good clinical practice (gcp) in clinical research suchart chongprasert, ph.d. food and drug...
TRANSCRIPT

Good Clinical Practice (GCP) in Clinical Research
Suchart Chongprasert, Ph.D.Food And Drug Administration
ICH GCP for Clinical Research investigators, 16 December 2013, DMSC

Presentation Outline Practice vs. Research
clinical research vs. clinical trial therapeutic vs. non-therapeutic research
GCP: Introduction and Principles Basic GCP Applications for Investigators
Ethical Principles in Clinical Research Informed Consent Process: Introduction

Research vs. Practice
“Practice” refers to interventions that are designed solely to enhance the well-being of an individual patient or client, and that have a reasonable expectation of success. standard interventions scientifically proven and
accepted
The purpose of medical or behavioral practice is to provide diagnosis, preventive treatment or therapy to particular individual.
BELMONT REPORT

Research vs. Practice (2) “Research” designates an activity
designed to test a hypothesis, permits conclusions to be drawn, and thereby to develop or contribute to generalizable knowledge expressed for example in theories, principles,
and statements or relationships) that can be corroborated by scientific observation and inference.
BELMONT REPORT

Research vs. Practice (3)
Research is usually described in a formal protocol that sets forth an objective, and a set of procedures designed to reach the objective.
BELMONT REPORT

Research vs. Practice (4) Research represents a systematic
investigation (formal), including research development, testing, evaluation, that is designed to develop or contribute generalizable knowledge. methodology
Practice gains more flexibilities compared with research
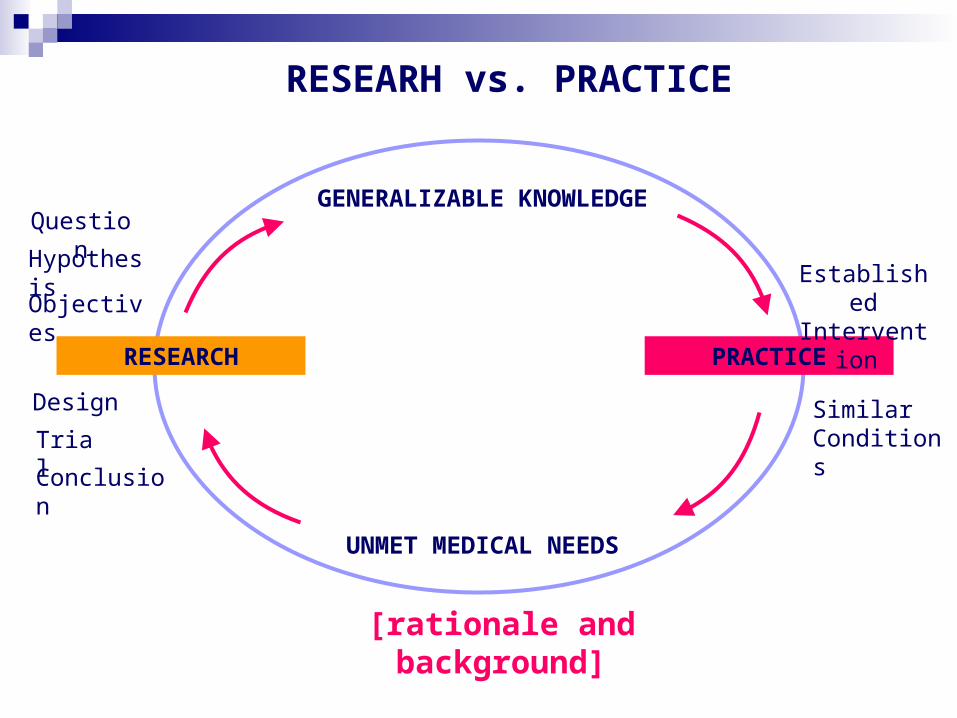
RESEARCH PRACTICE
UNMET MEDICAL NEEDS
Question
Design
Hypothesis
Trial
Objectives
Conclusion
GENERALIZABLE KNOWLEDGE
Established Intervention
Similar Conditions
RESEARH vs. PRACTICE
[rationale and background]

