group i metabotropic glutamate receptors in spinal cord injury: roles in neuroprotection and the...
TRANSCRIPT

JOURNAL OF NEUROTRAUMAVolume 19, Number 1, 2002Mary Ann Liebert, Inc.
Group I Metabotropic Glutamate Receptors in Spinal CordInjury: Roles in Neuroprotection and the Development of
Chronic Central Pain
CHARLES D. MILLS, KATHIA M. JOHNSON, and CLAIRE E. HULSEBOSCH
ABSTRACT
Spinal cord injury (SCI) initiates a cascade of biochemical events that leads to an increase in ex-tracellular excitatory amino acid (EAA) concentrations, which results in glutamate receptor–medi-ated excitotoxic events. An important division of these glutamate receptors is the metabotropic glu-tamate receptor (mGluR) class, which is divided into three groups. Of these three groups, group I(mGluR1 and mGluR5) activation can initiate a number of intracellular pathways that lead to in-creased extracellular EAA concentrations. To evaluate subtypes of group I mGluRs in SCI, we ad-ministered AIDA (group I antagonist), LY 367385 (mGluR1 specific antagonist), or MPEP (mGluR5specific antagonist) by interspinal injection to adult male Sprague-Dawley rats (175–200 g) imme-diately following injury at T10 with an NYU impactor (12.5-mm drop, 10-g rod, 2 mm in diame-ter). AIDA- and LY 367385-treated subjects had improved locomotor scores and demonstrated anattenuation in the development of mechanical allodynia as measured by von Frey stimulation of theforelimbs; however, LY 367385 potentiated the development of thermal hyperalgesia. MPEP hadno effect on locomotor recovery or mechanical allodynia, but attenuated the development of ther-mal hyperalgesia. AIDA and LY 367385 treatment resulted in a significant increase in tissue spar-ing compared to the vehicle-treated group at 4 weeks following SCI. These results suggest thatmGluRs play an important role in EAA toxicity and have different acute pathophysiological rolesfollowing spinal cord injury.
Key words: excitotoxicity; metabotropic glutamate receptors; neuroprotection; pain; spinal cord injury
23
Department of Anatomy and Neurosciences, University of Texas Medical Branch at Galveston, Texas.
INTRODUCTION
SPINAL CORD INJURY (SCI) is a devastating injury thatcan cause loss or impairment of motor and sensory
function below the level of injury. In addition to the lossof motor function, up to 75% of SCI victims may de-velop chronic central pain (CCP) syndromes (Balazy,1992; Beric et al. 1988; Boivie, 1984; Christensen and
Hulsebosch, 1997a; Davidoff and Roth, 1991; Richardset al., 1980; Rintala et al., 1998; Tasker and Dostrovsky,1989). Central pain syndromes in patients with SCI areoften recurring, spontaneous, wax and wane intermit-tently, and may be described as a numbness, burning, cut-ting, or piercing sensation (Davidoff and Roth, 1991).Additionally, patients may develop evoked pain syn-dromes, which are produced when a normally nonnox-

ious stimulus becomes noxious (allodynia) or when anoxious stimulus becomes more noxious (hyperalgesia;Merskey and Bogduk, 1994). The development of CCPcan so greatly affect the quality of life that depression isa major occurrence among SCI victims (Cairns et al.,1996; Lundqvist et al., 1991; Segatore, 1994).
There are a variety of mechanisms that may explainthe altered behavioral nociceptive states of CCP, includ-ing release of spinal cord nociceptive processing fromdescending inhibition (Sweet, 1991), deafferentation hy-perexcitability of spinal neurons and/or thalamic neurons(Lenz et al., 1994; Rinaldi et al., 1991; Tasker andDostrovsky, 1989), increased efficacy of previously in-effective synapses (Basbaum and Wall, 1976; Devor andWall, 1981) and structural alterations such as intraspinalsprouting (Christensen and Hulsebosch, 1997b; Hulse-bosch and Coggeshall, 1981a,b; Krenz and Weaver,1998; Krenz et al., 1999; McNeil et al., 1990, 1991), allof which contribute to the maintained hyperexcitabilityof dorsal horn neurons (Christensen and Hulsebosch,1997a). Another mechanism for maintained hyperex-citability of dorsal horn neurons involves EAA receptor-mediated changes (Bennett et al., 2000; Hulsebosch etal., 2000; Mills et al., 2000, 2001a; Woolf and Thomp-son, 1991) and acute changes in EAA transporters(McAdoo et al., 2000; Vera-Portocarrero et al., 1999).Following injury there is a brief, but large amount of glu-tamate released at the injury site (Liu et al., 1991;McAdoo et al., 1999; Panter, 1990). This increase in ex-tracellular glutamate activates glutamate receptors, whichare divided into two major types: (1) ionotropic recep-tors (iGluR): N-methyl-D-aspartate (NMDA), alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid(AMPA), kainate (KA) receptors and (2) metabotropicreceptors: metabotropic glutamate receptors (mGluR),groups I, II, and III. Since the mGluRs are G-protein cou-pled, their activation can initiate intracellular signalingpathways that have multiple long-lasting effects.
The separation of mGluRs into three groups (groups I,II, and III) is based on sequence homology, transductionmechanisms and pharmacological profiles (for a review,see Conn and Pin, 1997). mGluRs are key componentsin glutamate-initiated intracellular signaling transductioncascades that can potentiate iGluR responses (Aniksztejinet al., 1992; Bleakman et al., 1992; Fitzjohn et al., 1996;Glaum and Miller, 1993; Harvey and Collingride, 1993;Kelso et al., 1992), increase neurotransmitter release(Herrero et al., 1992), and raise intracellular calcium lev-els (Courtney et al., 1990; Llano et al., 1991; Manzoni etal., 1991; Murphy and Miller, 1988, 1989; Rainnie et al.,1994; Yuzaki and Mikoshia, 1992). Group I mGluRs(mGluR1 and mGluR5) stimulate phospholipase C (PLC)pathways, activate protein kinase C (PKC), and enhance
glutamate release (Conn and Pin, 1997). There is a ma-jor role for mGluRs in mediating nociceptive responses(Fisher and Coderre, 1996a,b; Fundytus et al., 1998;Neugebauer et al., 1994, 1999; Young et al., 1994, 1995,1997, 1998) and injury-induced excitotoxicity (Bruno etal., 1995, 1998, 1999; Buisson and Choi, 1995; Mukhinet al., 1997), but little is known about mGluRs in the de-velopment of CCP following SCI.
Recently it was demonstrated that a group I mGluR se-lective antagonist, 1-aminoindan-1,5-dicarboxylic acid(AIDA), decreased extracellular glutamate concentrationsand attenuated the development of mechanical allodyniafollowing SCI (Mills et al., 2000). Here we extend thesestudies by further examining a different dose of AIDAand determining the contributions of individual group ImGluRs to the development of CCP following SCI usingthe mGluR1 selective antagonist, (1)-2-methyl-4-car-boxyphenylglycine (LY 367385), and the mGluR5 selec-tive antagonist, 2-methy-6-(phenylethynyl)-pyridine(MPEP). Locomotor recovery, responses to mechanicaland thermal stimulation, and the amount of spared tissuefollowing contusion SCI were used to assess treatmenteffects.
MATERIALS AND METHODS
Experimental Animals
Subjects were male Sprague-Dawley rats, 175–200 g,obtained from Harlan Sprague-Dawley, Inc. and housedwith a light/dark cycle of 12 h/12 h. Experimental pro-cedures were in accordance with the NIH Guide for theCare and Use of Laboratory Animals. Twenty-eight ratswere randomly divided into four groups: vehicle-, AIDA-,LY 367385-, or MPEP-treated (n 5 7 for each group).
Injury Production
An incomplete spinal cord contusion injury was pro-duced as previously described (Huang and Young, 1994;Hulsebosch et al., 2000) on three separate days, with twoto three animals from each treatment group each day.Briefly, subjects were anesthetized by an intraperitonealinjection of pentobarbital (40 mg/kg). Anesthesia wasconsidered complete when there was no flexor with-drawal in response to noxious foot pinch. The subjects’backs were shaven, an incision made to expose the ver-tebral column, and a laminectomy was performed to ex-pose spinal segment T10. Spinal cord injury was pro-duced using the New York University (NYU) injurydevice (Constantini and Young 1994; Gruner, 1992). A10-g weight, 2.0 mm in diameter, was dropped from aheight of 12.5 mm onto the exposed cord. There was no
MILLS ET AL.
24

