highly-ordered mesoporous carbon nitride with ultrahigh surface area and pore volume as a superior...
TRANSCRIPT

Highly-Ordered Mesoporous Carbon Nitride with Ultrahigh SurfaceArea and Pore Volume as a Superior Dehydrogenation CatalystZhongkui Zhao,* Yitao Dai, Jinhan Lin, and Guiru Wang
State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, Liaoning,P. R. China
*S Supporting Information
ABSTRACT: In this work, a highly ordered mesoporouscarbon nitride nanorods with 971−1124 m2 g−1 of superhighspecific surface area, 1.31−1.79 cm3 g−1 of ultralarge porevolume, bimodal mesostructure, and 9.3−23 wt % of high Ncontent was prepared via a facile nanocasting approach usingSBA-15 as template and hexamethylenetetramine as carbonnitride precursor, and the specific surface area and porevolume as well as N content are strongly dependent on thechosen precursor and pyrolysis temperature. The as-preparedmaterials were well characterized by HRTEM, FESEM, XRD,BET, Raman, FT-IR, XPS, and the textural structure andmorphology were confirmed. The finding breaks through the bottleneck problems for fabricating mesoporous carbon nitride withboth ultrahigh surface area and super large pore volume by employing an unexplored hexamethylenetetramine as carbon nitrideprecursor. The current synthetic strategy can be extended to the preparation of various mesoporous carbon nitride with differenttextural characteristics by using diverse templates under changeable preparation conditions. The developed mesoporous carbonnitride material with 750 °C of pyrolysis temperature exhibits high superior catalytic performance, ascribed to the promotingeffect of nitrogen within the carbon matrix, the rich CO group and defect/edge feature on the surface, small size of graphiticcrystallite, as well as the ultrahigh surface area and pore volume. It can also be concluded that the microstructures including bulkand surface structure features and surface chemical properties of the carbon-based materials have a decisive influence on theircatalytic performance. The developed material can be employed in various organic transformations such as the base-catalyzedreactions, selective oxidation, dehydrogenation, photocatalysis, and electrocatalysis as well as acting as a novel and efficientcandidate for CO2 capture, supercapacitor, purification of contaminated water, and future drug-delivery systems.
■ INTRODUCTION
Carbon nitride is a well-known and fascinating material that hasattracted worldwide attention because the incorporation ofnitrogen atom into the carbon nanostructure can efficientlyenhance the mechanical, field emission, and energy-storageproperties.1−9 Accordingly, the attempted synthesis of bulkcarbon nitride is the subject of an even larger number of reportsin the literature.1,2,10−16 The history of carbon nitride and itsprecursors can be traced back to the very early days of theobservation of Berzelium described by Liebig in 1834.17 Carbonnitride is a potentially useful substitute for amorphous andgraphitic carbon in a variety of applications such as catalysis, gasstorage, and purification of contaminated water.18−23 Carbonnitride materials collected directly after self-condensation oforganic precursors are bulk materials with a very small surfacearea, normally below 10 m2 g−1. From the viewpoint of practicalapplications in the above fields especially in catalysis and gasstorage, the introduction of controlled porosity at the nanoscalein bulk carbon nitride is mandatory to enhance itsfunction.18−28 Mesoporous carbon nitride materials with two-dimension pore architecture possess higher specific surface areaand larger pore volume, which results in enhanced perform-
ances in such applications due to higher surface density ofactive sites exposed on the surface and their easier accessibilityby strengthened diffusion.24,26,29−37 Therefore, many scientistshave focused on the preparation of mesoporous carbon nitridematerials by using different strategies and precursors.29−48 Asoft template method generally produces the mesoporouscarbon nitride with lower surface area, while the nanocastingtechnique using mesoporous silica as a hard template is anadvisible strategy to synthesize mesoporous carbon nitride withhigher surface area and controlled mesoporous structures, andmany reports on synthesizing this kind of materials withtunable pore diameter, surface area, and nitrogen by thenanocasting approach can be found.18,24−48 Owing to a facileone-pot feature, the soft-template method also attracts muchattention. Some fruitful results have been achieved in improvingsurface area and pore volume by employing various templatesand precursors and also by controlling the pyrolysisprocess.49−52 However, the further improvement in surface
Received: February 17, 2014Revised: April 25, 2014Published: April 28, 2014
Article
pubs.acs.org/cm
© 2014 American Chemical Society 3151 dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−3161

and pore volume is highly desirable. Through post-treatment ofmesoporous carbon, well-ordered mesoporous carbon nitridewith a high surface area of 612−851 m2 g−1 can beobtained,18,53−57 but the N content is commonly less than 5wt %. Moreover, this method is generally subjected to anitrogen-containing atmosphere, most often highly toxic NH3at high temperatures.41,53−57 In contrast, in situ doping ofcarbons using nitrogen-containing precursors can realize ahomogeneous incorporation of nitrogen into the carbonmaterial with a controlled chemistry, and various precursorssuch as ethylenediamine with carbon tetrachloride, melamine,cyanamide, Dicyandiamide, gelatin, and N-containing ionicliquids38−48 and even the employment of melamine-cyanuricacid supramolecular as a precursor in the synthesis ofmesoporous carbon nitride has also been recently reported.58
Unfortunately, although many efforts have been put intofabricating various mesoporous carbon nitride materials, unlikemicroporous carbon nitride material,59,60 the synthesis ofmesoporous ones with an ultrahigh surface up to 1000 m2
g−1 area and large pore volume up to 1.0 cm3 g−1 still remainsan insurmountable challenge.29−48 However, in order toenhance the performance of this material in diverseapplications, the development of a facile and efficient methodto synthesize ordered mesoporous carbon nitride materials withultrahigh surface area, ultralarge pore volume, and higher Ncontent is highly desirable but remains a challenge.Herein we report for the first time on the preparation of
highly ordered hexagonal mesoporous carbon nitride materialwith 971−1124 m2 g−1 of ultrahigh specific surface area, 1.31−1.79 cm3 g−1 of superlarge pore volume, bimodal pores (4.8 and11.0 nm), and 9.2−23% of N content through the self-polymerization reaction of hexamethylenetetramine (HTM), acommercially available basic chemical material using SBA-15 asa hard template via a facile and efficient nanocasting approach(Scheme 1). In a typical synthesis, the calcined SBA-1561,62 was
added to a HTM solution. The resulting mixture was stirred atambient temperature for the desired time followed byevaporation using a rotary evaporator. The HTM filled SBA-15 was placed in a drying oven for 12 h and then heat-treated ina nitrogen flow to self-polymerize and subsequently carbonizethe formed polymer with the pores of SBA-15. Themesoporous carbon nitride was recovered after removal of
silica framework by HF treatment and then was washed severaltimes with water and followed by a drying process at 105 °Covernight. The resulting mesoporous carbon nitride materialwas labeled as DUT-1. The detailed procedure for the synthesisof DUT-1 is given in the Experimental Section. Forcomparison, the classical mesoporous carbon (CMK-3)63 andmesoporous carbon nitride (MCN-1)29 were also preparedaccording to the literature. The microstructure and surfacechemical properties of the obtained materials have beenunambiguously characterized by employing sophisticatedtechniques such as X-ray diffraction (XRD), high-resolutiontransmission electron microscopy (HRTEM), field-emissionscanning electron microscopy (FESEM), X-ray photoelectronspectroscopy (XPS), nitrogen adsorption, Fourier transforminfrared spectroscopy (FT-IR), Raman spectroscopy, andelemental analysis. We also demonstrate for the first time thecatalytic performance for direct dehydrogenation of ethyl-benzene to styrene, and DUT-1-750 shows an obviouslysuperior catalytic performance to CMK-3, MNC-1, and even tothe reported nanodiamond (ND) in this reaction with morethan 11.5% of styrene yield and 93.8% of selectivity, althoughonly 0.025 g of catalyst was loaded. The superior catalyticperformance of the as-synthesized carbon nitride materials isnotably dependent on their microstructure and surfacechemical properties. We expect that well-ordered DUT-1materials with bimodal pore architectures, ultrahigh surfacearea, and superlarge pore volume could be interesting for abroad range of applications including the separation andadsorption of large biomolecules such as proteins, enzymes,CO2 capture, H2 storage, catalysis, supercapacitors, andpurification of polluted water.