Clinical Research
research that either directly involves a particular person or group of people or uses materials derived from humans such as behaviors or samples of the tissues, that can be
linked to a particular living person (identifiable tissue or bio-specimens)

Declaration of Helsinki 2013
ethical principles for medical research involving humans subjects, including research on identifiable human materials and data

Clinical Research [2]: categories[1] Patient-oriented research: involves a
particular person or group of people or uses of materials from humansstudies of mechanisms of human disease;studies of therapies or interventions for
diseases;clinical trials; andstudies to develop new technology related to
diseases

Clinical Research [3] ]: categories[2] Epidemiological and behavior
studies:.. examining the distribution of diseases, the factors that affect health, and how people make health-related decisions
[3] Outcomes and health services research: seeking to identify the most effective and most efficient interventions, treatments, and services

Clinical Trials
one type of clinical research that involves a researcher or researchers who directly observes a person or people, and/or who collect data to answer a scientific or medical question about the safety or potential benefit of an intervention such as medication, device, teaching concept, training method or behavioral change

Clinical Trials[2]
a prospective biomedical or behavioral research study of human subjects that is designed to answer specific questions about biomedical or behavioral interventions (drugs, treatments, devices, or new ways of using known drugs, treatments, or devices) [US NIH]

Clinical Trials[3] a controlled study involving human subjects that
is designed to prospectively evaluate the safety and effectiveness of new drugs or devices or of behavioral interventions [US DHHS, IRB Handbook]
a systematic investigation, including research development, testing, and evaluation that is designed to develop or contribute to generalizable knowledge [US CFR Title 46 Part 102]

Clinical trial [4] any investigation in human subjects intended to
discover or verify the clinical, pharmacological, and/or other pharmacodynamic effects of an investigational product(s), and/or to identify any adverse reactions to an investigational product(s), and/or to study absorption, distribution, metabolism, and excretion of an investigational product(s) with the object of ascertaining its safety and/or efficacy. The terms clinical trial and clinical study are deemed synonymous. [ICH GCP]

Clinical Trial
Clinical Research

Where generalizable knowledge comes from

Data Quality vs. Data Integrity

Data Quality vs. Integrity Quality: a measure of the ability of a product,
process, or service to satisfy stated or implied needs
Data Quality: the essential characteristics of each piece of data, in particular, quality data should include: accurate; legible; complete and contemporaneous (recorded at the time
activity occurs; original; attributable to the person who generated the data

Data Quality vs. Integrity
Data Integrity: the soundness of the body of the data as a whole, in particular, the body of data should be 1credible, 2internally consistent, and 3verifiable
Both quality and integrity are essential for data to be relied upon for regulatory decision-making

Good Clinical Practice (GCP) in Clinical Research
Suchart Chongprasert, Ph.D.Food and Drug Administration
ICH GCP for Investigators, 31 July 2013, Chaingmai

o
[1] [2]

Special attentions paid on
Human subjects are protectedRights;Safety;Well being
Clinical trial data are credible for regulatory decision; for scientific use to improve interventions

GCP significance
..randomized controlled clinical trials form the foundation for “evidence-based medicine”, but such research can be relied upon only if it is conducted according to principles and standards collectively referred to as “Good Clinical Practice” (GCP)…
Source: WHO Handbook for GCP: Guidance for Implementation

International Conference on Harmonization GCP

Clinical trial
...any investigation in human subjects intended to discover or verify the clinical, pharmacological, and/or other pharmacodynamic effects of an investigational product(s), and/or to identify any adverse reactions to an investigational product(s), and/or to study absorption, distribution, metabolism, and excretion of an investigational product(s) with the object of ascertaining its safety and/or efficacy. The terms clinical trial and clinical study are deemed synonymous.
ICH GCP [1.12]

Clinical Trial
investigation/research in humans using pharmaceutical products
investigational phase or approved one
the objective is to discover or verify clinical / pharmacological / pharmacodynamic
effects absorption/ distribution / metabolism / excretion
so as to ascertain the safety and efficacy of investigational products

