hallazgos por resonancia magnetica de la...
TRANSCRIPT

REPÚBLICA BOLIVARIANA DE VENEZUELA UNIVERSIDAD DEL ZULIA FACULTAD DE MEDICINA ESCUELA DE MEDICINA
HALLAZGOS POR RESONANCIA MAGNETICA DE LA CEREIDOLIPOFUSCINOSIS EN NIÑOS
Trabajo Especial de Grado presentado ante el Consejo Técnico de la Facultad de Medicina de La Universidad del Zulia para optar al Título de Doctora en Ciencias
Médicas
Maracaibo, Noviembre 2011
Autor: Liliana Beatriz Mora López Especialista en Radiología
Tutor: Dr. Eduardo Mora La Cruz Profesor Titular de LUZ Doctor en Ciencias Médicas Especialista en Radiología

HALLAZGOS POR RESONANCIA MAGNETICA DE LA CEREIDOLIPOFUSCINOSIS EN NIÑOS

ÍNDICE GENERAL
Resumen………………………………………………………………………….. 7
Abstract…………………………………………………………………………… 8
Índice General……………………………………………………………………. 5
CAPITULO I. EL PROBLEMA
Planteamiento del problema…………………………………………………….. 10
Formulación del problema………………………………………………………... 12
Objetivos de la investigación…………………………………………………….. 12
Justificación e importancia de la investigación…………………………………… 13
Delimitación de la investigación…………………………………………………. 13
Factibilidad y viabilidad…………………………………………………………. 13
CAPITULO II. MARCO TEORICO
Antecedentes de la investigación………………………………………………… 16
Bases teóricas……………………………………………………………………… 17
Marco teórico operacional………………………………………………………… 31
Definición conceptual y operacional de las variables…………………………….. 31
Operacionalización de las variables………………………………………………. 32
CAPITULO III. MARCO METODOLOGICO
Tipo de Investigación……………………………………………………………... 34
Diseño de la Investigación………………………………………………………… 34
Material y Métodos………………………………………………………………… 35
Recolección de Datos……………………………………………………………… 36
Análisis de Datos………………………………………………………………….. 37

CAPITULO IV. ANALISIS Y DISCUSION DE LOS RESULTADOS
Análisis y Discusión de los Resultados……..…………………….……………..... 39
Conclusiones……………………….……………………………………………….. 43
Recomendaciones………………………..………………………………………….. 43
REFERENCIAS BIBLIOGRAFICAS…………………………………………….. 44

Mora López, Liliana Beatriz: “HALLAZGOS POR RESONANCIA MAGNETICA DE LA CEREIDOLIPOFUSCINOSIS EN NIÑOS” Trabajo de Investigación para optar al Título de Doctora en Ciencias Medicas. Universidad del Zulia. Facultad de Medicina. Maracaibo. Venezuela, 2011. 46 p.
RESUMEN El objetivo de la presente investigación fue identificar los hallazgos en la Resonancia Magnética de la cereidolipofuscinosis en pacientes pediátricos ya que esta es una patología que puede dejar secuelas de severidad variable, como déficit neurocognitivo, por lo cual es importante su diagnostico precoz para la instauración del tratamiento adecuado. Además hay muy pocos registros acerca de la incidencia y la prevalencia de esta enfermedad. La investigación fue de tipo descriptiva, prospectiva, transversal, pudiendo determinar los hallazgos más frecuentes en la Resonancia Magnética, la incidencia en niños Zulianos, así como la incidencia con respecto al sexo. La técnica de análisis fue la estadística descriptiva, mediante el uso de cifras absolutas y porcentajes, a través del paquete estadístico S.P.S.S versión 10.0. Concluyendo que los hallazgos más frecuentes observados en la Resonancia Magnética son la atrofia cerebral y cerebelosa, siendo esta patología es más frecuente en el sexo masculino. La Resonancia Magnética es la técnica de imagen de elección para la evaluación de esta patología Palabras claves: Cerereidolipofuscinosis, Resonancia Magnética. Correo Electrónico: [email protected]

Mora López, Liliana Beatriz: “FINDINGS BY MAGNETIC RESONANCE OF THE CEREIDOLIPOFUSCINOSIS IN CHILDREN”. Degree thesis presented to the Technical Council of the Faculty of Medicine, University of Zulia to obtain the Title of Doctor in Medical Sciences. Maracaibo, Zulia, Republica Bolivariana de Venezuela, 2011. 46 p.
ABSTRACT The objective of the present investigation was determine the findings by Magnetic Resonance of the Cereidolipufuscinosis in children since this is a pathology that can leave sequels of variable severity, like neurocognitive deficit, thus is important the fast diagnose for the restoration of the suitable treatment. In addition there is not a good register about the incidence and the prevalence of this disease. The research was descriptive, prospective, cross-sectional type. The following variables were evaluated: findings more frequently in the magnetic resonance, incidence in children and incidence by sex. The analysis technique was the descriptive statistic, using absolute numbers and percentage, through statistical package S.P.S.S version 10.0. Concluding that the most frequent findings were brain and cerebelar atrophy and is more frequent in boys than in girls. The Magnetic Resonance is the technique of choice to asses this disease. Key words: Cereidolipofuscinosis, Magnetic Resonance. Electronic Mail: [email protected]

CAPITULO I
EL PROBLEMA

10
PLANTEAMIENTO DEL PROBLEMA
La ceroidolipofuscinosis neuronal (CLN) representa un grupo de trastornos hereditarios,
progresivos en niños y adultos, caracterizados morfológicamente por dos procesos: sobrecarga
de lipopigmentos autofluorescentes en varios tipos celulares y pérdida de células, especialmente
en la corteza cerebral [1]. Las características clínicas de estos trastornos comprenden la aparición
de convulsiones, la regresión psicomotora y la pérdida visual, con edades y comienzo diferente
para cada tipo de CLN. El tratamiento disponible actualmente consiste en el manejo de los
síntomas, y los pacientes progresan hacia el estado vegetativo y el fallecimiento [2]. Si bien no
existen datos epidemiológicos mundiales, se ha observado una alta incidencia de CLN en los
países norteños [3]. Para todas las formas infantiles, se ha informado de una frecuencia de 1,2
por 100.000 nacidos vivos, pero formas clínicamente diferentes pueden darse con frecuencias
diferentes en regiones diversas.
Con el microscopio de luz, todas las CLN presentan lipopigmentos con tinción
característica similar al ceroide, que aparece en condiciones tales como la cirrosis hepática, y
lipofuscina, un pigmento que normalmente se acumula a una edad avanzada. Desde el punto de
vista ultraestructural, estos lipopigmentos se presentan como depósitos granulares osmiófilos,
como cuerpos curvilíneos o como estructuras en huella dactilar, objetivados en los linfocitos
circulantes y en las biopsias de piel o músculo.
En función de la edad de comienzo, el curso clínico y la morfología ultraestructural, las
CLN se han clasificado en cuatro tipos principales: la CLN infantil (INCL, enfermedad de
Haltia-Santavuori, CLN1), hallada predominantemente en Finlandia, la CLN infantil tardía
(LINCL, enfermedad de Jansky-Bielschowsky, CLN2), la CLN juvenil (JNCL, enfermedad de
Batten, enfermedad de Spielmeyer-Vogt-Sjorgen, CLN3) y la CLN del adulto (ANCL,
enfermedad de Kufs) [1,3-6]. Esta clasificación tradicional y sencilla se ha confundido con la
descripción de las variantes o formas atípicas, que exhiben síntomas clínicos o rasgos
patológicos diferentes y representan alrededor de un 20% de las CLN en diferentes poblaciones,
generalmente distribuidas entre los grupos infantil tardío y juvenil. Se han descrito dos variantes
de la forma infantil tardía (vLINCL): la variante finlandesa (CLN5) [4,6] y la variante CLN6