significant difference in impact parameters for depth ofcompression and impactor velocity between the treatmentgroups. Mean compression depths (6SEM) were 2.25 6
0.31, 2.23 6 0.23, 2.29 6 0.21, and 2.31 6 0.30 mm forvehicle-, AIDA-, LY 367385-, and MPEP-treatmentgroups, respectively. Mean velocities (6SEM) were0.50 6 0.02, 0.48 6 0.01, 0.47 6 0.01, and 0.48 6 0.02m/sec for vehicle-, AIDA-, LY 367385-, and MPEP-treat-ment groups, respectively.
Following injury, the muscle and fascia were sutured,the skin autoclipped, and the animals were allowed to re-cover from anesthesia. Postoperative treatments included0.9% saline (1.0 mL subcutaneously) for rehydration anda prophylactic antibiotic (Baytril, 30 mg/kg, subcuta-neously) was given twice daily until bladder control re-turned. Bladders were manually expressed twice daily un-til micturition control recovered, usually by 10 daysfollowing injury.
Drug and Vehicle Administration
AIDA, LY 367385, MPEP (Tocris; Ballwin, MO), orartificial cerebrospinal fluid [ACSF (vehicle control);containing, in mM: 151.1 Na21, 2.6 K1, 0.9 Mg21, 1.3Ca21, 122.7 Cl2, 21.0 HCO3
2 and HPO422] was injected
stereotaxically into the epicenter of impact through a 30-gauge needle coupled to a syringe pump (Surgical Neu-rology, NIH), at a depth of 1 mm immediately followingSCI (within 3 min). AIDA (pH 7.4) was administered byinjecting 4 mL of a 50 mM solution (200 nmol total) inACSF over a 20-min interval. LY 367385 (pH 7.4) andMPEP (pH 7.3) were both administered by injecting 5 mlof a 50 mM solution (250 nmol total) prepared in ACSFover a 25-min interval. Vehicle controls were injectedwith 5 ml of ACSF over a 25-min interval. The dose ofAIDA selected was based on previous works demon-strating AIDA’s effect on glutamate release and the de-velopment of mechanical allodynia and thermal hyperal-gesia (Mills et al., 2000; Moroni et al., 1997). For LY367385 and MPEP, we selected the lowest doses thatgave the maximum behavioral and neuroprotective ef-fects (Bruno et al., 1999, 2000; Chapman et al., 1999,2000).
Locomotor Function
Locomotor function was evaluated using the Basso,Beattie, and Bresnahan (BBB) open-field locomotor test(Basso et al. 1995). Briefly, the BBB scale ranges from0 (no hindlimb movement) to 21 (normal movement-co-ordinated gait with parallel paw placement). Scores from0-7 indicate the return of isolated movements in the threejoints (hip, knee, and ankle). Scores from 8-13 indicatethe return of paw placement and coordinated movements
with the forelimbs. Scores 14-21 show the return of toeclearance during stepping, predominant paw position,trunk stability, and tail position. BBB scores were mea-sured before surgeries (baseline) and on postcontusiondays (PCD) 1–14, 21, and 28. Since the BBB scale isnonlinear, the median and percentile points (10%, 25%,75%, and 90%) are reported to display distribution of thedata about the median; however, to allow comparisonwith previous studies we also report the means and SEMs.To control for possible asymmetric injuries, left and righthindlimbs were scored individually. Animals that hadhindlimbs scores that differed by 3 or more between eachhindlimb on PCD 1 were excluded (none in the presentstudy). Reported BBB scores for an individual subjectrepresent the average from both hindlimbs; therefore, anoninteger median is reported at some time points.
Responses to Mechanical Stimuli
Paw withdrawal frequency in response to repeated me-chanical stimuli to the glabrous surface of the forelimbswas used to quantify mechanical sensitivity. Prior to theonset of testing, all animals were handled daily for oneweek and were acclimated to clear Plexiglas testing cu-bicles (8 3 8 3 18 cm) 4 h daily for 3 days. The cubi-cles provide some freedom of movement allowing the an-imal to turn around within the cubicle. The occurrenceof paw withdrawals in response to graded mechanicalstimuli (4.79, 9.96, 204.1 mN von Frey filaments andpin), which were accompanied by supraspinal behavior(such as head turns, vocalizations, and biting the fila-ment), was recorded prior to injury and on PCD 7, 14,21, and 28 as previously described (Christensen andHulsebosch 1997a). The occurrence of forelimb pawwithdrawals to von Frey filament stimulation in each of10 trials was repeated three times for each forelimb, witha least 5 min between trials, and averaged for each testday. Data is expressed as a difference in response fre-quency (percent of paw withdrawals after injury minuspercent of paw withdrawals before injury) for betweengroup comparisons. An increase in the percent of with-drawals to von Frey filament stimulation is consistentwith an increase in mechanical sensitivity and is termedmechanical allodynia.
Thermal Stimuli
The latency of forelimb paw withdrawal to heat stim-uli was measured using methods previously described(Bennet and Xie 1988; Dirig et al., 1997; Hargreaves etal., 1988) with a commercially available paw thermalstimulating system (UARDG, Department of Anesthesi-ology, University of California, San Diego, La Jolla, CA)prior to surgery and at PCD 7, 14, 21, and 28. The rats
GROUP I mGluRs IN SCI
25

were placed on a glass plate over a light box and a radi-ant heat stimulus was applied by aiming a beam of lightthrough a hole in the light box onto the glabrous surfaceof the paw through the glass plate. The glass plate washeated from 30°C to 50°C, linearly, over 15 sec. The lightbeam was turned off automatically by a photocell whenthe rat lifted the limb, allowing the measurement of timebetween the start of the light beam and the paw with-drawal. This time is defined as the paw withdrawal la-tency and gives an objective measure of the time neededfor the stimulus to become noxious. The paw withdrawalmust be accompanied by supraspinal behaviors (e.g.,head turning and licking of the paw) to be recorded as aresponse. Five minutes were allowed between each trial,and three measurements were averaged for each limb oneach test day. Withdrawal latencies were normalized tozero for baseline and reported as change, in seconds, frombaseline.
Spared Tissue
On PCD 28 animals were deeply anesthetized withsodium pentobarbital and perfused intracardially with he-parinized 0.9% saline followed by 4% cold bufferedparaformaldehyde. After perfusion, the spinal cord waspostfixed in 4% paraformaldehyde at 4°C, processed forparaffin embedding, and then serially sectioned at 15 mm.
Sections were mounted on gelatin/potassium chromiumsulfate coated slides and stained with Luxol blue (0.1%)and cresyl violet (0.1%) to visualize myelinated whiteand gray matter, respectively. Every 20th section was an-alyzed by measuring the area of gray and white matterspared using Bioquant v3.50 software. Areas ofmacrophage infiltration and gliosis (glial scarring) thattypically surrounded cystic cavities were excluded.
Pre-injury gray and white matter volumes were deter-mined by calculating correction factors as previously de-scribed (Olby and Blakemore, 1996). Briefly, normalspinal cords (n 5 3) were sectioned, stained, and areaswere measured as described above. The section corre-sponding to 6.0 mm rostral to the center of T10 was usedas a reference section. The numerical factor by which thearea of this section had to be multiplied to arrive at themeasured areas of all subsequent blocks was establishedfor gray and white matter separately (Table 1). These cor-rection factors were then applied to the correspondingreference segment (6.0 mm rostral to the epicenter of in-jury) in subjects of the experimental groups to calculateindividual pre-injury gray and white matter volumes.
The Cavalieri method was used to obtain an unbiasedestimate of spared tissue from sequential serial sections(Gundersen et al., 1988; Michel and Cruz-Orive, 1988).The volume of spared tissue (Vsp) was calculated by mul-tiplying the measured area (a) of the tissue, gray or white,
MILLS ET AL.
26
TABLE 1. CORRECTION FACTORS FOR GRAY AND WHITE MATTER TISSUE AREAS
Distance from Distance fromcenter of T10 (mm) Gray matter White matter center of T10 (mm) Gray matter White matter
6.0 1.000 6 0.000 1.000 6 0.000 20.3 1.063 6 0.019 1.105 6 0.0185.7 1.002 6 0.008 0.997 6 0.010 20.6 1.064 6 0.017 1.104 6 0.0225.4 0.994 6 0.010 1.004 6 0.008 20.9 1.069 6 0.025 1.107 6 0.0255.1 1.006 6 0.008 0.947 6 0.009 21.2 1.069 6 0.013 1.106 6 0.0294.8 0.956 6 0.007 0.942 6 0.023 21.5 1.071 6 0.006 1.107 6 0.0334.5 1.011 6 0.006 0.950 6 0.019 21.8 1.077 6 0.018 1.110 6 0.0364.2 1.014 6 0.009 0.967 6 0.015 22.1 1.081 6 0.010 1.110 6 0.0323.9 1.017 6 0.010 0.941 6 0.009 22.4 1.086 6 0.013 1.115 6 0.0253.6 1.021 6 0.011 0.916 6 0.013 22.7 1.090 6 0.019 1.114 6 0.0073.3 1.025 6 0.015 1.000 6 0.019 23.0 1.090 6 0.017 1.116 6 0.0263.0 1.029 6 0.017 1.022 6 0.025 23.3 1.093 6 0.024 1.122 6 0.0332.7 1.032 6 0.031 1.029 6 0.023 23.6 1.095 6 0.029 1.124 6 0.0372.4 1.034 6 0.025 1.036 6 0.016 23.9 1.099 6 0.009 1.120 6 0.0252.1 1.039 6 0.016 1.045 6 0.019 24.2 1.104 6 0.012 1.126 6 0.0351.8 1.038 6 0.018 1.069 6 0.026 24.5 1.102 6 0.036 1.120 6 0.0271.5 1.040 6 0.030 1.078 6 0.030 24.8 1.106 6 0.032 1.121 6 0.0331.2 1.048 6 0.006 1.082 6 0.021 25.1 1.105 6 0.031 1.123 6 0.0420.9 1.053 6 0.011 1.086 6 0.015 25.4 1.108 6 0.023 1.125 6 0.0440.6 1.057 6 0.009 1.085 6 0.012 25.7 1.11 6 0.014 1.125 6 0.0500.3 1.052 6 0.019 1.095 6 0.011 26.0 1.113 6 0.025 1.123 6 0.0390 1.062 6 0.022 1.103 6 0.007
Mean CF 6 SEM (n 5 3) Mean CF 6 SEM (n 5 3)

