genetic heterogeneity of hepatocellular carcinoma
TRANSCRIPT

Proc. Natl. Acad. Sci. USAVol. 91, pp. 822-826, January 1994Medical Sciences
Genetic heterogeneity of hepatocellular carcinoma(p53 tumor suppressor gene/hepatitis B virus/aflatoxins/mutation/liver cancer)
HILAL UNSAL*, CENGIZ YAKICIERt, CHRISTOPHE MAR;AISt, MICHEAL KEWt, MARTIN VOLKMANN§,HANS ZENTGRAF1, KURT J. ISSELBACHER*, AND MEHMET OZTURKtII*Massachusetts General Hospital Cancer Center, Charlestown, MA 02129; tLaboratoire d'Oncologie Mol6culaire, Institut National de la Sant6 et de laRecherche M6dicale CJF 9302, Centre Leon Berard, 28 rue Laennec, 69008 Lyon, France; tUniversity of The Witwatersrand, Johannesburg, South Africa;§Department of Internal Medicine, University of Heidelberg, Heidelberg, Federal Republic of Germany; and 1Deutsches Krebsforschungszentrum, Heidelberg,Federal Republic of Germany
Contributed by Kurt J. Isselbacher, September 20, 1993
ABSTRACT We studied 80 hepatocellular carcinomasfrom three continents for p53 gene (TP53) mutations andhepatitis B virus (HBV) sequences. p53 mutations were fre-quent in tumors from Mozambique but not in tumors fromSouth Africa, China, and Germany. Independent of geo-graphic origin, most tumors were positive for HBV sequences.X gene coding sequences of HBV were detected in 78% oftumors, whereas viral sequences in the surface antigen- andcore antigen-encoding regions were present in less than 45% oftumors. These observations indicate that hepatocellular carci-nomas are genetically heterogeneous. Mozambican-type ofhepatocellular carcinomas are characterized by a high inci-dence of p53 mutations related to aflatoxins. In other tumors,the rarity of p53 mutations combined with the frequent pres-ence of viral X gene coding sequences suggests a possibleinterference of HBV with the wild-type p53 function.
Hepatocellular carcinoma (HCC) is the most common cancerof the liver. Because most HCC cells display biochemical andmorphological features of normal hepatocytes, it is assumedthat HCC results from malignant transformation of normalhepatocytes, although a "stem cell" origin is also suspected(1-3). Etiologically, human HCC is a complex and multifac-torial disease that is linked to many agents such as viral andchemical carcinogens or to conditions such as chronic hepa-titis and cirrhosis (2, 4). Moreover, patients with HCC may beexposed simultaneously to multiple etiologic factors, depend-ing on their living conditions, which may vary between coun-tries or even within a single geographical location. It is notknown whether the genetic events that occur during thedevelopment of HCC are the same, similar, or distinct indifferent populations. In this study, we compared the natureand the frequency of genetic alterations in HCCs from threecontinents selected on the basis ofgeographic location, ethnicorigin, and life style. The nature and frequency of somaticmutations of the p53 gene (designated TP53 for tumor protein53) were compared with the presence of surface (S), core (C),and X gene sequences of hepatitis B virus (HBV) in tumorsfrom southern Africa, southeastern Asia, and Europe.
MATERIAL AND METHODSTumor Specimens. In a previous study, we reported on the
frequency of the AGG -* AGT mutation in codon 249 of thep53 gene in 167 HCCs obtained from 14 countries (5). In thepresent study, we investigated 80 of these tumors obtainedfrom patients living in Mozambique (n = 16), South Africa (n= 23), China (n = 28), and Germany (n = 13).
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Single-Strand Conformation Polymorphism (SSCP) Analysisof p53 Gene Mutations. The following forward (F) and reverse(R) primers were used for exons 5 + 6, F: 5'-(CGGGATCCCG) TTCCT CTTCC TGCAG TACTC-3' and R: 5'-(CGGAA TTCCG) AGTTG CAAAC CAGAC CTCAG-3';for exon 7, F: 5'-(CGGGA TCCCG) GTGTT GCCTCCTAGG TTGGC-3' and R: 5'-(CGGAA TTCCG) CAAGTGGCTC CTGAC CTGGA-3'; and for exons 8 + 9, F:5'-(CGGGA TCCCG) CCTAT CCTGA GTAGT GGTAA-3'and R: 5'-(CGGAA TTCCG) CCAAG ACTTA GTACCTGAAG-3'.PCR for SSCP was done in the presence of 25 ,M dNTPs
and 0.1 mCi (1 Ci = 37 GBq) of [32P]dCTP (3000 Ci/mmol,Amersham) per ml with 35 cycles of 94°C (1 min), 55°C (2min), and 72°C (3 min). PCR-amplified material was analyzedon 6% polyacrylamide gel in the presence of 10% (vol/vol)glycerol, followed by autoradiography. For nucleic acidsequencing, PCR products were prepared in the presence of250 ,uM dNTP, digested with EcoRI and BamHI, and ligatedto pBluescript SK vector. At least five different plasmidsfrom each sample were sequenced to confirm mutations.PCR Analysis of HBV DNA Sequences in Tumor DNAs.
