genesis of the csiro polymer group and the discovery and significance of nitroxide-mediated living...
TRANSCRIPT

HIGHLIGHT
Genesis of the CSIRO Polymer Group and the Discoveryand Significance of Nitroxide-Mediated Living RadicalPolymerization
DAVID H. SOLOMONDepartment of Chemical & Biomolecular Engineering, University of Melbourne,Parkville, Vic 3010, Australia
Received 2 August 2005; accepted 5 August 2005DOI: 10.1002/pola.21067Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: The background to
the formation of the Commonwealth
Scientific and Industrial Research
Organization (CSIRO) polymer
group is discussed. In particular,
the challenges of working with high-
conversion polymerization, as found
in commercial systems, and the need
to explain variations in polymer
properties led to important advan-
ces in the theory of radical poly-
merization and control over both
the initiation and termination steps.
Studies on the fate of the macro-
monomer, formed in termination
by disproportionation, led to an
early form of addition/fragmenta-
tion now known as reversible addi-
tion–fragmentation chain transfer,
whereas detailed studies on initia-
tion pathways using nitroxide trap-
ping led to nitroxide-mediated liv-
ing radical polymerization. These
studies contributed to the renais-
sance in free-radical polymerization
studies. VVC 2005 Wiley Periodicals, Inc. J
Polym Sci Part A: Polym Chem 43: 5748–
5764, 2005
Keywords: initiation; nitroxide
mediated living radical polymeriza-
tion (NMP); radical trapping and
kinetic control
David H. Solomon is currently a professorial fellow at the University
of Melbourne, where he was previously the head of the School of
Chemistry. Before that, he was the foundation chief of the Common-
wealth Scientific and Industrial Research Organization (CSIRO) Divi-
sion of Applied Organic Chemistry (now Molecular Science), where he
established and led the polymer research group. His original employ-
ment at Dulux Paints was the foundation and pathway for his interest
in polymer science.
In 1990, he accepted an invitation to a Chair at the University of
Melbourne, where he formed the polymer research group.DAVID H. SOLOMON
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 43, 5748–5764 (2005)VVC 2005 Wiley Periodicals, Inc.
Correspondence to: D. H. Solomon (E-mail: [email protected])
5748

INTRODUCTION
In the 1960s and early 1970s, radical polymerization
was considered a mature science; the majority of poly-
mer scientists accepted that the definitive texts had
been written and that the detailed mechanisms for ini-
tiation, propagation, and termination were understood.
One leading researcher, Mayo,1 even went so far as to
suggest, in an article entitled Whither Free Radical Poly-merization, that
Those who want to pioneer on the borders of
polymer science will probably find better opportu-
nities than in free-radical polymerization.
Contrast this state of affairs with the renaissance in
radical polymerization that began in the late 1970s: today,
radical polymerization, particularly living polymeriza-
tion, is one of the most active areas in polymer science.
In this article, I will review the work carried out in
the Commonwealth Scientific and Industrial Research
(CSIRO) polymer group, which I formed in the Division of
Applied Organic Chemistry (later Chemicals and Polymers
and now Molecular Science) in 1974. In particular, I will
discuss how our work contributed to the significant change
in the approach to the theory of radical polymerization
(kinetic vs thermodynamic control), studies on macromo-
nomer copolymerization, which gave an early form of what
is now called reversible addition–fragmentation chain
transfer (RAFT), and the use of nitroxides as trapping
agents, which in turn led to nitroxide-mediated living
radical polymerization (NMP).
I will also indicate how the strong influence of
working with commercial polymer systems led to stud-
ies not normally undertaken in academic research.
BACKGROUND
I was fortunate to spend the early part of my career
(1959–1960) in the surface coating industry, and I was
actively researching new polymers at the time vinyl
and acrylic monomers became commercially available.
At that time, the choice of initiators for radical poly-
merizations was largely dictated by cost, critical tem-
perature, and availability. The different chemical end-
group structures expected from the individual initiators
were not considered significant; after all, they repre-
sented less than 0.2% of the polymer.2
He was appointed to the Order of Australia in 1990 for his services
to science and for his invention of the plastic banknote.
