harnessing stem cells as drug delivery …vc043cd5324/deveza... · harnessing stem cells as drug...
TRANSCRIPT

HARNESSING STEM CELLS AS DRUG DELIVERY VEHICLES FOR
THERAPEUTIC ANGIOGENESIS:
A BIOMATERIALS-MEDIATED APPROACH
BY
Lorenzo R. Deveza
A DISSERTATION SUBMITTED TO THE DEPARTMENT OF BIOENGINEERING AND THE
COMMITTEE ON GRADUATE STUDIES OF STANFORD UNIVERSITY
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY IN BIOENGINEERING
AT
STANFORD UNIVERSITY
August 2014

http://creativecommons.org/licenses/by-nc/3.0/us/
This dissertation is online at: http://purl.stanford.edu/vc043cd5324
© 2014 by Lorenzo De Rama Deveza. All Rights Reserved.
Re-distributed by Stanford University under license with the author.
This work is licensed under a Creative Commons Attribution-Noncommercial 3.0 United States License.
ii

I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Fan Yang, Primary Adviser
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Jennifer Cochran
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Ngan Huang
Approved for the Stanford University Committee on Graduate Studies.
Patricia J. Gumport, Vice Provost for Graduate Education
This signature page was generated electronically upon submission of this dissertation in electronic format. An original signed hard copy of the signature page is on file inUniversity Archives.
iii

Harnessing Stem Cells as Drug Delivery Vehicles for Therapeutic Angiogenesis:
A Biomaterials-Mediated Approach
By
Lorenzo R. Deveza
Abstract
Cardiovascular disease (CVD) represents a global medical and economic problem with
high morbidity and mortality rates. CVD is often associated with partial occlusion of the blood
vessels and tissue ischemia, and restoring blood supply to ischemic tissues is critical to prevent
irreversible tissue damage. Therapeutic angiogenesis aims to stimulate the growth of new blood
vessels from pre-existing vessels, which offers a valuable tool for treating CVD. Several
strategies have been developed to promote angiogenesis, including growth factor delivery and
gene therapy. Direct delivery of angiogenic growth factors has the potential to stimulate new
blood vessel growth. However, it is limited by short half-lives in vivo, and uncontrolled
diffusion of angiogenic factors may also cause undesirable side effects. Gene therapy offers an
alternative approach by delivering genes encoding angiogenic factors, but previous approaches
often require the use of viral vectors for efficient gene delivery, and are limited by safety
concerns such as immunogenicity.
The goal of this thesis research is to develop novel strategies for stimulating therapeutic
angiogenesis by harnessing stem cells as drug delivery vehicles and to validate the efficacy of
non-viral engineered stem cells in vivo using mouse models of hindlimb ischemia. Our strategy
takes advantage of the natural homing capacity of stem cells towards ischemic tissues in vivo,
and their ability to secrete paracrine signals to stimulate blood vessel growth. Specifically we
developed two strategies including: (1) isolating and transfecting stem cells ex vivo using
biodegradable polymeric nanoparticles to overexpress therapeutic genes, followed by
transplanting non-viral engineered stem cells back to ischemic tissues; and (2) recruiting and
programming endogenous stem cells in situ using biomaterials-mediated delivery of biologics.
In the first strategy, we have chosen adipose-derived stem cells (ADSCs), an abundantly
available autologous cell source that can be easily obtained in a minimally invasive manner. To
enhance the paracrine signaling of ADSCs for therapeutic angiogenesis, we transfected ADSCs
using in-house developed biodegradable polymeric nanoparticles, which eliminate the
dependence on viruses for efficient gene delivery. Using the optimized polymeric vectors, we
examined the efficacy of ADSCs overexpressing various angiogenic factors or homing factors
iv

on therapeutic angiogenesis in vitro and in vivo. Transplantation of non-viral engineered ADSCs
led to significantly enhanced tissue salvage in a murine model of hindlimb ischemia with faster
restoration of blood reperfusion and muscle regeneration. Our results suggest that stem cells
programmed with biodegradable polymeric nanoparticles can serve as delivery vehicles to
express therapeutic factors in situ to promote therapeutic angiogenesis.
In the second strategy, we seek to circumvent the need of isolating and manipulating
stem cells ex vivo by directly recruiting and transfecting endogenous progenitor cells in situ at
the site of ischemia. To achieve this, we developed a biomaterials-mediated delivery platform
for sequential release of stem cell homing factors and DNA encoding therapeutic genes. Our
results show that biomaterials-mediated release of homing factors followed by delayed DNA
delivery enhanced recruitment of endogenous progenitor cells and improved limb salvage in a
mouse model of hindlimb ischemia. Finally, we demonstrate the potential of using microfluidic-
synthesized microspheres to aid therapeutic angiogenesis using a biomaterials-mediated drug
delivery depot.
Thesis Advisor: Fan Yang, PhD
Title: Asst. Professor in Bioengineering and Orthopaedic Surgery
v

vi
Acknowledgments
I wish to dedicate this thesis to my wife and son, Shenna and Eli. We are in this together and
will continue to grow as we traverse life’s terrain. I am grateful to Fan Yang for her mentorship
and support in helping me to develop as a scientist and a professional; guiding me from Medical
Scholars research to Ph.D. research. I am thankful for all those that have worked with me on
this thesis, especially Jeffrey Choi who deserves wholeheartedly to be recognized for his
contributions. Also, I wish to acknowledge Jothikritka Ashoken and Gloria Castaneda, who
spent the last year working with us on the angiogenesis team! Of course, I also wish to
acknowledge all the members of the Fan Yang lab for all your help and great times.

Table of Contents
1. Introduction……………………………………………………………………………………..1
2. Background…………………………………………………………………………..................4
3. Overexpressing Angiogenic Growth Factors in Adipose-Derived Stem Cells using
Biodegradable Polymeric Nanoparticles………………………………………………………31
4. Paracrine Release from Non-viral Engineered Adipose-Derived Stem Cells Promotes
Endothelial Cell Survival and Migration in vitro……………………………………................49
5. CXCR4-overexpressing Stem Cells Enhance Tissue Regeneration in a Mouse Model of
Hindlimb Ischemia………………………………………….…………………………………70
6. Adipose-Derived Stem Cells Co-expressing Hepatocyte Growth Factor and CXCR4 Promotes
Ischemic Tissue Repair………………………………………………………………………..93
7. Recruiting and Programming Endogenous Progenitor Cells in situ for Therapeutic
Angiogenesis…………………………………………………………………………………115
8. Microfluidic Synthesis of Biodegradable Polyethylene-Glycol Microspheres for Controlled
Delivery of Growth Factors and DNA Nanoparticles…..…………………………………….135
9. Summary, Limitations and Future Work……………………………………………………..154
vii

List of Figures
Figure 2.1 Methods for incorporating biological signals into scaffolds for controlled release..24
Figure 2.2 Polymeric scaffolds for dual delivery of VEGF and PDGF……………...………....25
Figure 2.3 Effects of VEGF temporal presentation on angiogenesis…………………………..26
Figure 2.4 Environmental-responsive controlled release system………………………………27
Figure 2.5 Cell-triggered VEGF delivery using biomimetic hydrogels………………………..28
Figure 2.6 Combined stem cell and gene therapy approach for therapeutic angiogenesis……...29
Figure 2.7 Exploiting cell homing for treating acute myocardial infarction…………………...30
Figure 3.1. Synthesis of end-modified poly(β-amino esters) (PBAE)…………………………42
Figure 3.2. Effects of polymer on transfection and viability of human adipose-derived stem cells,
mouse adipose-derived stem cells, and human embryonic kidney cells….……………………43
Figure 3.3. Enhancement of hADSC proliferation and transfection in the presence of basic
fibroblast growth factor…………………………………………………………..……………44
Figure 3.4. Effects of culture conditions post-transfection…………………………………….45
Figure 3.5. Transfection of cells with different plasmids encoding for multiple genes………...46
Figure 3.6. Effects of transfected cell paracrine release on human umbilical vein endothelial
cell (HUVEC) and human aortic smooth muscle cell (HASMC) viability……………………..47
Figure 4.1. Efficiency of gene delivery to human adipose-derived stem cells (hADSC) utilizing
poly(β-amino esters) (PBAE) nanoparticles…………………………………………………...63
Figure 4.2 Effects of nanoparticle dose on transfection efficiency and cell viability…………..64
Figure 4.3. VEGF release from human adipose-derived stem cells (hADSCs) after transfection
using polymeric nanoparticles…………………………………………….…………………...65
Figure 4.4 Effects of hypoxia on VEGF production and cell viability of non-viral engineered
ADSCs………………………………………………………………………………………...66
Figure 4.5. Effects of paracrine signals from non-viral engineered ADSCs on endothelial cell
viability and apoptosis…………………………………………………………….…………...67
Figure 4.6. Paracrine release from PBAE/VEGF engineered human adipose-derived stem cells
(hADSCs) enhanced endothelial cell migration……………………………………………….68
Figure 4.7. Paracrine release from PBAE/VEGF engineered human adipose-derived stem cells
(hADSCs) enhanced endothelial cell tube formation………………………………………….69
Figure 5.1. Genetic upregulation of CXCR4 and/or VEGF in ADSCs by PBAE nanoparticles.86
Figure 5.2. Improvement of cell fate and therapeutic efficacy due to CXCR4-overexpressing
ADSCs………………………………………………………………………………………...87
viii

Figure 5.3. CXCR4-overexpressing ADSCs enhanced muscle regeneration and mediated host
angiogenic and inflammatory response………………………………………………………..88
Figure 5.4. CXCR4-overexpression in ADSCs promotes a pro-angiogenic and anti-
inflammatory phenotype under hypoxia………………………………………………..……...89
Figure S5.1. Improvement of transfection efficiency and low cytotoxicity by PBAE
nanoparticles…………………………………………………………………………………..90
Figure S5.2. Cell engraftment of transfected ADSCs………………………………………….90
Figure S5.3. Reperfusion of the ischemic hindlimb at one week post-procedure………………91
Figure 6.1. Non-viral gene delivery to ADSCs via DNA polyplexes composed of a
biodegradable poly(β-amino ester) and pCK-HGF-IRES-CXCR4 plasmid………………….108
Figure 6.2. Production of functional therapeutic protein from genetically modified ADSCs...109
Figure 6.3. Survival of genetically modified ADSCs post-transplantation in vivo…………...110
Figure 6.4. Angiogenic response in ischemic hindlimbs post-transplantation in vivo………111
Figure 6.5. Analysis of limb status and muscle health………………………………………..112
Figure S6.1 – Plasmid Design of bi-directional and bicistronic vectors………………………113
Figure S6.2 – Optimization of D32-122 transfection by polymer to DNA weight ratio………114
Figure 7.1 SDF-1α Enhances Cell Recruitment Post-ischemic Induction……………………129
Figure 7.2 SDF-1α Promotes In Situ Transfection of recruited endogenous cells……………130
Figure 7.3 Timing of Plasmid DNA Delivery Affects the Level and Duration of Transfection131
Figure 7.4 Enhanced HGF Transfection In Situ by SDF-1α Delivery………………………...132
Figure 7.5 Angiogenic Improvement in the Ischemic Hindlimb by HGF overexpression…...133
Figure 7.6 Promotion of limb salvage………………………………………………………...134
Figure 8.1. Schematics of microfluidic chip design for microsphere synthesis………………148
Figure 8.2. Control of microsphere size by adjusting microchip in put channel dimensions…149
Figure 8.3. Effect of varying PEG concentration on microsphere property including swelling
ratio and microgel mesh size…………………………………………………………………150
Figure 8.4. Protein encapsulation in biodegradable PEG microspheres of differing size and
polymer composition……………………………………………………………...…………151
Figure 8.5. Release kinetics and bioactivity of bFGF from biodegradable PEG microspheres of
varying size and PEG concentration………………………………………………………….152
Figure 8.6. Release kinetics and bioactivity of bFGF from biodegradable PEG microspheres of
varying size and PEG concentration………………………………………………………….153
ix

List of Tables
Table 3.1: List of plasmids used for transfection………………………………………………48
Table 3.2: List of primers used for quantitative real time-polymerase chain reaction………….48
Table S5.1: List of primers used for quantitative real time-polymerase chain reaction…….…..92
x

1. Introduction
Peripheral arterial disease (PAD) is an important medical problem caused by a blockage
of blood flow to the limbs. The prevalence of PAD increases with age and those newly
diagnosed are six-times more likely to die within the next 10 years; this is because the diagnosis
of PAD often coincides with underlying advanced atherosclerosis or Diabetes Mellitus.
Symptomatic PAD can cause pain when walking, and in severe cases, lead to non-healing ulcers
or limb amputation. Treatment options often involve surgical revascularization, such as surgical
bypass or percutaneous angioplasty. However, many patients are unable to withstand surgery
due to the presence of surgically-prohibitive co-morbidities, thereby leaving them without
treatment.
Therapeutic angiogenesis offers an alternative treatment option for PAD, which relies
on the delivery of biologics or stem cells to promote host blood vessel reperfusion. In particular,
stem cell-based therapeutics may offer advantages to biologics by having the potential to
respond to environmental cues and secrete a multitude of paracrine signals. Despite this
potential, many stem cell therapeutics remain deficient, which is partly related to poor stem cell
engraftment and insufficient paracrine signaling. Gene therapy has the potential to overcome
these issues by strategically overexpressing therapeutic factors involved in stem cell mediated
therapeutic angiogenesis, such as angiogenic growth factors, cell survival factors, or cell homing
factors. Indeed, various genes encoding angiogenic factors along with various cell types have
been studied showing promising results.
The goal of this thesis is to develop a clinically translatable platform for treating PAD
using autologous stem cells genetically engineered with non-viral vectors. To achieve such a
platform several criteria were applied, including: 1) selection of a readily and abundantly
available autologous stem cell source; 2) use of a safe and efficient method for gene delivery to
primary cells in vivo; and, 3) the proper selection of therapeutic factors for genetic upregulation.
To meet these criteria, we developed two different strategies for achieving a genetically
engineered stem-cell based therapy for PAD. These were: 1) ex vivo engineered stem cells; and
2) endogenous progenitor cell recruitment and programming in situ.
With regards to the first approach, we achieve non-viral engineered stem cells ex vivo
using the following: 1) adipose-derived stem cells (ADSCs), which are a readily available
autologous stem cell source; and 2) biodegradable polymeric vectors, which are a non-viral
transfection agent. Using this platform, we assessed several genes for therapeutic enhancement.
1

In particular, we assessed CXC chemokine receptor 4 (CXCR4) for its role in stem cell homing
to ischemia. We also assessed vascular endothelial growth factor (VEGF) and hepatocyte
growth factor (HGF); where the latter is explored as an angiogenic factor that has shown the
most clinical potential to date.
With regards to the second approach, we attempt to achieve non-viral engineered stem
cells in situ using: 1) endogenous progenitor cells, which are recruited cell populations to sites
of ischemia under the action of chemokines; and 2) plasmid DNA, for therapeutic enhancement
of the recruited populations. This strategy is meant to circumvent the need for isolating stem
cells, which otherwise requires cumbersome and expensive manipulation in the laboratory, and
subsequent transplantation back to the patient. Effectively, this approach eliminates the need to
perform multi-step procedures by achieving genetically engineered stem cells wholly in situ.
We attempted to achieve this approach by controlled release of stromal derived factor-1α (SDF-
1α), a chemokine involved in cell homing to ischemia, and subsequent plasmid DNA delivery
encoding therapeutic factors.
These two approaches are described over 7 chapters (Chapters 2-8), which are
organized as follows:
Chapter 2 describes the main background for using stem cells and biomaterials for
therapeutic angiogenesis
Chapter 3 focuses on the optimization of ADSC transfection using poly(β-amino esters)
(PBAE), a biodegradable polymeric vector. Several aspects are explored for their effects on
ADSC transfection including: cell source and isolation; PBAE type; cell culture conditions; and,
plasmid design and gene.
Chapter 4 assesses the effects of paracrine release from non-virally engineered ADSCs
on endothelial cell (EC) behavior in vitro.
Chapter 5 explores the effects of CXCR4-overexpressing ADSCs on promoting cell
survival and engraftment, and subsequent therapeutic response in vivo. Further study on the
phenotypic behavior of CXCR4-overexpressing ADSCs cultured under hypoxia is also
performed.
Chapter 6 demonstrates the development of ADSCs co-overexpressing CXCR4 and
hepatocyte growth factor (HGF) using a bicistronic plasmid designed to enable dual expression
in the same ADSC. The therapeutic effects of these cells were assessed in an aged murine model
of hindlimb ischemia induced with severe ischemia to better reflect severe PAD.
2

Chapter 7 shows the potential of endogenous cell recruitment and programming as an
alternative to stem cell-based therapeutics. The effects of controlled SDF-1α release on cell
recruitment and in situ transgene expression are demonstrated. Timing of DNA delivery post-
surgery is further assessed. Therapeutic potential is validated in a murine model of hindlimb
ischemia using HGF.
Chapter 8 is about the development of monodisperse, biodegradable polyethylene
glycol (PEG) microspheres for tuning the release of biologics using a microfluidic platform.
The goal of this platform is to enable controlled delivery of protein or DNA, which may be used
to achieve sequential release of protein and DNA for recruitment and programming strategy.
Chapter 9 summarizes the results of this thesis and also discusses limitations and future
directions.
3

2. Background
Therapeutic Angiogenesis for Treating Cardiovascular Diseases
Lorenzo Deveza1, 2, Jeffrey Choi3, Fan Yang2,4 ,*
1 School of Medicine, Stanford University, Stanford, CA, 94305, USA; 2 Department of Bioengineering,
Stanford University, Stanford, CA, 94305, USA; 3 Department of Biology, Stanford University, Stanford,
CA, 94305, USA; 4 Department of Orthopaedic Surgery, Stanford University, Stanford, CA, 94305, USA
Abstract
Cardiovascular disease is the leading cause of death worldwide and is often associated with
partial or full occlusion of the blood vessel network in the affected organs. Restoring blood
supply is critical for the successful treatment of cardiovascular diseases. Therapeutic
angiogenesis provides a valuable tool for treating cardiovascular diseases by stimulating the
growth of new blood vessels from pre-existing vessels. In this chapter, we discuss strategies
developed for therapeutic angiogenesis using single or combinations of biological signals, cells
and polymeric biomaterials. Recent progress in exploiting genetically engineered stem cells and
endogenous cell homing mechanisms for therapeutic angiogenesis is also discussed.
2.1 Clinical significance
Cardiovascular disease (CVD) represents a global medical and economic problem with
high morbidity and mortality rates. The World Health Organization (WHO) has listed CVD as
the number one cause of death worldwide. In the United States alone, CVD accounted for
32.8% of the approximately 2.5 million deaths in 2008 [1]. The prevalence of CVD is similarly
staggering, with an estimated 82.6 million American adults (1 in 3) having one or more types
of CVD. Of these, 40.4 million are estimated to be older than 60 years of age with the average
annual rates of first time CVD events increasing with age [1]. For those that have experienced
one CVD event, CVD has been listed as one of the 15 leading conditions that cause functional
disability, thereby affecting quality of life and the ability to work.
Depending on the organs affected, CVD can be classified into coronary artery disease,
cerebrovascular disease, peripheral arterial disease and aortic (thoracic or abdominal)
atherosclerosis. CVD is generally characterized by narrowing or occlusion of the blood supply
of these vascular beds, and is most commonly caused by atherosclerosis [2, 3]. Treatment
options for CVD generally aim to re-establish blood flow through the affected vascular beds,
4

and are administered based on the severity of the disease [2, 4]. For early-stage disease,
management focuses on lifestyle modifications to reduce the number of modifiable risk factors.
As the disease progresses, pharmacological or surgical interventions may be needed to increase
the blood flow through the affected tissue or to reduce the energy requirements.
Pharmacological therapy acts to decrease oxygen demand by applying drugs to decrease the
heart rate; or to increase the blood supply by applying drugs that cause vascular smooth muscle
dilation. In cases of acute disease or full vascular occlusion, vascular stents may be used to
expand vessels when one or a few vessels are affected, while surgical bypass is necessary when
multiple vascular beds are occluded [4]. Despite the set of currently available treatment options
for patients with CVD, there is a subset of patients with advanced disease for whom surgical
revascularization is not an option due to the existence of various co-morbidities that prohibit
them from undergoing surgical procedures.
Driven by the clinical demands, therapeutic angiogenesis aims to stimulate and augment
the growth of new blood vessels from pre-existing vessels in order to re-supply blood flow to
affected ischemic tissues. In this chapter, strategies for therapeutic angiogenesis are discussed,
including direct delivery of angiogenic growth factors and the delivery of cells to ischemic
tissues. Recent progress on therapeutic angiogenesis utilizing polymeric biomaterials, combined
stem cell and gene therapy and regulation of endogenous stem cell homing are also discussed.
2.2 Biology of Angiogenesis
There are several mechanisms by which blood vessel formation occurs including
vasculogenesis, angiogenesis, and arteriogenesis [5, 6]. Each of these processes is interrelated
and leads to the formation of the vasculature of the body. The earliest blood vessel formation
in a developing embryo arises via vasculogenesis, in which endothelial progenitor cells coalesce
to form solid cords. These initially lumenless cords then transform into patent vessels in the
process of tubulogenesis [6]. Unlike the de novo blood vessel formation process associated with
vasculogenesis, angiogenesis is defined as sprouting new blood vessels from pre-existing blood
vessel networks and is important for expanding the vascular bed initially formed via
vasculogenesis. Arteriogenesis is the maturation of arterio-arteriolar anastomoses by the
recruitment and coating of pre-formed vessels with pericytes or vascular smooth muscle cells.
Arteriogenesis results in completely developed, functional arteries [7]. In the post-natal period,
angiogenesis and arteriogenesis play the major role in re-vascularizing under-supplied tissues
[5].
5

Under normal physiological conditions, there is a steady state in which quiescent
endothelial cells are maintained by autocrine signaling including vascular endothelial growth
factor (VEGF), NOTCH, angiopoietin-1 and fibroblast growth factor (FGF) [5, 6]. This steady
state may become disrupted under conditions of low oxygen, inflammation, wound healing, or
within a tumor. Such hypoxic conditions can be sensed by endothelial cells and other stromal
cells, which express oxygen sensors and hypoxia-inducible factors, such as prolyl hydroxylase
domain 2 (PHD2) and hypoxia-inducible factor 2α (HIF-2α) [5, 6]. When hypoxia is sensed,
angiogenic signals are released, such as VEGF, angiopoietin-2, and FGF [5, 6]. Initially, these
signals cause pericytes to detach from the vessel walls and endothelial cells to loosen their cell-
cell junctions, which enable the blood vessels to dilate. This increases vascular permeability
and allows plasma proteins to extravasate and form a provisional extracellular matrix (ECM)
through which ECs can migrate. Simultaneously, proteases liberate angiogenic molecules from
the ECM, which further potentiate the process. The migrating ECs differentiate to become
guiding tip cells or proliferating stalk cells. The tip cells lead the direction of sprouting, while
the stalk cells proliferate to elongate the sprouting vessels. Blood flow is initiated when two
growing vessels meet and fuse. Maturation later ensues as pericytes are stimulated to cover
endothelial cells under the action of platelet-derived growth factor-B (PDGF-B), angiopoietin-
1 (ANG-1), and transforming growth factor-β (TGF-β) [5]. It is known that bone-marrow
derived endothelial progenitor cells (EPCs) also participate in angiogenesis and become
incorporated into the vascular wall, but their importance is not well understood [8].
2.3 Therapeutic angiogenesis
Therapeutic angiogenesis aims to induce, augment and control the host angiogenic
response in order to re-vascularize ischemic tissues, and often involves delivery of growth
factors or stem/progenitor cells. Growth factors may be delivered in the form of proteins or
genes encoding target proteins. The premise behind this approach is to apply well-studied
growth factors (VEGF, FGF) in ischemic tissues to guide angiogenic cellular and tissue
behavior. Cell therapy may similarly act to induce the angiogenic response by the release of
paracrine signals. Delivered cells may also act by becoming incorporated into the growing
vascular supply, thereby acting as building blocks to form new blood vessels. Both mechanisms
likely occur upon delivery of cells to ischemic tissues. Transmyocardial laser revascularization
offers another strategy for therapeutic angiogenesis, which is believed to stimulate host
angiogenic response by injuring the ischemic myocardium in specified locations [9].
6

2.3.1 Growth Factor Therapy
Various growth factors have been applied for therapeutic angiogenesis including
VEGF, bFGF, hepatocyte growth factor (HGF), PDGF, ANG-1 and insulin-like growth factor
(IGF-1). Among these, VEGF and bFGF are the most well-studied and have reached human
clinical trials. VEGF is the most important regulator of physiological angiogenesis during
growth, healing and in response to hypoxia [5, 10]. VEGF is upregulated 30-fold by hypoxia-
inducible transcription factor, which is more than any other inducible angiogenic factor.
However, when administered alone, VEGF may lead to the formation of leaky, unstable
capillaries. PDGF-B can help stabilize nascent blood vessels by recruiting mesenchymal
progenitors, and co-delivery of VEGF and PDGF has been shown to lead to early formation of
mature vessels [11]. bFGF is among the first discovered angiogenic factors to have both
angiogenic and arteriogenic properties, and FGF receptors are expressed on both endothelial
cells and smooth muscle cells, which may facilitate formation of a mature blood vessel network
[5, 10]. The HGF family induces potent angiogenic responses by binding to the c-MET receptor,
which is expressed on endothelial cells, vascular smooth muscle cells and hematopoietic stem
cells. HGF is known to have mitogenic, angiogenic, anti-apoptotic, and anti-fibrotic activities
in various cells [10].
To date, there have been several pre-clinical and clinical studies evaluating the safety
and efficacy of growth factors for inducing angiogenesis in ischemic heart disease and
peripheral arterial disease (PAD). Early phase I and II human clinical trials with VEGF, bFGF
and HGF were promising, but larger randomized placebo-controlled trials failed to demonstrate
significant benefits in the approved end-points. Preclinical data demonstrate that the delivery
of bFGF or VEGF eventually results in unstable vessel growth that resembles immature tumor
vasculature. The limited clinical response to growth factor therapy is likely due to a number of
factors. Growth factor delivery is generally limited by their rapid diffusion, poor biostability
and short half-lives in vivo. Furthermore, it often requires supraphysiological doses or multiple
injections, which could lead to excessive uncontrolled vascular formation in undesired
locations.
3.2. Cell-based therapy
Cell-based therapy is the most extensively studied approach so far for therapeutic
angiogenesis and may contribute via either directly participating in new vessel formation or
7

secreting paracrine signals. Excellent reviews have been published previously with detailed
discussion on this topic [12, 13]. In this review, we have chosen to focus on more recently
developed platforms for therapeutic angiogenesis including biomaterials-based approach,
combined stem cells and gene therapy and exploiting stem cell homing for angiogenesis. As a
therapeutic approach, cells have the potential to act as hypoxia-responsive paracrine release
vehicles by virtue of their diverse cytokine contents. As cells migrate and respond to the
environment, they may also lead to more dynamic paracrine release. The most extensively
studied cell populations for therapeutic angiogenesis are endothelial progenitor cells (EPC) and
bone marrow-mesenchymal stem cells (BMMSCs). EPCs have been found to mobilize in
response to ischemia and become incorporated into the growing vasculature [8, 14]. However,
it should be recognized that various studies have employed different protocols for isolating this
population of cells, and this has resulted in inconsistent expression profiles among different
studies, which consists of true EPCs and other cell types, such as hematopoietic stem cells [12].
For this reason, this cell population has been reported and identified according to the method of
cell isolation, e.g., “granulocyte colony stimulating factor (G-CSF) mobilized peripheral blood
cells,” or “bone marrow aspirate mononuclear cells” (BMNCs). These cell populations can
differentiate into endothelial cells, pericytes, or smooth muscle cells, thereby acting as building
blocks for angiogenesis, but may also potentiate vascular growth through paracrine
mechanisms. BMMSCs also have the potential for differentiation into vascular support cells,
but they more likely contribute to angiogenesis by the release of proangiogenic factors. Adipose-
derived stem cells (ADSC) exhibit similar differentiation potential and paracrine release
characteristics as BMMSCs, and is currently being evaluated in preclinical trials for therapeutic
angiogenesis. Compared to BMMSC, ADSCs are more abundant and easier to isolate, yet more
studies are still needed to fully assess their potential for therapeutic angiogenesis.
The main advantages of autologous stem cell therapy relate to immunogenicity and
ethical issues. Because these cells will be transplanted back into the same patient from which
they are harvested from, there should be no immune rejection, such as that seen in allogeneic
implants. Also, since these progenitor cells are isolated from adult tissues and are not derived
from fetal tissues, they face no major identified ethical concerns. Furthermore, as adult stem
cells, their differentiation potential is relatively limited with lesser risk for aberrant tissue
formation and tumorigenesis. On the other hand, cell-based therapies still face several
limitations. Upon isolation, the cell population is often heterogeneous, which may lead to varied
responses. To obtain the large cell numbers needed for transplantation, ex vivo cell expansion
8

is often required, which leads to regulatory concerns and increased cost and time, thereby
increasing the barriers for clinical translation. Furthermore, the cell engraftment efficiency is
typically very low upon transplantation, and may induce inflammatory responses. Still, cell-
based therapies have met with success in the clinic. In a meta-analysis of cell therapy for PAD,
it was found that autologous cell delivery (BMMSCs and G-CSF mobilized peripheral blood
cells) led to improved indices of ischemia and subjective symptoms (pain-free walking) [15].
However, the study also pointed out limitations of their analysis, such as publication bias and
the inclusion of unblinded study results. Further work is needed to determine the efficacy of
autologous stem cell therapy.
2.4 Polymeric Biomaterials for therapeutic angiogenesis
Polymeric biomaterials may serve as drug delivery depots for controlled release of
biological signals in a temporal and spatial-controlled manner. In the ideal situation, such
systems may release growth factor within a physiological range over the course of several days
to weeks, thereby overcoming the issues of short protein half-lives and rapid diffusion from the
site. Polymeric biomaterials have been applied in therapeutic angiogenesis to delay the release
of single growth factors, to release multiple biologics or to achieve environment-responsive
release of biologics in response to changes in pH or cell-mediated matrix metalloproteinase
activities.
2.4.1 Commonly utilized polymers for therapeutic angiogenesis
Polymers for therapeutic angiogenesis include synthetic-based polymers, such as
poly(lactic-co-glycolic acid) (PLGA) and poly(ethylene glycol) (PEG), as well as naturally-
derived polymers, such as alginate or gelatin. PLGA was among the first to be studied for drug
release and many initial systems for therapeutic angiogenesis utilized PLGA as a base polymer
for the controlled release of VEGF [16-18]. PLGA is a favorable polymer due to its
biodegradability and ability to control release, but when degraded it increases the local acidity,
which could cause undesirable inflammation. PEG is a biocompatible polymer that is inert by
nature, with easily tunable biochemical and mechanical properties [19]. PEG has been utilized
to create cell-responsive hydrogels for VEGF release [20]. Alginate and gelatin are composed
of natural polymers that can interact well with cells in vivo and may also help to control drug
release. However, natural-derived materials suffer from batch-to-batch variance, and they are
less tunable in chemical properties compared to synthetic polymers.
9

2.4.2 Strategies for modulating growth factor release using biomaterials
To achieve effective therapeutic angiogenesis, it would be desirable to have a sustained
release of biological signals over time. However, most drug delivery systems have an initial
“burst-release,” where the large-majority of loaded growth factors is released in the first few
hours. While the mechanisms underlying “burst-release” have not yet been entirely elucidated,
several parameters are believed to play a critical role in regulating matrix-controlled drug
release including processing conditions, surface characteristics, and geometry [21]. Several
strategies have been developed to delay such burst release by modulating physical interactions,
ionic/biochemical affinity interactions, and covalent binding between the loaded biologics and
the polymeric biomaterials (Figure 1).
2.4.2.1 Physical interactions. Physical interactions or physical entrapment can delay protein
release based on the size of the drug relative to the pore size of the scaffolds. Controlled release
of VEGF from PLGA scaffolds improved angiogenesis compared to free-VEGF administration
in a myocardial ischemia model or hindlimb ischemia model [17, 18]. These reports
demonstrate the benefit of prolonged release of VEGF over bolus growth factor injection.
Biological signals can also be loaded into composite scaffolds using both PLGA microspheres
containing alginate hydrogel, which also demonstrated statistically significant increases in
vessel density compared to recombinant human-VEGF (rh-VEGF) injection alone in vivo [22].
2.4.2.2 Ionic/biochemical interactions. Proteins can form hydrogen or ionic bonds with
macromolecules such as heparin sulfate. Such interactions can be utilized to slow down the
protein release by incorporating heparin, heparan sulfate or hyaluronic acid into the drug-
releasing scaffolds [23-27]. bFGF specifically binds to heparin and heparan sulfate, and loading
bFGF into heparin conjugated PLGA nanospheres led to linear release up to 15 days [23]. This
release can be further prolonged by encapsulating these PLGA nanospheres into fibrin hydrogels
[23]. Sustained release of bFGF resulted in significant improvements in capillary density and
cell proliferation in a murine hindlimb ischemia model, [23]. Enhanced angiogenesis was also
observed using heparin-containing chitosan hydrogels or fibrous matrices to release bFGF in
vivo, and release can be tuned by varying heparin concentration in the scaffold [24, 27]. Heparin
binding alginate hydrogels have also been developed for prolonged release of HGF, which
resulted in significant increases in blood flow and capillary density compared to bolus HGF
10

delivery [26]. These studies show that release of growth factor may be prolonged by exploiting
heparin-binding interactions and this has potential for improving growth factor approaches for
therapeutic angiogenesis.
2.4.2.3 Covalent linking. Lastly, some biologics can be covalently bound to the polymeric
matrix via a hydrolytically degradable or MMP-degradable linker [20, 28, 29]. It has been
demonstrated that matrices incorporating covalently attached VEGF via a MMP-degradable
linker can lead to the formation of a more controlled and stable vasculature [20, 29]. However,
covalent linkage requires chemical modification of the biologic which can affect its biological
activity. Also, the chemistry of the linker must be carefully designed to prevent unwanted
immune responses [19].
2.4.3 Co-delivery of multiple biologics.
Angiogenesis is a dynamic process that is regulated by complex biological signals.
Polymeric biomaterials offer the potential to simultaneously or sequentially deliver multiple
biologics better mimicking the complexity of angiogenesis [11, 30-35]. For example, VEGF is
a potent angiogenic factor, but when delivered alone is not sufficient for developing a mature
and stable vascular network. Dual delivery of VEGF and PDGF using PLGA or alginate
hydrogel resulted in the formation of larger and more mature vessels and improved blood flow,
thereby suggesting the potential benefits of co-delivering synergistic growth factors (Figure 2)
[11] [30]. Biodegradable hydrogels such as hyaluronan (HA) has also been examined to achieve
sequential delivery of VEGF and keratinocyte growth factor (KGF) [33]. When transplanted
into the ear pinna of a mouse they found significant increases in microvascular density in the
dual-delivery group from the HA hydrogel compared to dual growth factor bolus injections, or
single growth factor delivery from HA hydrogels [33]. Gelatin-based hydrogel has also been
shown as a promising depot for co-delivery of bFGF and G-CSF to enhance therapeutic
angiogenesis [31]. bFGF is a mitogen and chemoattractant of both fibroblasts and endothelial
cells, while G-CSF acts as a potent mobilizer of hematopoietic stem cells and bone marrow
stromal cells. Dual release of bFGF and G-CSF from a gelatin hydrogel enabled increased
reperfusion and capillary density by 2 weeks, which was sustained up to 8 weeks. In contrast,
bFGF alone led to a reduction in these parameters by 8 weeks [31]. Bolus delivery of these
factors led to improvements in capillary density, but no effects on blood reperfusion, which
further supports the benefit of controlled release [31]. Dextran-based hydrogels capable of
11

releasing multiple angiogenic factors (VEGF, SDF-1, IGF, Ang I) has been recently developed
[35]. Significant increases in vessel number and size were observed when all four growth
factors were delivered in a subcutaneous model compared to single or dual growth factor
delivery, again suggesting the potential synergistic benefit of delivering multiple biological
signals for therapeutic angiogenesis [35].
2.4.4 Temporal controlled release.
Angiogenic response to growth factors is time-dependent and the ability to control the
temporal release of growth factors is highly desirable to achieve maximal benefits. In support
of this concept, Silva and colleagues demonstrated that a gradual reduction in VEGF
concentration led to increases in vessel sprouts in an in vitro sprout assay compared to
consistently high VEGF concentration or increasing VEGF concentration over time (Figure 3)
[36]. Tengood and colleagues developed a cellulose hollow fiber capable of sequential release
of VEGF followed by sphingosine-1-phosphate (S1P) [34]. VEGF initiates angiogenesis by
increasing vascular permeability and recruits endothelial cells, while S1P acts to stabilize
intracellular junctions and decrease the permeability of endothelial cells but simultaneously
inhibits endothelial cells migration [34]. If delivered together, these two signals may inhibit
each other. Their results showed that a significant increase in angiogenic response was only
observed when VEGF and S1P were delivered sequentially. In contrast, low angiogenic
response was observed when the two factors were delivered simultaneously or if S1P was
delivered first. These results demonstrate that growth factors can act in temporal manner
depending on the stage of angiogenesis.
2.4.5 Environmental-responsive hydrogels for releasing biologics.
To aid releasing angiogenic growth factors specifically in ischemic tissues, pH and
temperature responsive hydrogels have been developed. pH-responsive heparin functionalized
chitosan/poly(γ-glutamic acid) nanoparticles have been developed to gel and enable sustained
release of bFGF under low pH, and then disintegrate and release heparin under normal
physiological pH (Figure 4) [37]. This design aimed to utilize bFGF to induce angiogenesis,
while releasing heparin as an anti-coagulant [37]. The efficacy of such a system for in vivo
therapeutic angiogenesis remains unknown and requires further testing. Another study reported
the development of a pH and temperature sensitive hydrogel capable of gelation at pH of 6.8
and at 37°C [38]. When transplanted in a rat model of myocardial ischemia, it led to statistically
12

significant improvements in blood flow, capillary density and arteriole density compared to
bolus injection and polymer only controls, and these parameters correlated with improvements
in myocardial functions [38]. These results demonstrate the feasibility and promise of applying
environmental-responsive polymers to achieve better control over growth factor release.
2.4.6 Biomimetic polymers to modulate cell response.
Biomimetic scaffolds containing cell-responsive domains can promote therapeutic
angiogenesis. For example, matrices can be designed to interact with cells via integrin binding
sites to facilitate cell migration and cell-cell interactions. Injecting RGD-modified alginate
hydrogels into a chronic rat myocardial infarction model led to significant improvements in
cardiac function 70 days post-infarction, with commensurate increases in arteriole density
compared to non-RGD containing gel control [39]. In another study, an MMP-sensitive PEG
hydrogel containing RGD and covalently linked VEGF was evaluated for therapeutic
angiogenesis in vivo (Figure 5) [20]. The cell-responsive hydrogel led to angiogenesis
specifically within the hydrogel, while soluble VEGF release led to increased vascular density
in the surrounding tissue [20]. These systems demonstrate the importance of cell-matrix
interactions and potential of exploring biomimetic polymers to modulate cell responses for
therapeutic angiogenesis.
2.5 Combined Stem Cell and Gene-based Therapy
The therapeutic potential of stem cells can be further enhanced by combining with gene
therapy. In this approach, stem cells can be genetically modified prior to transplantation in such
a way that particular cellular processes are strategically exploited (Figure 6). In particular,
researchers have focused on addressing the challenges associated with applying cells alone, such
as insufficient paracrine release, poor cell survival upon engraftment, and lack of cell homing.
Gene delivery can be used to over-express desired therapeutic factors to induce a biological
response. In contrast to growth factor delivery, gene delivery may also be utilized to up-regulate
expression of intracellular transcription factors or cell surface receptors. These strategies offer
the advantage of controlling cell behavior at the intracellular signaling level.
2.5.1 Viral vs. Nonviral
Both viral and non-viral vectors have been employed for gene delivery for therapeutic
angiogenesis, with most studies utilizing a viral-based approach due to its high efficiency [40-
13

