glycine metabolism
TRANSCRIPT


Simple amino acid.
Non essential amino acid.
Optically inactive due to absence of
asymetric carbon atom.

Glycine is actively involved in the synthesis
of many specialized products (heme, purines,
creatine etc.
Required for synthesis of serine & glucose.
Involved in one-carbon metabolism.
Glycine is the most abundant amino acid
normally excreted into urine (0.5-1.0 g/g
creatinine).

Nutritional - non-essential amino acid.
Metabolically - glucogenic amino acid.
Glycine is one among the commonest amino
acids found in protein structure.
Glycine is mostly present in the interior
structure of protein.
Collagen contains very high (about 30%)
content of glycine.


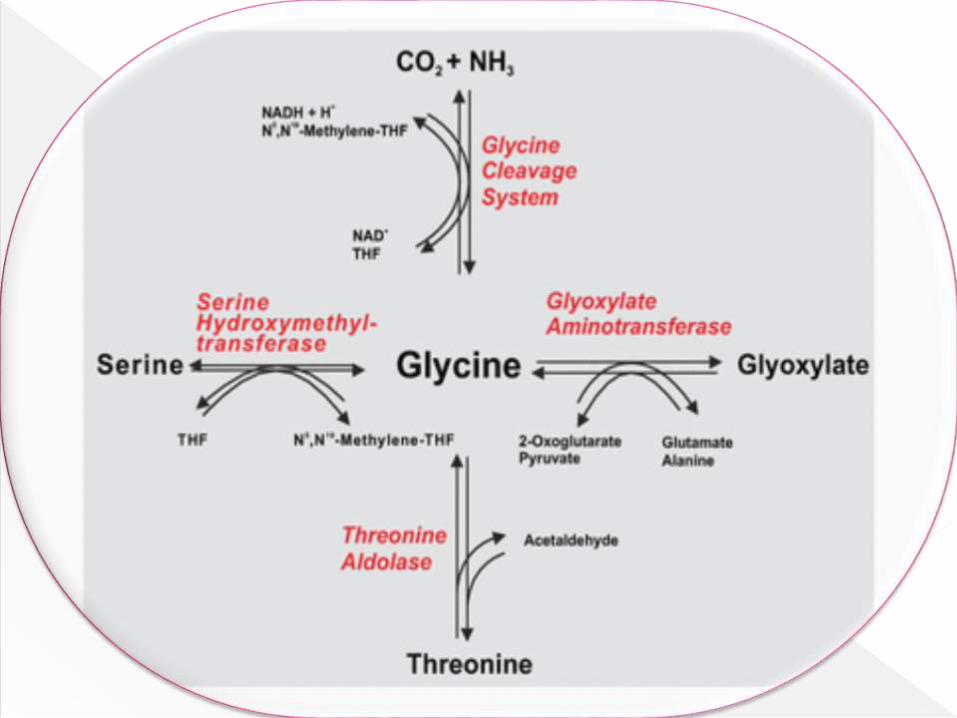
Glycine is synthesized from:
From Serine
From Threonine
From CO2, NH3.
From Glyoxalate.

