J Exp Med 2003;198:1415–25.
37. Bachmann MF, Rohrer UH, Kundig TM, Burki K, Hengartner H,Zinkernagel RM. The influence of antigen organization on B cellresponsiveness. Science 1993;262:1448–51.
38. Ferry H, Leung JC, Lewis G, Nijnik A, Silver K, Lambe T, et al. B-celltolerance. Transplantation 2006;81:308–15.
39. Jennings GT, Bachmann MF. The coming of age of virus-like particlevaccines. Biol Chem 2008;389:521–36.
40. Spohn G, Bachmann MF. Therapeutic vaccination to block receptor-ligand interactions. Expert Opin Biol Ther 2003;3:469–76.
41. Hunter Z, Smyth HD, Durfee P, Chackerian B. Induction of mucosaland systemic antibody responses against the HIV coreceptor CCR5upon intramuscular immunization and aerosol delivery of a virus-likeparticle based vaccine. Vaccine 2009;28:403–14.
42. Ramqvist T, Andreasson K, Dalianis T. Vaccination, immune and genetherapy based on virus-like particles against viral infections andcancer. Expert Opin Biol Ther 2007;7:997–1007.
43. Tegerstedt K, Lindencrona JA, Curcio C, Andreasson K, Tullus C,Forni G, et al. A single vaccination with polyomavirus VP1/VP2Her2virus-like particles prevents outgrowth of HER-2/neu-expressingtumors. Cancer Res 2005;65:5953–7.
44. Gathuru JK, Koide F, Ragupathi G, Adams JL, Kerns RT, ColemanTP, et al. Identification of DHBcAg as a potent carrier proteincomparable to KLH for augmenting MUC1 antigenicity. Vaccine2005;23:4727–33.
45. Kazaks A, Balmaks R, Voronkova T, Ose V, Pumpens P. Melanomavaccine candidates from chimeric hepatitis B core virus-like particlescarrying a tumor-associated MAGE-3 epitope. Biotechnol J2008;3:1429–36.
46. Zhang Y, Song S, Liu C, Wang Y, Xian X, He Y, et al. Generation ofchimeric HBc proteins with epitopes in E. coli: formation of virus-likeparticles and a potent inducer of antigen-specific cytotoxic immuneresponse and anti-tumor effect in vivo. Cell Immunol 2007;247:18–27.
47. Storni T, Ruedl C, Schwarz K, Schwendener RA, Renner WA, Bach-mann MF. Nonmethylated CG motifs packaged into virus-like parti-cles induce protective cytotoxic T cell responses in the absence ofsystemic side effects. J Immunol 2004;172:1777–85.
48. Kong J, Diao Z, Deng X, Zhong H, Yao W, Hu X. Anti-tumor effects ofimmunotherapeutic peptide on the treatment of hepatocellular carci-noma with HBc carrier. Oncol Rep 2007;18:279–85.
49. Dorn DC, Lawatscheck R, Zvirbliene A, Aleksaite E, Pecher G, Sas-nauskas K, et al. Cellular and humoral immunogenicity of hamsterpolyomavirus-derived virus-like particles harboring a mucin 1 cyto-toxic T-cell epitope. Viral Immunol 2008;21:12–27.
50. Li M, Bharadwaj U, Zhang R, Zhang S, Mu H, Fisher WE, et al.Mesothelin is a malignant factor and therapeutic vaccine target forpancreatic cancer. Mol Cancer Ther 2008;7:286–96.
Auto-Antibody Induction against CLDN18.2 by VLP Immunization
www.aacrjournals.org Cancer Res; 71(2) January 15, 2011 527
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292

2011;71:516-527. Published OnlineFirst January 11, 2011.Cancer Res Thorsten Klamp, Jens Schumacher, Georg Huber, et al. Kill Tumor Cells and Inhibit the Growth of Lung Metastases2 Induced by a Chimeric HBcAg Virus-Like Particle Vaccine Highly Specific Auto-Antibodies against Claudin-18 Isoform
Updated version
10.1158/0008-5472.CAN-10-2292doi:
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/71/2/516.full#ref-list-1
This article cites 50 articles, 18 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/71/2/516.full#related-urls
This article has been cited by 4 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/71/2/516To request permission to re-use all or part of this article, use this link
Research. on April 10, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 11, 2011; DOI: 10.1158/0008-5472.CAN-10-2292