50]. However, clinical translation of viral-based gene delivery is limited due to safety concerns
such as immunogenicity and insertional mutagenesis. Non-viral methods provide a potentially
safer alternative, and rely on physical methods such as electroporation and nucleofection, or
using cationic lipids or polymers [51]. However, most non-viral based gene delivery methods
suffer from low transfection efficiency and often high cytoxicity, and few studies for therapeutic
angiogenesis have applied such methods [52-54]. All of the following genetic approaches
described here utilized a viral-approach, except those described in the “Non-viral Approaches”
section.
2.5.2 Stem cells overexpressing growth factors
The efficacy of stem cell-based therapy may be augmented by overexpressing desirable
paracrine signals that promote angiogenesis. Overexpressing VEGF reduced the number of
EPCs needed by 30-fold to achieve a similar therapeutic effect as unmodified EPCs alone in
promoting angiogenesis and limb salvage [42]. By enhancing paracrine effects, fewer cells
would be necessary to achieve therapeutic effects. Similarly, virally transduced BMMSCs
overexpressing VEGF or ANG I led to significantly enhanced angiogenesis and improved
cardiac function in a rat myocardial infarction model compared to non-transfected controls [43,
44]. HGF and bFGF are known to stimulate both endothelial cells and smooth muscle cells, and
may stimulate both the initiation and maturation of angiogenesis. Virally transduced MSCs or
ADSCs overexpressing HGF resulted in improvement in cardiac performance and increasing
number of capillaries [41, 50]. The improvement in cardiac function can be detected as early as
day 7 and continued over time, supporting the benefits of HGF overexpression on both early
and late stages of angiogenesis. EPC overexpressing FGF1 also led to significant increases in
mature vessel density by α-smooth muscle actin staining in vivo [40]. Overall, these studies
demonstrate the potential of stem cells overexpressing angiogenic factors for accelerating blood
vessel growth and tissue functions.
2.5.3 Genetic modification to enhance cell survival
To enhance cell survival post-transplantion, ex vivo genetic modification of stem cells
with cell survival factors has been studied [55, 56]. Akt is a serine threonine kinase that is
known to be involved in several cellular processes, such as glucose metabolism and cell
migration. Foremost, among these processes is that it is a cell survival factor. Based on these
effects, Mangi and colleagues sought to enhance survival of MSCs upon transplantation by
14

overexpressing Akt1 by a retroviral method [55]. In comparison to GFP transfected controls,
they found significant increases (over two-fold) in c-kit+ cells (transplanted cells) and decreases
in c-kit+ cell apoptosis 24 hours post-transplantation, and this led to significant decreases in
fibrosis and increases in cardiac function [55]. Their results also demonstrated improvements
in cardiac function utilizing half the number of Akt-positive MSCs as LacZ controls (2.5x105
cells vs 5.0x105 cells) further supporting their hypothesis. Another report sought to build on
these results by positing an interesting approach to co-overexpress a cell-survival factor with a
growth factor. In this study, Ang1 and Akt co-overexpressing MSCs were developed utilizing
adenovirus and then were transplanted into a rat myocardial infarction model. They
demonstrated that Akt overexpression leads to enhanced cell engraftment as long as 3 months
compared to non-transfected controls with resulting increases in mature vessel density and
improvements in heart function [56]. However, it is unclear what the benefits of dual over-
expression were, since the study lacked single Akt or Ang1 controls [56]. More work will need
to be done to determine how these effects might synergize.
2.5.4 Genetic modification to enhance stem cell homing
Efficient cell homing to ischemic tissue is critical for achieving effective angiogenesis
as the therapeutic effects are largely dependent upon the engraftment efficiency of transplanted
cells. Current methods rely mostly on intramuscular or intravascular cell injection, which often
results in poor cell engraftment at the site of ischemia. Gene therapy has been applied to modify
stem cells to overexpress cell homing factors to enhance cell targeting to ischemic tissues [45,
47-49]. The hypoxia-inducible factor-1 (HIF-1)/stromal-derived factor-1 (SDF-1)/CXC
receptor 4 (CXCR4) is a well-known axis that enables host cells to home to sites of ischemia
[57]. Based on this understanding, both CXCR4 and SDF-1 have been studied for induction of
transplanted cell homing and host cell recruitment [45, 47-49]. Two studies have reported the
use of virally transduced CXCR4 overexpressing MSCs for therapeutic application in rat models
of myocardial ischemia [45, 49]. The method of delivery was IV injection for both studies and
each found significant increases in cell engraftment into the ischemic myocardium compared to
non-transfected controls. However, these studies reported conflicting results with regards to
increases in vessel density due to CXCR4 transduction in MSCs [45, 49]. This may be due to
differences in negative controls with one demonstrating significant increases compared to
EGFP-adenoviral transfected controls and one showing no difference compared to non-
transfected controls. In either case, CXCR4-overexpressing MSCs led to improvements in
15

cardiac function, thereby demonstrating that such increased cell homing may provide
therapeutic effects by another mechanism. Tang and colleagues took another approach seeking
to upregulate SDF-1 in MSCs for the purpose of recruiting host cells for therapeutic
vascularization [47, 48]. In their study, they chose to overexpress both VEGF and SDF-1 to
achieve the effect of initiation and maturation of new blood vessels in a rat model of myocardial
infarction [48]. Interestingly, they demonstrated that overexpression of SDF-1 leads to genetic
upregulation of the survival factor, Akt [47, 48]. They found that SDF-1 overexpression alone
was able to mediate significant increases in vascular density by vWF and α-smooth muscle actin
staining, but combination therapy led to significantly higher increases with corresponding
improvements in cardiac function [48]. These approaches may become more interesting as an
understanding of host cell recruitment for injury becomes better understood.
2.5.5 Non-viral approaches
Non-viral gene transfection approaches are important for clinical translation. However,
few studies have reported the use of non-viral vectors for ex vivo gene modification in
therapeutic angiogenesis. Many of the current approaches for non-viral gene transfer are
unsatisfactory due to low transfection efficiency or high cytotoxicty. To overcome some of
these issues, Yang and colleagues developed VEGF over-expressing MSCs utilizing novel non-
viral biodegradable polymeric nanoparticles called poly(β-amino esters) that were able to
achieve 4-5 times the transfection efficiency of the leading commercially available reagent,
Lipofectamine 2000, with minimal cytoxicity [54]. When transplanted into a murine model of
hindlimb ischemia, they found enhancement in angiogenesis and limb salvage compared to GFP
transfected controls, thereby demonstrating the clinical potential of non-viral engineered stem
cell-based therapy for therapeutic angiogenesis (Figure 6) [54]. In another study, a novel
plasmid for a hypoxia inducible VEGF was transfected into MSCs utilizing a non-degradable
polymer (linear poly-ethyliminine), which also demonstrated improved efficiency and
cytoxicity characteristics compared to Lipofectamine. This plasmid was designed to enable
better control over VEGF release, and led to improved cell engraftment, capillary density and
increased cardiac functions in a rat myocardial ischemia model. These results demonstrate the
promise of applying non-viral based gene delivery for therapeutic efficacy, which may provide
a safer alternative than the viral approach.
16

2.6 Exploiting stem cell homing for therapeutic angiogenesis
Stem cell therapy holds great potential for therapeutic angiogenesis, but in general,
clinical translation of stem cell-based approaches for therapeutic angiogenesis has been slow
due to several inherent limitations. First, most methods require lengthy procedures which
include cell isolation, in vitro cell modification, and subsequent transplantation back into the
patient. Such multi-step procedures lead to high cost and significant regulatory concerns.
Second, the vast majority (>90%) of transplanted cells die and few manage to migrate to the
ischemic site. Third, ex vivo stem cell modifications may be dependent on culture conditions
and be transient, and may thus lose effectiveness upon transplantation in vivo.
One strategy to overcome the aforementioned limitations is to exploit natural cell
homing mechanisms to attract host progenitor cells to sites of ischemic injury. In the normal
physiological state, bone marrow-derived populations are recruited to sites of hypoxia to
participate in the angiogenic process [8, 58]. However, during states of cardiovascular disease,
host cells often have reduced functionality and a compromised cell niche [58]. Various
researchers have proposed methods to increase cell homing to ischemic tissue for therapeutic
angiogenesis and for tissue engineering [58, 59]. The number of circulating EPCs from the
bone marrow has been shown to increase in response to ischemia [8], which led to improved
corneal neovascularization when ischemia was induced in the mouse hindlimb, and applying
cell mobilization factor GM-CSF led to further increases in angiogenesis. These results
demonstrate that ischemia induced at a distant site can have an effect on angiogenesis
throughout the body by cell mobilization [8]. The number of EPCs recruited to the site of
ischemia has been shown to be correlated to the level of hypoxia [57]. Increasing hypoxia led
to increasing SDF-1 expression and vessel density, which was attenuated using antibodies to
block the SDF-1/CXCR4 pathway [57]. Further study revealed that G-CSF induced
mobilization of bone marrow-derived cells by disrupting the SDF-1/CXCR4 interactions in the
bone marrow niche, with gradual decrease in SDF-1 signaling and increased CXCR4 expression
over 5 days [60]. When G-CSF mobilized cells were transplanted into a bone marrow transplant
model, cell engraftment was inhibited by blocking SDF-1 or CXCR4 pathway with antibodies.
In a later study, it was shown that HSCs, EPCs and MSCs could be differentially mobilized by
application of different combinations of G-CSF, CXCR4 antagonists and VEGF [61].
The potential of cell homing strategies for treating cardiovascular disease has been
demonstrated by the application of G-CSF in a mouse model of myocardial ischemia. In a study
performed by Orlic and colleagues, G-CSF was applied once daily for 5 days prior to inducing
17

myocardial infarction, followed by 3 more days of G-CSF injection post-myocardial infarction
[62]. This protocol resulted in over 70% mouse survival in comparison to about 20% mouse
survival in the untreated group [62]. This treatment also led to increased left ventricular wall
thickness and enhanced ejection fraction [62]. However, in practice, this type of protocol is
unrealistic since myocardial infarction gives few early warning signs. Despite some positive
results in animal models, clinical trials with G-CSF for acute myocardial infarction have been
inconclusive. One potential cause for the unsatisfactory results may be the lack of homing
efficiency of G-CSF mobilized cells to sites of injury due to the rapid cleavage of SDF-1 by
CD26/dipeptidylpeptidase-IV (DPP-IV) [63]. By applying both G-CSF and DPP-IV inhibitor
(Diprotin-A), researchers have shown significant increases in homing to the infarcted
myocardium, which correlated with improvements in capillary density and cardiac function and
mouse survival compared to G-CSF treatment alone (Figure 7) [63]. Since these factors are
administered at the time of injury, this therapy may be more realistic for clinical intervention.
The field of exploiting stem cell homing for therapeutic angiogenesis is young, and represents
a promising future direction for therapeutic angiogenesis. For future work, polymeric scaffolds
that are capable of in situ recruiting and programming endogenous stem cells in ischemic sites
would provide valuable tools for reducing the translational barrier for therapeutic angiogenesis.
2.7 Conclusions
Therapeutic angiogenesis is an important interventional approach for treating a broad
range of cardiovascular diseases, which represents a global challenge with high morbidity.
Conventional strategies involve delivering growth factors or stem cells, or a combination of
both. Polymeric biomaterials have the potential to enhance therapeutic angiogenesis by
prolonging the release of angiogenic signals, and co-delivery of synergistic signals may further
accelerate the formation of functional and mature blood vessel networks. Engineering
biomimetic scaffolds that can release biological signals in an environmental-responsive manner
would facilitate targeted angiogenesis, while mitigating the undesirable side effects caused by
uncontrolled diffusion. Controlled release systems with precise temporal and spatial control
will be of great value for therapeutic angiogenesis by mimicking the dynamic biological release
profiles characteristic of normal angiogenesis processes. Combined stem cell and gene-based
therapy may further improve the efficacy of using autologous cells alone by overexpressing
therapeutic factors or cell homing factors, and its translation relies largely on safe and effective
methods to deliver genes into stem cells. Finally, strategies exploiting cell homing may provide
18

a solution to reduce the translational barrier of cell-based therapeutic angiogenesis by reducing
the time and cost associated with ex vivo cell expansion and manipulation.
References
1. Roger, V.L., et al., Heart disease and stroke statistics--2012 update: a report from the
American Heart Association. Circulation, 2012. 125(1): p. e2-e220.
2. Libby, P. and P. Theroux, Pathophysiology of coronary artery disease. Circulation,
2005. 111(25): p. 3481-8.
3. Jawad, E. and R. Arora, Chronic stable angina pectoris. Dis Mon, 2008. 54(9): p. 671-
89.
4. McFalls, E.O., et al., Coronary-artery revascularization before elective major vascular
surgery. N Engl J Med, 2004. 351(27): p. 2795-804.
5. Carmeliet, P. and R.K. Jain, Molecular mechanisms and clinical applications of
angiogenesis. Nature, 2011. 473(7347): p. 298-307.
6. Xu, K. and O. Cleaver, Tubulogenesis during blood vessel formation. Semin Cell Dev
Biol, 2011. 22(9): p. 993-1004.
7. Heil, M., et al., Arteriogenesis versus angiogenesis: similarities and differences. J Cell
Mol Med, 2006. 10(1): p. 45-55.
8. Takahashi, T., et al., Ischemia- and cytokine-induced mobilization of bone marrow-
derived endothelial progenitor cells for neovascularization. Nat Med, 1999. 5(4): p.
434-8.
9. Horvath, K.A., Transmyocardial laser revascularization. J Card Surg, 2008. 23(3): p.
266-76.
10. Losordo, D.W. and S. Dimmeler, Therapeutic angiogenesis and vasculogenesis for
ischemic disease. Part I: angiogenic cytokines. Circulation, 2004. 109(21): p. 2487-91.
11. Richardson, T.P., et al., Polymeric system for dual growth factor delivery. Nat
Biotechnol, 2001. 19(11): p. 1029-34.
12. Leeper, N.J., A.L. Hunter, and J.P. Cooke, Stem cell therapy for vascular regeneration:
adult, embryonic, and induced pluripotent stem cells. Circulation, 2010. 122(5): p. 517-
26.
13. Rafii, S. and D. Lyden, Therapeutic stem and progenitor cell transplantation for organ
vascularization and regeneration. Nat Med, 2003. 9(6): p. 702-12.
19

14. Asahara, T., et al., VEGF contributes to postnatal neovascularization by mobilizing
bone marrow-derived endothelial progenitor cells. EMBO J, 1999. 18(14): p. 3964-72.
15. Fadini, G.P., C. Agostini, and A. Avogaro, Autologous stem cell therapy for peripheral
arterial disease meta-analysis and systematic review of the literature. Atherosclerosis,
2010. 209(1): p. 10-7.
16. Cleland, J.L., et al., Development of poly-(D,L-lactide--coglycolide) microsphere
formulations containing recombinant human vascular endothelial growth factor to
promote local angiogenesis. J Control Release, 2001. 72(1-3): p. 13-24.
17. Formiga, F.R., et al., Sustained release of VEGF through PLGA microparticles
improves vasculogenesis and tissue remodeling in an acute myocardial ischemia-
reperfusion model. J Control Release, 2010. 147(1): p. 30-7.
18. Golub, J.S., et al., Sustained VEGF delivery via PLGA nanoparticles promotes vascular
growth. Am J Physiol Heart Circ Physiol, 2010. 298(6): p. H1959-65.
19. Lin, C.C. and K.S. Anseth, PEG hydrogels for the controlled release of biomolecules in
regenerative medicine. Pharm Res, 2009. 26(3): p. 631-43.
20. Zisch, A.H., et al., Cell-demanded release of VEGF from synthetic, biointeractive cell
ingrowth matrices for vascularized tissue growth. FASEB J, 2003. 17(15): p. 2260-2.
21. Huang, X. and C.S. Brazel, On the importance and mechanisms of burst release in
matrix-controlled drug delivery systems. J Control Release, 2001. 73(2-3): p. 121-36.
22. Lee, J. and K.Y. Lee, Local and sustained vascular endothelial growth factor delivery
for angiogenesis using an injectable system. Pharm Res, 2009. 26(7): p. 1739-44.
23. Jeon, O., et al., Long-term and zero-order release of basic fibroblast growth factor from
heparin-conjugated poly(L-lactide-co-glycolide) nanospheres and fibrin gel.
Biomaterials, 2006. 27(8): p. 1598-607.
24. Kim, M.S., et al., Development of functional fibrous matrices for the controlled release
of basic fibroblast growth factor to improve therapeutic angiogenesis. Tissue Eng Part
A, 2010. 16(10): p. 2999-3010.
25. Pike, D.B., et al., Heparin-regulated release of growth factors in vitro and angiogenic
response in vivo to implanted hyaluronan hydrogels containing VEGF and bFGF.
Biomaterials, 2006. 27(30): p. 5242-51.
26. Ruvinov, E., J. Leor, and S. Cohen, The effects of controlled HGF delivery from an
affinity-binding alginate biomaterial on angiogenesis and blood perfusion in a hindlimb
ischemia model. Biomaterials, 2010. 31(16): p. 4573-82.
20

27. Fujita, M., et al., Therapeutic angiogenesis induced by controlled release of fibroblast
growth factor-2 from injectable chitosan/non-anticoagulant heparin hydrogel in a rat
hindlimb ischemia model. Wound Repair Regen, 2007. 15(1): p. 58-65.
28. DuBose, J.W., C. Cutshall, and A.T. Metters, Controlled release of tethered molecules
via engineered hydrogel degradation: model development and validation. J Biomed
Mater Res A, 2005. 74(1): p. 104-16.
29. Ehrbar, M., et al., Cell-demanded liberation of VEGF121 from fibrin implants induces
local and controlled blood vessel growth. Circ Res, 2004. 94(8): p. 1124-32.
30. Sun, Q., et al., Sustained release of multiple growth factors from injectable polymeric
system as a novel therapeutic approach towards angiogenesis. Pharm Res, 2010. 27(2):
p. 264-71.
31. Layman, H., et al., Co-delivery of FGF-2 and G-CSF from gelatin-based hydrogels as
angiogenic therapy in a murine critical limb ischemic model. Acta Biomater, 2009. 5(1):
p. 230-9.
32. Patil, S.D., F. Papadmitrakopoulos, and D.J. Burgess, Concurrent delivery of
dexamethasone and VEGF for localized inflammation control and angiogenesis. J
Control Release, 2007. 117(1): p. 68-79.
33. Peattie, R.A., et al., Dual growth factor-induced angiogenesis in vivo using hyaluronan
hydrogel implants. Biomaterials, 2006. 27(9): p. 1868-75.
34. Tengood, J.E., et al., Sequential delivery of vascular endothelial growth factor and
sphingosine 1-phosphate for angiogenesis. Biomaterials, 2010. 31(30): p. 7805-12.
35. Sun, G., et al., Functional neovascularization of biodegradable dextran hydrogels with
multiple angiogenic growth factors. Biomaterials, 2011. 32(1): p. 95-106.
36. Silva, E.A. and D.J. Mooney, Effects of VEGF temporal and spatial presentation on
angiogenesis. Biomaterials, 2010. 31(6): p. 1235-41.
37. Tang, D.W., et al., Heparinized chitosan/poly(gamma-glutamic acid) nanoparticles for
multi-functional delivery of fibroblast growth factor and heparin. Biomaterials, 2010.
31(35): p. 9320-32.
38. Garbern, J.C., et al., Delivery of basic fibroblast growth factor with a pH-responsive,
injectable hydrogel to improve angiogenesis in infarcted myocardium. Biomaterials,
2011. 32(9): p. 2407-16.
39. Yu, J., et al., The effect of injected RGD modified alginate on angiogenesis and left
ventricular function in a chronic rat infarct model. Biomaterials, 2009. 30(5): p. 751-6.
21

40. Chen, S.Y., et al., Autologous transplantation of EPCs encoding FGF1 gene promotes
neovascularization in a porcine model of chronic myocardial ischemia. Int J Cardiol,
2009. 135(2): p. 223-32.
41. Duan, H.F., et al., Treatment of myocardial ischemia with bone marrow-derived
mesenchymal stem cells overexpressing hepatocyte growth factor. Mol Ther, 2003.
8(3): p. 467-74.
42. Iwaguro, H., et al., Endothelial progenitor cell vascular endothelial growth factor gene
transfer for vascular regeneration. Circulation, 2002. 105(6): p. 732-8.
43. Matsumoto, R., et al., Vascular endothelial growth factor-expressing mesenchymal
stem cell transplantation for the treatment of acute myocardial infarction. Arterioscler
Thromb Vasc Biol, 2005. 25(6): p. 1168-73.
44. Sun, L., et al., Mesenchymal stem cells modified with angiopoietin-1 improve
remodeling in a rat model of acute myocardial infarction. Biochem Biophys Res
Commun, 2007. 357(3): p. 779-84.
45. Cheng, Z., et al., Targeted migration of mesenchymal stem cells modified with CXCR4
gene to infarcted myocardium improves cardiac performance. Mol Ther, 2008. 16(3):
p. 571-9.
46. Jabbarzadeh, E., et al., Induction of angiogenesis in tissue-engineered scaffolds
designed for bone repair: a combined gene therapy-cell transplantation approach. Proc
Natl Acad Sci U S A, 2008. 105(32): p. 11099-104.
47. Tang, J., et al., Mesenchymal stem cells over-expressing SDF-1 promote angiogenesis
and improve heart function in experimental myocardial infarction in rats. Eur J
Cardiothorac Surg, 2009. 36(4): p. 644-50.
48. Tang, J., et al., Combination of chemokine and angiogenic factor genes and
mesenchymal stem cells could enhance angiogenesis and improve cardiac function after
acute myocardial infarction in rats. Mol Cell Biochem, 2010. 339(1-2): p. 107-18.
49. Zhang, D., et al., Over-expression of CXCR4 on mesenchymal stem cells augments
myoangiogenesis in the infarcted myocardium. J Mol Cell Cardiol, 2008. 44(2): p. 281-
92.
50. Zhu, X.Y., et al., Transplantation of adipose-derived stem cells overexpressing hHGF
into cardiac tissue. Biochem Biophys Res Commun, 2009. 379(4): p. 1084-90.
51. Park, H.J., F. Yang, and S.W. Cho, Nonviral delivery of genetic medicine for
therapeutic angiogenesis. Adv Drug Deliv Rev, 2012. 64(1): p. 40-52.
22

52. Das, H., et al., Stem cell therapy with overexpressed VEGF and PDGF genes improves
cardiac function in a rat infarct model. PLoS One, 2009. 4(10): p. e7325.
53. Kim, S.H., et al., Hypoxia-inducible vascular endothelial growth factor-engineered
mesenchymal stem cells prevent myocardial ischemic injury. Mol Ther, 2011. 19(4): p.
741-50.
54. Yang, F., et al., Genetic engineering of human stem cells for enhanced angiogenesis
using biodegradable polymeric nanoparticles. Proc Natl Acad Sci U S A, 2010. 107(8):
p. 3317-22.
55. Mangi, A.A., et al., Mesenchymal stem cells modified with Akt prevent remodeling and
restore performance of infarcted hearts. Nat Med, 2003. 9(9): p. 1195-201.
56. Shujia, J., et al., Stable therapeutic effects of mesenchymal stem cell-based multiple
gene delivery for cardiac repair. Cardiovasc Res, 2008. 77(3): p. 525-33.
57. Ceradini, D.J., et al., Progenitor cell trafficking is regulated by hypoxic gradients
through HIF-1 induction of SDF-1. Nat Med, 2004. 10(8): p. 858-64.
58. Dimmeler, S., Regulation of bone marrow-derived vascular progenitor cell mobilization
and maintenance. Arterioscler Thromb Vasc Biol, 2010. 30(6): p. 1088-93.
59. Chen, F.M., et al., Homing of endogenous stem/progenitor cells for in situ tissue
regeneration: Promises, strategies, and translational perspectives. Biomaterials, 2011.
32(12): p. 3189-209.
60. Petit, I., et al., G-CSF induces stem cell mobilization by decreasing bone marrow SDF-
1 and up-regulating CXCR4. Nat Immunol, 2002. 3(7): p. 687-94.
61. Pitchford, S.C., et al., Differential mobilization of subsets of progenitor cells from the
bone marrow. Cell Stem Cell, 2009. 4(1): p. 62-72.
62. Orlic, D., et al., Mobilized bone marrow cells repair the infarcted heart, improving
function and survival. Proc Natl Acad Sci U S A, 2001. 98(18): p. 10344-9.
63. Zaruba, M.M., et al., Synergy between CD26/DPP-IV inhibition and G-CSF improves
cardiac function after acute myocardial infarction. Cell Stem Cell, 2009. 4(4): p. 313-
23.
23

Figure 2.1 Methods for incorporating biological signals into scaffolds for controlled release. A)
Biologics encapsulated directly via physical entrapment. B) Biologics covalently tethered to
polymer chains. C) Biologics loaded into microspheres and then encapsulated into hydrogel
scaffolds. Reproduced with permission from ref [19].
24

Figure 2.2 A) Polymeric scaffolds for dual delivery of VEGF and PDGF. (B) In vitro release
kinetics of VEGF from scaffolds fabricated from PLGA (85:15, lactide:glycolide), measured
using 125I-labeled tracers. (C) In vitro release kinetics of PDGF pre-encapsulated in PLGA
microspheres ( 75:25, intrinsic viscosity = 0.69 dl/g; 75:25, intrinsic viscosity = 0.2 dl/g),
before scaffold fabrication. D) Dual delivery led to early maturation of blood vessels as
demonstrated by α-smooth muscle actin staining at 2 weeks. E) Representative micro-CT
images after 5 weeks intramuscular injections of alginate (Blank), alginate containing VEGF165
(VEGF), or alginate containing VEGF165 combined with PLGA microspheres containing
PDGF-BB (VEGF/PDGF).(A-D) Reproduced with permission from ref [11] (E) Reproduced
with permission from ref [30].
25

Figure 2.3 Effects of VEGF temporal presentation on angiogenesis. Varying amounts of VEGF
is applied to human microvascular endothelial cells (HMVEC) over five days in a sprouting
assay. Each color bar represents different temporal presentations of VEGF. Red bars represent
the same concentration of VEGF applied daily over five days; blue bars represent decreasing
VEGF dose over five days; green bars represent gradual decrease in VEGF concentration; and,
brown bars represent increasing concentrations of VEGF applied. These are represented in the
lower graphs. As shown in the larger graph, decreasing the VEGF dose from an initial high
concentraion (blue bars) induced a greater number of endothelial cells sprouts, as compared to
constant VEGF doses (50 ng/ml day) (red bars), increasing VEGF doses (brown bars), or a
gradual VEGF dose decrease over time (green bars). Reproduced with permission from ref [36].
26

Figure 2.4 Environmental-responsive controlled release system for sustained bFGF delivery in
ischemic tissue triggered by pH. TEM micrography of heparinized chitosan/poly(gamma-
glutamic acid) (HP-CS/g-PGA) nanoparticles at a distinct pH value: (A) pH 6.0 (after 2 h), (B)
pH 7.4 (after 10 min), (C) pH 7.4 (after 30 min), (D) pH 7.4 (after 2 h). (E) Release of bFGF or
heparin from the smart nanoparticles, depending on the environmental pH variation.
Reproduced with permission from ref [37].
27

Figure 2.5 Cell-triggered VEGF delivery using biomimetic hydrogels. A) Schematic of MMP-
degradable PEG hydrogels containing RGD peptide and covalently linked VEGF. B) Cell-
triggered VEGF delivery led to targeted angiogenesis within the hydrogel region, whereas
soluble VEGF treatment resulted in diffusive and uncontrolled angiogenesis. Reproduced with
permission from ref [20].
28

Figure 2.6 Combined stem cell and gene therapy approach for therapeutic angiogenesis (A).
Transplantation of genetically modified stem cells into ischemic tissues led to enhanced
paracrine secretion and angiogenesis in situ. Reproduced with permission from ref [51] (B).
VEGF-overexpressing MSCs using biodegradable polymeric nanoparticles led to enhanced
angiogenesis and limb salvage in a murine hindlimb ischemia model (C). Reproduced with
permission from ref [54].
29

Figure 2.7 Exploiting cell homing for treating acute myocardial infarction. A) Signaling
pathway related to stem cell homing to ischemic tissue. G-CSF promoted cell mobilization and
Diprotin A (Dip) inhibited SDF-1 protease, thereby maintaining the stability of cell homing
factor SDF-1 released from ischemic tissue. B) G-CSF and Dip dual treatment led to
significantly increased myocardial stem cells detected in the ischemic tissue. C) G-CSF and Dip
dual treatment increased mouse survival after myocardial infarction. Reproduced with
permission from ref [63].
30

3. Overexpressing Angiogenic Growth Factors in Adipose-Derived Stem Cells using
Biodegradable Polymeric Nanoparticles
Lorenzo Deveza1, 2, Jeffrey Choi3, Fan Yang2,4 ,*
1 School of Medicine, Stanford University, Stanford, CA, 94305, USA; 2 Department of
Bioengineering, Stanford University, Stanford, CA, 94305, USA; 3 Department of Biology,
Stanford University, Stanford, CA, 94305, USA; 4 Department of Orthopaedic Surgery, Stanford
University, Stanford, CA, 94305, USA
Abstract
Stem cell-based therapies are promising candidates for treating ischemic cardiovascular
diseases by promoting angiogenic reperfusion of the affected tissues. Genetic modification of
stem cells may further promote their ability for ischemic tissue repair by strategically
overexpressing relevant therapeutic genes, and may also enable stem cells to present a more
consistent phenotype across various autologous sources. Amongst the available cell sources
adipose-derived stem cells (ADSCs) are a clinically abundant cell source that are easy to isolate
and show promise for ischemic tissue repair. However, non-viral transfection of ADSCs has
been difficult to achieve using commercially available polymeric vectors. In this study, we
assess and optimize non-viral transfection of ADSCs using optimized biodegradable polymeric
nanoparticles, known as poly(β-amino esters). We demonstrate that transfection varies across
multiple cell isolations of ADSCs, which can be partly rescued by optimizing culture conditions,
such as adding 10 ng/ml basic fibroblast growth factor (bFGF). Transfection of ADSCs with
therapeutic genes is then demonstrated using eight different plasmids encoding for three
different angiogenic factors, vascular endothelial growth factor (VEGF), hepatocyte growth
factor (HGF), and CXC chemokine receptor 4 (CXCR4). Paracrine release from transfected
ADSCs is then evaluated for promoting endothelial cell and smooth cell viability in vitro.
3.1 Introduction
Stem cells represent potential therapeutic options for tissue regeneration and repair,
which may act via two main mechanisms: 1) engraftment into the developing tissue; and, 2)
delivery of paracrine signals to promote repair. In particular, the second mechanism is
increasingly believed to be more likely in treating ischemic muscle and myocardium in a process
called therapeutic angiogenesis; as few cells are found to exhibit long-term cell engraftment and
31

a similar therapeutic response is achieved by delivering stem cell conditioned medium [1].
Despite the purported role of stem cells as delivery vehicles, stem cell therapies have had limited
clinical success, which may be due to issues of variability in autologous cell source and
insufficient paracrine release. Genetic engineering of stem cells may help to overcome these
issues by reinforcing the overexpression of desired therapeutic factors across multiple cell
sources, and in effect, make them more consistent. To achieve such an approach for clinical
usage, what is desired is a readily available cell source and a safe transfection system of relevant
therapeutic genes.
Adipose-derived stem cells (ADSCs) are a clinically abundant cell source that are easy
to isolate and have the potential to differentiate towards mesenchymal lineages [2]. Importantly,
ADSCs have been shown to secrete a number of angiogenic and anti-apoptotic growth factors
[3, 4] and have demonstrated the ability promote therapeutic angiogenesis in vivo [5]. Genetic
modification of ADSCs has also been shown to promote their therapeutic capabilities using
various angiogenic factors with beneficial effect, including vascular endothelial growth factor
(VEGF) [6], hepatocyte growth factor (HGF) [7], and CXC chemokine receptor 4 (CXCR4) [8].
While these approaches have shown promise for the therapeutic use of ADSCs, their clinical
usage remains hampered by the use of viruses to achieve gene transfection, due to issues of
immunogenicity and insertional mutagenesis.
There are several non-viral approaches for gene transfection, which may overcome the
concerns associated with viral-based vectors. Amongst such approaches, cationic polymers,
such as Lipofectamine (Lipo) and polyethylenimine (PEI) enable gene delivery by forming
complexes with plasmid DNA to generate nanoparticles, which protect plasmid DNA from
extracellular degradation and enable passage across cell membranes. However, in contrast to
viral-based vectors, many non-viral approaches suffer low transfection efficiency and high
cytotoxicity. Poly(β-amino esters) (PBAE) are a class of biodegradable cationic polymers for
gene delivery that were screened in a relatively high-throughput, combinatorial fashion for
achieving high transfection efficiency with low cytotoxicity in human cells [9, 10], and further
screened for transfection into stem cells [11]. Using PBAE, high transfection efficiencies were
also achieved in human ADSCs [11-13]. However, transfection was only explored in ADSCs
of one cell isolation in these studies. It is well known that transfection efficiency is cell type
and cell isolation specific and for clinical usage, it is important to understand how ADSCs of
various isolations might transfect using an optimal non-viral vector, such as PBAE.
32

In this study, we assess transfection of various ADSC cell isolations using PBAE
polymeric nanoparticles complexed with plasmids encoding for VEGF, HGF, and/or CXCR4.
We first assess two different PBAE polymers for transfection efficiency and cell cytoxicity. We
then optimize the culture conditions by exploring tranfection in the presence or absence of basic
fibroblast growth factor (bFGF) and fetal bovine serum (FBS). Lastly, ADSCs are transfected
with various angiogenic factors and assessed for mRNA expression, protein production, and
bioactivity of paracrine signals against human umbilical vein endothelial cells (HUVECs) and
human aortic smooth muscle cells (HASMCs).
3.2 Materials and Methods
3.2.1 Materials
Monomers were purchased from Sigma-Aldrich (St Louis, MO, USA), Scientific
Polymer Products Inc. (Ontario, NY, USA) and Molecular Biosciences (Boulder, CO, USA).
All chemicals were used as received. Anhydrous dimethyl sulfoxide was purchased from
(Sigma-Aldrich). A 25-mM sodium acetate buffer solution pH5.2 (NaAc buffer) was prepared
by diluting a 3-M stock (Molecular Biosciences). Plasmid DNAs pGluc and pcDNA3.1 was
obtained from Elim Biopharmaceuticals (Hayward, CA, USA). Plasmid DNA pBLAST-VEGF
was obtained from Aldeveron (Fargo, ND, USA). Plasmid DNA pCK-HGF was obtained from
Viromed (Seoul, Korea). Plasmid DNAs pCMV-VEGF, pCMV-CXCR4/VEGF, pCMV-
CXCR4, pCK-CXCR4/HGF were cloned in house.
3.2.2 Polymer Synthesis
Acrylate-terminated (C32-Ac and D32-Ac) polymer were synthesized by mixing
butanediol diacrylate (C) or hexanediol (D) with aminopentanol (32) in a 1.2:1.0
diacrylate/amine molar ratio at 90 ºC for 24 h to generate acrylate-terminated polymer chains
(Figure 1A, 1B). Amine-capped polymers (C32-122 and D32-122) were prepared by
subsequently reacting C32-Ac or D32-Ac with diamine monomers (122) in dimethyl sulfoxide
(Figure 1A, 1B). End-capping reactions were performed overnight at room temperature using a
1.6-fold molar excess of amine over acrylate end groups.
3.2.3 Cell Isolation and Culture
Human adipose-derived stem cells (hADSCs) were isolated from fat tissues that were
obtained from the abdominal fat of three separate female patients who had undergone a free flap
33

breast reconstruction surgery at Stanford University. All procedures were approved and guided
under Stanford IRB protocol. Mouse adipose-derived stem cells (mADSCs) were isolated from
the inguinal fat pad of FVB/NJ mice. Fat tissues were washed 2-3 times with Phosphate
buffered saline and digested at 37 °C for 30 min with 0.5 U/ml Blendzyme 3 (Roche
Diagnostics, Indianapolis, IN, USA). Enzyme activity was neutralized with DMEM, containing
10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA), 1% Penicillin/Streptomycin.
Cells were then filtered through a 100 µm nylon mesh to remove cellular debris before seeding.
Following the initial 48 hours of incubation at 37 °C and 5% CO2, cells were washed with PBS
and maintained in growth medium containing DMEM, 10% FBS, 1% penicillin/ streptomycin
and 10ng/ml of the basic fibroblast growth factor (FGF) (PeproTech, Rocky Hill, NJ, USA).
Once 85-95% confluent, the cells were passaged by trypsin (0.05%) digestion and plated at a
density of 5000-6000 cells per cm2 for further expansion. Human umbilical vein endothelial
cells (HUVECs) and human aortic smooth muscle cells (HASMCs) were obtained from Lonza
(Basal, Switzerland) and expanded utilizing endothelial growth medium (EGM-2) or smooth
muscle growth media (Lonza).
3.2.4 Transfection
Cells were transfected with the various DNA plasmids shown in Table 1. Passage 3-4
ADSCs were seeded at 12 000 cells per well in clear 96 well plates or 300 000 cells per well in
clear 6 well plates and incubated over night prior to transfection. Cells were then transfected
utilizing poly (β-amino esters) (PBAE) C32-122 or D32-122. Briefly, polyplexes were formed
by mixing pDNA and PBAE polymers in 25 mM sodium acetate at a 20:1 polymer to DNA
weight ratio. Polyplexes were added to the cell culture after 10 minutes of complexation. Cells
were then incubated with polyplexes for four hours, at which time cell medium was replaced.
Effects of culture media on transfection was assessed by using different culture conditions
during four hour incubation, as well as after replacement. The effects of basic fibroblast growth
factor (bFGF) on transfection was assessed by transfecting cells with and without 10 ng/ml
bFGF. Effects of replacement media was assessed by varying the serum concentration (0%,
1%, 5%, 10%) and bFGF concentration (0 ng/ml, 1 ng/ml, 5 ng/ml, 10 ng/ml) in the media used
for four hour replacement.
34