Glycine is synthesized from serine by the
enzyme serine hydroxymethyl transferase
which is dependent on tetrahydrofolate
(THF).
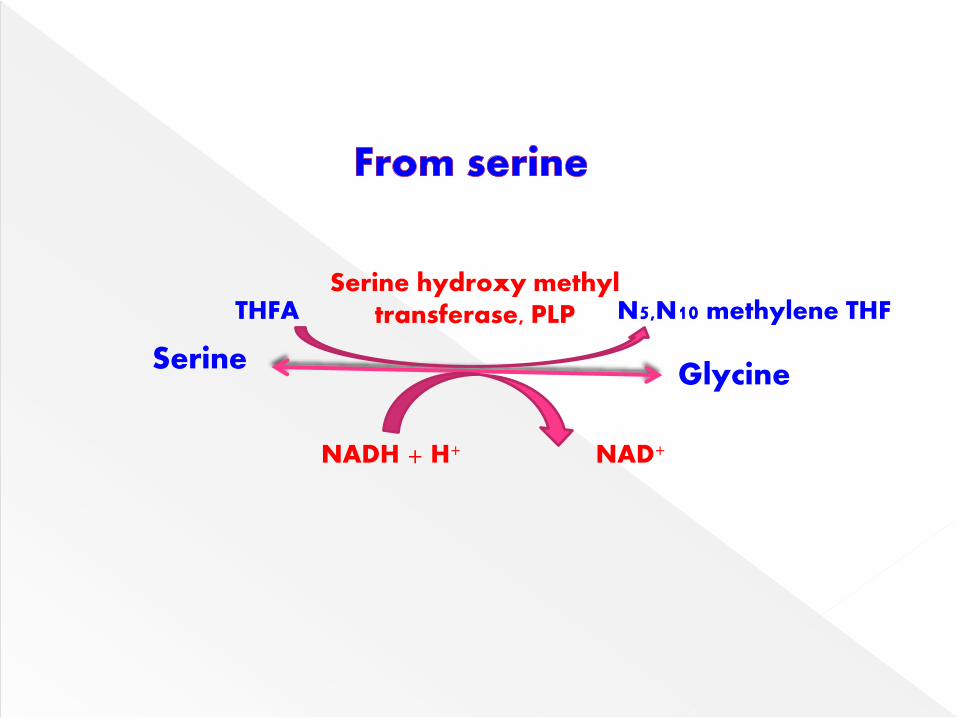
SerineGlycine
Serine hydroxy methyl transferase, PLPTHFA N5,N10 methylene THF
NAD+NADH + H+

Glycine can also be obtained from threonine,
catalysed by threonine aldolase.
Threonine Glycine + acetaldehydeThreonine aldolase

Glycine can be synthesized by the glycine
synthase reaction from CO2, NH3 & one
carbon unit.
This is the reversal of the glycine cleavage
system.
It is a multienzyme complex.
It needs the co-enzymes, NAD, lipoamide,
tetrahydrofolic acid & PLP.

CO2 + NH3 Glycine
Glycine synthasecomplex, PLP
N5,N10 Methylene FH4 FH4
NAD+NADH + H+

Glycine amino transferase can catalyze the
synthesis of glycine from glyoxylate &
glutamate or alanine.
This reaction strongly favors synthesis of
glycine.
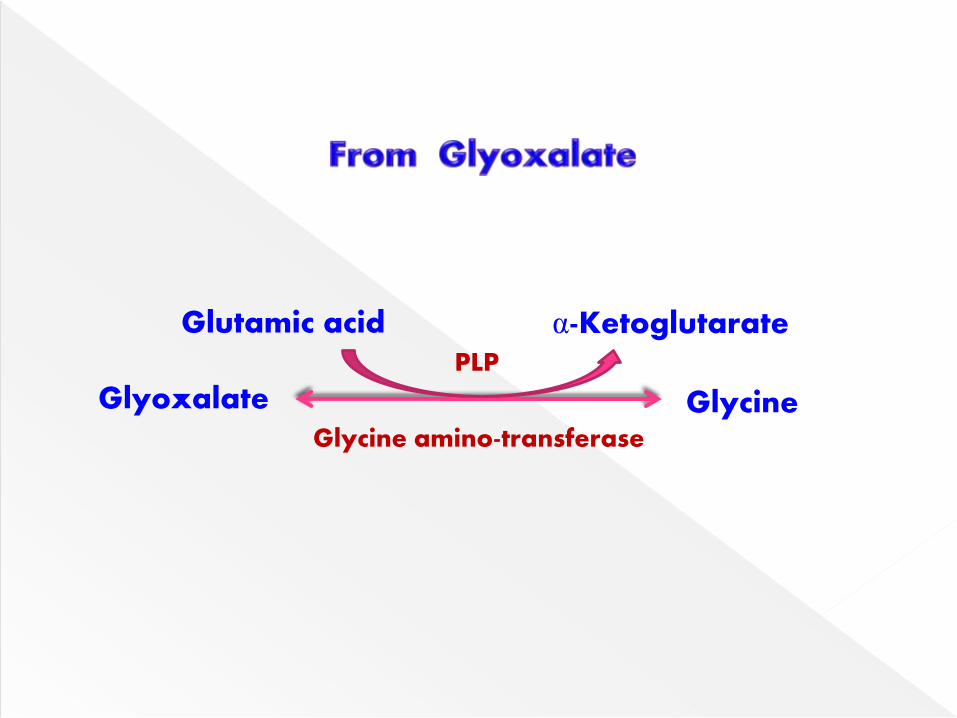
Glyoxalate Glycine
Glutamic acid α-Ketoglutarate
Glycine amino-transferase
PLP