■ EXPERIMENTAL SECTIONSynthesis of a SBA-15 Template. SBA-15 was prepared
according to the procedure reported by Zhao et al.61,62 In a typicalsynthesis, 4.0 g of Pluronic P123 purchased from Sigma-Aldrich Co.was dissolved in 30 g of deioned water and 120 g of 2 M HCl solutionwith stirring at 35 °C. Then 8.50 g of TEOS was added into the abovesolution with stirring at 35 °C for 20 h. The resulting mixture was agedat 80 °C overnight. The obtained solid product was recovered, washed,and then dried at room temperature. The final product was calcined at500 °C for 6 h.
Synthesis of Mesoporous Carbon Nitride DUT-1 and MCN-1As Well As Mesoporous Carbon CMK-3. DUT-1 was synthesizedas follows: 0.5 of calcined SBA-15 was dispersed in the deioned waterfollowed by adding HTM to the above solution. The resultant mixturewas stirred at room temperature and then was evaporated by usingrotary evaporator. The solid was dried in an oven overnight, followedby heated to 750 °C in nitrogen atmosphere. The mesoporous carbonnitride materials were recovered after removing silica framework by40% of HF treatment and then were washed several times with waterand subsequently dried. The resultant mesoporous carbon nitride waslabeled as DUT-1. The series of DUT-1 materials with differentpyrolysis temperatures (600, 750, and 900 °C) were prepared anddenoted as DUT-1-600, DUT-1-750, and DUT-1-900, respectively.MCN-1 was prepared according to the reported procedure:29 0.5 g ofcalcined SBA-15 was added to the mixture containing 2.2 g ofethylenediamine and 5.4 g of carbon tetrachloride and was stirred at 90°C for 6 h. The resultant solid mixture was dried for 12 h andsubsequently heated in a nitrogen flow at 600 °C for 5 h to carbonizethe polymer. The silica template was removed by 5 wt % HF solution.The template-free carbon nitride MCN-1 was obtained by the filtering,washing with ethanol, and drying at 100 °C. According to ref 63,CMK-3 was also prepared. Briefly, 1 g of calcined SBA-15 was addedto a solution containing 1.25 g of sucrose, 0.14 g of H2SO4, and 5 g ofH2O. The mixture was placed in a drying oven for 6 h at 100 °C, then
Scheme 1. Illustration of Synthetic Procedure for Highly-Ordered Mesoporous Carbon Nitride Material (DUT-1) byUsing SBA-15 as a Hard Template
Chemistry of Materials Article
dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−31613152

the temperature was increased to 160 °C, and the mixture was kept atthis temperature for 6 h. The resultant solid, containing silica template,partially polymerized, and carbonized sucrose, was treated again at 100°C and then at 160 °C using the same drying oven after the addition ofthe solution containing 0.8 g of sucrose, 0.09 g of H2SO4, and 5 g ofH2O. The carbonization was performed by pyrolysis at 900 °C innitrogen atmosphere. The obtained carbon-silica composite waswashed with 5 wt % HF at room temperature to remove the silicatemplate. The template-free carbon product was obtained by filtering,washing with ethanol, and drying at 120 °C.Characterization of Samples. X-ray diffraction (XRD) profiles
were collected from 10 to 80° at a step width of 0.02° using RigakuAutomatic X-ray Diffractometer (D/Max 2400) equipped with a CuKasource (λ = 1.5406 Å). Nitrogen adsorption and desorption isothermswere determined on a Micromeritics apparatus of a model ASAP-2050system at −196 °C. The specific surface areas were calculated by theBET method, and the pore size distributions were calculated from anadsorption branch of the isotherm by the BJH model. Scanningelectron microscope (SEM) experiments were performed on a JEOLJSM-5600LV SEM/EDX instrument. Transmission electron micros-copy (TEM) images were obtained by using a Tecnai F30 HRTEMinstrument (FEI Corp.) at an acceleration voltage of 300 kV. The XPSspectra were carried out on an ESCALAB 250 XPS system with amonochromatized Al Ka X-ray source (15 kV, 150 W, 500 μm, passenergy = 50 eV). The Raman spectra were measured using a laser withan excitation wavelength of 532 nm at room temperature on a ThermoScientific DXR Raman microscope. FT-IR spectroscopy character-ization of catalysts was performed at 150 °C under ultrahigh vacuumusing a Bruker EQUINOX55 infrared spectrometer. In situ FT-IRexperiments were conducted on the same IR instrument, the samplewas degassed at 150 °C under ultrahigh vacuum for 30 min, thenethylbenzene was injected into the environmental chamber with thetemperature of chamber increased from 150 to 400 °C at a heating rateof 5 °C min−1, and the infrared spectra were recorded per 5 min.Catalytic Performance Measurement. Direct dehydrogenation
of ethylbenzene was carried out at 550 °C for 20 h in a stainless steel,fixed bed flow microreactor. A catalyst of 25 mg was placed at thecenter of the reactor using quartz wool plugs. The system was heatedto 600 °C and kept for 30 min in Ar as the pretreatment of catalyst.After the system was cooled down to 550 °C and kept for 10 min, thereactant of 2.8% ethylbenzene with feed flow rate 10 mL min−1 and Aras balance was then fed into the reactor from a saturator kept at 40 °C.The effluent from the reactor was condensed in two traps containingethanol connected in a series. The condensed material was cooledexternally in an ice water bath. Quantitive analysis of the collectedreaction products (ethylbenzene, styrene, toluene, and benzene) wasperformed on a FULI 9790 II GC equipped with HP-5 column, 30 m× 0.32 mm × 0.25 μm, and FID detector. The resulting carbonbalance was above 100 ± 4% in all reactions. The ethylbenzeneconversion (XEB), yield of styrene (YEB), and the selectivity of styrene(SST) were calculated on the basis of the following eqs 1-3:
= − ×X n n(%) (1 / ) 100EB EB,outlet EB,inlet (1)
= ×Y n n(%) / 100EB ST,outlet EB,inlet (2)
= + + ×S n n n n(%) /( ) 100ST ST,outlet ST,outlet BZ,outlet TOL,outlet
(3)
EB is ethylbenzene, ST is styrene, BZ is benzene, and TOL istoluene.
■ RESULTS AND DISCUSSIONThe pore structural order of the DUT-1 material was analyzedby using powder XRD measurements, and the XRD patternsare presented in Figure 1. In small angle region of XRDpatterns, three distinct peaks assigned to the (100), (110), and(200) reflections of the highly ordered 2D hexagonal lattice(space group p6mm) with a lattice constant a100 = 10.41 nm are
clearly inherited from the parent SBA-15 silica template, whichconsists of a hexagonal arrangement of cylindrical poreinterlinked by the micropores existing in the wall.29,40,48 Thecorresponding unit-cell constants of DUT-1 together with thevalues toward parent SBA-15 are summarized in Table 1. Tocheck whether the carbon nitride wall structure of DUT-1 iscrystalline or amorphous, this material was further characterizedby wide angle XRD analysis. The wide angle XRD pattern ofthe as-prepared DUT-1 (inset in Figure 1) exhibits a broaddiffraction peak at 25.3° corresponding to interlayer d spacingof 0.352 nm, which is quite similar to the characteristic (002)basal plane diffraction peak obtained in the nonporous carbonnitrides. This indicates the graphitic wall structure of the DUT-1 sample is composed of carbon and nitride that are arranged ina turbostratic form.29,33,40,43 The formation is the turbostraticordering of carbon and nitride atoms in the wall structure ofcarbon nitride materials is extremely important, since itpresents information about their electrical, electronic, andbasic catalytic properties.33
The successful replication and the formed orderedmesostructures are also confirmed by FESEM. Figure 2b-cclearly demonstrates that the typical rod-like morphology ofSBA-15 is well replicated in the carbon nitride material DUT-1nanorod. The 8−20 nm of pores can also be observed in themagnified FESEM image (Figure 2d). HRTEM was employedto further examine the morphology and textural structure ofmesoporous carbon nitride DUT-1. Bright contrast strips onthe under focused images represent the pore wall images,whereas dark contrast cores display empty channels. Theporous structure of the DUT-1 sample, when viewed in thedirection perpendicular to their axis, displays a linear array ofmesopores arranged in a regular pattern (Figure 2e). However,when viewed down the pore axis, the image reveals ahexagonally ordered honeycomb-like structure with a uniformmesoporous channels (Figure 2f), similar to that of the highlyordered SBA-15.61,62 These well ordered mesopores originatedfrom the silica walls that are dissolved with 40 wt % of HF inwater. The pore diameter calculated from the HRTEM imageswhich show linear arrays of mesopores with a regular interval isabout 5 nm. It should be noted that a well-ordered mesoporous
Figure 1. Small angle XRD patterns of SBA-15 and the as-preparedDUT-1. Insets: Wide angle XRD pattern of DUT-1. XRD patternswere recorded on a Rigaku diffraction meter using Cu Kα (λ =0.154056 nm) radiation in the 2 theta range of 0.5 to 10° and 10 to80° with a 2 theta step size of 0.02°.
