henry a. boateng - research statementboateng/boateng_henry_research...henry a. boateng - research...
TRANSCRIPT

Henry A. Boateng - Research Statement
1. Introduction
I am interested in research problems at the intersection of applied mathematics, scientificcomputing and modeling of physical phenomena. Presently, I am working on problems inatomistic simulations with applications in materials science and computational chemistry.My research is concerned with modeling physical systems with fidelity to elucidate the prop-erties of the system and algorithm design to perform simulations efficiently. Here I give abrief overview of my work and the later sections will provide a more in-depth presentationof completed, current and future projects.
1.1. Project 1 - Efficient Algorithms for Advanced Potential Energy Surfaces.The paramount goal of this project is to efficiently implement advanced classical electrostaticpotential energy surfaces in DL POLY [27], the U.K.’s flagship massively parallel moleculardynamics (MD) simulation package. In basic MD simulations the time evolution of a systemof chemical species is modeled by solving Newton’s laws of motion. This is an iterative processof first finding the forces on the species as the negative of the gradient of the potential energy,followed by finding the acceleration from the forces, then the velocities from the accelerationand finally the new positions from the velocities.
When the chemical species are charged, the potential energy has terms which depend onthese charges. A typical example is the Coulomb potential energy. Primarily because of thelimitations of computing, the charge clouds for chemical species have been hitherto repre-sented as fixed point charges (monopole approximation) in order to make the simulationstractable at the cost of high accuracy. However, with increasing availability of more powerfulcomputers, its become more feasible to use advanced potential energy surfaces to achievehigher accuracy in reasonable time by representing charges with higher order multipoles.
Yet the use of higher order representations of charges has been limited to up to fourth orderbecause of the cumbersome form of the representation. As part of my current work I haveextended the work of Sagui et. al. [24] to provide an alternative Cartesian formulation ofelectrostatic multipolar interactions that enables the specification of an arbitrary order ofmultipoles and in addition derived a closed-form formula for the stress tensor, due to anarbitrary order multipole formulation of Ewald sums, required in constant pressure MDsimulations. This work [11] has been submitted to the Journal of Chemical Physics andimplemented in DL POLY.
1.2. Project 2 - Kinetic Monte Carlo Simulation of Heteroepitaxy. This projectis an extension of my work with my post-doctoral advisors, Profs. Tim Schulze and PeterSmereka. The long term goal is to develop an efficient method for studying heteroepitaxy onexperimental spatial and temporal scales. This problem is important to materials scientistswho manufacture semiconductors by depositing monolayers of crystals of a film on a substrateof a different crystal via molecular beam epitaxy or chemical vapor deposition.
Heteroepitaxy is the growth of a thin film crystal on a mismatched substrate crystal wherethe two crystals have different lattice separations, or different distances between neighboringatoms in their respective lattices. We call the relative difference in lattice separations of thefilm and substrate the misfit. A common example is the growth of a germanium film ona silicon substrate. The lattice separation in germanium is about four percent larger thanthat of silicon and this leads to growth modes that are different from what is observed when

2 Henry A. Boateng - Research Statement
silicon is grown on silicon. We are interested in developing fast algorithms to capture thechanges in the film and substrate as a result of the misfit, during the growth process. Thesechanges are affected by the rate of diffusion of surface atoms. The misfit controls the growthmodes of the film on the substrate, which in turn affects the optoelectronic properties ofthe system. Together with my post-doctoral advisors, we developed a fast off-lattice kineticMonte Carlo method for simulating heteroepitaxy in (1 + 1)-dimensions. The method retainsthe simplicity and speed of lattice based models but captures the essential features in themore natural off-lattice setting. We have performed simulations and verified that the methodcaptures experimental findings such as the dependence of the growth modes on the misfit,anti-correlation of quantum-dots grown on both sides of a substrate and the effect of therate of diffusion of surface atoms on the growth process. In addition, the method naturallyincorporates intermixing and unlike lattice models, nucleates dislocations. This work hasbeen published in SIAM Journal on Multiscale Modeling and Simulation [10].
1.3. Project 3 - Treecode Algorithms. Project 3 is concerned with speeding up manybody interactions in atomistic simulations and is a continuation of my doctoral work. Themain computational bottleneck in atomistic simulations like molecular dynamics or MonteCarlo comes from computing the energy and forces on atoms as a result of many bodyinteractions. The second project is aimed at easing this bottleneck. One direction of theproject has been in applications where the target and source particles are disjoint. Forsuch applications, we have developed and implemented a treecode algorithm we call cluster-particle treecode that is more efficient than the standard particle-cluster algorithm [4] whenthe target particles outnumber the source particles. The algorithm has potential applicationsin problems involving particle-mesh computations, where the targets are the particles andthe source are mesh points or vice versa. An example is in biophysical simulations wherethe effects of a solvent, perhaps modeled as a mesh, is needed on a solute molecule. Thiswork has been published in the Journal of Computational Chemistry [9] and the code madepublicly available[8] under GNU license. In general, treecode algorithms achieve speedup byseparating interactions into near-field and far-field interactions. The near-field interactionsare computed exactly while the far-field interactions are handled in a coarse-grained mannerby multipole approximations. We are able to reduce the cost of computing from O(N2) toO(N logN). Although the treecode algorithm is presented here in the context of atomisticsimulations, it has been applied in a wide range of fields including astrophysics and fluiddynamics [4, 25].
