hereditary cancer: ascertainment and management
TRANSCRIPT

colorectal cancers occur before age 45,iin genetically predisposed families withor without polyposis, the mean age atonset is 45 years,2-4 (2) a marked excess of bilateral cancer occurrence inpaired organs, e.g., breasts, adrenal(pheochromocytomas) and thyroidglands, carotid body, kidney (Wilms'tumor), acoustic neurinoma;5'6 (3) innonpaired organs, multiple primary ormulticentnc cancer occurs with a frequency many times greater than otherwise expected.7 Tumor registry datahave shown that the risk of other cancerin patients with certain histologic varieties is significantly higher than in cancerfree patients of the same age.8 Whilethese studies did not evaluate specificetiologies, many of the most frequentlyoccurring multiple primary tumor associations in cancer registry data were considered consistent with a genetic etiology; (4) although there are well established autosomal recessive, sex-linked,and cytogenetic cancer and precancerousdisorders, in many cases vertical transmission in consecutive generations offamilies has been identified with segregation patterns consistent with autosomal dominant inheritance.2
These characteristics can be utilizedas familial cancer selection criteria whenidentified in isolated patients and nuclearfamily cancer clusters, with or withoutan immediate impression as to the specific hereditary cancer or precancer syn
Generalities in Family Cancer
Virtually all hereditary cancers and precancerous diseases show the followingcharacteristics: (1) early age of canceronset, the identical histologic varietyoften occurring 20 or more years earlierthan in the general population. For example, while only four percent of all
Dr. Henry Lynch is Professor and Chairman, Department of Preventive Medicine/Public Health and the Oncology Clinic,Creighton School of Medicine, Omaha,Nebraska.Mr. Patrick Lynch is Instructor, Department of Preventive Medicine/Public Healthand the Oncology Clinic, Creighton Schoolof Medicine, Omaha, Nebraska.Dr. Albano is Assistant Professor, Departments of Surgery and Preventive Medicine/Public Health, Surgery, and the OncologyClinic, Creighton School of Medicine,Omaha, Nebraska.Dr. Edney is Surgical Oncology Fellow,American Cancer Society.Dr. Organ is Professor and Chairman, Department of Surgery, Creighton School ofMedicine, Omaha, Nebraska.Jane Lynch is Instructor, Department ofPreventive Medicine/Public Health, Creighton School of Medicine, Omaha, Nebraska.We gratefully acknowledge support from theAmerican Cancer Society, Nebraska andIowa Divisions, from Council for TobaccoResearch, USA, Inc., Grant #941 AR2, andfrom countless physicians, patients, andtheir families, whose unswerving dedicationand cooperation through the years havemade these cancer genetic investigationspossible.
CA-ACANCERJOURNALFORCLINICIANS216
HereditaryCancer:AscertainmentandManagement
Henry T. Lynch, M.D., Patrick M. Lynch,J.D., William A. Albano, M.D., JohnEdney, M.D., Claude H. Organ, M.D.and Jane F. Lynch, RN.

drome that may be involved. As seen inthe “¿�nuclearcomponents―of Fig. 1 (thebreast cancer-prone family), when a patient presents with breast cancer at anearly age (below age 45) and has a sister or mother similarly affected early inlife, the physician should immediatelyconsider the possibility of a hereditarybreast cancer syndrome. When bilateraldisease and/or associated malignant neoplasms (such as carcinoma of the ovary)are observed, the likelihood of a hereditary cancer syndrome is greater andshould provide a basis for extending thefamily history to include second degreerelatives. When these uncommon features continue to be expressed in collateral branches of the same family, theinitial impression of a familial/geneticpredisposition should be regarded as essentially confirmed. At this point, thedeveloped pedigree can be formally compared with known cancer genetic syndromes or previously reported familialtumor aggregations. In certain circumstances, cutaneous signs of one of themore than 50 cancer-associated genodermatoses might be identified, therebyproviding useful clues to hereditary cancer syndrome identification.9
Important Elements in CompilingFamily Cancer History
The ready accessibility, patent simplicity, and predictive capability of the family cancer history could reduce cancermorbidity and mortality perhaps moreeffectively than some of our most sophisticated and expensive diagnostic tools.In our Oncology Clinic, nurses with anorientation in cancer genetic theory andinterview techniques have assumed fullresponsibility for the compilation of cancer family histories.'0 Family history interviews taken by them have focused onfirst degree relatives and, in young patients, on second degree relatives.
In the Clinic, pedigree schematicshave helped structure the initial familyhistory interview. The patient, by seeingthe pedigree relationships, is better ableto recall a more complete genealogy and
medical history of his relatives. When,as is often the case, the patient is accompanied'by other relatives, they are askedto participate in the history collectionsince they may be able to provide moredetailed genealogic and medical factsabout the family. Patients are encouraged to discuss these historical matterswith other relatives and to bring anyof this information, including availablevital documents, to the Clinic at the timeof their next appointment.
In evaluating family history for thepresence of adult onset, autosomal dominant inherited disorders, certain secondand third degree relatives, because oftheir older ages, may be more “¿�genetically informative―th3n the patient's relatively young children, siblings, andperhaps even parents.
Our efforts have enabled Clinic physicians to devote more of their attentionto the evaluation of the pedigrees. A decision to pursue the history in greaterdetail involves referral to the familialcancer registry of the Institute forFamilial Cancer Management and Control (2500 California, Omaha, Nebraska68178). As syndromes are determined toexist in a particular family, appropriatemanagement protocols for the patientand his/her high-risk relatives are eitherinstituted or recommended.'0
In following up a family history, thepatient's report that a given relative hadcancer focuses critical attention on specific issues: age at onset; target organinvolved; the presence of significant nongenetic risk factors. Age and associatedrisk factors (smoking history, occupation) may be fairly well known to thepatient. The critical factors of organ andcell type are subject to extraordinaryinaccuracy, requiring documentationthrough primary pathologic recordswhenever possible.
Among the first 300 cancer patientsevaluated in our Oncology Clinic, approximately 10 percent have already reported striking familial aggregations ofcancer that on further inspection fulfilledmore rigorous criteria for hereditarycancer syndromes.1°
VOL. 29, NO. 4 JULY/AUGUST 1979 217

FamilialCancerRegistriesIt is not possible for the busy practitioneralone to devote the resources necessaryto elucidate the subtleties of cancer expression in an extended cancer-pronefamily, involving perhaps 100 to 500 ormore members. One possibility for thecentralized handling of cancer in genetically predisposed extended familiesanalogous to health department responsibilities in monitoring communicabledisease—would be more routine referralto highly visible centers charged withthe duties of evaluating such disordersand counselling patients at high risk ofor suffering from the disease.― Noncompulsory referrals by concerned patients and/or their physicians shouldminimize the potential for invasions ofprivacy, breaches of confidentiality, orimproper use of the information gathered.