Chemistry of Materials Article
dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−31613153

matrix is seen throughout the sample, revealing the purity andthe uniformity of the ordered porous matrix in the highlyordered mesoporous carbon nitride sample. Combing FESEMand HRTEM images (Figure 2c-f), we can observe bimodalpore architecture on the DUT-1 samples. Furthermore, duringcareful examination of the magnified HRTEM image of theDUT-1 (Figure S1), many crystalline domains, formed by anumber of parallel fringes that are composed of graphenesheets and self-align perpendicularly to the template wallsduring the synthesis process, are observed. The HRTEM imagealso clearly illustrates small crystalline domains with a d spacingof 0.33 nm which agrees well the high-angle XRD pattern(Figure 1 inset).The nitrogen-adsorption isotherms of DUT-1 and the parent
mesoporous silica template SBA-15 are presented in Figure 3a.Both of the isotherms are of type IV according to IUPACclassification and exhibit H1 hysteresis with a featured capillarycondensation in the mesopores, indicating the presence of wellordered mesopores in the two samples. It is interesting that theisotherm toward DUT-1 presents two capillary condensationsteps; one is at 0.40−0.75 of a lower relative pressure (p/p0)
and another is at 0.75−0.95 of a higher relative pressure region,suggesting the characteristics of highly ordered mesoporousmaterials with bimodal pores. The bimodal pores are alsoconfirmed by the BJH adsorption pore size distribution. As canbe seen in Figure 3b, the DUT-1 contains two types of poreswith small mesopores (the center of pore distribution from theadsorption branch is 4.8 nm) and large complementarymesopores (11 nm), which is also confirmed by FESEM andHRTEM images. Scheme S1 illustrates the plausible formationprocess of the bimodal pores of DUT-1. The smallermesopores are thought to be generated due to the removal ofthe silica template as in the usual hard templating process,
Table 1. Texture Parameters of Mesoporous Carbon Nitride DUT-1 and MCN-1 Prepared by Nanocasting Technique withSBA-15 as Template (SBA-15 Included for Comparison)
sample SBET (m2 g−1) pore volumea (cm3 g−1) pore sizeb (nm) d100c (nm) a0
d (nm) wall thicknesse (nm) Laf (nm)
SBA-15 843 1.10 (0.034) 7.8 10.03 11.59 3.8DUT-1 1116 1.45 (0.064) 4.8/11 9.01 10.41 5.6 7.39MCN-1g 505 0.55 (0.049) 4.0 9.15
aDetermined at p/p0 = 0.98, where p is the equilibrium pressure and p0 is the saturation pressure of nitrogen at 77 K, the value inside brackets is thet-plot micropore volume. bThe center of the pore distribution from the adsorption branch. cd (100) spacing or d value of characteristic reflection.dCalculated by 2d100/√3. eDetermined from a0-pore size. fThe size of the graphene layer calculated on the basis of the empirical relationship fromRaman spectra: La (nm) = (2.4 × 10−10)λ4(ID/IG)
−1. gFrom ref 29.
Figure 2. a-d) FESEM images of SBA-15 and DUT-1 and e,f)HRTEM images of DUT-1: a) SBA-15; b-d) DUT-1; e) longitudinalprojection (along the mseopores); f) cross-section projection (crossthe mesopores).
Figure 3. a) Nitrogen adsorption isotherms (closed symbols:adsorption; open symbols: desorption) and b) Barrett−Joyner−Halenda (BJH) adsorption pore-size distribution of DUT-1 and theparent SBA-15. Nitrogen adsorption and desorption isotherms weremeasured at 77 K on a Micromeritics apparatus of model ASAP-2050system.
Chemistry of Materials Article
dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−31613154

whereas the complementary mesopores are assumed to beoriginated due to the incomplete filling of the pores of thetemplate with the carbon nitride precursors that creates emptylarge pores before the silica removal.38,39,64−66 It is veryinteresting to note that the pore size (4.8 nm) of the DUT-1 isobviously larger than the wall thickness (3.8 nm) of parentSBA-15, ascribed to the shrinkage of filled carbon nitridepolymeric materials inside the pores of SBA-15.29
The specific Brunauer−Emmett−Teller (BET) surface area,specific pore volume, and the pore diameters of the DUT-1 andthe parent SBA-15 are summarized in Table 1. The DUT-1exhibits 1116 m2 g−1 of ultrahigh specific BET surface area and1.45 cm3 g−1 of ultralarge specific pore volume (mainly frommesoporous carbon nitride materials).29−48 Moreover, the poresize of DUT-1 is tunable by the choice of SBA-15 template witha mesopores since only 0.064 cm 3 g−1 of pore volume is frommicropores, which are the highest values of the reportedordered ordered mesoporous carbon nitride materials.29−48
Moreover, the pore size of DUT-1 is tunable by the choice ofthe SBA-15 template with an appropriate pore size and wallthickness, as well as by changing the molar ratio of HTM toSBA-15.24,29 The ultrahigh specific surface area, ultralarge porevolume, unique bimodal mesoporous architectures, and 16.1 wt% of high N content (CHN elemental analysis) of thedeveloped highly ordered mesoporous carbon nitride DUT-1allow it to be a fascinating carbon nitride material to be used inwide fields like catalysis (as support or metal-free catalyst),capacitor, energy storage.Raman spectroscopy is a powerful tool for identifying carbon
materials and detecting the doping effect of heteroatoms.67−73
Raman spectra of all samples exhibit two bands at around 1350and 1580 cm−1 corresponding to D (A1g mode) and G (E2gmode) bands, respectively (see Figure 4). The increasing
intensity of the Raman spectra band of DUT-1 and MCN-1 incontrast to CMK-3 provides evidence of the intensifiedgraphitic nature of the N-doped carbon materials,43,68−70
which is in agreement with XRD and HRTEM results inFigure 1 and Figure S1. Moreover, it is found that the G-bandshifts from 1597 to 1576 cm−1 and the D-band moves from1338 to 1354 cm−1 in comparison to those of the mesoporouscarbon CMK-3 which can be ascribed to the incorporation ofnitrogen into a carbon matrix.74−82 We calculated the in-planecrystallite sizes (La) of DUT-1 and MCN-1 by the following
equation,74,75 La (nm) = (2.4 × 10−10)λ4(ID/IG)−1 where λ is
the Raman excitation wavelength (532 nm), the ID/IG is thearea ratio of D and G peaks, respectively. The results are listedin Table 1. The crystallite size of the graphitic layer towardDUT-1 (7.39 nm) is a little smaller than that of the reportedMCN-1 in the literature (9.15 nm). The larger ID/IG of theRaman peaks toward DUT-1 than that of the other twomaterials is also an indicator of more defect or amorphouscarbon on graphitic carbon materials.74−78 In order to stablyexist the carbon atoms at the defective site may be saturated byan oxygen atom to produce more oxygen functional groups,serving as active sites for oxidation reaction and dehydrogen-aion.76,82 In contrast to that of CMK-3, the Raman spectra ofDUT-1, similar to MCN-1, present a broad peak in the range of2000−3000 cm−1 (Figure S2), which should be attributed toeither the overlap of several peaks corresponding to vibrationpeaks of various carbon nitride bonds,72 or the associatedovertone of D band, 2D band corresponding to small crystallitesizes and the presence of high proportions of edge planes intheir structures.73 Correlated to the observed intense D band aswell as its associated 2D band, we think the broad D bondmight be ascribed to small graphitic crystallite and highproportions of edge planes in the carbon nitride materials.The nature and coordination of the carbon, nitrogen, and
oxygen in the DUT-1 and MCN-1 as well as carbon and oxygenin the CMK-3 were examined by XPS.83−86 The XPS surveyspectrum (Figure 5a) of the DUT-1 shows strong signals from
C, N, and O elements. No Si signals can be observed,suggesting that silica species are almost completely removedduring the HF etching. The absence of the Si atom in theDUT-1 sample, confirming that the intense XRD peaks withp6mm symmetry, does not come from the SBA-15 mesoporoussilica template but from itself. The presence of O species mayarise from oxidation of the precursors during the process. Thecarbonaceous C 1s line (284.6 eV) was used as a reference tocalibrate the binding energies. The spectrum of C 1s in theDUT-1 (Figure 5b) can be deconvoluted into four single peaksthat correspond to CC (sp2 C, 284.6 eV), C−N or C−C (sp3
C, 286.0 eV), C−O (286.5 eV), and CN/CO (288.5 eV)
Figure 4. Raman spectra of DUT-1, MCN-1, and CMK-3.