Section 2 provides some results on the arbitrary order Cartesian multipolar electrostaticinteractions. I will elaborate on the project on simulating heteroepitaxial growth and providesome results in section 3. After that, section 4 will provide basic background and resultsfrom comparing the particle-cluster and cluster-particle treecode algorithms in the case ofdisjoint source and target particles. Both sections will point to possible directions for futurework and potential problems for undergraduate research.
2. Efficient Algorithms for Advanced Potential Energy Surfaces
Following Sagui et. al. [24], we consider a set of N interacting point multipoles and define
the multipolar operator, Li by
(1) Li = (qi + pi · ∇i + Qi : ∇i∇i + Oi :∇i∇i∇i + Hi :: ∇i∇i∇i∇i + . . . ),

3 Henry A. Boateng - Research Statement
where qi, pi, Qi, Oi, and Hi are the charge, dipole, quadrupole, octupole, and hexadecapole,respectively of atom i and the “dot” products stand for tensor contraction. For any functionψ(|ri − rj|) = ψ(rij), ∇jψ(rij) = −∇iψ(rij), thus the multipolar operator for atom j is
(2) Lj = (qj − pj · ∇i + Qi : ∇i∇i −Oi :∇i∇i∇i + Hi :: ∇i∇i∇i∇i + . . . ).
With these definitions we can rewrite the multipolar operators up to order p on atoms i andj respectively in a more compact form as
(3) Li =
p∑s=0
MsiD
si ,
and
(4) Lj =
p∑s=0
(−1)||s||MsjD
si ,
where s = (s1, s2, s3), ||s|| = s = s1 + s2 + s3, Dsi = Ds3
z Ds2y D
s1x , Ms is the multipole
corresponding to the (s1, s2, s3) index and p is an arbitrary multipole order.
The Ewald sum is a method to effectively evaluate the Coulomb energy of charged species,with zero net charge, in a periodic domain by casting the conditionally convergent infiniteCoulumb sum into two absolutely convergent sums, one in real space and the other inreciprocal space in order to be able to control the error in truncating the sums. In constantpressure simulations, the stress tensor, σ, needs to be monitored in order to verify that thesystem is indeed at constant pressure. While the stress tensor from the real space part isfairly straight forward, until now there has been no closed-form formula for the stress tensordue to an arbitrary order reciprocal space sum.
Given the reciprocal space sum as
(5) Urec =1
2V ε0ε
∑k 6=0
exp(−k2/4η2)k2
|S(k)|2 ,
with
(6) S(k) =N∑i=1
Liexp(ık · ri).
where η is a convergence parameter, k is a reciprocal space vector, V ε0ε is a product ofconstants, Nose and Klein [23] derived the formula for the elements of the stress tensor in thecase of point-charges for the reciprocal space Ewald sum. Following this work, stress tensorformulas have been derived for the case where in addition to point charges the particleshave dipoles [28], dipoles and quadrupoles [1], and dipoles, quadrupoles, octupoles andhexadecapoles [24]. One result from our work is the derivation of the stress tensor dueto an arbitrary order reciprocal space Ewald sum. For an arbitrary order p, by defining
J `i (k) =M`(i)D`ieık·ri and Sβi (−k) =
p∑`=0
`β
N∑i=1
J `i (−k), the stress tensor is given as
(7)
V σrecαβ =
1
2V ε0ε
∑k 6=0
exp(−k2/4η2)k2
{|S(k)|2
[δαβ − 2
(k2/4η2 + 1
k2
)kαkβ
]+ 2S(k)Sβi (−k)
kαkβ
}.

4 Henry A. Boateng - Research Statement
(a)
Cells
Unit
(b)
Figure 1. A depiction of two crystals with different lattice distances in (a)and the deformation that arises from deposition of one crystal on another (b).
(a) (b)
Wetting Layer
(c)
Figure 2. Schematic depiction of growth modes. Figure (a) is layer-by-layer(FM) growth, (b) is island-on-layer (SK) growth and (c) is island (VW) growth.
3. Kinetic Monte Carlo Simulation of Heteroepitaxy
3.1. A Broad View of Methods for Simulating Heteroepitaxy. As indicated earlier,the ultimate goal of this project is to develop a KMC algorithm that models heteroepitaxialgrowth on experimental space and time scales. There is active research spanning the spec-trum of scales. The most detailed of these are the quantum mechanical density functionalmethods which provide microscopic detail but access the smallest space and time scales. Onthe other end of the spectrum are continuum methods which model device scale but provideonly macroscopic detail. The KMC method I work on strikes a balance between atomicdetail and computational speed.
3.2. Background.
3.2.1. Growth Modes. The growth modes of a film deposited on a substrate depends on themisfit between the two crystals. Figure 1 (a) depicts two crystals with a misfit and Figure 1(b) shows a deformation in the film when deposited on the substrate. The deformation leadsto an elastic strain. The strength of the elastic strain depends on the magnitude of the misfit.Deformation is most severe when the film atoms cover the substrate. In order to minimizethe strain and hence the elastic energy, the film atoms coalesce to form islands or quantumdots at the expense of increasing the surface area and hence the surface energy. Thus, thegrowth modes are a result of competition between elastic energy and surface energy. Figure 2explains the three growth modes in heteroepitaxy.

5 Henry A. Boateng - Research Statement
When the misfit is low, elastic effects are negligible and the surface forces dominate. Theenergetically favorable state is to reduce the surface area and thus the surface energy, as suchthe film grows layer by layer on the substrate. This type of growth mode is called Frankvan-der-Merwe (FM) or layer-by-layer growth and is depicted in Figure 2 (a).