3.2.5 Luciferase Expression Assay
Cells transfected with plasmid DNA encoding for Gaussia Luciferase (pGLUC) were
assessed by collecting the cell supernatant at 48 hours post-transfection and assaying for
secreted luciferase. Cell supernatant was diluted 10-fold in PBS and added to opaque 96-well
plates (Falcon). The substrate Luciferin (New England Biolabs) was diluted in buffer according
to the manufacturers instructions, and added at 1:1 ratio to the diluted cell supernatant.
Luminescence was immediately measured using a plate reader.
3.2.6 Cell Viability and Proliferation
To measure cytotoxicity and cell viability, cellular metabolic activity was measured
using the Cell Titer 96 Aqueous One Solution assay kit (Promega). Metabolic activity was
measured using optical absorbance on a Multi-label plate reader (Perkin Elmer Life Sciences,
Boston, MA, USA). Measurements of treated cells were converted to percentage viability in
comparison with untreated controls
3.2.7 RNA Isolation, Reverse Transcription, and quantitative PCR
To confirm successful transfection by PBAE in vitro, total RNA was harvested from
cells at 48 hours post-transfection using RNeasy Kit (Qiagen, Germantown, MD, USA). cDNA
was synthesized by reverse transcription using the Superscript First-Strand Synthesis System
(Invitrogen) and 1 μg of total RNA. Quantitative PCR was performed using Power SYBR Green
PCR Master Mix following the manufacturer's protocol. Primers used are shown in Table 2.
All samples were run for 40 PCR cycles on Applied Biosystems 7900 Real-Time PCR System
(Carlsbad, CA, USA). The relative expression level of target genes was determined by the
ΔΔCT method, where target gene expression was first normalized to an endogenous
housekeeping gene, GAPDH, followed by a second normalization to the gene expression level
measured in untransfected control cells.
3.2.8 VEGF and HGF ELISA
VEGF and HGF production were assessed by collecting conditioned medium and
measuring using an ELISA kit for human VEGF (Peprotech, Rocky Hill, NJ) and human HGF
(R&D Systems, Minneapolis, MN) according to the manufacturer’s protocol. The level of
secreted VEGF and HGF was expressed as the amount of VEGF per milliliter of medium
supernatant.
35

3.2.9 HUVEC and HASMC viability assay
HUVECs or HASMCs were plated in gelatin-coated 96 well plates (11 200 cells/well).
HUVECs and HASMCs were cultured in low serum medium (DMEM + 2% FBS + 1% P/S) for
24 hours prior to switching to conditioned medium. HUVEC and HASMC viability was
determined by MTS assay as described above.
3.2.10 Statistical Analysis
All experiments were performed in triplicate and repeated for consistency. The results
were reported as mean ± standard error of the mean. Statistical analysis was performed using
the one way analysis of variance with Dunnett’s multiple comparison test as the post-test to
compare all the experimental groups.
3.3 Results
3.3.1 Polymer Synthesis
Poly(β-amino esters) were synthesized by the conjugate addition of amine monomers
to diacrylates. For the synthesis of acrylate-terminated C32 (C32-Ac) or D32 (D32-Ac) ,
butanediol diacrylate (C) or hexanediol diacrylate (D) was mixed in a 1.2:1.0 molar ratio with
aminopentanol (32; Figure 1). End-modified polymers were synthesized by subsequently
reacting C32-Ac or D32-Ac individually with one diamine monomer (122) to yield end-
modified C32-122 (Figure 1A) or D32-122 (Figure 1B) polymers.
3.3.2 Assessment of transfection of ADSCs using C32-122 or D32-122
Two end-modified PBAE polymers (C32-122 and D32-122) were compared for
transfection into five different cell cultures: 3 human ADSCs from different patients, 1 mouse
ADSC cell source, and HEK293T cells as a control. Transfections were assessed for cell
viability and transfection efficiency. A polymer to DNA weight ratio of 20:1 and a DNA dose
of 5 µg per 70 000 cells was used for all transfections. PBAE-mediated transfection of all cells
caused a change in cell morphology in comparison to non-transfected controls (Figure 2A).
Control images demonstrate that normal ADSC morphology was spindle shaped, while
transfected ADSCs had a cobblestone pattern, suggesting some change in cell behavior. When
assessing for cell viability, there was no significant difference at 48 hours post-transfection for
all cells in comparison to controls. Transfection efficiency, as assessed using a Gaussia
36

Luciferase reporter plasmid, demonstrated that C32-122 led to higher overall transfection
compared to D32-122. D32-122 was unable to efficiently and consistently transfect hADSCs.
However, transfection by D32-122 was successful in HEK293T cells. We found D32-122
transfection to be variable based on the batch. Transfection efficiency also varied by cell type
and culture. Human ADSCs transfected less efficiently than mouse ADSCs and HEK293T cells.
Amongst the hADSC isolations, hADSC-I3 transfected the most efficiently, while transfection
in hADSC-I2 was low or undetectable.
3.3.3 Enhancing ADSC proliferation and transfection with bFGF
Considering the difference in transfection potential in each hADSC cell isolation and
known reliance of non-viral transfection on cell proliferation, the various hADSC cell isolations
were assessed for their ability to proliferate (Figure 3A, 3B, 3C). We found that transfection
efficiency of each hADSC cell isolation correlated with their relative ability to proliferate, as
demonstrated by BrDu assay (Figure 1A), MTS assay (Figure 1B), and Picogreen DNA content
assay (Figure 1C). Furthermore, hADSC proliferation could be enhanced in the presence of 10
ng/ml basic fibroblast growth factor (a common component of cell growth medium) (Figure 1A,
1B, 1C). This treatment improved transfection efficiency in two of the three hADSC cell
isolations (hADSC-I1 and hADSC-I3), as demonstrated by green fluorescence protein
transfection (Figure 3D) and luciferase assay (Figure 3E). Transfection efficiency in hADSC-
I2 remained low despite the proliferation increase by 10 ng/ml bFGF, suggesting that promoting
proliferation does not wholly explain the ability to transfect.
3.3.4 Effects of post-transfection culture conditions
To determine the effects of culture media on ADSC transfection after incubating with
PBAE nanoparticles, ADSC-I3 was transfected and subsequently placed in media containing
varying amounts of FBS (0%, 1%, 5%, 10%) and bFGF (0 ng/ml, 1 ng/ml, 5 ng/ml, 10 ng/ml).
The amount of FBS had the largest effect on ADSC viability (Figure 4A) and transfection
(Figure 4B). The effects of bFGF on ADSC viability and transfection relied on the
concentration of FBS in the media. Interestingly, bFGF only affected viability and transfection
in the presence of 10% FBS, but did not lead to any significant effect otherwise (Figure 4A,
4B). In the presence of 10% FBS, as little as 1 ng/ml bFGF led to a significant increase in
transfection. Also, when bFGF was not added, there was no increase in transfection by
increasing FBS above 5%.
37

3.3.5 Upregulation of various angiogenic growth factors in ADSCs
To assess the ability to transfect ADSCs with various angiogenic factors, ADSCs were
transfected with seven different plasmids encoding functional genes and one backbone plasmid
(pcDNA3.1) (Table 1). All transfections were performed using C32-122 and in the presence of
10 ng/ml bFGF. Of the plasmids transfected, two encoded for vascular endothelial growth factor
(VEGF) which differed based on promoter, one encoded for CXC chemokine receptor 4
(CXCR4) only, one encoded for hepatocyte growth factor (HGF) only, one encoded for both
CXCR4 and VEGF, and one encoded for CXCR4 and HGF. The plasmids mainly differed in
promoters and size. Transfection with each of these plasmids were confirmed by RT-PCR
(Figures 5A, 5B, 5C) and ELISA (Figures 5D, 5E). As further confirmation of the above
luciferase data, human ADSCs transfected less well than mouse ADSCs and HEK293T cells.
Protein production from human ADSCs was low overall, and transfection led to increases that
were significant (p < 0.05). VEGF upregulation using all three plasmids encoding for VEGF
was comparable, suggesting little differences in transfection due to promoter and plasmid size
(Figure 5D). HGF upregulation using the two plasmids encoding for HGF was similarly
comparable (Figure 5E). The highest amounts of VEGF and HGF production occurred by
mouse ADSCs and HEK293T, reaching over 10 ng/ml and 50 ng/ml for VEGF and HGF,
respectively.
3.3.6 Effects of transfected ADSC paracrine release on HUVEC and HASMC viability
To determine whether growth factor production from ADSCs was bioactive,
conditioned media (CM) was collected from transfected ADSCs at 48 hours post-transfection
and applied to human umbilical vein endothelial cells (HUVECs) and human aortic smooth
muscle cells (HASMCs), then assessed for viability. CM from mADSCs and HEK293T cells
did increase HUVEC viability that was comparable to 10 ng/ml VEGF or 10 ng/ml HGF and
was significantly higher than transfection controls (pGLUC) and non-transfection controls (cells
only; Figure 6A). However, CM from hADSCs did not increase HUVEC (Figure 6A) or
HASMC (Figure 6B) viability as compared to basal media controls. No effects on HASMC
viability were observed due to VEGF and HGF protein controls, so as can be expected there
was no effect on HASMC viability due to CM from any transfected cell type (Figure 6B). The
ability of transfected mADSCs and HEK293T cells to elicit effects on HUVEC viability,
suggested that the effects of released growth factor was bioactive and concentration dependent.
38

3.4 Discussion
Herein, we demonstrated the transfection of various isolations of ADSC using PBAE
biodegradable nanoparticles with eight different plasmid DNAs encoding angiogenic factors or
reporter genes. We found that the PBAE polymer C32-122 achieved higher transfection in the
presence of 10 ng/ml bFGF, and further found that the culture conditions post-transfection also
affected transfection efficiencies and cell viabilities. Under these conditions, the transfection
of various angiogenic factors into the ADSCs using multiple plasmids was successfully
demonstrated. Notably, transfection efficiencies amongst various hADSC isolations varied, and
was overall lower than mADSCs and HEK293T cells. This was apparent when paracrine release
from transfected ADSCs were applied to HUVECs, where only mADSCs and HEK293T
paracrine release led to significant increases in viability.
Using the three different isolations of hADSCs, we determined that ADSCs are difficult
to transfect and further that their ability to transfect varied. At least one characteristic that
differed amongst these hADSCs was their proliferative potential, which correlated with their
ability to transfect. This is not surprising, as it is fairly well-established that non-viral
transfection relies on cell proliferation. [14] In lieu of this correlation, bFGF was used to
enhance ADSC proliferation, which also promoted ADSC transfection in two of three cell
isolations. Curiously, hADSC-I2 increased in proliferation in the presence of bFGF, but this
could not rescue their ability to transfect. This suggests that proliferation does not fully explain
transfection potential. Still, the idea of promoting proliferation to enhance transfection had not
yet been rigorously explored previously to our knowledge and does open up possibilities for
further exploration. Considering that two of three hADSCs responded well to bFGF suggests
that despite initial differences, select autologous hADSCs can be genetically modified to present
a similar phenotype.
Transfection into ADSCs was demonstrated using eight different plasmids, which
encoded for four different genes (VEGF, HGF, CXCR4, GLUC), that contained different
promoters, had different molecular weights, and were either singly expressing or dually
expressing. While each gene was able to be transfected into ADSCs, we found that there were
some differences in their level of expression. For example, pck-HGF led to much higher
increases in expression as compared to pBLAST-VEGF or pCDNA-VEGF. This is likely
plasmid specific, whereby pck-HGF is a highly engineered plasmid [15], whereas pBLAST-
39

VEGF and pCDNA-VEGF have not been fully optimized. This suggests that plasmid
development is another strategy to promote non-viral mediated transfection.
Considering that endothelial cell and smooth muscle cell viability are two cellular
aspects of angiogenesis, the effects of paracrine release from transfected cells were assessed
against HUVECs and HASMCs. The robust response of HUVECs by paracrine release from
transfected mADSC and HEK293T cells but not from the untransfected cells, suggested that
non-viral gene upregulation of VEGF and HGF can successfully promote angiogenic behavior.
However, considering the lower transfection efficiency of hADSCs, this effect was not observed
by transfected hADSCs, suggesting that the concentration of angiogenic growth factors were
too low. This could also be due to using conditioned medium, as opposed to a co-culture system,
which suffers from some limitations due to protein degradation and does not enable potential
cell-cell interactions. Further study may explored a co-culture system of transfected hADSCs
with HUVEC or HASMCs.
In conclusion, non-viral gene transfection of hADSCs can be achieved using PBAE
biodegradable nanoparticles and may promote more consistent behavior amongst various
hADSC cell isolations. Transfection of hADSCs is difficult, but could be improved by
polymeric vector, transfection conditions, and plasmid design. Overexpression of angiogenic
factors can promote endothelial cell survival in vitro. Overall, PBAE-mediated transfection of
hADSCs using angiogenic factors may be a strategy for promoting consistent hADSC behavior
towards therapeutic angiogenesis.
References
1. Ranganath, S.H., et al., Harnessing the mesenchymal stem cell secretome for the
treatment of cardiovascular disease. Cell Stem Cell, 2012. 10(3): p. 244-58.
2. Zuk, P.A., et al., Multilineage cells from human adipose tissue: implications for cell-
based therapies. Tissue Eng, 2001. 7(2): p. 211-28.
3. Thangarajah, H., et al., IFATS collection: Adipose stromal cells adopt a proangiogenic
phenotype under the influence of hypoxia. Stem Cells, 2009. 27(1): p. 266-74.
4. Rehman, J., et al., Secretion of angiogenic and antiapoptotic factors by human adipose
stromal cells. Circulation, 2004. 109(10): p. 1292-8.
5. Miranville, A., et al., Improvement of postnatal neovascularization by human adipose
tissue-derived stem cells. Circulation, 2004. 110(3): p. 349-55.
40

6. Jabbarzadeh, E., et al., Induction of angiogenesis in tissue-engineered scaffolds
designed for bone repair: a combined gene therapy-cell transplantation approach. Proc
Natl Acad Sci U S A, 2008. 105(32): p. 11099-104.
7. Zhu, X.Y., et al., Transplantation of adipose-derived stem cells overexpressing hHGF
into cardiac tissue. Biochem Biophys Res Commun, 2009. 379(4): p. 1084-90.
8. Cho, H.H., et al., Overexpression of CXCR4 increases migration and proliferation of
human adipose tissue stromal cells. Stem Cells Dev, 2006. 15(6): p. 853-64.
9. Green, J.J., et al., Combinatorial Modification of Degradable Polymers Enables
Transfection of Human Cells Comparable to Adenovirus. Advanced Materials, 2007.
19(19): p. 2836-2842.
10. Zugates, G.T., et al., Rapid optimization of gene delivery by parallel end-modification
of poly(beta-amino ester)s. Mol Ther, 2007. 15(7): p. 1306-12.
11. Yang, F., et al., Gene delivery to human adult and embryonic cell-derived stem cells
using biodegradable nanoparticulate polymeric vectors. Gene Ther, 2009. 16(4): p. 533-
46.
12. Nauta, A., et al., Adipose-derived stromal cells overexpressing vascular endothelial
growth factor accelerate mouse excisional wound healing. Mol Ther, 2013. 21(2): p.
445-55.
13. Deveza, L., et al., Paracrine release from nonviral engineered adipose-derived stem cells
promotes endothelial cell survival and migration in vitro. Stem Cells Dev, 2013. 22(3):
p. 483-91.
14. Brunner, S., et al., Cell cycle dependence of gene transfer by lipoplex, polyplex and
recombinant adenovirus. Gene Ther, 2000. 7(5): p. 401-7.
15. Pyun, W.B., et al., Naked DNA expressing two isoforms of hepatocyte growth factor
induces collateral artery augmentation in a rabbit model of limb ischemia. Gene Ther,
2010. 17(12): p. 1442-52.
41

Figures
Figure 3.1. Synthesis of end-modified poly(β-amino esters) (PBAE). (A) Shows the synthesis
of C32-Ac by mixing 1,4-butanediacrylate (C) with 1-aminopentanol in 1.2:1 molar ratio, then
shows the synthesis of C32-122 by the addition of 122 in excess molar ratio to C32-Ac. (B)
Shows the synthesis of D32-122, which is synthesized in the same manner as C32-122, except
using 1,6 hexanediacrylate (D).
42

Figure 3.2. Effects of polymer on transfection and viability of human adipose-derived stem
cells (hADSCs), mouse adipose-derived stem cells (mADSCs), and human embryonic kidney
cells (HEK293T). (A) Images demonstrating the morphology and observable health of the cells
at 48 hours post-transfection with either C32-122 or D32-122. (B) MTS cell viability assay of
transfected cells at 48 hours. (C) Luciferase release from transfected cells.
43

Figure 3.3. Enhancement of hADSC proliferation and transfection in the presence of basic
fibroblast growth factor. (A) BrDu cell proliferation assay, (B) MTS cell viability assay, and
(C) Picogreen DNA content assay, on various isolations of hADSC at 48 hours with and without
10 ng/ml bFGF. (D) Fluorescence images of various isolations of ADSCs transfected with
EGFP with and without 10 ng/ml bFGF. (E) Luciferase release from various isolations of
ADSCs transfected with Gaussia Luciferase with and without 10 ng/ml bFGF.
44

Figure 3.4. Effects of culture conditions post-transfection. (A) MTS cell viability assay and
(B) luciferase release from hADSC-I3 transfected with Gaussia Luciferase and then cultured
with various concentrations of fetal bovine serum (FBS; 0%, 1%, 5%, 10%) and various
concentrations of bFGF (0 ng/ml, 1 ng/ml 5 ng/ml 10 ng/ml).
45

Figure 3.5. Transfection of cells with different plasmids encoding for multiple genes, including
vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), CXC chemokine
receptor 4 (CXCR4). (A-C) RT-PCR mRNA expression assessment of transfected cells for
VEGF (A), HGF (B), and CXCR4 (C). (D-E) ELISA protein release assessment of transfected
cells for (D) VEGF, and (E) HGF.
46

Figure 3.6. Effects of transfected cell paracrine release on human umbilical vein endothelial
cell (HUVEC) and human aortic smooth muscle cell (HASMC) viability. Conditioned medium
was collected from cells transfected with different plasmids at 48 hours and then applied to
HUVEC and HASMCs. (A) HUVEC and (B) HASMC cell viability was then assessed at 48
hours by MTS assay.
47

Table 3.1: List of plasmids used for transfection
Plasmid ID Genes encoded
pBlast-VEGF Vascular Endothelial Growth Factor 165
pCMV-VEGF Vascular Endothelial Growth Factor 165
pCK-HGF Hepatocyte Growth Factor 723 & 728
pCMV-CXCR4 CXC Chemokine Receptor 4
pCMV-CXCR4/VEGF CXC Chemokine Receptor 4 + Vascular Endothelial Growth Factor 165
pCK-CXCR4/HGF CXC Chemokine Receptor 4 + Hepatocyte Growth Factor 723 & 728
pGLUC Gaussia Luciferase
pCDNA-3.1 Blank Vector
Table 3.2: List of primers used for quantitative real time-polymerase chain reaction
Gene
Name Species Primer Sequence
hGAPDH Human Forward (5'-ACAGTCAGCCGCATCTTCTT-3')
Reverse (5'-CGACCAAATCCGTTGACTC-3')
mGAPDH Mouse Forward (5'-AACGACCCCTTCATTGAC-3')
Reverse (5'-TCCACGACATACTCAGCAC-3')
hCXCR4 Human Forward (5’- TTCTACCCCAATGACTTGTG-3’)
Reverse (5’- ATGTAGTAAGGCAGCCAACA-3’)
hVEGF Human Forward (5’-TACCTCCACCATGCCAAGTG-3’)
Reverse (5’-TGATGATTCDTGCCCTCCTCC-3’)
hHGF Human Forward (5'-CAATAGCATGTCAGGTGGAG-3')
Reverse (5'-CTGTGTTCGTGTGGTATCAT-3')
48

4. Paracrine Release from Non-viral Engineered Adipose-Derived Stem Cells Promotes
Endothelial Cell Survival and Migration in vitro
Lorenzo Deveza1,2, Jeffrey Choi3, Galym Imanbayev 4, Fan Yang PhD2,5,*
1School of Medicine, Stanford University, Stanford, CA; 2 Department of Bioengineering,
Stanford University, Stanford, CA; 3Department of Biology, Stanford University, Stanford,
CA; 4Department of Economics, Stanford University, Stanford, CA; 5Department of
Orthopaedic Surgery, Stanford University, Stanford, CA
Abstract
Stem cells hold great potential for therapeutic angiogenesis due to their ability to directly
contribute to new vessel formation or secrete paracrine signals. Adipose-derived stem cells
(ADSCs) are particularly attractive autologous cell sources for therapeutic angiogenesis due to
their ease of isolation and relative abundance. Gene therapy may be used to further enhance
the therapeutic efficacy of ADSCs by over-expressing desired therapeutic factors. Here we
developed VEGF overexpressing ADSCs utilizing poly(β-amino esters) (PBAE), a
biodegradable polymeric nanoparticle, and examined the effects of paracrine release from
non-viral modified ADSCs on the angiogenic potential of human umbilical vein endothelial
cells (HUVEC) in vitro. PBAE polymeric vectors deliver DNA into ADSCs with high
efficiency with low cytotoxicity, and led to over 3-fold in VEGF production by ADSCs
compared to Lipofectamine 2000. Paracrine release from PBAE/VEGF transfected ADSCs
enhanced HUVECs viability and decreased HUVEC apoptosis under hypoxia. Furthermore,
paracrine release from PBAE/VEGF transfected ADSCs significantly enhanced HUVEC
migration and tube formation, two critical cellular processes for effective angiogenesis. Our
results demonstrate that genetically engineered ADSCs using biodegradable polymeric vectors
may provide a promising autologous cell source for therapeutic angiogenesis in treating
cardiovascular diseases.
4.1 Introduction
Therapeutic angiogenesis is a promising treatment approach for patients suffering
from ischemic cardiovascular disease. This approach involves methods of regenerative
medicine to augment the host angiogenic response. Angiogenesis is the physiological process
of blood vessel growth from pre-existing vessels and is the body’s natural mechanism for
49

restoring blood perfusion to sites of ischemia. Stem cell-based therapies hold great promise for
therapeutic angiogenesis by either acting as cellular building blocks for new blood vessels or
by secreting paracrine signals to promote blood vessel growth [1-2]. The most well studied
cell sources for therapeutic angiogenesis include endothelial progenitor cells (EPCs) and bone
marrow-derived mesenchymal stem cells (BMMSCs) [3]. Despite the therapeutic efficacy of
these cell sources [3-7], their broad clinical application is largely limited due to requiring an
invasive cell isolation procedure with low cell yield, and the need for ex vivo cell expansion
prior to transplanting back to the patient.
Adipose-derived stem cells (ADSCs) represent a promising alternative cell source due
to their relative abundance, ease of isolation, and demonstrated potential for therapeutic
angiogenesis [8-9]. Indeed, when directly compared to BMMSCs, ADSCs were reported to
possess higher angiogenic potency demonstrating improved blood perfusion and a reduction in
infarct size two weeks post-transplantation in a mouse model of hindlimb ischemia [10].
Among the mechanisms by which stem cells contribute to angiogenesis, paracrine signaling is
regarded is being a key contributor [5,11]. In support of this, it was shown that direct
injection of conditioned medium (CM) from BMMSC versus BMMSC transplantation into a
rat model of myocardial ischemia led to similar reductions in infarct size and improvements in
ventricular function by 72 hours [12]. A broad spectrum of angiogenic cytokines were found
to be present in the conditioned medium of BMMSCs including vascular endothelial growth
factor (VEGF), fibroblast growth factor-2 (FGF-2), interleukin-6, and monocyte chemotactic
protein-1 (MCP-1) [6]. Similarly, ADSCs have been shown to release a number of angiogenic
and anti-apoptotic paracrine factors [13], which lead to enhanced new blood vessel formation
in vivo [13-18]. Despite the promise of stem cell-based therapies for therapeutic angiogenesis,
the efficacy of using stem cells alone remains limited due to the insufficient production of
therapeutic factors in vivo, as well as low cell engraftment and survival in ischemic tissues.
Gene therapy has the potential to improve stem cell-mediated therapeutic
angiogenesis by increasing the paracrine release of specific angiogenic factors. In fact, it has
been shown that transplantation of virally transduced BMMSCs overexpressing VEGF into rat
models of myocardial ischemia lead to enhanced blood vessel density and heart function in
comparison to MSCs alone [19]. Furthermore, it was demonstrated that overexpressing VEGF
reduced the number EPCs needed by 30-fold to achieve a similar therapeutic effect as
unmodified EPCs in promoting angiogenesis and limb salvage [20]. However, most studies so
far have relied on viral vectors for efficient gene transfer, which are limited by potential risks
50

of mutagenesis, carcinogenesis, and immunogenicity. In the pursuit of a safer alternative
approach, strategies utilizing non-viral vectors for therapeutic angiogenesis are beginning to
emerge [21]. Conventional non-viral delivery systems involve using positively charged
polymers which can condense negatively charged DNA into nanoparticles for cellular uptake.
Polyethylenimine and Lipofectamine are the most widely used polymer-based transfection
reagents for gene delivery, but their clinical application is limited due to non-degradability,
poor transfection efficiency and high toxicity [21]. Poly(β-amino esters) (PBAE) are a family
of hydrolytically biodegradable polymers, which can deliver DNA into human stem cells with
high efficiency and low toxicity [11,22]. We have previously reported that MSCs modified
using PBAE/VEGF nanoparticles lead to substantially enhanced angiogenesis and limb
salvage in a mouse hindlimb ischemia model [22]. However, the efficacy of non-viral
engineered ADSCs for therapeutic angiogenesis remains largely unknown.
In this study, we assess the effects of paracrine release from ADSCs modified
utilizing PBAE/VEGF nanoparticles on human umbilical vein endothelial cell (HUVEC)
behavior in vitro. We first developed biodegradable PBAE/VEGF nanoparticles that can
transfect ADSCs with high efficiency for VEGF overexpression. Conditioned medium from
transfected ADSCs were then applied to HUVECs to examine the effects of such paracrine
release on HUVEC viability and apoptosis under low oxygen (1% O2), HUVEC migration and
tube formation.
4.2 Materials and Methods
4.2.1 Cell isolation and culture
Fat tissue was obtained from the abdominal fat of a female patient who had undergone
a free flap breast reconstruction surgery at Stanford University. All procedures were approved
and guided under Stanford IRB protocol. The fat tissue was washed 2-3 times with Phosphate
buffered saline and digested at 37 °C for 30 min with 0.5 U/ml Blendzyme 3 (Roche
Diagnostics, Indianapolis, IN, USA). Enzyme activity was neutralized with DMEM,
containing 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA), 1%
Penicillin/Streptomycin. Cells were then filtered through a 100 µm nylon mesh to remove
cellular debris before seeding. Following the initial 48 hours of incubation at 37 °C and 5%
CO2, cells were washed with PBS and maintained in growth medium containing DMEM, 10%
FBS, 1% penicillin/ streptomycin and 10ng/ml of the basic fibroblast growth factor (FGF)
(PeproTech, Rocky Hill, NJ, USA). Once 85-95% confluent, the cells were passaged by
51

trypsin (0.05%) digestion and plated at a density of 5000-6000 cells per cm2 for further
expansion. Human umbilical vein endothelial cells (HUVECs) were obtained from Lonza
(Basal, Switzerland) and expanded utilizing endothelial growth medium (EGM-2) (Lonza).
Passage 4 HUVECs were utilized for all experiments.
4.2.2 Transfection
Human adipose-derived stem cells (ADSCs) were transfected with DNA plasmids
encoding vascular endothelial growth factor (pVEGF) (Aldevron, Fargo, ND, USA) or
enhanced green fluorescence protein (pEGFP) (ELIM Biopharm, Hayward, CA, USA).
Passage 4 ADSCs were seeded at 65 000 cells per well in clear 24-well plates or 10 400 cells
per well in clear 96 well plates and incubated over night prior to transfection. Cells were then
transfected utilizing poly (β-amino esters) (PBAE) C32-122 as previously reported [11].
Briefly, polyplexes were formed by mixing pDNA and PBAE polymers in 25 mM sodium
acetate. Polyplexes were added to the cell culture after 10 minutes of complexation. Cells
were then incubated with polyplexes for four hours, at which time cell medium was replaced.
Transfection parameters were optimized by varying pDNA loading dose (1 µg, 2 µg, 3 µg, 4.5
µg, 6 µg) and GFP expression was examined at 48 hours. Lipofectamine 2000 (Invitrogen)
was used as a positive control and non-transfected cells were included as a negative control.
GFP expression by transfected ADSCs was measured at 48 hours post-transfection utilizing
fluorescence-activated cell sorting on a BD Facscalibur (BD Biosciences, San Jose, CA,
USA). Dead cells were excluded from the analysis by staining with propidium iodide and
10,000 live cells per sample were included for analyses. The percentage of GFP positive cells
was quantified using Flowjo 7.6 software (Tree Star, Inc. Ashland, OR, USA). Cell viability
was determined 48 hours post-transfection using Cell Titer 96 Aqueous One Solution Cell
Proliferation Assay (MTS) (Promega, Madison, WI, USA), and the value was normalized
using non-treated cells as a control.
4.2.3 Preparation of conditioned medium
Conditioned medium (CM) was prepared by culturing ADSCs in 24 well plates (65
000/well) for 48 hours in serum-free endothelial basal medium (EBM) (Lonza). Three groups
were examined including: 1) PBAE-VEGF transfected ADSCs, 2) Lipofectamine-VEGF
transfected ADSCs and 3) non-transfected ADSCs. CM was collected at 48 hours and stored
at -80 °C. To examine the effects of hypoxia on VEGF release, ADSCs were cultured in
52

either low oxygen (1% O2) or normoxia (20% O2). CM from both groups was collected at 48
hours, and CM from ADSCs cultured under normoxia was also collected on days 4, 6, and 8 to
quantify VEGF release kinetics. VEGF in the supernatant was determined by enzyme linked
immunosorbent assay (ELISA) (Peprotech).
4.2.4 HUVEC survival under hypoxia
Passage 5 HUVECs were plated in either 96 well plates (11 200 cells/well) for MTS
assay or in gelatin-coated 8-well chamber slides (28 000 cells/well) for TUNEL staining.
HUVECs were cultured in serum-free medium for 24 hours prior to switching to conditioned
medium, when they were placed in a low oxygen chamber (1% O2) for 48 hours. HUVEC
viability was determined by MTS assay as described above. HUVEC apoptosis was
determined utilizing the DeadEnd Fluorometric TUNEL System (Promega), and mounted with
Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA, USA) for
counterstain. Fluorescent microscopic images were analyzed utilizing ImageJ software (NIH,
USA). Apoptosis was analyzed by determining the ratio of TUNEL positive cells to DAPI
positive cells.
4.2.5 HUVEC Migration
HUVEC migration was assessed in a modified boyden chamber (E&K Scientific,
Santa Clara, CA, USA). The upper wells were pre-treated with 50 µl of 10 µg/ml fibronectin
(Sigma-Aldrich, St. Louis, MO) and incubated at 4oC for 24 hours prior to HUVEC plating.
HUVECs were seeded onto the upper chamber (20 000 cells/well) and EBM was added to the
bottom chamber. Cells were allowed to settle for 24 hours at 37oC. EBM in the bottom wells
was then replaced with hADSC-CM in quadruplicate and cells were allowed to migrate for 6
hours. The cells were stained with Hemacolor (Merck, Darmstadt, Germany) and cell number
was quantified by microscopy per high powered field. Results were reported as a ratio of
migrated cells over total number of cells.
4.2.6 Tube Formation Assay
Tube formation was assessed by culturing HUVECs on BD Matrigel (BD
Biosciences). Prior to HUVEC seeding, 96-well microtiter plates were coated with 80 µl of
Matrigel per well and incubated for 1 hour. Passage 5 HUVECs were suspended in hADSC-
CM or serum-free EBM and plated in triplicate at 12 000 cells per well. The formation of
53

tube-like structures was examined at each hour up to 16 hours. Representative images were
taken and analyzed utilizing ImageJ software (NIH, USA). Tube formation was analyzed by
quantifying the number of branches per high powered field.
4.2.7 Statistical Analysis
All experiments were performed in triplicate and repeated for consistency. The results
were reported as mean ± standard error of the mean. Statistical analysis was performed using
the one way analysis of variance with Dunnett’s multiple comparison test as the post-test to
compare all the experimental groups with Lipofectamine 2000 as a positive control and non-
transfected cells as negative controls.
4.3 Results
4.3.1 Gene delivery efficiency into ADSCs using PBAE nanoparticles
Transfection efficiency of poly(β-amino esters) (PBAE) was first examined in human
adipose-derived stem cells (ADSCs) using a leading PBAE polymer C32-122 [11] and a
reporter DNA encoding enhanced-green fluorescence protein (pEGFP). Fluorescence
microscopy and flow cytometry confirmed gene delivery into ADSCs 48 hours post-
transfection (Figure 1). Increasing DNA doses from 1 µg to 2 µg significantly increased the
transfection efficiency from 6% to 26%, and further increase in DNA doses up to 6 µg did not
significantly change the transfection efficiency. The PBAE-mediated transfection also showed
4-fold higher efficiency compared to the positive control transfected using Lipofectamine
2000 (5.27%±0.7%) (Figure 2a). Meanwhile, all groups transfected with PBAE demonstrated
much higher cell viability compared to the Lipofectamine 2000 transfected group (88.6±6%
vs. 70.4±4%, p < 0.05) (Figure 2b). Varying DNA dose led to no statistically significant
change in cell viability among PBAE-transfected groups.
4.3.2 VEGF release from PBAE/VEGF transfected ADSCs
The effect of DNA dose on VEGF release from PBAE/VEGF transfected ADSCs was
examined by VEGF ELISA 48 hours post-transfection (Figure 3). ADSCs transfected using
PBAE with low DNA doses (1-3 µg DNA) or lipofectamine 2000 did not show significant
increase in VEGF production compared to non-transfected ADSC control. Increasing
PBAE/DNA dose to 4.5 µg led to enhanced VEGF production (5290±563 pg/106 cells) and
further increase was observed when DNA dose increased to 6 µg (6308±1810 pg/106 cells),
54

which was over 3-fold higher than Lipofectamine control (1726.25±46.25 pg/106 cells, p <
0.05) and non-transfected ADSC control (1188±133.75 pg/106 cells, p < 0.05) (Figure 3a).
Accumulated VEGF release overtime showed that the optimized PBAE transfected group
produced over 3-fold higher amount of VEGF than that of non-transfected ADSC controls,
while Lipofectamine only slightly increased VEGF production.
4.3.3 Effect of hypoxia on VEGF release and ADSC proliferation
To assess the effects of ischemia on ADSC fate, ADSCs were cultured in a hypoxia
chamber (1% O2) for 48 hours to mimic ischemic conditions in vivo. Outcomes were analyzed
for VEGF release and ADSC cell viability (Figure 4). Hypoxia treatment enhanced VEGF
release in non-transfected ADSC controls by over 200% when compared to culture under
normoxia. PBAE/VEGF transfection led to further increase in VEGF production, with the
highest VEGF concentrations observed for the 4.5 µg and the 6 µg DNA doses (Figure 4a).
The lowest cell number was observed in the Lipofectamine transfected group (~70%). Groups
transfected using PBAE demonstrated high viability (85%-95%) overall. Hypoxia treatment
led to further increase in cell number in all PBAE transfected groups except the 6 µg dose.
This increase is likely a combination of improved cell survival and increased cell proliferation.
All PBAE-transfected groups increased in cell number under hypoxia in comparison to the
normoxia-cultured, non-transfected ADSC controls (Figure 4b).
4.3.4 Effects of paracrine release on HUVEC viability and apoptosis under hypoxia
Conditioned medium from ADSCs (transfected or control) were collected and added
to human umbilical vein endothelial cell (HUVEC) culture under hypoxia to evaluate the
effects of paracrine signals from ADSCs on HUVEC viability and apoptosis (Figure 5).
Conditioned medium from all ADSC groups led to an increase in HUVEC viability
(87.3±3.9%) compared with EBM (69.6±5.1%, p < 0.05) (Figure 5a). However, no significant
difference in HUVEC viability was observed between groups treated with conditioned
medium from VEGF-overexpressing ADSCs or untransfected ADSCs. The effects of
paracrine release from ADSCs on HUVEC apoptosis was analyzed using TUNEL assay.
Conditioned medium from PBAE-transfected ADSCs led to a significant decrease in HUVEC
apoptosis (13.7±2.6%) compared to Lipo (33.2±2.4%, p < 0.05) and non-transfected controls
(39±5.3%, p < 0.05).
55