11
[7,8]. Hayotra variante conocida como CLN8, epilepsia norteña o epilepsia progresiva con
retardo mental (EPMR) [9].
El estudio de la CLN puede orientarse tomando como referencias para su identificación
sus manifestaciones clínicas, su neurofisiología, la neuroimagen o su caracterización genética, de
los cuales la neuroimagen tiene una alta especificidad y sensibilidad para establecer el
diagnostico debido a la identificación de lesiones especificas, constituyéndose en un para clínico
gold estándar en esta patología.
Los cambios más significativos en los estudios de neuroimagen (RM), en todos los tipos
de CLN están representados por atrofia cerebral progresiva. En el tipo clásico en la LINCL, sin
embargo, la atrofia es más pronunciada en las estructuras infratentoriales, particularmente en el
cerebelo [12,13]. En los subtipos variantes de la LINCL, los hallazgos de RM, especialmente en
las etapas tempranas de la enfermedad, pueden variar desde la normalidad hasta áreas
discernibles de densidad aumentada en la sustancia blanca periventricular, alrededor de los
ventrículos laterales, intensidad disminuida en el tálamo y el putamen, y atrofia cerebelosa
[10,11,14]. Se han descrito anormalidades en el tálamo y los ganglios basales, tanto para las
variantes de LINCL como para la CLN infantil [12,15,16]. También se han comunicado señales
anormales hiperintensas en la sustancia blanca, tanto para la CLN clásica infantil tardía como
para la juvenil. En la fase tardía de las variantes de la LINCL, la atrofia cerebral difusa y las
áreas de hipointensidad que incluyen los ganglios basales, dominan las imágenes de la RM
[12,16].
En la actualidad los estudios de RM con espectroscopia han revelado cambios
progresivos, con reducción del ácido N-acetil aspártico, incremento de mioinositol y de la
porción lutamato-glutamina, en relación con la pérdida neuroaxonal generalizada. Más
recientemente, se ha enfatizado el valor de los estudios seriados con RM convencional y
espectroscópica, que revelan la pérdida progresiva del volumen cerebral lo cual testimonia la
degeneración neural. La importancia que reviste la utilización de la RM y espectroscopia en
pacientes con CLN permite plantear la caracterización de los hallazgos más significativos en

12
estos pacientes que acuden al Servicio de Neuropediatria del Servicio Autónomo Hospital
Universitario de Maracaibo, Estado Zulia.
FORMULACION DEL PROBLEMA
En función de lo planteado, se formula la siguiente interrogante:
¿Cuáles son los hallazgos más comunes en la Resonancia Magnética de la
ceroidolipofuscinosis en niños del Servicio de Neurología del Servicio de Radiología del
Servicio Autónomo Hospital Universitario de Maracaibo?
OBJETIVOS DE LA INVESTIGACIÓN
OBJETIVO GENERAL
Determinar la frecuencia de los hallazgos por Resonancia Magnética de la
ceroidolipofuscinosis en niños del Servicio de Neuropediatria y del Servicio de Radiología del
Hospital Universitario de Maracaibo.
OBJETIVOS ESPECÍFICOS.
Identificar los hallazgos en la RM de la ceroidolipofuscinosis en niños del Servicio de
Neuropediatria y del Servicio de Radiología del Servicio Autónomo Hospital Universitario de
Maracaibo.
Determinar la frecuencia de la ceroidolipofuscinosis en niños del Servicio de
Neuropediatria y del Servicio de Radiología del Hospital Universitario de Maracaibo
Determinar la frecuencia por sexo de la ceroidolipofuscinosis en niños del Servicio de
Neuropediatria y del Servicio de Radiología del Hospital Universitario de Maracaibo

13
JUSTIFICACIÓN DE LA INVESTIGACIÓN
La evaluación por neuro imágenes de patologías del sistema nervioso central en la
actualidad se ha convertido en estudios de rutina, debido a la especificidad y sensibilidad para
determinar la presencia de lesiones cerebrales, por lo tanto toda enfermedad del sistema nervioso
central que evidencie lesiones estructurales son susceptibles de ser visualizadas, demostrando
que el uso de estas técnicas de imagen revisten una gran importancia en el manejo de estas
patologías. La ceroidolipofuscinosis en niños es una de estas, en la cual se pueden observar a
través de la RM las características de las lesiones con la finalidad de aplicar las herramientas
terapéuticas oportunas y mejorar la calidad de vida de estos pacientes.
El manejo de la ceroidolipofuscinosis en niños con un seguimiento por RM constituye
una alternativa diagnostica para evaluar la incidencia de esta patología en la población infantil
venezolana y lograr el seguimiento de estas lesiones y lograr el control de la enfermedad.
DELIMITACIÓN DE LA INVESTIGACIÓN
La investigación se desarrollo en el Servicio de Neurología Pediátrica y Servicio de
Radiología del Hospital Universitario de Maracaibo, Estado Zulia, para determinar las
características en la Resonancia Magnética de la ceroidolipofuscinosis en niños, durante el
período de Marzo 2009 hasta Diciembre 2009
FACTIBILIDAD Y VIABILIDAD DE LA INVESTIGACION
La realización de esta investigación fue posible en el Servicio de Radiología e Imágenes
del Servicio Autónomo Hospital Universitario de Maracaibo y en la región contamos con un
Resonador, así mismo existe un servicio de Neuropediatria, el cual maneja una alta frecuencia de
pacientes pediátricos con posible diagnostico de ceroidolipofuscinosis, los cuales son referidos al
servicio de Radiología, para corroborar diagnostico. Así mismo está disponible material
bibliográfico y fichas técnicas referentes a esta patología y la autora, tiene la autorización del jefe
del Departamento de Radiología e Imágenes y del Director de la referida institución asistencial,

14
y dispone de los recursos económicos para cubrir los gastos que ocasione la investigación, así
como también el tiempo necesario para llevar a cabo los objetivos.

CAPITULO II
MARCO TEÓRICO

16
2.1 ANTECEDENTES DE LA INVESTIGACION
R. Caraballo, S. Monges, Carmen Medina Monzón, Víctor Luis Ruggieri, A.
Sologuestua, A. L. Taratuto, N. Fejerman, R. Cersósimo. (2005) en su investigación titulada
Lipofuscinosis neuronal ceroidea infantil tardía: aspectos clínicos y electroencefalográficos, la
cual se propone como objetivo describir las características clínicas, particularmente las crisis
epilépticas y los hallazgos electroencefalográficos, en 15 pacientes con diagnóstico
anatomopatológico de lipofuscinosis neuronal ceroidea (LNC) infantil tardía. Se estudiaron y se
analizaron las historias clínicas de nueve pacientes del sexo femenino y seis del masculino
durante el período comprendido entre febrero de 1990 y junio de 2003. En todos los casos se
realizaron neuroimágenes, estudios neurometabólicos, ERG, PE y repetidos EEG. La edad
mediana de comienzo de la enfermedad fue de 3 años (intervalo: 1-5 años). La manifestación
inicial fue la epilepsia en todos los casos. Las crisis más frecuentes fueron las mioclonías
masivas y las crisis mioclonicoatónicas. Se observaron mioclonías focales en seis pacientes.
Otros tipos de crisis epilépticas observados fueron tonicoclónicas generalizadas, ausencias,
focales motoras y focales complejas. Las crisis epilépticas fueron refractarias al tratamiento. En
todos los casos se presentaron deterioro neurológico y visual progresivo, signos piramidales y
cerebelosos y retraso mental. Los EEG intercríticos mostraron paroxismos de punta y polipunta
onda difusos, espigas multifocales y, menos frecuentemente, espigas focales predominantes en
las regiones posteriores. La fotoestimulación mostró espigas occipitales de elevada amplitud
(300-450) durante el estímulo lumínico entre 1 y 8 Hz. El ERG, los PE visuales y los PE
somatosensoriales fueron patológicos. Las imágenes evidenciaron signos de atrofia cerebral y
cerebelosa. Siete de los pacientes fallecieron entre los 8,5 y los 11 años. En conclusión, en un
niño de 1-5 años que comienza con convulsiones, predominantemente mioclonías generalizadas
y mioclonicoatónicas asociadas a deterioro neurológico progresivo que incluye signos
piramidales, cerebelosos y visuales con un EEG con paroxismos occipitales desencadenados por
la fotoestimulación a baja frecuencia, debemos pensar en una LNC infantil tardía.(17).
J.A. Peña, C. Montiel-Nava, W. Delgado, M.L. Hernández ,J.J. Cardozo , E. Mora, L.
Soto-Faneite. (2004) realizaron una investigación denominada Caracterización de la
ceroidolipofuscinosis en niños venezolanos considerando que la lipofuscinosis ceroide (NCL),