Figure 2 (b) shows the growth mode for moderate misfit. Elastic effects are significantbut surface forces dominate initially. As a result the film grows layer-by-layer to a criticalheight at which the elastic strain dominates, then the film forms islands. This is calledStranski-Krastanov (SK) or island-on-layer growth.
In a high misfit regime, the elastic strain is overwhelming and dominates from the onset ofdeposition, thus the film forms islands immediately in order to minimize elastic energy. Thisgrowth mode, depicted in Figure 2 (c) is called Volmer-Weber or island growth.
3.2.2. Off Lattice KMC. A configuration of atoms at a local minimum oscillates randomlyin configuration space about the minimum. After a very long time, the configuration mighttransition from its present minimum to another minimum. The off-lattice KMC model forsimulating crystal growth is aimed at capturing the infrequent jumps of a dynamic atomicconfiguration in equilibrium from one basin of attraction, i.e. a local potential energy min-imum, to another. The observation that a configuration spends a long time oscillating in alocal basin implies that memory of how the configuration got to the basin in the first place islost. As such, the transitions from one minimum to another is modeled as a Markov Chainwhere each possible transition path from a minima i to a neighboring minima j has a rateRij given by transition state theory [29].
Our method seeks to have the same state space as other fully off-lattice KMC methods, butcomputes the rates in the spirit of lattice-based, bond-counting KMC methods. The modelis approximate but captures a lot of the physics of the more involved off-lattice schemeswhile providing speed on the order of those provided by lattice-based schemes.
3.2.3. Results. We have applied our approximate off-lattice KMC to a two-dimensionalmodel system where film atoms are deposited on a substrate and the interactions betweenatoms is governed by a Lennard Jones [2] interatomic potential. Figure 3 shows some experi-mental results and corresponding results from our simulations. Figure 3(a) is an experimentalresult from[21] showing Stranski-Krastanov growth. The substrate is Silicon and the filmis Germanium. Figure 3(b) is experimental results from[19] showing anti-correlation of Gequantum-dots grown on a Silicon nanowire. Figures 3(c) and (d) are results from our sim-ulations capturing Stranski-Krastanov growth and anti-correlation of film atoms grown onboth sides of a substrate. This work has been published in the SIAM Journal on MultiscaleModeling and Simulation[10].
3.3. Current and Future Work.
3.3.1. There are several interesting directions to explore from this project, most of whichwill be suitable for students. An immediate and ongoing direction is an extension of themodel to three dimensions. Here we use the same Lennard Jones potential as before andthe main challenge is devising and implementing algorithms that ameliorate the increasedcomputational demands due to the increase in the dimensionality of the problem.

6 Henry A. Boateng - Research Statement
NATURE PHOTONICS | VOL 4 | AUGUST 2010 | www.nature.com/naturephotonics 527
FOCUS | REVIEW ARTICLESPUBLISHED ONLINE: 30 JULY 2010!|!DOI: 10.1038/NPHOTON.2010.157
For many years, high-quality Germanium (Ge) crystals have been used as the primary material for highly sensitive near-infrared detectors. Such detectors are usually cooled to around 77 K to
reduce dark currents, making them expensive and of limited use, par-ticularly for spectroscopy. However, within the past ten years, the use of Ge in detector applications has changed dramatically. ! is develop-ment was triggered by the ability to grow Ge epitaxially on silicon (Si). Rather than using single-crystal bulk Ge, the use of Si as a substrate not only reduced device cost but also enabled completely new appli-cations for Ge in optical communications, which until then had typi-cally been covered by compound semiconductors such as InGaAs.
! e epitaxial growth of Ge on Si is di" cult because of the 4.2% lattice mismatch between the two elements. In the 1990s, the # rst attempts were made to epitaxially grow Ge on Si at reason-ably low dislocation densities, primarily motivated by the higher mobilities for electrons and holes in Ge than in Si. ! is Review # rst describes the techniques that eventually yielded high-quality Ge-on-Si devices. Initially this progress was used to develop free-space Ge detectors with responsivities and speeds compa-rable to group $$$–% semiconductor detectors. ! e development of the free-space Ge detector, and the following evolution to the waveguide-integrated Ge detector, is discussed. ! is evolution not only enabled electronic–photonic integration on silicon but also achieved higher performance by eliminating the trade-o& between bandwidth and quantum e" ciency. Di& erent coupling schemes and their corresponding performances are also reviewed. Finally, the recent progress of Ge-based avalanche photodetectors is discussed, as these important devices are currently competing with group $$$–% avalanche photodetectors for a market share in optical communications.
Choice of materials Growth of high-quality Ge epitaxial ! lms on Si. ! e great-est challenge for high-quality Ge epitaxy on Si is the 4.2% lat-tice mismatch between the two materials. ! is mismatch causes two serious issues: high surface roughness resulting from the Stransky–Krastanov growth, and a high density of threading dis-locations in the Ge epitaxial layer. High surface roughness hinders the process of integrating Ge devices with Si electronics because complementary metal–oxide–semiconductor (CMOS) devices require planar processing, whereas a high density of thread-ing dislocations severely a& ects the performance of Ge devices because of the recombination centres that are introduced along these dislocations.