Figure 5. XPS spectra of the DUT-1 material: a) Survey spectra(CMK-3 was included for comparison), b-d) C 1s, N 1s, and O 1sspectra, respectively. The XPS measurements were performed on anEscalab 250 spectrometer using a monochromatized Al Ka radiation asthe excitation source.
Chemistry of Materials Article
dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−31613155

functional groups,83,84 The N 1s spectrum (Figure 5c) arecurvefitted into three peaks with binding energies of 397.7,399.7, and 403 eV that correspond to pyridinic N, pyrrolic N,and oxidized N, respectively. We cannot see the peakcorresponding to graphitic N.85,86 The spectrum of O 1s inthe DUT-1 (Figure 5d) can be deconvoluted into two singlepeaks with binding energies of 531.5 and 533.0 eV thatcorrespond to CO and C−O, respectively.84 The shape andposition of both the C 1s and N 1 s spectra of DUT-1 (FigureS3) are similar to those of MCN-1 prepared by the reportedmethod29 (the similar N content of various N species),suggesting that the nature and coordination of the DUT-1 arenot affected by different carbon nitride precursors althoughthey have the obvious difference in structure and morphologyconfirmed by XRD, HRTEM, and nitrogen adsorption.However, the shape of the O 1s spectrum of DUT-1 is quitedifferent from that of MCN-1, although the position (bingingenergy) is similar (Figures 5d and S3). CO is the mainexisting form of the oxygen atom in the DUT-1 but C−O in theMCN-1 (Table 2). However, the shape and position of the O
1s spectrum of MCN-1 is similar to that of CMK-3 (Figures S3and S4), and the amount of CO in MCN-1 is comparable tothat of C−O in both MCN-1 and CMK-3. The dominant CO in the developed DUT-1 may allow it to exhibit superiorcatalytic performance for CO group-activated reactions likeoxidation, dehydrogenation, and hydrogen-transferring reac-tions, besides metal-free basic catalysis due to high N contentand high surface area and pore volume.Styrene, an important basic chemical material for polymer
production, is mostly produced by direct dehydrogenation ofethylbenzene. Unfortunately, a great deal of carbonaceousspecies inevitably deposit on the surface of multiply promotedFe catalysts and cause a heavy loss of activity. Therefore, excesssteam must be introduced into the feed as a remediationmethod to remove the resulting coke deposited on the catalyst,and thus much energy is wasted. Recently, a fascinating methodfor direct dehydrogenation of ethylbenzene to styrene underoxygen- and steam-free conditions was reported by usingnanodiamond as a metal-free catalyst,87 which makes sail thestudies on developing a carbon-based catalyst for this reaction.Herein we present the first approach for the nitrogen-doped
carbon as a metal-free catalyst to enhance the directdehydrogenation of enthylbenzene to styrene under oxygen-and steam-free conditions. The catalytic performance of thedeveloped mesoporous carbon nitride DUT-1 as well as theclassical mesoporous carbon nitride MCN-1 and mesoporouscarbon CMK-3 was examined, and the reported best carboncatalyst nanodiamond was also included for comparison (Figure6). The dehydrogenation conditions are the same as thosereported in the literature,87 except for 0.025 g of halved catalystdosage. The developed mesoporous carbon nitride DUT-1
exhibits superior catalytic performance than the othersincluding the established ND catalyst in the reference. The11.6%, 9.1%, 6.8%, and 5.6% of styrene yield with 93.6%,95.4%, 96.0%, and 84.3% of selectivity toward the desiredstyrene are achieved over DUT-1, ND, MCN-1, and CMK-3,respectively. The main byproducts are benzene and tolueneresulting from the cracking of ethylbenzene, which consistswith the results reported in the literature.87 In comparison ofthe N-doped and undoped carbon materials, the former exhibitssignificantly superior catalytic performance especially theselectivity. The CO serving as Lewis bases to activatesaturated hydrocarbon for this dehydrogenation reaction hasbeen confirmed,87,88 the surface phenolic hydroxyl group mayenhance the cracking of ethylbenzene due to its acidity, sinceacid sites are active for cracking reaction of hydrocarbon.89−91
The introduction of a nitrogen atom into a carbon matrix canincrease the electron density of carbon materials and thereforestrengthen the basicity but weaken the acidity of the catalyst,which may result in an improvement in catalytic activity forstyrene production but compressing the benzene and tolueneformation. As a result, the superior catalytic activity andselectivity over N-doped carbon to mesoporous carbon CMK-1can be demonstrated. The catalytic activity of MCN-1 is lowerbut that of DUT-1 is higher than that of the nanodiamondalthough they have the same N content, suggesting ultrahighsurface area and ultralarge pore volume are essential forenhancing the direct dehydrogenation reaction under oxygen-and steam-free conditions.In order to further illustrate the reason for the superior
catalytic performance of DUT-1, the FT-IR experiments on theDUT-1, CMK-3, and MCN-1 as well as the in situ FT-IRexperiments on the ethylbenzene adsorbed samples as afunction of temperature from 150 to 400 °C were performed.From Figure 7a, in contrast of CMK-3, the red shift of the peakat around 1740 cm−1 corresponding to the CO group on theFT-IR of DUT-1 and MCN-1 can be clearly observed,suggesting the electron-rich atmosphere around CO, whichimproves the catalytic performance of mesoporous carbonnitride. In the in situ FT-IR spectra, the peak at around 910cm−1 can be assigned to the C−H out-of-plane mode of thevinyl group CH2,
92 which can be considered as an indicationfor the styrene formation. From Figures 7b, S5, S6, thetemperature for the initial formation of styrene over DUT-1,
Table 2. N Content from CHN Elemental Analysis As WellAs Relative Integrated Intensity of the Deconvoluted N 1sand O 1s XPS Spectra for the DUT-1 and MCN-1
sampleNa
(wt %)pyridinicNb (%)
pyrrolicNb (%)
oxidizedNb (%)
CO(%)
C−O(%)
DUT-1 16.1 40.0 51.5 8.5 80.6 19.4MCN-1 16.0 40.9 51.1 8.0 48.0 52.0
aThe total N content of the carbon nitride materials. bPercentage ofvarious nitrogen species occupying in the total N concent.
Figure 6. Catalytic performance of DUT-1, MCN-1, CMK-3, andnanodiamond for direct dehydrogenation of ethylbenzene to styreneunder oxygen- and steam-free conditions. Reaction conditions: 0.025 gcatalyst, 550 °C, 2.8% of ethylbenzene in argon, 10 mL min−1.