17
representa un grupo de trastornos hereditarios neurodegenerativos. Con base en la edad del
paciente en el inicio, curso clínico y la morfología ultraestructural se ha identificado tres tipos
clínicos para el grupo pediátrico: 1) NCL infantil (INCL), 2) NCL infantil tardía (LINCL), y 3)
menores de NCL ( JNCL). Otras variantes o formas atípicas representan alrededor del 20% de la
NCL en diferentes poblaciones, avances genéticos han permitido una mejor caracterización,
diagnóstico y clasificación de estos trastornos. Los informes de casos. Se presentan los datos
clínicos, neurofisiológicos, neurorradiológicos y morfológicos de 6 pacientes con NCL, que
fueron evaluados en el departamento de neurología pediátrica del Hospital Universitario de
Maracaibo durante un período de diez años (1993-2003). Todos los 6 casos correspondieron a la
forma infantil tardía. La edad de comienzo varió entre 2 a 5 años. Para la mayoría de los
pacientes los síntomas iníciales incluyen convulsiones, retraso psicomotor, acompañado por la
degeneración macular y atrofia óptica. El EEG se ha caracterizado por picos de alta tensión
provocada por la estimulación fótica de baja frecuencia, en 5 casos hallazgos de neuroimagen
fueron característicos de la forma infantil tardía de la NCL. En tres pacientes una disminución de
la intensidad de la señal se vio en los tálamos y putamine en las imágenes ponderadas en T2. El
examen ultraestructural de las muestras obtenidas a través de una biopsia mostró cuerpos
curvilíneos en todos los pacientes. Conclusión. No hay datos epidemiológicos de la NCL en
Venezuela, que se presume la presencia de formas clínicas y variantes en el grupo pediátrico.
Este primer estudio podría contribuir al conocimiento y una mejor investigación de este grupo de
trastornos en nuestra población. (18).
2.2 BASES TEORICAS
Niñez es, por definición, es una etapa de crecimiento y desarrollo. Muchas consultas en
pediatría y neurología pediátrica se relacionan con el retraso en la adquisición de nuevas
habilidades, estimándose que los trastornos del desarrollo afectan a alrededor del 10% de la
población infantil. Mucho menos frecuentes, pero más dramáticos, son los casos en que existe
pérdida progresiva de habilidades. Frente a esta situación, habiéndose descartado tumor o
hidrocefalia, deben investigarse causas genéticas, alteraciones metabólicas, infecciones y acción
de tóxicos. En ese contexto, las lipofuscinosis ceroides neuronales se consideran el trastorno
neurodegenerativo de depósito más frecuente de la infancia (1, 2).

18
Las lipofuscinosis ceroideas neuronales (LCN, y en inglés NCL) son un grupo de
enfermedades neurodegenerativas con herencia autosómica recesiva (3), que se presentan
principalmente en la infancia y adolescencia, caracterizadas por síntomatología variable que
incluye convulsiones, deterioro cognitivo, pérdida visual y/o atrofia cerebral. El curso es
habitualmente progresivo, con desarrollo de demencia que lleva a la muerte (4). Desde el punto
de vista neuropatológico se caracterizan por la acumulación progresiva de lipofuscina, un
lipopigmento autofluorescente, en neuronas y otros tejidos.
Producidas por mutaciones en distintos genes, estas enfermedades presentan en conjunto
una prevalencia de 0,1 a 7 por cada 100.000 recién nacidos vivos (3). En la población finlandesa
se ha observado una prevalencia de 1 por cada 12.500 recién nacidos vivos (5).
Historia
La descripción original fue realizada en 1826 por Stengel en Noruega, y la primera
sistematización y correlación anatomoclínica la hizo el médico francés Frederick Batten en 1903,
por lo que el cuadro llevó inicialmente su nombre. Se consideró en un principio como una
variante de la enfermedad reportada por Sachs en 1887. Sin embargo, fue el mismo Batten quien
en 1914 concluyó que este cuadro no tenía relación con la enfermedad de Tay-Sachs, sino que
correspondía a una entidad clínica nueva (6).
Las lipofuscinosis fueron agrupadas inicialmente bajo el nombre de “idiocia familiar
amaurótica”, intentando destacar así sus dos principales características: compromiso intelectual y
visual. En el año 1969, Zeman y Dyken acuñaron el término “lipofuscinosis ceroidea neuronal” y
luego de múltiples descripciones de otras patologías que compartían características similares, se
propuso la primera clasificación realizada por Norman y Wood. Con posterioridad se han
planteado otras, a medida que se han identificado los materiales de depósito intracelular. En
1998, Wisniewski (7) propuso una clasificación en la que distingue cinco grupos principales,
separados de acuerdo a las características clínicas y a la edad de inicio de los síntomas de cada
patología. Esta clasificación se ha ido actualizando con el aporte de la genética y de la
morfología microscópica (4).

19
El término lipofuscinosis ceroidea neuronal se ha mantenido, enfatizando la presencia de
acumulación de lipofuscina, pese a que no es la causa del daño celular.
Patogenia
La lipofuscina es un polímero intralisosomal compuesto de lipoproteínas residuales de
procesos oxidativos. Frecuentemente llamada el “pigmento de la edad”, es considerada el
marcador del envejecimiento, ya que aumenta con la edad de un modo casi lineal, observándose
frecuentemente en células parenquimatosas de órganos o tejidos con atrofia normal o patológica,
neuronas del sistema nervioso central y de ganglios simpáticos, en la zona fascicular de la
corteza suprarrenal y en el epitelio de las vesículas seminales (8). Su nombre viene del griego
lipo (grasa) y del latín fuscus (oscuro). Los productos de desecho celular se acumulan en
autofagosomas. A éstos se unen lisosomas constituyéndose los autofagolisosomas, en los que se
realiza la degradación a productos que vuelven a ser utilizados por la célula. Este es un proceso
fisiológico donde teóricamente no debería sobrar nada, sin embargo se produce una desviación
hacia la peroxidación de lípidos con formación de ácidos grasos insaturados, que se acumulan
como residuos visibles al microscopio de luz bajo la forma de gránulos de lipofuscina. La
sudanofilia se va perdiendo en estos gránulos a medida que los ácidos grasos no saturados se van
transformando, dando origen a este pigmento autofluorescente amarillo-café (8).
En las lipofuscinosis el depósito de material proteico en lisosomas de distintos grupos
celulares toma una forma característica según el tipo de LCN estudiada. Pese a que estos
cúmulos se producen en distintas células, la muerte celular aparece específicamente en neuronas
del sistema nervioso central y de la retina, aparentemente por la inexistencia de actividad
mitótica de éstas. Se han obtenido distintos modelos animales (en especial ratas) que presentan
las mutaciones descritas para las variantes de LCN. Al estudiar estos modelos se aprecia que la
aparición del cuadro clínico es similar entre los distintos modelos y se correlaciona
estrechamente con el humano, lo que hace plantear que existe un momento crítico en que se
activan los mecanismos de destrucción celular que llevan a la expresión clínica, hipótesis que en
la actualidad es objeto de estudio, dado que tiene la potencialidad de convertirse en una ventana
terapéutica (9).

20
Genética
Las lipofuscinosis son cuadros de herencia autosómica recesiva, a excepción del tipo 4
(LCN4), que podría heredarse de forma autosómica dominante (10). En los años 90 fueron
identificados cinco genes asociados a estas patologías, como también la secuencia y función de
los productos proteicos de los genes para LCN1 y LCN2. Posteriormente se completó la
identificación de seis genes (LCN1, LCN2, LCN3, LCN5, LCN6 y LCN8) (11), se identificaron
los productos de LCN3, LCN5 y LCN8, y se avanzó en la identificación de al menos 150
mutaciones diferentes en los genes descritos, asociadas a diversos fenotipos (12).
A pesar de que la investigación ha aportado nuevos conocimientos en torno a las LCN,
queda aún mucho por dilucidar tanto en relación a otros genes y mutaciones implicados, como a
sus productos proteicos.
Cuadro clínico
En función de la edad de comienzo, el curso clínico y la morfología ultraestructural, las
LCN se han clasificado en cuatro tipos principales:
1. LCN infantil (enfermedad de Haltia-Santavuori, LCN1), hallada predominantemente
en Finlandia.
2. LCN infantil tardía (enfermedad de Jansky-Bielschowsky, LCN2).
3. LCN juvenil (enfermedad de Batten, enfermedad de Spielmeyer-Vogt-Sjorgen, LCN3).
4. LCN del adulto (enfermedad de Kufs)
Se han descrito además variantes o formas atípicas que representan alrededor de un 20%
de las LCN en diferentes poblaciones, generalmente distribuidas entre los grupos infantil tardío y
juvenil. Para la forma infantil tardía se han descrito la variante finlandesa (LCN5) (13,14) y la
variante LCN6 (15). Existe otra variante conocida como LCN8, epilepsia del norte o epilepsia
progresiva con retardo mental (EPRM). Los casos de la forma turca infantil tardía pueden
representar variantes de la EPRM.

