genomic big data management, integration and mining - emanuel weitschek
TRANSCRIPT

Genomic Big Data Management, Integration, and Mining
E. Weitschek1,2
1 Department of Engineering, Uninettuno International University, Italy 2 Institute of Systems Analysis and Computer Science, National Research Council, Italy
Joint work with P. Bertolazzi, G. Felici , F. Cumbo, G. Fiscon, E. Cappelli

2
Outline
• Growth of biological data
• Next generation sequencing
• Biological data sources
• Biological data management
• Biological data integration
• Big data bioinformatics
• Knowledge extraction
• Supervised Learning
• Biomedical applications
• Conclusions and future directions
3
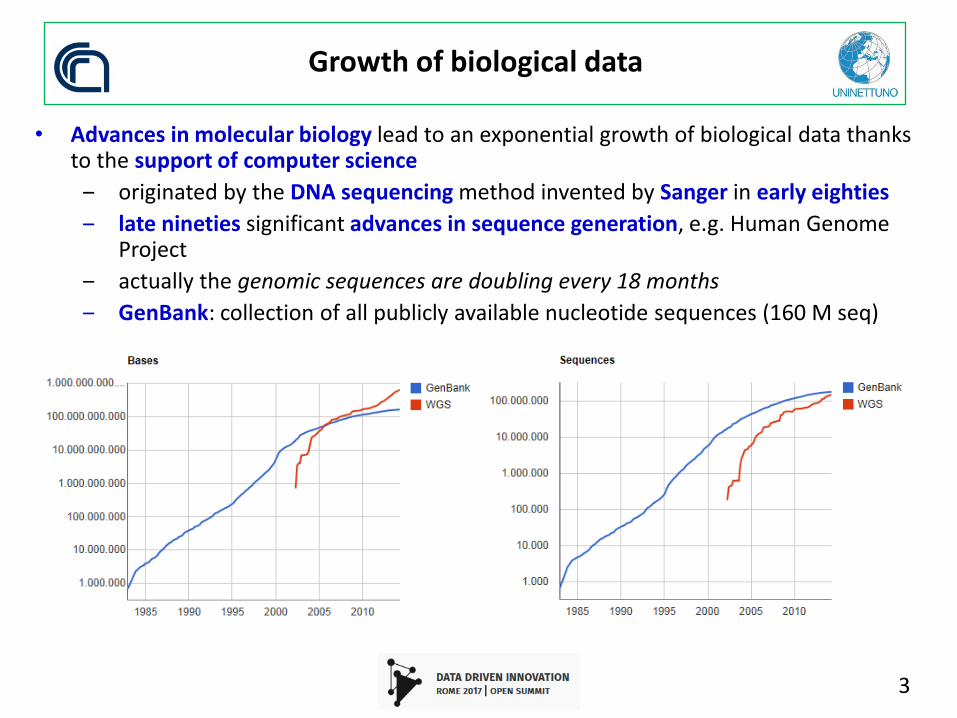
Growth of biological data
• Advances in molecular biology lead to an exponential growth of biological data thanks to the support of computer science
‒ originated by the DNA sequencing method invented by Sanger in early eighties
‒ late nineties significant advances in sequence generation, e.g. Human Genome Project
‒ actually the genomic sequences are doubling every 18 months
‒ GenBank: collection of all publicly available nucleotide sequences (160 M seq)

4
Growth of biological data
• Advances in molecular biology lead to an exponential growth of biological data thanks to the support of computer science
‒ Today next generation high throughput data from modern parallel sequencing machines, are collected and huge amounts of biological data are currently available on public and private sources
‒ 10000 Human Genomes project (3000 Mbp)
‒ Nowadays: 1000$ genome
• Very large data sets, that are generated by several different biological experiments, need to be automatically processed and analyzed with computer science methods

5
DNA Sequencing
• DNA (deoxyribonucleic acid) is the hereditary material in almost all organisms
• DNA sequencing is the process of determining the order of nucleotides within a DNA molecule
• It includes any method or technology that is used to determine the order of the four bases—adenine (A), cytosine (C), guanine (G), and thymine (T)
• Originated by the DNA sequencing method invented by Sanger in early eighties
• In late nineties significant advances in sequence generation techniques, largely inspired by massive projects such as the Human Genome Project
• High costs and time, e.g., for the Human Genome Project 5 billions $ and 13 years