HBV DNA sequences were analyzed by separate amplifica-tion of S, C, andX gene coding regions by using primers andconditions as described by Liang et al. (6) and Paterlini et al.(7). In addition to general rules to be respected when using thePCR technique (8), other measures were taken. All DNAsamples were analyzed separately at least three times foreach region of the virus by Southern blotting of PCR-amplified material. Genomic DNAs from Hep G2 cells (9) andHep G2/2.2.1.5 cells (10) were systematically used as nega-tive and positive controls, respectively.
RESULTSEighty HCCs obtained from four distant geographic locationswere studied for the status of the p53 gene and HBV DNA.Sixteen tumors were from Mozambique, a country with thehighest recorded incidence of HCC in the world. Chronicinfection with HBV and oral exposure to aflatoxins are majorrisk factors in Mozambique (11-13). Twenty-three tumorswere from South Africa where the incidence of HCC is highand the major risk factor is chronic infection with HBV (13).Twenty-eight of the tumors were from different geographiclocations in China where the overall incidence ofHCC is highand chronic infection with hepatitis B virus is the major riskfactor (14). Oral exposure to aflatoxins is also an important
Abbreviations: HCC, hepatocellular carcinoma; HBV, hepatitis Bvirus; HCV, hepatitis C virus; HBsAg, HBxAg, and HBcAg, HBVsurface, X, and core antigens; S, C, and X genes, HBV genesencoding these antigens; SSCP, single-strand conformation poly-morphism; wt, wild type.IlTo whom reprint requests should be addressed.
822

Proc. Natl. Acad. Sci. USA 91 (1994) 823
Table 1. Descriptive list of the status of p53 and HBV DNA in HCCs from Mozambique, South Africa, China, and GermanyChange
Amino HBV genep53 Mut. Nucleic acid acid
Val-}+PheArg--SerArg--SerArg--SerArg-.SerArg--SerArg--SerArg-.SerArg--Ser
AGG-+AGT Arg--*Ser8 bp-del. Frameshift
GTC--TTC Val-.PheAGG-*AGT Arg-*Ser
S C X Any- - + +
++ - - +_ - + +_ - + +
++ + ++ +
++ + ++ +++ + ++ +- + ++ +
+ - ++ +++ - + +_ - + +
_ - + +
++ + + +
+++- +
++ ++ ++ +
_ - + +
_ _ + +
_ - + +
++ ++ + +
++ ++ + +NT NT NT NTNT NT NT NT
DNA(country) p53 Mut.ClO (C) 249C4 (C) 251C14 (C) 281C25 (C) wtC26-1 (C) wtC12(C) wtC2(C) wtC28-1 (C) wtC5(C) wtC18(C) wtC22 (C) wtC9 (C) wtC13 (C) wtC17(C) wtC6 (C) wtC7(C) wtC8(C) wtCll(C) wtC16 (C) wtC20 (C) wtC23 (C) wtC24 (C) wtC29-1 (C) wtC3 (C) wtC1 (C) wt(E5-7)C15 (C) wt(E7-9)G6 (G) 137T78 (G) 273G4 (G) 273T75(G) wtT80 (G) wtT82 (G) wtG1(G) wtG2(G) wtGS (G) wt(E5-7)G7 (G) wt(E7-9)T83 (G) wt(E7-9)T73 (G) wt(E7-9)T74 (G) wt(E7-9)
ChangeAmino
Nucleic acid acidAGG- 'AGTATC del.