21
LCN1 o lipofuscinosis aguda infantil (Santavuori-Haltia).
Su primera descripción fue realizada por Muldegrer en 1903. Sin embargo, fue
Santavuori en 1973 quien sistematizó y dio su nombre al cuadro (15). Se inicia habitualmente
entre los 6 meses y los 2 años de vida con aparición relativamente aguda de déficit motor,
hipotonía e irritabilidad, a los que siguen crisis convulsivas, generalmente mioclonías, ataxia y
finalmente ceguera y grave compromiso cognitivo. Se acompaña de microcefalia adquirida y
atrofia cerebral severa con pérdida neuronal en la corteza cerebral y cerebelosa, en la médula
espinal y tronco cerebral (4). La mayoría de los pacientes fallece entre los 5 y 10 años.
La enfermedad de Santavuori-Haltia se relaciona con diversas mutaciones en el gen
LCN1, en el cromosoma 1p32, de las cuales hay al menos 41 descritas hasta ahora (11) y que
determinan el déficit de una enzima lisosomal denominada tioesterasa proteinopalmitoil 1 (o
PPT1). El diagnóstico se establece midiendo la actividad de esta enzima en leucocitos y cultivos
de fibroblastos, aunque también se puede realizar mediante la genética molecular, identificando
las mutaciones del caso índice. En la resonancia magnética de encéfalo lo más característico es la
pérdida de intensidad de señal talámica en secuencias T2, adelgazamiento del cuerpo calloso y
aumento de señal periventricular, seguidos de atrofia cerebelar y atrofia cerebral difusa que se
estabiliza alrededor de los 4 años. Posteriormente se describe un aumento difuso de señal de
sustancia blanca y en la espectroscopía se aprecia pérdida de N-acetil-aspartato y reducción de
creatina con aumento de mioinositol y lactato en sustancia gris y blanca. En el SPECT (Single
Photon Emission Computed Tomography) se describe hipoperfusión cerebelar y cerebral
progresiva con conservación de la perfusión de ganglios basales (10).
El estudio enzimático se recomienda como primer análisis frente a la sospecha clínica. Si hay
deficiencia de actividad enzimática, se hace estudio molecular. El diagnóstico de aproximación
se establece con el análisis histológico de muestra de piel - donde se observan
predominantemente depósitos osmofílicos granulares en la microscopía electrónica - y se
confirma midiendo la actividad de esta enzima en leucocitos y cultivos de fibroblastos. También
puede realizarse mediante genética molecular (10).
LCN2 o lipofuscinosis infantil tardía. (Bielschowsky-Jansky).

22
Conocida como enfermedad de Bielschowsky, su primera descripción fue realizada en 1908 por
Jansky. Se caracteriza por iniciarse entre los 2 y 4 años con crisis tónico-clónicas generalizadas,
ausencias o crisis parciales secundariamente generalizadas, aunque su característica típica es la
mioclonía, que aparece posteriormente junto a pérdida cognitiva, ataxia, mioclonías, signos
extrapiramidales y piramidales, y deterioro rápidamente progresivo de la visión por atrofia
óptica. Las crisis convulsivas se asocian a pérdida o falta de adquisición de habilidades del
desarrollo psicomotor. El paciente puede fallecer en este primer episodio o derivar a estado
vegetativo permanente por años. Las neuroimágenes muestran atrofia cerebral y cerebelosa
progresiva con indemnidad de tálamos y ganglios basales (12). La tomografía por emisión de
positrones (PET) muestra hipometabolismo generalizado de la glucosa (10) y en el análisis
histológico se observan depósitos de citosomas curvilíneos, aunque puede haber inclusiones
lisosomales mixtas (12). La lipofuscinosis infantil tardía se debe a diversas mutaciones en el gen
LCN2 ubicado en el cromosoma 11p15, que determinan la deficiencia de una enzima lisosomal,
la tripeptidil-peptidasa o TPP1. Hasta ahora hay al menos 52 mutaciones descritas (11). Los
estudios neurofisiológicos son importantes para el diagnóstico. El electroencefalograma (EEG)
se altera precozmente, presentando una disfunción lenta difusa asociada a actividad epileptiforme
multifocal y descargas de punta-onda y poliespiga-onda interictales e ictales. Con la estimulación
fótica de baja frecuencia (1 a 2 Hz) se obtiene una respuesta característica de puntas de alto
voltaje (mayor a 200 mV) en regiones posteriores, seguidas de una onda lenta que corresponde a
un potencial evocado visual gigante El electrorretinograma es usualmente anormal desde el
inicio del cuadro con extinción precoz de la respuesta por alteración de conos y bastones (16, 17,
18).El diagnóstico de LCN2 se puede confirmar a través de la medición de la actividad de la
TPP1 en linfocitos o fibroblastos. El diagnóstico prenatal se realiza mediante el análisis de la
actividad enzimática en vellosidades coriales o líquido amniótico, o mediante la búsqueda de
mutaciones de los casos índice y los portadores (12).
LCN3 o lipofuscinosis juvenil crónica (Spielmayer-Vogt-Batten)
Conocida como enfermedad de Batten, en honor al responsable de la primera descripción
realizada en 1903, es la más común de las lipofuscinosis en la población norteamericana,
correspondiendo al 45 a 52% de los casos. Tiene característicamente dos formas fenotípicas, la
clásica (OMIM 204200) y la retardada o tardía (12). En la forma clásica los primeros síntomas

23
aparecen entre los 4 y 7 años de vida, con pérdida progresiva de la capacidad visual,
apreciándose degeneración pigmentaria de la retina y atrofia óptica. Posteriormente se hace
evidente el deterioro intelectual progresivo, con trastornos del habla, pérdida de las funciones
cognitivas y crisis convulsivas tónico-clónicas generalizadas o parciales complejas, raramente
mioclónicas (10). En la segunda década de la vida se hacen más prominentes los trastornos de
comportamiento, signos extrapiramidales y trastornos del sueño. En el estudio histológico de
tejido rectal, piel y conjuntivas son característicos los linfocitos vacuolados y citosomas en forma
de huella digital (10). Las neuroimágenes pueden mostrar atrofia cerebral y sólo tardíamente
cerebelosa. No existe aún una alteración enzimática identificada pero se sabe que el gen LCN3 se
encuentra en el cromosoma 16p12 y codifica para una proteína estructural del lisosoma (4),
habiéndose descrito hasta ahora 31 mutaciones asociadas a distinta presentación clínica (11).
LCN4, lipofuscinosis crónica del adulto (Enfermedad de Kufs).
Descrita por Kufs en 1925, los síntomas iniciales aparecen alrededor de los 30 años,
siendo una de sus características relevantes la indemnidad ocular. Clásicamente existen 2
fenotipos: un cuadro caracterizado por epilepsia mioclónica progresiva con demencia, ataxia y
signos piramidales y extrapiramidales tardíos, y otro con trastornos del comportamiento y
demencia que se pueden asociar a ataxia y signos extrapiramidales (9). En la microscopía
electrónica se observan inclusiones mixtas lisosomales. Su genotipo es desconocido ya que el
gen LCN4 aún no se ha tipificado.
Formas atípicas o variantes
LCN5, lipofuscinosis infantil tardía (variante finlandesa).
Se manifiesta más tardíamente que la forma infantil tardía (Jansky-Bielchowsky) clásica,
entre los 4 y 7 años de edad, con características clínicas muy similares a esta forma, pero a
diferencia de ella el compromiso ocular es el síntoma más relevante. En el análisis por
microscopía electrónica se observa material de inclusión del tipo cuerpos rectilíneos y en huella
digital. El gen, cuyo defecto causa la enfermedad, se encuentra en el cromosoma 13q22 y,
aunque ya se han descrito al menos cuatro mutaciones, aún se desconoce la función de su
producto (11).

24
LCN6, lipofuscinosis infantil tardía (variante checa o gitana o india).
Esta forma de lipofuscinosis es más frecuente en poblaciones gitanas europeas. El gen
asociado, LCN6, está ubicado en el cromosoma 15q21-23, existiendo 18 mutaciones conocidas
hasta ahora. Su producto proteico está siendo caracterizado (11). Respecto al cuadro clínico,
predominan los síntomas motores sobre los oculares. En la microscopía electrónica es posible
observar inclusiones celulares mixtas: citosomas curvilíneos, en huella digital y cuerpos
rectilíneos.
LCN7, lipofuscinosis infantil tardía (variante turca).
Descrita el año 1999, se inicia entre el primer y sexto año de vida, con pérdida de las
habilidades motrices, deterioro visual y deterioro cognitivo. Se han registrado 14 pacientes, todos
turcos y la mayoría con relación de parentesco (19).El gen LCN7 no ha sido aún mapeado ni su
producto proteico definido. En algunos de los pacientes inicialmente descritos se ha observado
una mutación alélica de LCN8 y deben, por lo tanto, realizarse estudios para demostrar si esta
patología existe como una entidad clínica separada (10). En la histología se observan inclusiones
celulares mixtas: citosomas curvilíneos, en huella digital y cuerpos rectilíneos.
LCN8, LCN infantil tardía (epilepsia del norte).
Descrita en 1999, se presenta clínicamente con epilepsia tónico-clónica o parcial
compleja con retardo mental y disfunción motriz. Los síntomas se presentan entre los 5 y 10 años
de vida (20). El gen LCN8 se encuentra en el cromosoma 8p23 y codifica para una proteína
estructural del retículo endoplásmico (21). Se han descrito 5 mutaciones con gran variabilidad
fenotípica (7). El diagnóstico se realiza por medio del análisis histológico, en que se detectan
inclusiones celulares o depósitos granulares osmofílicos, y se confirma por medio del análisis de
las mutaciones del gen LCN8.
La lipofuscinosis neuronal ceroidea (NCLS, por sus siglas en inglés) es un tipo de
trastorno neurodegenerativo que involucra una acumulación de un material anormal llamado
lipofuscina en el cerebro. La evidencia sugiere que este trastorno es causado por problemas con
la capacidad del cerebro para eliminar y reciclar proteínas.