Chemistry of Materials Article
dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−31613156

MCN-1, and CMK-3 is 200, 245, and 265 °C, respectively,suggesting the lower activation energy for the directdehydrogenation of ethylbenzene to styrene, which is inagreement with the reaction results. Due to the ultrahighsurface area, ultralarge pore volume, the ordered bimodalmesoporous architecture as well as high N content, thedeveloped DUT-1 material by a facile and efficient nanocastingapproach using SBA-15 as a template and low cost HTM as acarbon nitride precursor demonstrates the unprecedentedcatalytic performance, and therefore it could be a promisingcatalyst for industrial applications.Correlating the XPS and IR results to the catalytic
performance, although the structure of mesoprous carbonnitride is different from that of nanodiamond, the activationmechanism of ethylbenzene over nanodiamond or DUT-1 isnot different. The surface ketone-/diketone-type carbonylgroups (CO) possess substantial electron density at theoxygen atom and therefore serve as a Lewis base to efficientlyactivate saturated hydrocarbons, through which styrene isproduced and the CO is transformed to be hydroxyl group(C−OH) performing as an intermediate. The catalytic cycle isrealized by the thermal decomposition of C−OH to CO and
H2 molecule.18 The N doping and the change in carbonstructures may adjust the properties, existing environment, andtheir accessibility of catalytic active sites. The amount ofcatalytic active sites exposed on the carbon surface and theiraccessibility can be efficiently enhanced by the larger surfacearea and pore volume, which can cut down the activationenergy and therefore improve catalytic activity. Althoughsimilar N content and chemical states to those of MCN-1,the rich defective structure and the high proportion of edgeplanes of DUT-1 can contain more oxygen atoms, which allowmore oxygen functional groups to be formed on the surface ofDUT-1, especially the larger percentage of CO in the totaloxygen-containing groups on the DUT-1 surface confirmed byXPS analysis. The introduction of an N atom into the matrixcan also increase the electron density and basicity of carbonmaterials, besides further changing the surface edge-corner anddefective structure composed of oxygen-containing functionalgroups, which is responsible for the higher activity. Moreover,the microstructure feature of DUT-1 with larger surface areaand pore volume can efficiently enhance accessibility ofcatalytic active sites (CO). The properties and existingenvironment of active sites and enhanced accessibility lead tosuperior catalytic performance in direct dehydrogenation ofethylbenzene to styrene but depresses the formation of benzeneand toluene of those produced by a cracking reaction.Generally, the texture feature and surface properties are
strongly dependent on the pyrolysis temperature, besides thedifferent precursors of carbon nitride, which may further affectthe catalytic performance of the as-synthesized mesoporouscarbon nitride materials. Therefore, we prepared threemesoporous carbon nitride materials DUT-1-600, DUT-1-750, and DUT-1-900 and study the structure-performancerelationship by employing various characterization techniquessuch as XRD, N2 adsorption−desorption, Raman, XPS, andelemental analysis.XRD patterns of the developed mesoporous carbon nitride
materials with diverse pyrolysis temperatures are presented inFigure 8. Both DUT-1-600 and DUT-1-750 samples exhibitthree clear reflection peaks corresponding to (100), (110), and
Figure 7. a) FT-IR of the mesoporous CMK-3, MCN-1 and thedeveloped DUT-1 in this work and b) in situ FT-IR spectra of DUT-1catalyst adsorbed ethylbenzene as a function of temperature. Initialtemperature for styrene formation is 200 °C. The spectra wererecorded on a Bruker EQUINOX55 infrared spectrometer, thedegassed DUT-1 under ultrahigh vacuum was sufferred from ainjection of ethylbenzene, and then the chamber was heated from 150to 400 °C at a ramp rate of 5 °C min−1.
Figure 8. Small angle XRD patterns of the DUT-1-600, DUT-1-750,and DUT-1-900. Insets: Wide angle XRD patterns. XRD patterns wererecorded on a Rigaku diffraction meter using Cu Kα (λ = 0.154056nm) radiation in the 2 theta range of 0.5 to 10° and 10 to 80° with a 2theta step size of 0.02°.
Chemistry of Materials Article
dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−31613157
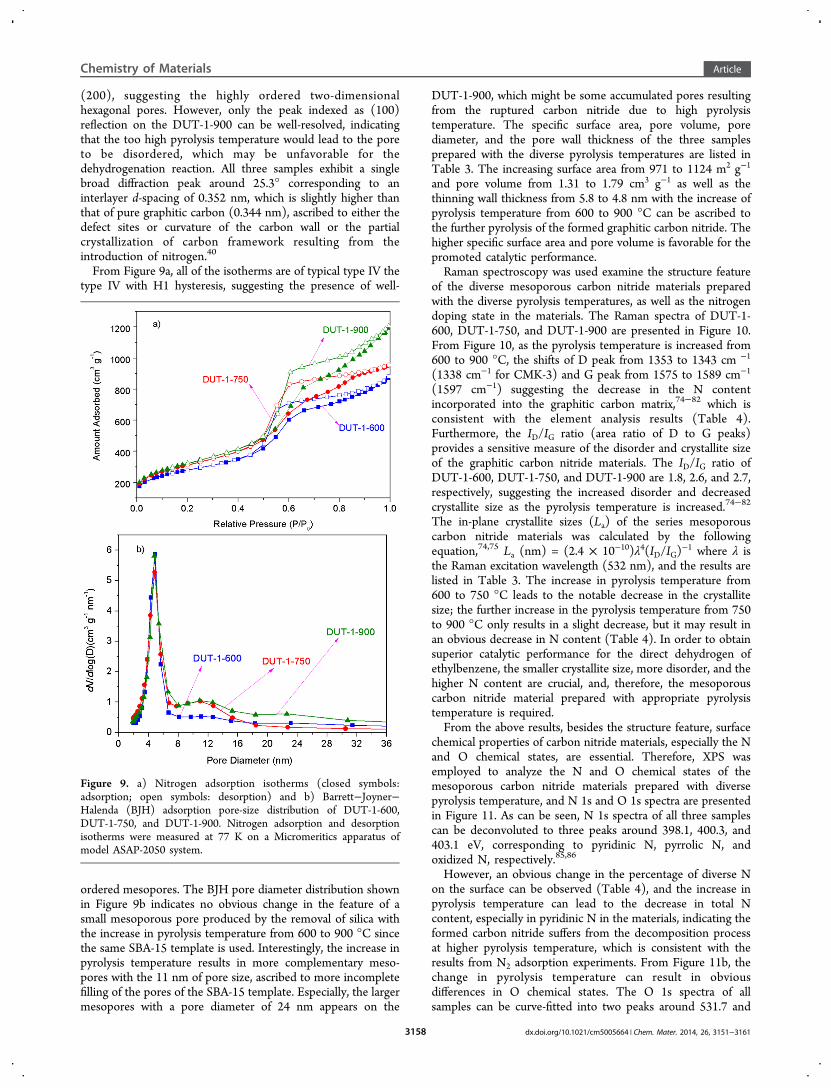
(200), suggesting the highly ordered two-dimensionalhexagonal pores. However, only the peak indexed as (100)reflection on the DUT-1-900 can be well-resolved, indicatingthat the too high pyrolysis temperature would lead to the poreto be disordered, which may be unfavorable for thedehydrogenation reaction. All three samples exhibit a singlebroad diffraction peak around 25.3° corresponding to aninterlayer d-spacing of 0.352 nm, which is slightly higher thanthat of pure graphitic carbon (0.344 nm), ascribed to either thedefect sites or curvature of the carbon wall or the partialcrystallization of carbon framework resulting from theintroduction of nitrogen.40
From Figure 9a, all of the isotherms are of typical type IV thetype IV with H1 hysteresis, suggesting the presence of well-
ordered mesopores. The BJH pore diameter distribution shownin Figure 9b indicates no obvious change in the feature of asmall mesoporous pore produced by the removal of silica withthe increase in pyrolysis temperature from 600 to 900 °C sincethe same SBA-15 template is used. Interestingly, the increase inpyrolysis temperature results in more complementary meso-pores with the 11 nm of pore size, ascribed to more incompletefilling of the pores of the SBA-15 template. Especially, the largermesopores with a pore diameter of 24 nm appears on the
DUT-1-900, which might be some accumulated pores resultingfrom the ruptured carbon nitride due to high pyrolysistemperature. The specific surface area, pore volume, porediameter, and the pore wall thickness of the three samplesprepared with the diverse pyrolysis temperatures are listed inTable 3. The increasing surface area from 971 to 1124 m2 g−1
and pore volume from 1.31 to 1.79 cm3 g−1 as well as thethinning wall thickness from 5.8 to 4.8 nm with the increase ofpyrolysis temperature from 600 to 900 °C can be ascribed tothe further pyrolysis of the formed graphitic carbon nitride. Thehigher specific surface area and pore volume is favorable for thepromoted catalytic performance.Raman spectroscopy was used examine the structure feature
of the diverse mesoporous carbon nitride materials preparedwith the diverse pyrolysis temperatures, as well as the nitrogendoping state in the materials. The Raman spectra of DUT-1-600, DUT-1-750, and DUT-1-900 are presented in Figure 10.From Figure 10, as the pyrolysis temperature is increased from600 to 900 °C, the shifts of D peak from 1353 to 1343 cm −1