GROUP I mGluRs IN SCI
27
on a single section and by the distance (d) between thenext section (Vsp 5 a � d). The volumes of spared tissuewere summed across the entire measured area to give thetotal volume of spared tissue (VTsp 5 Vsp1 1 Vsp2 1
. . . 1 Vsp41) for gray (VTspg) and white (VTspw) matter.The total volume of spared tissue (VT(sp)) was defined asspared gray matter 1 spared white matter (VT(sp) 5
VTspg 1 VTspw). The amount of spared tissue was ex-pressed as a percent of the pre-injury volume using theformula (VT(sp)/VTpi) � 100, where VTpi is the calculatedpreinjury volume. Reporting volume of spared tissue asa percentage of preinjury volume avoids overestimationof the spared tissue volume (Rabchevsky et al., 2001).
Statistical Analysis
One-way analysis of variance (ANOVA) was per-formed to assess changes between groups for impact pa-rameters (depth of cord compression and impactor ve-locity), and for comparison of spared tissue measures. Toexamine specific differences between time points,Tukey’s HSD test was used for post hoc comparisons.The Mann-Whitney U test was used for nonparametricanalysis of BBB scores. One way repeated measuresANOVA was used to assess changes over time for re-sponses to mechanical and thermal stimulation, followedby the Bonferroni t test for post hoc pairwise compar-isons. An alpha level of significance at 0.05 was used forall statistical tests. Data are expressed as means 6 stan-dard error of the mean (SEM). BBB scores are addition-ally reported as medians and percentile ranges.
RESULTS
Locomotor Function
All groups had presurgical mean baseline BBB scoresof 21.0 6 0.0 (Figs. 1 and 2). On PCD 1, vehicle-, AIDA,LY 367385-, and MPEP-treated had mean combinedhindlimb BBB scores of 0.5 6 0.2, 1.3 6 0.8, 4.2 6 1.8,and 0.8 6 0.2, respectively. BBB scores rose until reach-ing plateau values on PCD 14. AIDA- and LY 367385scores were consistently higher than vehicle-treated;however, MPEP-treated animals were not significantlydifferent compared to controls at anytime point measured.LY 367385-treated animals were significantly higherthan vehicle-treated on PCD 2 through PCD 8. On PCD28 there was no significant difference between any of thetreatment groups. Mean BBB scores on PCD 28 were:15.6 6 0.8, 17.1 6 0.9, 17.1 6 1.1, and 15.6 6 1.1 forvehicle-, AIDA-, LY 367385-, and MPEP-treated ani-mals, respectively. BBB scores reported here are consis-tent with previous reports from this laboratory for these
injury parameters (Hulsebosch et al., 2000, Mills et al.,2001b,c). Medians and percentile ranges (10%, 25%,75%, and 90%) for BBB scores are reported in Figure 2.
Mechanical Allodynia
Following SCI, vehicle-treated animals demon-strated a temporal increase in number of forelimb pawwithdrawals to von Frey filament stimulation, whichwas statistically significant compared to presurgicalvalues by PSD 14 (p , 0.05; Fig. 3). AIDA signifi-cantly attenuated the increase in paw withdrawals to4.79 mN von Frey stimulation through PCD 21 com-pared to the vehicle-treated group (p , 0.05); however,by PCD 28 there was no significant difference betweenvehicle or AIDA-treated groups (Fig. 3A–C) for anystrength of von Frey stimulation. LY 367385 displayeda similar pattern as AIDA by significantly attenuatingthe increase in number of paw withdrawals to allstrengths of von Frey stimulation tested through PCD21 compared to vehicle controls (p , 0.05; Fig. 3D–F).However, by PCD 28 there was no difference betweenLY 367385-treated and vehicle-treated groups. MPEPadministration did not have a significant effect on pawwithdrawals to mechanical stimulation at any strengthtested compared to the vehicle-treated group (Fig.3G–I). Each group responded 100 percent of the timeto pin stimulus, before and at each time point after in-jury (data not shown).
Thermal Hyperalgesia
The vehicle-treated group demonstrated a significanttemporal decrease in withdrawal latencies to a thermalstimulus from 21.3 6 0.5 sec on PCD 7 to 23.3 6 0.6sec, 23.8 6 .7 sec, and 24.9 6 0.9 sec on 14, 21, and28, respectively (p , 0.05 on PSD 14–28; Fig. 4). TheAIDA-treated group did not display a significant differ-ence compared to the vehicle-treated group at any timepoint measured; however, there was a trend for a decreasein withdrawal latencies, which was significantly differ-ent compared to baseline values by PSD 7 (p , 0.05; Fig.4A). Interestingly, LY 367385 significantly potentiated,at all time points, the decrease in withdrawal latenciescompared to the vehicle-treated group (p , 0.05; Fig.4B). Values for the LY 367385-treated group were:23.9 6 0.9 sec, 26.1 6 0.8 sec, 26.6 6 0.7 sec, and27.8 6 0.8 sec on PCD 7, 14, 21, and 28 respectively.MPEP significantly attenuated the decrease in withdrawallatencies until PCD 21 compared to the vehicle-treatedgroup (p , 0.05; Fig. 4C). Values for the MPEP-treatedgroup were: 0.4 6 0.3 sec, 21.4 6 0.5 sec, 22.2 6 0.7sec, and 25.2 6 1.1 sec on PCD 7, 14, 21, and 28, re-spectively.
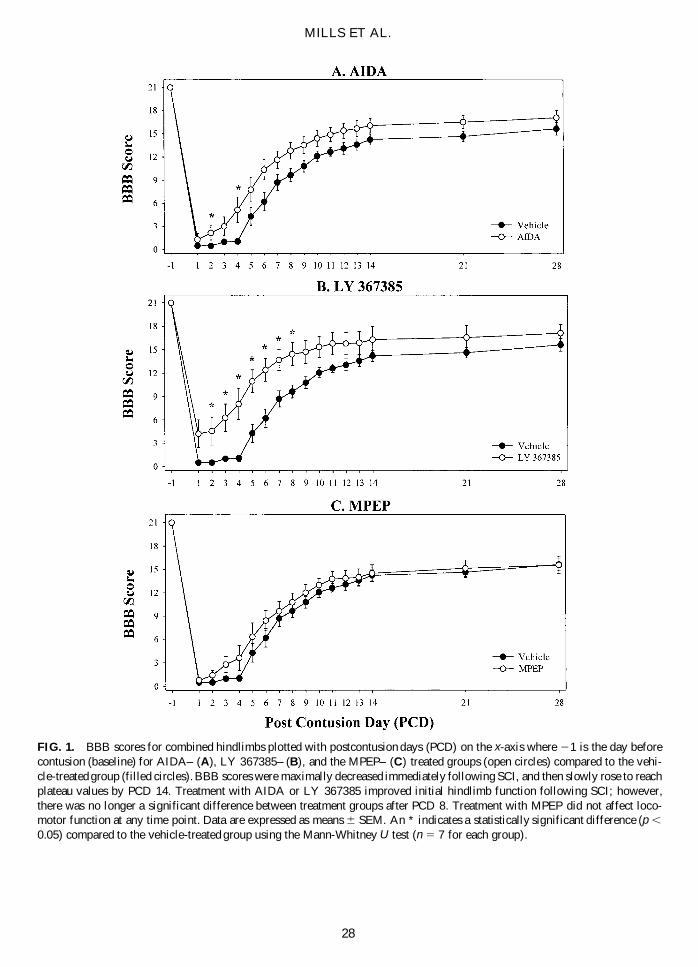
MILLS ET AL.
28
FIG. 1. BBB scores for combined hindlimbs plotted with postcontusion days (PCD) on the x-axis where 21 is the day beforecontusion (baseline) for AIDA– (A), LY 367385– (B), and the MPEP– (C) treated groups (open circles) compared to the vehi-cle-treated group (filled circles). BBB scores were maximally decreased immediately following SCI, and then slowly rose to reachplateau values by PCD 14. Treatment with AIDA or LY 367385 improved initial hindlimb function following SCI; however,there was no longer a significant difference between treatment groups after PCD 8. Treatment with MPEP did not affect loco-motor function at any time point. Data are expressed as means 6 SEM. An * indicates a statistically significant difference (p ,
0.05) compared to the vehicle-treated group using the Mann-Whitney U test (n 5 7 for each group).

GROUP I mGluRs IN SCI
29
FIG. 2. Box plot of BBB scores for combined hindlimbs with postcontusion days (PCD) on the x-axis where 21 is the day be-fore contusion (baseline) for AIDA– (A), LY 367385– (B), and the MPEP– (C) treated groups (shaded boxes) compared to thevehicle-treated group (open boxes). Data are expressed as box plots with median (horizontal bar) and percentile points [25–75%(box) and 10–90% (error bars); see key in A for a description of percentile points]. The box plot gives the distribution of the dataabout the median. All baseline BBB scores were 21 and therefore appear as a single line. An * indicates a statistically signifi-cant difference (p , 0.05) compared to the vehicle-treated group using the Mann-Whitney U test (n 5 7 for each group).

MILLS ET AL.
30
FIG
. 3.
For
elim
b pa
w w
ithd
raw
al r
espo
nses
fol
low
ing
SC
I, e
xpre
ssed
as
perc
ent
chan
ge f
rom
bas
elin
e, t
o vo
n F
rey
fila
men
t (4
.79,
9.9
6, a
nd 2
04.1
mN
) st
imul
atio
nin
the
AID
A–
(A–C
), L
Y 3
6738
5– (
D–F
), a
nd M
PE
P–
(G–I
) tr
eate
d gr
oups
. F
ille
d ci
rcle
s re
pres
ent
the
vehi
cle-
trea
ted
grou
p an
d op
en c
ircl
es i
ndic
ate
AID
A,
LY
3673
85,
or M
PE
P t
reat
men
t gr
oups
. T
he t
empo
ral
incr
ease
in
wit
hdra
wal
res
pons
es s
ugge
sts
the
deve
lopm
ent
of m
echa
nica
l al
lody
nia
afte
r S
CI.
A s
ingl
e tr
eatm
ent
with
AID
A o
r L
Y 3
6738
5 im
med
iate
ly a
fter
SC
I at
tenu
ated
the
dev
elop
men
t of
mec
hani
cal
allo
dyni
a th
roug
h P
CD
21.
Tre
atm
ent
wit
h M
PE
P d
id n
ot a
ffec
t th
e de
-ve
lopm
ent
of m
echa
nica
l al
lody
nia
follo
win
g S
CI.
Dat
a w
ere
anal
yzed
with
rep
eate
d m
easu
res
AN
OV
A f
ollo
wed
by
pair
wis
e co
mpa
riso
ns u
sing
the
Bon
ferr
oni
tte
st.
An
1in
dica
tes
a st
atis
tical
ly s
igni
fica
nt d
iffe
renc
e (p
,0.
05)
com
pare
d to
bas
elin
e va
lues
. An
* in
dica
tes
a st
atis
tical
ly s
igni
fica
nt d
iffe
renc
e (p
,0.
05)
com
pare
d to
the
vehi
cle-
trea
ted
grou
p. D
ata
are
expr
esse
d as
mea
ns6
SE
M (
n5
7 fo
r ea
ch g
roup
).