25
Este trastorno se puede observar al nacer, pero generalmente se diagnostica mucho más
tarde. Los niños desarrollan descoordinación muscular (ataxia), problemas para caminar,
problemas visuales, retraso mental y convulsiones. Cuanto más joven sea la persona cuando
aparece la enfermedad, mayor será el riesgo de discapacidad y de muerte temprana.
Las lipofuscinosis se heredan como rasgos autosómicos recesivos. Esto significa que si
ambos padres son portadores del rasgo, cada hijo tiene:
• Una posibilidad de uno entre cuatro de tener la enfermedad.
• Una posibilidad de dos entre cuatro de no tener la enfermedad pero portar el rasgo.
• Una posibilidad de uno entre cuatro de no tener la enfermedad y tampoco ser portador
Entre las enfermedades neurodegenerativas que los especialistas consideran raras se
encuentra una que, en la Argentina, ya se diagnostica con la misma precisión y excelencia que en
los centros científicos internacionales más importantes dedicados al estudio de esos males.
Es la lipofuscinosis neuronal ceroidea (LNC), un conjunto de desórdenes genéticos
terminales que afectan en el mundo a entre cuatro y ocho personas por cada 100.000 nacimientos
de padres aparentemente sanos.
Desde hace dos años, un grupo de investigadores del Centro de Estudios de
Metabolopatías Congénitas (Cemeco) del Hospital de Niños de la ciudad de Córdoba estudia los
ocho tipos genéticos de la LNC que, en general, se subdiagnostican debido a que poseen
síntomas comunes a otras afecciones.
En la LNC, un pigmento anormal denominado lipofuscina se acumula inadecuadamente
en las células cerebrales y produce un daño neuronal progresivo hasta la muerte del paciente.
Cuanto más tarde se despierta la enfermedad, puntualizó la especialista, más tiempo de vida tiene
el paciente.
En cambio, en los más chiquitos, el tiempo de sobrevida puede no superar los dos a nueve
años si se tiene en cuenta que algunas formas de la lipofuscinosis neuronal ceroidea pueden

26
aparecer en los recién nacidos y, otras, a partir de los dos años.
Los síntomas más comunes son los problemas visuales, de comportamiento, de
aprendizaje, de lenguaje y de marcha. "Los chicos, por ejemplo, empiezan a tener problemas de
visión nocturna o presentan un atraso psicomotor o tienen convulsiones que afectan a todo el
cuerpo y no responden a las drogas anticonvulsivas", explicó la titular del programa.
Aunque hasta el momento no existe la cura del mal, "en el mundo se está investigando la
posibilidad de usar terapias génicas, de reemplazo enzimático o de disolución, como ocurre con
otras enfermedades con una falla enzimática en las células del cuerpo".
Los síntomas pueden aparecer en los primeros años de vida, en la juventud o en la edad
adulta. "Las formas más tempranas son las más severas", indicó Halac.
Según la edad del paciente en la que se manifiesta, es el tipo clínico del mal. En total
existen cuatro tipos clínicos (infantil, tardío infantil, juvenil y adulto) que, según el cromosoma y
el gen afectados, forman un conjunto de ocho variantes o tipos genéticos. Su detección se realiza
con estudios microscópicos, moleculares y enzimáticos.
Llegar al diagnóstico de esta afección, que suele aparecer en más de un hermano en una
familia, afirma Halac, es difícil. ¿El motivo? Concurren varios exámenes: la microscopia
electrónica, el diagnóstico molecular, el estudio de la retina y las reacciones enzimáticas (tipos I
y II).
Estas últimas son un estudio de excelencia y único en el país realizado en un hospital
público, según los estándares internacionales para la detección de la acumulación irregular de
lipofuscinas en las neuronas.
La calidad de sobrevida de los pacientes dependerá de la ayuda familiar para cumplir con
el tratamiento, que incluye fisioterapia, actividad física y fármacos para aliviar los síntomas,

27
además de continuar con el disfrute de las actividades cotidianas.
Lipofuscinosis Ceroideas Neuronales (Enfermedad de Batten; Lipofuscinosis Neuronal
Ceroide; Enfermedad de Jansky-Bielschowsky; Enfermedad de Kufs; Enfermedad de
Santavuori-Haltia; Enfermedad de Spielmeyer-Vogt) enfermedad degenerativa hereditaria que se
caracteriza por inclusiones neuronales citoplasmáticas que se tiñen positivamente con ceroide y
lipofuscina. Los individuos afectados desarrollan degeneración de la retina, convulsiones,
mioclonias, ataxia, rigidez, y demencia progresiva. Clinicamente hay cuatro subtipos, divididos
por la edad del comienzo de los síntomas: infantil (tipo Santavuori-Haltia), infantil tardío (tipo
Jansky-Bielschowsky), juvenil (tipo Spielmeyer-Vogt), y adulto (enfermedad de Kuf). Las
formas infantil tardía y juvenil pueden referirse ambas como la enfermedad de Batten y
enfermedad de Batten-Mayou.
La enfermedad de Batten es la forma más común de un grupo de raros trastornos
conocidos como lipofuscinosis ceroides neuronales (NCL). La enfermedad de Batten es un
trastorno genético heredado que causa una acumulación de lipopigmentos en el tejido corporal.
Enfermedad de Batten se refiere a la forma juvenil de NCL, pero otras formas de NCL también
se pueden referir como enfermedad de Batten. Aproximadamente están afectados 2 a 4 de cada
100.000 nacimientos. Las formas de NCL incluyen:
• NCL infantil
• NCL infantil tardía
• NCL juvenil
• NCL adulta
La enfermedad de Batten es causada por anormalidades en los genes que se involucran
con la producción y uso de ciertas proteínas corporales. Esta enfermedad provoca la acumulación
de grasas y proteínas llamadas lipopigmentos en las células del cerebro, ojos, piel, y otros
tejidos.
Investigadores han hecho avance en la identificación de enzimas defectuosas y genes mutados
que subyacen estos trastornos, pero aún no se sabe exactamente cómo es que las mutaciones de
genes causan esta acumulación de lipopigmentos.

28
Un factor de riesgo es aquello que incrementa las probabilidades de contraer una
enfermedad o afección. Debido a que la enfermedad de Batten es una condición hereditaria, las
personas en riesgo incluyen:
• Hijos de padres con enfermedad de Batten
• Hijos de padres no afectados con enfermedad de Batten, pero que llevan los genes
anormales que causan la enfermedad
Los síntomas de enfermedad de Batten incluyen los siguientes:
• Pérdida de la visión (una señal temprana) y ceguera
• Descoordinación muscular
• Retraso mental o reducción de las funciones mentales
• Alteraciones o dificultades emocionales
• Convulsiones
• Espasmos musculares
• Deterioro del tono muscular
• Problemas con el movimiento
Los síntomas de la enfermedad de Batten son similares en cada tipo de la enfermedad. Sin
embargo, el tiempo de aparición, severidad, e índice de avance de los síntomas pueden variar
dependiendo del tipo de la enfermedad. Por ejemplo:
NCL infantil (enfermedad de Santavuori-Haltia): los síntomas comienzan entre los 6
meses y los 2 años y progresan con rapidez. Los niños con este tipo generalmente viven hasta la
etapa media de la niñez (aproximadamente cinco años de edad), aunque algunos sobreviven en
un estado vegetativo unos cuantos años más.
NCL infantil tardía (enfermedad de Jansky-Bielschowsky): los síntomas comienzan a
aparecer entre los 2 y 4 años y progresan con rapidez. Los niños con este tipo por lo general
viven hasta los 8-12 años de edad.