Glycine undergoes oxidative deaminaion by
glycine synthase to liberate NH4+, CO2 & one
carbon fragment as N5, N10 methylene THF.
This provides a major route for glycine
breakdown in mammals.
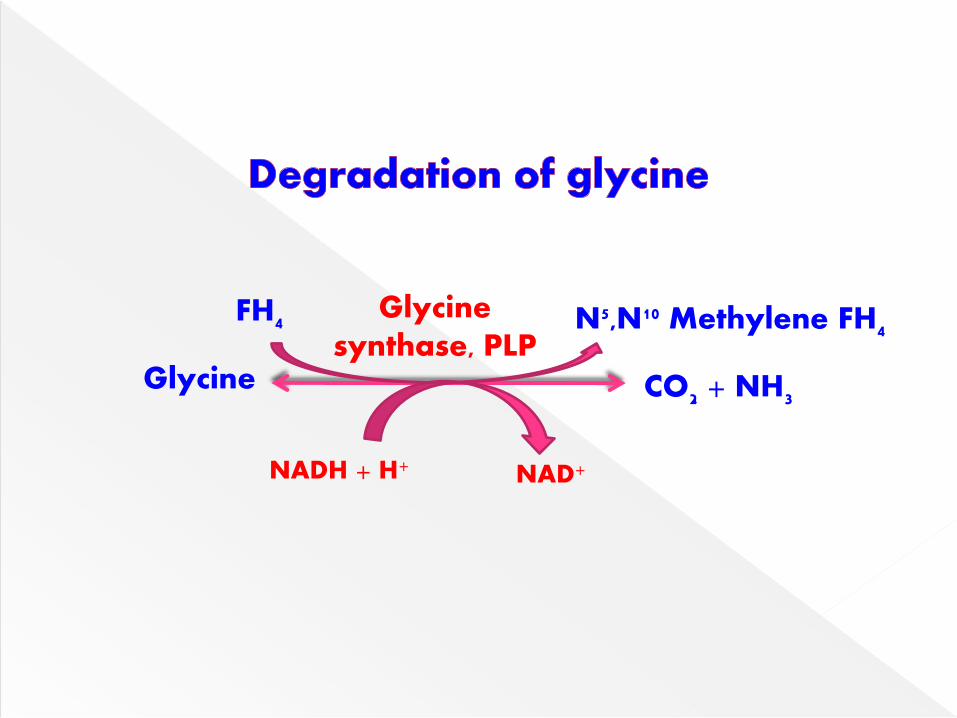
Glycine
Glycinesynthase, PLP
N5,N10 Methylene FH4FH4
NAD+NADH + H+
CO2 + NH3

It is a multienzyme complex.
It requires co-enzymes - PLP, NAD, THFA.
PLP-dependent glycine decarboxylase.
Lipoamide containing amino
methyltransferase
Methylene THFA synthesizing enzyme.
NAD+ dependent lipoamide dehydrogenase.