GAC-+GAA
Arg-.Ser1 aa del.Asp-.Glu
CTG-+CTA Leu-*LeuCGT--TGT Arg-+CysCGT--*TGT Arg--'Cys
HBV gene
S C X Any_ ++ +
+ - ++ +
+ + ++ +
NT NT NTNT NT NT+ + ++
NTNT+
++ + ++ +
NT NT NT NT_
+ +
NT NT NT NTNT NT NT NT+ + + +
+ + ++ +
NT NT NT NT_ ++ +
+ + ++ +
++ + ++ +
+ ++ +
NT NT NT NTNT NT NT NT++ + ++ +
NT NT NT NTNT NT NT NT
_ ++ +
- - NT NT
+ +
NT NT NT NT
_ + +
++++ ++ +
+ + ++ +
NT ++ ++ +
NT + NT +
_ ++ +
_ _ + +
p53 mutations were studied by SSCP analysis followed by nucleic acid sequencing of exons 5 to 9 as described in the text. Geographic originand the race of patients from South Africa are shown in parenthesis. All but six mutations (Mut.) have been previously reported in refs. 5 and18. Mutations described for the first time are those found at codons 157, 251, and 281 in tumors from China and those found at codons 137 and273 (two cases) in tumors from Germany. wt, wild-type p53 sequences between exons 5 and 9; wt(E5-7), wt(E7), and wt(E7-9), wt p53 sequencesin exons 5-7, 7, and 7-9, respectively. The presence of HBV in tumors was tested by PCR with three sets of primers allowing amplificationof the C (core) gene, the S (surface) gene (including pre-SI, pre-S2, and S), and the X gene repetitively. The values reported in the last column(Any HBV DNA) were derived from the compilation of the columns HBV S, C, and X. NT, Not tested or no data available; aa, amino acid;-, absence of repetitively detectable DNA sequences; + and + +, samples with low and high levels of detectable DNA sequences, respectively;del., deletion; M, Mozambique; SA-T, South Africa (Transkei); SA-C, South Africa (Caucasian); SA-L, South Africa (Lesotho); SA-N, SouthAfrica (Natal); C, China; G, Germany.
risk factor in distinct geographic locations within Chinawhere the incidence of HCC sharply increases (15, 16). Thelast group of tumors was from Germany (n = 13) with a lowincidence ofHCC (14), low risk ofexposure to aflatoxins, andlow rate of chronic infection with HBV (17). Another knownrisk factor, infection with hepatitis C virus (HCV), mayadditionally contribute to the development ofHCC in all fourcountries studied. There is, however, no clear evidencedemonstrating a major difference between Mozambique,South Africa, China, and Germany in the rates of chronicinfection with hepatitis C virus.p53 Mutations in HCC. Table 1 describes individually the
state of p53 and HBV DNA in 80 tumors studied. Completeanalysis of exons 5-9 of the p53 gene was possible with 60
tumors; 32% had a mutation. Both the frequency and thespectra ofp53 mutations varied geographically. Ofthe tumorsfrom Mozambique, 69o showed a "band shift" in SSCPassay. All of the p53 mutations corresponded to previouslyreported guanine-to-thymine (G -* T) transversions; eight, toan arginine-to-serine amino acid change at codon 249; andone, to a valine-to-phenylalanine change at codon 157 (5, 18).Of 13 tumors from South Africa, 15% (2) had mutations, bothof which were reported previously (5, 18): a G -+ T trans-
version at codon 249 leading to an arginine-to-serine changeand a 8 base pair (bp) deletion starting at codon 286 andleading to a frameshift. Of26 tumors from China, 19% (5) hadp53 mutations; two had G -* T transversions at codon 249
changing arginine to serine as reported previously (5), one
GTC--TTCAGG--oAGTAGG-+AGTAGG--AGTAGG---AGTAGG-*AGTAGG-+AGTAGG-*AGTAGG-+AGT
DNA(country)
T47 (M)T1S (M)K3T (M)KilT (M)K15T (M)T27 (M)T29 (M)T37 (M)T55 (M)T13 (M)T51 (M)T53 (M)T9(M)K8T (M)K9T (M)K17T (M)K7T (SA-T)T43 (SA-T)T25 (SA-T)T31 (SA-T)T33 (SA-T)T23 (SA-T)T12 (SA-T)T19 (SA-T)T49 (SA-C)T41 (SA-L)T39 (SA-S)T8 (SA-L)T17 (SA-L)T35 (SA-T)K16T (SA-C)KiT (SA-N)K2T (SA-N)K5T (SA-N)K12T (SA-N)KiOT (SA-C)K6T (SA-T)K13T (SA-T)C19 (C)C21 (C)
157249249249249249249249249wtwtwtwt
wt(E7)wt(E7)wt(E7)249286wtwtwtwtwtwtwtwtwtwtwt
wt(E5-7)wt(E7-9)wt(E7-9)wt(E7)wt(E7)wt(E7)wt(E7)wt(E7)wt(E7)157249
Medical Sciences: Unsal et al.

824 Medical Sciences: Unsal et al.
Table 2. Comparison of p53 status with clinicopathological dataof HCCs from Mozambique and South Africa
Mutant p53/TotalHCC tissue Southcomparisons Mozambique Africa Total (%)
Serum HBsAgNegative 6/8 0/5 6/13 (46)Positive 3/5 2/6 5/11 (45)
Liver histologyNo cirrhosis 5/6 1/9 6/15 (40)Cirrhosis 3/6 0/3 3/9 (33)
Tumor histologyAcinar 0/1 0/1 0/2 (0)Trabecular 4/6 0/8 4/12 (33)Solid 2/3 1/1 3/4 (75)
Tumor differentiationWD 0/3 0/6 0/9 (0)MWD 4/5 0/3 4/8 (50)PD 2/2 1/2 3/4 (75)
Tumor sizeSmall 2/2 0/4 2/6 (33)Large 7/11 1/8 8/19 (42)
MetastasisNo 4/7 0/7 4/14 (29)Yes 2/3 0/3 2/6 (33)Serum HBV surface antigen (HBsAg) status was determined as
reported (18). Differentiation is indicated as well differentiated (WD),moderately well differentiated (MWD), and poorly differentiated(PD) tumors. Massive tumors and tumors larger than 10 cm wereclassified as Large. Metastasis always refers to distant metastasis/metastases.