29
NCL juvenil (enfermedad de Spielmeyer-Vogt-Sjogren-Batten): los síntomas comienzan
a aparecer entre los 5 y los 8 años y progresan con menos rapidez. Las personas afectadas por lo
general viven hasta los últimos años de la adolescencia o la primera etapa de los 20 años; y en
algunos casos, hasta los 30 años.
NCL del adulto (enfermedad de Kufs o de Party): los síntomas generalmente comienzan a
aparecer antes de los 40 años, progresan lentamente y normalmente son más leves. Sin embargo,
esta forma de la enfermedad generalmente sí acorta el periodo de vida de una persona.
Diagnóstico
El diagnóstico clínico se sospecha por la edad de inicio, el compromiso visual, las
convulsiones y la regresión de las funciones psicomotoras y se confirma con los hallazgos en las
pruebas complementarias.
El electroencefalograma revela salvas de ondas lentas, de puntas lentas y ondas agudas,
sobre un trazado de fondo desorganizado y de baja amplitud.
Los estudios de neuroimagen, especialmente la resonancia magnética nuclear, revelan
atrofia cerebral grave y progresiva y puede observarse disminución de la sustancia blanca, así
como atrofia cerebelosa, aunque nunca tan grave como en la forma infantil tardía.
La biopsia de piel o músculo y en los linfocitos muestra inclusiones vacuolares o las
granulaciones azuráfilas en los polinucleares. Las inclusiones típicas en huella dactilar o
curvilíneas se evidencian mediante el estudio ultraestructural (visibles al microscopio
electrónico).
Neuroimagen
El uso de la neuroimagen es extremadamente importante para establecer una mejor
correlación clínica y esclarecer el diagnóstico. La tomografía axial computarizada (TAC)
cerebral es generalmente normal al inicio y usualmente aparecen anormalidades hasta 5-14 días

30
después, limitando su utilidad para confirmar el diagnóstico, donde se observarán lesiones de
baja atenuación y lesiones multifocales en la sustancia blanca subcortical. Definitivamente, las
lesiones logran apreciarse tempranamente y mejor con la resonancia magnética (RM) cerebral.
La afección del cerebro y cerebelo. Las características de las lesiones vistas en la RM cerebral
aparecen como áreas de atrofia cerebral, con pérdida de la sustancia blanca y atrofia cerebelosa.
(2).
Mientras los hallazgos neurofisiológicos que aparecen durante las etapas tempranas son
esenciales para el diagnóstico de la LINCL, los estudios de resonancia magnética (RM) son muy
útiles tanto en la diferenciación de las diversas formas clínicas, así como por su capacidad para
seguir la evolución (20-22). Los cambios más significativos en los estudios de neuroimagen
(RM), en todos los tipos de CLN están representados por atrofia cerebral progresiva. En el tipo
clásico de la LINCL, sin embargo, la atrofia es más pronunciada en las estructuras
infratentoriales, particularmente en el cerebelo (22,23). En los subtipos variantes de la LINCL,
los hallazgos de RM, especialmente en las etapas tempranas de la enfermedad, pueden variar
desde la normalidad hasta áreas discernibles de densidad aumentada en la sustancia blanca
periventricular, alrededor de los ventrículos laterales, intensidad disminuida en el tálamo y el
putamen, y atrofia cerebelosa. Se han descrito anormalidades en el tálamo y los ganglios basales,
tanto para las variantes de LINCL como para la CLN infantil (26). También se han comunicado
señales anormales hiperintensas en la sustancia blanca, tanto para la CLN clásica infantil tardía
como para la juvenil [15,31]. En la fase tardía de las variantes de la LINCL, la atrofia cerebral
difusa y las áreas de hipointensidad que incluyen los ganglios basales, dominan las imágenes de
la RM (22, 26,).
Los estudios de RM con espectroscopia han revelado cambios progresivos, con reducción
del ácido N-acetil aspártico, incremento de mioinositol y de la porción glutamato-glutamina, en
relación con la pérdida neuroaxonal generalizada. Más recientemente, se ha enfatizado el valor
de los estudios seriados con RM
convencional y espectroscópica, que revelan la pérdida progresiva del volumen cerebral lo cual
testimonia la degeneración neural.

31
2.3 MARCO TEÓRICO OPERACIONAL
2.3.1 DEFINICIÓN CONCEPTUAL DE LA VARIABLE.-
Ceroidolipofuscinosis en niños: son un grupo de enfermedades neurodegenerativas con
herencia autosómica recesiva, que se presentan principalmente en la infancia y adolescencia,
caracterizadas por síntomatología variable que incluye convulsiones, deterioro cognitivo, pérdida
visual y/o atrofia cerebral. El curso es habitualmente progresivo, con desarrollo de demencia que
lleva a la muerte. Desde el punto de vista neuropatológico se caracterizan por la acumulación
progresiva de lipofuscina, un lipopigmento autofluorescente, en neuronas y otros tejidos. (3, 4).
2.3.2 DEFINICIÓN OPERACIONAL DE LA VARIABLE.-
La variable del estudio, características de la ceoidolipofuscinosis en niños en la
resonancia magnética se evaluó a través de sus dimensiones hallazgos en la resonancia
magnética, y su incidencia en niños con sus respectivos indicadores
2.4 OPERACIONALIZACIÓN DE LAS VARIABLES
OBJETIVO GENERAL: Determinar las características en la Resonancia Magnética de
la ceroidolipofuscinosis en niños del Servicio de Neuropediatria del Servicio de Radiología del
Servicio Autónomo Hospital Universitario de Maracaibo.
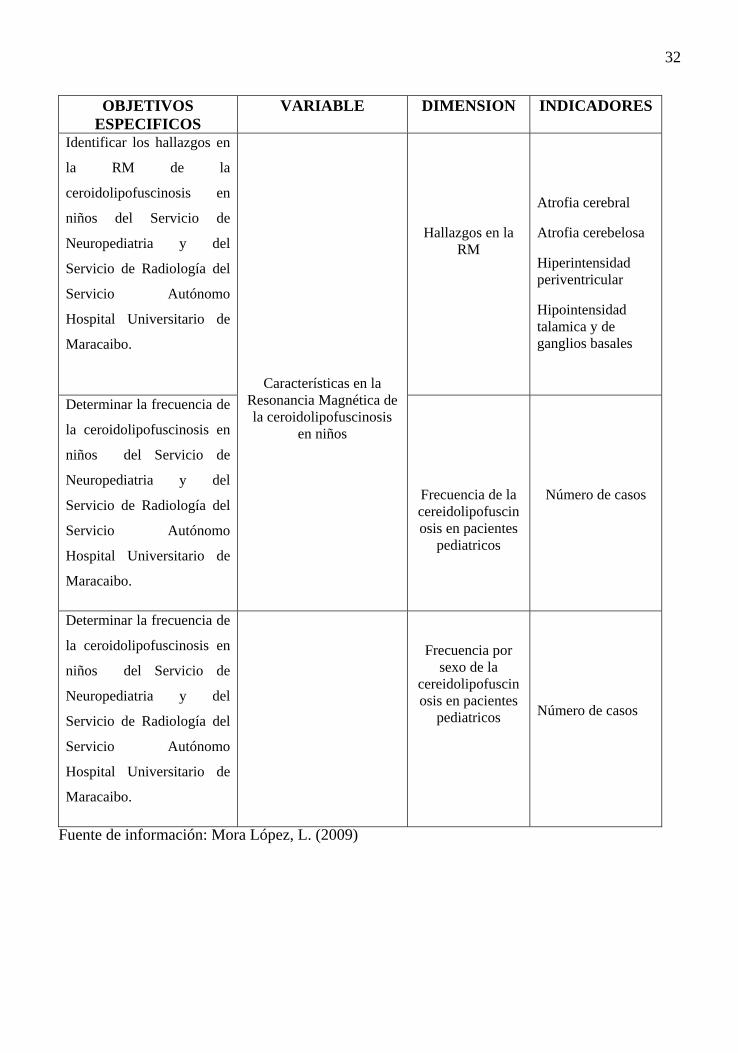
32
OBJETIVOS ESPECIFICOS
VARIABLE DIMENSION INDICADORES
Identificar los hallazgos en
la RM de la
ceroidolipofuscinosis en
niños del Servicio de
Neuropediatria y del
Servicio de Radiología del
Servicio Autónomo
Hospital Universitario de
Maracaibo.
Características en la Resonancia Magnética de la ceroidolipofuscinosis
en niños
Hallazgos en la RM
Atrofia cerebral
Atrofia cerebelosa
Hiperintensidad periventricular
Hipointensidad talamica y de ganglios basales
Determinar la frecuencia de
la ceroidolipofuscinosis en
niños del Servicio de
Neuropediatria y del
Servicio de Radiología del
Servicio Autónomo
Hospital Universitario de
Maracaibo.
Frecuencia de la cereidolipofuscinosis en pacientes
pediatricos
Número de casos
Determinar la frecuencia de
la ceroidolipofuscinosis en
niños del Servicio de
Neuropediatria y del
Servicio de Radiología del
Servicio Autónomo
Hospital Universitario de
Maracaibo.
Frecuencia por sexo de la
cereidolipofuscinosis en pacientes
pediatricos
Número de casos
Fuente de información: Mora López, L. (2009)