GROUP I mGluRs IN SCI
31
FIG. 4. Forelimb paw withdrawal latencies to thermal stimulation following SCI, reported as change (in seconds) from base-line, in the AIDA– (A), LY 367385– (B), and MPEP– (C) treated groups. Filled circles represent the vehicle-treated group andopen circles indicate AIDA, LY 367385, or MPEP treatment groups. The temporal decrease in paw withdrawal latencies is con-sistent with the development of thermal hyperalgesia after SCI. Treatment with AIDA did not have a significant effect on with-drawal latencies; however, LY 367385 treatment potentiates the development of thermal hyperalgesia. MPEP treatment resultedin an attenuation of withdrawal latencies through PCD 14. Data were analyzed with repeated measures ANOVA followed bypairwise comparisons using the Bonferroni t test. An 1 indicates a statistically significant difference (p , 0.05) compared tobaseline values. An * indicates a statistically significant difference (p , 0.05) compared to the vehicle-treated group. Data areexpressed as means 6 SEM (n 5 7 for each group).

MILLS ET AL.
32
FIG. 5. Representative photomicrographs of spinal cord cross-sections from each treatment group stained with Luoxol blue andcresyl violet at sections 2.4 mm rostral (left), 2.4 mm caudal (right), and through the epicenter of injury (middle) for the vehi-cle– (row 1), AIDA– (row 2), LY 367385– (row 3), and MPEP– (row 4) treated groups. All sections at the epicenter are reducedin diameter and are characterized by a small rim of white matter sparing. A rostro-caudal tapering of tissue loss is seen in alltreatment groups with the ventral portion of the dorsal funiculus (corticospinal region) and central gray matter being the most af-fected. AIDA and LY 367385 treatments produced an increase in the amount of spared white matter at the epicenter of injury.
Spared Tissue
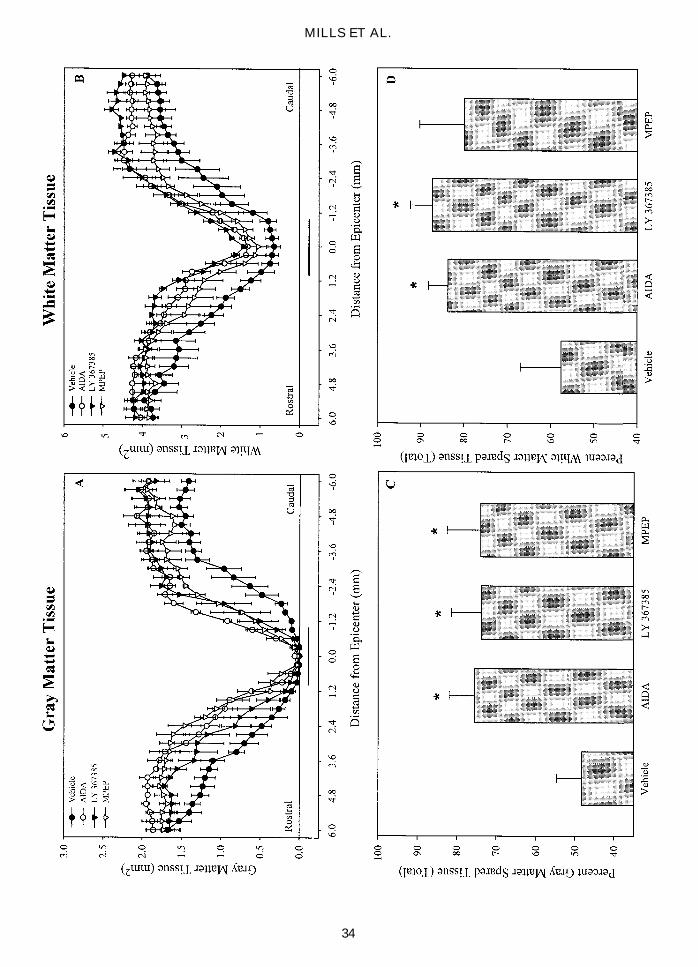
SCI in all treatment groups produced maximum tissueloss at the epicenter of injury (Figs. 5–7). The epicenterof injury was characterized by a total loss of gray mat-ter with a small, incomplete rim of white matter preser-vation, and a decrease in spinal cord diameter comparedto sections rostral and caudal. The loss of both gray andwhite matter was spatially reduced away from the epi-center of injury. Moving away from the epicenter of in-jury, the lesion tapers with the ventral portion of the dor-
sal funiculus (corticospinal region) and central gray mat-ter being the most affected. Treatment with AIDA andLY 367385 led to a significant increase in the totalamount of gray and white matter over the 6.0 mm mea-sured rostral and caudal to the injury (p , 0.05; Fig.6C,D). MPEP treatment had a significant effect on totalgray matter, but was less effective at preserving whitematter (p , 0.05; Fig. 6C,D). Treatment with AIDA ledto significant gray matter tissue sparing at the epicenterof injury (p , 0.05; Fig. 7A); whereas, treatment withboth AIDA and LY 367385 resulted in increased sparing

GROUP I mGluRs IN SCI
33
of white matter at the epicenter of injury (p , 0.05; Fig.7A). AIDA and LY 367385, but not MPEP, led to a sig-nificant increase in overall total tissue sparing (p , 0.05;Fig. 7B).
DISCUSSION
The present study demonstrates that acute inhibition ofdifferent subtypes of group I metabotropic glutamate re-ceptors (mGluR1 and mGluR5) differentially affects (1)locomotor recovery, (2) development of chronic centralpain (CCP)-like behaviors, and (3) amount of spared tis-sue following spinal cord injury. The group I antagonistAIDA attenuated the development of mechanical allody-nia, was neuroprotecitve in gray and white matter, buthad no effect on the development of thermal hyperalge-sia following SCI. A single treatment with the mGluR1specific antagonist LY 367385 improved initial locomo-tor recovery, attenuated the development of mechanicalallodynia, was neuroprotective in gray and white matter,but potentiated the development of thermal hyperalgesiafollowing SCI. Treatment with the mGluR5 specific an-tagonist MPEP had no effect on locomotor recovery orresponses to mechanical stimulation, was neuroprotectivein gray matter, and attenuated the development of ther-mal hyperalgesia after SCI. The different effects ofmGluR1 and mGluR5 antagonists suggest different acutepathophysiological roles for subtypes of group I mGluRsin the spinal cord following injury.
Neuroprotection
The neuroprotective action of group I mGluR antago-nists is well established (Bruno et al., 1999; Buisson andChoi, 1995; Cozzi et al., 1997; Strasser et al., 1998).AIDA has been shown to confer neuroprotection whenadministered up to 60 min following oxygen-glucose de-privation in cortical cultures and in hippocampal neuronsfollowing transient global ischemia (Pellegrini-Giampi-etro et al., 1999). MPEP has been shown to protect CA1neurons following ischemia and reperfusion (Rao et al.,2000) and is neuroprotective in cortical cultures chal-lenged with NMDA or intra-striatal injection of NMDAor quinolinic acid (Bruno et al., 2000). LY 367385 is neu-roprotective against NMDA-induced excitotoxicity invitro and in vivo (Bruno et al., 1999; Kingston et al.,1999). However, the specific role of mGluR1 andmGluR5 in trauma induced excitotoxicity remains un-clear. Neither pharmacological blockade nor disruptionof the mGluR1 gene in mice had an effect on the extentof ischemic brain injury or kainic acid–induced excito-toxicity (Ferraguti et al., 1997). Furthermore, overex-pression of mGluR5 protected against apoptotic death
and treatment with mGluR5 antisense oligonucleotidesaccelerated the development of apoptosis in culturedgranule cells (Copani et al., 1998). Similarly, exposureto ambient glutamate reduces susceptibility of culturedhippocampal cells to injury from excessive NMDA re-ceptor activation and this protection is inhibited bymGluR5 antisense oligonucleotide treatment (Adamchikand Baskys, 2000), suggesting that mGluR5 activationprior to injury may protect neurons from excitotoxicity.Taken together, these studies suggest that activation ofgroup I mGluRs has both neuroprotective and neurotoxiceffects (Nicoletti et al., 1999).
The present study is in agreement with a protectiverole for group I antagonists. AIDA, LY 367385, andMPEP are neuroprotective in the gray matter followingSCI; however, only AIDA and LY 367385 are neuro-protective in the white matter at the epicenter of injuryand increase overall tissue sparing. The protective effectsof inhibiting group I mGluRs in the white matter fol-lowing SCI is in agreement with a previous report demon-strating a preservation of compound action potentials inan in vitro model of white matter SCI (Agrawal et al.,1998). The fact that mGluR5 is localized almost com-pletely in the gray matter (Alvarez et al., 2000; Jia et al.,1999; Mills et al., 2001a) may account for MPEP’s lackof effect on white matter sparing. The protective effectsof inhibition of group I mGluRs following SCI may arisefrom several nonexclusive mechanisms, including af-fecting EAA and GABA release (Jones et al., 1998; Millset al., 2000; Pellegrini-Giampiertro et al., 1999; Thomaset al., 2000), inhibition of apoptotic cascades (Copani etal., 1995; Martin et al., 2000), and interactions withiGluRs (Alagrsamy et al., 1999; Bruno et al., 1995, 2000;Mukhin et al., 1997; Nicoletti et al., 1999).
Contributions to Chronic Central Pain FollowingSpinal Cord Injury
Group I mGluRs may mediate the excitability of dor-sal horn projection neurons in the pain pathway, and thusaffect the development and/or maintenance of CCP by anumber of nonexclusive mechanisms: (1) activation ofphospholipase C (PLC), which stimulates synthesis of in-ositol 1,4,5-triphosphate leading to release of Ca21 frominternal stores, (2) activation of Ca21 channels, (3) inhi-bition of IKM and IKAHP, (4) potentiating iGluR re-sponses, (5) a potentiation of mGluR responses byiGluRs, and (6) activation of extracellular signal–regu-lated kinases (Fagni et al., 2000; Karim et al., 2001; Pinand Dovoisin, 1995). Recently it was shown that a sin-gle treatment with the group I antagonist, AIDA (0.1nmol), reduced glutamate release and attenuated the de-velopment of mechanical allodynia following SCI (Millset al., 2000). Here the group I–specific antagonist AIDA
MILLS ET AL.
34