What is ICH GCP
…. international ethical and scientific quality standard for the designing, conducting, recording, performing, monitoring, auditing, reporting a clinical trial that involves human participation...
ICH GCP: Introduction

ICH GCP quality system being used in a clinical trial
from the beginning to the end quality standard
encompassing both [1] scientific, and [2] ethical aspects

Why GCP
…. compliance with this guideline assures the public that the rights, safety, and well-being of trial subjects are protected, and that trial data are credible .
ICH GCP : Introduction

Importance is given to
Human subjects are protectedRightsSafety,Well being
Clinical trial data are credible for regulatory decision for scientific use to improve interventions

ICH GCP - Objective
…….to provide a unified standard for European Union, United States, and Japan to facilitate mutual acceptance clinical data by regulatory authorities in the three jurisdictions no repetition of quality clinical trials conducted
previously in any of these regions ensuring the quality for the protection of humans
as well as the quality and integrity of clinical data

ICH GCP - Development
...developed with the consideration of the current GCP of the EU, Japan, US as well as Australia, Canada, the Nordic Countries, and the WHO.
combined together principles and guidelines implemented in advanced countries or organization
promoting global acceptance of clinical data generated
ICH GCP: Introduction

Applicability
When to apply the ICH GCP?
….should follow when generating clinical data that are intended to be submitted to regulatory authorities in the three regions. support a regulatory decision for marketing
authorization of pharmaceutical products
ICH GCP: Introduction

other applicabilities
.. the principles established in this guideline can also be used for other clinical investigations that may affect safety and well-being of trial subjects. can be applied to broader than a clinical trial

considerations
..some principles of GCP may not apply to all types of research on human subjects; consideration of these principles is strongly encouraged wherever applicable as a means of ensuring the ethical, methodologically sound and accurate conduct of human subject’s research..
Source: WHO Handbook for GCP: Guidance for Implementation

Applications of GCP
studies of a physiological, biochemical, or pathological process, or of a specific intervention
controlled studies of diagnostic, preventive or therapeutic measures
studies designed to determine the consequence for individuals and communities of specific preventive or therapeutic measures
Source: WHO Handbook for GCP: Guidance for Implementation

Applications of GCP [2] studies concerning human health-related
behavior in a variety of circumstances and environments
studies that employ either observation or physical, chemical, or psychological intervention may generate records or make use of existing
records containing biomedical or other information about individuals who may or may not be identifiable from the records or information

GCP Implementation:Shared Responsibilities



ICH GCP: STRUCTURE & CONTENT
Glossary
Principles of ICH GCP
IRB/IEC
Investigator
Sponsor
Clinical Trial Protocol
Investigator’s Brochure (IB)
Essential Documents for the
conduct of Clinical Trials
Documents
Personnel
Standard arrangement

Fundamental Concepts of ICH G CP


ICH GCP Principles[1] ethical principles established in the
Declaration of Helsinki (updated 2013)
[2] favorable risk and benefits ratio at the beginning and continuing of a clinical trial
[3] rights, safety, and well-being of trial subjects most important consideration, and precede the interests of science and society
[4] nonclinical and clinical data (if any) available to support the proposed trial

ICH GCP Principles [2]
[5] trial scientifically sound and written in a clear protocol
[6] protocol receives prior approval from IEC/IRB
[7] medial care and medical decision made be responsible by qualified physicians or dentists
[8] investigator and staff qualified by education, training, and experience
[9] freely informed consent obtained from individual subject prior to his/her participation

ICH GCP Principles [3]
[10] clinical trial information be managed in a way that allows accurate reporting, interpretation, and verification
[11] confidentiality of trial subjects
[12] investigational product manufactured, handles, and stored according to GMP
[13] QA system and procedures implemented throughout the trial