4.3.5 Effects of paracrine release on HUVEC migration and tube formation
We lastly assessed the effects of VEGF-overexpression on endothelial cell migration
and tube formation, two key steps involved in angiogenesis. HUVEC migration towards
ADSC paracrine release was assessed in a modified Boyden chamber (Figure 6). Conditioned
medium from non-transfected ADSC increased HUVEC migration (12.7±1%, p < 0.05)
compared to EBM (6.4±2%, p < 0.05), and conditioned medium from PBAE/VEGF-
transfected ADSCs led to further increase in HUVEC migration (16.8±1%). Similarly,
HUVEC formed homogeneous and interconnected tubular structures when exposed to
conditioned medium from PBAE/VEGF transfected ADSCs (49±4 branches per high powered
field) (Figure 7), whereas significantly fewer tubular structures were observed in HUVECs
exposed to conditioned medium from Lipo/VEGF-transfected ADSCs (31.7±1 branches per
high powered field, p < 0.05) or ADSCs alone (32.0±2 branches per high powered field, p <
0.05).
4.4 Discussion
Here, we demonstrated that poly(β-amino esters) (PBAE) are an effective non-viral,
polymeric vector for gene delivery to adipose-derived stem cells (ADSCs). VEGF-
overexpressing ADSCs utilizing biodegradable nanoparticles enhanced angiogenesis-related
cellular behavior, including endothelial cell survival under hypoxia, migration and tube
formation. Although a previous study has shown beneficial effects of VEGF-overexpressing
ADSCs on angiogenesis using a viral-based approach [23], the broad clinical translation of
viral-mediated gene delivery remains limited by safety concerns. Non-viral based gene
delivery is potentially safer, but often suffers from low transfection efficiency and high
toxicity. Our results showed that PBAE-nanoparticles are promising non-viral polymeric
vectors that could transfect ADSCs with 4-5 times higher transfection efficiency than
Lipofectamine 2000, a commercially available transfection reagent, and with higher cell
viability (Figure 2). Polyethylenimine (PEI) [24], nucleofection, and electroporation [25-26]
are other commonly used non-viral gene delivery methods, which may achieve higher
transfection efficiency than Lipofectamine 2000, but these suffer from high toxicity. PBAE
nanoparticles are able to achieve high transfection at little expense to cell viability, which
makes them a more attractive option to over-express desired therapeutic genes. PBAE-
mediated VEGF overexpression is transient (Figure 3), which may limit any negative effects
due to constitutive release of VEGF, such as excessive and aberrant vascularity. This
56

demonstrates that PBAE nanoparticles could be utilized to strategically overexpress known
therapeutic factors, thereby manipulating stem cells to augment their natural angiogenic
responses.
Given that ADSCs will be exposed to hypoxia when transplanted into ischemic
tissues, it is important to understand ADSC behavior under hypoxia to determine their
response post-transplantation. ADSCs are known to release several paracrine factors with
angiogenic effects [13,27]. Hypoxia has been shown to enhance paracrine release of
angiogenic factors by ADSCs or BMMSCs, which may induce therapeutic benefit both in
vitro and in vivo [27-30]. Paracrine release from ADSCs cultured under hypoxia were shown
to improve endothelial cell (EC) survival and such effects were attenuated by addition of anti-
VEGF blocking antibodies [30]. We examined the effects of hypoxia on ADSC VEGF release
and cell viability, and how such cellular responses changed upon up-regulating VEGF by
PBAE. Consistent with previous reports, our results showed that hypoxia led to an increase in
both ADSC VEGF release and cell proliferation compared with normoxia culture. VEGF-
overexpression further improved VEGF release by ADSCs due to hypoxia, demonstrating that
gene therapy and hypoxia can act synergistically. We also found that hypoxia enhanced
ADSC viability, which has been shown in several prior reports utilizing different assays
[13,27,30]. Endogenous ADSCs have been found to proliferate in situ due to ischemia at a
distant site, suggesting that ADSCs respond favorably to low oxygen tension [27]. These
findings support ADSCs as an excellent cell source for treating ischemic disease, due to their
ability to survive under hypoxia and enhance paracrine release of angiogenic factors. Though
most previous work has focused on the use of bone marrow-derived mesenchymal stem cells
(BMMSCs) for angiogenesis [6-7,12,22,31], ADSCs represent a more attractive alternative
cell source due to their relative abundance and ease of isolation. Both cell sources have been
reported to improve angiogenesis in vivo, but their paracrine release profiles are different.
While both cell types secrete VEGF, BMMSCs secrete more fibroblast growth factor (bFGF)
[6] and ADSCs more prominently secrete hepatocyte growth factor (HGF) and transforming
growth factor-β (TGF-β) [13].
Angiogenesis is the formation of new blood vessels from pre-existing vessels, which
involves endothelial cell proliferation, migration, and new tube formation. To determine the
effects of paracrine release from VEGF-overexpressing ADSCs on angiogenesis, we applied
conditioned medium from PBAE/VEGF transfected ADSCs to human umbilical vein
endothelial cells (HUVEC) and examined HUVEC cellular processes related to angiogenesis.
57

Prior studies assessed endothelial cell survival under low-serum (2-5%) containing medium
[4,7,28]. In our study, endothelial cells were cultured in serum-free EBM, which represents a
more challenging culture condition for endothelial cells. Our results confirmed hypoxia
exposure had detrimental effects on HUVECs, with decreased cell viability and markedly
increased cell apoptosis (Figure 5). Consistent with previous reports, here we found that
paracrine release from ADSCs alone enhanced HUVEC viability,[13,16] HUVEC migration
[6,30], and HUVEC tube formation [28,30]. PBAE/VEGF overexpression led to a marked
decrease in HUVEC apoptosis, with improvements in HUVEC migration and more
homogeneous tube formation. These results confirmed the beneficial effects of VEGF-
overexpression on HUVEC behavior. Addition of anti-VEGF blocking antibodies was
previously reported to reduce endothelial cell migration [30], again suggesting the functional
importance of VEGF. While previous study has also shown protective effects on endothelial
cells using VEGF supplementation, the dose of VEGF applied was two orders of magnitude
higher than that secreted by cells alone (4-10 ng/ml versus 0.08-0.1 ng/ml VEGF from cells
alone) [6], which may lead to undesirable excessive vascular formation. It is also worth noting
that Lipofectamine/VEGF transfection did not improve tube formation or HUVEC apoptosis
compared to ADSCs alone, which is likely the result of the low transfection efficiency using
Lipofectamine compared with PBAE nanoparticles (Figure 1). Overall, these results show
that PBAE-mediated non-viral gene delivery provides an effective strategy for overexpressing
therapeutic factors to induce HUVEC cellular processes related to angiogenesis.
4.5 Conclusion
In conclusion, we have shown that non-virally modified ADSCs using biodegradable
polymeric vectors have beneficial effects on HUVEC behavior in vitro. ADSCs are promising
autologous cell sources that can serve as delivery vehicles for therapeutic paracrine signals.
Biodegradable poly(β-amino esters) nanoparticles can deliver DNA into ADSCs with high
efficiency and low cytotoxicty. Paracrine release from VEGF-overexpressing ADSC improved
HUVEC survival, migration and tube formation, and markedly reduced HUVEC apoptosis.
The results of this study suggest that non-virally engineered ADSCs may be a promising
autologous cell source for therapeutic angiogenesis in cardiovascular disease.
58

Acknowledgements
The authors would like to thank American Heart Association National Scientist Development
Grant (10SDG2600001), Stanford Bio-X Interdisciplinary Initiative Program, Donald E. and
Delia B. Baxter Foundation and Stanford Medical Scholars Research Program for funding.
References
1. Potente M, H Gerhardt and P Carmeliet. (2011). Basic and therapeutic aspects of
angiogenesis. Cell 146:873-87.
2. Losordo DW and S Dimmeler. (2004). Therapeutic angiogenesis and vasculogenesis
for ischemic disease. Part I: angiogenic cytokines. Circulation 109:2487-91.
3. Losordo DW and S Dimmeler. (2004). Therapeutic angiogenesis and vasculogenesis
for ischemic disease: part II: cell-based therapies. Circulation 109:2692-7.
4. Gruber R, B Kandler, P Holzmann, M Vogele-Kadletz, U Losert, MB Fischer and G
Watzek. (2005). Bone marrow stromal cells can provide a local environment that
favors migration and formation of tubular structures of endothelial cells. Tissue Eng
11:896-903.
5. Kamihata H, H Matsubara, T Nishiue, S Fujiyama, Y Tsutsumi, R Ozono, H Masaki,
Y Mori, O Iba, E Tateishi, A Kosaki, S Shintani, T Murohara, T Imaizumi and T
Iwasaka. (2001). Implantation of bone marrow mononuclear cells into ischemic
myocardium enhances collateral perfusion and regional function via side supply of
angioblasts, angiogenic ligands, and cytokines. Circulation 104:1046-52.
6. Kinnaird T, E Stabile, MS Burnett, CW Lee, S Barr, S Fuchs and SE Epstein. (2004).
Marrow-derived stromal cells express genes encoding a broad spectrum of
arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through
paracrine mechanisms. Circ Res 94:678-85.
7. Potapova IA, GR Gaudette, PR Brink, RB Robinson, MR Rosen, IS Cohen and SV
Doronin. (2007). Mesenchymal Stem Cells Support Migration, Extracellular Matrix
Invasion, Proliferation, and Survival of Endothelial Cells In Vitro. STEM CELLS
25:1761-1768.
8. Zuk PA, M Zhu, H Mizuno, J Huang, JW Futrell, AJ Katz, P Benhaim, HP Lorenz
and MH Hedrick. (2001). Multilineage cells from human adipose tissue: implications
for cell-based therapies. Tissue Eng 7:211-28.
59

9. Schäffler A and C Büchler. (2007). Concise Review: Adipose Tissue-Derived Stromal
Cells—Basic and Clinical Implications for Novel Cell-Based Therapies. STEM
CELLS 25:818-827.
10. Kim YJ, HK Kim, HH Cho, YC Bae, KT Suh and JS Jung. (2007). Direct Comparison
of Human Mesenchymal Stem Cells Derived from Adipose Tissues and Bone Marrow
in Mediating Neovascularization in Response to Vascular Ischemia. Cellular
Physiology and Biochemistry 20:867-876.
11. Yang F, JJ Green, T Dinio, L Keung, SW Cho, H Park, R Langer and DG Anderson.
(2009). Gene delivery to human adult and embryonic cell-derived stem cells using
biodegradable nanoparticulate polymeric vectors. Gene Ther 16:533-46.
12. Gnecchi M, H He, OD Liang, LG Melo, F Morello, H Mu, N Noiseux, L Zhang, RE
Pratt, JS Ingwall and VJ Dzau. (2005). Paracrine action accounts for marked
protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med
11:367-8.
13. Rehman J, D Traktuev, J Li, S Merfeld-Clauss, CJ Temm-Grove, JE Bovenkerk, CL
Pell, BH Johnstone, RV Considine and KL March. (2004). Secretion of angiogenic
and antiapoptotic factors by human adipose stromal cells. Circulation 109:1292-8.
14. Cao Y, Z Sun, L Liao, Y Meng, Q Han and RC Zhao. (2005). Human adipose tissue-
derived stem cells differentiate into endothelial cells in vitro and improve postnatal
neovascularization in vivo. Biochemical and Biophysical Research Communications
332:370-379.
15. Kondo K, S Shintani, R Shibata, H Murakami, R Murakami, M Imaizumi, Y
Kitagawa and T Murohara. (2009). Implantation of Adipose-Derived Regenerative
Cells Enhances Ischemia-Induced Angiogenesis. Arteriosclerosis, Thrombosis, and
Vascular Biology 29:61-66.
16. Miranville A, C Heeschen, C Sengenes, CA Curat, R Busse and A Bouloumie. (2004).
Improvement of postnatal neovascularization by human adipose tissue-derived stem
cells. Circulation 110:349-55.
17. Moon MH, SY Kim, YJ Kim, SJ Kim, JB Lee, YC Bae, SM Sung and JS Jung.
(2006). Human adipose tissue-derived mesenchymal stem cells improve postnatal
neovascularization in a mouse model of hindlimb ischemia. Cell Physiol Biochem
17:279-90.
60

18. Rubina K, N Kalinina, A Efimenko, T Lopatina, V Melikhova, Z Tsokolaeva, V
Sysoeva, V Tkachuk and Y Parfyonova. (2009). Adipose stromal cells stimulate
angiogenesis via promoting progenitor cell differentiation, secretion of angiogenic
factors, and enhancing vessel maturation. Tissue Eng Part A 15:2039-50.
19. Matsumoto R, T Omura, M Yoshiyama, T Hayashi, S Inamoto, KR Koh, K Ohta, Y
Izumi, Y Nakamura, K Akioka, Y Kitaura, K Takeuchi and J Yoshikawa. (2005).
Vascular endothelial growth factor-expressing mesenchymal stem cell transplantation
for the treatment of acute myocardial infarction. Arterioscler Thromb Vasc Biol
25:1168-73.
20. Iwaguro H, J Yamaguchi, C Kalka, S Murasawa, H Masuda, S Hayashi, M Silver, T
Li, JM Isner and T Asahara. (2002). Endothelial progenitor cell vascular endothelial
growth factor gene transfer for vascular regeneration. Circulation 105:732-8.
21. Park HJ, F Yang and SW Cho. (2012). Nonviral delivery of genetic medicine for
therapeutic angiogenesis. Adv Drug Deliv Rev 64:40-52.
22. Yang F, S-W Cho, SM Son, SR Bogatyrev, D Singh, JJ Green, Y Mei, S Park, SH
Bhang, B-S Kim, R Langer and DG Anderson. (2010). Genetic engineering of human
stem cells for enhanced angiogenesis using biodegradable polymeric nanoparticles.
Proceedings of the National Academy of Sciences 107:3317-3322.
23. Jabbarzadeh E, T Starnes, YM Khan, T Jiang, AJ Wirtel, M Deng, Q Lv, LS Nair, SB
Doty and CT Laurencin. (2008). Induction of angiogenesis in tissue-engineered
scaffolds designed for bone repair: a combined gene therapy-cell transplantation
approach. Proc Natl Acad Sci U S A 105:11099-104.
24. Ahn HH, JH Lee, KS Kim, JY Lee, MS Kim, G Khang, IW Lee and HB Lee. (2008).
Polyethyleneimine-mediated gene delivery into human adipose derived stem cells.
Biomaterials 29:2415-2422.
25. Lopatina T, N Kalinina and E Parfyonova. (2009). Nonviral Transfection of Adipose
Tissue Stromal Cells: An Experimental Study. Bulletin of Experimental Biology and
Medicine 147:509-512.
26. Zaragosi L-E, N Billon, G Ailhaud and C Dani. (2007). Nucleofection Is a Valuable
Transfection Method for Transient and Stable Transgene Expression in Adipose
Tissue-Derived Stem Cells. STEM CELLS 25:790-797.
27. Thangarajah H, IN Vial, E Chang, S El-Ftesi, M Januszyk, EI Chang, J Paterno, E
Neofytou, MT Longaker and GC Gurtner. (2009). IFATS Collection: Adipose Stromal
61

Cells Adopt a Proangiogenic Phenotype Under the Influence of Hypoxia. STEM
CELLS 27:266-274.
28. Hung S-C, RR Pochampally, S-C Chen, S-C Hsu and DJ Prockop. (2007). Angiogenic
Effects of Human Multipotent Stromal Cell Conditioned Medium Activate the PI3K-
Akt Pathway in Hypoxic Endothelial Cells to Inhibit Apoptosis, Increase Survival,
and Stimulate Angiogenesis. STEM CELLS 25:2363-2370.
29. Kinnaird T, E Stabile, MS Burnett, M Shou, CW Lee, S Barr, S Fuchs and SE
Epstein. (2004). Local delivery of marrow-derived stromal cells augments collateral
perfusion through paracrine mechanisms. Circulation 109:1543-9.
30. Nakagami H, K Maeda, R Morishita, S Iguchi, T Nishikawa, Y Takami, Y Kikuchi, Y
Saito, K Tamai, T Ogihara and Y Kaneda. (2005). Novel Autologous Cell Therapy in
Ischemic Limb Disease Through Growth Factor Secretion by Cultured Adipose
Tissue–Derived Stromal Cells. Arteriosclerosis, Thrombosis, and Vascular Biology
25:2542-2547.
31. Burdon TJ, A Paul, N Noiseux, S Prakash and D Shum-Tim. (2011). Bone marrow
stem cell derived paracrine factors for regenerative medicine: current perspectives and
therapeutic potential. Bone Marrow Res 2011:207326.
62

Figures
Figure 4.1. Efficiency of gene delivery to human adipose-derived stem cells (hADSC)
utilizing poly(β-amino esters) (PBAE) nanoparticles. Under leading transfection conditions,
PBAE (a) delivered eGFP DNA into hADSCs with 4-times the efficiency of Lipofectamine
2000 (b). Fluorescence microscopy showed markedly increased GFP signals in hADSCs
transfected using PBAE nanoparticles (d) compared to cells transfected using Lipofectamine
(e). Scale bar: 500 µm.
63

Figure 4.2 Effects of nanoparticle dose on transfection efficiency and cell viability. (a)
Poly(β-amino esters) (PBAE) led to significantly increased transfection efficiency in human
adipose-derived stem cells (hADSCs) using doses as low as 2 µg/well compared to
Lipofectamine 2000 (Lipo) (# p < 0.001 compared to Lipo). (b) Cells transfected using PBAE
at all doses demonstrated high cell viability, which were about 1 fold higher compared to the
cells transfected using Lipo (# p < 0.01 versus Lipo, * p < 0.05 versus Cells only).
64

Figure 4.3. VEGF release from human adipose-derived stem cells (hADSCs) after
transfection using polymeric nanoparticles. (a) VEGF release from PBAE-modified hADSCs
increased with DNA dose and reached a maximum at DNA dose of 6 µg, which was
significantly higher than groups transfected using Lipofectamine 2000 (Lipo) (# p < 0.05) and
cells alone (* p < 0.05). (b) Accumulated VEGF release showed continuously enhanced
VEGF release from cells transfected using PBAE/VEGF nanoparticles compared to Lipo
control.
65

Figure 4.4 Effects of hypoxia on VEGF production and cell viability of non-viral engineered
ADSCs. (a) Hypoxia (1% O2 for 48 hrs) enhanced VEGF release from non-viral engineered
human adipose-derived stem cells in a dose-dependent manner. (b) Hypoxia treatment led to
an increase in ADSC viability in most PBAE transfected groups, except in the groups
transfected with 6 µg PBAE/DNA or Lipofectamine 2000 (Lipo).
66

Figure 4.5. Effects of paracrine signals from non-viral engineered ADSCs on endothelial cell
viability and apoptosis. (a) Paracrine release from hADSCs significantly improved HUVEC
viability under hypoxia (* p < 0.05 versus EBM), with no significant difference in cell
viability due to VEGF overexpression. (b) Paracrine release from hADSCs markedly
decreased HUVEC apoptosis under hypoxia, and PBAE/VEGF transfected group led to the
greatest decrease in cell apoptosis (* p < 0.05 versus EBM, # p < 0.05 versus Lipo
VEGF/hADSCS-CM). (c-f) Representative images of TUNEL stained endothelial cells
undergoing apoptosis. Scale bar 200 µm.
67

Figure 4.6. Paracrine release from PBAE/VEGF engineered human adipose-derived stem
cells (hADSCs) enhanced endothelial cell migration. (a-c) Representative images of migrated
endothelial cells in response to conditioned medium from hADSCs or EBM (negative
control). (d). Percentage of migrated endothelial cells towards conditioned medium (* p < 0.05
versus EBM only, # p < 0.05 versus hADSC-CM only).
68

Figure 4.7. Paracrine release from PBAE/VEGF engineered human adipose-derived stem
cells (hADSCs) enhanced endothelial cell tube formation. (a-d). Conditioned medium from
PBAE/VEGF transfected hADSCs led to formation of interconnected tubular structures by
endothelial cells (scale bar: 500 µm). (e) The number of branch points formed by endothelial
cells in response to conditioned medium from all groups. (* p < 0.05 versus EBM, # p < 0.05
compared to Lipo).
69

70
5. CXCR4-overexpressing Stem Cells Enhance Tissue Regeneration
in a Mouse Model of Hindlimb Ischemia
Lorenzo Deveza1,2, Jeffrey Choi3, Jerry Lee4, Ngan Huang5, John Cooke4, Fan Yang1,6*
1Department of Bioengineering, Stanford University, Stanford, CA; 2Stanford University School
of Medicine, Stanford, CA; 3Department of Biology, Stanford University, Stanford, CA;
4Division of Cardiovascular Medicine, Stanford University, Stanford, CA; 5Department of
Cardiovascular Surgery, Stanford University, Stanford, CA;6Department of Orthopaedic
Surgery, Stanford University, Stanford, CA.
Abstract
Peripheral arterial disease (PAD) is an important disease caused by a blockage of blood flow to
the limbs. Treatment often involves surgical revascularization, but many patients possess co-
morbidities which prohibit their treatment. Stem cell-based therapeutics offer an alternative,
which may promote angiogenesis in ischemic tissues. However, current approaches have met
with variable clinical success, which might be attributed to insufficient paracrine signals or poor
cellular engraftment into regenerating tissues. To overcome these limitations, we developed
CXC chemokine receptor 4 (CXCR4) overexpressing adipose-derived stem cells (ADSCs)
using biodegradable nanoparticles. CXCR4 is a well-known receptor involved in stem cell
targeting to sites of ischemia, which we hypothesized would promote ADSCs as drug delivery
vehicles to ischemia. CXCR4-overexpressing ADSCs led to a robust healing response in murine
models of hindlimb ischemia, leading to 100% limb salvage that was associated with early
reperfusion and muscle repair. CXCR4- overexpressing ADSCs also improved cell engraftment
and survival over 2 weeks compared to transfection controls. Assessement of CXCR4-ADSCs
in vitro revealed that CXCR4-overexpression induced a pro-angiogenic and anti-inflammatory
phenotype in ADSCs. These results suggest that non-virally engineered ADSCs overexpressing
CXCR4 are a promising strategy for treating PAD.
5.1 Introduction
Peripheral arterial disease (PAD) represents a large, yet underrepresented medical
problem worldwide. It is caused by a reduction in blood flow to the limbs, and usually signifies
advanced underlying atherosclerotic disease [1, 2]. Poor blood flow by PAD can cause
significant leg pain when walking, and in severe cases lead to non-healing ulcers or limb

71
amputation. Treating PAD generally involves surgical revascularization [1, 2]. Unfortunately,
many patients suffering from PAD are unable to withstand surgery. Therapeutic angiogenesis
using stem cells offers a valuable alternative for treating PAD by secreting paracrine signals to
promote blood vessel growth in situ [3]. However, stem cell-based approaches have met with
variable clinical success, which might be attributed to poor cell engraftment and an insufficient
paracrine response.
Combination stem cell-gene therapy can overcome many of these limitations by
overexpressing genes to enhance relevant cellular processes, such as cell migration and
angiogenic growth factor release. Previously, we demonstrated that vascular endothelial growth
factor (VEGF)-transfected bone marrow-derived mesenchymal stem cells (BM-MSCs) could
improve therapeutic responses in a murine model of hindlimb ischemia [4]. Notably,
transfection was achieved utilizing an optimized biodegradable polymeric nanoparticle for non-
viral gene transfection, namely poly(β-amino ester) (PBAE), which is an important
characteristic for clinical translation [5]. However, the use of VEGF and BM-MSCs may not
be ideal, as VEGF can lead to leaky vasculature and promote inflammation, while BM-MSCs
are difficult to obtain from the patient and suffer low yields and donor site morbidity. Further
developments of combined stem cell-gene therapy for clinical translation include: 1) the
identification of therapeutic gene targets that can promote cell engraftment and enhance
paracrine signaling; as well as, 2) the proper selection of a reliably abundant autologous cell
population.
CXC chemokine receptor 4 (CXCR4) is a G-protein coupled receptor known to be
involved in cell homing towards ischemia, and may potentially be involved in other cellular
processes, such as cell survival and growth factor release [6]. Physiologically, CXCR4 is
expressed on progenitor and inflammatory cells, which localize to hypoxic tissues to participate
in physiologic revascularization and tissue repair [7]. The ligand for CXCR4 is stromal derived
factor-1α (SDF-1α), which is secreted under the action of the hypoxia inducible factor-1α
transcription factor and is mainly expressed in hypoxic tissues [8-10]. Due to its physiologic
involvement in hypoxic signaling, CXCR4 is a promising homing factor to overexpress in stem
cells to promote their role as drug delivery vehicles to ischemia.
With regards to cell source, adipose derived stem cells (ADSCs) represent a relatively
abundant, autologous cell population with similar lineage differentiation potential to BM-
MSCs. Clinically, ADSCs are easier to isolate and have high yields. For therapeutic purposes,
ADSCs have shown benefits in models of hindlimb ischemia [11, 12] and myocardial ischemia

72
[13], and are known to secrete a number of proangiogenic and anti-apoptotic cytokines [14].
Previously, we engineered ADSCs using PBAE nanoparticles to overexpress VEGF and found
that their paracrine release could enhance endothelial cell processes of angiogenesis in vitro
[15].
In the present study, we aimed to evaluate the potential of CXCR4-overexpressing
ADSCs using PBAE nanoparticles for enhancing therapeutic angiogenesis in a murine model
of hindlimb ischemia (figure 1A). We hypothesized that CXCR4 overexpression would enable
cell survival and localization to the ischemic environment. Therapeutic efficiency of genetically
engineered ADSCs was assessed by evaluating cell survival and engraftment, angiogenic and
inflammatory response, and muscle morphology.
5.2 Materials and Methods
5.2.1 Materials
Monomers for synthesizing poly(β-amino esters)were purchased from Sigma–Aldrich,
Scientific Polymer Products, and Molecular Biosciences. Anhydrous dimethyl sulfoxide
(DMSO) was purchased from Sigma–Aldrich. A 25 mM sodium acetate (NaOAc) buffer
solution (pH 5.2) was prepared by diluting a 3 M stock solution (Sigma-Aldrich, St Louis, MO).
Plasmids (pEGFP-N1 and pcDNA3.1(+)-CXCR4) were purchased from Elim
Biopharmaceuticals and pVEGF165 plasmid was obtained from Aldevron.
5.2.2 Poly(β-Amino Esters) Synthesis
PBAE polymers were synthesized as previously described [4]. Briefly, acrylate-
terminated C32 polymer (C32-Ac) was prepared by mixing butanediol (C) and aminopentanol
(32) monomers using a 1.2:1.0 diacrylate/amine monomer molar ratio at 90 °C for 24 h.
Subsequently, amine-terminated C32 polymers were generated by reacting C32-Ac with
diamine monomer (122) in DMSO. End-capping reactions were performed overnight at room
temperature by using a 1.6-fold molar excess of amine over acrylate end groups.
5.2.3 ADSC isolation and culture
ADSCs were collected by harvesting the inguinal fat pads from 5 to 6 weeks old
luciferase-positive/GFP-positive transgenic male mice (Joseph Wu, MD, PhD laboratory,
Stanford University) and FVB wild-type male mice (purchased from Charles River, Willington,
MA). Following serial betadine washes, the fat pads were finely minced in cold, sterile PBS,

73
followed by digestion in 0.075% Collagenase II (Sigma-Aldrich) at 37 °C for 30 minutes in a
shaking water bath. Digestion was neutralized with Dulbecco’s modified Eagle’s medium
(Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen) and 1%
penicillin-streptomycin (Sigma-Aldrich). Neutralized cells were centrifuged at 4 °C and 1,000
rpm for 5 minutes, and floating adipocytes and media were removed before pelleted ADSCs
from the stomal-vascular portion were resuspended in culture media, passed through a 100 μm
Falcon nylon cell strainer (Becton Dickinson, Bedford, MA) and plated on a 10 cm dish. Cells
were incubated at 37 °C and 5% atmospheric CO2. Twenty-four hours after isolation, the cells
were washed in PBS to remove debris and nonadherent cells, and fresh medium was added to
the cells. Medium was changed every 2 days, and adherent spindle-shaped cells were grown to
subconfluence, passaged using standard methods of trypsinization, and propagated through
passage 2.
5.2.4 Transfection
ADSCs were transfected with CXCR4 plasmid, VEGF plasmid, or control plasmid
(EGFP) in DMEM containing 10% FBS using optimized PBAE transfection conditions [15].
PBAE nanoparticles were formulated by mixing C32-122 PBAE polymers with plasmids at 30:1
polymer weight to DNA weight ratio in 25 mM NaAc and incubuating for 10 minutes. PBAE
nanoparticles were then added to ADSCs cultured in DMEM containing 10% FBS. After 4 h
incubation, the medium was replaced with fresh growth medium. For transfection
characterization assays, ADSCs were transfected at 3.0 x 105 cells per well in 6 well plates. For
therapeutic animal studies, ADSCs were transfected in T150 tissue culture flasks at 3.5 x 106
cells.
5.2.5 Flow cytometric analysis of ADSC transfection efficiency and CXCR4 cell surface
expression
To verify transfection efficiency by PBAE nanoparticles, GFP expression by
transfected WT ADSCs was measured at 48 h post transfection utilizing fluorescence-activated
cell sorting (FACS) on a BD FACScalibur (BD Biosciences, San Jose, CA). Dead cells were
excluded from the analysis by staining with propidium iodide and 10,000 live cells per sample
were included for the analysis. The percentage of GFP-positive cells was quantified using
Flowjo 7.6 software (Tree Star, Inc.,Ashland, OR).

74
5.2.6 Quantitative real-time polymerase chain reaction (RT-PCR) in vitro
To confirm successful transfection of human CXCR4 and human VEGF by PBAE in
vitro, total RNA was harvested from ADSCs at 48 hours post-transfection using RNeasy Kit
(Qiagen, Germantown, MD, USA). cDNA was synthesized by reverse transcription using the
Superscript First-Strand Synthesis System (Invitrogen) and 1 μg of total RNA. Quantitative
PCR was performed using Power SYBR Green PCR Master Mix following the manufacturer's
protocol and running all samples for 40 PCR cycles on Applied Biosystems 7900 Real-Time
PCR System (Carlsbad, CA, USA). The relative expression level of target genes was
determined by the ΔΔCT method, where target gene expression was first normalized to an
endogenous housekeeping gene, GAPDH, followed by a second normalization to the gene
expression level measured in untransfected control cells. Genetic upregulation of mouse
angiogenic and inflammatory genes was confirmed by isolating total RNA from ADSCs
cultured under 1% O2 conditions for 48 hours then proceeding through cDNA synthesis and RT-
PCR. Measurement of mRNA extracted from ischemic muscle was performed by digesting
muscle utilizing TRIzol Reagent (Invitrogen) and proceeding through RNA extraction, cDNA
synthesis, and RT-PCR as described above. The utilized primers are listed in Table S1.
5.2.7 Measurement of VEGF production by Enzyme-Linked Immunosorbent Assay
(ELISA)
VEGF production from ADSCs was assessed by collecting conditioned medium and
measured using an ELISA kit for human VEGF (Peprotech, Rocky Hill, NJ) according to the
manufacturer’s protocol. VEGF production was measured on days 2, 4, 6, 8. Accumulated
VEGF release was calculated by quantifying the total amount of VEGF released from all
previous time points. The level of secreted VEGF in vitro was expressed as the amount of
VEGF per milliliter of medium supernatant.
5.2.8 Determination of CXCR4 upregulation by Western Blot
CXCR4 protein production was determined by Western blot method. Total protein was
isolated utilizing RIPA buffer (Thermo Scientific, Waltham, MA). Proteins were then separated
by gel electrophoresis utilizing NuPage Novex Bis-Tris Mini Gels (Invitrogen) at 200 V for 35
minutes. Separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane
(Invitrogen) utilizing the XCell II Blot Module (Invitrogen) at 30 V for 1 hour. To assess for
CXCR4, blotted PVDF membranes were stained anti-human CXCR4 (Abcam, Cambridge,

75
MA), utilizing a HRP-labeled secondary antibody (Abcam). Staining for anti-β actin (Abcam)
served as a loading control. X-ray films were developed utilizing the chemiluminescent reagent,
SuperSignal West Pico (Thermo Scientific).
5.2.9 Cell viability/proliferation assay
Cell viability or proliferation following transfection was evaluated at 48 hours post-
transfection using the Cell Titer 96 Aqueous One Solution (MTS) assay kit (Promega,
Fitchburg, WI) to measure metabolic activity. Results were expressed as a percentage of optical
absorbance values obtained for nontreated cells.
5.2.10 Transplantation of stem cells in mouse model of hindlimb ischemia
All experiments were performed in accordance with the Stanford University Animal
Care and Use Committee Guidelines and approved APLAC protocols. Hindlimb ischemia was
induced in female FVB mice age 10-12 weeks (Charles River) as previously described [16].
Briefly, the femoral artery was ligated at two sites (distal to the external iliac artery and proximal
to the popliteal artery) through a skin incision with 5-0 silk suture (Ethicon). The femoral artery
was excised between the points of ligation. After arterial dissection, cells (1.0 x 106 cells per
injection) were suspended in 100 µL of PBS and injected intramuscularly into two sites (the
adductor muscle region in the medial thigh and the gastrocnemius) using a 29-gauge tuberculin
syringes. Four experimental groups (n = 10 per group) were examined as follows: (i) CXCR4-
ADSC, (ii) CXCR4/VEGF-ADSC, (iii) CXCR4/VEGF-ADSC, and (iv) GFP-ADSC
transfection control. Physiological status of ischemic limbs was followed up to 4 weeks after
treatment. All of the animals were killed at the 4-week time point, and limbs of the ischemic
sides were retrieved for analyses.
5.2.11 Bioluminescent Imaging (BLI)
ADSC survival was monitored post-injection by BLI over the course of two weeks. BLI
was performed on the day of surgery, and on days 1, 3, 4, 7, 10, 14. Animals were anesthetized
by 2–3% inhaled isoflurane and injected intraperitoneally with the reporter probe D-Luciferin
at 100 mg/kg body weight. IVIS Spectrum system (Xenogen, Alameda, CA) was used to image
the animals while under 2% inhaled isoflurane anesthesia. Each animal was scanned until the
peak signal was reached, recording radiance of regions of interest in photons. Radiance was
quantified in photons per second per centimeter squared per steridian.

76
5.2.12 In vivo assessment of clinical limb salvage and limb reperfusion
Ischemic limbs were evaluated by direct observation of clinical limb status and by
measurement of blood reperfusion. Limbs were assessed every week up to 4 weeks. Grading
of limb ischemia was based on the severity of limb ischemia utilizing following categories: 1)
limb salvage; 2) toe necrosis; 3) foot necrosis; 4) foot loss; and 5) limb loss. Blood reperfusion
in ischemic limbs was measured using a PeriScan PIM3 laser Doppler system (Perimed AB,
Sweden), as previously described [16]. Briefly, each animal was pre-warmebd to a body
temperature of 36°C. ROIs were drawn around the entire hindlimbs and the level of perfusion
in ischemic and normal hindlimbs were quantified using the mean pixel value within each ROI.
Relative changes in blood flow were expressed as the ratio of the ischemic limb to the normal
limbs.
5.2.13 Histology, Immunofluorescence, Capillary Densitometry, and Macrophage
Quantification
Mice were euthanized at 4 days and 28 days to retrieve muscle tissue from ischemic
limbs for histological analysis. Specimens were embedded in optimal cutting temperature
(O.C.T.) for cryosectioning. Sections were cut at 8 µm thickness. General tissue morphology
was assessed with Masson’s Trichrome Staining. Immunofluorescence was performed to assess
morphologic features of angiogenesis and inflammation. Transplanted GFP-positive/luciferase-
positive cells were detected in day 28 sections with anti-GFP (Invitrogen). Capillaries were
detected with anti-CD31 (BD Biosciences, San Jose, CA). Macrophages were detected with
anti-CD68 (AbD Serotec, Oxford, UK). Sections were mounted with Vectashield Mounting
Medium with 4’,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA)
for counterstain. Sections were imaged using a fluorescent microscope. The number of CD31-
positive vessels was quantified to estimate capillary density per high powered field (hpf) using
ImageJ software (NIH). Quantitative analysis of macrophages was performed by determining
the number of DAPI-positive cells that overlap CD68-positive signals and reported as a
percentage of total DAPI-positive cells utilizing MATLAB software (MathWorks, Natick, MA).
5.2.14 Statistical Analysis
GraphPad Prism (Graphpad Software, San Diego, CA, USA) was used to perform all
statistical analyses. One-way analysis of variance with Tukey's multiple comparison test as a

77
post-hoc test was performed to compare all experimental groups and determine statistical
significance. All quantitative data were expressed as mean ± standard error of the mean.
5.3 Results
5.3.1 Successful overexpression of CXCR4 and VEGF in ADSCs utilizing biodegradable
polymeric nanoparticles
To generate CXCR4- and/or VEGF- overexpressing ADSCs, PBAE nanoparticles were
applied to ADSCs ex vivo (figure 1A). Genetic overexpression was then verified by RT-PCR
and ELISA or Western blot. Initially, transfection was performed using WT mouse ADSCs
with the reporter gene enhanced green fluorescent protein (EGFP), which verified that PBAE
nanoparticles improved transfection efficiency (19.9±2.0%) compared to Lipo controls
(5.4±0.8%, p < 0.05) (figure S1A). For all remaining experiments, transfections were carried
out in luciferase positive/GFP positive (luc+/GFP+) mouse ADSCs; these cells enabled live cell
tracking during animal experiments.
RT-PCR and Western blot confirmed successful CXCR4-overexpression in ADSCs
using PBAE nanoparticles (figure 1B, 1C). As CXCR4 is a transmembrane receptor, cell
surface expression was quantified by flow cytometry (FACS). Cell surface expression of
CXCR4 was statistically greater than GFP-transfected controls (figure 1D); however, the
increased cell surface expression was subtle (1.41±0.02% vs 0.9±0.11%, p < 0.05) suggesting
that CXCR4 is predominantly intracellular as supported by previous studies [17]. To understand
the kinetics of CXCR4 overexpression, CXCR4 mRNA levels were quantified over 10 days.
This demonstrated that CXCR4 expression peaked at 48 hours post-transfection and remained
high over 8 days (figure 1E). VEGF-overexpression was similarly confirmed by RT-PCR
(figure 1F) and VEGF ELISA (figure 1G). As with CXCR4 expression, VEGF release peaked
at 48 hours and was significantly higher in VEGF-transfected ADSCs (2.41±0.19 ng/ml) and
CXCR4/VEGF- co-transfected ADSCs (0.69±0.09 ng/ml) compared to GFP-transfected
controls (0.25±0.4 ng/ml, p < 0.05) (figure 1G). By 10 days post-transfection, accumulated
VEGF release was over three-times greater than non-transfected controls (figure 1H). Finally,
it was demonstrated that cell viability due to PBAE-mediated transfection was not significantly
different compared to non-transfected controls.