(200 nmol) and the mGluR1 specific antagonist LY367385, but not the mGluR5-specific antagonist MPEP,attenuated the development of mechanical allodynia fol-lowing SCI, suggesting that mGluR1 is involved in me-diating mechanical responses after SCI. This is consis-tent with the spatial distribution of mGluR1, which isfound mainly in laminae III and IV of the rat thoracicdorsal horn (Alvarez et al., 2000; Mills et al., 2001a),which receives input from laminae I and II and from largemyelinated sensory fibers (such as the Aa and Ab fibers)that carry information from muscle, skeletal, cutaneous,and subcutaneous mechanoreceptors. The expression ofmGluR1 in deeper laminae suggests involvement in theprocessing of mechanical information and integration ofnociceptive information from the upper laminae. Thus,regulation of mechanical responses, but not thermal re-sponses, by mGluR1 is consistent with the localizationof mGluR1 to the deeper laminae of the dorsal horn. An-tagonism of mGluR1 immediately after injury may blockthe development of mechanical allodynia by inhibitingsynaptic glutamate release that contributes to excitotox-icity and/or by delaying activation of mGluR1-mediatedintracellular pathways that lead to hyperexcitability.
In the present study, treatment with a single dose ofthe mGluR5 specific antagonist MPEP has no effect onthe development of mechanical allodynia but attenuatesthe development of thermal hyperalgesia through PCD21. Previous reports show that MPEP is ineffective in thetreatment of mechanical hyperalgesia associated with sci-atic nerve ligation and has no effect on locomotion(Walker et al., 2001). Furthermore, the selective mGluR5antagonist SIB-1757 attenuates thermal hyperalgesia, butnot mechanical allodynia following spinal nerve ligation(Dogrul et al., 2000). These results suggest that mGluR5mediates thermal responses, without affecting mechani-cal responses or locomotor ability, which agrees with theresults reported here. Modulation of thermal responsesby mGluR5 is consistent with the circuitry and distribu-tion of mGluR5 in the spinal cord. Laminae I and II ofthe dorsal horn receive the central projections from pri-
mary afferent fibers carrying nociceptive (including ther-mal) information, and highest expression levels ofmGluR5 are in lamina II of the dorsal horn (Alveraz etal., 2000; Jia et al., 1999; Mills et al., 2001a); however,tactile information is mediated through Ab fibers, whichproject to deeper laminae, and are not thought to expressmGluR5 (Dogrul, et al., 2000; Jia et al., 1999; Valero etal., 1997). The differential localization of mGluR5 is con-sistent with the effects of MPEP on thermal but not me-chanical responses.
Although there were significant acute behavioralchanges following treatment with group I mGluR an-tagonists, a surprising finding was the lack of signifi-cant differences between all groups in any behavioralmeasure by PCD 28, despite the significant differencein the amount of tissue sparing. There are three possi-ble explanations for this observation: (1) Since the epi-center is in a thoracic segment, preservation of graymatter after injury would be reflected in local circuitsinnervating trunk musculature. Thus, the use of behav-ioral measures that require trunk musculature such asspontaneous measures of activity (e.g., rearing; Millset al., 2001c), or mapping the girdle zone of hyper-pathia at the level of injury (Hulsebosch et al., 2000),may provide better behavioral discrimination betweengroups with significant gray matter tissue sparing. (2)The overall preservation of white matter by LY 367385,and to a lesser extent AIDA, was evident in the initialphase of locomotor recovery using the BBB scale. Per-haps a more sensitive locomotor rating scale, such asthe BBB subscale (Lankhorst et al., 1999), or a com-bination of several neurological assessments, such asthe combined behavioral score (Noble and Wrathall,1985, 1989; Panjabi and Wrathall, 1988) would haveshown a more profound difference in recovery betweenthe treatment groups. (3) The development of CCP af-ter SCI may be dependent on the increase in expres-sion of mGluR1 (Mills et al. 2001a). The single doseof an mGluR1 antagonist (AIDA or LY 367385) mayhave delayed the injury-induced increase in mGluR1
GROUP I mGluRs IN SCI
35
FIG. 6. Quantification of spared tissue for gray and white matter 28 days following contusion SCI. Area measurements of gray(A) and white (B) matter as a function of distance from the epicenter of injury. Spinal cords were sectioned at 15 mm and every20th section (0.3 mm between sections) was measured for area in the vehicle– (filled circles), AIDA– (open circles), LY 367385–(filled triangles), and MPEP– (open triangles) treated groups. The horizontal bar above the x-axis in A and B indicate the widthof the impactor (2 mm). The total volume of gray (C) and white (D) matter was calculated over the entire region shown in Aand B (6 mm rostral to 6 mm caudal) using the Cavalieri method and reported as a percentage of calculated preinjury volume.SCI produced maximum tissue loss at the epicenter of impact. Treatment with AIDA and LY 367385 resulted in significant spar-ing of both gray and white matter. However, treatment with MPEP resulted in an increase in total gray matter sparing without asignificant effect on white matter sparing. Data were analyzed with one way ANOVA followed by pairwise comparisons usingTukey’s HSD test. An * indicates a statistically significant difference (p , 0.05) compared to the vehicle-treated group. Data areexpressed as means 6 SEM.

expression. However, after this initial delay, mechani-cal allodynia would develop as expression of mGluR1increased to normal pathophysiological levels. Thus, byPCD 28 there was no difference in responses to me-chanical stimulation between the treatment groups.
mGluR5 may contribute to the acute release of gluta-mate resulting in excitotoxicity; however, there is notemporal change in expression of mGluR5 after SCI,suggesting a more limited role in the development ofCCP (Mills et al., 2001a).
MILLS ET AL.
36
FIG. 7. Quantification of tissue sparing at the epicenter of injury (area under the impactor) and the overall amount of tissuespared (gray 1 white matter) reported as a percent of pre-injury volume. Treatment with AIDA resulted in a significant (p ,
0.05) amount of gray matter sparing at the epicenter of injury (A). Treatment with MPEP resulted in increased gray matter at theepicenter of injury, but was not statistically significant compared to vehicle-treated. Only AIDA and LY 367385 treatments pro-duced an increase in white matter sparing at the epicenter of injury (A) and in the total amount of spared tissue (B). Data wereanalyzed with one way ANOVA followed by pairwise comparisons using Tukey’s HSD test. An * indicates a statistically sig-nificant difference (p , 0.05) compared to the vehicle-treated group. Data are expressed as means 6 SEM.