Advantages of GCP importance is given to (1) the protection of human sub
jects and (2) the quality and integrity of clinical data
clear assignment of key individuals involving in a clinical trial i.e., investigator, EC, sponsor
standardized glossary of terms
standardized elements of informed consent
standardized IRB/IEC membership
emphasize on principles, de-emphasize on “how to”

Limitations of GCP very high standards to be applied in for the Thai
context (in general);
subject to easy deviation;
require intensive resources to implement; sponsor QA
may not be practical in a resource-poor setting;
require adequate resources and investment in a regulatory authority

Ethics in Clinical Research and Independent Ethics Committee
Suchart Chongprasert, Ph.D.Food and Drug Administration
ICH GCP for Investigators, 16 December 2013, DMSC

Presentation Outline
Morality vs. Ethics Basic Ethical Principles in Clinical
ResearchRBJ Principles
Practical Applications Independent Ethics Committee (IEC) /
Institutional Review board (IRB)

Clinical trials should be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and that are consistent with GCP and the applicable regulatory requirements
ICH GCP Principle 1

Research involving humans should be scientifically sound and conducted in accordance with the basic ethical principles, which have their origin in the Declaration of Helsinki.
Three basic ethical principles of equal
importance, namely respect for person, beneficence, and justice permeate all GCP principles.
WHO GCP Principle 1

“… the two more important things needed for ethical clinical research i.e., informed consent and virtuous investigators.”
summarized from Dr. Henry Beecher’s Article

Basic Ethical Principles Respect for person
Autonomy (independence)
Beneficence and [non-maleficence]maximizing benefit; minimizing harms; do no harm
Justice distributive justice

Respect for Person
Respect for person requires that subjects, to the degree that they are capable, be given opportunities to choose what shall or shall not happen to them.
Belmont Report

Respect for Person
People should be treated as an autonomous agentsubject autonomy
Subjects with diminished autonomy need special protection vulnerable subjects

Respect for Person
autonomous agent means individuals who are able to make their own decision regarding their goal and the process to achieve such goals focus paid on choice and opinion of each
autonomous individual upon one’s determination
right to self-determination

People with Diminished Autonomy
Individuals whose willingness to volunteer in a clinical trial may be unduly influenced by the expectation, whether justified or not, of benefits associated with participation or of a retaliatory response from senior members of a hierarchy in case of refusal to participate

People with Diminished Autonomy (2)
Examples are members of a group with a hierarchical structure such as medical, pharmaceutical, and nursing students, subordinate hospital and laboratory personnel, employees of pharmaceutical industry, members of the arm forces, and persons kept in detention.

People with Diminished Autonomy (3)
Other vulnerable subjects include patients with incurable diseases, persons in nursing home, unemployed or impoverished persons, refugees, minors, and those incapable of giving own consent

special safeguard measures additional safeguards to protect the rights,
safety, and well-being of subjects with diminished autonomy special justification to the ethics committee
that research could not be carried out equally well with less vulnerable subjects
seeking permission of a legal guardian or other legally authorized representatives
impartial witness monitoring the conduct of the study

Respect for Person
right for self-determination; human’s dignity; informed consent; vulnerable subjects; privacy and confidentiality

Beneficence moral obligation to maximize the benefits
and to minimize harm requiring that
risks of research be reasonable in the light of the expected benefits
research design be sound instigators be competent both to conduct the
research and to safeguard the welfare of the subjects

Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefits for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks
ICH GCP 2.2

Clinical trials should be scientifically sound and described in clear detailed protocol
ICH GCP 2.5

The medical care given to, and medical decision made on behalf of, subjects should always be the responsibility of a qualified physician or, when appropriate, of a dentist
ICH GCP 2.7

Beneficence
in addition to maximizing benefits and minimizing risks or harm
beneficence sometimes expressed as a separate principle of non-maleficence (do no harm)

“Risks and benefits of research may affect the individual subjects… and society at large (or special groups of subjects in society).” “In balancing these different elements, the risks and benefits affecting the immediate research subjects will normally carry special weight.”
Belmont Report

The rights, safety, and well-being of the trial subjects are the most important considerations, and should prevail over the interests of science and society.
ICH GCP 2.3