Prof. Solomon holds Ph.D. and D.Sc. degrees from the University
of New South Wales and a diploma in chemistry from the Sydney
Technical College.
He has worked across the field of polymer science and published
reference books on Step-Growth Polymerization, The Chemistry of
Organic Film Formers, The Chemistry of Pigments and Fillers, and The
Chemistry of Free-Radical Polymerization. The last was a significant
contribution to our present views on radical polymerization. In
addition, he has published 253 papers and holds 43 patents, includ-
ing that which describes the original nitroxide-mediated living
radical polymerization.
He has always been active in the Royal Australian Chemical Insti-
tute, being national president in 1979–1980. He was a prime mover
in the formation of the polymer division of the institute and was
foundation secretary and then chairman in 1966–1967.
Prof. Solomon is a Fellow of the Royal Society (London), the Australian
Academy of Science, the Australian Academy of Technological Sciences
and Engineering, and the Royal Australian Chemical Institute. He was
the sole recipient of the Australian Bicentennial Science Achievement
Award for his role in the invention and then commercialization
of the world’s first plastic banknote with greater security against
counterfeiting.
HIGHLIGHT 5749
Hence polymers were represented by general for-
mula 1:
This was satisfactory when physical properties were
evaluated but, as I will show, was not adequate when
chemical properties were considered.
From practical observations of poly(methyl meth-
acrylate) (PMMA) and polystyrene (PS), I knew that
polymers of the same molecular weight within each
series showed significant differences in their resistance
to weathering and that these differences could be
related to the polymerization conditions, including the
choice of initiator. Others had published similar obser-
vations.
There was growing evidence that anomalous ordefect structures could be formed at each of the initia-
tion, propagation, and termination steps. Examples arereported in our book2 and in the listed references.3,4 I
will illustrate the significant effect of one defect group
per polymer chain by using the example of PMMA.Radical-polymerized PMMA is less thermally stable
than a similar molecular weight polymer prepared byan anionic mechanism. Obvious differences between
the two polymers are the groups formed in the termi-
nation step: the head–head linkage and the unsaturatedend group in the radical polymer. We later showed that
such groupings or defects are the weak links at which
depolymerization is initiated in the radical polymer, as
reported by Cacioli et al.5 and Moad et al.6
There was also building evidence both from obser-
vations of polymers and from model systems that pro-vided strong grounds for questioning the exclusivity of
each of the steps in the traditional radical mechanism.
Of equal importance was the growing recognition thatstereoelectronic and polar effects were the dominant
factors in determining the outcome of radical reactions(kinetic control), not the product radical stability (ther-
modynamic control). I published on this as early as
1975.7 Our work on the structure of the cyclopolymersformed from 1,6-diallylamines was to prove the ex-
treme case demonstration that radical stability was notthe dominant factor.