High-performance Ge-on-Si photodetectorsJurgen Michel*, Jifeng Liu and Lionel C. Kimerling
The past decade has seen rapid progress in research into high-performance Ge-on-Si photodetectors. Owing to their excel-lent optoelectronic properties, which include high responsivity from visible to near-infrared wavelengths, high bandwidths and compatibility with silicon complementary metal–oxide–semiconductor circuits, these devices can be monolithically integrated with silicon-based read-out circuits for applications such as high-performance photonic data links and infrared imaging at low cost and low power consumption. This Review summarizes the major developments in Ge-on-Si photodetectors, including epi-taxial growth and strain engineering, free-space and waveguide-integrated devices, as well as recent progress in Ge-on-Si ava-lanche photodetectors.
! e # rst successful approach for growing high-quality epi-taxial Ge layers on Si was reported in a forward-looking paper by Luryi et al. in 19841. In this study, a graded SiGe bu& er layer, grown in a molecular beam epitaxy chamber, was used to reduce the threading dislocation density in the Ge layer. ! is method was later improved by Fitzgerald et al. using optimized SiGe graded bu& er layers2–5. A low grading rate of ~10% Ge per micrometre was adopted to minimize dislocation nucleation rates. Choosing ade-quate growth temperatures for di& erent SiGe compositions gave a high dislocation glide velocity but slow dislocation nucleation kinet-ics, allowing the # lm to relax mainly by gliding existing threading dislocations instead of generating additional dislocations. At 50% Ge composition, an ex situ chemical mechanical polishing step was used to remove crosshatch surface roughness and greatly reduce the dislocation pile-up formation that hinders dislocation gliding. For SiGe growth with Ge content of more than 50%, the growth
MIT Microphotonics Center, Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. *e-mail: [email protected]
Ge
Ge
Si
Si
a
b
Figure 1 | Transmission electron microscope cross-sections of Ge-on-Si layers for Ge epitaxial growth at di" erent temperatures. a, Stransky–Krastanov growth of Ge-on-Si at 550 °C in an ultrahigh vacuum CVD reactor. b, Ge-on-Si grown at 335 °C in an ultrahigh vacuum CVD reactor. The thickness of the Ge layer shown here is 60 nm.
nphoton_.2010.157_AUG10.indd 527nphoton_.2010.157_AUG10.indd 527 10.7.16 3:02:00 PM10.7.16 3:02:00 PM
© 20 Macmillan Publishers Limited. All rights reserved10
(a)
circumference in an alternating fashion corresponding to the n= 1 mode in Figure 1b. The average wavelengths of Ge QDarrays (as de�ned in Figure 1) are L = 56.6 and 53.3 nm,respectively. These results are strongly indicative of thepreviously proposed anticorrelated organization of Ge dotson nanoscale Si NW substrates due to the periodic correlationof strain �eld in the nanowires.In thecaseof nanowiresgrown with smaller, 10 nmdiameter
catalysts, however, the morphology of the Ge deposition isdistinctively di�erent. Figure2eshowstwo thin nanowireswith13 and 11 nm diameter backboneon which instead of discreteGeQDscovering both sides, wider Ge islands are formed andthey are found on a single side of Si surface only. In addition,the nanowires exhibit a bending curvature with the Ge on theconvex surfaces (Figure2e). Such bending can beattributed tothehigh /exibility of these thin nanowirecores.36 At such smalldiameter, the core substrate itself is close to or even less inthickness than the thickness of the Ge shell deposition, hencemost of the strain energy is accommodated by bending stresswithin the core.At theother end of thespectrum, aswefocusonmuch larger
diameter Si NW cores (Figure 2d), the arrangement, anddensity of Gedotsresemble island growth on planar substrates,covering all exposed surfaces around the circumference of thecoreandwithout clear angular or axial correlation among islandlocation distributions. These represent higher order growthmodes (n 1 2) in Figure 1d because with large core diameterthe correlating e�ect of strain �eld across the core NWdiameter is no longer signi�cant enough to produce thecorrelated pattern of Ge dots.To gain quantitative insight into theself-organization of QDs
on theSi nanowiresubstrates,wecollected andanalyzed imagesfrom a variety of diameter-dependent morphologies of QDarrays. For this purpose, two key growth parameters, corediameter and Ge deposition time, were extensively varied andTEM analysis was used to perform a statistical study on thewavelength of QD arrays.Table I summarizes the core diameter dependence of Ge
shell growth morphologies. Three groups of nanowires wereincluded based on initial growth of Si NW with catalyst Aucolloids of 10, 20, and 40 nm. For samples with the smallestdiameter (D < 15 nm), single-side Ge growth mode with Gedeposition on only one side of the Si NW is found on amajority (90%) of the nanowires. Among these single-sidegrowth samples most (80%) exhibit bending curvature similarto that in Figure2e.Only onesmall diameter samplewasfound
with GeQDson opposite sideof thecircumference in either n= 0or n= 1geometry. For medium rangediameter samples(15= D = 30 nm), however, we �nd a predominant distribution(84%) of n= 1modegrowth, whilenoneshowsn= 0behavior.Lastly, the largest diameter nanowires in this study (D > 30nm) uniformly exhibit higher indexmode (n 1 2) growth withthe core surrounded by Ge dots on all sides. The fact thatsurface morphologies or growth modes of the Ge QDs aresensitively de�ned by the core diameter is strong indication ofthe role of growth substrate diameter in directing thecorrelation of mis�t-strain across the circumference and isdirectly supportive of previous theoretical calculations.21,25,31The diameter dependence can be qualitatively understood byconsidering that the ability to form regular arraysof correlatedQDs on the nanowire surface closely relies on the strain �eldcorrelation across the nanowire backbone (Figure 1 bottompanel), therefore the larger the core diameter, the weaker thee�ect of strain on one side of the nanowire will propagate toa�ect theopposite side. At 84, 90, and 100%for each diametergroups, such large, near-uniform distribution of certain growthmodes suggests a novel route toward self-organization of QDnanostructures driven by minimization of strain energy innanoscale heterostructures.Among each of the di�erent growth modes, we also