Integrated familial cancer registrysystems could pooi data on suspectedcancer-prone families. Registry staffcould provide information on hereditarycancer syndromes to physicians caringfor high-risk relatives. In addition to riskfigures pertaining to cancers of specificanatomic sites, the most current methodsof surveillance for and management ofa particular disorder could be rapidlydisseminated to these physicians in amanner similar to that of most healthdepartments that keep physicians informed of control methods for communicable disease in their communities(Table 1).
Familial Cancer Diagnosis andManagementFamilial Polyposis Coli
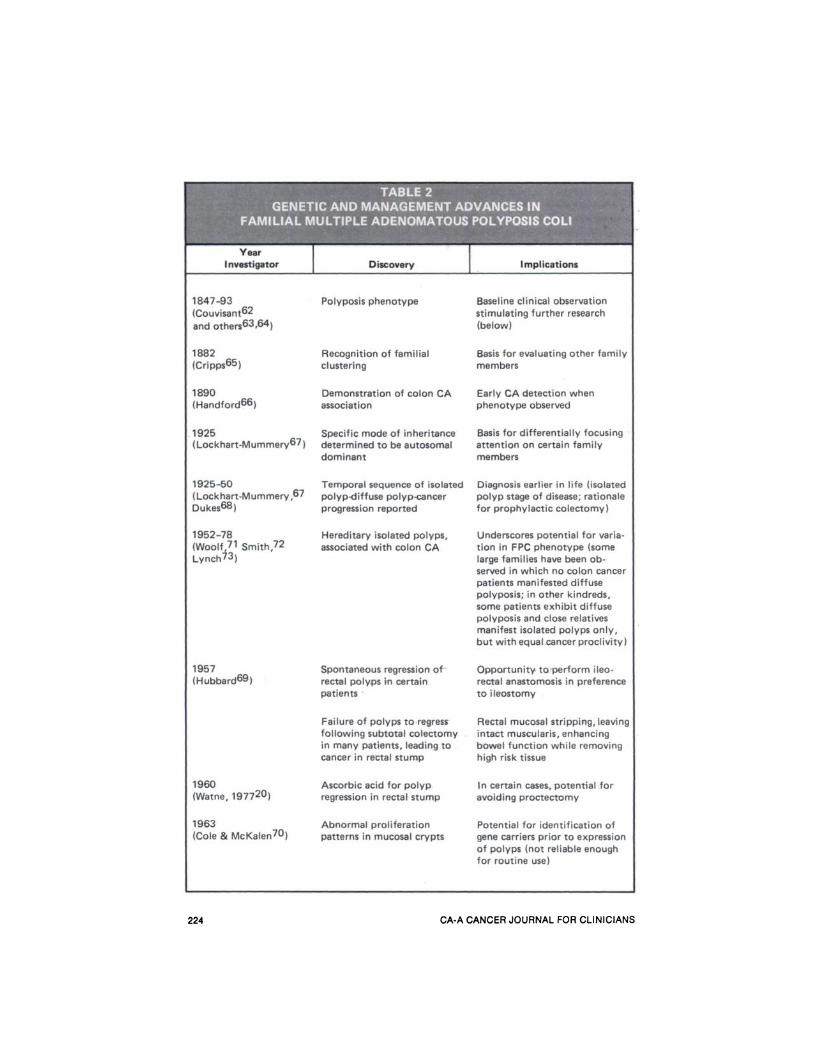
Familial multiple adenomatous polyposis of the colon (FPC), is frequentlycited as the classical prototype of ahereditary cancer-predisposing disorder.This is due to its supposed unvaryingprogression from isolated to diffusecolonic polyps to cancer of the colon,and because its genetic segregationclearly follows a Mendelian inheritancepattern, autosomal dominant.'2 Table 2
provides a cursory sketch of the chronology of FPC and of the clinical and basicresearch into its natural history, pathogenesis, and treatment.
Estimates of the frequency of thepolyposis gene range from approximatelyone in 7ØØ(J13to one in 24,00014 livebirths (applied to the population of theUnited States this would mean that between 150 and 500 newborns would beaffected each year). Because this diseasehas been so extensively investigated, itssurgical management (Table 2) is perhaps the most familiar and generally accepted cancer prophylaxis known inhereditary precancerous disease.'5 Recent evidence of heterogeneity in its expression and advances made in its management and control warrant review.
Families have been observed inwhich apparent carriers of the FPC gene(i.e., patients having both a parent andchildren with the classical phenotype)have developed colon cancer despite having no more than a few isolated polypsby age 20@3016 (Fig. 2). Because ofthis heterogeneity, the traditionally accepted rule of thumb—that at least 100polyps are required to establish the diagnosis,'7—occasionally may not apply;thus, failure by offspring of affected parents to manifest multiple polyposis ofthe colon by age 20 or 30 should notresult in relaxed surveillance, nor shouldit be supposed that such offspring cannot themselves have children affectedwith the “¿�complete―phenotype.
The traditional distinctions betweenclassical FPC and the related, but allegedly discrete (genotypically) polyposis syndromes18-2' (Table 2) have beenblurred by the meticulous clinical studiesof several investigators.22'23 For example, occult osteomatous and cutaneouschanges have been observed in FPCfamilies thought to lack the stigmatagenerally associated with Gardner's syndrome.@ The notion that the adenomatous polyposis in FPC is limited to thecolon has been criticized by clinicianswho have identified adenomatous or hyperplastic polyps in the small bowel andstomach.23'@ The earlier views are at
218 CA-A CANCER JOURNAL FOR CLINICIANS
C
6>
Cng.C
0@@
a)Ca)
@.0a)- •¿�o
0Cl)Cl)Co
C c...@
Eu@vo @.-C
1@ 0
zOEo(Cl
C,).9(Cl
.0
oC
-c .@ (00@t Cl)
.2@
8@ °@2
eo@@2c(00
(1)
oEE
o2-
20 0 ,@.—
t;@@@i .9
@ @o.C t —¿� Ea)
a) a) #@ COO@ < 2@
EEa) a)@ULL C 0(01.. @.@ .@
oa)@a) @o 0 (DO
.j@cC @(D
> o•; cc
D.@@3 it2
>

Co
E C0 .c Ca@ .@ .2o0 @0 .@ CE c0 - 0@ @‘¿�E
—¿� C ‘¿�-.co= a) ii0._a) 0 a)@
@HD-aoE>E 5
@i@h_@Da)9 >.
.C a)u@a)
@ @9
C'..— a)@—¿�°oo Co>@o
fl2@ @.a@@ ,..@@ 2 ..- .E
2@ @Jc -@@@
C@J C@)@
.c
@0 ‘¿�@ -D@ E @‘¿�o
9 -@ >. [email protected]@ 0 •¿�EC
.2― E 2 o@
@“¿� o-@ Cl)
C @O I-@ c@2@ 0. 2 22@a @.- .E@ a .@0
c@E@a>.@ g@
—¿�@ E@
E―'a).E@2r0@
ccoa.CoCEwoa CC@E@@
0—¿�
2 O@@°.c co ‘¿�@@
CD
a@
0 ._9 @2 o@a) >.
o.9 0oa @0 C-@@ COO—¿�0 E @- —¿�c@D U) E E
@?2.@@@@ @v0@
IL@ coo CO -
@a) —¿�.2@co —¿�.2,n—@.Ec0
<a@ z8@awtC W.@C0 W °W@@0
CC,
C,a..na)
0*1CC,
EC,
C,CC,
*
a)
4.'a)
CC,4.'a)
0.