had a G -+ T transversion at codon 157 changing valine tophenylalanine, another had a G -- T transversion at codon281 changing aspartic acid to glutamic acid, and the fifthmutation was a 3-bp inframe deletion of codon 251 (ATCdeleted), resulting in the loss of the isoleucine residue. Of 8tumors from Germany, 37.5% (3) displayed mutations: onewas a silent G -- A mutation at codon 137 (detected only ina minor fraction oftumor DNA), and two were missense CT mutations at codon 273, changing arginine to cysteine.The relationship between p53 gene mutations and clinico-
pathological features was studied in HCCs from Mozambiqueand South Africa. As summarized in Table 2, there was nocorrelation between p53 mutations and cirrhosis, tumor size,metastasis, or the presence of HBsAg in patient sera. How-ever, there was a good correlation between p53 mutationsand tumor cell differentiation. None of nine well-differenti-ated tumors had a p53 mutation. In contrast, p53 mutationswere found in 50% and 75% of moderately and poorlydifferentiated tumors, respectively.HBV DNA in HCC. Sixty-nine tumors were analyzed for
HBV DNA sequences by PCR. Viral DNA sequences weredetectable in 79% of completely analyzed tumors (Table 1).HBV has been rarely found as a replicative complete virus inHCCs because viral DNA integrates into host genome andundergoes multiple rearrangements during tumor develop-ment (19). In this study, it was not possible to analyze directlythe structure of viral DNA in tumors. But the analysis ofthreeseparate sequences ofthe viralgenome demonstrated that only34% of tumors contained all three viral genes studied, sug-gesting that most tumors contained only partial or rearrangedviral DNA sequences. Therefore, it was important to knowwhich viral genes were retained in the tumor cells. Viral DNAsequences coding for HBsAg and core antigen (HBcAg) werefound in 45% and 42% of tumors, respectively. In contrast tothese two regions, X gene sequences were found at very highfrequency (78%). Xgene codes fora small protein of 154 aminoacids, whereas C and S genes code for larger polypeptides (20,
21). Thus, the probability of a random loss of HBcAg- andHBsAg-encoding sequences from the integrated viral DNA ishigh. Or alternatively, X gene, which is known to display alimited transforming activity, may be selectively retained bytumor cells during tumor development.We also analyzed our data according to the geographic
origin of the tumors. Oftumors from Mozambique, 87.5% haddetectable HBV DNA sequences. X gene sequences weredetectable in 81% of tumors, in contrast to C and S genes,which were detectable in 31% and 37.5% of tumors, respec-tively. OfHCCs from South Africa, 65% had detectable HBVsequences; 61% of tumors had X sequences, but C and Ssequences were present in 39%o and 48% of tumors, respec-tively. All of the tumors from China had detectable X genesequences. S and C gene sequences were found with equalfrequency (62.5%). Surprisingly, a large fraction (67%) oftumors from Germany also had detectable HBV DNA. Thiswas due mostly to the detection ofX gene sequences in 64%of tumors. In contrast, only 20% of tumors had detectable Sgene sequences, whereas C sequences were detected in 33%of tumors.Comparative Analysis of p53 and HBV Status in Tumors.
Feitelson et al. (22) have recently reported on complexformation between p53 protein and the X antigen of HBV(HBxAg), similar to interactions ofp53 with the E6 protein ofhuman papillomaviruses (23-25). Therefore, it was importantto compare the status of p53 gene in X-positive and X-neg-ative HCCs. p53 mutations (one silent) were detected in 27%of X-negative and 33% of X-positive tumors, respectively,suggesting the absence of direct relation between the twofactors studied. It is, however, important to emphasize that,in HBV-related HCCs, viral integration is an early event (19,26), whereas p53 mutations appear to occur later on, duringtumor progression (27). A rare exception to this situationcould be seen in patients exposed to both aflatoxins andHBV, where aflatoxins as powerful mutagens could causemutations in the p53 gene in parallel to viral integration at anearly stage ofHCC. In this regard, we noticed that, in tumorsfrom Mozambique and South Africa, p53 mutations weredetected in both early and late tumors with similar frequen-cies (Table 2). Furthermore, 74% of mutations in our serieswere G -> T transversions detected mostly in high-aflatoxinareas (Fig. 1). Ifwe separate this type of mutation apart fromthe analysis, the overall frequency of p53 mutations drops to10%. The copresence of HBV X DNA with the wt p53 genein a high proportion of these tumors may indicate an inverserelation between viral integration and mutational inactivationof the p53 gene (Fig. 1). In support of this hypothesis, two offour X-negative HCCs from Germany had a p53 mutation(one being silent), whereas all of three X-positive HCCs hadwt p53 sequences (Table 1).