investigated the e�ect of core diameter on the wavelength ofGe QDs. In particular, Figure 3a shows in the case ofanticorrelated (n = 1) nanowires the wavelength L is nearlyinsensitive to core diameter at 20?50 nm but increases rapidlywith smaller D. Theseobservationsarealso in good agreementwith theoretical calculation (solid line, Figure 3a) of the shellinstability using �rst order perturbation25 (see SupportingInformation). Therefore with careful control of the nanowirecore diameter, one can rationally tune theQD wavelength asanew route toward nanomaterial design.Besidessimplen= 1mode, wecollected alargeset of dataof
Ge QD wavelength versus core diameter from all observedgrowth modes (n = 0 and above) that also include variousGegrowth time from 3.5 to 20 min. Here the Si core nanowireswere grown with 10?40 nm gold catalyst. The resulting LversusD trend is plotted in Figure 3b and reveals interestinglyan inverse relationship between L and D. The trend isconsistent with that from previous theoretical calcula-tions21,25,31 and strongly reinforces the picture of thecontrollability of self-organized QDs by strain engineeringwithin speci�c core diameters.
Figure 4. Evolution of GeQDsasa function of growth time. (a?c) TEM images showing anticorrelated array of GeQDsat di�erent Ge growthtime; 3.5, 5, and 10min from left to right. (d?e) Summary of width, height, and wavelength of GeQDsasa function of growth time. Inset of (e)TEM imageof 20 min growth of GeQDs. Error bars indicate the standard deviation frommean size of QDson each nanowire. Sale bars, 20 nm.
Nano Letters Letter
dx.doi.org/10.1021/nl302190e | Nano Lett. 2012, 12, 4757?47624760
(b)
9
9.5
(c)
(d)
Figure 3. Figure (a) is an experimental result from Michel et al. [21]showing SK growth. Figure (b) is experimental results from Kwon et al. [19]showing anti-correlated growth of Ge on Si nanowire. Figures (c) and (d) aresimulation results from our approximate KMC method showing SK growthand anti-correlated growth respectively.
3.3.2. After understanding the 3D problem using a Lennard Jones pair potential, the nextstep will be to implement the method with a more realistic potential like Stillinger-Weber[26], which incorporates three-body terms or a semi-empirical embedded-atom potential [13].The complicated form of these potentials will most likely lead to interesting computationalproblems on ways to achieve a fast implementation to reach device scale.
3.3.3. Another direction will be to study the process of island-on-layer (SK) growth in twodimensions as has been previously done with other KMC models [5, 22].
3.3.4. The long term goal to reach device scale will most likely require a multiscale modelwhere the 3-D approximate KMC method is coupled to a continuum model. The field ofatomistic and continuum coupling is an active area of research [30]. I intend to push theproject in this direction in the next 5-10 years.
3.3.5. Research For Undergraduates. One possible problem out of this project will be forstudents to simulate and compute physical quantities of systems by both MD and MonteCarlo methods to compare the two methods on accuracy and speed. This will be a goodopportunity for students to learn about the strengths and drawbacks of both methods, togain deeper knowledge of physical systems and to strengthen programming skills.
4. Treecode Algorithms for N-body Interactions
4.1. N-body Interactions. N−body interactions are of the form
(8) s(xi) =N∑j=1
λjφ(xi,yj),
where xi is a target site or particle, yj is a source particle, λj is a weight and N is thenumber of sources. Methods like the particle-particle-particle-mesh (P3M) method [18], thefast multipole method (FMM) [17] and the treecode method (TM) [4], which is the focus ofour work, are used to reduce the cost of N -body interactions from O(N2) to O(N logN).

7 Henry A. Boateng - Research Statement
All these methods divide the interactions into near-field interactions and far-field interac-tions. The near-field interactions are computed directly and the far-field interactions arecomputed approximately. Treecode methods as well as FMM are grid free methods whichhierarchically restructure the interacting system into a tree of clusters of particles and usemultipole expansions to approximate the effect of far away clusters on target particles to adesired accuracy. One difference between FMM and TM is that the 3D FMM uses sphericalharmonics expansions while TM uses a Cartesian Taylor series expansion.
Treecode methods have broad implementations in fields where summations of the form inEquation 8 are present. Some of these implementations are in astrophysics [25] to com-pute gravitational interactions, atomistic simulations in condensed matter physics [7, 15] tocompute pair potentials, computational fluid dynamics [20] to compute the Biot-Savart inte-gral for vortex sheet motion in 3D, and for computing radial basis functions [14, 31], whichare used in scattered data interpolation and in solving differential equations. Our specificimplementation is in the context of atomic simulations.