Glycine is mainly channelled into the
glucogenic pathway by getting first
converted to serine.
This is the reversal of serine hydroxy
methyltransferase reaction.
The serine is then converted to pyruvate by
serine dehydratase

Glycine is reversibly converted to serine by THF
dependent serine hydroxymethyl transferase.
Pyruvate produced from serine by serine
dehydratase, serves as a precursor for glucose.
Glycine Serine
Serine hydroxy methyl transferase, PLP
THFAN5,N10 methylene THF
NAD+NADH + H+

Serine is degraded to glyoxylate which
undergoes transamination to give back to
glycine.
Glyoxylate is also converted to oxalate, an
excretory product & formate enter one
carbon pool.
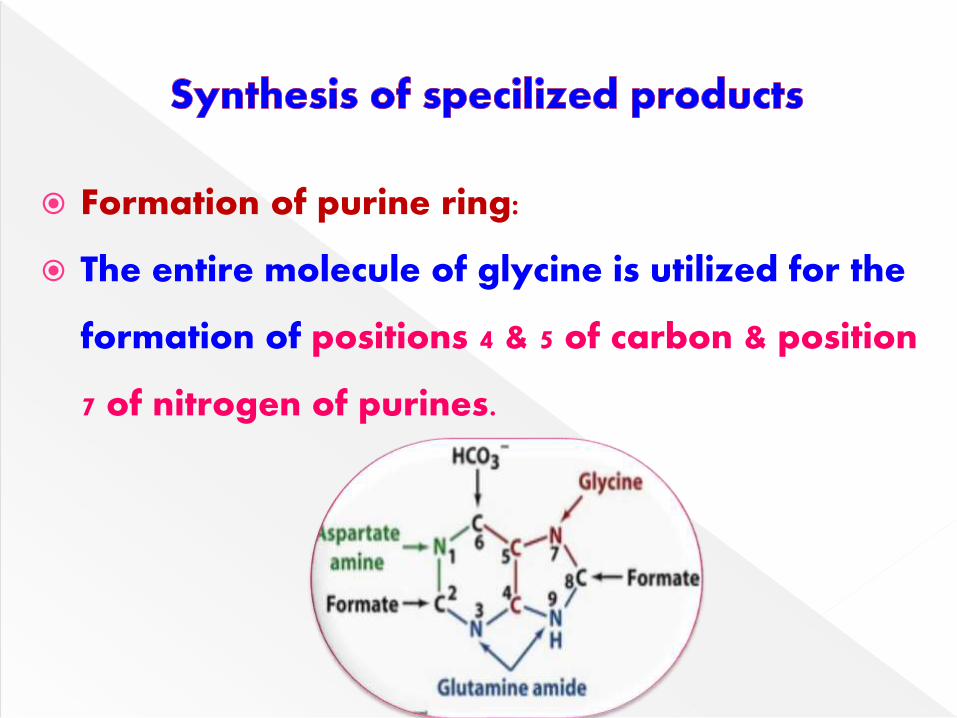
Formation of purine ring:
The entire molecule of glycine is utilized for the
formation of positions 4 & 5 of carbon & position
7 of nitrogen of purines.

It is a tri-peptide, containing glutamic acid,
cysteine, glycine.
Present as reduced form (GSH) & oxidized
form (GSSG).


Conjugating agent, glycine performs two
important functions.
The bile acids - cholic acid & chenodeoxy
cholic acid- are conjugated with glycine.
Cholic acid + glycine Glycocholic acid
Chenodeoxy cholic acid + glycine Glycochenodeoxycholic acid

Benzoic acid is used in small amounts as
preservative in foods.
Glycine is used for detoxification of
benzoic acid to form hippuric acid.
Benzoic acid + glycine Hippuric acid

Glycine condenses with succinyl CoA to δ-
amino levulinate which serves as a
precursor for heme synthesis.
Glycine + Succinyl CoA Amino levulinate (ALA)ALA Synthase

Creatine is present in the tissues as a high
energy compound, phosphocreatine & as free
creatine.
Three amino acids glycine, arginine &
methionine are required for creatine formation.