DISCUSSIONDuring the past decade, systematic analysis of geneticchanges in different tumors has made it possible to uncovermolecular pathways leading to malignant transformation ofdifferent cell types in the human body (28). It appears fromthese studies that major differences in the susceptibility ofdifferent cells to malignant transformation are related tospecific changes in the expression of their genetic program.This is probably true for most human cancers. However, alarge body of data also indicates that tumors of the same cellorigin may differ in terms of their epidemiological, clinical,and pathologic manifestations. HCC is one such tumor thatshows striking differences according to its geographic loca-tion. Our major goal was to study whether phenotypicdifferences between tumors of the same cellular origin wereassociated with distinct genetic changes.
Like other solid tumors, HCCs appear to occur as a resultof multiple structural and functional changes affecting cellu-
Proc. Natl. Acad. Sci. USA 91 (1994)
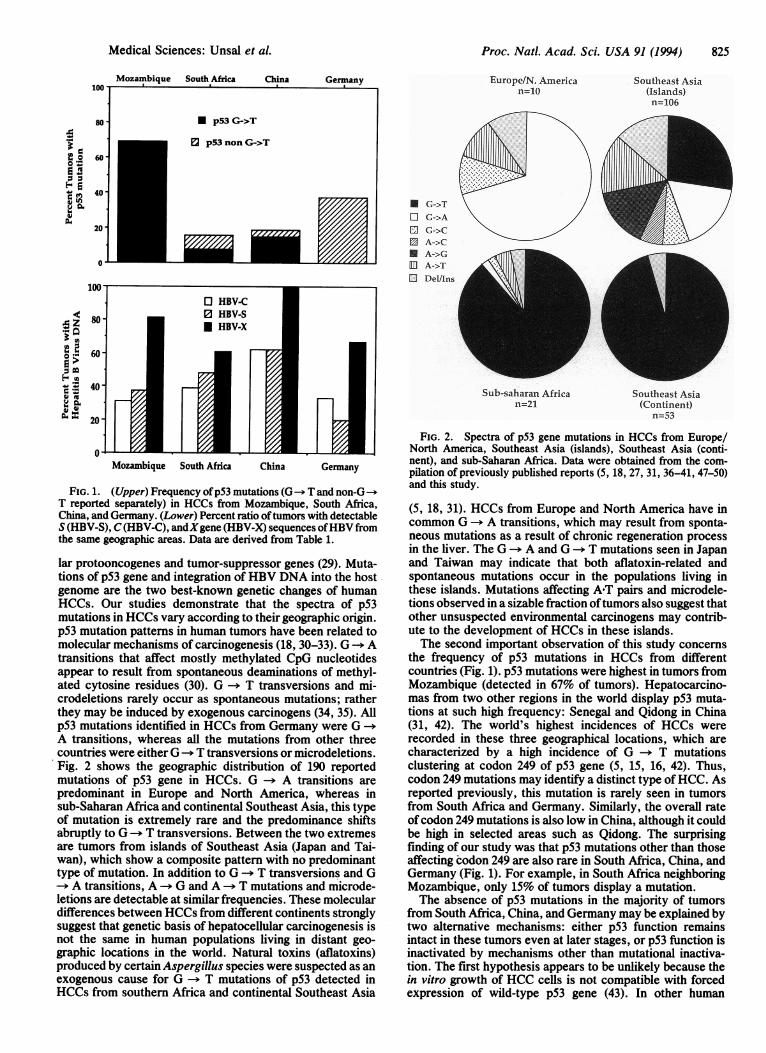
Proc. Natl. Acad. Sci. USA 91 (1994) 825
Europe/N. American=10
U
IDU
S<(HBVS),(HV-C)andgen0 HBV-Stions 80p53geneandintegratHBV-X
60
40
20
0Mozambique South Africa China Germany
FIG. 1. (Upper) Frequency of p53 mutations (G T and non-GT reported separately) in HCCs from Mozambique, South Africa,China, and Germany. (Lower) Percent ratio oftumors with detectableS (HBV-S), C(HBV-C), andrXgene (HBV-X) sequences ofHBV fromthe same geographic areas. Data are derived from Table 1.