Justice
“distributive” justicewho bears the burden/risk of research who takes benefits
giving rise to moral requirements that there be fair procedures and outcomes in the selection of trial subjects

Justice [2] justice in the selection of research
subjects requires attention in two respects individualsocial
equity requires that no group or class of persons should bear more than its fair share of the burdens of participation in research

Justice [3]
Subject should be drawn from qualifying population in the general geographic area of the trial without regard to race, ethnicity, economic status, or gender unless there is a sound scientific reason to do otherwise
CIOMS Guideline 12

Practical Applications
Informed consent [Respect for Person] informed decision making
Risk/benefit assessment [Beneficence] favorable ratio
Selection of trial subjects [Justice] fair and equitable procedures and outcomes

Suitability of Informed Consent
sufficiency of information given;basic minimum elements
understanding;understood consent
voluntariness; freely given without undue influence

Remember!
The three ethical principles are originally derived from the Western culture
In applying these principles, please take into account local norms, cultures, and traditions as appropriate
Consult IEC, if needed

Ethics Committee

A trial should be conducted in compliance with the protocol that has received prior institutional review board (IRB) /independent ethics committee (IEC) approval or/favorable opinion.
ICH GCP Principle 2.5

A trial should be conducted in compliance with the protocol that has received prior IRB/IEC approval/favorable opinion.
ICH GCP Principle [2.6]

Institutional Review BoardAn independent body constituted of medical,
scientific, and non-scientific members whose responsibility is to ensure the protection of rights [1], safety [2], and well-being [3] of human subjects involved in a trial by, among other things, reviewing, approving, and providing continuing review of trial protocol and amendment (s) and of the methods and material to be used in obtaining and documenting informed consent of the trial subjects
ICH GCP [1.31]

Independent Ethics CommitteeAn independent body (a review board or a committee,
institutional, regional, national or supranational) constituted of medical professionals and non-medical members whose responsibility is to ensure the protection of the rights [1], safety [2], and well-being [3] of human subjects involved in a trial, and to provide public assurance of that protection by, among other things, reviewing and approving / providing favorable opinion on, the trial protocol, the suitability of the investigator(s), facilities, and the methods and material to be used in obtaining informed consent of the trial subjects
ICH GCP [1.27]

IRB/IEC
Institutional Review Board (IRB) used in the US
Independent Ethics Committee (IEC) used in
Europe
deem synonymous
in Thailand, both are used interchangeablyERC, REC

IRB/IEC independent board / committee (free of any undue
influence to perform their task and make the decision)
exist in several levels depending on situations e.g., institutional (CU, MU) , national (MOPH), regional (EU), or supranational
members with medical and non-medical professions
responsible for protection of human subjects in a trial (rights, safety, and well being)
84

IRB/IEC
assuring the public of their achieved responsibilities by performing the following tasks
reviewing and approving the trial protocol; suitability of the investigator and the team
(through most update CVs); adequate facilities to conduct the trial; methods and materials to be used in obtaining
informed consento etc.
85

IRB/IEC interface between the investigator and the
subjects
possessing a formal authority for the approval of the trial and/or suspending/terminating the trial based upon the applicable laws and regulations
86

Why IEC Review? required independent review of a research
protocolo one of the criteria for determining if the
conduct of a clinical trial is ethical
protection of subjects
ensure safety and well being of subjects
87
Declaration of Helsinki !!

Policy IRB/IEC constituting and operating in
accordance with the ICH GCP
written approval received prior to initiating the trial
if no constituted / appropriate IEC, use an independent IEC such as MOPH IEC
88

IEC Composition
3.2.1 consist of a reasonable number of members, who collectively have the qualifications and experiences to review and evaluate the science[1], medical[2] aspects, and ethics[3] of the proposed trial
If it is not scientifically valid, it is unethical
89

Preparation and Submission
investigator needs to obtain confirmation of IEC members present
and occupation
inform IEC of any payments to subjects and advertisements, if any
obtain a list of IEC members
90