0a))CCC,C,
UCl)
CA-A CANCER JOURNAL FOR CLINICIANS220
Ca)
a)a
(0
IL)
C')
w0z0I-Cl)
w
-j
-J
00w
wI
a)to
0) a).4-
@0>C i-@a) .C
D.Ca)OCiC- .@ @_
..U'C0‘¿�-@ OC
8 8C C .9
8@ .@
.0 @i.9@
@ 0@
0 @E@
>2@::.@1 a) a) —¿�a) = Cw.0@
0
Va)
E-(U)
a).a),-
a) 0.Noa.C (00)
C CL@
OV We.,.@ =@4L
EEa)0.2@2 .E 8
@Co0a) i
c 8a)
w
0
0z>-Cl)(@4
u-C%1-
C@ 2
z@2w 8
VOL. 29,NO.4 JULY/AUGUST 1979 221
.2@
@F@C @.2'
ill@@a)
a)C>a)0―'@
a)a)0@a).0
@. ‘¿�V@
a) @a)C
.CCCOC..a)00)O
—¿� a C@>CO
.@ .2@Ea@
.c
•¿�g@E —¿�
0 Ca)0@—¿� X a)>[email protected]
V 2c
@ .2 2i―a)><
2‘¿�I)@ >a..'a)E
a)@ a)
—¿� —¿� a)Va)a)
Cl)@ E co0a) Ca)a),..@.
o.@o 8i@@@ 228
Vi00 —¿� a)u0@ Co
—¿�@ a) @a)a) 00)
.c'.-8@—¿� C —¿�c
a) C0o0 .c .!C
@ .@ @I
2@ E@js.@ a)a)=a)
OC@- a)>@._a)
@@ >@@
.E a @Ca a―;a)o.c a)@>0 ._ a)
.—@W [email protected]— ,,, C
£@
a)0
C@@
“¿�‘a)a)
.—Eo@ oV
xEa)a)ii-.c 00@,
-0@0o @u.
(@14
0C0
.c8
.@
V22―' a.98 =00 a)
C
Co a) U'N a)@ •¿�g
a) a)a)[email protected] •¿�5@0a)5@
I-
@ ....C 0.C@0―@ C@
a).Ca)@ I.@
o.@a@C

0a)0)
a)Ca)
Ca)Ea)a)
-DCa)0)C
Ca)a)0
a)0C
8a)a)
.0
a)
Ea)
6Va)
a).cC
a)
a)0C
8a)a)
.0
.2u—¿�LLCOa)E .E
V0Ca)8
@ C.E oa)0
a)0Ca)0@—¿�C
C-04-0
0CCa)a)C
a)@
0
>0)
“¿�[email protected]
a)4-o.@
C-.—
8@C.@
>._@a)
E@•¿�c@.@oC
@“¿�.
2ia) C―.
V4.._
g@aa)C
@!;0—¿�
V2-@
.@—V@
E=a)0x.—
@C0.a)
ia@E
c28
.?i•¿�i@—¿�
C
I.0@4-
@)
—¿�
VCCO
0).EC>
C')@. @.
a).c a)V—¿�
EC@toOa)—
V@V
1L@8g‘¿�CC-C.C
•¿�@0.!!@
E@@>
.8@2
COO.--ca)VC'@Ea)a)C
—¿� —¿�
:@i@ .2a)gCC a)ga)
U' @a)_@a)@
@ .1@V!@C aa)9@a)@
a)o@―'.!ECO>@
OCa)Ca)@a).2>.a)aO0@C-@4-a)a)
VEU'@C'.'
CDa)
c.9 @‘¿�VE
@2-@ .V
@Ca)@>..J0)CJ)
.C...a)>.a)E@E0-JOo .CC-0)•_
—¿�@a)(‘)_lO‘¿�@4t@
@a)C-
C/)@8—¿�0E
.9
C
20)
coE
V
i-
@3CC@
a)a)
w.2
—¿� a)-Jcr a
=a)
C/)< —¿� a)@2<°' .@0.@E@
0 D.,- C- a)I—ZV C<<C 0.
J—@Cl)@
•¿� a)
CC,
4.'C,
0.
C,
t
I*-@
a)
4'C,
CC,4.'a)a0
CCC,C,
Ufd')
C,
0a)
C,.,I—w
wo@cc
2(1@
<cx:—¿�@w
@ 2.@ <0
-iwo
<ocr@_C'00—¿�w
@ ww'J)OLL.
0
a>.
—¿�C
8 E—¿� 0Cl) C
.0i.0 C-—¿� oua) .4-U)
>a)
E ECa) 0.@
C4;. .@>
a) a)[email protected] oC- .— > a)a>
‘¿�,@-@; 00
0.0—0 a)
E c
C0
a)0.4.'V 2
w U'CO 2@ a) .EZ C- -@<@ .2@O@I—L@ E@ Cu)C'E @a)a)0w@ C a)@@
@ —¿� 0
@ C@ .@ -@
.9 o —¿�“¿�‘a)@0a)Ca)
@ >-@
@j OV
LL@ @0a)―@a@ aO
0 a)O@@V0) a)V@@
@ E2
CA-ACANCERJOURNALFORCLINICIANS222

tributed primarily to a failure to routinely explore the upper gastrointestinaltract. However, the malignant potentialof extra-colonic polyps in affected patients appears not to be as great as forpolyps of the colon proper.24
In a remarkable kindred reported byBinder et al,25 medulloblastoma characteristic of Turcot's syndrome (more descriptively called the polyposis-gliomasyndrome and believed to be autosomalrecessively inherited) was reported in apatient whose relatives manifested sebaceous cysts and gastric polyps compatible with Gardner's syndrome. Thus, features of several supposedly distinct syndromes were witnessed in a single familyunit.
Screening Patients withUnexpressed FPC Phenotype
Management of FPC has heretoforeconsisted primarily of proctoscopicscreening of patients until polyps appeared in sufficient numbers, at whichtime either subtotal colectomy with ileorectal anastomosis or total proctocolectomy with ileostomy was performed.The now apparent heterogeneity in thisdisorder requires, and advances in diagnostic and treatment techniques allow,a more flexible approach to its management.
Screening for polyps should be commenced at age 10 in high-risk patients(those with one or more affected firstdegree relatives) or earlier if there aresymptoms or signs, such as rectal bleeding or cutaneous and/or osseous manifestations of Gardner's syndrome. Thedifficulty of performing adequate proctosigmoidoscopy in youngsters coupledwith frequent noncompliance, necessitates biannual screening for occult fecalblood. Hemoccult screening should becontinued biannually through age 45,and annually thereafter.