33
CAPITULO III
MARCO METODOLOGICO

34
MARCO METODOLOGICO
En este capítulo se describen los aspectos metodológicos de la investigación, señalando el
tipo y diseño de la misma, los sujetos y la forma a través de la cual se recogieron los datos, así
mismo las características de sus análisis estadísticos para la interpretación de los resultados.
TIPO DE INVESTIGACIÓN
La investigación que se realizo es de tipo descriptiva, ya que permitió determinar la
frecuencia de los hallazgos por Resonancia Magnética de la Cereidolipofuscinosis en los
pacientes del Servicio de Neuropediatria y del Servicio de Radiología del Servicio Autónomo
Hospital Universitario de Maracaibo. Al respecto Tamayo y Tamayo, afirman que ``los estudios
descriptivos comprenden la descripción, registro, análisis e interpretación de la naturaleza actual
y la composición o proceso de los fenómenos”(20).
Hernández, Fernández y Baptista, señalan que ``los estudios descriptivos buscan
especificar las propiedades importantes de personas, grupos, comunidades o cualquier otro
fenómeno que sean sometidos a análisis, miden o evalúan diversos aspectos, dimensiones o
compromisos del fenómeno o fenómenos a investigar”(21).
DISEÑO DE INVESTIGACIÓN
El diseño de investigación plantea la estrategia general que adopta el investigador para
responder al problema. Para Tamayo y Tamayo, ``el diseño plantea una serie de actividades
sucesivas y organizadas, que deben adaptarse a las particularidades de cada investigación y que
indican los pasos y pruebas a efectuar y las técnicas a utilizar para recolectar y analizar datos”.
(20).
De acuerdo a esto, el diseño fue considerado como no experimental en virtud de que no
hay “manipulación deliberada de las variables objeto a evaluación, sino que se observan los
fenómenos tal y como se dan en su contexto natural para poder analizarlo y dar respuesta y
solución”. Hernández et al.

35
Según el tiempo en el cual se desarrollo la investigación, el estudio fue descriptivo
transeccional puesto que según Hernández et al. “la recolección de la información se hizo solo
una vez en el tiempo”. (21).
SUJETOS DE LA INVESTIGACIÓN
POBLACIÓN
Tamayo y Tamayo, señalan que una población es la “totalidad de un fenómeno de
estudio, incluye la totalidad de las unidades de análisis o entidades de la población que integran
dicho fenómeno”. (20).
La población estudiada estuvo constituida por 50 pacientes que acudieron al Servicio de
Neuropediatria y Servicio de Radiología del Servicio Autónomo Hospital Universitario de
Maracaibo con hallazgos clínicamente sugestivos de cereidolipofuscinosis, durante el período
de Marzo 2010 hasta Diciembre 2010
MUESTREO
La técnica de muestreo para seleccionar la muestra fue de tipo no probabilística,
intencional debido a las características de la variable utilizada. Padua, lo describe como “ el
producto de una selección de casos según el criterio de algún experto; por medio de esto se
seleccionan algunos casos que resultan ser típicos´´. (22).
Para la selección de la muestra se consideraron como criterios de inclusión a todos los
pacientes pediátricos que presentaron hallazgos en la resonancia magnética de
cereidolipofuscinosis.
MUESTRA
La muestra se establecio realizando el muestreo, y la aplicación de los criterios de
inclusión y exclusión a la población, quedando finalmente la muestra constituida por 50

36
pacientes. Sierra Bravo, señala que una muestra es simplemente “una parte representativa de la
población debidamente elegida, que se somete a observación científica en representación del
conjunto, con el propósito de obtener resultados validos” (23), Igualmente Fidias. A, define la
muestra es un subconjunto representativo y finito que se extrae de la población accesible. (24).
INVESTIGADORES/OBSERVADORES/ENCUESTADORES
El equipo de trabajo que colaborara para la realización de esta investigación estuvo
constituido por la autora, el tutor académico, el personal del servicio de radiología y del servicio
de Neuropediatria, quienes además fueron observadores del proceso. Los encuestadores
estuvieron constituidos por la autora y el equipo que trabajo durante la realización de cada
estudio en el área respectiva.
EQUIPOS:
El equipo utilizado durante la evaluación de cada paciente fue:
Resonador GE 1.5 Teslas, gradiente 77 MiliTeslas, modelo Signa Excite.
PROCEDIMIENTOS
Se evaluaron los pacientes que acudieron al servicio de radiología, provenientes del
servicio de Neuropediatria del Servicio Autónomo Hospital Universitario de Maracaibo, donde
se les realizo una historia clínica detallada que incluye los antecedentes perinatales, examen
físico así como resultados de estudios de imágenes anteriores.
A cada paciente pediátrico se le administro hidrato de cloral por vía oral a una dosis de 1
cc x Kg de peso, para que sedarlo y no se mueva durante el estudio.
Luego el paciente se coloca en el Resonador, en posición supino y se procede a realizar
cortes axiales, sagitales y coronales, en secuencias de pulso a predominio T1 y T2 con técnica de

37
Fast Flair y Ecogradiente. Luego las imágenes son procesadas en la Workstation GE, modelo
4.2
PLAN DE ANÁLISIS DE DATOS
La recolección de los datos se realizo a través de la historia clínica donde se especifican
los datos de cada uno de los pacientes evaluados a través de la realización de la resonancia
magnética, para posterior aplicar las técnicas de análisis. Se utilizo la estadística descriptiva
mediante el uso de cifras absolutas y porcentajes, a través del paquete estadístico SPSS Versión
10.0 y los datos se presentan en tablas.

38
CAPITULO IV
ANALISIS Y DISCUSION DE
LOS RESULTADOS

39
En el siguiente capítulo se hace el análisis y discusión de los resultados que permitieron
determinar las características por Resonancia Magnética de la ceroidolipofuscinosis en niños del
Servicio de Neuropediatria y Servicio de Radiología del Hospital Universitario de Maracaibo, a
través de la identificación de los hallazgos y la frecuencia de aparición en la RM de la
ceroidolipofuscinosis en estos pacientes.
Se evaluó la frecuencia de incidencia de casos de ceredolipofuscinosis en niños y se
observo que en la muestra evaluada el 34% de los niños presentaron hallazgos en la resonancia
magnética de atrofia cerebral, atrofia cerebelosa, hipointensidad periventricular, hipointensidad
talamica y de los ganglios basales, los cuales establecen el diagnostico, así mismo se evidencio
que en el 66% no hubo presencia de alguna lesión compatible con la presencia de esta patología.
El análisis descriptivo evidencio una media de 1.66 con una mediana de 2, lo que expresa una
tendencia de la muestra a la alternativa negativa para la presencia de ceredolipofuscinosis en
niños.
TABLA 1
Análisis frecuencial y descriptivo de la presencia de Cereidolipofuscinosis en niños
ANALISIS/ALTERNATIVA
Fr % Media + DE Mediana
Cereidolipofuscinosis 17 34 1.66±0.47 2.00
Fuente: Elaboración propia.

40
Como vemos en la tabla 2 la distribución frecuencial y porcentual por sexo de los
niños con hallazgos de ceredolipofuscinosis en la resonancia magnética, evidenciándose un
predominio del sexo masculino correspondiente al 66% de los casos y en el femenino solo en el
34% respectivamente.
TABLA 2
Análisis frecuencial y descriptivo de la presencia por sexo de
Cereidolipofuscinosis en niños
ANALISIS/ALTERNATIVA
Fr %
Femenino 5 29
Masculino 12 71
Fuente: Elaboración propia.

41
En la tabla numero 3 se vemos que la presencia de hallazgos en la RM cerebral de la muestra
evaluada evidencio que del total de la población evaluada en el 34% se observaron lesiones tipo
atrofia cerebral y cerebelosa, con una media de 1.66; en el 18% de los casos, se observaron
cambios correspondientes a hiperintensidad periventricular y solo en el 4% con una media de
1.96 los casos hipointensidad talamica y de ganglios basales cada una respectivamente.
TABLA 3
Análisis frecuencial y descriptivo de los hallazgos de ceredolipofuscinosis en la Resonancia Magnética cerebral en niños
ANALISIS/ALTERNATIVA
Fr % Media + DE Mediana
Atrofia cerebral 17 34 1.66±0.47 2.00
Atrofia cerebelosa 17 34 1.66±0.47 2.00
Hiperintensidad periventricular 9 18 1.82±0.38 2.00
Hipointensidad talamica 2 4 1.96±0.19 2.00
Hipointensidad de ganglios basales 2 4 1.96±0.19 2.00
Fuente: Elaboración propia.