(1338 cm−1 for CMK-3) and G peak from 1575 to 1589 cm−1
(1597 cm−1) suggesting the decrease in the N contentincorporated into the graphitic carbon matrix,74−82 which isconsistent with the element analysis results (Table 4).Furthermore, the ID/IG ratio (area ratio of D to G peaks)provides a sensitive measure of the disorder and crystallite sizeof the graphitic carbon nitride materials. The ID/IG ratio ofDUT-1-600, DUT-1-750, and DUT-1-900 are 1.8, 2.6, and 2.7,respectively, suggesting the increased disorder and decreasedcrystallite size as the pyrolysis temperature is increased.74−82
The in-plane crystallite sizes (La) of the series mesoporouscarbon nitride materials was calculated by the followingequation,74,75 La (nm) = (2.4 × 10−10)λ4(ID/IG)
−1 where λ isthe Raman excitation wavelength (532 nm), and the results arelisted in Table 3. The increase in pyrolysis temperature from600 to 750 °C leads to the notable decrease in the crystallitesize; the further increase in the pyrolysis temperature from 750to 900 °C only results in a slight decrease, but it may result inan obvious decrease in N content (Table 4). In order to obtainsuperior catalytic performance for the direct dehydrogen ofethylbenzene, the smaller crystallite size, more disorder, and thehigher N content are crucial, and, therefore, the mesoporouscarbon nitride material prepared with appropriate pyrolysistemperature is required.From the above results, besides the structure feature, surface
chemical properties of carbon nitride materials, especially the Nand O chemical states, are essential. Therefore, XPS wasemployed to analyze the N and O chemical states of themesoporous carbon nitride materials prepared with diversepyrolysis temperature, and N 1s and O 1s spectra are presentedin Figure 11. As can be seen, N 1s spectra of all three samplescan be deconvoluted to three peaks around 398.1, 400.3, and403.1 eV, corresponding to pyridinic N, pyrrolic N, andoxidized N, respectively.85,86
However, an obvious change in the percentage of diverse Non the surface can be observed (Table 4), and the increase inpyrolysis temperature can lead to the decrease in total Ncontent, especially in pyridinic N in the materials, indicating theformed carbon nitride suffers from the decomposition processat higher pyrolysis temperature, which is consistent with theresults from N2 adsorption experiments. From Figure 11b, thechange in pyrolysis temperature can result in obviousdifferences in O chemical states. The O 1s spectra of allsamples can be curve-fitted into two peaks around 531.7 and
Figure 9. a) Nitrogen adsorption isotherms (closed symbols:adsorption; open symbols: desorption) and b) Barrett−Joyner−Halenda (BJH) adsorption pore-size distribution of DUT-1-600,DUT-1-750, and DUT-1-900. Nitrogen adsorption and desorptionisotherms were measured at 77 K on a Micromeritics apparatus ofmodel ASAP-2050 system.
Chemistry of Materials Article
dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−31613158

533.0 corresponding to CO and C−O−C/C−OH, respec-tively. From Table 4, the 80.2% of CO percentage on theDUT-1-750 can be obtained, the too high or too low pyrolysistemperature can lead to an obvious decrease in the percentageof the surface CO group. The reason would be furtherexplored in future work. The surface CO is the active site forthe C−H activation, which performs a catalysis role in thedirect dehydrogenation reaction of ethylbenzene.We measure the catalytic performance of the three
mesoporous carbon nitride materials with diverse pyrolysistemperatures for the direct dehydrogenation of ethylbenzene asa model reaction, and the reaction results are demonstrated inFigure 12. It can be observed that the catalytic performance ofthe mesoporous carbon nitride materials is dramatically
dependent on the pyrolysis temperature, and the DUT-1-750sample exhibits superior catalytic performance than the othertwo. In combination of the reaction results with thecharacterization results of structure and surface chemicalproperties of the materials, it can be concluded that the surfaceCO is crucial for catalyzing the direct dehydrogenation, andthe introduction of N can increase the electronic density,electron conductivity, and the basicity of the carbon materialsas well as the high surface area and large pore volume and canpromote the reaction by enhanced mass transfer. Themicrostructure and surface chemical properties are dependenton the pyrolysis temperature, besides the carbon nitrideprecursor shown as above.
Table 3. Texture Parameters of the Developed Mesoporous Carbon Nitride DUT-1, DUT-2, and DUT-3 Prepared byNanocasting Technique with Diverse Pyrolysis Temperatures
sample SBET (m2 g−1) pore volumea (cm3 g−1) pore sizeb (nm) d100c (nm) a0
d (nm) wall thicknesse (nm) Laf (nm)
DUT-1-600 971 1.31 (0.076) 4.8/11.1 9.20 10.62 5.8 10.68DUT-1-750 1116 1.45 (0.064) 4.8/11.0 9.01 10.41 5.6 7.39DUT-1-900 1124 1.79 (0.049) 4.8/10.9 8.33 9.62 4.8 7.12
aDetermined at p/p0 = 0.98, where p is the equilibrium pressure and p0 is the saturation pressure of nitrogen at 77 K, the value inside the brackets isthe t-plot micropore volume. bThe center of the pore distribution from the adsorption branch. cd(100) spacing or d value of characteristic reflection.dCalculated by 2d100/√3. eDetermined from a0-pore size.
fThe size of the graphene layer calculated on the basis of the empirical relationship fromRaman spectra: La (nm) = (2.4 × 10−10)λ4(ID/IG)
−1.
Figure 10. Raman spectra of the various mesoporous carbon nitridematerials DUT-1-600, DUT-1-750, and DUT-1-900 prepared bynanocasting technique with diverse prrolysis temperatures using HTMas carbon nitride precursor.
Table 4. N Content from CHN Elemental Analysis As WellAs Relative Integrated Intensity of the Deconvoluted N 1sand O 1s XPS Spectra for the DUT-1-600, DUT-1-750, andDUT-1-900
sampleNa
(wt %)pyridinicNb (%)
pyrrolicNb (%)
oxidizedNb (%)
CO(%)
C−O(%)
DUT-1-600
23.0 43.5 49.7 6.8 49.5 51.5
DUT-1-750
16.1 40.0 51.5 8.5 80.2 19.8
DUT-1-900
9.3 28.0 60.0 12.0 44.1 55.9
aThe total N content of the carbon nitride materials. bPercentage ofvarious nitrogen species occupying in the total N concent.
Figure 11. XPS spectra of the DUT-1-600, DUT-1-750, and DUT-1-900: a) N 1s and b) O 1s spectra. The XPS measurements wereperformed on an Escalab 250 spectrometer using a monochromatizedAl Ka radiation as the excitation source.
Figure 12. Catalytic performance of DUT-1-600, DUT-1-750, andDUT-1-900 for direct dehydrogenation of ethylbenzene to styreneunder oxygen- and steam-free conditions. Reaction conditions: 0.025 gcatalyst, 550 °C, 2.8% of ethylbenzene in argon, 10 mL min−1.