4.2. Our Contributions. Although in most N -body interactions the target (T ) and source(S) particles are the same, there are instances where these are disjoint. Two examples are(i) interpolation of data from mesh points to a set of particles or vise versa and (ii) boundaryintegral simulations of solvated biomolecules with the solvent molecules being either targetsor sources and the biomolecule as the other. Our main contribution is the observation thatin applications where T and S are disjoint, it is important to adapt the algorithm to attainbetter performance. The potential on a target particle i, where T = {xi, i = 1 : M}, due toa set of source particles S = {yj, j = 1 : N} with associated weights qj is
(9) V (xi) =N∑j=1
qjφ(xi,yj).
The cost of computing Equation 9 directly for all target particles is O(MN).
The standard treecode method which we call particle-cluster treecode (PC) [4] approximatesthe potential V C , on a target particle xi, due to a far-field cluster C of the source as a Taylorseries expansion around the center yc, of cluster C
(10) V C(xi) =∑yj∈C
qjφ(xi,yj) ≈∑yj∈C
qj
p∑||k||=0
1
k!∂kyφ(xi,yc)(yj − yc)
k,
where ||k|| = k1 + k2 + k3, k! = k1!k2!k3!, ∂ky = ∂yk11 ∂y
k22 ∂y
k33 , (yj −yc)
k = (yj1 − yc1)k1(yj2 −yc2)
k2(yj3 − yc3)k3 and p is the order of the Taylor approximation. The formulation of PCenforces a restriction that only the source particles can be restructured into a tree. This isoptimal if N > M . However, when M > N , the optimal choice is to instead restructure thetarget particles and to expand the Taylor series around the center of clusters xc, of the targetparticles to create a new algorithm we call cluster-particle treecode (CP). This results in adifferent approximation which we derived and has also been derived independently in anotherapplication [14]. In CP the potential V C , on a target particle xi, in cluster C due to all the

8 Henry A. Boateng - Research Statement
source particles yj in the far-field of the cluster C which we will call Cf is approximated as
(11) V C(xi) =∑
yj∈Cf
qjφ(xi,yj) ≈∑
yj∈Cf
qj
p∑||k||=0
1
k!∂kyφ(xc,yj)(xi − xc)
k.
4.2.1. Results. We compared (PC) and (CP) for the Coulomb potential [9],
(12) φ(x,y) =1
4πε0|x− y|,
in terms of the root mean square error, E2, in the potential
(13) E2 =
√∑Mi=1 |V (xi)− V (xi)|2∑M
i=1 |V (xi)|2,
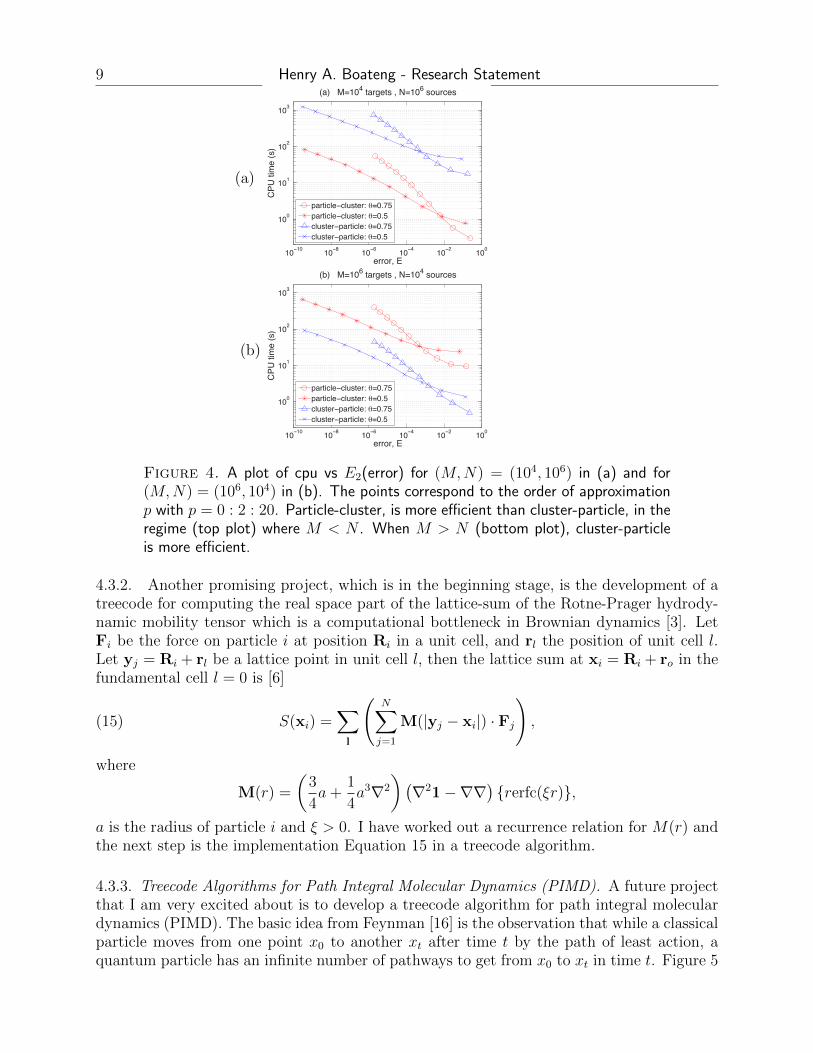
cpu time and memory requirement, where V (xi) is the exact potential and V (xi) is theapproximation. Figure 4 shows a plot of cpu time versus error for different M and Nand different multipole acceptance criterion θ, for determining the far-field. The multipoleacceptance criterion is r/R < θ, where r is the radius of a cluster and R is the distance fromthe center of the cluster to the target particle. The smaller θ is, the bigger the near-fieldand more direct summations are performed. We see that for (M,N) = (104, 106) shown inFigure 4 (a) PC is more efficient and for (M,N) = (106, 104) shown in Figure 4 (b) CP ismore efficient. We have tested the algorithms and seen the same behavior for other valuesof M and N . The two algorithms have similar memory requirements. This work has beenpublished in the Journal of Computational Chemistry.