78
5.3.2 Transplanted ADSCs overexpressing CXCR4 exhibited prolonged cell survival
To assess the effects of non-viral engineered, CXCR4- overexpressing ADSCs on cell
survival, GFP+/luc+ ADSCs were injected into the medial muscles of a murine model of
hindlimb ischemia and monitored by bioluminescence imaging (BLI) (figure 2A, 2B). GFP-
transfected ADSCs were utilized as a control for PBAE-mediated transfection. BLI
demonstrated that cell engraftment of CXCR4-, VEGF-, and both CXCR4/VEGF- transfected
ADSCs was significantly greater than that of GFP-transfected controls (13.00±6% combined
for CXCR4, VEGF and CXCR4/VEGF groups vs 6.11±3% for GFP group, p < 0.05) (figure
S2A). For all groups, BLI signal reached a peak at day 4, indicating that the transplanted cells
were able to recover and proliferate (figure 2B). At day 7, BLI signal for all groups began to
decline, but CXCR4- and CXCR4/VEGF- transfected groups remained relatively high
(11.20±8.8% combined for CXCR4 and CXCR4/VEGF) compared to the VEGF- and GFP-
transfected groups (2.96±2.6% combined for VEGF and GFP, p < 0.05) (figure 2B, S2B). By
day 10, BLI signal was only detected in CXCR4 and CXCR4/VEGF groups (figure 2A, 2B),
thereby indicating improved cell survival due to CXCR4-overexpression. To confirm genetic
upregulation of CXCR4 or VEGF in transplanted ADSCs, ischemic tissue sections were
harvested on day 4 and analyzed by RT-PCR. Genetic upregulation of CXCR4 and/or VEGF
was confirmed in their respective groups (figure 2C), which verified the cell survival of
transplanted ADSCs to day 4.
5.3.3 Improved therapeutic efficacy due to CXCR4-overexpressing ADSCs
Therapeutic efficacy was monitored by observation of clinical limb outcome and
functional blood reperfusion. Importantly, CXCR4-overexpression led to 100% clinical limb
salvage (n = 8), which was the best therapeutic response of all groups (figure 2D, 2I). Co-
transfection with CXCR4/VEGF also led to improved clinical limb salvage compared to VEGF-
and GFP-transfected groups, albeit with two mice suffering toe necrosis and one mouse
suffering foot necrosis. Interestingly, VEGF-transfection led to 50% limb salvage, which was
greater than controls, but also led to the highest amount of limb loss. With regards to functional
blood reperfusion, laser Doppler perfusion imaging (LDPI) was performed weekly (figure 2E,
2G). LDPI measurements performed on the day of surgery confirmed successful induction of
hindlimb ischemia (figure 2G). By day 7, the CXCR4-overexpressing groups demonstrated a
robust reperfusion response, including CXCR4- and CXCR4/VEGF- groups, compared to
VEGF- and GFP- transfected groups (p < 0.05) (figure 2G, S3). Thereafter, relative reperfusion

79
levels were not statistically different from each other, indicating that the host regenerative
response may have taken over.
5.3.4 Improvement of muscle regeneration in ischemic hindlimbs due to CXCR4-
overexpressing ADSCs
Muscle histology was performed to determine the health of the ischemic hindlimbs at 4
weeks post-ischemic induction (figure 3A). Sections were stained with Masson’s Trichrome
and assessed for muscle regeneration or damage (fibrosis and atrophy). Several regenerating
muscle fibers were observed in the CXCR4- and CXCR4/VEGF- groups, as indicated by the
presence of centrally located nuclei. This suggested that these tissues had reached late stage
muscle repair. In contrast, there were few regenerating muscle fibers noted in the VEGF-
groups; instead, frank fibrosis was observed throughout. Similarly, much fibrosis and muscle
atrophy was observed in GFP- groups. Considering that this was a 4 week time point, it is likely
that these tissues had undergone chronic fibrosis.
5.3.5 Effects of CXCR4- and/or VEGF- overexpressing ADSCs on angiogenic and
inflammatory response
As angiogenesis and inflammation are important processes in muscle repair, ischemic
tissue sections were immunostained for capillaries (anti-CD31/PECAM) and macrophages
(anti-CD68). Transplanted cells were immunostained with anti-GFP antibody to determine the
location of their long-term engraftment (figure 3B). In day 28 tissue sections, the few remaining
transplanted ADSCs identified had become incorporated into nascent capillaries (figure 3B).
Their engraftment location suggested a potential role for these transplanted cells in the
angiogenic process. To gauge the magnitude of the angiogenic response, capillaries were
quantified by high powered field (hpf) at 4 weeks. As expected, VEGF-overexpressing ADSCs
led to the largest increase in vessel density (48.9±10 vessels per hpf) compared to GFP-
transfected controls (42.7±2 vessels per hpf, p < 0.05) (figure 3D). Co-transfection with
CXCR4/VEGF also resulted in increased vessel density, but this was not statistically greater
than GFP-controls. Interestingly, vessel density in the CXCR4- group was not statistically
different compared to GFP- controls.
Because the initial inflammatory response generally peaks around days 3-7, day 4 tissue
sections were stained for macrophages, the expected dominant inflammatory cell population at
this time point. This analysis revealed a large inflammatory response in ischemic muscles

80
transplanted with VEGF- overexpressing ADSCs (figure 3C). Indeed, both VEGF- and
CXCR4/VEGF- groups led to a statistically significant increase in CD68 macrophages
compared to GFP- controls (p < 0.05) (figure 3E). CXCR4- overexpressing ADSCs led to an
apparent increase in CD68 macrophages, but this was not statistically greater than GFP-
controls. RT-PCR for inflammatory cytokines, including IL-1β, TNF-α, IL-6, and IL-10, was
performed on day 4 tissue sections to complement immunostaining assessment for
inflammation. Consistent with CD68 staining, the VEGF group led to significant increases in
pro-inflammatory cytokines IL-1β (p < 0.001) (figure 3F) and TNF-α (p < 0.001) compared to
controls (figure 3G), while co-transfection by CXCR4/VEGF led to significant increases in
TNF-α only (p < 0.001). IL-6 was upregulated in both CXCR4- and CXCR4/VEGF- groups
compared to VEGF- and GFP- groups (p < 0.001), indicating that CXCR4 had a role in
upregulating this cytokine. The anti-inflammatory cytokine IL-10 followed similar trends to
that of TNF-α, suggesting that its upregulation may have occurred in response to the increase in
pro-inflammatory cytokines (figure 3I).
5.3.6 CXCR4-overexpression in ADSCs promotes a pro-angiogenic and anti-inflammatory
phenotype in vitro hypoxia culture
To better understand how CXCR4-overexpression might be promoting therapeutic
response into ischemic tissues, transfected ADSCs were cultured under hypoxia in vitro and
assessed for genetic markers of angiogenesis and inflammation. Interestingly, CXCR4-
overexpression led to genetic upregulation of angiogenic factors compared to GFP- and cells
only controls, including mouse VEGF (p < 0.05), basic fibroblast growth factor (bFGF) (p <
0.05), and hepatocyte growth factor (HGF) (p < 0.05) (figure 4A). With regards to inflammatory
cytokines, CXCR4-overexpression led to significant decreases in IL-1β (p < 0.05) and IL-6 (p
< 0.05) compared to GFP- and cells only controls (figure 4B). These trends were not observed
for CXCR4/VEGF- or VEGF- transfected groups.
Paracrine release from transfected ADSCs was assessed against human umbilical vein
endothelial cells (HUVECs) and THP-1 macrophages in assays of HUVEC proliferation and
THP-1 migration in vitro. Both CXCR4-overexpressing ADSCs and VEGF-overexpressing
ADSCs increased HUVEC proliferation compared to controls (p < 0.05), which was not seen
by co-transfection with CXCR4-VEGF. THP-1 migration was significantly enhanced by
CXCR4-VEGF and VEGF-transfected ADSCs compared to controls (p < 0.05), which was not

81
observed by CXCR4-ADSCs. These results indicated that CXCR4-overexpression might be
promoting a pro-angiogenic and anti-inflammatory in response to hypoxia (figure 4C).
5.4 Discussion
PAD remains a significant problem in the U.S. that affects roughly 12% of U.S. adults.
For many patients, treatment remains lacking due to the presence of co-morbidities that prohibit
them from undergoing surgical revascularization. Therapeutic angiogenesis using stem cells
has potential for treating PAD, but is hindered by poor cell engraftment and insufficient
paracrine signaling. Genetic engineering can improve stem cell therapy by enhancing cellular
processes related to cell survival, migration, and paracrine signal release [3]. One interesting
target known to mediate these processes is CXCR4, which is involved in host stem cell homing
and survival in relation to ischemia and injury [6]. In this study, we found that ADSCs
engineered to overexpress CXCR4 using biodegradable polymers enhanced cell engraftment
and survival, which subsequently led to a quicker reperfusion response and complete limb
salvage. Because this approach used a readily available stem cell population and a non-viral
transfection agent, it represents a potentially clinically translatable method for treating PAD.
As one of the main goals for treating PAD is the prevention of ischemic tissue damage,
the most interesting result in our study was that CXCR4-overexpressing ADSCs led to complete
limb salvage in all hindlimb ischemia murine models studied (100% limb survival). In
comparison, VEGF-overexpressing ADSCs resulted in less than 60% limb survival. Muscle
histology further demonstrated that CXCR4-overexpression improved healing response by
indicating wide-spread muscle regeneration that was not observed in the VEGF- and GFP-
overexpressing groups. Together, these findings clearly demonstrate that CXCR4-
overexpressing ADSCs have a beneficial effect and promote tissue salvage. Furthermore,
because our findings for VEGF-overexpressing ADSCs were similar to results obtained in prior
studies using VEGF-transfected EPCs [18] and VEGF-transfected MSCs [4], it is reasonable to
compare our results to prior work.
In considering the mechanism of repair, CXCR4-overexpressing ADSCs likely acted
by promoting an early angiogenic response and moderating inflammation. We found that
CXCR4-overexpressing ADSCs led to a robust reperfusion response by day 7 which then
aligned with the host healing response by day 14 (figure 2G). Histological sections at day 28
revealed a low relative number of capillaries compared to VEGF-overexpressing ADSCs, which
suggested that the tissue repair process had returned to baseline. This phenomenon can be

82
explained by the natural response to ischemic injury, where the blood supply increases above
normal levels during healing and then gradually returns to pre-ischemic levels upon complete
repair [19]. The other aspect of tissue repair is the early inflammatory phase, which is a
necessary process that can become damaging to the tissue if excessive. We found that CXCR4-
ADSCs tended to moderate inflammation, while VEGF-ADSCs led to an exuberant and likely
damaging inflammatory response as indicated by the increase in inflammatory cytokines (IL-1
and TNF-α) and CD68+ tissue macrophages. These findings were further supported in vitro,
where CXCR4-ADSCs cultured under hypoxia promoted endothelial cell proliferation and
decreased THP-1 macrophage migration.
Cell engraftment and survival remain challenges for stem cell-based therapeutics. Our
study suggests that CXCR4-overexpresson may help both of these processes. On BLI, we found
that CXCR4-overexpressing ADSCs, including CXCR4- and CXCR4/VEGF- ADSCs,
demonstrated high BLI signal compared to controls at 24 hours, a trend which persisted over 14
days (figure 2B). As CXCR4 is a G-protein coupled receptor known to activate multiple
intracellular signaling cascades leading to cell migration and cell survival [6], these findings
suggest a role for utilizing CXCR4 to overcome issues related to cell engraftment and survival.
The improvements in cell engraftment and survival likely contributed to the early angiogenic
response achieved by CXCR4- overexpressing ADSCs, which then promoted tissue salvage.
Two important characteristics for successful clinical translation of combined stem cell-
gene therapy include: 1) an available and abundant cell population and 2) the use of a non-viral
gene transfection vector. With regards to cell population, ADSCs represent a relatively
abundant, autologous cell source that are easy to isolate. While this is clear, the therapeutic
potential for PAD is still being explored. Indeed, multiple studies have demonstrated their
therapeutic effects in murine models of a hindlimb ischemia [11, 12, 14, 20]. We further studied
the effects of VEGF-overexpressing ADSCs on endothelial cells in vitro, finding enhancement
in cellular process of angiogenesis, in a prior report [15]. Carrying this forward here, we have
demonstrated that CXCR4-overexpressing ADSCs support angiogenesis and tissue repair in a
murine model of hindlimb ischemia. Overall, these findings, in combination with their ready
availability, support ADSCs as a clinically applicable therapeutic stem cell source.
With regards to non-viral gene transfection vectors, such vectors are clinically preferred
because they may overcome many of the issues related to viral vectors, such as immunogenicity
and insertional mutagenesis. However, many non-viral vectors suffer low transfection
efficiency and high cytotoxicity. In our platform, we used a biodegradable polymer for non-

83
viral gene transfection, poly(β-amino esters) (PBAE), which had been previously optimized
based on combinatorial polymer synthesis and high-throughput screening [21, 22]. We further
screened these polymers for transfection in stem cells, where we identified PBAE C32-122 for
efficiently transfecting multiple stem cell types with low cytotoxicity, including embryonic stem
cells, BM-MSCs, and ADSCs [5]. [20]. In the present study, PBAE C32-122 was used to
achieve successful overexpression of CXCR4 and VEGF. Protein expression of each gene was
clearly demonstrated by Western blot or ELISA for CXCR4 or VEGF, respectively.
Furthermore, genetic upregulation by PBAE C32-122 is transient, suggesting that the plasmid
DNA is not integrated into the genome and would not lead to insertional mutagenesis. Thus,
the use of PBAE C32-122 for transfection into ADSCs constitutes a platform that may meet
clinical standards for combined stem cell-gene therapy.
While our study shows promise for non-viral engineered CXCR4-overexpressing
ADSCs for treating PAD, some limitations of the study include: 1) the use of small rodent model
to characterize therapeutic response and 2) the reliance on ex vivo genetic engineering. Use of
a large animal of a similar size to humans would better represent ischemic tissue damage and
repair because of the dependence of diffusion distance for oxygen and nutrient delivery.
Therefore, further study in large animals is warranted that may discover beneficial modifications
to the present platform for human therapy. The next limitation relates to using a multi-step
procedure to transfect stem cells, whereby ADSCs would be isolated in one procedure, then ex
vivo expanded and transfected, and then delivered back to the patient in a second procedure.
The process would increase cost and regulatory concerns, and may perhaps cause variability in
ADSC phenotype. To overcome this issue, platforms based on a one-step approach (i.e.
enabling cell isolation and transfection in the same procedure) would overcome many of the
concerns related to regulatory and cost, and may thus be more readily applied in the clinic.
In summary, this study suggests that CXCR4-overexpressing ADSCs using
biodegradable polymers can promote therapeutic angiogenesis and treat PAD. CXCR4
overexpression supports cell engraftment and survival and promotes an appropriate healing
response to ischemic injury. This approach has clinical potential for the engineering and
regeneration of many other tissue types, as well as in treating other types of ischemic diseases
such as myocardial infarction and cerebral ischemia.

84
Acknowledgments: Thank others for any contributions. Funding: This work was funded by
the American Heart Association (10SDG2600001) supported by Stanford Medical Scholars
Research funding and Stanford Medical Scientist Training Program.
Supplementary Materials
Fig. S5.1. Improvement of transfection efficiency and low cytotoxicity by PBAE nanoparticles.
Fig. S5.2. Cell engraftment of transfected ADSCs.
Fig. S5.3. Reperfusion of the ischemic hindlimb at one week post-procedure.
Table S5.1: List of primers used for quantitative real time-polymerase chain reaction
References:
1. Selvin, E. and T.P. Erlinger, Prevalence of and risk factors for peripheral arterial disease
in the united states results from the national health and nutrition examination survey,
1999–2000. Circulation, 2004. 110(6): p. 738-743.
2. Criqui, M.H., et al., Mortality over a period of 10 years in patients with peripheral
arterial disease. New England Journal of Medicine, 1992. 326(6): p. 381-386.
3. Fischbach, M.A., J.A. Bluestone, and W.A. Lim, Cell-based therapeutics: the next pillar
of medicine. Sci Transl Med, 2013. 5(179): p. 179ps7.
4. Yang, F., et al., Genetic engineering of human stem cells for enhanced angiogenesis
using biodegradable polymeric nanoparticles. Proc Natl Acad Sci U S A, 2010. 107(8):
p. 3317-22.
5. Yang, F., et al., Gene delivery to human adult and embryonic cell-derived stem cells
using biodegradable nanoparticulate polymeric vectors. Gene Ther, 2009. 16(4): p. 533-
46.
6. Busillo, J.M. and J.L. Benovic, Regulation of CXCR4 signaling. Biochim Biophys
Acta, 2007. 1768(4): p. 952-63.
7. Takahashi, T., et al., Ischemia- and cytokine-induced mobilization of bone marrow-
derived endothelial progenitor cells for neovascularization. Nat Med, 1999. 5(4): p.
434-8.
8. Ceradini, D.J., et al., Progenitor cell trafficking is regulated by hypoxic gradients
through HIF-1 induction of SDF-1. Nat Med, 2004. 10(8): p. 858-64.
9. Jin, D.K., et al., Cytokine-mediated deployment of SDF-1 induces revascularization
through recruitment of CXCR4+ hemangiocytes. Nat Med, 2006. 12(5): p. 557-67.

85
10. Petit, I., D. Jin, and S. Rafii, The SDF-1–CXCR4 signaling pathway: a molecular hub
modulating neo-angiogenesis. Trends in Immunology, 2007. 28(7): p. 299-307.
11. Miranville, A., et al., Improvement of postnatal neovascularization by human adipose
tissue-derived stem cells. Circulation, 2004. 110(3): p. 349-55.
12. Kim, Y., et al., Direct comparison of human mesenchymal stem cells derived from
adipose tissues and bone marrow in mediating neovascularization in response to
vascular ischemia. Cell Physiol Biochem, 2007. 20(6): p. 867-76.
13. Cai, L., et al., IFATS collection: Human adipose tissue-derived stem cells induce
angiogenesis and nerve sprouting following myocardial infarction, in conjunction with
potent preservation of cardiac function. Stem Cells, 2009. 27(1): p. 230-7.
14. Rehman, J., et al., Secretion of angiogenic and antiapoptotic factors by human adipose
stromal cells. Circulation, 2004. 109(10): p. 1292-8.
15. Deveza, L., et al., Paracrine release from nonviral engineered adipose-derived stem cells
promotes endothelial cell survival and migration in vitro. Stem Cells Dev, 2013. 22(3):
p. 483-91.
16. Niiyama, H., et al., Murine model of hindlimb ischemia. J Vis Exp, 2009(23).
17. Schantz, J.T., H. Chim, and M. Whiteman, Cell guidance in tissue engineering: SDF-1
mediates site-directed homing of mesenchymal stem cells within three-dimensional
polycaprolactone scaffolds. Tissue Eng, 2007. 13(11): p. 2615-24.
18. Iwaguro, H., et al., Endothelial progenitor cell vascular endothelial growth factor gene
transfer for vascular regeneration. Circulation, 2002. 105(6): p. 732-8.
19. Hong, G., et al., Near-Infrared II Fluorescence for Imaging Hindlimb Vessel
Regeneration with Dynamic Tissue Perfusion Measurement. Circulation:
Cardiovascular Imaging, 2014.
20. Cao, Y., et al., Human adipose tissue-derived stem cells differentiate into endothelial
cells in vitro and improve postnatal neovascularization in vivo. Biochem Biophys Res
Commun, 2005. 332(2): p. 370-9.
21. Green, J.J., et al., Combinatorial modification of degradable polymers enables
transfection of human cells comparable to adenovirus. Advanced Materials, 2007.
19(19): p. 2836-+.
22. Zugates, G.T., et al., Rapid optimization of gene delivery by parallel end-modification
of poly(beta-amino ester)s. Mol Ther, 2007. 15(7): p. 1306-12.

86
Figures:
Figure 5.1. Genetic upregulation of CXCR4 and/or VEGF in ADSCs by PBAE nanoparticles.
(A) Schematic showing experimental design. Briefly, ADSCs were transfected ex vivo by first
generating PBAE nanoparticles utilizing plasmid DNA encoding CXCR4 or VEGF and then
applying to cells. Gene transfected ADSCs were transplanted into murine models of hindlimb
ischemia. CXCR4-overexpression was verified by RT-PCR (B) and by western blot (C) at 48
hours post-transfection. (D) FACS quantification of CXCR4 cell surface expression. (E)
Kinetics of CXCR4 mRNA expression over 10 days. VEGF overexpression was verified by RT-
PCR (F) and ELISA (G) at 48 hours. (H) VEGF release over time quantifying release every 48
hours. (I) Effects of PBAE transfection on ADSC cell viability. (* p < 0.05, ** p < 0.01
compared with GFP-ADSC controls)

87
Figure 5.2. Improvement of cell fate and therapeutic efficacy due to CXCR4-overexpressing
ADSCs. (A) Representative BLI images at day 10. (B) Percentage of initial BLI signal
remaining over the first 14 days post-procedure. (C) RT-PCR analysis of harvested tissue at 4
days post-procedure measuring human CXCR4 mRNA and human VEGF mRNA (relative to
limbs transplanted with CXCR4-ADSC and limbs transplanted with VEGF-ADSC,
respectively). Representative images of clinical limb outcome (D) and laser doppler perfusion
imaging (E) at day 14 post-procedure. Ischemic limbs are indicated by red circles.
Quantification of clinical limb outcome at day 21 (F) and relative reperfusion over 21 days (G).
(* p < 0.05 compared to GFP-ADSC controls)

88
Figure 5.3. CXCR4-overexpressing ADSCs enhanced muscle regeneration and mediated host
angiogenic and inflammatory response. (A) Representative tissue sections at 28 days post-
procedure stained with Masson’s Trichrome. (B) Immunostaining of tissue sections at 28 days
post-procedure for capillaries (anti-CD31: red), transplanted cells (anti-GFP: green), and
cellular counterstain (Dapi: blue). (C) Immunostaining of tissue sections at 4 days post-
procedure for macrophages (anti-CD68: red) and cellular counterstain (Dapi: blue). (D)
Quantification of CD31-positive capillaries per high powered field (hpf). (E) Percentage of
CD68-positive macrophages to all cells. RT-PCR analysis on harvested tissue at day 4 for
inflammatory cytokines, including IL-1β (F), TNF-α (G), IL-6 (H), and IL-10 (I). (* p < 0.05,
** p < 0.01, *** p < 0.001 compared to GFP-ADSC controls; # p < 0.05 compared to VEGF-
ADSCs)

89
Figure 5.4. CXCR4-overexpression in ADSCs promotes a pro-angiogenic and anti-
inflammatory phenotype under hypoxia. Transfected ADSCs were cultured under hypoxia and
assessed at 48 hours by RT-PCR for markers of angiogenesis (A) and inflammation (B) in
comparison to normoxia culture. (C) Paracrine release from transfected ADSCs applied to
HUVECs and assessed for MTS viability. (D) Paracrine release from transfected ADSCs
assessed for THP-1 macrophage migration. (E) Proposed model for CXCR4- mediated
therapeutic response to tissue ischemia. SDF-1α is secreted from sites of ischemia, which
activates CXCR4 on transfected ADSCs to promote an ischemia specific therapeutic response.
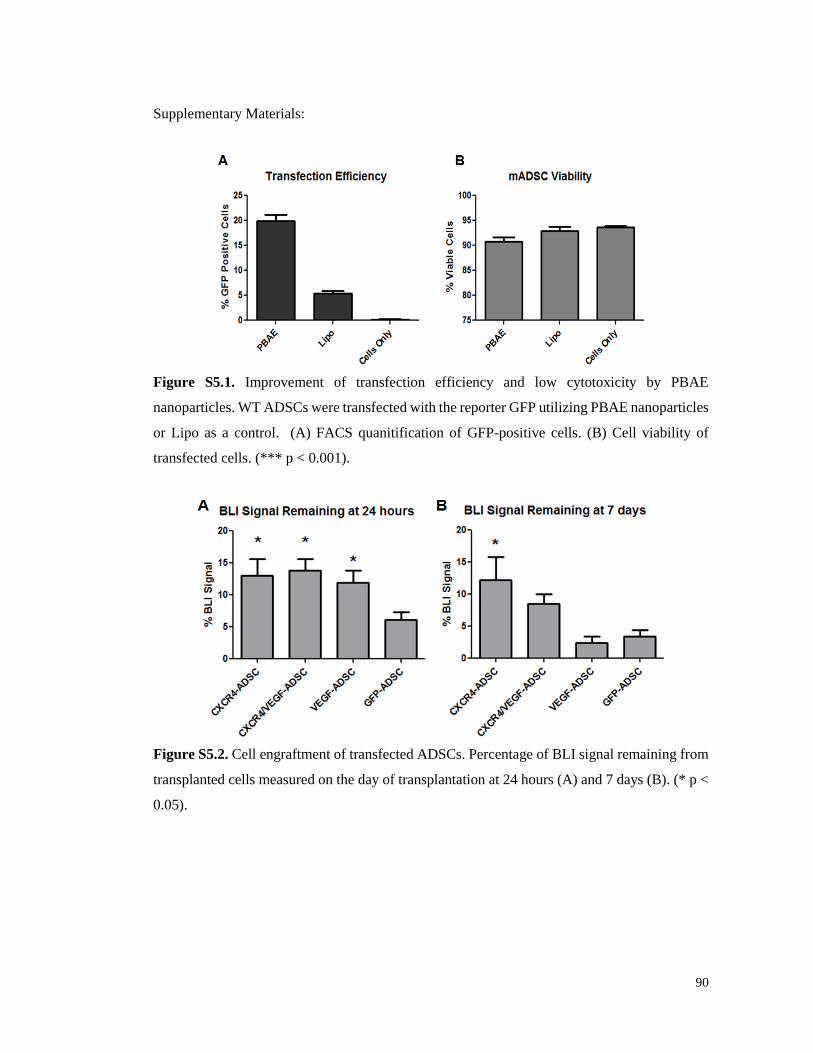
90
Supplementary Materials:
Figure S5.1. Improvement of transfection efficiency and low cytotoxicity by PBAE
nanoparticles. WT ADSCs were transfected with the reporter GFP utilizing PBAE nanoparticles
or Lipo as a control. (A) FACS quanitification of GFP-positive cells. (B) Cell viability of
transfected cells. (*** p < 0.001).
Figure S5.2. Cell engraftment of transfected ADSCs. Percentage of BLI signal remaining from
transplanted cells measured on the day of transplantation at 24 hours (A) and 7 days (B). (* p <
0.05).

91
Figure S5.3. Reperfusion of the ischemic hindlimb at one week post-procedure. Relative
reperfusion as assessed by LDPI at one week. (* p < 0.05)

92
Table S5.1: List of primers used for quantitative real time-polymerase chain reaction
Gene
Name
Species Primer Sequence
GAPDH Mouse Forward (5'-AACGACCCCTTCATTGAC-3')
Reverse (5'-TCCACGACATACTCAGCAC-3')
CXCR4 Human Forward (5’- TTCTACCCCAATGACTTGTG-3’)
Reverse (5’- ATGTAGTAAGGCAGCCAACA-3’)
VEGF Human Forward (5’-TACCTCCACCATGCCAAGTG-3’)
Reverse (5’-TGATGATTCDTGCCCTCCTCC-3’)
VEGF Mouse
Forward (5'-
GCCCTGGAGTGCGTGCCCACGTCAGAGAGCA-3')
Reverse (5'-TGGCGATTTAGCAGCAGATA-3')
bFGF Mouse Forward (5'-GCCAGCGGCATCACCTCGCT-3')
Reverse (5'-TATGGCCTTCTGTCCAGGTCCCGT-3')
HGF Mouse Forward (5'-TTGGCCATGAATTTGACCTC-3')
Reverse (5'-ACATCAGTCTCATTCACAGC-3')
IL-1β Mouse Forward (5'-CAACCAACAAGTGATATTCTCCAT G-3')
Reverse (5'-GATCCACACTCTCCAGCTGCA-3')
TNF-α Mouse Forward (5'-CATCTTCTCAAAATTCGAGTGACAA-3')
Reverse (5'-TGGGAGTAGACAAGGTACAACCC-3')
IL-6 Mouse Forward (5'-TGGCTAAGGACCAAGACCATCCAA-3')
Reverse (5'-AACGCACTAGGTTTGCCGAGTAGA-3')
IL-10 Mouse Forward (5'-GGTTGCCAAGCCTTATCGGA-3')
Reverse (5'-ACCTGCTCCACTGCCTTGCT-3')

6. Adipose-Derived Stem Cells Co-expressing Hepatocyte Growth Factor and CXCR4
Promotes Ischemic Tissue Repair
Jeffrey Choi1*, Lorenzo Deveza2,3*, Sungwon Lim2, Fan Yang2,4
1Department of Biology, Stanford University, Stanford, CA; 2Department of Bioengineering,
Stanford University, Stanford, CA; 3Stanford University School of Medicine, Stanford, CA; 4Department of Orthopaedic Surgery, Stanford University, Stanford, CA.
Abstract
Stem cells hold great potential for therapeutic angiogenesis due to their ability to respond to
environmental cues and secrete beneficial growth factors. However, stem cell therapies have
been largely unsuccessful in clinical trials, which is in part related to issues of poor cell
engraftment and cell homing, as well as insufficient paracrine signaling. To overcome these
issues, we engineered adipose-derived stem cells (ADSCs), a clinically abundant cell source, to
dually express CXC chemokine receptor 4 (CXCR4) and hepatocyte growth factor (HGF);
based on the involvement of CXCR4 in stem cell homing to sites of ischemia, as well as the
potent angiogenic effects of HGF. To achieve this, we developed a bicistronic plasmid encoding
both genes and then assembled polymeric nanoparticles using poly(β-amino esters) (PBAE), a
non-viral transfection vector, prior to delivery to ADSCs ex vivo. Engineered syngeneic ADSCs
were transplanted into aged mice surgically induced with severe hindlimb ischemia to evaluate
their therapeutic efficacy. HGF overexpression resulted in increased overall angiogenesis in the
ischemic hindlimb based on blood reperfusion and density of vascular structures. CXCR4
overexpression prolonged ADSC retention in vivo and led to improved muscle integrity with a
muted inflammatory response. Notably, ADSCs co-expressing HGF and CXCR4 not only
exhibited increased cell survival, but also quenched early inflammation and promoted functional
angiogenesis, leading to improved limb salvage. These results demonstrate that non-viral
transfer of multiple genes via a single plasmid to ADSCs can tailor their behavior for treating
ischemic skeletal muscle, and perpetuate the larger goal of engineering cell-based therapeutic
tools that can be controlled and tuned for different diseases.
6.1 Introduction
Peripheral arterial disease (PAD) represents an important, yet underrepresented medical
problem, caused by decreased blood flow to the limbs. It may signify severe underlying
93

atherosclerosis and often occurs with advanced Diabetes Mellitus. Treatment options for PAD
resemble those of other ischemic diseases, including surgical revascularization via bypass
surgery or percutaneous angioplasty. However, because of the presence of surgically
prohibitive co-morbidities, many patients are without treatment.
Stem cell-based therapeutics are an alternative treatment option for patients unable to
withstand surgery [1]. It involves transplanting autologous stem cells to ischemic regions to
induce host-blood vessel growth in a process known as therapeutic angiogenesis, and likely acts
via secreting beneficial paracrine factors [2]. Multiple stem cell sources have shown therapeutic
effect in pre-clinical animal models [3-6], but overall success in human clinical trials has been
limited [7]. This lack of response in human trials might be attributed to insufficient paracrine
signaling, poor stem cell engraftment, or an inappropriate response to the disease state [2, 8].
Ex vivo genetic manipulation of stem cells may improve their behavior by enhancing
those cellular process involved in therapeutic angiogenesis [8]. Prior strategies to enhance stem
cell-therapeutics have included genetic modification with angiogenic growth factors [9, 10],
survival factors [11], and homing factors [12]. However, as stem cells have complex
intracellular signaling pathways and respond to environmental stimuli, single genetic
modifications with paracrine signals might underappreciate the potential for stem cell
therapeutics. Stem cells could be engineered to overexpress appropriate cellular receptors to
respond to environmental cues, as well as overexpress paracrine signals to induce a therapeutic
response. In particular, CXC chemokine receptor 4 (CXCR4) is a receptor involved in stem cell
homing and is known to localize cells to sites of ischemia in response to stromal derived factor-
1α (SDF-1α) [13]. Hepatocyte growth factor (HGF) is a potent angiogenic growth factor that
has shown great promise in human clinical trials for treating clinical limb ischemia [14, 15].
HGF is also known to have activity against both endothelial cells and smooth muscle cells, and
may thereby stimulate both angiogenic initiation and maturation. Dual overexpression of
CXCR4 and HGF may thus promote stem cell-based therapeutics by localizing stem cells to
sites of ischemia, where they may appropriately deliver HGF to induce an angiogenic response.
Methods to achieve genetic overexpression of two or more genes, such as CXCR4 and
HGF, typically use as many plasmids to transfect cells ex vivo. While this approach can be
satisfactory, there is no guarantee that the same cell is transfected with both genes; and, dually
transfected cells may vary in the relative expression of each gene due to variations in the number
of plasmid copies or by the promoter regulating each gene. An alternative approach is to
recombine both genes into a single plasmid and design the genes to be regulated by a single
94

promoter. Engineering both CXCR4 and HGF into a single bicistronic plasmid could thus
ensure that both genes are transfected into the same cell and expressed under a single promoter.
Clinically, a successful approach for combined stem cell/gene therapy would be safe
and broadly applicable. To meet this criteria, our approach has been to utilize non-virally
transfected adipose-derived stem cells (ADSCs) [16, 17]. Poly(β-amino esters) (PBAE) are a
class of non-viral gene transfection vectors that have been optimized for efficiently transfecting
stem cells for therapeutic angiogenesis, including ADSCs [9, 18]. ADSCs are an easy to isolate
and clinically abundant cell source [19] that naturally possesses angiogenic potential [6, 20],
making them excellent candidates for treating ischemic disease. Indeed, we have demonstrated
that ADSCs can be efficiently engineered using PBAE and that paracrine release from VEGF-
transfected ADSCs improves therapeutic response both in vitro [16] and in vivo [17].
In this study, we genetically engineered ADSCs with a bicistronic plasmid encoding
both HGF and CXCR4 using PBAE biodegradable polymers, and then assessed their ability for
therapeutic efficacy in an aged murine model of hindlimb ischemia. Two types of dually
expressing plasmids for HGF and CXCR4 were designed and assessed for genetic upregulation;
using an optimized HGF gene sequence that expresses two isoforms of HGF [21]. PBAE-
mediated transfection of ADSCs was then optimized using the bicistronic plasmid. Therapeutic
efficacy was assessed in retired breeder mice induced with hindlimb ischemia to simulate a
difficult healing situation in an immunocompetent mouse (many previous studies have focused
on younger mice and/or nude mice). Outcomes analysis in vivo included evaluating cell survival
and engraftment, angiogenic and inflammatory response, and muscle morphology.
6.2 Materials and Methods
6.2.1 Materials
Monomers for synthesizing poly(β-amino esters)were purchased from Sigma–Aldrich,
Scientific Polymer Products, and Molecular Biosciences. Anhydrous dimethyl sulfoxide
(DMSO) was purchased from Sigma–Aldrich. A 25 mM sodium acetate (NaOAc) buffer
solution (pH 5.2) was prepared by diluting a 3 M stock solution (Sigma-Aldrich, St Louis, MO).
Plasmids (pEGFP-N1 and pcDNA3.1(+)-CXCR4) were purchased from Elim
Biopharmaceuticals and pck-HGF plasmid was obtained from Viromed (Seoul, Korea).
95

6.2.2 Poly(β-Amino Esters) Synthesis
PBAE polymers were synthesized as previously described [9]. Briefly, acrylate-
terminated D32 polymer (C32-Ac) was prepared by mixing hexanediol (D) and aminopentanol
(32) monomers using a 1.2:1.0 diacrylate/amine monomer molar ratio at 90 °C for 24 h.
Subsequently, amine-terminated D32 polymers were generated by reacting D32-Ac with
diamine monomer (122) in DMSO. End-capping reactions were performed overnight at room
temperature by using a 1.6-fold molar excess of amine over acrylate end groups.
6.2.3 ADSC isolation and culture
ADSCs were collected by harvesting the inguinal fat pads from 5 to 6 weeks old
luciferase-positive/GFP-positive transgenic male mice (Joseph Wu, MD, PhD laboratory,
Stanford University) and FVB wild-type male mice (purchased from Charles River, Willington,
MA). Following serial betadine washes, the fat pads were finely minced in cold, sterile PBS,
followed by digestion in 0.075% Collagenase II (Sigma-Aldrich) at 37 °C for 30 minutes in a
shaking water bath. Digestion was neutralized with Dulbecco’s modified Eagle’s medium
(Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen) and 1%
penicillin-streptomycin (Sigma-Aldrich). Neutralized cells were centrifuged at 4 °C and 1,000
rpm for 5 minutes, and floating adipocytes and media were removed before pelleted ADSCs
from the stomal-vascular portion were resuspended in culture media, passed through a 100 μm
Falcon nylon cell strainer (Becton Dickinson, Bedford, MA) and plated on a 10 cm dish. Cells
were incubated at 37 °C and 5% atmospheric CO2. Twenty-four hours after isolation, the cells
were washed in PBS to remove debris and nonadherent cells, and fresh medium was added to
the cells. Medium was changed every 2 days, and adherent spindle-shaped cells were grown to
subconfluence, passaged using standard methods of trypsinization, and propagated through
passage 2.
6.2.4 Transfection
ADSCs were transfected with pCK-HGF-X7, pCK-HGF-X7-IRES-CXCR4,
pcDNA3.1-CXCR4, or the pcDNA3.1 empty plasmid as a negative control for transfection.
ADSCs were seeded at 65 000 cells per well in clear 24-well plates and incubated over night at
37 °C. Cells were then transfected utilizing novel end-modified PBAE polymers (D32-122).
Transfection efficiency was optimized by varying vector parameters including polymer/DNA
weight ratios (10:1, 12.5:1, and 15:1) and plasmid DNA (pDNA) loading dose (4, 5, 6, and 7 μg
96

per well in a 24-well plate). Polyplexes were formed by mixing pDNA with PBAE polymers in
25 mM sodium acetate buffer and waiting 10 minutes for complexes to form. Polyplexes were
added to the cells cultured in growth medium containing 10% serum. Cells were then incubated
with polyplexes for 4 hours, at which time complexes were removed and replaced with fresh
medium.
6.2.5 Quantitative real-time polymerase chain reaction (qRT-PCR)
To evaluate transfection efficiency, total RNA was harvested from cells using RNeasy
Kit (Qiagen, Germantown, MD, USA). cDNA was synthesized by reverse transcription using
the Superscript First-Strand Synthesis System (Invitrogen) and 1 μg of total RNA. Quantitative
PCR was performed using Power SYBR Green PCR Master Mix () following the manufacturer's
protocol and running all samples for 40 PCR cycles on Applied Biosystems 7900 Real-Time
PCR System (Carlsbad, CA, USA). The expression of the transfected genes – HGF and CXCR4
– was examined. Relative expression level of target genes was determined by the comparative
CT method, where target gene expression was first normalized to an endogenous housekeeping
gene, GAPDH, followed by a second normalization to the gene expression level measured in
untransfected control cells.
6.2.6 Cell Viability/Proliferation Assay
To evaluate possible cytotoxic effects of transfection, cell viability or proliferation was
evaluated 48 hours post-transfection using the Cell Titer 96 Aqueous One Solution assay kit
(Promega) to measure metabolic activity by optical absorbance on a plate reader. Measurements
of cells transfected with polymer-plasmid polyplexes were converted to relative absorbance
values in comparison to untransfected control cells.
6.2.7 Mouse Hindlimb Ischemia Model and Cell Transplantation
FVB female retired breeder mice were housed five per cage in a 12-hour light/dark
cycle and provided ad libitum with standard food and water. Aged mice were used to better
distinguish the effect of therapeutic intervention from the inherent robust recovery of young
mice. Hindlimb ischemia was induced as previously described (28). Briefly, the femoral artery
and its branches were ligated through a skin incision with 5-0 silk suture (Ethicon). The external
iliac artery and all the above arteries were then ligated. The femoral artery was excised from its
proximal origin as a branch of the external iliac artery to the distal point where it bifurcates into
97

the saphenous and popliteal arteries. Laser Doppler imaging of blood perfusion was used to
confirm induction of hindlimb ischemia post-operatively.
After inducing hindlimb ischemia, cells (1.0 x 106 cells per injection) were suspended
in 100 μL of PBS and injected intramuscularly into two sites of the gracilis muscle in the medial
thigh by using 27-gauge tuberculin syringes. Five experimental groups (n=5 per group) were
examined as follows: (i) PBS, (ii) ADSC-pcDNA3.1, (iii) ADSC-HGF, (iv) ADSC-CXCR4,
and (v) ADSC-HGF-IRES-CXCR4. All experiments were performed in accordance with the
Stanford University Animal Care and Use Committee Guidelines and approved APLAC
protocols.
6.2.8 Bioluminescence Imaging
ADSC survival was monitored post-injection with bioluminescence imaging
technology. Bioluminescence imaging (BLI) was performed on the day of surgery, day 1, day
4, day 7, day 10, and day 14. Animals were anesthetized by 2–3% inhaled isoflurane and injected
intraperitoneally with D-Luciferin reporter probe at 100 mg/kg body weight. IVIS Spectrum
system (Xenogen, Alameda, CA) was used to image the animals while under 2% inhaled
isoflurane anesthesia. Each animal was scanned until the peak signal was reached, and using
Living Image 4.3.1 software (Caliper Life Sciences), radiance of regions of interest (ROIs) in
photons was recorded. Radiance was quantified in photons per second per centimeter squared
per steridian.
6.2.9 In Vivo Assessment of Ischemic Damage and Limb Perfusion
Physiological status of ischemic limbs was followed up to 3 weeks after treatment.
Hindlimb blood perfusion was assessed using a PeriScan PIM3 laser Doppler system (Perimed
AB, Sweden)on day 0, 1, 4, 7, 14, and 21, as previously described (30). Each animal was pre-
warmed to a body temperature of 37°C to avoid data variations raised from confounding factors.
ROIs were drawn around the entire hindlimbs, and the level of perfusion in ischemic and normal
hindlimbs were quantified using the mean pixel value within each ROI. Relative changes in
blood flow were expressed as the ratio of the left (ischemic) over the right (normal) laser
Doppler blood perfusion.
98