Paradoxical Roles
Acute administration of a specific mGluR5 antagonistafter SCI protects against the development of thermal hy-peralgesia but not mechanical allodynia. While acute ad-ministration of a mGluR1 antagonist attenuates mechan-ical allodynia, but potentiates thermal hperalgesia. Bothreceptors belong to group I mGluRs, which are coupledto the intracellular pathway of PLC activation. It is pos-sible that individual subtypes within group I activate dif-ferent intracellular pathways resulting in different be-havioral outcomes following SCI. The hypotheses thatseparate pathways mediate mechanical and thermal re-sponses is consistent with other models of central pain(e.g. capsaicin-induced central sensitization; Sluka andWillis, 1997; Sluka et al., 1997). One possible explana-tion is that different group I mGluRs activate differentisoforms of PKC (Polgar et al., 1999). For example, neu-rons that contain PKCg are found mainly in lamina IIi,which contains no mGluR1 positive neurons; however,lamina IIi contains a large number of mGluR5 positiveneurons. Thus, in the spinal cord it is likely that mGluR5,but not mGluR1, activates PKCg. Additionally, mGluR1may be less efficiently coupled to polyphosphoinisitidehydrolysis (PI; Casabona et al., 1997).
The contrasting effects of mGluR1 and mGluR5 an-tagonists may be dependent on the functional status ofindividual receptor subtypes. For example, activation ofgroup I mGluRs has been shown to increase or inhibitneurotransmitter release depending on neuronal activity(Herrero et al., 1998; Rodriguez-Moreno et al., 1998; Sis-tiaga and Sanchez-Prieto, 2000). A functional switch, me-diated by receptor phosphorylation, has been proposed toaccount for the transition from facilitation to inhibition(Herrero et al., 1998). The naïve, unphosphorylated re-ceptor is coupled to PI hydrolysis and facilitates neuro-transmitter release and upon activation becomes phos-phorylated and couples to a different pathway thatinhibits Ca21 entry and glutamate release (Herrero et al.,1998; Nicoletti et al., 1999). mGluR5 is known to un-dergo PKC-mediated receptor phosphorylation, which isreversed by NMDA-mediated activation of a proteinphosphatase (Alagarsamy et al, 1999). If mGluR5 un-dergoes the switch from facilitation to inhibition, butmGluR1 remains in a facilitory state, it is possible thatpotentiation of glutamate release in the dorsal horn is me-diated by mGluR1, rather than mGluR5. Thus, mGluR1antagonists may have a greater effect than mGluR5 an-tagonists on outcome measures dependent on excitotox-icity. Furthermore, it is possible that the switch from ei-ther facilitation or inhibition of neurotransmitter releasefollowing SCI is not only subtype specific, but also fiber-type dependent (e.g., Ab-, Aa-, Ad-, or C-fiber), which
could lead to different thermal and mechanical responses.While it is known that different in vitro and in vivo mod-els show opposing roles for group I mGluRs (Nicolettiet al., 1999), the specific mechanisms mediating these ef-fects are unknown and demand further study.
However, the different behavioral effects might sim-ply be a function of the amount and specificity of sparedtissue. Since, mGluR1 and mGluR5 are differentially lo-cated in the dorsal horn, it is possible that antagonists toeach specific subtype are protective for specific areas ofthe dorsal horn. However, we found no evidence for thisusing the methodology of the current study. Additionally,the differences in tissue sparing between the treatmentgroups may affect the expression levels of both mGluRsand iGluRs, which could differentially affect recoveryand the development of CCP following SCI. Future ex-periments will help to determine if treatments with groupI mGluR antagonists following SCI affect specific celltype survival, alter mGluR and iGluR expression levels,change receptor activation states, or affect different in-tracellular pathways.
CONCLUSION
This study confirms previous work demonstrating thatantagonism of group I mGluRs modulates behavioral out-comes following SCI (Mills et al., 2000). Furthermore,specific antagonists of group I mGluR subtypes differ-entially affect locomotor recovery, development of me-chanical allodynia and thermal hyperalgesia, and amountof spared tissue following SCI. The mGluR1 specific an-tagonist LY 367385 improves initial locomotor recovery,attenuates the development of mechanical allodynia, andis neuroprotective in gray and white matter. However, in-hibition of mGluR1 following SCI potentiates the devel-opment of thermal hyperalgesia. Treatment with themGluR5 specific antagonist MPEP has no effect on lo-comotor recovery or responses to mechanical stimulation,but is neuroprotective in gray matter and attenuates thedevelopment of thermal hyperalgesia. The different ef-fects of mGluR1 and mGluR5 antagonists on outcomemeasures following SCI suggests different pathophysio-logical roles for subtypes of group I mGluRs in the spinalcord following injury.
ACKNOWLEDGEMENTS
We would like to thank Debbie Pavlu for her secre-tarial assistance. This work was supported by the KentWaldrep, RGK and Spinal Cord Research Foundations,Mission Connect of TIRR, and NIH grants NS 11255 andNS 39161.
GROUP I mGluRs IN SCI
37

REFERENCES
ADAMCHIK, Y., and BASKYS, A. (2000). Glutamate-medi-ated neuroprotection against N-methyl-D-aspartate toxicity:a role for metabotropic glutamate receptors. Neuroscience 99,731–736.
AGRAWAL, S.K., THERIAULT, E., and FEHLINGS, M.G.(1998). Role of group I metabotropic glutamate receptors intraumatic spinal cord white matter injury. J Neurotrauma 15,929–941.
ALGARSAMY, S., MARINO, M.J., ROUSE, S.T., et al.(1999). Activation of NMDA receptors reverses desensitiza-tion of mGluR5 in native and recombinant systems. Nat. Neu-rosci. 2, 234–240.
ALVAREZ, F.J., VILLALBA, R.M., CARR, P.A., et al. (2000).Differential distribution of metabotropic glutamate receptors1a, 1b, and 5 in the rat spinal cord. J. Comp. Neurol. 422,464–487.
ANIKSZTEJIN, L., OTANI, S., and BEN-ARI, Y. (1992).Quisqualate metabotropic receptors modulate NMDA cur-rents and facilitate induction of long-term potentiationthrough protein kinase C. Eur. J. Neurosci. 4, 500–505.
BALAZY, T.E. (1992). Clinical management of chronic painin spinal cord injury. Clin. J. Pain 8, 102–110.
BASBAUM, A.I., and WALL, P.D. (1976). Chronic changesin the response of cells in adult cat dorsal horn following par-tial deafferentation: the appearance of responding cells in apreviously non-responding region. Brain Res. 116, 181–204.
BASSO, M., BEATTIE, M.S., and BRESNAHAN, J.C. (1995).A sensitive and reliable locomotor rating scale for open fieldtesting in rats. J. Neurotrauma 12, 1–21.
BENNETT, G.J., and XIE, Y.K. (1988). A peripheral mononeu-ropathy in rat that produces disorders of pain sensation likethose seen in man. Pain 33, 87–107.
BENNETT, A.D., EVERAHART, A.W., and HULSEBOSCH,C.E. (2000). Intrathecal NMDA and non NMDA receptor an-tagonists reduce mechanical but not thermal allodynia in arodent model of chronic central pain after spinal cord injury.Brain Res. 859, 72–82.
BERIC, A., DIMITRJEVIC, M.R., and LINDBLOOM, U.(1988). Central dysesthesias syndrome in spinal cord injurypatients. Pain 34, 39–48.
BLEAKMAN, D., RUSIN, K. I., CHARD, P. S., et al. (1992).Metabotropic glutamate receptors potentiate ionotropic glu-tamate responses in the rat dorsal horn. Molec. Pharmac. 42,192–196.
BOIVIE, J. (1984). Disturbances in cutaneous sensibility in pa-tients with central pain caused by the spinal cord lesions ofsyringomyelia. Pain Suppl. 2, s82.
BRUNO, V., COPANI, A., KNOPFEL, T., et al. (1995). Acti-vation of metabotropic glutamate receptors coupled to
inositol phospholipid hydrolysis amplifies NMDA-inducedneuronal degeneration in cultured cortical cells. Neurophar-macology 34, 1089–1098.
BRUNO, V., BATTAGLIA, G., COPANI, A., et al. (1998).Metabotropic glutamate receptors and neurodegeneration.Prog. Brain Res. 116, 209–221.
BRUNO, V., BATTAGLIA, G., KINGSTON, A., et al. (1999).Neuroprotective activity of the potent and selective mGlu1ametabotropic glutamate receptor antagonist, (1)-2–methyl-4carboxyphenyglycine (LY367385): comparison withLY357366, a broader spectrum antagonist with equal affin-ity for mGlu1a and mGlu5 receptors. Neurophamacology 38,199–207.
BRUNO, V., KSIAZEK, I., BATTAGLIA, G., et al. (2000).Selective blockade of metabotropic glutamate receptor sub-type 5 is neuroprotective. Neuropharmacology 39,2223–2230.
BUISSON, A., and CHOI, D.W. (1995). The inhibitory mGluRagonist, S-4–carboxy-3–hydroxy-phenylglycine selectivelyattenuates NMDA neurotoxicity and oxygen-glucose depri-vation-induced neuronal death. Neuropharmacology 34,1081–1087.
CAIRNS, D.M., ADKINS, R.H., and SCOTT, M.D. (1996).Pain and depression in acute traumatic spinal cord injury: ori-gins of chronic problematic pain? Arch. Phys. Med. Reha-bil. 77, 329–335.
CASABONA, G., KNOPFEL, T., KUHN, R., et al. (1997). Ex-pression and coupling to polyphosphoinositide hydrolysis ofgroup-1 metabotropic glutamate receptors in early postnataland adult rat brain. J. Neurosci. 9, 12–17.
CHAPMAN, A.G., YIP, R.K., YAP, J.S., et al. (1999). Anti-convulsant actions of LY 367685 ([1]-2-methyl-4-car-boxyphenylglycine) and AIDA ([RS]-1-aminoindan-1,5–di-carboxylic acid). Eur. J. Pharm. 368, 17–24.
CHAPMAN, A.G., NANAN, K., WILLIAMS, M., et al. (2000).Anticonvulsant activity of two metabotropic glutamateGroup I antagonists selective for the mGlu5 receptor:2–methyl-6–(phenylethynyl)-pyridine (MPEP), and (E)-6–methyl-2–styryl-pyridine (SIB 1893). Neuropharmacology39, 1567–1574.
CHRISTENSEN, M.D., and HULSEBOSCH, C.E. (1997a).Chronic central pain after spinal cord injury. J. Neurotrauma14, 517–537.
CHRISTENSEN, M.D., and HULSEBOSCH, C.E. (1997b).Spinal cord injury and anti-NGF treatment results in changesin CGRP density and distribution in the dorsal horn in therat. Exp. Neurol. 147, 463–475.
CONN, P.J., and PIN, J.-P. (1997). Pharmacology and func-tions of metabotropic glutamate receptors. Annu. Rev. Phar-macol. Toxicol. 37, 205–237.
CONSTANTINI, S., and YOUNG W. (1994) The effects ofmethylprednisolone and the ganglioside GM1 on acute spinalcord injury in rats. J. Neurosurg. 80, 97–111.
MILLS ET AL.
38