4.3. Current and Future Work.
4.3.1. I am currently developing a cluster-cluster (CC) algorithm with the goal of makingit more efficient than both the particle-cluster (PC) and cluster-particle (CP) algorithms.While the PC and CP algorithms compute Taylor coefficients for each particle interactingwith a cluster, the CC algorithm computes fewer Taylor coefficients because interactions arebetween clusters. In this formulation, the Taylor approximation of the interaction betweencluster A and cluster B is centered at x = xc−yc, where xc and yc are the centers of clusterA and B respectively. Then, the potential on a particle at xi in cluster A due to cluster Bis given by
(14) V (xi) =∑yj∈B
qjφ(xi,yj) ≈p∑
||k||=0
bk(xc,yc)(xi − xc)k,
with
bk(xc,yc) =
p∑n=k
(n
k
)an(x)Mn−k
B and Mn−kB =
∑yj∈B
qj(yc − yj)n−k.
9 Henry A. Boateng - Research Statement
!"!"
!"#
!"$
!"%
!"&
!""
!""
!"!
!"&
!"'
())*)+,-./0,123(,456
476,,,89!"%,17):(15,+,;9!"
$,5*<)=(5
,
,
>7)12=?( =?<51()@, 9"ABC
>7)12=?( =?<51()@, 9"AC
=?<51() >7)12=?(@, 9"ABC
=?<51() >7)12=?(@, 9"AC
!"!"
!"#
!"$
!"%
!"&
!""
!""
!"!
!"&
!"'
())*)+,-
./0,123(,456
476,,,89!"$,1:);(15,+,<9!"
%,5*=)>(5
,
,
?:)12>@( >@=51()A, 9"BCD
?:)12>@( >@=51()A, 9"BD
>@=51() ?:)12>@(A, 9"BCD
>@=51() ?:)12>@(A, 9"BD
(a)
(b)
Figure 4. A plot of cpu vs E2(error) for (M,N) = (104, 106) in (a) and for(M,N) = (106, 104) in (b). The points correspond to the order of approximationp with p = 0 : 2 : 20. Particle-cluster, is more efficient than cluster-particle, in theregime (top plot) where M < N . When M > N (bottom plot), cluster-particleis more efficient.
4.3.2. Another promising project, which is in the beginning stage, is the development of atreecode for computing the real space part of the lattice-sum of the Rotne-Prager hydrody-namic mobility tensor which is a computational bottleneck in Brownian dynamics [3]. LetFi be the force on particle i at position Ri in a unit cell, and rl the position of unit cell l.Let yj = Ri + rl be a lattice point in unit cell l, then the lattice sum at xi = Ri + ro in thefundamental cell l = 0 is [6]
(15) S(xi) =∑l
(N∑j=1
M(|yj − xi|) · Fj
),
where
M(r) =
(3
4a+
1
4a3∇2
)(∇21−∇∇
){rerfc(ξr)},
a is the radius of particle i and ξ > 0. I have worked out a recurrence relation for M(r) andthe next step is the implementation Equation 15 in a treecode algorithm.
4.3.3. Treecode Algorithms for Path Integral Molecular Dynamics (PIMD). A future projectthat I am very excited about is to develop a treecode algorithm for path integral moleculardynamics (PIMD). The basic idea from Feynman [16] is the observation that while a classicalparticle moves from one point x0 to another xt after time t by the path of least action, aquantum particle has an infinite number of pathways to get from x0 to xt in time t. Figure 5

10 Henry A. Boateng - Research Statement
depicts three possible paths for the quantum particle. A time slice at ti intersects all thepossible paths.
1
2
3
t
3
2
1
x0
xt
ti
Figure 5. Three possible paths to go from x0 to xt. The inset is a ring polymerisomorphic to the quantum particle at time ti.
In this view, the quantum mechanical partition function is isomorphic to the classical parti-tion function of a fictitious higher dimensional classical system where each quantum particleis replaced by a closed chain of an infinite number of classical particles connected by springs[12]. Each classical particle corresponds to a point on a unique path at ti. In applicationsa finite number of springs are used. The inset in Figure 5 depicts the ring polymer of threeparticles that correspond to an approximation of one quantum particle. The cost to modela quantum particle with N particles where each particle is represented by a ring polymer ofP classical particles is equivalent to the cost to model a classical system with NP particles.The computational cost is immense, but I believe there is a possibility for speedup with atreecode algorithm.
Because of the wide applicability of treecode methods, I am hopeful of developing collabo-rations in different fields where they can be implemented.
4.3.4. Research For Undergraduates. A good problem for undergraduates will be to investi-gate potential applications of the treecode across several disciplines and to implement thealgorithm in these contexts. I envisage this problem appealing to both mathematics andnon-mathematics majors.
References
[1] A. Aguado and P. A. Madden, Ewald summation of electrostatic multipole interactions up to thequadrupolar level, J. Chem. Phys., 119 (2003), pp. 7471–7483.
[2] M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Oxford University Press, NewYork, 1987.