Step-1:
The first reaction occurs in the mitochondria
of kidney & pancreas.
It involves the transfer of guanidino group of
arginine to glycine, catalysed by arginine-
glycine transamidinase to produce
guanidoacetate (glycocyamine).

Step-2:
S-Adenosylmethionine (active methionine)
donates methyl group to guanidoacetate
(glycocyamine) to produce creatine.
This reaction occurs in liver.
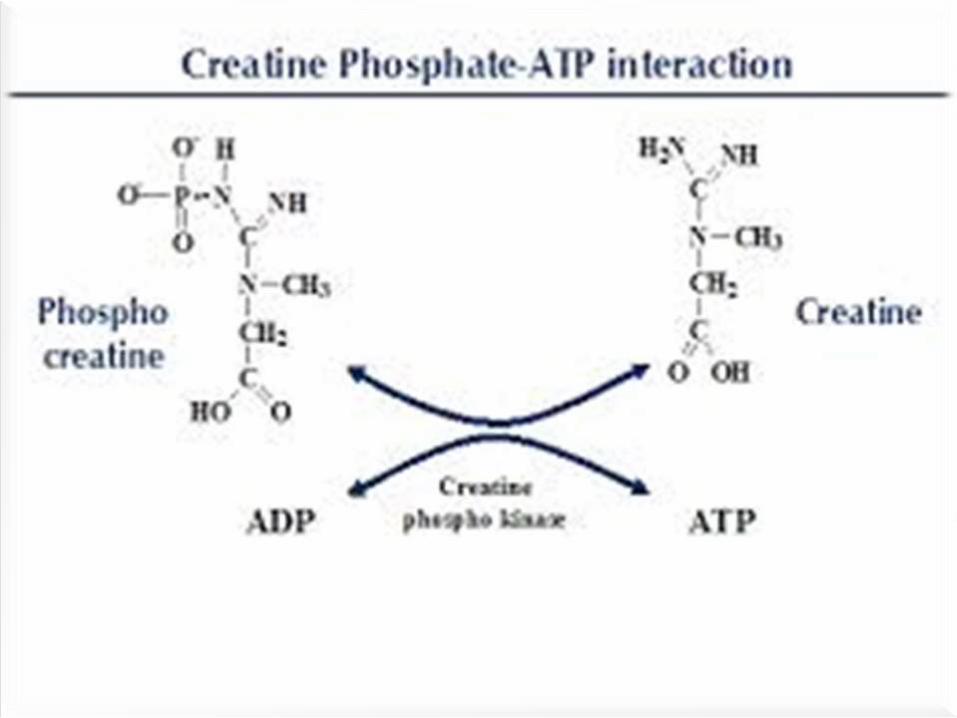
Step-3:
Creatine is reversibly phosphorylated to
phosphocreatine (creatine phosphate) by
creatine kinase.

It is stored in muscle as high energy
phosphate.
Serves as an immediate store of energy in the
muscle
Step-4:
The creatine phosphate may be converted to
its anhydride, creatinine.
It is a non-enzymatic spontaneous reaction.
Creatinine is excreted in urine.