6.2.10 Histology and Immunofluorescence staining
Mice were euthanized on day 7 and day 21 post-surgery and cell transplantation to
retrieve muscle tissues from ischemic sides and normal controls for analyses. Hindlimbs were
embedded in optimal cutting temperature compound and snap frozen for cryosectioning. Lateral
cross-sections of the muscle tissues were collected at 8 μm thickness. Tissue sections were
stained with hematoxylin and eosin to assess muscle integrity in ischemic regions. Muscle
tissue from normal limbs with no surgical alterations was used as a positive control.
For immunofluorescence staining, tissue sections were fixed in 4% paraformaldehyde
for 10 minutes, then blocked with 1% BSA in PBS and permeabilized with 0.1% Triton X-100
in PBS. To detect capillaries in ischemic regions, tissue sections were immunofluorescently
stained with rat monoclonal antibody to CD31 overnight at 4°C, followed by secondary
antibody (Alexa Fluor 594 goat anti-rat) incubation for 1 hour at room temperature. Nuclei
were counterstained with DAPI mounting medium (Vectashield) and images were taken with a
Zeiss fluorescence microscope.
6.2.11 Statistical Analysis
GraphPad Prism (Graphpad Software, San Diego, CA, USA) was used to perform all
statistical analyses. One-way analysis of variance with Tukey's multiple comparison test as a
post-hoc test was performed to compare all experimental groups and determine statistical
significance. All quantitative data were expressed as mean ± standard error of the mean.
6.3 Results
6.3.1 PBAE-mediated Gene Transfer of bicistronic plasmid co-encoding CXCR4 and
HGF to ADSCs
Non-virally engineered ADSCs were prepared by generating biodegradable polymeric
nanoparticles using poly(β-amino esters) (PBAE) and plasmid DNA, and then delivering the
resulting nanoparticles to ADSCs ex vivo as shown in Figure 1A. To ensure co-delivery of
CXCR4 and HGF to ADSCs, a plasmid was designed to encode for both genes (Figure 1B).
Two types of designs were initially evaluated: a bi-directional plasmid and a bicistronic plasmid
(pCK-HGF-IRES-CXCR4). Based on a preliminary evaluation, the bicistronic was chosen
because it led to higher genetic expression of both HGF and CXCR4 in HEK293T cells (Figure
S1). The PBAE polymer was synthesized as described in the methods (Figure 1C). Transfection
with the bicistronic plasmid was optimized in ADSCs using PBAE by varying polymer to DNA
99

weight ratio (Figure S2) and subsequently exploring DNA dose (Figure 1D, 1E). A polymer to
DNA weight ratio of 10:1 and DNA dose of 7 µg led to the highest gene expression and was
used for all subsequent experiments. The co-expressing bicistronic plasmid led to similar
genetic upregulation of HGF (Figure 1D) and CXCR4 (Figure 1E) as compared to single
expressing plasmids for the corresponding genes. ADSC viability was not significantly affected
by DNA dose as compared to non-transfected controls (Figure 1F).
6.3.2 Validation of Paracrine Release from Transfected ADSCs In Vitro
Paracrine release from transfected ADSCs was evaluated by HGF ELISA and by
assessing bioactivity against human umbilical vein endothelial cells (HUVECs) in vitro. HGF
ELISA demonstrated that ADSCs transfected with either the bicistronic HGF-CXCR4 plasmid
or the single HGF plasmid significantly increased HGF release compared to ADSCs transfected
with CXCR4 or the backbone plasmid (vehicle, p < 0.05; Figure 2A). The HUVEC viability
assay further confirmed that the paracrine release was bioactive, where conditioned medium
from HGF/CXCR4-, HGF- and CXCR4- overexpressing ADSCs led to significant increases in
HUVEC viability compared to vehicle and basal media controls (p < 0.05; Figure 2B). Notably,
CXCR4- overexpressing ADSCs led to similar increases in HUVEC viability as the co-
transfected HGF/CXCR4-ADSCs and the singly transfect HGF- ADSCs, suggesting additional
effects on ADSC paracrine release by CXCR4-overexpression. These data demonstrated that
transfected ADSCs exhibited potential therapeutic behavior.
6.3.3 Confirmation of HGF and CXCR4 Overexpression In Vivo
ADSCs transfected with HGF- and/or CXCR4- ex vivo were transplanted into the
adductor and gastrocnemius muscles of a murine model of hindlimb ischemia. To confirm that
HGF- and/or CXCR4 were upregulated in ADSCs in vivo, the ischemic hindlimb was harvested
at 4 days post-transplantation and assessed for human CXCR4 and human HGF mRNA
expression. Both hHGF and hCXCR4 were overexpressed in the corresponding ischemic
tissues after 4 days in vivo, which confirmed that the transplanted ADSCs were still expressing
these genes (Figure 3A, 3B). This also gave evidence that many transplanted cells remained
viable after 4 days post-transplantation.
100

6.3.4 Enhanced Cell Survival in the Ischemic Hindlimb
For all in vivo studies, ADSCs that constitutively expressed GFP and Luciferase
(GFP+/Luc+) were used, which enabled us to assess ADSC survival and engraftment in vivo by
bioluminescence imaging (BLI). At day 0, BLI demonstrated no significant differences in BLI
signal (data not shown), indicating that each animal had received a nearly equivalent number of
cells at the outset. By Day 1, much BLI signal was lost with roughly 10% of the original BLI
signal remaining; however, higher signal was observed in mice transplanted with HGF and/or
CXCR4- overexpressing ADSCs, compared to ADSCs transfected with the vehicle control
(Figure 3C). On kinetic read, BLI signal either increased or remained stable from day 1 to day
4, but dropped by day 7 and was undetectable by day 14. Only ADSCs transfected with CXCR4
(i.e. HGF/CXCR4 and CXCR4 only) were able to recover from day 1 to day 4. This indicated
that CXCR4 promoted cell survival post-transplantation compared to vehicle transfection,
whether by single transfection or co-transfection with HGF (p < 0.05; Figure 3D).
6.3.5 Increased Angiogenic Response in the Aged Mouse
Angiogenesis and reperfusion in the murine hindlimb ischemia model was assessed by
Laser Doppler Reperfusion Imaging (LDPI), as well as by immunofluorescence staining of
CD31 (capillaries) and alpha-smooth muscle actin (α-SMA; arterioles) (Figure 4). LDPI
confirmed that ischemic induction was successfully achieved (Day 0 images; Figure 4A), with
progressive reperfusion that occurred over several weeks. The largest changes in reperfusion
occurred between days 7 and 14 (Figure 4B). At day 7, a trend emerged where the HGF
transfected ADSCs led to the highest reperfusion response, followed by the HGF/CXCR4-
ADSCs and CXCR4-ADSCs, then finally, the vehicle-transfected ADSCs (Figure 4B). By day
14, reperfusion by HGF/CXCR4-ADSCs had caught up with the HGF-ADSCs, indicating that
both groups led to a robust reperfusion response (Figure 4B). These findings were further
supported by immunohistochemical staining at day 14, where CD31 capillary densitometry
demonstrated that HGF-ADSCs and HGF/CXCR4-ADSCs resulted in a significantly greater
number of capillaries than vehicle controls (p < 0.05; Figure 4C, 4E). Arteriole assessement
also demonstrated that these groups led to a statistically significant increase in the number of
arterioles compared to vehicle controls (p < 0.05; Figure 4D, 4F). Overall, these data
demonstrated that HGF was associated with an increased angiogenic response, which could be
achieved with either the single plasmid or the dual-expressing plasmid.
101

6.3.6 Promotion of Ischemic Limb Health
Limb health was assessed by gross limb status and muscle histology. Hindlimb
ischemia models in the aged mouse were found to be very severe in controls receiving only
PBS, which led to significant limb loss and necrosis (data not shown). Transplantation of
ADSCs that overexpressed either HGF or CXCR4 (or both) was able to rescue limb necrosis,
and led to complete limb salvage for many mice (Figure 5A, 5D). The healthiest limb status
was achieved by both HGF/CXCR4-ADSCs and CXCR4 only-ADSCs (Figure 5A). Day 4 limb
status measurements demonstrated that HGF/CXCR4-ADSCs and HGF-ADSCs caused some
early toe necrosis, which was less prevalent with CXCR4-ADSCs, but that both had the same
amount of limb salvage and toe necrosis by the final time point. Some foot necrosis was
observed by HGF-ADSC transplantation (Figure 5D), but was overall better than vehicle-ADSC
controls. Masson’s Trichrome staining of day 7 tissue samples was consistent with limb status
measurements, demonstrating good overall health of the muscle by HGF and/or CXCR4-
overexpressing ADSCs, but not by vehicle-ADSC controls (Figure 5B). Large areas of muscle
necrosis and inflammation were observed in vehicle-ADSC controls, which was not observed
in the other groups. Inflammatory assessement was further performed by immunostaining for
CD68+ macrophages on day 7 tissue samples (Figure 5C). As with Masson’s Trichrome
staining, much inflammation was observed in vehicle-ADSC controls. Notably, more
inflammation was observed in HGF-ADSC transplanted tissues than HGF/CXCR4-ADSC or
CXCR4-ADSC transplanted tissues. Overall, HGF and/or CXCR4-ADSCs were able to rescue
ischemic limb necrosis in the aged mouse model, with a less robust response by HGF-ADSCs.
6.4 Discussion
Herein, we developed HGF and CXCR4 overexpressing ADSCs using a bicistronic
plasmid for ischemic tissue repair. To develop this platform, a clinically applicable strategy
was employed by using PBAE biodegradable polymers for non-viral gene transfection, and an
abundant, autologous cell type with angiogenic potential. We hypothesized that dual
overexpression would synergize to enhance the behavior of ADSCs as delivery vehicles for
HGF paracrine release, which were evaluated in an aged murine model of hindlimb ischemia.
Overall, we found that dual HGF and CXCR4 overexpressing ADSCs enhanced ischemic tissue
repair by promoting stem cell survival and angiogenic response, as well as by attenuating
inflammation. Given the complex nature of stem cells, this approach demonstrates the ability
102

for exploiting multiple cellular processes involved in the therapeutic response to promote tissue
repair.
Two cellular processes involved in stem cell-based therapies for ischemic tissue repair
are: 1) stem cell engraftment and homing; and 2) angiogenic growth factor secretion. We found
that dual overexpression of CXCR4 and HGF could enhance both of these processes in ADSCs,
while single overexpression only promoted one process or the other. With regards to stem cell
engraftment and homing, BLI and RT-PCR data demonstrated that CXCR4-overexpression in
ADSCs could enhance this process, whether single or dual expression with HGF. This is further
supported by prior reports demonstrating that CXCR4 is required for stem cell engraftment in
the bone marrow [22] and promotes stem cell engraftment in the ischemic myocardium [23]. In
our own work, we have previously found that CXCR4-overexpressing ADSCs could promote
stem cell survival in the ischemic hindlimb in a similar manner as that demonstrated here. There
is some indication that dual overexpression by HGF and CXCR4 might even improve cell
survival in comparison to CXCR4-only overexpressing ADSCs, but the increase was not
significant (Figure 3D). Indeed, promoting stem cell survival may be important for long-term
therapeutic effect, as both CXCR4-ADSCs and HGF/CXCR4-ADSCs led to similarly improved
limb salvage.
With regards to the angiogenic response, HGF/CXCR4-ADSCs and HGF-ADSCs
promoted revascularization while CXCR4-ADSCs did not, as demonstrated by LDPI, CD31
capillary densitometry, and α-SMA arteriole assessment. This important difference between
CXCR4-ADSCs and HGF/CXCR4-ADSCs should be noted because a healthy angiogenic
response is important in promoting the health of the neuromuscular tissues. In fact, the effect
of HGF on the angiogenic response is well-known [15, 21]. Also, ADSCs are known to
naturally secrete HGF, in addition to a number of angiogenic growth factors, including vascular
endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF). While many of
these factors are upregulated in ADSCs by hypoxic pre-conditioning, HGF is less responsive
[20], which makes it an excellent candidate for genetic upregulation. Curiously, as HGF is also
known for its anti-inflammatory properties, we found that HGF overexpression could not
attenuate inflammation to levels observed by HGF/CXCR4- overexpression. This indicated that
CXCR4 may promote limb salvage by attenuating a damaging inflammatory response; which
generally acts to remove necrotic tissue and debris and could lead to widespread tissue loss, as
that observed by vehicle-ADSCs. Taken together, these data supported HGF and CXCR4
103

overexpression as acting synergistically for promoting long term tissue salvage and repair in
vivo.
The plasmid designed in this study was important in enabling therapeutic upregulation
of both HGF and CXCR4. After exploring both a bi-directional plasmid and a bicistronic
plasmid, we found that the bicistronic plasmid led to higher gene expression of both CXCR4
and HGF. This plasmid was organized with CXCR4 and HGF in sequence under a single CMV
promoter and separated by an internal ribosomal entry site (IRES). Further engineering on the
plasmid may be achieved by focusing on the genetic sequence of each respective gene. Here,
we have used a HGF gene sequence that has already been optimized for dual expression of two
HGF isoforms by alternative splicing [21]. Previously, the alternatively spliced HGF led to
enhanced therapeutic response when injected IM into a rabbit hindlimb ischemia model, in
comparison to single isoforms [21]. The potential for using this dually expressing plasmid was
further demonstrated in our study, where we found that HGF only-ADSCs led to a good
therapeutic response in a severe mouse model of hindlimb ischemia in comparison to vehicle-
ADSC controls. Designing a bicistronic plasmid that co-expressed HGF and CXCR4 using this
gene sequence, effectively enabled the overexpression of three different proteins, one isoform
of CXCR4 and two isoforms of HGF.
Two important criteria for successful clinical translation of genetically engineered stem
cells include: 1) the use of a non-viral gene transfection vector; and 2) an available and abundant
cell population. Non-viral gene transfection vectors may overcome many of the issues
associated with viral vectors, such as immunogenicity and insertional mutagenesis, but tend to
suffer low transfection efficiency and high cytotoxicity. Poly(β-amino esters) (PBAE) were
developed and optimized in response to these shortcomings by combinatorial polymer synthesis
and high-throughput screening [24, 25]. Indeed, we were able to successfully tranfect HGF and
CXCR4 into ADSCs using PBAE with minimal effects on cytotoxicity (Figure 1). An
additional benefit by PBAE transfection is the transient genetic upregulation, suggesting that
the plasmid DNA is not integrated into the genome and would not lead to insertional
mutagenesis.
In terms of cell population, ADSCs represent a relatively abundant, autologous cell
source that are easy to isolate. While this is clear, the therapeutic potential for PAD is still being
explored. Indeed, multiple studies have demonstrated their therapeutic effects in murine models
of a hindlimb ischemia [6, 20, 26, 27]. We further studied the effects of VEGF-overexpressing
ADSCs in vitro and CXCR4-overexpressing ADSCs in vivo [16, 17]. Carrying this forward
104

here, we have demonstrated that HGF/CXCR4-overexpressing ADSCs may further promote
ischemic tissue repair. Overall, these findings, in combination with their ready availability,
support ADSCs as a clinically applicable therapeutic stem cell source.
In summary, this study demonstrates that HGF/CXCR4-overexpressing ADSCs can be
developed using a bicistronic plasmid and biodegradable polymeric vectors. HGF and CXCR4
may synergize to promote ischemic tissue repair, by enhancing stem cell survival, angiogenic
response and ability to attenuate inflammation. Similar strategies may be employed to enhance
other cellular processes for therapeutic response, which may broaden the area of cellular-based
engineering. Overall, this approach has clinical potential for the engineering and regeneration
of many other tissue types, as well as in treating other types of ischemic diseases such as
myocardial infarction and cerebral ischemia.
Supplementary Materials
Fig. S6.1. Plasmid design optimization
Fig. S6.2. Optimization of bicistronic plasmid in ADSCs using poly(β-amino esters) (PBAE)
References
1. Losordo, D.W. and S. Dimmeler, Therapeutic angiogenesis and vasculogenesis for
ischemic disease: part II: cell-based therapies. Circulation, 2004. 109(22): p. 2692-7.
2. Ranganath, S.H., et al., Harnessing the mesenchymal stem cell secretome for the
treatment of cardiovascular disease. Cell Stem Cell, 2012. 10(3): p. 244-58.
3. Kalka, C., et al., Transplantation of ex vivo expanded endothelial progenitor cells for
therapeutic neovascularization. Proceedings of the National Academy of Sciences,
2000. 97(7): p. 3422-3427.
4. Katare, R., et al., Clinical-Grade Human Neural Stem Cells Promote Reparative
Neovascularization in Mouse Models of Hindlimb Ischemia. Arteriosclerosis,
Thrombosis, and Vascular Biology, 2014. 34(2): p. 408-418.
5. Kinnaird, T., et al., Local Delivery of Marrow-Derived Stromal Cells Augments
Collateral Perfusion Through Paracrine Mechanisms. Circulation, 2004. 109(12): p.
1543-1549.
6. Miranville, A., et al., Improvement of postnatal neovascularization by human adipose
tissue-derived stem cells. Circulation, 2004. 110(3): p. 349-55.
105

7. Fadini, G.P., C. Agostini, and A. Avogaro, Autologous stem cell therapy for peripheral
arterial disease: Meta-analysis and systematic review of the literature. Atherosclerosis,
2010. 209(1): p. 10-17.
8. Fischbach, M.A., J.A. Bluestone, and W.A. Lim, Cell-based therapeutics: the next pillar
of medicine. Sci Transl Med, 2013. 5(179): p. 179ps7.
9. Yang, F., et al., Genetic engineering of human stem cells for enhanced angiogenesis
using biodegradable polymeric nanoparticles. Proc Natl Acad Sci U S A, 2010. 107(8):
p. 3317-22.
10. Iwaguro, H., et al., Endothelial Progenitor Cell Vascular Endothelial Growth Factor
Gene Transfer for Vascular Regeneration. Circulation, 2002. 105(6): p. 732-738.
11. Mangi, A.A., et al., Mesenchymal stem cells modified with Akt prevent remodeling and
restore performance of infarcted hearts. Nat Med, 2003. 9(9): p. 1195-201.
12. Yu, J.-X., et al., Combination of stromal-derived factor-1α and vascular endothelial
growth factor gene-modified endothelial progenitor cells is more effective for ischemic
neovascularization. Journal of Vascular Surgery, 2009. 50(3): p. 608-616.
13. Ceradini, D.J., et al., Progenitor cell trafficking is regulated by hypoxic gradients
through HIF-1 induction of SDF-1. Nat Med, 2004. 10(8): p. 858-64.
14. Morishita, R., et al., Phase I/IIa clinical trial of therapeutic angiogenesis using
hepatocyte growth factor gene transfer to treat critical limb ischemia. Arterioscler
Thromb Vasc Biol, 2011. 31(3): p. 713-20.
15. Shigematsu, H., et al., Randomized, double-blind, placebo-controlled clinical trial of
hepatocyte growth factor plasmid for critical limb ischemia. Gene Ther, 2010. 17(9): p.
1152-61.
16. Deveza, L., et al., Paracrine release from nonviral engineered adipose-derived stem cells
promotes endothelial cell survival and migration in vitro. Stem Cells Dev, 2013. 22(3):
p. 483-91.
17. Nauta, A., et al., Adipose-derived stromal cells overexpressing vascular endothelial
growth factor accelerate mouse excisional wound healing. Mol Ther, 2013. 21(2): p.
445-55.
18. Yang, F., et al., Gene delivery to human adult and embryonic cell-derived stem cells
using biodegradable nanoparticulate polymeric vectors. Gene Ther, 2009. 16(4): p. 533-
46.
106

19. Zuk, P.A., et al., Multilineage cells from human adipose tissue: implications for cell-
based therapies. Tissue Eng, 2001. 7(2): p. 211-28.
20. Rehman, J., et al., Secretion of angiogenic and antiapoptotic factors by human adipose
stromal cells. Circulation, 2004. 109(10): p. 1292-8.
21. Pyun, W.B., et al., Naked DNA expressing two isoforms of hepatocyte growth factor
induces collateral artery augmentation in a rabbit model of limb ischemia. Gene Ther,
2010. 17(12): p. 1442-52.
22. Peled, A., et al., Dependence of Human Stem Cell Engraftment and Repopulation of
NOD/SCID Mice on CXCR4. Science, 1999. 283(5403): p. 845-848.
23. Tang, Y.L., et al., Hypoxic Preconditioning Enhances the Benefit of Cardiac Progenitor
Cell Therapy for Treatment of Myocardial Infarction by Inducing CXCR4 Expression.
Circulation Research, 2009. 104(10): p. 1209-1216.
24. Green, J.J., et al., Combinatorial modification of degradable polymers enables
transfection of human cells comparable to adenovirus. Advanced Materials, 2007.
19(19): p. 2836-+.
25. Zugates, G.T., et al., Rapid optimization of gene delivery by parallel end-modification
of poly(beta-amino ester)s. Mol Ther, 2007. 15(7): p. 1306-12.
26. Cao, Y., et al., Human adipose tissue-derived stem cells differentiate into endothelial
cells in vitro and improve postnatal neovascularization in vivo. Biochem Biophys Res
Commun, 2005. 332(2): p. 370-9.
27. Kim, Y., et al., Direct comparison of human mesenchymal stem cells derived from
adipose tissues and bone marrow in mediating neovascularization in response to
vascular ischemia. Cell Physiol Biochem, 2007. 20(6): p. 867-76.
107
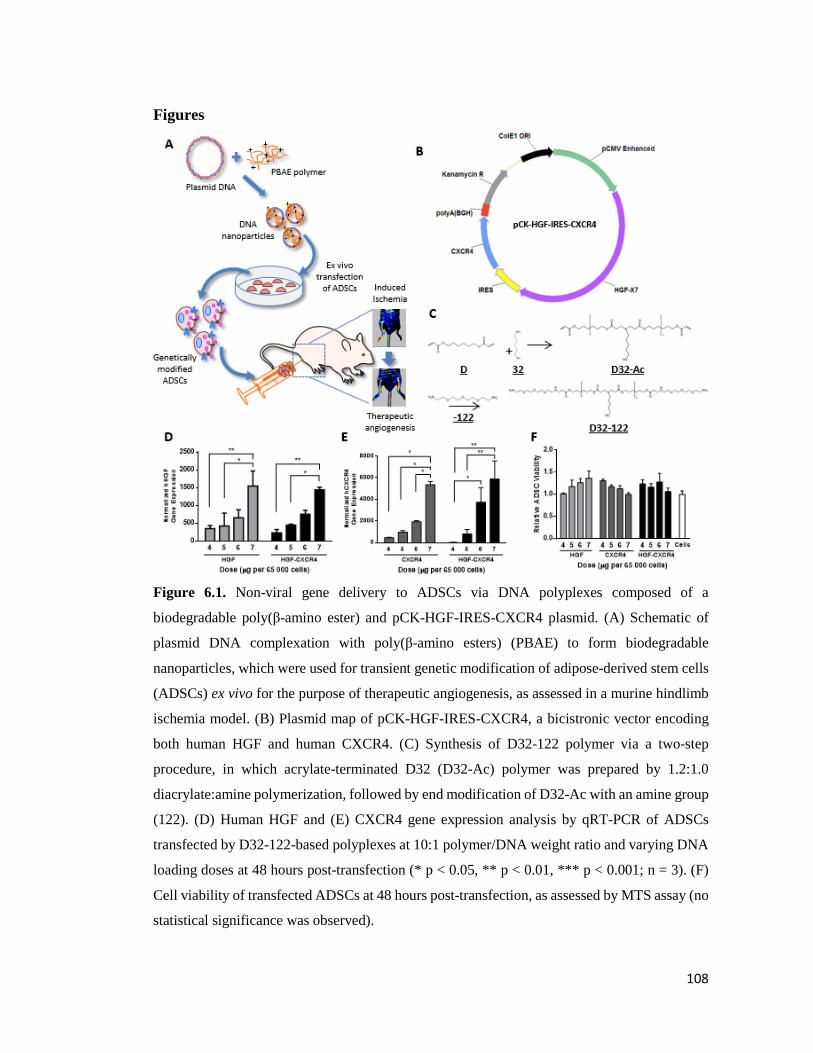
Figures
Figure 6.1. Non-viral gene delivery to ADSCs via DNA polyplexes composed of a
biodegradable poly(β-amino ester) and pCK-HGF-IRES-CXCR4 plasmid. (A) Schematic of
plasmid DNA complexation with poly(β-amino esters) (PBAE) to form biodegradable
nanoparticles, which were used for transient genetic modification of adipose-derived stem cells
(ADSCs) ex vivo for the purpose of therapeutic angiogenesis, as assessed in a murine hindlimb
ischemia model. (B) Plasmid map of pCK-HGF-IRES-CXCR4, a bicistronic vector encoding
both human HGF and human CXCR4. (C) Synthesis of D32-122 polymer via a two-step
procedure, in which acrylate-terminated D32 (D32-Ac) polymer was prepared by 1.2:1.0
diacrylate:amine polymerization, followed by end modification of D32-Ac with an amine group
(122). (D) Human HGF and (E) CXCR4 gene expression analysis by qRT-PCR of ADSCs
transfected by D32-122-based polyplexes at 10:1 polymer/DNA weight ratio and varying DNA
loading doses at 48 hours post-transfection (* p < 0.05, ** p < 0.01, *** p < 0.001; n = 3). (F)
Cell viability of transfected ADSCs at 48 hours post-transfection, as assessed by MTS assay (no
statistical significance was observed).
108

Figure 6.2. Production of functional therapeutic protein from genetically modified ADSCs.
(A) HGF ELISA of conditioned media harvested from genetically modified ADSCs 48 hours
post-transfection (n = 3). (B) Viability of human umbilical cord vein endothelial cells
(HUVECs) cultured in transfected ADSC-conditioned medium as assessed by MTS assay (* p
< 0.05 vs basal media control, n = 4).
109

Figure 6.3. Survival of genetically modified ADSCs post-transplantation in vivo. (A) Human
HGF and (B) human CXCR4 gene expression analysis by qRT-PCR of total mRNA extracted
from gastrocnemius tissue lysate harvested at 48 hours post-transplantation (n = 2). (C)
Representative images from bioluminescence imaging (BLI) to noninvasively track survival of
ADSCs transfected with HGF-CXCR4, HGF, CXCR4, or vehicle control by PBAE polymer
over two weeks. (D) Quantification of BLI signal observed in mice treated with ADSCs at day
4 post-transplantation (* p < 0.05, ** p < 0.01, *** p < 0.001; n = 4-5).
110

Figure 6.4. Angiogenic response in ischemic hindlimbs post-transplantation in vivo. (A)
Representative laser Doppler blood reperfusion images of treatment groups at day 0, 7, and 14.
(B) Quantification of mean blood perfusion ratio of the ischemic foot over the control foot of
ADSC-treated mice (* p < 0.05 vs Vehicle, # p < 0.05 vs. CXCR4, n = 4-5). (C) CD31
immunofluorescent staining of ischemic muscle sections 21 days post-transplantation. Scale
bar: 200 µm. (D) Double immunofluorescent staining of α-SMA and laminin in ischemic muscle
sections 21 days post-transplantation. Scale bar: 200 µm. (E) Quantification of CD31+
capillaries and (F) α-SMA+ vessels in the adductor muscle of the ischemic hindlimb at day 21
(* p < 0.05, *** p < 0.001 compared to vehicle control; # p < 0.05, ## p < 0.01 compared to
CXCR4).
111
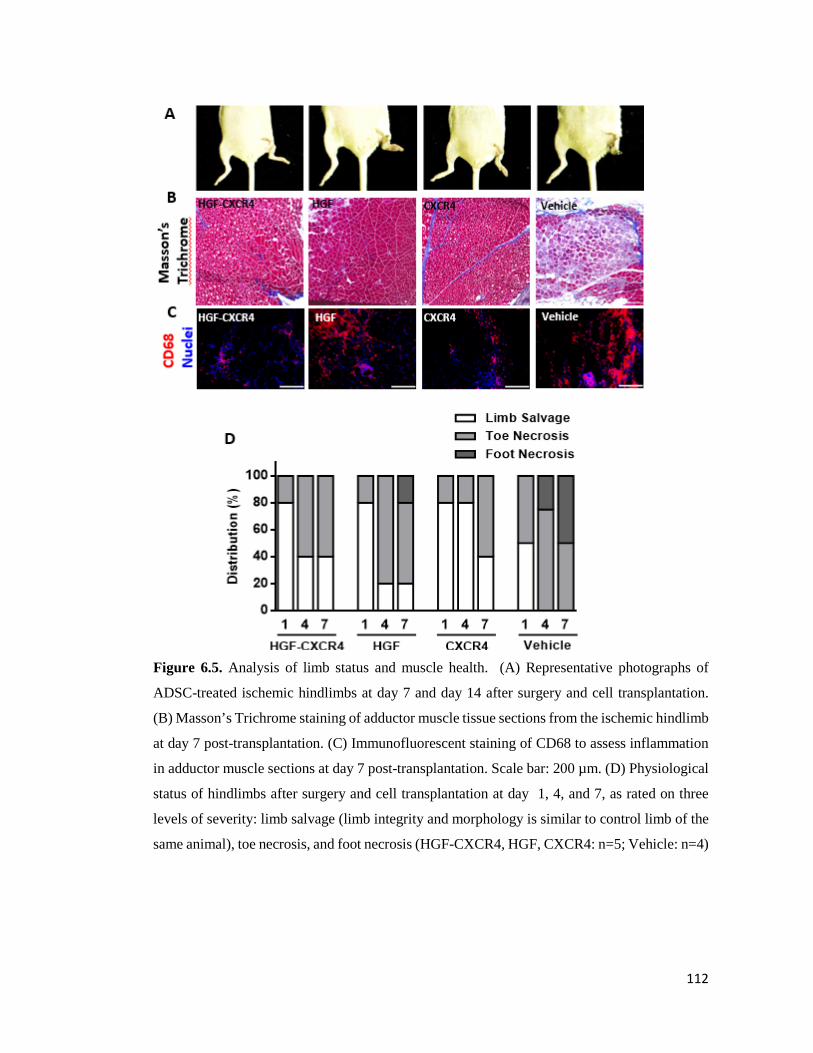
Figure 6.5. Analysis of limb status and muscle health. (A) Representative photographs of
ADSC-treated ischemic hindlimbs at day 7 and day 14 after surgery and cell transplantation.
(B) Masson’s Trichrome staining of adductor muscle tissue sections from the ischemic hindlimb
at day 7 post-transplantation. (C) Immunofluorescent staining of CD68 to assess inflammation
in adductor muscle sections at day 7 post-transplantation. Scale bar: 200 µm. (D) Physiological
status of hindlimbs after surgery and cell transplantation at day 1, 4, and 7, as rated on three
levels of severity: limb salvage (limb integrity and morphology is similar to control limb of the
same animal), toe necrosis, and foot necrosis (HGF-CXCR4, HGF, CXCR4: n=5; Vehicle: n=4)
112

Figure S6.1 – Plasmid Design of bi-directional and bicistronic vectors. Plasmid maps of bi-
directional (A) and bicistronic (B) plasmids. Transfection of each in HEK 293T cells showing
RT-PCR for CXCR4 (C) and HGF (D).
113

Figure S6.2 – Optimization of D32-122 transfection by polymer to DNA weight ratio. RT-
PCR for HGF (A) and CXCR4 (B). MTS cell viability at 48 hours post transfection.
114

7. Recruiting and Programming Endogenous Progenitor Cells in situ
for Therapeutic Angiogenesis
Lorenzo Deveza1,2, Jeffrey Choi3, Jothikritika Ashoken4, Fan Yang2,5
1Department of Bioengineering, Stanford University, Stanford, CA; 2Stanford University
School of Medicine, Stanford, CA; 3Department of Biology, Stanford University, Stanford, CA; 4Department of Biological Sciences, San Jose State University, San Jose, CA; 5 Department of
Orthopaedic Surgery, Stanford University, Stanford, CA.
Abstract
Stem cell therapy holds great promise for therapeutic angiogenesis via secreting a broad
spectrum of paracrine signals to promote new vessel growth. However, current methods often
require cumbersome and expensive procedures of isolating and modifying cells in vitro.
Endogeneous progenitor cells are known to possess the capacity to migrate towards ischemia in
situ under the action of stromal derived factor-1α (SDF-1α). Herein, we assess the potential for
sequential delivery of SDF-1α and DNA for recruiting and programming endogeneous
progenitor cells in situ for therapeutic angiogenesis. We further examine the effects of timing
of plasmid DNA delivery on transfection efficiency in situ. The outcomes of recruiting and
transfecting endogenous cells were evaluated using a murine model of hindlimb ischemia. We
found that controlled SDF-1α release enhanced the recruitment of CXCR4+ and c-kit+
endogenous cells by FACS, which led to improvements in the level and duration of luciferase
transgene expression. Timing of DNA delivery was important, which demonstrated only DNA
delivery at 7 days post-surgery led to enhanced transgene expression. Lastly, we found that the
therapeutic effects on cell recruitment were improved by HGF DNA delivery, which led to
improvements on the angiogenic response and ischemic muscle repair. Overall, this study
demonstrates that the strategy of recruitment and programming may offer an alternative
strategy for promoting angiogenesis by circumventing the need for isolating and
manipulating cells ex vivo.
7.1 Introduction
Peripheral arterial disease (PAD) represents an important medical problem caused by
decreased blood flow to the limbs. Treating PAD usually involves surgical revascularization,
but many patients suffering PAD carry surgically prohibitive co-morbidities. For such patients,
115

stem cell-based therapeutics are a promising, alternative treatment option [1]. This approach
involves stem cell injection into ischemic regions to promote host blood vessel growth in a
process known as therapeutic angiogenesis [2, 3]. Further enhancement on stem cell-based
therapeutics can be achieved by ex vivo genetic manipulation, which has shown promise by
strategically promoting cellular processes involved in angiogenic induction [2-4], such as
angiogenic growth factor release [5-7].
While these approaches have shown potential, clinical translation of stem cell-based
therapeutics has been slow due to several inherent limitations. Firstly, implementing most cell-
based therapies requires multi-step procedures which include: 1) autologous cell isolation; 2) ex
vivo cell expansion and transfection; and 3) transplantation back to the patient. In practice, this
requires multiple facilities with good laboratory practices (GLP), as well as multiple skilled
professionals, thereby leading to high cost and significant regulatory concerns. Secondly, the
technical aspects can be challenging, as ex vivo genetic manipulation of stem cells can be
inefficient and cytotoxic, and the majority of the cells die during re-injection.
One strategy to overcome these limitations is to exploit natural cell homing mechanisms
to attract endogeneous progenitor cells to sites of ischemic injury in order to replenish the stem
cell pool [8]. This is a process that has been shown to occur naturally, whereby bone marrow-
derived cell populations are recruited to sites of hypoxia to participate in the reparative process
[9-11]. Many researchers have proposed methods to augment systemic cell homing to ischemic
tissue as a way of supporting the local niche for therapeutic angiogenesis and tissue engineering
[8, 9]. The potential of using cell homing strategies for treating cardiovascular disease has been
successfully demonstrated using mobilization agents and chemokines in mouse models of
ischemic heart disease [12-14]. However, during states of cardiovascular disease, host cells
often have reduced functionality and a compromised cell niche that affects their ability for
therapeutic response [9].
In a similar manner that genetic manipulation of stem cells ex vivo can improve their
therapeutic effects, endogenous progenitor cell therapeutics might be enhanced by genetic
modification. Intramuscular (IM) injection of plasmid DNA to promote therapeutic transgene
expression has long been a strategy for therapeutic angiogenesis [15]. Indeed, multiple clinical
trials are currently being conducted to evaluate the potential of gene therapy for ischemic heart
disease and peripheral arterial disease; and are evaluating growth factors such as: basic
fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF) and hepatocyte
growth factor (HGF). In terms of the cells transfected in situ, plasmid DNA mediated delivery
116

to skeletal muscle has been shown to preferentially transfect regenerating muscle fibers [16, 17]
and non-inflammatory, infiltrating mononuclear cells [18]. In the setting of ischemic injury,
both of these cells increase in number; which are contributed by multiple sources, including
interstitial muscle satellite cells [19] and circulating progenitor cells [19, 20]. As this is the
case, it is very likely then that recruited endogenous progenitor cells can be transfected by IM
plasmid DNA delivery during ischemia.
Based on these findings, we hypothesize that endogenous progenitor cells could be
recruited to sites of ischemic injury and subsequently transfected to promote therapeutic
angiogenesis. Specifically, we propose to develop a biomaterials depot for sequential delivery
of stromal derived factor-1α (SDF-1α) and plasmid DNA for recruiting and programming
endogeneous progenitor cells in situ. The platform is evaluated in a murine model of hindlimb
ischemia. We initially evaluate cell recruitment by FACS and assess in situ transfection by
bioluminescence imaging (BLI). We further examine the effects of timing of plasmid DNA
delivery on transfection efficiency in situ. We lastly evaluate the potential for therapeutic
efficacy using a plasmid encoding for HGF.
7.2 Materials and Methods
7.2.1 Materials
Fibrinogen and thrombin were purchased from Sigma–Aldrich (St Louis, MO).
Recombinant murine stromal derived factor-1α (SDF-1α) was purchased from Peprotech
(Rocky Hill, NJ). Plasmids (pEGFP-N1 and pCMV-LUC) were purchased from Elim
Biopharmaceuticals; pCK-HGF plasmid was obtained from Viromed (Seoul, Korea).
7.2.2 Preparation of SDF-1α Encapsulated Fibrin Gels
SDF-1α encapsulated fibrin gels were prepared by premixing two solutions: 1)
Fibrinogen (50 mg/ml) + SDF-1α (5 µg/ml); and 2) Thrombin (2 U/ml). The two solutions were
then mixed together to create a solution with final concentrations of 25 mg/ml Fibrinogen, 2.5
µg/ml SDF-1α, and 1 U/ml Thrombin. The resulting solution (200 µl) was injected into mice
prior to polymerization. SDF-1α release from varying concentration Fibrin gels (10 mg/ml, 25
mg/ml, and 50 mg/ml) was assessed in vitro by preparing gels in a 50 µl gel mold and incubating
in a 48 well plate at 4 ºC. SDF-1α was released in a collection media containing 1 mg/ml BSA
and quantified by SDF-1α ELISA (Peprotech).
117