In asymptomatic patients, a baselinebarium enema with air contrast is performed at age 20. In symptomatic patients or those with a positive Hemocculttest, double air contract barium studiesof the lower gastrointestinal tract should
be performed. Proctosigmoidoscopy isbegun on a routine basis at age 15 if itcan be accomplished easily and effectively. This is continued on an annualbasis until age 45 and repeated at oneto two year intervals as long as the patient remains asymptomatic.
Treatment
In patients with more than 100 polyps,total proctocolectomy remains the treatment of choice. Utilization of a continent ileostomy or performance ofsphincter-preserving procedures warrantcareful consideration. While such techniques have led to an acceptable continency rate, patients contemplating theseprocedures must be told that an ileostomymay eventually be required.26'27
The frequently employed and moreconservative total abdominal colectomywith ileorectal anastomosis requires diligent evaluation for and fulguration ofrectal polyps on a three-month basis forlife (though the possibility exists of polypregression due to the action of “¿�ilealcontents―)- Because of the magnitude ofcancer risk to the rectal stump in patients with rectal polyps (59 percent at23 years)28 elective proctectomy at 10years postresection must be considered;the development of retroperitoneal fibrosis following total abdominal colectomyin Gardner's syndrome may make subsequent proctectomy technically difficult.Follow-up must include screening forpolyps of the upper gastrointestinal tract(see Table 2).
In FPC families, in patients having10 to 100 polyps and in whom the rectum is not extensively involved (six to 10polyps), a total abdominal colectomywith ileorectal anastomosis and fulguration of subsequent rectal polyps on athree-month basis for life is the procedure of choice. In those patients whoserectum is extensively involved, total coloproctectomy should be performed withconsideration for a continent ileostomyor pull-through procedure.
In patients with fewer than 10 polyps, biannual colonoscopic removal ofall polyps may be accomplished. If the
VflI 2q NC) 4 .IIJIY/AIJ(IJSTIQ7Q
224 CA-A CANCER JOURNAL FOR CLINICIANS

endometrium29 (and less frequently, thestomach, breast, and ovary30'31), occurring at much earlier ages (@ 45 years)than the same histologic varieties in thegeneral population.32 Lesions involvingthe colon show a proximal colonic predilection (65 percent are so situated) ,33in contrast to the 20 to 30 percent solocated in the general population. Initially affected patients have an extraordinary risk of multiple primary cancer, typically involving the end organscited above.7
The tumor expression pattern istransmitted vertically, and segregationratios are consistent with an autosomaldominant mode of inheritance.2 A number of investigators are actively searching for markers of the precancerous
polyps cannot be removed via the colonoscope or the frequency of recurrence appears to be increasing, totalabdominal colectomy with ileorectalanastomosis followed by proctoscopicfulguration of polyps should be considered. Factors such as the age of thepatient (polyps at an early age are morelikely genetic) and a history of coloncancer in relatives with isolated polypsmay aid in distinguishing the hereditary(colectomy required) from sporadic(polypectomy adequate) nature of theisolated polyps that are encountered.
The Cancer Family Syndrome
Criteria for the Cancer FamilySyndrome(CFS) include a familial excess of adenocarcinoma involving the colon and
VOL. 29, NO. 4 JULY/AUGUST 1979 225

state in the CFS,34-36though none havebeen definitively identified to date. Consequently, diagnosis of the syndrome ina given family must still be predicatedon the pedigree's consistency with theabove criteria.
Management of the CFS patientwith colonic cancer is much like that ofFPC, namely subtotal colectomy, continued surveillance of the rectal stump,and a high index of suspicion for noncolonic cancer.37-41 The proximal colonic excess precludes exclusive relianceon proctosigmoidoscopy as a screeningmeasure. Other diagnostic and therapeutic approaches that differ from thoseutilized in FPC are described in Table 1.
Familial Breast Cancer
That familial breast cancer is a heterogeneous disease is evidenced by its association with other malignant neoplastic lesions in certain families (Table1) 3,42,43 With the exception of Cowden's disease (multiple hamartoma syndrome) there are no reliable preclinicalmarkers of high breast cancer risk5(Table 1). Therefore, the familial riskfor this disease can only be estimatedin relation to the available family history. The occurrence of two or morefirst degree relatives with histologicallyverified breast cancer constitutes a reasonable basis for more intensive investigation of an extended family; studies ofconsecutive series of breast cancer probands have shown that approximately 15to 20 percent will have another first degree relative similarly affected.@ Premenopausal expression and bilateralityrepresent significant selection criteria intheir own right.3.5'6'8'42-45
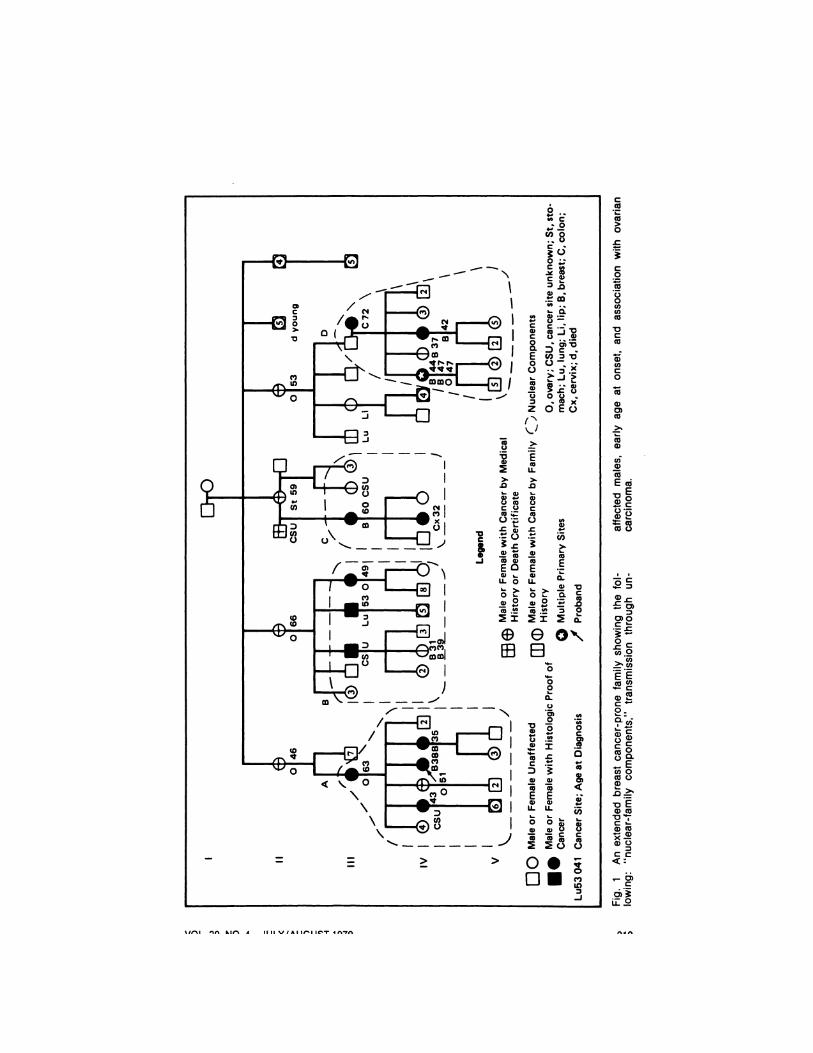
Fig. 1, a pedigree of an extendedbreast/ovarian cancer-prone family, illustrates four key issues in breast cancergenetics. Note the early ages at onset andassociated cancer (ovarian) - The pedigree includes an example of apparenttransmission through an unaffected male(nuclear component D), a phenomenonobserved in other families. The term“¿�nuclearcomponents― of the kindred
emphasizes the somewhat limited natureof the information typically available toa given family physician. This isstrengthened significantly when it can beincorporated into the larger unit of anextended kindred whose other branchesmay show similar expression and collectively enable the identification of amore complex hereditary cancersyndrome.