6
Next Generation Sequencing (NGS)
• Today: next generation high throughput data from modern parallel sequencing machines ‒ Roche 454, Illumina, Applied Biosystems SOLiD,
Helicos Heliscope, Complete Genomics, Pacific Biosciences SMRT, ION Torrent
‒ Next generation sequencing (NGS) machines output a large amount of short DNA sequences, called reads (in fastq format)
‒ Cannot read entire genome one nucleotides at a time from beginning to end
‒ shred the genome and generate shorts reads
‒ Low cost per base (1000$ for a whole human genome)
‒ High speed (24h to sequence a whole human genome )
‒ Large number of reads
‒ Problems: data storage and analysis, high costs for IT infrastructure

7
Next Generation Sequencing (NGS)
• Data dimension, time and cost of Next Generation Sequencing
Seq type Data Price $ Time
Human Genome 90 GB 1000 1 day
Human Gene Expression
9 GB 500 12 h
Plant Genome 150 GB 2000 5 days
Bacterial Genome 1 GB 300 6 h

8
Biological data sources
• Several heterogeneous sources of biomedical data are available
• Sequence Read Archive
• The Gene Expression Omnibus
• NCBI
• ELIXIR
• The Cancer Genome Atlas (TCGA)

9
Biological data management
10
Biological data integration
• Challenge for the research community
• Allow everyone to store, organize, access, and analyze the information available on the web and/or on private repositories
• Integration of data: providing a unified access to heterogeneous and independent data sources as a single source
• Many solutions from the I.T. and from the bioinformatics community, e.g.
− Heterogeneous Database Systems
− Distributed Database Systems
− SRS
− NCBI Entrez
− Federated databases (BioKleisli)
− Multi-databases (TAMBIS),
− Mediator-based (Bio-DataServer)
− Data warehousing (BioWarehouse)
• Integration of clinical and genomic data

11
Bioinformatics
• New methods are demanded able to extract relevant information from biological data sets
• Effective and efficient computer science methods are needed to support the analysis of complex biological data sets
• Modern biology is frequently combined with computer science, leading to Bioinformatics
• Bioinformatics is a discipline where biology and computer science merge together in order to design and develop efficient methods for analyzing biological data, for supporting in vivo, in vitro and in silicio experiments and for automatically solving complex life science problems
• Bioinformatician: a computer scientist and biology domain expert, who is able to deal with the computer aided resolution of life science problems

12
• The attention to Big Data in bioinformatics is steadily increasing, proportionally to the growth of the amount of biological data obtained through sequencing
• Dealing with such an amount of data, recorded at different stages during the life of a person and stored for dynamic analysis studies, requires scalable systems suitable for the collection, management, and analysis
• Biological Big Data Bases
Big Data Bioinformatics

13
• Comprehensive genomic characterization and analysis of more than 30 cancer type
• National Cancer Institute (NCI), National Human Genome Research Institute (NHGRI), and National Institute of Health (NIH)
• Aim: improve the ability to diagnose, treat and prevent cancer
• A free-available platform to search, download, and analyze data sets
• 33 tumors with more than 10000 patients
• Public data distributed with the open access paradigm
• Genomic experiments
– Copy Number Variation (CNV)
– DNA-methylation
– DNA-sequencing (whole genome, whole exome, mutations)
– Gene expression data (RNA-Seq V1, V2)
– MicroRNA sequencing
– Meta data (Clinical and Biospecimen)
• Contains more than 15 TB of genomic and clinical data, whose analysis and interpretation are posing great challenges to the bioinformatics community
The Cancer Genome Atlas (TCGA)

14
TCGA2BED
Data integration from external dbs

15
data set: DNA-Methylation
data set: RNA-sequencing
Genomic data integration
Typical problem in Bioinformatics:
• More than 1000 samples (patients), 450 000 features (genes, sites, clinical variables, proteins, )
• Aim: distinguish healthy vs diseased samples
• Not addressable by a classic machine learning algorithm
• Big Data solutions

16
• Aims: distinguish the diseased from the healthy samples and prediction
• Input: a training set (reference library) containing samples with a priori known class membership
• Model building: based on this training set the software computes the classification model
• The classification model can be applied to a test set (query set) which contains samples that require classification:
− query samples with unknown species membership or
− samples that also have a priori known species membership, allowing verification of the classifications
Classification and supervised machine learning