Documents for Submission
Informed Consent/Patient
Information Sheet
Diary cards
payment scheduleany
advertisements
protocol and amendments
Investigator’s Brochure/drug
information
EC submission


Roles of Investigator
interface between sponsor and ethics committee
satisfies documentation requirements
updates ethics committee as requiredsafety progress;safety, SAEs;progress report, etc.
93

Roles of Investigator [2] Investigator must
not be involved in the approval process
not influence / apply pressure to committee members
communicate with the IEC in an appropriate manner (e.g., respect for their decision, appeal the decision with supportive new scientific evidence for the protocol etc.)
94

IRB/EC Decision unconditional approval
documented in writing
be signed and dated by Chairman / Secretariat
approval letter bears full the protocol number and title and/or version
Note: a trial must not be begun prior to receiving the IEC approval letter !!
95

Summary independent review is mandatory in biomedical
research, including a clinical trial
competent IRB/IEC fulfills the requirement of the Declaration of Helsinki, ICH GCP, and applicable regulations
investigator should ensure that the trial protocol is reviewed and approved by a competent ethics committee
96

Summary [2]
investigator should obtain an EC approval letter before commencing the trial
investigator should ensure a regular contact with the EC in case of new information, amendments, progress report, or safety update etc.

Informed Consent Process
Suchart Chongprasert, Ph.D.Food and Drug Administration
ICH GCP for Clinical Research investigators, 31July 2013, Chiangmai

Presentation Outline
What does it means? Basic Elements of Informed Consent Certain Important Aspect of Informed
Consent Exercise

Freely given informed consent should be obtained from every subject prior to clinical trial participation
ICH GCP Principle [2.9]

“… each potential subject must be adequately informed of the aims, methods, anticipated benefits and potentials hazards of the study and the discomforts it may entail.”
“…the physician should obtain the subject’s freely given informed consent, preferably in writing.”
Declaration of Helsinki

A process1 by which a subject voluntarily2 confirms his or her willingness to participate in a particular trial, after having been informed3 of all aspects4 of the trial that are relevant to the subject’s decision5 to participate. Informed consent is documented by means of a written6 signed7 and dated8 informed consent form.
ICH 1.28

Informed consent
A process by which a subject voluntarily2 confirms his or her willingness to participate in a particular trial, after having been informed3 of all aspects4 of the trial that are relevant to the subject’s decision5 to participate. Informed consent is documented by means of a written6 signed7 and dated8 informed consent form.
..an ongoing process, not a form for just signing

Informed consent
A process1 by which a subject voluntarily confirms his or her willingness to participate in a particular trial, after having been informed3 of all aspects4 of the trial that are relevant to the subject’s decision5 to participate. Informed consent is documented by means of a written6 signed7 and dated8 informed consent form.
no coerce or use undue influence

Informed consent
A process1 by which a subject voluntarily2 confirms his or her willingness to participate in a particular trial, after having been informed3 of all aspects4 of the trial that are relevant to the subject’s decision5 to participate. Informed consent is documented by means of a written6 signed7 and dated8 informed consent form.
thoroughly about the trial e.g.,
purpose, benefits, payment etc.

Informed consent A process1 by which a subject voluntarily2
confirms his or her willingness to participate in a particular trial, after having been informed3 of all aspects4 of the trial that are relevant to the subject’s decision5 to participate. Informed consent is documented by means of a written6 signed7 and dated8 informed consent form.
subject’s autonomy to make their own
judgment

Informed consentA process1 by which a subject voluntarily2
confirms his or her willingness to participate in a particular trial, after having been informed3 of all aspects4 of the trial that are relevant to the subject’s decision5 to participate. Informed consent is documented by means of a written6 signed7 and dated8 informed consent form.
….acceptable way under the ICH GCP
context


Informed consent
Desirable informed consento adequate information disclosedo language suitable to the recipient
o not too technicalo avoid a non-mother languageo remember that the readers are not scientific
expert/ethics committee members