[3] T. Ando, E. Chow, Y. Saad, and J. Skolnick, Krylov subspace methods for computing hydrody-namic interactions in Brownian dynamics simulations, J. Chem. Phys., 137 (2012).
[4] J. Barnes and P. Hut, A hierarchical O(NlogN) force calculation algorithm, Nature, 324 (1986),pp. 446–449.
[5] A. Baskaran and P. Smereka, Mechanisms of Stranski-Krastanov growth, J. Appl. Phys., 111 (2012),p. 044321.
[6] C. W. J. Beenaker, Ewald sum of the Rotne-Prager tensor, J. Chem. Phys., 85 (1986), p. 1581.[7] H. A. Boateng, Cartesian Treecode Algorithms for Electrostatic Intercations in Molecular Dynamics
Simulations, PhD thesis, University of Michigan, Ann Arbor, MI, 2010.[8] H. A. Boateng, Comparison of cartesian treecodes: sourceforge.net/projects/compare-pc-cp, May 2013.

11 Henry A. Boateng - Research Statement
[9] H. A. Boateng and R. Krasny, A comparison of particle-cluster and cluster-particle treecodesfor Coulomb systems with different sets of source and target particles, J. Comput. Chem., 34 (2013),pp. 2159–2167.
[10] H. A. Boateng, T. P. Schulze, and P. Smereka, Approximate off-lattice kinetic monte carlo,Multiscale Model. Simu., 12 (2014), pp. 181–199.
[11] H. A. Boateng and I. T. Todorov, Arbitrary order permanent Cartesian multipolar electrostaticinteractions, J. Chem. Phys., (Submitted).
[12] D. Chandler and P. G. Wolynes, Exploiting the isomorphism between quantum theory and classicalstatistical mechanics of polyatomic fluids, J. Chem. Phys., 74 (1981), pp. 4078–95.
[13] M. S. Daw, S. M. Foiles, and M. I. Baskes, The embedded-atom method: a review of theory andapplications, Mat. Sci. Rep, 9 (1993), pp. 251–310.
[14] Q. Deng and T. A. Driscoll, A fast treecode for multiquadric interpolation with varying shapeparameters, SIAM J. Sci. Comput., 34 (2012), pp. A1126–A1140.
[15] Z. H. Duan and R. Krasny, An ewald summation based multipole method, J. Chem. Phys., 113(2000), pp. 3492–3495.
[16] R. Feynman and A. Hibbs, Quantum Mechanics and Path Integrals, McGraw-Hill Book Company,1965.
[17] L. Greengard and V. Rokhlin, A fast algorithm for particle simulation, J. Comput. Phys., 73(1987), p. 325.
[18] R. W. Hockney and J. W. Eastwood, Computer Simulation Using Particles, McGraw-Hill, NewYork, 1981.
[19] S. Kwon, Z. C. Y. Chen, J.-H. Kim, and J. Xiang, Misfit-guided self-organization of anticorrelatedGe quantum dot arrays on Si nanowires, Nano Lett, 12 (2012), pp. 4747–4762.
[20] K. Lindsay and R. Krasny, A particle method and adaptive treecode for vortex sheet motion inthree-dimensional flow, J. Comput. Phys., 172 (2001), pp. 879–907.
[21] J. Michel, J. Liu, and C. Kimerling, High-performance Ge-on-Si photodetectors, Nature Photonics,4 (2010), pp. 527–534.
[22] F. Much and M. Biehl, Simulation of wetting-layer and island formation in heteroepitaxial growth,Europhysics Lett., 63 (2003), pp. 14–20.
[23] S. Nose and M. Klein, Constant pressure molecular dynamics for molecular systems, Mol. Phys., 50(1983), pp. 1055–1076.
[24] C. Sagui, L. Pedersen, and T. Darden, Towards an accurate representation of electrostatics inclassical for fields: Efficient implementation of multipolar interactions in biomolecular simulations, J.Chem. Phys., 120 (2004), pp. 73–87.
[25] J. K. Salmon, M. S. Warren, and G. S. Winckelmans, Fast parallel tree codes for gravitationaland fluid dynamical n-body problems, Intl. J. of Sup. App., 8 (1994), pp. 129–142.
[26] F. Stillinger and T. A. Weber, Computer simulation of local order in condensed phases of Silicon,Phys. Rev. B, 31 (1985), pp. 5262–5271.
[27] I. T. Todorov, W. Smith, K. Trachenko, and M. T. Dove, DL POLY 3: New dimensions inmolecular dynamics simulations via massive parallelism, J. Mater. Chem., 16 (2006), pp. 1611–1618.
[28] A. Toukmaji, C. Sagui, J. Board, and T. Darden, Efficient particle-mesh Ewald based approachto fixed and induced dipolar interactions, J. Chem. Phys., 113 (2000), pp. 10913–10927.
[29] A. F. Voter, Introduction to the kinetic Monte Carlo method, in Radiation Effects in Solids, K. Sickafusand E. A. Kotomin, eds., Springer, NATO Publishing Unit, Dordrecht, The Netherlands, 2005.
[30] G. J. Wagner and W. K. Liu, Coupling of atomistic and continuum simulations using a bridgingscale decomposition, J. Comput. Phys., 190 (2003), pp. 249–274.
[31] L. Wang and R. Krasny, Fast evaluation of multiquadric RBF sums by a Cartesian treecode, SIAMJ. Sci. Comput., 33 (2011), pp. 2341–2355.