7.2.3 Induction of hindlimb ischemia and therapeutic injections
All experiments were performed in accordance with the Stanford University Animal
Care and Use Committee Guidelines and approved APLAC protocols. Hindlimb ischemia was
induced in female C57/Bl mice aged 7-9 weeks (Jackson Laboratory) as previously described
[21]. Briefly, the femoral artery was ligated at two sites (distal to the external iliac artery and
proximal to the popliteal artery) with 5-0 silk suture (Ethicon). The femoral artery was excised
between the points of ligation. One day after arterial dissection, 200 µL of Fibrin/SDF-1α
solution (25 mg/ml Fibrin, 500 ng SDF-1α) was injected intramuscularly into two sites (the
adductor muscle region and the gastrocnemius) using a 27-gauge tuberculin syringes. After a
specified time delay post-Fibrin/SDF-1α injection (0 days, 2 days, 6 days), 10 µg plasmid DNA
(pEGFP-N1, pCMV-LUC, or pCK-HGF) was injected into the same muscles. These time points
effectively represented 1 day, 3 days, and 7 days post-surgery.
7.2.4 Tissue harvest and FACS assessement of recruited cell populations
To assess recruited cell populations, cells were isolated from hindlimbs and analyzed
by flow cytometry (FACS). Cells were isolated by harvesting hindlimbs and then removing and
mincing all limb muscles. Minced limb muscles were then digested using 0.1 mg/ml
Collagenase type II (Invitrogen, Carlsbad, CA) on a shaker at 37 ºC for 45 min. Digestion was
then continued using 1 mg/ml Collagenase/Dispase (Roche Life Sciences, Branford, CT) on
shaker at 37ºC for another 90 min. The resulting digested tissue was then serially passed through
three cell strainers (100 µm, 70 µm, 40 µm) to remove debris and non-digested tissue lysate.
Cells were then centrifuged to remove the supernatant. Upon re-suspending the cells were split
into separate round bottom tubes for FACS analysis. Cell staining was performed by incubating
cells with one or three antibodies: CD45-PerCP, CD34-FITC, c-kit-PE, CXCR4-PE (all from
BD Pharmingen, San Jose, CA). Triple color staining was performed for either: 1) CD34, CD45,
CXCR4; or 2) CD34, CD45, c-kit. Single color staining or unstained cells were used as controls.
After incubating for 30 minutes, antibodies were removed and replaced with 300 µl Hank’s
Balanced Salt Solution (HBSS, Life Technologies, Carlsbad, CA). Stained cells were subjected
to flow cytometry using a LSR II.UV flow cytometer. Post-FACS analysis was performed using
FlowJo v7.6.5 software.
118

7.2.5 Bioluminescence Imaging
In vivo transfection with pCMV-LUC reporter plasmid was assessed by
bioluminescence imaging (BLI). Animals were anesthetized by 2–3% inhaled isoflurane and
injected intraperitoneally with the reporter probe D-Luciferin at 100 mg/kg body weight. IVIS
Spectrum system (Xenogen, Alameda, CA) was used to image the animals while under 2%
inhaled isoflurane anesthesia. Each animal was scanned until the peak signal was reached,
recording radiance of regions of interest in photons. Radiance was quantified in photons per
second per centimeter squared per steridian.
7.2.6 Protein extraction, ELISA and Western Blot
Protein was extracted from the gastrocnemius muscle of the ischemic hindlimb, which
was harvested at 5 or 14 days post-surgery. Harvested muscle was snap-frozen by collecting
into an 1.5 ml Eppendorf tube and subsequently submerging into liquid nitrogen. Samples were
then stored at -80 ºC. Protein was then extracted by directly submerging the muscle into liquid
nitrogen and then grinding it using a mortar and pestle. At least 50-80 mg of the ground muscle
was then collected into a new Eppendorf, into which 500 µl of RIPA Lysis and Extraction buffer
(Thermoscientific, Waltham, VA) containing 1x Halt Protease and Phosphatase Inhibitor
Cocktail (Thermoscientific) was added. The tissue was vortexed and pipetted to further break
up the muscle. The tissue was then centrifuged, and the supernatant was collected and stored
for protein analysis. Total protein content was measured using Bio-Rad Protein assay (Bio-Rad
Laboratories, Hercules, CA). HGF ELISA was performed by diluting the sample 2x and then
assaying according to the manufacturer’s protocol (R&D Systems, Minneapolis, MN). Western
Blot was performed by loading 6 µg of protein per sample and separating proteins using sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, Invitrogen). Proteins were
then transferred to polyvinylidine fluoride (PVDF) (Invitrogen) membranes overnight. PVDF
membranes were stained with antibodies for CD31, alpha smooth muscle actin (α-SMA) or beta
actin (β-actin) as a loading control (all from Abcam, Cambridge, England). Horse radish
peroxidase (HRP) conjugated secondary antibodies (Abcam) were used for labeling.
Supersignal West Pico Chemiluminescent substrate (Thermoscientific) was applied to the
washed membranes, which were then assessed by X-ray film using CL-XPosure Film
(Thermoscientific).
119

7.2.7 Histology and Immunohistochemistry
Mice were euthanized at 14 days to retrieve muscle tissue from ischemic limbs for
histological analysis. Specimens were embedded in optimal cutting temperature (O.C.T.) for
cryosectioning. Sections were cut at 8 µm thickness. General tissue morphology was assessed
with Masson’s Trichrome Staining and immunofluorescence staining for Laminin (Abcam),
using a FITC-labeled secondary antibody. Angiogenesis was assessed by staining for capillaries
(CD31, Abcam) and arterioles (α-SMA, Abcam) Cells transfected with pEGFP-N1 were
assessed by immunostaining with anti-GFP (Abcam), using a FITC-labeled secondary antibody.
Sections were mounted with Vectashield Mounting Medium with 4’,6-diamidino-2-
phenylindole (DAPI) (Vector Laboratories, Burlingame, CA) for counterstain. Sections were
imaged using a fluorescent microscope. The number of CD31-positive vessels was quantified
to estimate capillary density per number of cell nuclei using MATLAB software (MathWorks,
Natick, MA).
7.2.9 Assessment of Limb Status
Ischemic limbs were evaluated by direct observation of limb status. Limbs were
assessed on days 1, 3, 7, 10, 14 post-ischemic induction. Grading of limb ischemia was based
on the severity of limb ischemia utilizing following categories: 1) limb salvage; 2) toe necrosis;
3) foot necrosis; 4) foot loss; and 5) limb loss. Limb salvage is plotted by showing the fraction
of each category per group (n = 5).
7.2.10 Statistical Analysis
GraphPad Prism (Graphpad Software, San Diego, CA, USA) was used to perform all
statistical analyses. One-way analysis of variance with Tukey's multiple comparison test as a
post-hoc test was performed to compare all experimental groups and determine statistical
significance. Number samples per study was as follows: FACS analyses (n = 3); BLI
assessements (n = 3); therapeutic studies (n = 5). Each study was repeated to confirm results.
All quantitative data were expressed as mean ± standard error of the mean.
7.3 Results
7.3.1 Controlled Release of SDF-1α from Fibrin Gels
To protect and control the release of SDF-1α in vivo, SDF-1α was encapsulated into
injectable fibrin gels prior to delivery to the ischemic hindlimb (Figure 1A). The in vitro release
120

kinetics of SDF-1α from fibrin gels were studied to correlate timing of release with subsequent
cell recruitment and transfections. This demonstrated that the release was bi-phasic, showing a
burst release of nearly 70% on the first day, followed by a more gradual release over the next
3-4 days (Figure 1B). As the fibrin concentration did not significantly affect release, fibrin gels
composed of 25 mg/ml were used for all remaining experiments.
7.3.2 SDF-1α Enhances Cell Recruitment Post-ischemic Induction
The effect of SDF-1α treatment on in vivo cell recruitment was studied by injecting
SDF-1α/fibrin gels into ischemic hindlimbs at day 1 post-surgery and performing FACS
analysis on harvested tissues at days 1, 3, 7 (Figure 1C-1E). Recruited populations were
analyzed by staining for CD34, CD45, CXCR4 and c-kit. CD34 and CD45 were chosen as
general markers for progenitor and inflammatory cells, while CXCR4 and c-kit were chosen
because these populations have been shown to respond to SDF-1α (Figure 1C-1E). Overall,
ischemia was a strong stimulus for recruiting cells, which demonstrated increases in CXCR4+,
c-kit+, CD34-/CD45+/CXCR4+, and CD34-/CD45+/c-kit+ cell populations by days 3 and 7
compared to sham surgery controls (p < 0.05; Figure 1D, 1E). SDF-1α treatment further
improved recruitment of these populations compared to ischemia only and sham surgery
controls, which demonstrated increases at day 3 and 7; with a persistently high percentage of
CXCR4+ cells by day 7 (p < 0.05). We also observed an increase of CD45+ cells by SDF-1α
treatment at days 3 and 7 compared to ischemia only and sham surgery controls (p < 0.05). In
terms of cell recruitment kinetics, these data demonstrated that the peak cell recruitment was
around 3 days, but persisted towards 7 days with SDF-1α treatment. Overall, this indicated that
SDF-1α could enhance cell recruitment.
7.3.3 SDF-1α Promotes In Situ Transfection by Plasmid DNA Delivery
The effect of SDF-1α treatment on in situ plasmid DNA transfection was studied by
injecting SDF-1α/fibrin gels at day 1 post-surgery and then injecting plasmid DNA encoding
for either GFP or Luciferase at day 7 post-surgery (Figure 2A). Day 7 was chosen because cell
recruitment by SDF-1α treatment was enhanced compared to ischemia at this time point.
Analysis of GFP or Luciferase transfection was performed the day after plasmid DNA injection
by FACS or BLI assessment, respectively. Both FACS and BLI demonstrated increases in
transfection in situ by SDF-1α treatment compared to ischemia only (Figure 2B, 2C).
Specifically, FACS analysis demonstrated a ~2% increase in GFP+ cells by SDF-1α treatment
121

compared to ischemia only (p < 0.05; Figure 2B), while BLI data demonstrated a ~2-fold log
increase in luminescence by SDF-1α treatment compared to ischemia only (p < 0.05; Figure
2C). These data indicated that there were increases in both the number of cells transfected and
the amount of protein produced in ischemic hindlimbs treated with SDF-1α/fibrin gels, thereby
providing evidence that SDF-1α could promote plasmid DNA transfection in situ.
7.3.4 In Situ Transfected Cells are Largely Represented by the Recruited Cells
To assess whether the cell populations transfected were the same as those recruited, the
in situ transfected GFP+ populations were analyzed to determine their relative expression of
CD34, CD45, CXCR4, and c-kit (Figure 2D, 2E). We found that the transfected GFP+ cell
populations expressed many of the same markers as those assessed for cell recruitment (Figure
2D). Specifically, the GFP+ populations expressed the following: 50-60% CXCR4+; 50-60%
c-kit+; 30-40% CD34-/CD45+/CXCR4+, and 30-40% CD34-/CD45+/c-kit+ (Figure 2E). We
also determined that there was no significant difference in GFP+ transfected populations
whether SDF-1α was delivered or not (Figure 2E). Immunostaining was performed to determine
the relative location of transfected cells, which demonstrated that many of the transfected cells
were mononuclear cells that were located around regenerating muscle fibers (Figure 2F). This
was consistent with the FACS analysis, as c-kit+ or CXCR4+ cells represent mononuclear
inflammatory or progenitor cell populations. Overall, this provided evidence that the in situ
transfected cell populations were largely represented by the recruited cells.
7.3.5 Timing of Plasmid DNA Delivery Affects the Level and Duration of Transfection
The timing of plasmid DNA delivery was optimized by injecting plasmid DNA
encoding luciferase into ischemic hindlimbs at one of three different time points (days 1, 3, 7)
and then assessing for the level and duration of luciferase expression (Figure 3A). We found
that the timing of plasmid DNA delivery had a significant effect on luciferase expression, which
was lowest when delivered at day 1 and highest when delivered at day 7 (p < 0.05; Figure 3D,
3E). SDF-1α treatment was able to improve luciferase expression at each time point compared
to ischemia only (p < 0.05; Figure 3D, 3E), thereby further demonstrating the ability to use
SDF-1α to improve in situ transfection. In terms of the duration of transfections, we found that
plasmid DNA delivered at day 1 post-surgery only lasted one week, which was slightly
improved by SDF-1α treatment (Figure 3F). On the other hand, plasmid DNA that was
delivered at day 7 led to a robust and prolonged transfection response that persisted over 2 weeks
122

regardless of whether SDF-1α was delivered. Importantly, day 3 plasmid DNA injection was
strongly affected by SDF-1α treatment, where we found that the duration of luciferase
expression only persisted when SDF-1α was used (Figure 3F). Furthermore, the level of
luciferase expression from day 3 plasmid DNA injected hindlimbs treated with SDF-1α was
similar to the levels achieved by day 7 plasmid DNA injection. This suggested that SDF-1α
treatment could be used to enable earlier plasmid DNA injections for persistent transgene
expression.
7.3.6 Enhanced HGF Transfection In Situ by SDF-1α Delivery
To assess whether SDF-1α treatment could promote transgene expression of a
therapeutic growth factor, plasmid DNA encoding HGF (pHGF) was injected into mice at 3
days post-surgery and assessed for transfection on the fifth day (Figure 4A). Compared to
ischemia only, we found that SDF-1α treatment increased HGF release in ischemic hindlimbs
(p < 0.05; Figure 4B). Injecting pHGF alone at day 3 also led to increases in HGF release which
were significant compared to SDF-1α only and PBS controls (p < 0.05; Figure 4B). This
indicated that both treatment approaches, whether by timing plasmid DNA delivery or applying
SDF-1α were able to promote transgene expression of a therapeutic growth factor.
7.3.7 Angiogenic Improvement in the Ischemic Hindlimb
To assess whether promoting HGF transgene expression by SDF-1α treatment would
lead to an angiogenic response, day 14 tissue samples were analyzed for capillaries and
arterioles (Figure 5A). Compared to SDF-1α only and PBS controls, HGF transgene expression
led to an observable increase in both CD31+ capillaries (Figure 5B) and α-SMA+ arterioles
(Figure 5C) as demonstrated by immunofluorescence staining. CD31 quantification
demonstrated that the increase in capillaries due to HGF overexpression was statistically
significant compared to SDF-1α only and PBS controls (p < 0.05; Figure 5D). Western blotting
analysis of proteins extracted from muscle further confirmed that CD31 and α-SMA were
increased by HGF overexpression compared to SDF-1α only and PBS controls (Figure 5E). This
demonstrated the HGF transgene expression by SDF-1α treatment enhanced the angiogenic
response.
123

7.3.8 Promotion of Limb Health
The therapeutic effects of HGF transgene expression by SDF-1α pre-treatment were
assessed by evaluating limb status and by evaluating histological features of muscle health.
Limb status assessment demonstrated that the baseline response to ischemic induction was
moderate toe and foot necrosis, which presented around day 7 post-surgery and became
progressively worse over the next 7 days (PBS group; Figure 6B, 6E). Progressive limb damage
beyond day 7 was prevented by injecting SDF-1α (F/SDF; Figure 6E) or by HGF transgene
expression (F/SDF + HGF; Figure 6E), which were comparable. Notably, HGF transgene
expression led to improved muscle health and morphology compared to SDF-1α and PBS
controls, as demonstrated by Masson’s Trichrome staining and anti-Laminin
immunofluorescence staining (Figure 6C, 6D). This was clearly observable by anti-Laminin
staining, which demonstrated excellent overall muscle morphology with intact basement
membranes (Figure 6D). Overall, this suggested that HGF transgene expression by SDF-1α
could promote muscle health and lead to therapeutic effects.
7.4 Discussion
Herein, we demonstrated that scaffold-mediated release of SDF-1α followed by delayed
plasmid DNA injection offers a promising platform for recruiting and programming endogenous
progenitor cells in situ. Cell recruitment was strongly enhanced by ischemia and SDF-1α
delivery, which led to improvements in transgene expression in situ. The timing of plasmid
DNA delivery post-ischemia had a large effect on the level and duration of transgene expression,
where we found that SDF-1α had the potential to enable earlier transfection. As validation for
this approach, transfection with HGF was shown to promote therapeutic efficacy in a murine
model of hindlimb ischemia. These results demonstrate that such platforms may offer an
alternative strategy for promoting angiogenesis by circumventing the need for isolating and
manipulating cells ex vivo.
The strategy of recruiting and programming endogenous progenitor cells in situ relies
on two sequential processes: 1) initial cell homing; and 2) delayed transgene expression. In
many ways, the potential for using this approach was highlighted by the ability to improve
transgene expression by firstly inducing ischemia and secondly delivering DNA at a later time.
Here, the initial cell homing response is achieved by ischemia, which is a well-known stimulus
for recruiting inflammatory and progenitor cells [9]. This is further supported by our findings
that the CXCR4+ and c-kit+ populations increased over time due to ischemia compared to sham
124

surgery controls. As these populations peaked around the third to seventh day post-ischemic
induction, this suggested that it was the optimal time to deliver DNA for transfecting these cells.
Indeed, delayed transgene expression was achieved by delivering DNA at seven days post-
ischemic induction, which led to significant increases in transgene expression in situ. Further
analysis demonstrated that many of the transfected cells expressed CXCR4 and c-kit, suggesting
these were among the recruited populations. Overall, the finding that ischemic injury improves
transgene expression was consistent with other studies showing the same in skeletal muscle
injured by other mechanisms, such as bupivacaine injection [17] or cardiotoxin injection [16].
Each of these injuries may similarly lead to enhanced transfection by promotion of endogenous
cell recruitment and stem cell niche activation.
In our study, we exogenously delivered SDF-1α protein as a way for further promoting
cell recruitment and subsequent transfection. SDF-1α was chosen because it is one of the most
well-known chemokines for mediating cell homing to ischemia [22]. We found that
exogenously delivering SDF-1α one day post-ischemic induction was able to further promote
endogenous cell recruitment compared to with ischemia alone, demonstrating increases in the
CXCR4+ and c-kit+ populations. Indeed, these are known responders to SDF-1α [11, 13, 14,
23, 24]. These improvements were associated with better transfection of both luciferase and
HGF transfection, thereby supporting the strategy for promoting transfection by enhancing
endogenous cell recruitment using SDF-1α. This finding also opens many possibilities for
exploring other chemokines.
One of the major findings in this study was the effect of DNA delivery timing on the
ability for long-term transgene expression after inducing ischemia. In particular, we found that
delivering DNA too early after ischemic-injury led to short-term transgene expression, while
delivering DNA one week later enhanced transfection beyond that achieved in non-injured
muscles. Furthermore, pre-treatment of the ischemic muscle with SDF-1α enabled earlier
transgene expression (i.e. day 3 DNA delivery), which did not last when SDF-1α was not
delivered. These findings are pertinent for multiple reasons. Firstly, this suggests that there is
an optimal time to deliver DNA to treat acute injury (i.e. acute myocardial infarction or bone
fracture). Secondly, it demonstrates that DNA could be delivered earlier by SDF-1α treatment,
which may be important for therapeutic gene therapy when earlier treatment is warranted.
Thirdly, it suggests that injury may be a mechanism to enhance gene therapy, which may be
useful in therapeutic situations in which injury is purposely induced (i.e. transmyocardial laser
revascularization or microfractures for cartilage repair).
125

The therapeutic potential for genetically manipulating endogenous progenitor cells was
evaluated in a murine model of hindlimb ischemia. As with other studies, cell recruitment by
SDF-1α release could promote limb salvage. Further therapeutic effects were achieved by
programming endogenous cells with HGF, which is an angiogenic factor known to act on both
endothelial cells and smooth muscle cells; thereby promoting both capillaries and arterioles [25,
26]. Indeed, we found that this was the case, as HGF-modified endogenous cells increased both
types of vessels, which also led to therapeutic enhancement in the overall muscle health of the
ischemic hindlimb. This demonstrated that programming endogenous stem cells has promise
for treating peripheral arterial disease by therapeutic angiogenesis.
This study opens up the potential for many future studies and directions. Some of these
future directions, include: exploring other chemokines or therapeutic genes; developing dual
and sequential controlled release platform; and, studying the mechanism to better understand
the observations of this study. With regards to exploring other chemokines or therapeutic genes,
we chose to focus on SDF-1α, but many other proteins may achieve a similar enhancement on
transgene expression in situ, opening up the possibility for exploring these proteins. Also, the
field of gene therapy for inducing angiogenesis has demonstrated multiple factors for
upregulation, which could be explored upon initial cytokine delivery. With regards to a
controlled release platform, we achieved sequential release by controlled SDF-1α release and
delayed plasmid DNA injection, but a dual, sequential delivery platform would be more ideal
to truly enable a one-step procedure. Lastly, further study on mechanism would help us to more
specifically define which cell populations are transfected and why early DNA delivery post-
injury does not lead to prolonged transgene expression. This would enable us to design a better
strategy for achieving transgene expression in situ.
In conclusion, this study demonstrates the potential for using scaffold-mediated release
of SDF-1α and delayed DNA injection to program endogenous cells in situ. This strategy may
be useful for inducing therapeutic angiogenesis to treat ischemic diseases, such as PAD, which
can be an alternative to stem cell-based therapeutics. Additionally, it shows promise for other
tissue engineering approaches, such as wound or bone repair.
126

References
1. Losordo, D.W. and S. Dimmeler, Therapeutic angiogenesis and vasculogenesis for
ischemic disease: part II: cell-based therapies. Circulation, 2004. 109(22): p. 2692-7.
2. Ranganath, S.H., et al., Harnessing the mesenchymal stem cell secretome for the
treatment of cardiovascular disease. Cell Stem Cell, 2012. 10(3): p. 244-58.
3. Deveza, L., J. Choi, and F. Yang, Therapeutic angiogenesis for treating cardiovascular
diseases. Theranostics, 2012. 2(8): p. 801-14.
4. Fischbach, M.A., J.A. Bluestone, and W.A. Lim, Cell-based therapeutics: the next pillar
of medicine. Sci Transl Med, 2013. 5(179): p. 179ps7.
5. Deveza, L., et al., Paracrine release from nonviral engineered adipose-derived stem cells
promotes endothelial cell survival and migration in vitro. Stem Cells Dev, 2013. 22(3):
p. 483-91.
6. Nauta, A., et al., Adipose-derived stromal cells overexpressing vascular endothelial
growth factor accelerate mouse excisional wound healing. Mol Ther, 2013. 21(2): p.
445-55.
7. Yang, F., et al., Genetic engineering of human stem cells for enhanced angiogenesis
using biodegradable polymeric nanoparticles. Proc Natl Acad Sci U S A, 2010. 107(8):
p. 3317-22.
8. Chen, F.M., et al., Homing of endogenous stem/progenitor cells for in situ tissue
regeneration: Promises, strategies, and translational perspectives. Biomaterials, 2011.
32(12): p. 3189-209.
9. Dimmeler, S., Regulation of bone marrow-derived vascular progenitor cell mobilization
and maintenance. Arterioscler Thromb Vasc Biol, 2010. 30(6): p. 1088-93.
10. Takahashi, T., et al., Ischemia- and cytokine-induced mobilization of bone marrow-
derived endothelial progenitor cells for neovascularization. Nat Med, 1999. 5(4): p.
434-8.
11. Ceradini, D.J., et al., Progenitor cell trafficking is regulated by hypoxic gradients
through HIF-1 induction of SDF-1. Nat Med, 2004. 10(8): p. 858-64.
12. Orlic, D., et al., Mobilized bone marrow cells repair the infarcted heart, improving
function and survival. Proc Natl Acad Sci U S A, 2001. 98(18): p. 10344-9.
13. Zaruba, M.M., et al., Synergy between CD26/DPP-IV inhibition and G-CSF improves
cardiac function after acute myocardial infarction. Cell Stem Cell, 2009. 4(4): p. 313-
23.
127

14. Zhang, G., et al., Controlled release of stromal cell-derived factor-1 alpha in situ
increases c-kit+ cell homing to the infarcted heart. Tissue Eng, 2007. 13(8): p. 2063-
71.
15. Losordo, D.W. and S. Dimmeler, Therapeutic angiogenesis and vasculogenesis for
ischemic disease. Part I: angiogenic cytokines. Circulation, 2004. 109(21): p. 2487-91.
16. Davis, H.L., C.L. Millan, and S.C. Watkins, Immune-mediated destruction of
transfected muscle fibers after direct gene transfer with antigen-expressing plasmid
DNA. Gene Ther, 1997. 4(3): p. 181-8.
17. Vitadello, M., et al., Gene transfer in regenerating muscle. Hum Gene Ther, 1994. 5(1):
p. 11-8.
18. Ratanamart, J., C.G. Huggins, and J.A. Shaw, Transgene expression in mononuclear
muscle cells not infiltrating inflammatory cells following intramuscular plasmid gene
electrotransfer. J Gene Med, 2010. 12(4): p. 377-84.
19. Sherwood, R.I., et al., Determinants of skeletal muscle contributions from circulating
cells, bone marrow cells, and hematopoietic stem cells. Stem Cells, 2004. 22(7): p.
1292-304.
20. Cerletti, M., et al., Highly efficient, functional engraftment of skeletal muscle stem cells
in dystrophic muscles. Cell, 2008. 134(1): p. 37-47.
21. Niiyama, H., et al., Murine model of hindlimb ischemia. J Vis Exp, 2009(23).
22. Petit, I., D. Jin, and S. Rafii, The SDF-1-CXCR4 signaling pathway: a molecular hub
modulating neo-angiogenesis. Trends Immunol, 2007. 28(7): p. 299-307.
23. Askari, A.T., et al., Effect of stromal-cell-derived factor 1 on stem-cell homing and
tissue regeneration in ischaemic cardiomyopathy. The Lancet, 2003. 362(9385): p. 697-
703.
24. Jin, D.K., et al., Cytokine-mediated deployment of SDF-1 induces revascularization
through recruitment of CXCR4+ hemangiocytes. Nat Med, 2006. 12(5): p. 557-67.
25. Morishita, R., et al., Phase I/IIa clinical trial of therapeutic angiogenesis using
hepatocyte growth factor gene transfer to treat critical limb ischemia. Arterioscler
Thromb Vasc Biol, 2011. 31(3): p. 713-20.
26. Pyun, W.B., et al., Naked DNA expressing two isoforms of hepatocyte growth factor
induces collateral artery augmentation in a rabbit model of limb ischemia. Gene Ther,
2010. 17(12): p. 1442-52.
128

Figures
Figure 7.1 SDF-1α Enhances Cell Recruitment Post-ischemic Induction. (A) Study design
showing injection of SDF-1α encapsulated in fibrin gels at one day post-ischemic induction,
followed by delayed plasmid DNA injection (10 µg). (B) Controlled release of SDF-1α from
synthesized fibrin gels in vitro. (C-E) FACS assessment of endogenous progenitor cell
recruitment to ischemic limbs at (C) day 1, (D) day 3, and (E) day 7 (* p < 0.05, n = 3).
129

Figure 7.2 SDF-1α Promotes In Situ Transfection of recruited endogenous cells (A) Study
design showing injection of SDF-1α encapsulated in fibrin gels at one day post-ischemic
induction, followed by day 7 plasmid DNA injection (10 µg EGFP or luciferase). (B) FACS
analysis of GFP+ transfected cells in harvested muscle at one day post-GFP injection. (C)
BLI assessment of luciferase expression in ischemic limbs at one day post-luciferase injection.
(D) FACS plots demonstrating further analysis and gating of GFP+ populations for CD34,
CD45, CXCR4 and c-kit. (E) Quantitative analysis of GFP+ populations. (F)
Immunostaining for GFP, Laminin to demonstrate location of transfected cells in situ. Dapi is
used as a counter-stain. (* p < 0.05, n = 3).
130

Figure 7.3 Timing of Plasmid DNA Delivery Affects the Level and Duration of Transfection.
(A-C) Schematic demonstrating study design for (A) day 1 luciferase injection, (B) day 3
luciferase injection, and (C) day 7 luciferase injection. (D) BLI images and (E) quantitative
lumuninescence in ischemic limbs one day post-luciferase injection, with and without SDF-1α.
(F) Kinetics of luciferase transgene expression when injected at specified time points post-
ischemic induction, with and without SDF-1α. (* p < 0.05, n = 3-5)
131

Figure 7.4 Enhanced HGF Transfection In Situ by SDF-1α Delivery. (A) Schematics
demonstrating timing of SDF-1α delivery and plasmid HGF injection. Mice were harvested
two-days post-HGF injection for analysis. (B) ELISA analysis of HGF release in harvested
muscle. (* p < 0.05, n = 3)
132

Figure 7.5 Angiogenic Improvement in the Ischemic Hindlimb by HGF overexpression. (A)
Schematic of therapeutic model study design demonstrating timing of SDF-1α and HGF
delivery. Tissue were harvested at day 14 post-surgery for analysis. (B) CD31
immunostaining and (C) α-SMA immunostaining for capillaries and arterioles, respectively.
(D) Capillary densitometry assessment represented as the ratio of CD31+ capillaries to DAPI+
cell nuclei (p < 0.05). (E) Representative western blot analysis of CD31 and α-SMA in
ischemic hindlimbs (n= 3; H: HGF + Fibrin/SDF; FS: Fibrin/SDF only; P: PBS treatment
only).
133

Figure 7.6 Promotion of limb salvage. (A) Schematic of therapeutic model study design
demonstrating timing of SDF-1α delivery and subsequent HGF injections. Tissue were
harvested at day 14 post-surgery for analysis. (B) Images showing foot and limb morphology
of ischemic limbs. (C) Masson’s Trichrome staining of harvested ischemic muscle. (D)
Laminin (green) staining on harvested muscle demonstrating muscle architecture and integrity,
DAPI (blue) was used as counterstain. (E) Quantitative limb status over two weeks,
demonstrating fraction of total mice with foot necrosis, toe necrosis, or limb salvage (n = 9).
134

135
8. Microfluidic Synthesis of Biodegradable Polyethylene-Glycol Microspheres for
Controlled Delivery of Growth Factors and DNA Nanoparticles
Lorenzo Deveza1,2, Jothikritika Ashoken3, Gloria Castaneda4,
Xinming Tong4, Li-Hsin Han4, Fan Yang1,4
1Department of Bioengineering, Stanford University, Stanford, CA; 2Stanford University School
of Medicine, Stanford, CA; 3Department of Biological Sciences, San Jose State University, San
Jose, CA; 4Department of Orthopaedic Surgery, Stanford University, Stanford, CA.
Abstract
Polymeric microparticles represent an injectable platform for controlled release of biologics.
The ability to synthesize uniform and homogeneous polymeric microparticles under mild
conditions is important for tuning release in a reliable manner. Microparticle diameter or
polymer character could then be varied to control release of the encapsulated biologics. In this
study, we report the development of monodisperse, biodegradable poly(ethylene glycol) (PEG)-
based microparticles for controlled release of growth factors or DNA nanoparticles using a
microfluidic platform. We found that simple changes in microfluidic design could speed the
rate of microparticle formation (>10-fold increase), as well as control the size of the
microparticles ranging from ~50-150 µm. By using thiol-ene crosslinkable PEG, microparticles
with homogeneous matrices were synthesized. Mesh size and degradation rate could be
controlled by varying PEG concentration from 7.5% to 15 % (W/V). Both microparticle
diameter and mesh size were varied to tune the release of basic fibroblast growth factor, which
retained its biological activity of stimulating cell proliferation when applied to human adipose-
derived stem cells. Controlled release of DNA nanoparticles encoding vascular endothelial
growth factor demonstrated the ability to transfect HEK293T cells. This platform may provide
a useful tool for synthesizing monodisperse microparticles as injectable carriers to achieve
coordinated growth factor or DNA nanoparticle release, which may be broadly useful for
guiding desirable cell fates and tissue regeneration in situ.
8.1 Introduction
In tissue regeneration and repair, soluble cues such as growth factors play an
important role in guiding cell fate, requiring well-orchestrated doses and timing. To
achieve such delivery characteristics, polymeric microparticles containing different

136
biologics, such as protein or DNA nanoparticles, with well-tuned release kinetics
represent a facile method for temporal release in vivo. While a number of factors are
known and employed to tune the release of biologics from polymeric microparticles, the
ability to generate homogeneous polymer matrices and the ability to synthesize uniform
microparticles are fundamental to reliably generate microparticles with desired release
kinetics. Once established, changes in polymer physical and chemical characteristics, as
well as changes in microparticle size and morphology may be exploited to tune growth
factor release.
Amongst the microfabrication approaches for synthesizing polymeric microparticles,
droplet microfluidics represents a relatively simple and consistent method for generating
uniform microparticles.[1] With this approach, polymeric microparticles containing multiple
reagents and biologics can be synthesized by adding polymer and biomolecules to the
discontinuous phase and polymerizing the resulting droplets.[2-7] Microparticle size and
morphology can be easily controlled by adjusting the input flow rates or the microchannel
dimensions.[6] Monodisperse drug releasing microparticles synthesized in this manner have
demonstrated better control over drug release than microparticles generated via simple water-
in-oil emulsion, where microparticle size has been shown to have an effect on drug release
characteristics.[4, 6] Of the systems previously studied, [2, 4-7] most focus on encapsulating
and releasing a small molecule. Those that have focused on releasing a protein have been mainly
polysaccharide-based microparticles and have generally used a model protein (i.e. BSA) that is
not assessed for bioactivity. As polysaccharide-based hydrogel systems lead to heterogeneous
networks, many monodisperse polysaccharide-based microparticles fail to achieve consistent
and prolonged release characteristics.
Polyethylene glycol (PEG) based hydrogel matrices are potentially more
homogeneous than other biodegradable hydrogels owing to being a synthetic polymer
with minimal biomolecule affinity and being easily modified chemically. As a drug
delivery platform, various approaches have been explored, such as physical entrapment,
inclusion of a protein binding molecule (i.e. heparin sulfate), and covalently linking the
protein to the gel. Amongst these, physical entrapment represents a low-cost and less-
complicated approach, which generally relies on encapsulating protein within a PEG
mesh diameter of comparable or smaller size and releasing the protein upon degradation
of the PEG network. PEG matrices that have these characteristics can be polymerized
via step-growth or condensation reactions using PEG precursors that have been

137
functionalized with a hydrolytically degradable moiety. Step-growth polymerized PEG
matrices can generate more homogeneous network structures, which may lead to more
consistent and reliable release kinetics and may have further in vivo benefit due to having
less renal-toxicity owing to smaller molecular weight degradation products. [8]
Furthermore, degradation rates can be tuned by simply varying PEG concentration,
which can effectively tune release kinetics.
In this study, we synthesized biodegradable PEG-based, monodisperse
microparticles for controlled release of growth factors or DNA nanoparticles using a
microfluidic device. We hypothesize that release of each from PEG microparticles could
be tuned by varying the polymer concentration in order to tune the mesh size and
degradability of the PEG matrix, as well as by using monodisperse microparticles with
controlled diameter. To study this, various microfluidic devices were designed for
rapidly synthesizing microparticles of different sizes. The effects of varying microsphere
size and PEG concentration on release kinetics were then assessed. Lastly, the biological
activity of released basic fibroblast growth factor (bFGF) was assessed using a cell
proliferation assay, while the ability for DNA nanoparticles to transfect were tested
against HEK293T cells.
8.2 Experimental Section
8.2.1 Materials
Polyethylene glycol (PEG, 8-arm, 10 kDa) was purchased from Jenkem
Technology USA (Allen, TX). Thioglycolic acid, norbornene acid, and L-cysteine were
purchased from Sigma-Aldrich (St. Louis, MO). Lithium arylphosphanate (LAP) was
synthesized as previously described. [9] Novec 7500 engineered fluid and Novec 7000
engineered fluid were purchased from 3M (St Paul, MN). Dupont Krytox 157 FSH was
purchased from Miller-Stephenson (Danbury, CT). The PEG-PFPE-PEG tri-block
surfactant was synthesized as previously described. [10] Fluorescein-conjugated bovine
serum albumin (FITC-BSA) was purchased from Life Technologies (Carlsbad, CA).
Recombinant basic fibroblast growth factor (bFGF) and bFGF ELISA kit were purchased
from Peprotech (Rocky Hill, NJ). Cell Titer Aqueous One Solution Cell Proliferation
Assay (MTS) was purchased from Promega (Madison, WI).

138
8.2.2 Microfluidic Device Fabrication
The microfluidic devices were fabricated using standard soft lithography and
micromolding techniques at the Stanford Microfluidics Foundry clean room. A
transparency mask was printed using a high-resolution printer (Fineline Imaging,
Colorado Springs, CO). Master molds were fabricated with one layer of negative SU8
photoresist (MicroChem, Newton, MA) on a silicon wafer (100 µm). The microfluidic
device was fabricated using PDMS (Sylgard 184, Midland, MI) micromolding against
the master mold. Sharpened needles (20 gauge) were used to punch inlets and outlets.
The molded PDMS layer was plasma bonded to a glass slide that was coated with an
additional layer of PDMS.
8.2.3 PEG functionalization
8-arm-PEG-Norbornene (8 arm PEG-NB) and 8-arm-PEG-Mercaptoacetic acid
(8 arm PEG-MAA) were synthesized as previously described.[11] Briefly, 8-arm PEG-
MAA was synthesized by reacting 8-arm PEG with mercaptoacetic acid in toluene. 5.0
g 8-arm PEG was dissolved in 150 mL toluene, followed by addition of 34 mg p-
toluenesulfonic acid. After adding 1.74 mL 3-mercaptopropionic, the solution was
refluxed overnight. The azeotropic mixture was collected periodically. After cooling the
solution to room temperature, the product was precipitated in ice-cold diethyl ether. 8-
arm PEG-NB was synthesized by reacting 8-arm PEG with norbornene acid following
the same procedure of 8-arm PEG MAA.
8.2.4 Microparticle Synthesis
Microparticles were synthesized using a T-junction droplet break-up microfluidic
device designed with various size input channels (50 μm, 100 μm, 150 μm, 200 μm).
The discontinuous phase consisted of 8 arm PEG-NB and 8 arm PEG-MAA in 1:1
stochiometric ratio and dissolved at various ratios (7.5 wt%, 10 wt%, 12.5 wt%, 15 wt%).
The final solutions incorporated 0.1% LAP photointiator in the discontinuous phase. The
continuous phase consisted of 2% PEG-PFPE-PEG in Novec 7500. The solutions were
run through the microfluidic channels at ~1.0 psi / ~1.0 psi (water phase / oil phase).
Synthesized spheres were collected in an oil bath with the same composition as the
continuous phase positioned under a broad spectrum, high intensity UV lamp. Novec
7500 was extracted by mixing with Novec 7000 (5 mL per wash) then removing the oil

139
phase after each wash cycle. This process was repeated five times. The residual Novec
7000 was allowed to evaporate in a water bath at 37 ºC. The resulting microparticles
were diluted in 2 mg/ml L-cysteine and then lyophilized. Prior to use, microparticles
were resolubilized in PBS at 2.5% (W/V).
8.2.5 Swelling Ratio and Mesh Size Determination
The equilibrium degree of swelling for each hydrogel was acquired in order to
calculate PEG mesh size. PEG hydrogels (100 μL) were polymerized under UV light and
their mass after swelling (MS) was measured. The hydrogels were then lyophilized and
their dry mass (MD) was measured. The mass swell ratio (QM) was determined by taking
the ratio of the swollen hydrogel mass to dry hydrogel mass. The mass swell ratio was
determined for hydrogels composed of various PEG wt% (7.5 wt%, 10 wt%, 12.5 wt%,
15 wt%).
Hydrogel mesh size (ξ) was calculated from the mass swell ratio using Flory-Rehner
calculations as previously described.[12] Firstly, the mass swell ratio was converted to a
volumetric swell ratio (QV) using Equation 1.
𝑄𝑉 = 1 +𝜌𝑝
𝜌𝑆(𝑄𝑀 − 1) (1)
where ρp is the density of the dry hydrogel (1.12 g/cm3 for PEG)3 and ρs is the density of the
solvent (1 g/cm3 for water).
Secondly, molecular weight between cross-links (MC) was calculated using Equation 2.
1
𝑀𝑐̅̅ ̅̅=
2
𝑀𝑛̅̅ ̅̅̅−
�̅�
𝑉1(ln(1−𝑣2)+𝑣2+𝜒1𝑣2
2)
𝑣2
13⁄ −
𝑣24
(2)
where 𝑀𝑛̅̅ ̅̅ is the number-average molecular weight of the un-cross-linked hydrogel (the
molecular weight of the polymer), V1 is the molar volume of the solvent (18 cm3/mol for
water), ν2 is the polymer volume fraction in the equilibrium swollen hydrogel (1/QV), �̅�
is the specific volume of the polymer (ρs / ρp), and χ1 is the polymer-solvent interaction
parameter (0.426 for PEG-water).[12]
Lastly, the mesh size was calculated using Equation 3.
𝜉 = 𝑣213⁄ 𝑙𝐶𝑛
12⁄ (2
𝑀𝐶̅̅ ̅̅ ̅
𝑀𝑟)12⁄
(3)
Where l is the average PEG bond length (0.146 nm),[12] Cn is the characteristic ratio of
PEG (4.0), and Mr is the molecular weight of the PEG repeat unit (44 for PEG).