Methods for Documenting Informed Consent
written informed consent In accordance with ICH GCP
oral witnessed consentsno documented evidencebeyond the scope of ICH GCP

Elements of IC (ICH GCP) 1. involve research;
2. research purpose;
3. choice of treatments, including randomization;
4. trial procedures, including invasive ones;
5. subject’s responsibilities;
6. trial aspects that are experimental;
7. foreseeable risks/inconvenience

Elements of IC [2]
8. anticipated benefits, if not any, inform as well;
9. treatment alternative if not willing to participate and related risks/benefit;
10. compensation/treatment in case of trial-related injury;
11. prorated payment, if any;
12. expected expenses, if any, for participating;

Elements of IC [3] 13. voluntary statement;
14. statement for direct access by monitor, auditor, IEC/IRB, authority upon signing the consent form;
15. confidentiality of the subject’s private info. kept;
16. be informed timely available new information affecting the willingness to continue in the trial;

Elements of IC [4]
17. contact person for more information or in case of injury;
18. circumstances for withdrawal/ termination;
19. expected duration;
20. approximate number of trial subjects (local vs. global)

Important Considerations consent must be informed;
careful about the uninformed informed consent ensure subjects’ understanding (understood consent)
subjects given ample time; all questions answered to the subject's
satisfaction; no undue pressure/influence; language clearly understood by the subjects
recommended not higher than grade 8

Important Considerations [2]
no language causing the subjects or the legally acceptable representatives to waive their legal rights; IC not a legal contract / agreement
ICF personally signed and dated by the subject and the person obtaining the ICF;
a copy of ICF given to subject prior to participation;

Suitability Considerations for IC informed and [ understood ] consent;
respect for subject’s right to self-determination (autonomy)
disclose sufficient information; all aspects about the trial
suitable for subject’s capacity (normal vs. diminished); age; educational status; sex; culture, geosocial factors etc.

Suitability Considerations for IC [2]
voluntariness; and
practicalityamount of the given info. vs. time for
consideration in reality short form??
depending on investigator’s explanation

Importantly!
In principle, obtaining informed consent in a clinical research is a must
Waiving of informed consent is an exception which should be well-justified and received prior approval from IEC/IRB before implementation

IC Exercise

Practical Considerations
Q: Who should obtain informed consent ?
A: In general, the investigator or authorized person obtains the informed consent. According to the Helsinki Declaration, the physician who treats the potential subject should not obtain the consent by him/herself, instead other physician who understands the protocol well does. Avoid doctor/patient dependent relationship.

Practical Considerations
Q: Obtaining informed consent in children / minor ?
A: For children with legal incompetence (i.e., age <20 yrs), but able to give assent to the research protocol, an assent form should be obtained besides parent’s informed consent
[assent: affirmative agreement to participate in the trial]

Practical Considerations Q: When to conduct the trial in relation to
the time to obtain the informed consent ?
A: The study cannot start prior to receiving a written informed consent from the subjects. Also, an unconditional written ethics committee’s approval letter must be obtained before beginning the trial.

Practical Considerations Q: Informed consent in a clinical
pharmacology trials (PK, phase I study)? (i.e., non therapeutic trial)
A: Should be conducted in the subjects who can give informed consent by themselves by personally signing and dating on the form.

Practical Considerations Q: When should the informed consent be
amended ?
A: When new information that may affect the safety and well being of the subject becomes available, and it may affect the subject’s decision to continue participating in the trial.

Practical Considerations Q: How long should it take to obtain the
informed consent?
A: Depending on the situation. Remember that informed consent is an ongoing process, not finished upon the signature of the subject. Specifically, how long it takes until the subject signs and date on the form is what the investigator needs to know. Exact time cannot be established, but the investigator needs to achieve efficient project management and obligation under the GCP on obtaining informed consent.

Summary o Informed consent is a continuing process
to confirm subject's voluntary willingness to participate in the trial.
o Informed consent should be obtained from
each patient prior to enrolling the subject into the trial.
o Children’s assent form is required in most clinical trials involving children (below majority age) participation

Thank you for your attention