Prophylactic Surgery
Because of the great risk of cancer inthe contralateral breast among patientsfrom high risk families,6'8 patients in ourinstitution are considered for contralateral mastectomy approximately one yearfollowing initial mastectomy. The procedure is deferred somewhat in patientshaving had high-risk initial lesions. Allpatients are carefully evaluated for metastatic disease and are considered forsimultaneous reconstruction of the primary site. In all cases, the tissue removedat contralateral prophylactic mastectomyis carefully evaluated by both radiographic and histologic techniques, withappropriate treatment undertaken if cancer is found.
In highly selected unaffected patientsat risk for familial breast cancer, prophylactic bilateral subcutaneous mastectomy with reconstruction is worthy ofconsideration and has been performed ina growing number of cases.45@46
Familial Atypical MultipleMole-Melanoma Syndrome (FAMMM)
Familial clustering of malignant melanoma has been described frequentlyenough to suggest that in about three to10 percent of occurrences, primary genetic factors may predispose to thisdisease.47
As in the Cancer Family Syndromeand heredo-familial breast cancer, physical markers of the preneoplastic statehave been lacking. Recently, however, ahereditary, premelanotic cutaneous phenotype (characterized by multiple largemoles, irregularly shaped, reddish-brownto pink in color, and with evidence of
rA.A (@AM(@PR .1C1 IRNAI FoR (@.I INIOIANS
l.—Q p2 p3 4 5d29 d64 d68
II@
BT 71 69 * d74 77 65 d69 d65 d59Cx CSU 73 B L L Thr Lym
1 2 3 4 5
II hOPC 147 43 d36 32
T C C
* 21 20 18 17 16 14 12
PC 2325
1 2 Legend
Code number; male or female unaffected
4718 Age
Male or female with histologic proof of cancer
tJ@KDMaleorfemalewithcancerbyhistoryMale or female with cancer by death certificate
/Proband
* Isolated intestinal polyps
‘¿�“Diffusecolonic polyposis
PC, Prophylactic colectomy; B, Breast; BT, Brain Tumor;C, Colon; CSU, Cancer Site Unknown; Cx, Cervical;L, Leukemia; Li, Liver; Lu, Lung; Lym, Lymphoma;T, Testicular; Thr, Throat; d, died
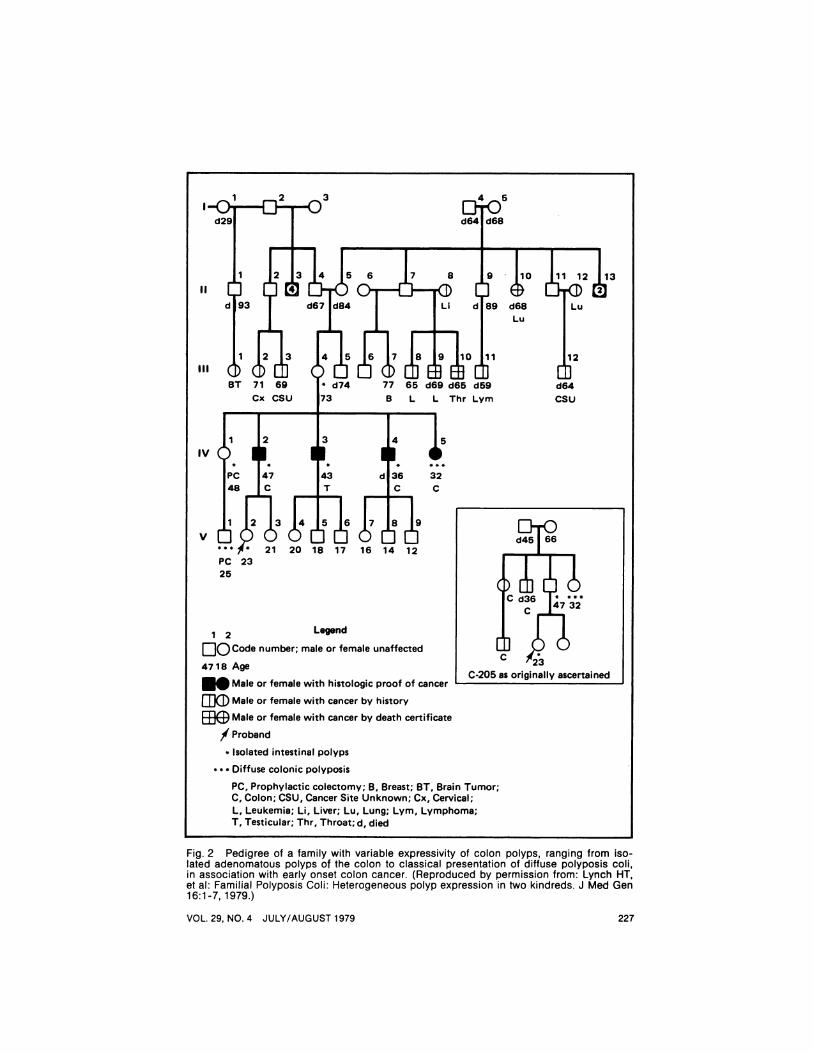
Fig. 2 Pedigree of a family with variable expressivity of colon polyps, ranging from isolated adenomatous polyps of the colon to classical presentation of diffuse polyposis coli,in association with early onset colon cancer. (Reproduced by permission from: Lynch HT,et al: Familial Polyposis Coli: Heterogeneous polyp expression in two kindreds. J Med Gen16:1-7, 1979.)