Chemistry of Materials Article
dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−31613159

■ CONCLUSIONSIn summary, we have demonstrated for the first time thefabrication of highly ordered mesoprous carbon nitride nanorodDUT-1 with 971−1124 m2 g−1 of superhigh specific surfacearea, 1.31−1.79 cm3 g−1 of ultralarge pore volume, bimodalmesostructure, and 9.3−23.0 wt % of high N content through afacile and efficient nanocasting approach by using SBA-15 as atemplate and HTM as a carbon nitride precursor, and thestructure feature and surface chemical properties are dependenton the carbon nitride precursor and pyrolysis temperature. Weexamined the catalytic performance of the as-preparedmesoporous carbon nitride materials in the direct dehydrogen-ation of ethylbenzene to styrene under oxygen- and steam-freeconditions. The developed mesoporous carbon nitride DUT-1-750 exhibits superior catalytic performance, ascribed tooptimum chemical properties, existing environment, and theaccessibility of the catalytic active sites (CO), which isstrongly dependent on microstructures and surface chemicalproper of materials including surface area, pore volume, surfaceedge-corner and defective structure, N content, affected bycarbon nitride precursors and pyrolysis temperatures. Theincreased electron density and basicity for CO resulting fromthe incorporation of nitrogen atom into a carbon martrix canefficiently promote the direct dehydrogenation of ethylbenzeneto styrene but depress the formation of cracking sideproductslike benzene and toluene. Moreover, we believe that the currentsynthetic strategy can be extended to the preparation of variousmesoporous carbon nitrides with different textural character-istics by using diverse templates under changeable preparationconditions. Owing to its ultrahigh surface and pore volume andhigh N content as well as unique surface structure, the DUT-1could be employed in various organic transformations such asthe base-catalyzed reactions, selective oxidation, dehydrogen-ation, photocatalysis, and electrocatalysis as well as acting as anovel and efficient candidate for CO2 capture, supercapacitor,purification of contaminated water, and future drug-deliverysystems.
■ ASSOCIATED CONTENT*S Supporting InformationExtra HRTEM image, Raman spectra, in situ FT-IR spectra.This material is available free of charge via the Internet athttp://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe acknowledge financial support by the National NaturalScience Foundation of China (grant no. 21276041, U1261104,and 20803006), also by the Chinese Ministry of Education viathe Program for New Century Excellent Talents in Universities(grant no. NCET-12-0079) and the Fundamental ResearchFunds for the Central Universities (grant no. DUT12LK51).
■ REFERENCES(1) Kroke, E.; Schwarz, M. Coord. Chem. Rev. 2004, 248, 493−532.(2) Liu, A. Y.; Cohen, M. L. Science 1989, 245, 841−842.(3) Qiu, Y.; Gao, L. Chem. Commun. 2003, 2378−2379.
(4) Dibandjo, P.; Bois, L.; Chassagneux, F.; Cornu, D.; Letoffe, J.-M.;Toury, B.; Babonneau, F.; Miele, P. Adv. Mater. 2005, 17, 571−574.(5) Kawaguchi, M.; Yagi, S.; Enomoto, H. Carbon 2004, 42, 345−350.(6) Zimmerman, J. L.; Williams, R.; Khabashesku, V. N.; Margrave, J.L. Nano Lett. 2001, 1, 731−734.(7) Kim, M.; Hwang, S.; Yu, J. S. J. Mater. Chem. 2007, 17, 1656−1659.(8) Bai, Y. J.; Lu, B.; Liu, Z. G.; Li, L.; Cui, D. L.; Xu, X. G.; Wang, Q.L. J. Cryst. Growth 2003, 247, 505−508.(9) Guo, Q.; Yang, Q.; Zhu, L.; Yi, C.; Zhang, S.; Xie, Y. Solid StateCommun. 2004, 132, 369−374.(10) Miller, D. R.; Wang, J.; Gillan, E. G. J. Mater. Chem. 2002, 12,2463−2469.(11) Jurgens, B.; Irran, E.; Senker, J.; Kroll, P.; Muller, H.; Schnick,W. J. Am. Chem. Soc. 2003, 125, 10288−10300.(12) Holst, J. R.; Gillan, E. G. J. Am. Chem. Soc. 2008, 130, 7373−7379.(13) Niu, C. M.; Lu, Y. Z.; Lieber, C. M. Science 1993, 261, 334−338.(14) Bojdys, M. J.; Muller, J.; Antonietti, M.; Thomas, A. Chem.Eur. J. 2008, 14, 8177−8182.(15) Zhang, Z.; Leinenweber, K.; Bauer, M.; Garvie, L. A. J.;McMillan, P. F.; Wolf, G. H. J. Am. Chem. Soc. 2001, 123, 7788−7796.(16) Teter, D. M.; Hemley, R. J. Science 1996, 271, 53−55.(17) Liebig, J. Ann. Pharm. 1834, 10, 10−21.(18) Wang, Y.; Wang, X.; Antonietti, M. Angew. Chem., Int. Ed. 2012,51, 68−89.(19) Tahir, M.; Cao, C.; Butt, F. K.; Idrees, F.; Mahmood, N.; Ali, Z.;Aslam, I.; Tanveer, M.; Rizwan, M.; Mahmood, T. J. Mater. Chem. A2013, 1, 13949−13955.(20) Osterloh, F. E. Chem. Soc. Rev. 2013, 42, 2294−2320.(21) Zheng, Y.; Liu, J.; Liang, J.; Jaroniec, M.; Qiao, S. Z. EnergyEnviron. Sci. 2012, 5, 6717−6731.(22) Zhu, C.; Dong, S. Nanoscale 2013, 5, 1753−1767.(23) Su, F.; Mathew, S. C.; Mohlmann, L.; Antonietti, M.; Wang, X.;Blechert, S. Angew. Chem., Int. Ed. 2011, 50, 657−660.(24) Vinu, A. Adv. Funct. Mater. 2008, 18, 816−827.(25) Vinu, A.; Murugesan, V.; Tangermann, O.; Hartmann, M. Chem.Mater. 2004, 16, 3056−3065.(26) Li, X. H.; Antonietti, M. Chem. Soc. Rev. 2013, 42, 6593−6604.(27) Hao, G. P.; Li, W. C.; Qian, D.; Lu, A. H. Adv. Mater. 2010, 22,853−857.(28) Portehault, D.; Giordano, C.; Gervais, C.; Senkovska, I.; Kaskel,S.; Sanchez, C.; Antonietti, M. Adv. Funct. Mater. 2010, 20, 1827−1833.(29) Vinu, A.; Ariga, K.; Mori, T.; Nakanishi, T.; Hishita, S.; Golberg,G.; Bando, Y. Adv. Mater. 2005, 17, 1648−1652.(30) Yang, W.; Fellinger, T. P.; Antonietti, M. J. Am. Chem. Soc. 2011,133, 206−209.(31) Wang, Y.; Zhang, J.; Wang, X.; Antonietti, M.; Li, H. Angew.Chem., Int. Ed. 2010, 49, 3356−3359.(32) Wang, Y.; Yao, J.; Li, H.; Su, D.; Antonietti, M. J. Am. Chem. Soc.2011, 133, 2362−3365.(33) Jin, X.; Selvan, S. T.; Sawant, D. P.; Chari, M. A.; Lu, G. Q.;Vinu, A. Angew. Chem., Int. Ed. 2009, 48, 7884−7887.(34) Fellinger, T. P.; Hasche, F.; Strasser, P.; Antonietti, M. J. Am.Chem. Soc. 2012, 134, 4072−4075.(35) Silva, R.; Voiry, D.; Chhowalla, M.; Asefa, T. J. Am. Chem. Soc.2013, 135, 7823−7826.(36) Lee, E. Z.; Jun, Y. S.; Hong, W. H.; Thomas, A.; Jin, M. M.Angew. Chem., Int. Ed. 2010, 49, 9706−9710.(37) Datta, K. K. R.; Reddy, B. V. S.; Ariga, K.; Vinu, A. Angew.Chem., Int. Ed. 2010, 49, 5961−5065.(38) Li, Q.; Yang, J.; Feng, D.; Wu, Z.; Wu, Q.; Park, S. S.; Ha, C. S.;Zhao, D. Nano Res. 2010, 3, 632−642.(39) Jun, Y. S.; Hong, W. H.; Antonietti, M.; Thomas, A. Adv. Mater.2009, 21, 4270−4274.