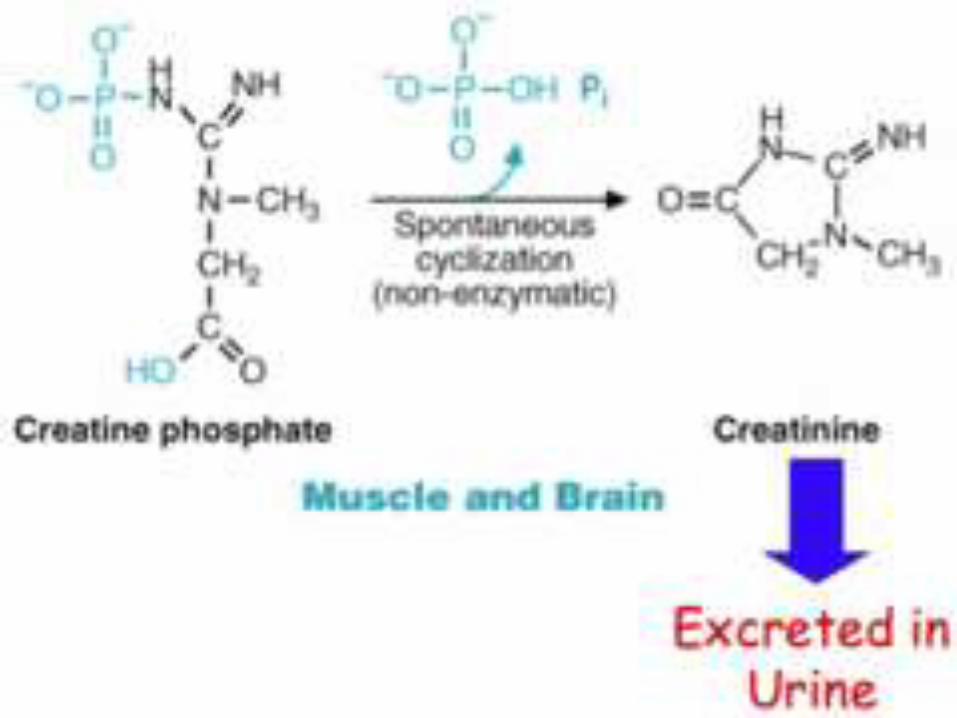

Normal serum creatinine level: 0.7 - 1.4 mg/dl.
Serum creatine level: 0.2 - 0.4 mg/dl.
Creatinine level in blood is a sensitive indicator
of renal function.
In muscular dystrophies, blood creatine &
urinary creatinine are increased.
The enzyme CK is elevated in MI.

Urine level:
Creatinine: 1 - 2 gm/day.
Creatine: 0 - 50 mg/day.
Creatinine coefficient:
Males: 20 - 30 mg/kg/24hrs.
Females: 10 - 20 mg/kg/24hrs.
Creatinine Clearance:
Males: 90 - 130 ml/min.
Females: 80 - 120 ml/min.

Excretion of creatinine is constant for an
individual depends on muscle mass.
Creatinine Clearance – measure of GFR.
Normally , urine contains – creatine (less).
Creatinuria – increased excretion of creatine
in urine.
Muscular dystrophy, Hypogonadism,
Hyperthyroidism, DM & Stravation.

Glycine is seen in the brainstem & spinal
cord.
Glycine opens chloride specific channels.
In moderate levels glycine inhibits neuronal
traffic; but at high levels it causes over-
excitation.

Glycine is seen where the polypeptide chain
bends or turns (beta bends or loops).
In collagen, every 3rd amino acid is glycine.

It is due to defect in glycine cleavage system.
Glycine level is increased in blood, urine & CSF.
Severe mental retardation & seizures are seen.
There is no effective management.

This is a rare disorder.
Serum glycine concentration is normal, but very
high amount (normal 0.5-1 g/day) is excreted in
urine.
It is due to defective renal reabsorption.
It is characterized by increased tendency for
formation of oxalate renal stones.
Urinary oxalate level is normal in these patients

It is due to protein targetting defect.
Normally, the enzyme alanine glyoxalate
amino transferase is located in peroxisomes;
but in these patients the enzyme is present in
mitochondria.
So, enzyme is inactive.
It characterized by increased urinary oxalate
resulting in oxalate stones.

Deposition of oxalate (oxalosis) in various
tissues is observed.
The urinary oxalate is of endogenous origin
& not due to dietary consumption of oxalate.
Primary hyperoxaluria is due to a defect in
glycine transaminase coupled with
impairment in glyoxalate oxidation to
formate.

It is a milder condition causing only
urolithiasis & results from deficient activity of
cytoplasmic glyoxalate reductase.
The management is to increase oxalate
excretion by increased water intake.
Minimise dietary intake of oxalates by
restricting the intake of leafy vegetables, tea,
beet-root etc.

Textbook of Biochemistry-u Satyanarayana
Textbook of Biochemistry-DM Vasudevan