C-205 as originally ascertained
IV
V
VOL. 29, NO. 4 JULY/AUGUST 1979 227
j 4@Ld68 LuLu
c112d64CsU
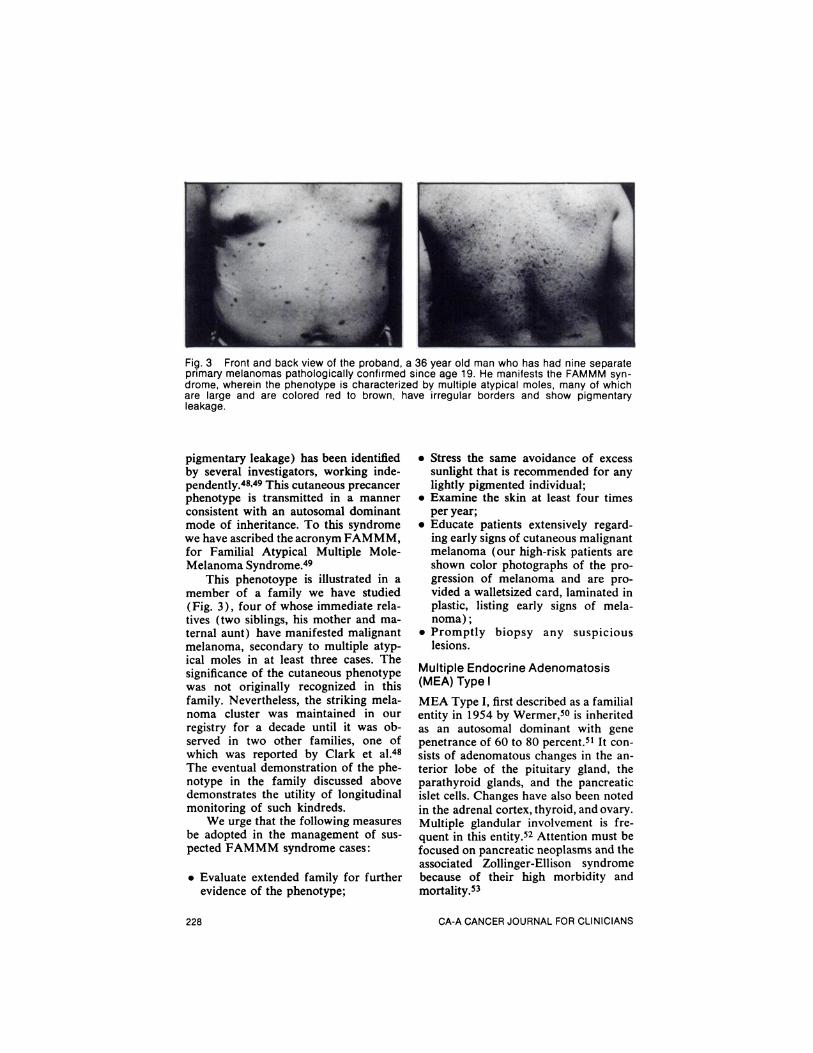
Fig. 3 Front and back view of the proband, a 36 year old man who has had nine separateprimary melanomas pathologically confirmed since age 19. He manifests the FAMMM syndrome, wherein the phenotype is characterized by multiple atypical moles, many of whichare large and are colored red to brown, have irregular borders and show pigmentaryleakage.
pigmentary leakage) has been identifiedby several investigators, working independently.48'49 This cutaneous precancerphenotype is transmitted in a mannerconsistent with an autosomal dominantmode of inheritance. To this syndromewe have ascribed the acronym FAMMM,for Familial Atypical Multiple MoleMelanoma Syndrome.49
This phenotoype is illustrated in amember of a family we have studied(Fig. 3), four of whose immediate relatives (two siblings, his mother and maternal aunt) have manifested malignantmelanoma, secondary to multiple atypical moles in at least three cases. Thesignificance of the cutaneous phenotypewas not originally recognized in thisfamily. Nevertheless, the striking melanoma cluster was maintained in ourregistry for a decade until it was observed in two other families, one ofwhich was reported by Clark et al.48The eventual demonstration of the phenotype in the family discussed abovedemonstrates the utility of longitudinalmonitoring of such kindreds.
We urge that the following measuresbe adopted in the management of suspected FAMMM syndrome cases:
•¿�Evaluate extended family for furtherevidence of the phenotype;
•¿�Stress the same avoidance of excesssunlight that is recommended for anylightly pigmented individual;
•¿�Examine the skin at least four timesper year;
•¿�Educate patients extensively regarding early signs of cutaneous malignantmelanoma (our high-risk patients areshown color photographs of the progression of melanoma and are provided a walletsized card, laminated inplastic, listing early signs of melanoma);
•¿�Promptly biopsy any suspiciouslesions.
Multiple Endocrine Adenomatosis(MEA) Type I
MEA Type I, first described as a familialentity in 1954 by Wermer,50 is inheritedas an autosomal dominant with genepenetrance of 60 to 80 percent.5' It consists of adenomatous changes in the anterior lobe of the pituitary gland, theparathyroid glands, and the pancreaticislet cells. Changes have also been notedin the adrenal cortex, thyroid, and ovary.Multiple glandular involvement is frequent in this entity.52 Attention must befocused on pancreatic neoplasms and theassociated Zollinger-Ellison syndromebecause of their high morbidity andmortality.53
228 CA-ACANCERJOURNALFORCLINICIANS

Asymptomatic patients in MEA-Ifamilies should be screened via annualpentagastrin radioimmunoassay betweenages 10 and 65. Emphasis should beplaced on signs and symptoms of pepticulcer disease, hypoglycemia, watery diarrhea, pituitary disorders, thyroidnodules, hyperparathyroidism, andCushing's syndrome. A treatment protocot has been included in Table 1•¿�50-53
MEA-Il and MEA-Ill Syndromes
MEA-II (also known as MEA-Ila orSipple's Syndrome) is characterized bythe association of medullary thyroid cancer, pheochromocytoma, and parathyroid neoplasm.51 The MEA-Ill (Il-b,multiple mucosal neuroma syndrome)variant is, in addition, associated withmarfanoid features and mucosal neuromas that may expedite its diagnosis. Central to the management of these syndromes is the early treatment of medullary thyroid cancer, since it is this component that most frequently leads to theaffected patient's early demise.
Although sporadic occurrences ofthe disease are relatively more frequent,the autosomal dominant mode of inheritance has been well established forthe hereditary variety. Familial medullary thyroid carcinoma bears a highlyspecific marker—calcitonin—that allowsfor the screening of high-risk relatives.54'55 Such screening should beginat age five and continue until age 60.56In high-risk patients, hoarseness, dysphagia, watery diarrhea, or a neck massshould alert the physician to the possibility of a medullary thyroid carcinoma.Symptoms of hypertension, diaphoresis,or headache may suggest pheochromocytoma. Patients should undergo annualparathormone assay, serum calcium andphosphorous, to rule out the possibilityof hyperparathyroidism.56
Elevated calcitonin in these patientsis diagnostic of medullary carcinoma ofthe thyroid.57-60 Early screening mayallow for the detection of C-cell hyperplasia prior to the onset of malignantchange.