17
Rule-based classification
A rule-based classifier is a technique for classifying samples by using a collection of “if… then rules”, named logic formulas:
– Antecedent Consequent
– (Condition1) or (Condition2) or … or (Conditionn) Class
– Conditioni: (A1 op v1) and (A2 op v2) and … and (Am op vm)
– A = attribute; v = value; op = operator {=, ≠, <, >, ≤, ≥}
• Example of logic classification formula is
• The evaluation of the logic formulas and the classification of the samples to the right class is performed according :
– Percentage split or cross validation sampling
– Accuracy
– F-measure
“IF Aph1b<0.507 then the experimental sample is CONTROL”

18
CAMUR
• Classifier with Alternative and Multiple Rule-based models (CAMUR)
• New method for classifying RNA-seq case-control samples, which is able to compute multiple human readable classification models
• Aims of CAMUR:
1) To classify RNA-seq experiments
2) To extract several alternative and equivalent rule-based models, which represent relevant sets of genes related to the case and control samples
• CAMUR extracts multiple classification models by adopting a feature elimination technique and by iterating the classification procedure
• Prerequisite: Gene expression normalization (RPKM or RSEM )
• Available at: http://dmb.iasi.cnr.it/camur.php

19
CAMUR: method
• CAMUR is based on:
1) a rule-based classifier (i.e., in this work RIPPER)
2) an iterative feature elimination technique
3) a repeated classification procedure
4) an ad-hoc storage structure for the classification rules (CAMUR database)
• In brief, CAMUR:
• iteratively computes a rule-based classification model through the supervised RIPPER algorithm,
• calculates the power set (or a partial combination) of the features present in the rules,
• iteratively eliminates those combinations from the data set, and
• performs again the classification procedure until a stopping criterion is verified: F-measure < threshold
Maximum number of iterations reached

20
Experimentation and results

21
Experimentation and results

22
(MAMDC2_dMet >= 6.63) and
(ACACB_rnaSeq >= 887.80)
=> class=normal (19.0/3.0)
[ ] => class=tumoral (1102.0/1.0)
Correctly Classified Instances 98.11 %
Incorrectly Classified Instances 1.88 %
Gene occurrences
FIGF_rnaSeq 44
SPRY2_dMet 37
SCN3A_rnaSeq 25
PAMR1_dMet 20
MMP11_rnaSeq 20
Class rule accuracy
Normal (FIGF_rnaSeq >= 184.15) and
(CLEC5A_dMet <= 5.44) ||
(TSHZ2_rnaSeq >= 471.04) and
(DLGAP2_dMet >= 10.06)
9.800
Normal (SPRY2_dMet >= 0.55) and
(CD300LG_rnaSeq >= 454.24) ||
(PAMR1_rnaSeq >= 712.17) and
(PARP8_dMet >= 2.17)
9.700
Camur: occurrences
Classification models for breast cancer
CAMUR: rules
Supervised model extraction

23
Aim: To extract relevant features from the ever-increasing amount of
biological data and to apply supervised learning to classify them
Biology Issue Features Software Data source
Clinical patient classification
Clinical variables (blood, imaging, psicosometric tests…)
DMB, Weka Heterogeneous health care facilities
Gene Expression Analysis
Discretize gene expression profiles
Gela, CAMUR TCGA, EBRI
DNA barcoding Nucleotide sequences of DNA-barcode
Blog, Fasta2Weka Barcode of Life Consortium
Polyoma/Rhyno Viruses
Nucleotide sequences of Polyoma/Rhyno viruses
DMB, MISSAL Istituto Superiore di Sanità
EEG signals processing
Fourier Coefficients extracted from EEG recordings
Matlab, Weka, DMB IRCCS Centro di Neurolesi “Bonino-Pulejo” of Messina
Biomedical image processing
Oriented Fast and Rotated BRIEF
Matlab, Weka, DMB
Alzheimer's Disease Neuroimaging Initiative
Other applications on biomedical data

24
Conclusions and future directions
• Exponential growth of biomedical data
• Release of many public data bases, data collection and data management projects
• Data integration
• Supervised classification analysis
• Advanced systems for data integration
• New big data approaches

25
Acknowledgments
Emanuel Weitschek
Department of Engineering
Uninettuno International University
www.iasi.cnr.it/~eweitschek