COPANI, A., BRUNO, V., BATTAGLIA, G., et al. (1995). Ac-tivation of metabotropic glutamate receptors prevents cul-tured neurons against apoptosis induced by beta-amyloidpeptide. Mol. Pharmacol. 47, 890–897.
COPANI, A., CASABONA, G., BRUNO, V., et al. (1998). Themetabotropic glutamate receptor mGlu5 controls the onset ofdevelopmental apoptosis in cultured cerebellar neurons. Eur.J. Neurosci. 10, 2173–2184.
COURTNEY, M.J., LAMBERT, J.J., and NICHOLLS, D.G.(1990). The interaction between plasma membrane depolar-ization and glutamate receptor activation in the regulation ofcytoplasmic free calcium in cultured cerebellar granule cells.J. Neurosci. 10, 3873–3879.
COZZI, A., LOMBARDI, G., LEONARDI, P., et al. (1997).AIDA, a group I metabotropic glutamate receptor antagonist,reduces the ischemia-induced neuronal loss. Soc. Neurosci.Abs. 23, 788.
DAVIDOFF, G., and ROTH, E.J. (1991). Clinical characteris-tics of central (Dysesthetic) pain in spinal cord injury pa-tients, in: Pain and Central Nervous System Disease: TheCentral Pain Syndromes. K.L. Casey (ed.), Raven Press: NewYork, pp. 77–83.
DEVOR, M., and WALL, P.D. (1981). Plasticity in the spinalcord sensory map following peripheral nerve injury in rats.J. Neuorsci. 1, 679–984.
DIRIG, D.M., SALAMI, A., RATHBUN, M.L., et al. (1997).Characterization of variables defining hindpaw withdrawallatency evoked by radiant thermal stimuli. J. Neurosci. Meth-ods 76, 183–191.
DOGRUL, A., OSSIPOV, M.H., LAI, J., et al. (2000). Periph-eral and spinal antihyperalgesic activity of SIB-1757, ametabotropic glutamate receptor (mGluR5) antagonist, in ex-perimental neuropathic pain in rats. Neurosci. Lett. 292,115–118.
FAGNI, L., CHAVIS, P., ANGO, F., et al. (2000). Complexinteractions between mGluRs, intracellular Ca21 stores andion channels in neurons. Trends Neurosci. 23, 80–88.
FERRAGUTI, F., PIETRA, C., VALERIO, C., et al. (1997).Evidence against a permissive role of metabotropic glutamatereceptor 1 in acute excitotoxicity. Neuroscience 79, 1–5.
FISHER, K., and CODERRE, T.J. (1996a). The contribution ofmetabotropic glutamate receptors to formalin-induced noci-ception. Pain 68, 255–263.
FISHER, K., and CODERRE, T.J. (1996b). Comparison of no-ciceptive effects produced by intrathecal administration ofmGluR agonists. NeuroReport 7, 2743–2747.
FITZJOHN, S.M., IRVING, A.J., PALMER, M.J., et al. (1996).Activation of group I mGluRs potentiates NMDA responsesin rat hippocampal slices. Neurosci. Lett. 203, 211–213.
FUNDYTUS, M.E., FISHER, K., DRAY, A., et al. (1998). In
vivo antinociceptive activity of anti-rat mGluR1 and mGluR5antibodies in rats. NeuroReport 9, 731–735.
GLAUM, S.R., and MILLER, R.J. (1993). Activation ofmetabotropic glutamate receptors produces reciprocal regu-lation of ionotropic glutamate and GABA responses in thenucleus of the tractus solitarius of the rat. J. Neurosci. 13,1636–1641.
GRUNER, J.A. (1992). A monitored contusion model of spinalcord injury in the rat. J. Neurotrauma 9, 123–128.
GUNDERSEN, H.J., BENDTSEN, T.F., KORBO, L., et al.(1988). Some new, simple and efficient stereological meth-ods and their use in pathological research and diagnosis. AP-MIS 96, 379–394.
HARGREAVES, K., DUBNER, R., BROWN, F., et al. (1988).A new and sensitive method for measuring thermal nocicep-tion in cutaneous hyperalgesia. Pain 32, 77–88.
HARVEY, J.C., and COLLINGRIDGE, G.L. (1993) Signaltransduction pathways involved in the acute potentiation ofNMDA responses by 1S,3R-ACPD in rat hippocampal slices.Br. J. Pharmacol. 109, 1085–1090.
HERRERO, I., MIRAS-PORTUGAL, M.T., and SANCHEZ-PRIETO, J. (1992). Positive feedback of glutamate exocyto-sis by metabotropic presynaptic receptor stimulation. Nature360, 163–166.
HERRERO, I., MIRAS-PORTUGAL, M.T., and SANCHEZ-PRIETO, J. (1998). Functional switch from facilitation to in-hibition in the control of glutamate release by metabotropicglutamate receptors. J. Biol. Chem. 273, 1951–1958.
HUANG, P.P., and YOUNG, W. (1994). The effects of arter-ial blood gas values on lesion volumes in a graded rat spinalcord contusion model. J. Neurotrauma 11, 547–562.
HULSEBOSCH, C.E., and COGGESHALL, R.E. (1981a).Quantitation of sprouting of dorsal root axons. Science 213,1020–1021.
HULSEBOSCH, C.E., and COGGESHALL, R.E. (1981b).Sprouting of dorsal root axons. Brain Res. 224, 170–174.
HULSEBOSCH, C.E., XU, G.-Y., PEREZ-POLO, J.R., et al.(2000). Rodent model of chronic central pain after spinal cordcontusion injury. J. Neurotrauma 17, 1205–1217.
JIA, H., RUSTIONI, A., and VALTSCHANOFF, J.G. (1999).Metabotropic glutamate receptors in superficial laminae ofthe rat dorsal horn. J. Comp. Neurol. 410, 627–642.
JONES, N.M., MONN, J.A., and BEART, P.M. (1998). TypeI and II metabotropic glutamate receptors regulate the out-flow of [3H]D-aspartate and [14C]g-aminobutyric acid in ratsolitary nucleus. Eur. J. Pharm. 353, 43–51.
KARIM, F., WANG, C.-C., and GEREAU, R.W. (2001).Metabotropic glutamate receptor subtypes 1 and 5 are acti-vators of extracellular signal-regulated kinase signaling re-quired for inflammatory pain in mice. J Neurosci. 21,3771–3779.
GROUP I mGluRs IN SCI
39

KELSO, S. R., NELSON, T. E., and LEONARD, J. P. (1992).Protein kinase C–mediated enhancement of NMDA currentsby metabotropic glutamate receptors in Xenopus oocytes. J.Physiol. Lond. 449, 705–718.
KINGSTON, A.E., O’NEILL, M.J., BOND, A., et al. (1999).Neuroprotective actions of novel and potent ligands of groupI and group II metabotropic glutamate receptors. Ann. N.Y.Acad. Sci. 890, 438–449.
KRENZ, N.R., and WEAVER, L.C. (1998). Sprouting of pri-mary afferent fibers after spinal cord transection in the rat.Neuroscience 85, 443–458.
KRENZ, N.R., MEAKIN, S.O., KRASSIOUKOUV, A.V., etal. (1999). Neutralizing intraspinal nerve growth factorblocks autonomic dysreflexia caused by spinal cord injury.J. Neurosci. 19, 7405–7414.
LANKHORST, A.J., VERZIJL, M.R., and HAMERS, F.P.T.(1999). Experimental spinal cord contusion injury: compar-ison of different outcome parameters. Neurosci. Res. Com-mun. 24, 135–148.
LENZ, F.A., KWAN, H.C., MARTIN, R., et al. (1994). Char-acteristics of somatotopic organization and spontaneous neu-ronal activity in the region of the thalamic principal sensorynucleus in patients with spinal cord transection. J. Neuro-physiol. 72, 1570–1587.
LIU, D., THANGNIPON W., and MCADOO, D.J. (1991). Ex-citatory amino acids rise to toxic levels upon impact injuryto the rat spinal cord. Brain Res. 547, 344–348.
LLANO, I., DREESEN, J., KANO, M., et al. (1991). In-tradendritic release of calcium induced by glutamate in cere-bellar Purkinje cells. Neuron 7, 577–583.
LUNDQVIST, C., SIOSTEEN, A., BLOMSTRAND, C., et al.(1991). Spinal cord injuries-clinical, functional, and emo-tional status. Spine 16, 78–83.
MANZONI, O. J., POULAT, F., DO, E., et al. (1991). Phar-macological characterization of the quisqualate receptor cou-pled to phospholipase C (Qp) in striatal neurons. Eur. J. Phar-mac. Molec. Pharmac. Sec. 207, 231–241.
MARTIN, L.J., SIEBER, F.E., and TRAYSTMAN, R.J. (2000).Apotosis and necrosis occur in separate neuronal populationsin hippocampus and cerebellum after ischemia and are asso-ciated with differential alterations in metabotropic glutamatereceptor signaling pathways. J. Cereb. Blood Flow Metab.20, 153–167.
MCADOO, D. J., XU, G.-Y., ROBACK, G., et al. (1999).Changes in amino acid concentrations over time and spacearound an impact injury and their diffusion through the ratspinal cord. Exp. Neurol. 159, 538–544.
MCADOO, D.J., XU, G.-Y., ROBAK, G., et al. (2000). Evi-dence that reversed glutamate uptake contributes signifi-cantly to glutamate release following experimental injury tothe rat spinal cord. Brain Res. 865, 283–285.
MCNEILL, D.L., CARLTON, S.M., COGGESHALL, R.E., et
al. (1990). Denervation induced intraspinal synaptogenesisof calcitonin gene-related peptide containing primary affer-ent terminals. J. Comp. Neurol. 296, 263–268.
MCNEILL, D.L., CARLTON, D.M., and HULSEBOSCH, C.E.(1991). Intraspinal sprouting of calcitonin gene-related pep-tide containing primary afferents after deafferentation in therat. Exp. Neurol. 114, 321–329.
MERSKEY, H., and BOGDUK, N. (1994). Classification ofChronic Pain. IASP Press: Seattle.
MICHEL, R.P., and CRUZ-ORIVE, L.M. (1988). Applicationof the Cavalieri principle and vertical sections method tolung: estimation of volume and pleural surface area. J. Mi-crosc. 150, 117–136.
MILLS, C.D., XU, G.-Y., JOHNSON, K.M., et al. (2000).AIDA reduces glutamate release and attenuates mechanicalallodynia after spinal cord injury. NeuroReport 11,3067–3070.
MILLS, C.D., FULLWOOD, S.D., and HULSEBOSCH, C.E.(2001a). Changes in metabotropic glutamate receptor ex-pression following spinal cord injury. Exp. Neurol. 170,244–257.
MILLS, C.D., HAINS, B.C., JOHNSON, K.M., et al. (2001b).Strain and model differences in behavioral outcomes fol-lowing spinal cord injury in rat. J Neurotrauma 18, 743–756.
MILLS, C.D., GRADY, J.G., and HULSEBOSCH, C.E.(2001c). Changes in exploratory behavior as a measure ofchronic central pain following spinal cord injury. J Neuro-trauma 18, 1091–1105.
MORONI, F., LOMBARDI, G., THOMSEN, C., et al. (1997).Pharmacolgical characterization of 1–aminoindan-1,5–dicar-boxylic acid, a potent mGluR1 antagonist. J. Pharm. Exp.Ther. 281, 721–729.
MUKHIN, A.G., IVANOVA, S.A., and FADEN, A.I. (1997).mGluR modulation of post-traumatic neuronal death: role ofNMDA receptors. NeuroReport 8, 2561–2566.
MURPHY, S., and MILLER, R.J. (1988). A glutamate recep-tor regulates Ca21 mobilization in hippocampal neurosn.Proc. Natl. Acad. Sci. USA 85, 8737–8741.
MURPHY, S., and MILLER, R.J. (1989). Two quisqualate re-ceptors regulate Ca21 homeostasis in hippocampal neurons.Molec. Pharmac. 35, 671–680.
NEUGEBAUER, V., LUCKE, T., and SCHAIBLE, H.-G.(1994). Requirement of metabotropic glutamate receports forthe generation of inflamamation-evoked hyperexcitability inrat spinal cord neurons. Eur. J. Neurosci. 6, 1179–1186.
NEUGEBAUER, V., CHEN, P.-S., and WILLIS, W.D. (1999).Role of metabotropic glutamate receptor subtype mGluR1 inbrief nociception and central sensitization of primate STTcells. J. Neurophysiol. 82, 272–282.
NICOLETTI, F., BRUNO, V., CATANIA, M.V., et al. (1999).Group-I metabotropic glutamate receptors: hypotheses to ex-
MILLS ET AL.
40