lar protooncogenes and tumor-suppressor genes (29). Muta-tions of p53 gene and integration of HBV DNA into the hostgenome are the two best-known genetic changes of humanHCCs. Our studies demonstrate that the spectra of p53mutations in HCCs vary according to their geographic origin.p53 mutation pattens in human tumors have been related tomolecular mechanisms of carcinogenesis (18, 30-33). G --* Atransitions that affect mostly methylated CpG nucleotidesappear to result from spontaneous deaminations of methyl-ated cytosine residues (30). G -- T transversions and mi-crodeletions rarely occur as spontaneous mutations; ratherthey may be induced by exogenous carcinogens (34, 35). Allp53 mutations identified in HCCs from Germany were G -aA transitions, whereas all the mutations from other threecountries were either G-a T transversions or microdeletions.Fig. 2 shows the geographic distribution of 190 reportedmutations of p53 gene in HCCs. G-t A transitions arepredominant in Europe and North America, whereas insub-Saharan Africa and continental Southeast Asia, this typeof mutation is extremely rare and the predominance shiftsabruptly to G -+) T transversions. Between the two extremesare tumors from islands of Southeast Asia (Japan and Tai-wan), which show a composite pattern with no predominanttype of mutation. In addition to G --* T transversions and G--> A transitions, A --* G and A --> T mutations and microde-letions are detectable at similar frequencies. These moleculardifferences between HCCs from different continents stronglysuggest that genetic basis of hepatocellular carcinogenesis isnot the same in human populations living in distant geo-graphic locations in the world. Natural toxins (aflatoxins)produced by certain Aspergillus species were suspected as anexogenous cause for G --+ T mutations of p53 detected inHCCs from southern Africa and continental Southeast Asia
Southeast Asia(Islands)n=106
G->TG->AG->CA->CA->GA->TDel/ins
Sub-saharan Africa Southeast Asian=21 (Continent)
n=53
FIG. 2. Spectra of p53 gene mutations in HCCs from Europe/North America, Southeast Asia (islands), Southeast Asia (conti-nent), and sub-Saharan Africa. Data were obtained from the com-pilation of previously published reports (5, 18, 27, 31, 36-41, 47-50)and this study.
(5, 18, 31). HCCs from Europe and North America have incommon G -- A transitions, which may result from sponta-neous mutations as a result of chronic regeneration processin the liver. The G -+ A and G -* T mutations seen in Japanand Taiwan may indicate that both aflatoxin-related andspontaneous mutations occur in the populations living inthese islands. Mutations affecting A-T pairs and microdele-tions observed in a sizable fraction oftumors also suggest thatother unsuspected environmental carcinogens may contrib-ute to the development of HCCs in these islands.The second important observation of this study concerns
the frequency of p53 mutations in HCCs from differentcountries (Fig. 1). p53 mutations were highest in tumors fromMozambique (detected in 67% of tumors). Hepatocarcino-mas from two other regions in the world display p53 muta-tions at such high frequency: Senegal and Qidong in China(31, 42). The world's highest incidences of HCCs wererecorded in these three geographical locations, which arecharacterized by a high incidence of G -- T mutationsclustering at codon 249 of p53 gene (5, 15, 16, 42). Thus,codon 249 mutations may identify a distinct type ofHCC. Asreported previously, this mutation is rarely seen in tumorsfrom South Africa and Germany. Similarly, the overall rateof codon 249 mutations is also low in China, although it couldbe high in selected areas such as Qidong. The surprisingfinding of our study was that p53 mutations other than thoseaffecting codon 249 are also rare in South Africa, China, andGermany (Fig. 1). For example, in South Africa neighboringMozambique, only 15% of tumors display a mutation.The absence of p53 mutations in the majority of tumors
from South Africa, China, and Germany may be explained bytwo alternative mechanisms: either p53 function remainsintact in these tumors even at later stages, or p53 function isinactivated by mechanisms other than mutational inactiva-tion. The first hypothesis appears to be unlikely because thein vitro growth of HCC cells is not compatible with forcedexpression of wild-type p53 gene (43). In other human
Medical Sciences: Unsal et al.