In patients manifesting familial medullary thyroid cancer, it is imperative torule out the associated pheochromocytom'a prior to neck exploration. If apheochromocytoma is diagnosed, patients should undergo bilateral adrenalectomy due to the high frequency ofbilateral involvement in this population,even when there are no palpable abnormalities within the glands at exploration.A treatment protocol for MEA-Il andIII is included in Table 1.@°.5461
Conclusion
The historical perspective provided byfamilial multiple adenomatous polyposisof the colon (Table 2) provides an excellent example of the advances in cancer epidemiology and characterizationof hereditary precancerous disease(s),through multidisciplinary study over along period of time. Concomitant advances in surgical management and prophylaxis have been witnessed. However,it has also been seen that even in thismost intensively studied disorder(s), agreat deal remains unknown about thevariability in clinical features.
Those disorders lacking the readilydiscernable markers of the FPC,FAMMM and the MEA syndromes havebeen a particular concern in this discussion. Clinical research into these syndromes has been similar to the approachused in the study of FPC. This researchhas arrived at varying stages in the development of differential diagnostic andtherapeutic protocols.
Due to the early ages of high-riskpatients and their unfamiliarity with therole of genetic factors in cancer etiology,most would not otherwise be subject toscreening for the diseases involved. Aregistry of such patients and their families could facilitate a greater recognitionof their medical genetic significance. OurRegistry-based program has clearly demonstrated the manner in which a carefully obtained family history can lead tothe recognition of patients with exceedingly high cancer risk.
VOL. 29, NO. 4 JULY/AUGUST 1979 229

References
1. Third National Cancer Survey: IncidenceData. Publication 75-787, NC! monograph41, 1975.2. Lynch HT, et al: Clinical, genetic, andbiostatistical progress in the cancer familysyndrome. Front Gastrointest Res, to bepublished.3. Lynch lIT, et a!: Genetic heterogeneityand familial carcinoma of the breast. SurgGynec Obstet 142:693-699, 1976.4. Mulvihill JJ: Congenital and genetic diseases, in Fraumeni JF Jr, (ed): Persons atHigh Risk of Cancer: An approach to Cancer Etiology and Control. New York, Academic Press, 1975, pp 3-35.5. Anderson DE: Genetic study of breastcancer: identification of a high risk group.Cancer 34:1090-1097, 1974.6. Harris RE, Lynch HT, Guirgis HA:Familial breast cancer: risk to the contralateral breast. J Natl Cancer Inst 60:955-960, 1978.7. Lynch HT, et al: Role of heredity inmultiple primary cancer. Cancer 40 (suppi4):1849-1854, 1977.8. SchottenfeldD, Berg J: Incidenceofmultiple primary cancers. IV. Cancers ofthe female breast and genital organs J NatlCancer Inst 46:161-170, 1971.9. Lynch HT, Frichot BC ifi: Skin, heredity, and cancer. Semin Oncol 5:67-84, 1978.10. Lynch HT, et al: Family history in anoncology clinic: implications concerningcancer genetics. JAMA, to be published.11. Lynch HT, Lynch PH, Lynch JF: Management and control of familial cancer, inMulvihill JJ, Miller RW, Fraumeni JF Jr(eds): Genetics of Human Cancer. NewYork, Raven Press, 1977, pp 235-252.12. Dukes C: The hereditary factor in polyposis intestini or multiple adenomata. Cancer Rev 5:241-256, 1930.13. Pierce ER: Some genetic aspects offamilial multiple polyposis of the colon ina kindred of 1422 members. Dis ColonRectum 11:321-329, 1968.14. Veale AMO: Intestinal polyposis, EugenLab Memoirs Series, 40. London, Cambridge Univ Press, 1965.15. DeCosse JJ, Adams MB, Condon RE:Familial polyposis. Cancer 39:267-273,1977.16, Lynch HT, Lynch PM, Follett KL, Harris RE: Familial polyposis coli: heterogeneous expression in two kindreds. J MedGenet 16:1-7, 1979.17. Bussey HJR: Familial Polyposis Coli.Baltimore. Johns Hopkins Univ Press, 1975,pp 19-23,86-89.18. Gardner El: Follow-up study of a family group exhibiting dominant inheritancefor a syndrome including intestinal polyps,osteomas, fibromas and epidermal cysts.
Amer J Hum Genet 14:376-390, 1962.19. Oldfleld MC: The association of familial polyposis of the colon with multiplesebaceous cysts, Br J Surg 41:534-541,1954.20. Watne AL, et a!: The diagnosis andsurgical treatment of patients with Gardner's syndrome. Surgery 82:327-333, 1977.21. Turcot J, Despres J, St Pierre F: Malignant tumors of the central nervous systemassociated with familial polyposis of thecolon: report of two cases. Dis Colon Rectum 2:465-468, 1959.22. Utsunomiya J, Nakamura T: The occultosteomatous changes in the mandible inpatients with familial polyposis coli. Br JSurg62:45-5!, 1975.23.UshioK, Cta!:Lesionsassociatedwithfamilial polyposis coil: studies of lesions ofthe stomach, duodenum, bones, and teeth.GastrointestRadio!1:67-80,1976.24. Denzler Th, Harned RK, Pergam CJ:Gastric polyps in familial polyposis coli.Radiology 130:63-66, 1979.25. Binder MK, et a!: Colon polyps, sebaceous cysts, gastric polyps, and malignantbrain tumor in a family. Am J Dig Dis23:460-466,1978.26. Kock NG, et a!: Ileostomy. Curr Prob!Surg 14:1-52, 1977.27. Heimann T, Beck AR, Greenstein AJ:Familial po!yposis co!i. Management bytotal colectomy with preservation of continence. Arch Surg 113:1104-1106, 1978.28. Wolfstein IH, Dreznik ZJ, Avigad IS:Total colectomy and anal ileostomy in multiple polyposis coli. Arch Surg 113:1101-1103, 1978.29. Lynch HT, Krush AJ: Heredity andadenocarcinoma of the colon. Gastroentero!ogy 53:517-527, 1967.30. Lynch HT, Krush AJ, Guirgis H:Genetic factors in families with combinedgastrointestinal and breast cancer. Am JGastroenterol 59:31-40, 1973.31. Lynch HT, Lynch PM: Heterogeneoustumor expression in the cancer family syndrome: ovarian carcinoma. Am J Surg, tobepublished.32. Lynch lIT, Krush AJ, Thomas RJ,Lynch J: Cancer family syndrome, inLynch HT (ed): Cancer Genetics. Springfield, Charles C. Thomas Pub, 1976, pp555-588.33. Lynch PM, Lynch HT, Harris RE: Hereditary proximal colonic cancer. Dis ColonRect 20:661-668, 1977.34. Lipkin M, Deschner E: Early proliferative changes in intestinal cells. CancerRes 36:2665-2668, 1976.35. Berlinger NT, et al: Defective recognitive immunity in family aggregates ofcolon carcinoma. J Clin Invest 59:761-769,1977.36. Guirgis HA, Lynch HT, Harris RE,