140
8.2.6 Microparticle Diameter Measurements
The average microparticle diameter was measured by image processing.
Microparticles were synthesized using varying size input channels (50 μm, 100 μm, 150
μm, 200 μm). Images were taken at 10x magnification. Images were analysed utilizing
MatLab Software (Natick, MA). Briefly, images were converted to black and white
based upon contiguous pixels. Microsphere area was then determined by quantifying the
number of pixels per microsphere and converting to micrometers. Sphere diameter was
then calculated. Ten images per group were assessed.
8.2.7 Protein Encapsulation and Release:
Protein encapsulation was assessed by adding FITC-labelled bovine serum
albumin (BSA) (0.1 mg/ml) or basic fibroblast growth factor (bFGF) (1.0 mg/ml) into
the discontinuous phase prior to microparticle synthesis. To assess protein encapsulation
efficiency, total BSA encapsulated was collected by dissolving the microparticles with
0.1 N NaOH and balancing the pH with 0.1 N HCL and phosphate buffered saline (PBS).
To determine the release kinetics of bFGF, microparticles were plated in the upper well
of a transwell plate (Corning, NY) with 1.0 mL PBS containing 1mg/ml BSA in the lower
well which comprised the collection medium. Released bFGF into the lower well was
collected at various time intervals over 10 days and replenished with fresh collection
medium. The amount of bFGF released was quantified by bFGF ELISA.
8.2.8 Cell Proliferation
The bioactivity of bFGF released from microparticles was determined by
assessing the mitogenic response of adipose-derived stem cells (ADSCs). ADSCs were
plated in a 96-well plate at a seeding density of 10,000 cells per well. After 24 hours,
the culture medium was replaced with 100 µl of collected released medium from various
time points, mixed with 50 µl of Dulbecco’s Modified Eagle Medium (DMEM,
Invitrogen, Carlsbad, CA) containing 2% fetal bovine serum (FBS, Invitrogen) per well,
and cultured for another 48 hours in a humidified chamber at 37 ºC. Cell proliferation
was assessed by MTS assay.

141
8.3 Results
8.3.1 Microfluidic Fabrication of PEG Microparticles
Three types of droplet microfluidic designs were evaluated for their ability to
efficiently synthesize monodisperse PEG microparticles in a repeatable and consistent
fashion (Figure 1). Essentially, the designs were composed of two main parts: 1) a
water-in-oil junction (i.e. either a t-junction or a flow-focusing design); and 2) an
extended output channel. This latter part was either a straight channel of the same
channel dimensions as the input channels, or an open channel that widens to roughly 7x
the dimension of the input channels (Figure 1A, 1B). We found that the volume of
spheres synthesized per hour increased from ~200 µl/hr to ~1400 µl/hr for the straight
channel and open channel designs, respectively (input channel width of 100 µm and input
pressures ~1.0 psi / ~1.0 psi, water/oil). To assess whether increasing the height of the
channel would further increase rates, we also compared microchannels of 100 µm height
with microchannels 150 µm height, but found the increased height led to jetting and could
not efficiently generate droplets. When comparing the t-junction to the flow-focusing
design (Figure 1B, 1C), we did not observe a significant difference in their ability to
efficiently synthesize monodisperse microparticles. However, due to the relative ease of
using and re-using the t-junction design, we chose to use the t-junction design with the
open extended output channel for the remaining experiments (Figure 1C).
The procedure for synthesizing monodisperse PEG-based microparticles is shown in
Figure 1D. The full description is provided in the experimental section. Briefly, end-
functionalized 8-arm PEG polymers were solubilized in water, which composed the
discontinuous phase. The generated droplets fell directly into an oil bath and were polymerized
under UV light. The synthesized microparticles underwent an oil-extraction procedure and then
were re-solubilized in an L-cysteine solution to prevent microparticle flocculation (i.e. covalent
bond formation between microparticles). The resulting solution was freeze-dried and then re-
solubilized in PBS prior to use.
8.3.2 Controlling microparticle diameter and mesh size
To control the microparticle diameter, four microfluidic devices were fabricated based
on the same design, which varied by input channel dimensions (i.e. 50 µm, 100 µm, 150 µm,
200 µm) (Figure 2). Using these four input channel dimensions and running the microfluidic
with input pressures of ~1.0 psi / ~1.0 psi (water pressure / oil pressure), microparticles with the

142
following average diameters were synthesized: ~42.4 µm, ~77.6 µm, ~120.1 µm, ~142.1 µm,
respectively (Figure 2C, 2D). As demonstrated in Figure 2C, the synthesized microparticles
were homogeneous and were easily controlled by varying input channel dimensions. Additional
tuning of microparticle diameter was achieved by varying the input pressures or flow rates.
Applying both of these strategies could lead to a broad range of microparticle size from ~25 μm
to ~200 μm.
To control PEG mesh size and degradation, the PEG concentration was varied in the
precursor solution ranging from 7.5% W/V to 15.0% W/V (Figure 3). Two types of end-
functionalized 8-arm PEG polymers with ester containing functional groups were used.
Polymerization of these two PEG polymers occurred via a thiol-ene reaction, which proceeds
as a step-growth reaction. As opposed to the radical chain polymerization scheme, step-growth
polymerization is expected to yield more homogeneous networks. We utilized this property to
synthesize PEG matrices with predicted mesh sizes that are on the order of protein
hydrodynamic radii (~2 nm). As demonstrated in Figure 3B, increasing PEG concentration
caused a progressive decrease in swell ratio, which corresponded with calculated mesh sizes of
~2.2 nm to ~0.9 nm (Figure 3B). To confirm degradation of the PEG matrices, swell ratio was
measured after incubating for 4 days in serum-free cell culture medium. At 4 days, the swell
ratio of each PEG hydrogel increased, which is consistent with a bulk degradation process and
would be expected for hydrolytically degradable hydrogels. At these swell ratios, the theoretical
mesh size for each group increased above 2 nm, except for that of 15 PEG wt%. Considering
the relative size of many growth factors, many growth factors would be free to diffuse through
these PEG matrices by this time point.
8.3.3 Protein encapsulation, release and bioactivity
To assess protein encapsulation, fluorescein-conjugated bovine serum albumin
(FITC-BSA) was encapsulated into PEG microparticles and confirmed by fluorescence
microscopy (Figure 4). It was found that the relative amount of FITC-BSA encapsulated
per microsphere did vary, which is demonstrated by the higher intensity of FITC in some
microparticles relative to the others. Protein encapsulation efficiency was then
quantified by digesting the PEG microspheres and measuring the amount of FITC-BSA
encapsulated compared to the amount used in the precursor solution. We found that
encapsulation efficiencies of varying concentration ranged from 19.7 ± 5.8 % to 23.2 ±

143
4.3% (Figure 4B). Furthermore, we found that varying microsphere size did not
significantly change protein encapsulation efficiency.
To assess growth factor release kinetics, we encapsulated basic fibroblast growth
factor (bFGF) into PEG microparticles with varying size or concentration and quantified
the accumulated release using ELISA (Figure 5). Microparticles fabricated using the 100
µm channel resulted in the highest level of protein release, with over 60% of encapsulated
growth factors released by day 10. In contrast, microparticles fabricated using 150 or 200
µm channels showed comparable slowed down protein release, with ~20% protein
released by day 10 (Fig. 5A). Decreasing PEG concentration generally resulted in more
rapid release, with 7.5% (W/V) PEG-based microspheres leading to ~70% accumulated
protein release by day 10 (Fig. 5B). In general, smaller microparticles and those
composed of lower PEG wt% released their contents most quickly. Of further note,
because of the type of degradation process (i.e. bulk degradation), it was interesting to
observe for any sudden boosts in release. Upon inspection, it was noted that around day
4, some of the release curves did experience a subtle increase in release rates. This
correlated with the increase in swell ratio by day 4 as shown in Figure 3. Still, much of
the bFGF was still retained in PEG matrices beyond this time point as suggested by the
release curves.
To confirm that the bioactivity of encapsulated bFGF was still retained upon
synthesis of PEG microparticles and subsequent release, the released bFGF was applied
to adipose-derived stem cells (ADSCs) and assayed for cell proliferation over 48 hours.
Compared to ADSCs with no bFGF treatment, ADSCs treated with the released
supernatant from all time points led to an increase in ADSC proliferation that was
comparable to positive controls treated with 10 ng/ml bFGF. The effect of timing on
bFGF release and ADSC proliferation was subtle, and might be due to the potency of
bFGF on ADSC. Consistent with the observation of a small increase in bFGF release
around day 4, we see that ADSC proliferation also increased around this time point. This
provided further evidence that the release of bFGF was delayed and remained bioactive
during processing and release.
8.3.4 DNA nanoparticle encapsulation and transfection
DNA nanoparticle encapsulated microparticles were synthesized by preparing
nanoparticles in a precursor solution containing various PEG wt % and flowing the

144
resulting solution through the microfluidic chip (Figure 6A). DNA nanoparticles were
prepared using D32-122 poly(β-amino ester) (PBAE) at a polymer to DNA weight ratio
of 20:1. Upon synthesis and re-solubilization, the resulting DNA nanoparticle
concentration was 70 µg per ml of microspheres.
The ability of encapsulated DNA nanoparticle to transfect was tested using DNA
nanoparticles encoding luciferase and vascular endothelial growth factor (VEGF).
Microparticles (200 µl or 14 µg DNA nanoparticles) were co-incubated with HEK293T
cells in transwell culture (Microparticles in the upper well and HEK293T cells in the
lower well). Medium was collected over 8 days and assayed for luciferase expression
(Figure 6B) and VEGF release (Figure 6C). Luciferase expression and VEGF release
correlated with PEG wt%, demonstrating more rapid increase in transfection by 7.5 wt%
PEG and slower transfection using 12.5 wt% PEG. This demonstrated that DNA
nanoparticles remained bioactive during encapsulation and release, and further that
release of DNA nanoparticles could be controlled by PEG wt%.
8.4 Discussion
Herein, we report the synthesis of biodegradable PEG-based, monodisperse
microparticles for controlled growth factor delivery and DNA nanoparticles with tunable release
kinetics using microfluidics. Due to the uniformity of the microparticles and the homogeneity
of the PEG matrices, this approach may serve as a reliable and consistent method for preparing
injectable growth factor delivery depots. Of particular interest is that controlling microparticle
properties was relatively simple and robust, and throughout the processing conditions the
encapsulated biologics remained bioactive (Figure 5C). Here, two microparticle properties were
controlled: 1) the mesh size of the hydrogel; and 2) the microparticle diameter. The first was
controlled by varying PEG concentration in the precursor solution; and, the second was
controlled by using microfluidic channels of differing dimensions. Varying these two
parameters considerably varied the rate of growth factor release and DNA nanoparticle release.
Given the bio-inertness of PEG and the mildness of the processing conditions, such a platform
may be useful for encapsulating and releasing a broad range of growth factors, as well as various
other biological molecules or cells.
Microparticle mesh size and degradation rate were easily controlled by varying PEG
concentration in the precursor solution, which correspondingly affected the rate of growth factor
release and DNA nanoparticle release. Controlling mesh size by varying polymer concentration

145
is a common strategy for controlling release, which has been demonstrated in many other studies
using bulk PEG hydrogels. Using this strategy in a microparticle formulation enables this
property to be used as an injectable formulation, in which separate microparticles could be
designed with different PEG concentrations. Indeed, we found that lower concentration PEG
wt% led to increased biologic release of both basic fibroblast growth factor and DNA
nanoparticles, while higher PEG wt% led to the opposite. Importantly, this led to differing rates
in transfection of HEK293T cells. These findings demonstrate the potential for generating
mixed formulations to control the release of multiple biologics, which otherwise cannot be
achieved by a bulk hydrogel.
PEG has many advantages to other polymers because it may be easily chemically
modified, which would provide another level of control over biologic encapsulation and release
[8]. Indeed, thiol-ene chemistry enables the easy incorporation of multiple types of functionality
by simply incorporating biomolecules containing a free thiol. For example, L-cysteine was
actually required in this experiment because of the spontaenous covalent crosslinking between
neighboring microparticles due to unreacted thiol groups. This piece of development also
demonstrated microparticles could be easily surface-modified by re-solubilizing the
microparticles with any other type of molecule that contained a thiol, such as a bio-functional
peptide. By the same token, functionality can be incorporated within the PEG microparticle by
adding a functional peptide or biomolecule containing a thiol to the precursor solution. By
taking advantage of this property, growth factor release could be further modulated by
incorporating protein binding biomolecules or even covalently linking the growth factor directly
to the microparticle.
Simple modifications to microfluidic design can be used to increase the rate of
microparticle synthesis and enable more robust control of size. To increase rate of microparticle
synthesis, the resistance of the output channel post-droplet formation was decreased by
expanding its width. This simple change led to increased rates of microparticle formation by
over ten-fold. To control microparticle size, multiple microfluidic chips were designed with
varying input channel dimensions. In comparison to adjusting input flow rates, we found this
to be a more robust approach. This is because relying on input flow rates requires more accuracy
between the water and oil channels. While accuracy can be easily achieved by a syringe pump,
a pneumatic controller might be a more efficient approach to use when running multiple
microfluidic devices in parallel, which would be subject to subtle changes in air pressure. We
demonstrated that such an approach was able to rapidly synthesize monodisperse microparticles

146
of varying sizes, which may support the use of the microfluidic device as a manufacturing
platform.
In summary, here we report a droplet microfluidics platform that allows synthesis of
biodegradable PEG-based microspheres for controlled release of proteins and DNA
nanoparticles. We further demonstrated our ability to synthesize monodisperse microspheres
by tuning the width of microfluidics channel width. PEG microsphere compositions can be
easily modulated by varying PEG concentration in the precursor solution. We demonstrated
gradual protein release from PEG-based microspheres, and the release kinetics can be further
turned by varying PEG composition and microsphere size. Given the bio-inertness of PEG, such
platform may be useful for encapsulating and releasing a broad range of growth factors or DNA
nanoparticles. Further chemical modification to the PEG composition may provide another level
of control over biologic encapsulation and release, and may be easily incorporated into the
present system. To achieve temporal release of multiple biologics to enhance tissue
regeneration, microparticles with different release kinetics or loaded with different cargos may
be easily combined to provide an injectable drug delivery system for treating a broad range of
diseases.
Acknowledgements
The authors would like to thank American Heart Association National Scientist
Development Grant (10SDG2600001) and Stanford Medical Scholars Research Program
for funding. The authors would also like to thank Stanford Microfluidics Foundry for
technical assistance with microchip fabrication.
References
1. Thorsen, T., et al., Dynamic pattern formation in a vesicle-generating microfluidic
device. Phys Rev Lett, 2001. 86(18): p. 4163-6.
2. De Geest, B.G., et al., Synthesis of monodisperse biodegradable microgels in
microfluidic devices. Langmuir, 2005. 21(23): p. 10275-9.
3. He, T., et al., A modified microfluidic chip for fabrication of paclitaxel-loaded poly(l-
lactic acid) microspheres. Microfluidics and Nanofluidics, 2011. 10(6): p. 1289-1298.
4. Huang, K.S., et al., Microfluidic controlling monodisperse microdroplet for 5-
fluorouracil loaded genipin-gelatin microcapsules. J Control Release, 2009. 137(1): p.
15-9.

147
5. Kesselman, L.R.B., et al., Synthesis of Monodisperse, Covalently Cross-Linked,
Degradable “Smart” Microgels Using Microfluidics. Small, 2012. 8(7): p. 1092-1098.
6. Xu, Q., et al., Preparation of monodisperse biodegradable polymer microparticles using
a microfluidic flow-focusing device for controlled drug delivery. Small, 2009. 5(13): p.
1575-81.
7. Yang, C.-H., K.-S. Huang, and J.-Y. Chang, Manufacturing monodisperse chitosan
microparticles containing ampicillin using a microchannel chip. Biomedical
Microdevices, 2007. 9(2): p. 253-259.
8. Lin, C.-C. and K. Anseth, PEG Hydrogels for the Controlled Release of Biomolecules
in Regenerative Medicine. Pharmaceutical Research, 2009. 26(3): p. 631-643.
9. Fairbanks, B.D., et al., Photoinitiated polymerization of PEG-diacrylate with lithium
phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility.
Biomaterials, 2009. 30(35): p. 6702-7.
10. Holtze, C., et al., Biocompatible surfactants for water-in-fluorocarbon emulsions. Lab
Chip, 2008. 8(10): p. 1632-9.
11. Fairbanks, B.D., et al., A Versatile Synthetic Extracellular Matrix Mimic via Thiol-
Norbornene Photopolymerization. Advanced Materials, 2009. 21(48): p. 5005-5010.
12. Zustiak, S.P. and J.B. Leach, Hydrolytically degradable poly(ethylene glycol) hydrogel
scaffolds with tunable degradation and mechanical properties. Biomacromolecules,
2010. 11(5): p. 1348-57.

148
Figures
Figure 8.1. Schematics of microfluidic chip design for microsphere synthesis. (A-C) Design of
the three droplet microfluidic chips assessed. (A, C) Two t-junction designs with differing
output channel widths. (B) A flow-focusing device with an wide output channel. A snapshot
of microsphere formation in each microfluidic chip is shown on the right. (D) Schematic of
fabrication process of polyethylene glycol (PEG)-based microspheres. 8-arm PEG-norbornene
(PEG-NB) and 8-arm PEG-mercaptoacetic acid (PEG-MAA) were added to the continuous
phase in 1:1 stochiometric ratio. Upon exiting the microfluidics chip, the PEG microspheres
were collected in oil bath and photocrosslinked using UV light. Microspheres are further
processed by an oil extraction procedure followed by lyophilization. Image shows microsphere
formation after re-solubilizing.

149
Figure 8.2. Control of microsphere size by adjusting microchip in put channel dimensions (50
µm, 100 µm, 150 µm and 200 µm). (A, B) Schematic of the t-junction open-channel design and
corresponding dimensions. (C) Microspheres synthesized based on the four different input
dimensions. Scale Bar: 200 µm. (D) Averaged diameters of microspheres synthesized using
microfluidics chip with varying microchannel width. (* p < 0.05)

150
Figure 8.3. Effect of varying PEG concentration on microsphere property including swelling
ratio and microgel mesh size at days 0 and 4. Increasing PEG concentration led to decreased
mass swelling ratio (A) and decreased mesh size (B), which increased by day 4 indicating
hydrogel degradation. (*** p < 0.001)

151
Figure 8.4. Protein encapsulation in biodegradable PEG microspheres of differing size and
polymer composition. (A) Protein encapsulation was confirmed using fluorescein-conjugated
bovine serum albumin (FITC-BSA). Images demonstrate FITC-BSA encapsulation in two
different sized microparticles. (B) Protein encapsulation efficiency was about 19.7 ± 5.8 % to
23.2 ± 4.3%, and did not significantly vary in microparticles of different PEG composition.

152
Figure 8.5. Release kinetics and bioactivity of bFGF from biodegradable PEG microspheres of
varying size and PEG concentration. (A) Effects of varying microsphere size on bFGF release
over 10 days. (B) Effects of varying PEG concentration on bFGF release over 10 days. (C)
Bioactivity of released bFGF over 10 days was retained, as shown by the cell proliferation assay
using adipose-derived stem cell (ADSC). Cells without FGF (-) or with fresh FGF (+) were
included as controls. (* p < 0.05).

153
Figure 8.6. Transfection by DNA nanoparticles encapsulated and released from biodegradable
PEG microspheres of varying polymer weight percent (W/V). (A) Schematic illustrating the
formation of DNA nanoparticles in a PEG precursor solution prior to synthesis of microspheres.
(B) Transfection of HEK 293T cells by released DNA nanoparticles encoding luciferase from
PEG microspheres of varying polymer weight percent (W/V). (C) VEGF transfection of
HEK293T cells by released microspheres encapsulating DNA nanoparticles encoding VEGF.

9. Summary, Limitations, and Future Directions
9.1 Summary
Over the course of this thesis, we explored two different strategies to harness stem cells
as drug delivery vehicles for therapeutic angiogenesis. The first platform explored the potential
for genetically modifying adipose-derived stem cells (ADSCs) with angiogenic factors ex vivo
using poly(β-amino esters) (PBAE). The second platform explored the potential for sequential
release of chemokine and DNA to recruit and program endogenous progenitor cells in situ.
9.1.1 Summary of the non-viral engineered ADSCs approach
With regards to the ex vivo modified stem cell platform, we focused on optimizing the
following components: 1) cell source; 2) non-viral transfection agent; and, 3) selection of an
appropriate therapeutic gene. In terms of cell source, we focused on ADSCs due to their relative
abundance, ease of isolation and demonstrated angiogenic potential. In our studies, we found
that paracrine release from ADSCs alone could promote multiple endothelial cell processes of
angiogenesis in vitro. We further found that ADSCs behaved well under hypoxia, whereby their
proliferation potential and angiogenic growth factor production increase. Our results are
supported by many others that have also demonstrated the therapeutic potential of ADSCs,
which indicate that ADSCs are potentially useful for treating ischemic disease.
ADSCs were genetically modified using poly(β-amino esters) (PBAE), a non-viral
transfection vector. PBAE/DNA nanoparticles were optimized for transfection into ADSCs,
including human and mouse ADSCs of multiple cell isolations. We found that transfection was
dependent on the cell type and culture conditions, and could also be optimized by varying
transfection parameters such as polymer to DNA weight ratio and DNA dose. We also found
that ADSC proliferation correlated with their ability to transfect and that promoting proliferation
could enhance their transfection efficiency. Under the optimized conditions, PBAE led to high
transfection efficiency and low cytoxicity in ADSCs compared to the commercially available
gold standard, Lipofectamine 2000. These transfection conditions enabled us to transfect
multiple therapeutic genes on different plasmid constructs into ADSCs, which demonstrated the
versatility of using PBAE for genetic upregulation in ADSCs.
The main therapeutic genes we explored were vascular endothelial growth factor
(VEGF), CXC chemokine receptor 4 (CXCR4), and hepatocyte growth factor (HGF). Initially,
VEGF was chosen because it had been the most well-studied angiogenic growth factor. We
154

found that VEGF-overexpressing ADSCs could enhance the therapeutic effects of ADSCs
compared to ADSCs alone in vitro (chapter 4), but did not perform as well in vivo (chapter 5).
VEGF-overexpressing ADSCs led to enhanced inflammation, which proved to be quite
damaging and led to detrimental consequences on ischemic limb salvage. CXCR4 was explored
for its ability to enhance ADSCs as drug delivery vehicles to ischemia. When CXCR4 was co-
expressed with VEGF, we found that this improved ischemic limb salvage compared to VEGF-
overexpression alone in vivo, but overall the two genes did not seem to synergize. However,
CXCR4-overexpression alone was enough to enhance ischemic tissue repair, promote early
blood reperfusion, and attenuate some of the early inflammation in vivo. The therapeutic
potential of CXCR4-overexpressing ADSCs was confirmed in a second in vivo study in a more
severe animal model.
HGF was explored as an alternative angiogenic factor to VEGF (chapter 6). HGF-
overexpressing ADSCs promoted both endothelial cell and smooth muscle cell responses in
vitro and in vivo, which led to better therapeutic effects compared to VEGF-overexpressing
ADSCs. In this study, we also developed a plasmid containing both CXCR4 and HGF genes,
which was used to overexpress both in ADSCs in one-to-one correspondence. The combined
overexpression of CXCR4 and HGF promoted both ADSC survival and angiogenic potential.
9.1.2 Summary of the recruitment and programming endogenous cells approach
With regards to the recruitment and programming endogenous progenitor cells in situ
strategy, we sought to assess the feasibility for using endogenous progenitor cells as a cell source
for gene therapy. This approach was partly motivated by our studies on CXCR4-overexpressing
ADSCs and the overall objective of eliminating ex vivo cell culture. Recruitment and
programming endogenous progenitor cells was evaluated by considering each process
separately (chapter 7), including: 1) endogenous progenitor cell recruitment; and, 2) in situ
programming of endogenous progenitor cells. Endogenous cell recruitment was assessed by
FACS analysis, which demonstrated that ischemia was a potent stimulus for recruiting
endogenous cells; and further, that controlled release of stromal derived factor-1α (SDF-1α)
could increase recruitment compared to ischemia at each time point. Endogenous progenitor
cell programming in situ was assessed by bioluminescence imaging, which demonstrated that
in situ programming correlated with endogenous progenitor cell recruitment. Controlled SDF-
1α delivery further promoted in situ programming.
155

The duration of in situ transgene expression depended largely on the timing of DNA
delivery post-ischemic induction. Our main findings were that delivering DNA too early post-
ischemia without SDF-1α could not last, while delivering DNA later enabled prolonged
transgene expression. Controlled release of SDF-1α enabled earlier DNA delivery. This
approach was validated for therapeutic efficacy using HGF, which demonstrated improved limb
salvage and angiogenic response.
Development on a drug delivery platform was done by using a microfluidic platform to
generate biodegradable PEG-based microspheres with tunable release profiles (Chapter 8).
Microspheres synthesized using microfluidics were more uniform and homogeneous. With this
platform, we were able to achieve controlled release of bioactive growth factors and DNA
nanoparticles. The successful development of these microspheres enable the sequential delivery
of SDF-1α and DNA in a single injection and set the stage for further designing one-step
platforms for ADSC transfection.
9.2 Limitations
9.2.1 Limitations on the non-viral engineered ADSCs approach
Some of the limitations that should be noted in relation to the non-viral engineered
ADSC platform, include: variability of cell sources, animal model and mild genetic
overexpression. In terms of cell sources, we have found differences in ADSCs from different
cell sources, at different passage numbers, and over long-term storage. As an autologous cell
source, this should be noted, which suggests that there could be more work on isolating and
normalizing ADSCs from different sources. Some possibilities for differences include: different
anatomical locations of the fat pads, demographics of the patient (i.e. race, age, gender, etc), as
well as disease state of the patient. The cell isolation procedure is fairly basic, simply relying
on fat pad dissection, enzymatic digestion, and cell adherence to the tissue culture plate. This
process of negative/positive selection could be improved to help normalize the cell type. Other
methods for overcoming this limitation, include: genetic modifications ex vivo and use of
allogeneic cell sources. Genetic modifications may overcome some of the important
deficiencies per cell type by overexpressing relevant genes, which is the focus of this thesis.
Allogeneic cell sources, such as bone marrow-derived mesenchymal stem cells, are known to
exhibit low immunogenicity and are currently being used as an allogeneic source for cell
therapy.
156

In terms of the animal model, the models used in this study were generated by acutely
ligating the femoral artery in an otherwise healthy mouse. Since this is the case, many of these
mice possessed strong inherent regenerative capacity, which tended to override the healing
process beyond a certain time point. This could mask some of the therapeutic responses
afforded by the cell sources and could also lead to over-interpretation of the healing responses
caused by the gene modified cells, if additional measures are not also assessed. In an effort to
overcome these issues, retired breeder mice were used, which are older and possess less
regenerative potential. This model is a more difficult model to achieve a therapeutic response.
Other models of the general co-morbid state of patients with peripheral arterial disease (PAD)
include using mice with genetic backgrounds of being non-obese diabetic (NOD) or ApoE-
deficient mice.
With regards to transfection, we found that human ADSCs were a challenging cell
source to transfect in comparison to mouse ADSCs and human embryonic kidney cells
(HEK293T). Transfection efficiencies in human ADSCs generally reached 15-25%. Although
these were higher than that achieved by commercially available non-viral vectors, this generally
led to a mild genetic overexpression of therapeutic factors. To help overcome some of these
challenges, we have found that transfecting hADSCs in growth conditions could enhance their
transfection. Other methods to improve transfection efficiency include: further promoting cell
proliferation; synthetic modifications to the polymer; designing high-expressing plasmid
constructs; and, also better selection of ADSC sources. Promoting cell proliferation can
improve transfection because the nuclear membrane dissolves during cell proliferation, which
enables plasmid DNA to traffic from the cytoplasm to the nucleus. Modifications to the polymer
to incorporate nuclear localization signals are another strategy to enable trafficking from the
cytoplasm to the nucleus. Designing plasmids with high expressing promoters or as smaller
constructs, such as minicircle DNA, are methods to promote DNA entry and gene expression.
As far as cell sources, some of our recent ADSC isolations have enabled about 3x greater
transfection efficiencies (~60%) compared to those used in our earlier studies. This
demonstrates that certain ADSC populations have more transfection potential.
9.2.2 Limitations on the recruitment and programming endogenous cells approach
Some of the limitations of the recruitment and programming endogenous progenitor
cells approach include: 1) use of only one mouse strain; 2) reliance on acute injury, and, 3) lack
of control over gene delivery. The mouse strain used in our study was C57/Black, which was
157

chosen based on other similar studies of endogenous cell recruitment. Although we may
speculate that the results of our study extend to other mouse strains, as well as into other species,
further study is needed to demonstrate that this is the case. Importantly, endogenous progenitor
cell recruitment might vary according to disease state. This raises the question if similar
recruitment would be achieved in a more diseased animal model.
Another limitation in our study is that the hindlimb ischemia mouse model actually
simulated an acute ischemic episode rather than a chronic ischemic state. In this context, we
may conclude that acute ischemia is a potent stimulus for cell recruitment, which could be
further improved by SDF-1α delivery. It is unclear whether chronic ischemia would promote
endogenous progenitor cell recruitment to the same degree. Still, the acute ischemia model is
relevant in the clinical setting, because this reflects diseases such as acute myocardial infarction.
Also, causing an acute injury is often employed to promote tissue regeneration, such as
transmyocardial laser revascularization to promote angiogenesis or microfracture to promote
cartilage growth. These factors should be considered when further studying or designing
treatments based on the recruitment and programming approach.
In terms of the inability to control transgene expression by DNA delivery into tissues,
this is a concern because of the potential for unregulated gene expression, as well as the
possibility for causing an immune response. Several studies have suggested that plasmid DNA
mainly transfects regenerating muscle fibers, as well as non-inflammatory mononuclear cells.
Despite these findings, the precise fate of plasmid DNA is not entirely known. Further study
should be done to help determine which cell populations are transfected by plasmid DNA and
the mechanism of its removal. To help control gene expression, additional genes might be
engineered into the plasmid, which could be used for plasmid DNA regulation.
9.2.3 Limitations on the synthesis of biodegradable PEG microspheres using microfluidics
In the use of microfluidics to synthesize biodegradable polyethylene glycol (PEG)
microspheres using microfluidics, some of the limitations include: 1) ultraviolet (UV) light
initiated polymerization; and 2) requirement of high cross-linking efficiency. The use of UV
light to initiate polymerization is a potential limitation that might cause denaturation of the
encapsulated protein or DNA. While UV light is a standard approach in biomaterial research,
other methods that might be more biologic-friendly would be preferred, such as covalent cross-
linking. The requirement of high cross-linking polymers to effectively polymerize
microspheres limits versatility of the polymers used. For example, many designed biomaterials
158

might incorporate multiple signals or peptides that might not otherwise be available for use due
to their low cross-linking efficiency. Increasing concentration of the precursor solution may
overcome this issue.
9.3 Future Directions
There are many future directions that will help better understand the mechanism of each
approach, as well as further advance these approaches towards the clinic. These are discussed
separately for each platform.
9.3.1 Future directions of the non-viral engineered ADSCs approach
For the non-viral engineered ADSC platform, the directions that could be pursued
include: 1) understanding the therapeutic mechanism of CXCR4-overexpressing ADSCs; 2)
determining the fate of ADSCs upon transplantation; 3) using a biomaterial cell carrier for
delivery; and, 4) studying the therapeutic effects in a larger animal model.
Towards understanding the mechanism of CXCR4-overexpressing ADSCs, we have
begun to understand some of the changes in the growth factor and cytokine profile due to
CXCR4-overexpression, as well as the effects of paracrine release on endothelial cells and
macrophages. This may help to explain some of the therapeutic effects we see in vivo, but
further study is warranted to understand how CXCR4-overexpressing ADSCs lead to their
therapeutic effects. This may also enable us to identify and potentially exploit the relevant
process in future strategies.
When we transplant the ADSCs into hindlimbs of the mice, it is not completely clear
what happens to the fate of the cells with regards to location of engraftment and whether these
might persist over time. From bioluminescence imaging (BLI) and immunostaining, we have
an idea of the cell survival time and the location of cell engraftment. However, a direct
correlation of BLI signal and immunostained transplanted cells is inconsistent. For example,
despite the high BLI signal observed, immunostaining in the same limbs only identifies few
transplantated cells. This suggests that further study and potentially better experimental
methods would be important to understand the cell fate, which will also be important for clinical
translation.
To better improve cell survival upon transplantation, one approach is to include a
biomaterial cell carrier. A biomaterial might be able to protect the cells during injection and
may also provide a substrate to which the cells could attach in vivo. This direction could also
159

open up several avenues for tailoring the biomaterial to provide additional cues to the cells to
promote their therapeutic effects.
For clinical translation, it will be important to continue assessing the therapeutic effects
of non-viral engineered ADSCs in larger animal models. Performing studies in larger animal
models may better reflect the scale of ischemia in human patients and help to identify other
considerations not represented in the mouse.
9.3.2 Future directions of the recruitment and programming approach
For the recruitment and programming endogenous progenitor cells platform, future
studies would include: 1) deeper characterization of the recruited cell populations; 2)
assessement of inflammation; 3) modulating inflammation to promote endogenous progenitor
cell programming; and, 4) exploring additional cues for recruitment.
In our studies, we mainly focused on characterizing populations that are known to
respond to SDF-1α. However, there was a large increase in recruited cells due to ischemic
injury, and our current characterization only extended to a small proportion of these cells.
Overall, our observation was that the number of isolated cells increased nearly 10-fold due to
ischemia compared to sham controls. Further characterization on these cell sources may enable
us to know which cells are transfected and participate in therapeutic responses.
Inflammation is an important process that occurs post-ischemic injury that should be
studied in our experimental model. A proper understanding the dynamics of inflammation and
progenitor cell recruitment will help us to understand the effects of timing on gene delivery and
subsequent transfection. One finding was that early gene delivery did not last, which was likely
due to the inflammatory process. Considering that there was some transgene expression, it is
unlikely that the DNA was simply removed. More work should be done to understand how
inflammation might extinguish transgene expression in the transfected cells.
To potentially promote early gene delivery post-ischemic injury, one potential strategy
is to modulate inflammation. With a better understanding of the inflammatory cells that are
responsible for DNA removal, more targeted therapeutics might be designed to modulate the
relevant cell populations, while continuing to promote the recruitment of progenitor cells.
To further promote endogenous progenitor cell recruitment and activation, additional
recruitment cues could be studied. We found that ischemia was a potent stimulus for
endogenous cell recruitment, which could be further promoted by SDF-1α delivery. However,
there is a wide variety of stimuli that could be explored, including: homing factors, mobilization
160

factors, inflammatory factors, hypoxia stimuli, and inducing injury. Other homing factors
include: VEGF and sphingosine-1-phosphate (S1P). Inflammatory factors might include:
interleukin-1 (IL-1) and tumor necrosis factor alpha (TNF-α). Mobilization factors include:
granulocyte colony stimulating factor (G-CSF); granulocyte-macrophage colony stimulating
factor (GM-CSF), CXCR4 antagonists (AMD3100). Hypoxia mimicking agents include cobalt,
which promotes hypoxia inducible factor-1α (HIF-1α) stabilization. Also, other injurious
stimuli might initiate cell responses, such as transmyocardial laser revascularization or subtle
damaging agents.
9.3.3 Future directions of the biodegradable PEG microspheres using microfluidics
Moving forward with the biodegradable PEG microspheres for drug release, some of
the next steps include: 1) covalently cross-linking microspheres during microfluidic synthesis;
and, 2) application of the sequential delivery approach for biologics. Covalent cross-linking
during microfluidic synthesis could be done to obviate the need for UV light-based
polymerization. This would be a more mild synthesis condition for encapsulation of biologics
and even cells. Applying microspheres for temporal delivery of two or more biologics is now
possible based on the currently developed platform. Drug delivery systems should be designed
that may deliver protein and DNA to achieve recruitment and programming of endogenous
progenitor cells. Another possibility is the co-delivery of DNA-encapsulated microspheres and
ADSCs, which might enable transfection of ADSCs during injections.
9.4 Concluding remarks
For patients with PAD and other ischemic diseases, surgical revascularization will
remain the standard of care. Therapeutic angiogenesis will help treat the subset of patients that
are unable to withstand surgery. However, current clinical approaches for therapeutic
angiogenesis have led to variable results. To overcome these limitations, the work presented in
this thesis advances the strategies for both cell-based therapeutics and biologic-based
therapeutics. Overall, we have added new tools in the repertoire of therapeutic angiogenesis by
demonstrating the potential for non-viral engineered ADSCs with CXCR4 and/or HGF as
therapeutic targets; and also demonstrating the promise for recruiting and programming
endogenous progenitor cells in situ.
161