826 Medical Sciences: Unsal et al.
tumors, there are examples of p53 protein inactivation bycomplex formation with products of cellular (mdm2) and viral(E6 gene of human papillomavirus) oncogenes (44). Thepresence of these oncogenes might therefore be sufficient tounlock p53-mediated growth control in the absence of astructural alteration in the p53 gene. Accordingly, p53 genemutations were found to be rare or absent in mdm2-positiveosteosarcomas and papillomavirus-positive cervical cancers(23-25, 44). It is not known whether HCC cells expresscellular transforming proteins capable of inactivating the p53protein, but HBxAg appears to form complexes with p53protein (22). In this regard, our comparative analysis failed toreveal any striking correlation between the presence of Xgene sequences and the absence of p53 mutations in HCCs.Although p53 mutations were infrequent and X gene se-quences were present in a high proportion of HCCs, we diddetect p53 mutations in X DNA-positive tumors. However,these observations are not sufficient to dismiss a possible roleof HBxAg in the inactivation of p53 protein. Indeed, p53mutations were occasionally found in papillomavirus-positive cervical cancers (45). Interestingly, all of the p53mutations detected in papillomavirus-positive cervical can-cers were missense mutations resulting in the expression offaulty proteins. Such mutant p53 proteins may display "gainof function," in addition to the loss of wild-type p53 function(45). In this regard, 10 of 12 p53 mutations that we detectedinXDNA-positive HCCs were missense mutations (Table 1).Two remaining mutations were deletions. One of the dele-tions resulted in the loss of a single amino acid at codon 251.In the tumor displaying this mutation, increased levels of amutant p53 protein were detected by Western immunoblot-ting (data not shown). The other deletion caused a frame-shiftat codon 286 which should lead to the synthesis ofa truncatedp53 protein lacking 107 amino acids at the carboxyl terminus(Table 1). Therefore, it remains possible that in HBV-relatedHCC, wild-type p53 function is inactivated by a viral protein(i.e., HBxAg) and that "gain of function" mutations mayoccur during tumor progression.HCV is a newly identified RNA virus associated with
non-A, non-B hepatitis and HCC (46). As an RNA virus, HCVis unlikely to disturb the host genome in a way similar to HBV.It is also unknown whether HCV carries any transformingprotein. In this study, we did not analyze our tumors for thepresence ofHCV RNA. However, our study ofp53 mutationsand HBV DNA was sufficiently informative in demonstratingthe genetic heterogeneity of HCCs with different etiologies.Genetic heterogeneity in tumors from the same cell origin maybe a more common feature ofhuman cancers, as suggested bytheir clonality and phenotypic heterogeneity.H.U. and C.Y. have equally contributed to this work. This work
was supported by grants from the National Institutes of Health(CA-54567), the Departemental Commitees of AIN and RHONE ofthe Ligue de Lutte Contre le Cancer, and the TumorzentrumHeidelberg/Mannheim (Grant I/I.2). C.M. was supported by afellowship from the Fondation pour la Recherche Medicale.
1. Craig, J. R., Peters, R. L. & Edmondson, H. A. (1989) Atlas ofTumor Pathology, Second series, Fascicle 26 (Armed Forces Inst.Pathol., Washington, DC).
2. Okuda, K. (1992) Hepatology 15, 948-963.3. Sell, S. (1993) Int. J. Dev. Biol. 37, 189-201.4. Beasley, R. P. (1982) Hepatology 2, 21S-26S.5. Ozturk, M., Bressac, B., Puisieux, A., Kew, M., Volkmann, M., et
al. (1991) Lancet 338, 1356-1359.6. Liang, T. J., Blum, H. E. & Wands, J. R. (1990) Hepatology 12,
204-212.7. Paterlini, P., Gerken, G., Nakajima, E., Terre, S., D'Errico, A.,
Grigioni, W., Nalpas, B., Franco, D., Wands, J., Kew, M., Pisi, E.,Tiollais, P. & Brechot, C. (1990) N. Engl. J. Med. 323, 80-85.
8. Wright, D. K. & Manos, M. M. (1990) inPCR Protocols, eds. Innis,M. A., Gelfand, D. H., Sninsky, J. J. & White, T. J. (Academic,San Diego), pp. 153-158.
9.
10.
11.
12.
13.
14.
15.
16.17.
18.
19.
20.
21.22.
23.
24.
25.
26.
27.
28.29.30.
31.
32.
33.
34.35.
36.
37.
38.
39.
40.
41.
42.
43.44.45.46.
47.
48.
49.
50.