230 CA-A CANCER JOURNAL FOR CLINICIANS

Vandevoorde JP: Genetic and communicable effects on carcinoembryonic antigen cxpressivity in the cancer family syndrome.Cancer Res 38:2523-2528, 1978.37. Winawer SJ, Sherlock P: Approach toscreening and diagnosis in cob-rectal cancer. Semin Oncol 3:387-397, 1976.38. Creasman WT, Weed JC Jr: Screeningtechniques in endometria! cancer. Cancer38 (suppl 1):436-440, 1976.39. Lynch HT, et al: Management of hereditary site-specific colon cancer. Arch Surg112:170-174, 1977.40. Greegor DH: Occult blood testing fordetection of asymptomatic colon cancer.Cancer 28:131-134, 197!.41. Tenney JB, Graney MJ: The quest forcontinence: a morphologic survey of approaches to a continent colostomy. DisColon Rect 21:522-533, 1978.42. Lynch HT, et a!: Familial associationof breast/ovarian carcinoma. Cancer 41:1543-1549, 1978.43. Lynch HT, et al: Genetic and pathologic findings in a kindred with hereditarysarcoma, breast cancer, brain tumors, leukemia, lung, laryngeal, and adrenal cortical carcinoma. Cancer 41:2055-2064, 1978.44. Lynch HT, et a!, unpublished data.45. Lynch HT, Harris RE, Organ CH Jr,Lynch JF: Management of familial breastcancer. I. Biostatistical-genetic aspects andtheir limitations as derived from a familialbreast cancer resource. II. Case reports,pedigrees, genetic counseling, and teamconcept.ArchSurg 113:1053-1067,1978.46.LeisHP Jr:Selectiveand reconstructivesurgicalproceduresforcarcinomaofthebreast.Surg Gynec Obstet148:27-32,1979.47. Cawley EP, Kruse WT, Pinkus HK:Genetic aspects of malignant melanoma.AMA Arch Derm Syph 65:440-450, 1952.48. Clark WH, et a!: Origin of familialmalignant melanomas from heritable melanocytic lesions. ‘¿�TheB-K mole syndrome.'Arch Dermatol 114:732-738, 1978.49. Lynch HT, Frichot BC, Lynch JF:Familial atypical multiple mole-melanomasyndrome. J Med Genet 15:352-356, 1978.50. Wermer D: Genetic aspects of adenomatosis of endocrine glands. Am J Med16:363,1954.51. Harrison TS, Thompson NW: Multipleendocrine adenomatosis—I and II. CurrProbl Surg Aug: 1-51, 1975.52. Tateishi R, et al: Coexistence of bilateral pheochromocytoma and pancreatic isletcells. Cancer 42:2928-2934, 1978.53. Friesen SR: Zo!linger-El!ison Syndrome.Curr Probl Surg Apr: 1-52, 1972.54. Goltzman D, Ct a!: Calcitonin as atumor marker. Use of the radioimmunoassay for calcitonin in the postoperativeevaluation of patients with medullary thy
roid carcinoma. N Eng! J Med 290:1035-1039, 1974.55. Graze K, et a!: Natural history offamilial medullary thyroid carcinoma. Effect of a program for early diagnosis. NEngl J Med 299:980-985, 1978.56. Freitas JE, Sisson JC, Freier DT,Thompson NW: Type Ila syndrome. Dilemmas in modern management. SeminNuc! Med 8:73, 1979.57. Chong GC, et a!: Medullary carcinomaof the thyroid gland. Cancer 35:695-704,1975.58. Freier DT, et a!: Dilemmas in the earlydiagnosis and treatment of multiple endocrine adenomatosis, type II. Surgery 82:407-413, 1977.59.WellsSA,eta!:Provocativeagentsandthe diagnosis of medul!ary carcinoma ofthe thyroid gland. Ann Surg 188: 139-14 1,1978.60. King HR, et al: Sipple's syndrome:medu!lary carcinoma, pheochromocytoma,and parathyroid disease, NIH Conferencediscussions. Ann Intern Med 78:561-579,1973.61. Block MA, Jackson CE, Tashjian AHJr: Management of parathyroid glands insurgery for medullary thyroid carcinoma.Arch Surg 110:617-624, 1975.62. Couvisant L: Hypertrophie partielle dela muqueuse intestinale. Bul Soc Anat22:400,1847.63. Chargelaigue A: Des poiyps du rectum.Thesis, Paris, 1859.64. Virchow R: Die krankhaften geschwubste. Berlin, A Hirschwabd, 1863.65. Cripps WH: Two cases of disseminatedpolyps of the rectum. Trans Path Soc Lond33:165-168,1882.66. Handford H: Disseminated pobyps ofthe large intestine becoming malignant.Trans Path Soc Lond 41:133, 1890.67. Lockhart-Mummery JP: Cancer andheredity. Lancet 1:427-429, 1925.68. Dukes CE: Familial intestinal polyposis.Ann R Coll Surgeons 10:293-304, 1952.69. Hubbard TB Jr: Familial pobyposis ofthe colon: the fate of the retained rectumafter cobectomy in children. Am Surg 23:577-586, 1957.70. Cole JW, McKalen A: Studies on themorphogenesis of adenomatous polyps inthe human colon. Cancer 16:998-1002,1963.7!. Woolf CM, Richards RC, Gardner EJ:Occasional discrete polyps of the colon andrectum showing an inherited tendency in akindred. Cancer 8:403-408, 1955.72. Smith WG: The cancer family syndrome and heritable solitary colonic polyps.Dis Colon Rect 13:362-367, 1970.73. Lynch PM, et a!: Heritable colon cancer and solitary adenomatous polyps, inNieburgs HE (ed): Cancer Detection and
VOL.29, NO.4 JULY/AUGUST1979 231

Prevention, Part I. New York, Marcel Dekker, 1978, vol 3, pp 1573-1589.74. Port K: Multiple polypenbildung intractus intestinalis; Deutsche Ztschr Chir42:181, 1896.75. Bo!ey SJ, .McKinnon WMP, MarzulliVF: The management of @familialgastrointestinal polyposis involving stomach andcolon. Surgery 50:691-696, 1961.
76. Yonemoto RH, et a!: Familial polyposisof the ‘¿�entiregastrointestinal tract. ArchSurg 99:427-434, 1969.77. Baughman FA Jr, et a!: The gliomapo!yposis syndrome. N Engl J Med 281:1345-1346, 1969.78. Weary PE, et al: Multiple hamartomasyndrome (Cowden's disease). Arch Dermato! 106:682-690, 1972.
THE LANGUAGE OF. MEDICINEErrors and misconceptions of former times are embalmed in terms like cholera, socalled because the diarrhea and vomiting characteristic of the disease were believedto be a discharge of malignant bilious humor (chole-bi!e), and gonorrhea, whichmeans literally a flow of semen. Hysteria is so named because in ancient times theuterus (hyster) was considered a seat of mental afflictions. There is another allusionto this notion in globus hystericus, which refers to the primitive belief that the “¿�lumpin the throat―of a distraught woman was the uterine fundus.
From: The Language of Medicine: its Evolution, Structure and Dynamics, by John H.Dirckx, M.D., published by Harper and Row, inc., 1976. Page 66.
232 CA-A CANCER JOURNAL FOR CLINICIANS