Chemistry of Materials Article
dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−31613160

(40) Mane, G. P.; Talapaneni, S. N.; Anand, C.; Varghese, S.; Iwai,H.; Ji, Q.; Ariga, K.; Mori, T.; Vinu, A. Adv. Funct. Mater. 2012, 22,3596−3604.(41) Paraknowitsch, J. P.; Zhang, J.; Su, D.; Thomas, A.; Antonietti,M. Adv. Mater. 2010, 22, 87−92.(42) Zhang, J.; Chen, X.; Takanabe, K.; Maeda, K.; Domen, K.;Epping, J. D.; Fu, X.; Antonietti, M.; Wang, X. Angew. Chem., Int. Ed.2010, 49, 441−444.(43) Xia, Y.; Mokaya, R. Adv. Mater. 2004, 16, 1553−1558.(44) Groenewolt, M.; Antonietti, M. Adv. Mater. 2005, 17, 1789−1792.(45) Wei, J.; Zhou, D.; Sun, Z.; Deng, Y.; Xia, Y.; Zhao, D. Adv. Funct.Mater. 2013, 23, 2322−2328.(46) Liu, R.; Wu, D.; Feng, X.; Mullen, K. Angew. Chem., Int. Ed.2010, 49, 2565−2569.(47) Zhang, J.; Guo, F.; Wang, X. Adv. Funct. Mater. 2013, 23, 3008−3014.(48) Goettmann, F.; Fischer, A.; Antonietti, M.; Thomas, A. Angew.Chem., Int. Ed. 2006, 45, 4467−4471.(49) Zhang, Y.; Schnepp, Z.; Cao, J.; Ouyang, S.; Li, Y.; Liu, S. Sci.Report 2013, 3, 2163.(50) Wang, Y.; Wang, X.; Antonietti, M.; Zhang, Y. ChemSusChem2010, 3, 435−439.(51) Mane, G. P.; Dhawale, D. S.; Anan, C.; Arig, K.; Ji, Q.; Wahab,M. A.; Mori, T.; Vinu, A. J. Mater. Chem. A 2013, 1, 2913−2920.(52) Yan, H. Chem. Commun. 2012, 48, 3430−3432.(53) Liu, L.; Deng, Q. F.; Ma, T. Y.; Lin, X. Z.; Hou, X. X.; Liu, Y. P.;Yuan, Z. Y. J. Mater. Chem. 2011, 21, 1600−1606.(54) Roy, S. C.; Harding, A. W.; Russell, A. E.; Thomas, K. M. J.Electrochem. Soc. 1997, 144, 2323−2328.(55) Sidik, R. A.; Anderson, A. B.; Subramanian, N. P.; Kumaraguru,S. P.; Popov, B. N. J. Phys. Chem. B 2006, 110, 1787−1793.(56) Jiang, L. Q.; Gao, L. Carbon 2003, 41, 2923−2929.(57) Jaouen, F.; Lefevre, M.; Dodelet, J. P.; Cai, M. J. Phys. Chem. B2006, 110, 5553−5558.(58) Jun, Y. S.; Lee, E. Z.; Wang, X.; Hong, W. H.; Stucky, G. D.;Thomas, A. Adv. Funct. Mater. 2013, 23, 3661−3667.(59) Xia, Y.; Mokaya, R.; Walker, G. S.; Zhu, Y. Adv. Energy Mater.2011, 1, 678−683.(60) Xia, Y.; Walker, G. S.; Grant, D. M.; Mokaya, R. J. Am. Chem.Soc. 2009, 131, 16493−16499.(61) Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B. F.; Stucky, G. D. J. Am.Chem. Soc. 1998, 120, 6024−6036.(62) Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G. H.;Chmelka, B. F.; Stucky, G. D. Science 1998, 279, 548−552.(63) Jun, S.; Joo, S. H.; Ryoo, R.; Kruk, M.; Jaroniec, M.; Liu, Z.;Ohsuna, T.; Terasaki, O. J. Am. Chem. Soc. 2000, 122, 10712−10713.(64) Lu, A. H.; Schmidt, W.; Spliethoff, B.; Schuth, F. Adv. Mater.2003, 15, 1602−1606.(65) Talapaneni, S. N.; Anandan, S.; Mane, G. P.; Anand, C.;Dhawale, D. S.; Varghese, S.; Mano, A.; Mori, T.; Vinu, A. J. Mater.Chem. 2012, 22, 9831−9833.(66) Shen, W.; Ren, L.; Zhou, H.; Zhang, S.; Fan, W. J. Mater. Chem.2011, 21, 3890−3894.(67) Zhang, P.; Gong, Y.; Li, H.; Chen, Z.; Wang, Y. Nat. Commun.2013, 4, 1593.(68) Ansari, M. B.; Min, B. H.; Mo, Y. H.; Park, S. E. Green Chem.2011, 13, 1416−1421.(69) Xia, Y.; Mokaya, R. Chem. Mater. 2005, 17, 1553−1560.(70) Khabashesku, V. N.; Zimmerman, J. L.; Margrave, J. L. Chem.Mater. 2000, 12, 3264−3270.(71) Mao, Y.; Duan, H.; Xu, B.; Zhang, L.; Hu, Y.; Zhao, C.; Wang,Z.; Chen, L.; Yang, Y. Energy Environ. Sci. 2012, 5, 7950−7955.(72) Yang, S.; Gong, Y.; Zhang, J.; Zhan, L.; Ma, L.; Fang, Z.; Vajtai,R.; Wang, X.; Ajayan, P. M. Adv. Mater. 2013, 25, 2452−2456.(73) Silva, R.; Al-Sharab, J.; Asefa, T. Angew. Chem., Int. Ed. 2012, 51,7171−7175.(74) Zhang, C.; Fu, L.; Liu, N.; Liu, M.; Wang, Y.; Liu, Z. Adv. Mater.2011, 23, 1020−1024.
(75) Wu, P.; Qian, Y.; Du, P.; Zhang, H.; Cai, C. J. Mater. Chem.2012, 22, 6402−6412.(76) Qi, W.; Liu, W.; Zhang, B.; Gu, X.; Guo, X.; Su, D. S. Angew.Chem., Int. Ed. 2013, 52, 14224−14228.(77) Tuinstra, F.; Koenig, J. J. Chem. Phys. 1970, 53, 1126−1130.(78) Maciel, I.; Anderson, N.; Pimenta, M.; Hartschuh, A.; Qian, H.;Terrones, M.; Terrones, H.; Campos-Delgado, J.; Rao, A.; Novotny,L.; Jorio, A. Nat. Mater. 2008, 7, 878−883.(79) Liang, J.; Jiao, Y.; Jaroniec, M.; Qiao, S. Z. Angew. Chem., Int. Ed.2012, 51, 11496−11500.(80) Sheng, Z. H.; Shao, L.; Chen, J. J.; Bao, W. J.; Wang, F. B.; Xi, X.H. ACS Nano 2011, 5, 4350−4358.(81) Jun, G. H.; Jin, S. H.; Lee, B.; Kim, B. H.; Chae, W. S. S.; Hong,H.; Jeon, S. Energy Environ. Sci. 2013, 6, 3000−3006.(82) Sun, X.; Eang, R.; Su, D. S. Chin. J. Catal. 2013, 34, 508−523.(83) Zheng, Y.; Jiao, Y.; Ge, L.; Jaroniec, M.; Qiao, S. Z. Angew.Chem., Int. Ed. 2013, 52, 3110−3116.(84) Li, W.; Zhang, Z.; Kong, B.; Feng, S.; Wang, J.; Wang, L.; Yang,J.; Zhang, F.; Wu, P.; Zhao, D. Angew. Chem., Int. Ed. 2013, 52, 8151−8155.(85) Sun, F.; Liu, J.; Chen, H.; Zhang, Z.; Qiao, W.; Long, D.; Ling,L. ACS Catal. 2013, 3, 862−870.(86) Lin, Z.; Waller, G.; Liu, Y.; Liu, M.; Wong, C. P. Adv. EnergyMater. 2012, 2, 884−888.(87) Zhang, J.; Su, D. S.; Blume, R.; Schlogl, R.; Wang, R.; Yang, X.;Gajovic, A. Angew. Chem., Int. Ed. 2010, 49, 8640−8644.(88) Zhang, J.; Liu, X.; Blume, R.; Zhang, A.; Schlogl, R.; Su, D. S.Science 2008, 322, 73−77.(89) Gounder, R.; Iglesia, E. J. Am. Chem. Soc. 2009, 131, 1958−1971.(90) Serrano, D. P.; Aguado, J.; Escola, J. M. ACS Catal. 2012, 2,1924−1941.(91) Kissin, Y. V. Catal. Rev. 2001, 43, 85−146.(92) Addiego, W. P.; Estrada, C. A.; Goodman, D. W.; Rosynek, M.P.; Windham, R. G. J. Catal. 1994, 147, 407−416.
Chemistry of Materials Article
dx.doi.org/10.1021/cm5005664 | Chem. Mater. 2014, 26, 3151−31613161