Aden, D. P., Fogel, A., Plotkin, S., Damjanov, I. & Knowles, B. B.(1979) Nature (London) 282, 615-616.Sells, M. A., Chen, M. L. & Acs, G. (1987) Proc. Natl. Acad. Sci.USA 84, 1005-1009.Dusheiko, G. M., Brink, B. A., Conradie, J. D., Marimuthu, T. &Sher, R. (1989) Am. J. Epidemiol. 129, 138-145.Van Rensburg, S. J., Cook-Mozaffari, P., Van Schalkwyk, D. J.,Van Der Watt, J. J., Vincent, T. J. & Purchase, I. F. (1985) Br. J.Cancer 51, 713-726.Kew, M. C. (1984) in Advances in Hepatitis Research, ed. Chisari,F. V. (Masson, New York), pp. 203-215.Parkin, D. M., Laara, E. & Muir, C. S. (1988) Int. J. Cancer 41,184-197.Yu, S.-Z. (1985) in Subclinical Hepatocellular Carcinoma, ed.Tang, Z.-Y. (China Acad. Publishers, Beijing and Springer, Berlin),pp. 189-212.Yeh, F.-S. & Shen, K.-N. (1986) Adv. Cancer Res. 47, 297-329.Langa, W. & Masihi, K. N. (1987) Med. J. (Engl. Transl.) 63 (S2),21-26.Bressac, B., Kew, M., Wands, J. & Ozturk, M. (1991) Nature(London) 350, 429-431.Matsubara, K. (1991) in MolecularBiology ofHepatitis B Virus, ed.McLachlan, A. (CRC, Boca Raton, FL).Robinson, W. S., Klote, L. & Aoki, N. (1990) J. Med. Virol. 31,18-32.Robinson, W. S. (1992) J. Gastroenterol. Hepatol. 7, 622-638.Feitelson, M. A., Zhu, M., Duan, L. X. & London, W. T. (1993)Oncogene 8, 1109-1117.Scheffner, M., Werness, B. A., Huibregtse, J. M., Levine, A. J. &Howley, P. (1990) Cell 63, 1129-1136.Scheffner, M., Munger, K., Bryne, J. C. & Howley, P. (1991) Proc.Natl. Acad. Sci. USA 88, 5523-5527.Crook, T., Wrede, D., Tidy, J. A., Mason, W. P., Evans, D. &Vousden, K. H. (1992) Lancet 339, 1070-1073.Takada, S., Gotoh, Y., Hayashi, S., Yoshida, M. & Koike, K.(1990) J. Virol. 64, 822-828.Tanaka, S., Toh, Y., Adachi, E., Matsumata, T., Mori, R. &Sugimachi, K. (1993) Cancer Res. 53, 2884-2887.Vogelstein, B. & Kinzler, K. W. (1993) Trends Genet. 9, 138-141.Ozturk, M. (1994) CRC Crit. Rev., in press.Rideout, W. M., Coetzee, G. A., Olumi, A. F. & Jones, P. A.(1990) Science 249, 1288-1290.Hsu, I. C., Metcalf, R. A., Sun, T., Welsh, J. A., Wang, N. J. &Harris, C. C. (1991) Nature (London) 350, 427-428.Hollstein, M., Sidransky, D., Vogelstein, B. & Harris, C. C. (1991)Science 253, 49-53.Puisieux, A., Lim, S., Groopman, J. & Ozturk, M. (1991) CancerRes. 51, 6185-6189.Miller, J. H. (1983) Annu. Rev. Genet. 17, 215-238.Harwood, J., Tachibana, A. & Meuth, M. (1991) Mol. Cell. Biol. 11,3163-3170.Buetow, K. H., Sheffield, V. C., Zhu, M., Zhou, T., Shen, F.-M.,Hino, O., Smith, M., McMahon, B. J., Lanier, A. P., London,W. T., Redeker, A. G. & Govindarajan (1992) Proc. Natl. Acad.Sci. USA 89, 9622-9626.Challen, C., Lunec, J., Warren, W., Collier, J. & Bassendine, M. F.(1992) Hepatology 6, 1362-1366.Hosono, S., Chou, M. J., Lee, C. S. & Shih, C. (1993) Oncogene 8,491-496.Kress, S., Jahn, U. R., Buchmann, A., Bannasch, P. & Schwarz,M. (1992) Cancer Res. 52, 3220-3223.Li, D., Cao, Y., He, L., Wang, N. J. & Gu, J. R. (1993) Carcino-genesis 14, 169-173.Murakami, Y., Hayashi, K., Hirohashi, S. & Sekiya, T. (1991)Cancer Res. 51, 5520-5525.Coursaget, P., Depril, N., Chabaud, M., Nandi, R., Mayelo, V.,LeCann, P. & Yvonnet, B. (1993) Br. J. Cancer 67, 1395-1397.Puisieux, A., Ponchel, F. & Ozturk, M. (1993) Oncogene 8, 487-490.Vogelstein, B. & Kinzler, K. W. (1992) Cell 70, 523-526.Crook, T. & Vousden, K. H. (1992) EMBO J. 11, 3935-3940.Choo, Q., Kuo, C., Veiner, A., Overby, L., Bradley, D. & Hough-ton, M. (1989) Science 244, 359-362.Nishida, N., Fukuda, Y., Kokuryu, H., Toguchida, J., Yandell,D. W., Ikenega, M., Imura, H. & Ishizaki, K. (1993) Cancer Res.53, 368-372.Oda, T., Tsuda, H., Scarpa, A., Sakamoto, M. & Hirohashi, S.(1992) Cancer Res. 52, 6358-6364.Scorsone, K. A., Zhou, Y. Z., Butel, J. S. & Slagle, B. L. (1992)Cancer Res. 52, 1635-1638.Sheu, J. C., Huang, G. T., Lee, P. H., Chung, J. C., Chou, H. C.,Lai, M. Y., Wang, J. T., Lee, H. S., Shih, L. N., Yang, P. M.,Wang, T. H. & Chen, D. S. (1992) Cancer Res. 52, 6098-6100.
Proc. Natl. Acad. Sci. USA 91 (1994)