plain their dual role in neurotoxicity and neuroprotection.Neuropharmacology 38, 1477–1484.
NOBLE, L.J., and WRATHALL, J.R. (1985). Spinal cord con-tusion in the rat: morphometric analyses of alterations in thespinal cord. Exp. Neurol. 88, 135–140.
NOBLE, L.J., and WRATHALL, J.R. (1989). Correlativeanalysis of lesion development and functional status aftergraded spinal cord contusive injuries in the rat. Exp. Neurol.103, 34–40.
OLBY, N.J., and BLAKEMORE, W.F. (1996). A new methodof quantifying the extent of tissue loss following spinal cordinjury in the rat. Exp. Neurol. 138, 82–92.
PANJABI, M., and WRATHALL, J.R. (1988). Biomechanicalanalysis of spinal cord injury and functional loss. Spine 13,1365–1370.
PANTER S.C., YUM, S. W., and FADEN, A. I. (1990). Al-teration in extracellular amino acids after traumatic spinalcord injury. Ann. Neurol. 27, 96–99.
PELLEGRINI-GIAMPIETRO, D.E., PERUGINELLI, F.,MELI, E., et al. (1999). Protection with metabotropic gluta-mate 1 receptor antagonists in models of ischemic neuronaldeath: time-course and mechanisms. Neuropharmacology 38,1607–1619.
PIN, J.-P., and DUVOISIN, R. (1995). Review: Neurotrans-mitter receptors I—the metabotropic glutamate receptors:structure and functions. Neuropharmacology 34, 1–26.
POLGAR, E., FOWLER, J.H., MCGILL, M.M., et al. (1999).The types of neuron which contain protein kinase C gammain rat spinal cord. Brain Res. 833, 71–80.
RABCHEVSKY, A.G., FUGACCIA, I., SULLIVAN, P.G., etal. (2001). Cyclosporin A treatment following spinal cord in-jury to the rat: behavioral effects and stereological assess-ment of tissue sparing. J. Neurotrauma 18, 513–522.
RAINNIE D.G., HOLMES, K.H., and SHINNICK-GALLAGHER, P. (1994). Activation of postsynapticmetabotropic glutamate receptors by trans-ACPD hyperpo-larizes neurons of the basolateral amygdala. J. Neurosci. 14,7208–7220.
RAO, A.M., HATCHER, J.F., and DEMPSEY, R.J. (2000).Neuroprotection by group I metabotropic glutamate receptorantagonists in forebrain ischemia of gerbil. Neurosci. Lett.293, 1–4.
RICHARDS, J.S., MEREDITH, R.L., NEPOMUCENO, C., etal. (1980). Psycho-social aspects of chronic pain in spinalcord injury. Pain 8, 355–366.
RINALDI, P.C., YOUNG, R.F., ALBE-FESARD, D., et al.(1991). Spontaneous hyperactivity in the medial and in-tralaminar thalamic nuclei of patients with deafferentationpain. J. Neurosurg. 74, 415–421.
RINTALA, D.H., LOUBSER, P.G., CASTRO, J., et al. (1998).Chronic pain in a community-based sample of men with
spinal cord injury: prevalence, severity, and relationship withimpairment, disability, handicap and subjective well-being.Arch Phys. Med. Rehabil. 79, 604–614.
RODRIQUEZ-MORENO, A., SISTIAGA, A., LERMA, J., etal. (1998). Switch from facilitation to inhibition of excitatorysynaptic transmission by group I mGluR desensitization.Neuron 21, 1477–1486.
SEGATORE, M. (1994). Understanding chronic pain afterspinal cord injury. J. Neurosci. Nurs. 26, 230–236.
SISTIAGA, A., and SANCEHZ-PRIETO, J. (2000). Proteinphosphatase 1 and 2A inhibitors prolong the switch in thecontrol of glutamate release by group I metabotropic gluta-mate receptors: characterization of the inhibitory pathway. J.Neurochem. 75, 1566–1574.
SLUKA, K.A., and WILLIS, W.D. (1997). The effects of G-protein kinase inhibitors on the behavioral responses of ratsto intradermal injection of capsaicin. Pain 71, 165–178.
SLUKA, K.A., REES, H., CHEN, P.-S., et al. (1997). Cap-saicin-induced sensitization of primate spinothalamic tractcells is prevented by a protein kinase C inhibitor. Brain Res.772, 82–86.
STRASSER, U., LOBNER, D., BEHRENS, M.M., et al. (1998).Antagonists for group-I mGluRs attenuate excitotoxic neu-ronal death in cortical cultures. J. Neurochem. 10,2848–2855.
SWEET, W.H. (1991). Deafferentation syndromes in humans:a general discussion, in: Deafferentation Pain Syndromes:Pathophysiology and Treatment. B.S. Nashold, Jr. and J.Ovelmen-Levitt (eds), Raven Press: New York, pps.259–283.
TASKER, R.R., and DOSTROVSKY, J.O. (1989). Deaf-ferentation and central pain, in: Textbook of Pain, 2nd ed.P.D. Wall and R. Melzack (eds), Churchill Livingstone: NewYork, pps 154–180.
THOMAS, L.S., JANE, D.E., HARRIS, J.R., et al. (2000).Metabotropic glutamate autoreceptors of the mGlu5 subtypepositively modulate neuronal glutamate release in the ratforebrain in vitro. Neuropharmacology 39, 1554–1566.
VALERO, A., RIZZONELLI, P., MORETTO, M., et al. (1997).mGluR5 metabotropic glutamate receptor distribution in ratand human spinal cord: a developmental study. Neurosci.Res. 28, 49–57.
VERA-PORTOCARRERO, L., YE, Z., XU, G.-Y., et al.(1999). Spinal contusion induces changes in immunodensityof neuronal and glial glutamate transporters. J. Neurotrauma16, 968.
WALKER, K., BOWES, M., PANESAR, M., et al. (2001).Metabotropic glutamate receptor subtype 5 (mGlu5) and no-ciceptive function I. Selective blockade of mGlu5 receptorsin models of acute, persistent and chronic pain. Neurophar-macology 40, 1–9.
WOOLF, C.J., and THOMPSON, S.W.N. (1991). The induc-
GROUP I mGluRs IN SCI
41

tion and maintenance of central sensitization is dependent onN-methyl-D-aspartic acid receptor activation; implications forthe treatment of post-injury pain hypersensitivity states. Pain44, 293–299.
YOUNG, M.R., FLEETWOOD-WALKER, S.M., MITCHELL,R., et al. (1994). Evidence for a role of metabotropic gluta-mate receptors in sustained nociceptive inputs to rat dorsalhorn neurons. Neuropharmacology 33, 141–144.
YOUNG, M. R., FLEETWOOD-WALKER, S.M., MITCHELL,R., et al. (1995). The involvement of metabotropic glutamatereceptors and their intracellular signaling pathways in sus-tained nociceptive transmission in rat dorsal horn neurons.Neuropharmacology 34, 1033–1041.
YOUNG, M.R., FLEETWOOD-WALKER, S.M., DICKIN-SON, T., et al. (1997). Behavioral and electrophysiologicalevidence supporting a role for group 1 metabotropic gluta-mate receptors in mediation of nociceptive inputs to the ratspinal cord. Brain Res. 777, 161–167.
YOUNG, M.R., BLACKBURN-MUNRO, G., DICKINSON,T., et al. (1998). Antisense ablation of type I metabotropicglutamate receptor mGluR1 inhibits spinal nociceptive trans-mission. J. Neurosci. 18, 10180–10188.
YUZAKI, M., and MIKOSHIA, K. (1992). Pharmacologicaland immunocytochemical characterization of metabotropicglutamate receptors in cultured Purkinje cells. J. Neurosci.12, 4253–4263.
Address reprint requests to:
Claire E. Hulsebosch, Ph.D.Department of Anatomy and Neurosciences
University of Texas Medical Branch at GalvestonGalveston, Texas 77555-1043
E-mail: [email protected]
MILLS ET AL.
42