42
Discusión.-
La cereidolipofuscinosis se consideran el trastorno neurodegenerativo de depósito más
frecuente de la infancia, como lo plantea Boustany y Santavuori (1996-1974), considerando esta
afirmación, la incidencia de casos de ceredolipofuscinosis en niños observada en la muestra
evaluada es considerable ya que el 34% de los niños mostraron hallazgos con hallazgos en la
resonancia magnética
La distribución según sexo de los niños con hallazgos de ceredolipofuscinosis en la
resonancia magnética, evidenciándose un predominio del sexo masculino en la muestra evaluada.
Con respecto a la presencia de hallazgos en la RM cerebral de la muestra evaluada
evidencio que en total de la población con cambios en la resonancia magnetica se observaron
lesiones tipo atrofia cerebral y cerebelosa, en el 18% de los casos, se observaron cambios
correspondientes a hiperintensidad periventricular y solo en el 4%, los casos hipointensidad
talamica y de ganglios basales cada una respectivamente. Estos resultados apoyan a lo planteado
por T, Raininko R, Launes J, Nuutila A, Santavuori P. Jansky-Bielschowski (1992) los que
afirman este tipo de imagen son muy útiles tanto en la diferenciación de las diversas formas
clínicas, así como por su capacidad para seguir la evolución. Los cambios más significativos en
los estudios de neuroimagen (RM), en todos los tipos de CLN están representados por atrofia
cerebral progresiva, especialmente en las etapas tempranas de la enfermedad, pueden variar
desde la normalidad hasta áreas discernibles de densidad aumentada en la sustancia blanca
periventricular, alrededor de los ventrículos laterales, intensidad disminuida en el tálamo y el
putamen, y atrofia cerebelosa. También se han comunicado señales anormales hiperintensas en la
sustancia blanca, En la fase tardía se observan la atrofia cerebral difusa y las áreas de
hipointensidad que incluyen los ganglios basales. Todos estos cambios evidenciados en la
neuroimagenes de los pacientes evaluados en la investigación.

43
Conclusiones y Recomendaciones.-
Conclusiones.-
El análisis y discusión de los resultados permitió establecer las siguientes conclusiones:
Se identificaron los hallazgos en la RM de la ceroidolipofuscinosis en los niños
estudiados, siendo la atrofia cerebral y cerebelosa los mas frecuentes.
Se determino la incidencia de la ceroidolipofuscinosis en niños según el sexo,
determinando que la enfermedad es mas frecuente en el sexo masculino.
Finalmente se concluye que la Resonancia Magnética Cerebral es útil para identificar las
lesiones características de la ceroidolipofuscinosis y ayudar a establecer basado en el análisis o
evaluación clínica el diagnostico en niños, enfatizando el valor de la RM para el diagnostico,
control y manejo de los pacientes con esta enfermedad.
Recomendaciones.-
Se recomienda el uso de la RM cerebral en todo paciente pediátrico como técnica de
imágenes cuando exista regresión psicomotora y el posible diagnostico de cereidolipofuscinosis.
Promover la utilización de las técnicas de imágenes radiológicas como herramienta para
facilitar la realización de diagnósticos en Neuropediatria.
Utilizar esta técnica de imágenes como Gold estándar en todo paciente con sospecha
clínica de un proceso degenerativo del SNC en los pacientes pediátricos.

44
Bibliografia
1.- Boustany RM. Batten disease or neuronal ceroid lipofuscinosis. In Moser H, ed. Handbook of
clinical neurology: neurodystrophies and neurolipidoses. Amsterdam: Elsevier Science; 1996. p.
671-700.
2.- Santavuori P, Haltia M, Rapola J. Infantile type of so-called neuronal ceroid-lipofuscinoses.
Dev Med Child Neurol 1974; 16: 644-53.
3.- Mole S, Gardiner M. Molecular genetics of the neuronal ceroid lipofuscinoses. Epilepsia
1999; 40 (Suppl 3): 29-32.
4.- Haltia M. The neuronal ceroid-lipofuscinoses. J Neuropathol Exp Neurol 2003; 62: 1-13.
5.- Santavuori P, Haltia M, Rapola J. Infantile type of so-called neuronal ceroid-lipofuscinoses.
Dev Med Child Neurol 1974; 16: 644-53.
6.- Savukoski M, Klockars T, Holmberg V, Santavuori P, Lander ES, Peltonen L. Cln5, A novel
gene encoding a putative transmembrane proREVNEUROL 2004; 38 (1): 42-48 J.A. PEÑA, ET
AL 48 tein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat Genet
1998; 19: 286-8.
7.- Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L, Antonellis KA, et al. Mutations
in a novel Cln6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in
man and mouse. Am J Hum Genet 2002; 70: 324-35.
8.- Teixeira C, Espinola J, Huo L, Kohlschutter J, Persaud-Sawin DA, Minassian B, et al. Novel
mutations in the Cnl6 gene causing a variant late infantile neuronal ceroid lipofuscinosis. Human
Mutation 2003; 21: 502-8.

45
9.- Lonka L, Kyttala A, Ranta S, Jalanko A, Lehesjoki AE. The neuronal ceroid lipofuscinosis
Cln8 membrane protein is a resident of the endoplasmic reticulum. Hum Mol Genet 2000; 9:
1691-7.
10.- Autti T, Raininko R, Launes J, Nuutila A, Santavuori P. Jansky-Bielschowski variant
disease: CT, MRI, and SPECT findings. Pediatr Neurol 1992; 8: 121-6.
11.- Holmberg V, Lauronen L, Autti T, Santavuori P, Savukoski M, Uvebrant P, et al.
Phenotype-genotype correlation in eight patients with Finnish variant late infantile NCL (CLN5).
Neurology 2000; 55: 579-81.
12.- Pettersen B, Handwerker M, Huppertz H. Neuroradiological findings in classical late
infantile neuronal ceroid-lipofuscinosis. Pediatr Neurol 1996; 15: 344-7.
13.- Machen BC, Williams JP, Lum GB, Dyken P, Joslyn JN, Harpen MD. Magnetic resonance
imaging in neuronal ceroid-lipofuscinosis. J Comput Tomogr 1987; 2: 160-6.
14.- Peña JA, Cardozo J, González S, Luna D. Aspectos neurológicos de la
ceroidolipofuscinosis. Rev Neurol 2000; 31: 283-7.
15. Santavuori P, Vanhanen SL, Autti T. Clinical and neuroradiological diagnostic aspects of
neuronal ceroid lipofuscinoses disorders. Eur J Paediatr Neurol 2001; 5 (Suppl): 157-61.
16.- Vanhanen SL, Raininko R, Santavuori P. Early differential diagnosis of infantile neuronal
ceroid lipofuscinosis, Rett-syndrome and Krabbe disease by CT. Am J Neuroradiol 1994; 15:
1443-53.
17.- R. Caraballo, S. Monges, Carmen Medina Monzón, Víctor Luis Ruggieri. Lipofuscinosis
neuronal ceroidea infantil tardía: aspectos clínicos y electroencefalográficos .Revista de
neurología, Vol. 40, Nun. 3, 2005. , pags. 135-140

46
19.- J.A. Peña, C. Montiel-Nava, W. Delgado, M.L. Hernández ,J.J. Cardozo , E. Mora, L. Soto-
Faneite. Caracterización de la Cereidolipofuscinosis en niños Venezolanos. Nota Clínica. 2004
20.-Tamayo y Tamayo. (2003). El Proceso de la Investigación Científica. Editorial Limusa.
Mexico. Pag.- 46-187-176.
21.-.- Hernandez, Fernandez y Batispta. (1998). Metodología de la Investigación. Mc Graw Hill.
Tercera Edición. Mexico. Pag.- 60-74.24.- Padua, J.(1998). Técnicas de Investigación Aplicadas
a la Ciencias Sociales. Editorial Fondo de Cultura Económica. México. Pág 68-70.
22.- Padua, J.(1998). Técnicas de Investigación Aplicadas a la Ciencias Sociales. Editorial Fondo
de Cultura Económica. México. Pág 68-70.
23.- Sierra Bravo. (1997). Metodología de la Investigación Segunda edición. Pág 98.
24.- Fidias G. Arias. (1997). El Proyecto de Investigación. Quinta Edición. Editorial Episteme.
Pág 83.

47
ANEXOS

48
Anexo 1. Instrumento de Recolección de Datos.
HALLAZGOS POR RESONANCIA MAGNETICA DE LA CEREIDOLIPOFUSCINOSIS
EN NINOS
Fecha # de
historia
NOMBRE Y APELLIDO
EDAD
DIRECCION
TELEFONO
ANTECEDENTES FAMILIARES
ANTECEDENTES PERSONALES
cambios en comportamiento
retardo psicomotor
regresion de destrezas alcanzadas
MANIFESTACIONES CLINICAS
FIEBRE CEFALEA
CONVULSIONES OTROS
CARACTERISTICAS DE LA LESION POR RM
EXISTE ATROFIA CEREBRAL
EXISTE HIPERINTENSIDAD PV
EXISTE VENTICULOMEGALIA
ATROFIA CEREBELOSA
OTROS