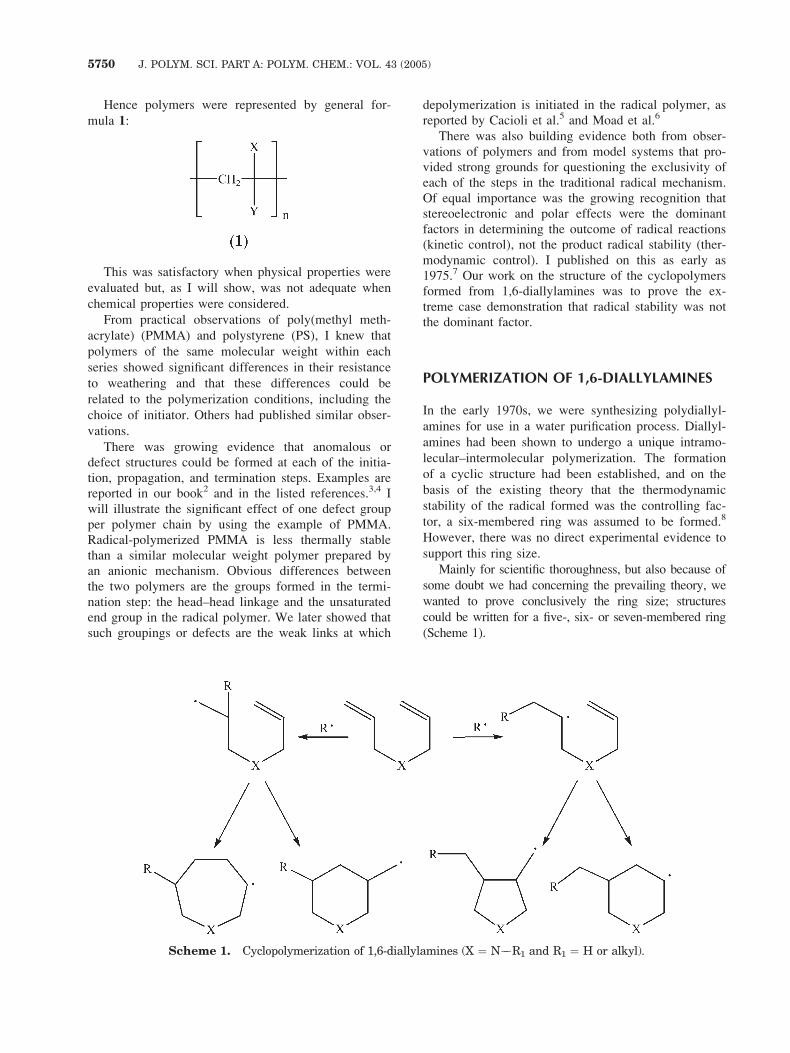
POLYMERIZATION OF 1,6-DIALLYLAMINES
In the early 1970s, we were synthesizing polydiallyl-
amines for use in a water purification process. Diallyl-
amines had been shown to undergo a unique intramo-
lecular–intermolecular polymerization. The formation
of a cyclic structure had been established, and on the
basis of the existing theory that the thermodynamic
stability of the radical formed was the controlling fac-
tor, a six-membered ring was assumed to be formed.8
However, there was no direct experimental evidence to
support this ring size.
Mainly for scientific thoroughness, but also because of
some doubt we had concerning the prevailing theory, we
wanted to prove conclusively the ring size; structures
could be written for a five-, six- or seven-membered ring
(Scheme 1).
Scheme 1. Cyclopolymerization of 1,6-diallylamines (X ¼ N��R1 and R1 ¼ H or alkyl).
5750 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 43 (2005)

In a series of publications, we showed by the use
of models, by trapping with excess initiator, by elec-
tron spin resonance (ESR), and then by NMR of
the polymers that exclusive formation of the kinet-
ically preferred five-membered rings occurred.9,10
The NMR study included the use of a 13C recovery
technique to identify the end groups. Later, we
extended the use of NMR by using 13C-enriched ini-
tiators.11–16
This finding was a major step on the path to rewrit-
ing the theory of radical polymerization. Thermody-
namic control was not dominant: kinetic factors were
of greater significance. We had published our views on
kinetic control versus thermodynamic control and con-
tinued to do so over the years.17
In 1990, Graeme Moad and I wrote a review on this
fundamental change to our understanding of radical
polymerization,18 and this was a precursor to our
books, The Chemistry of Free Radical Polymeriza-tion19 and the revised edition, The Chemistry of Radi-cal Polymerization,20 due out this year.
This realization of the importance of kinetic factors
was an added reason for doubting the exclusive addi-
tion to the tail of the monomer and provided a theoret-
ical reason why we expected other pathways in the ini-
tiation step.
INDUSTRIAL CHALLENGES
In the commercial systems on which I had worked in
industry, we were concerned with a high conversion of
monomer to polymer. A high monomer conversion
raises questions not addressed in most academic stud-
ies, in which a low conversion is deliberately chosen
to simplify the analysis and interpretation of results.
For example, what is the initiator efficiency, does it
vary throughout the polymerization, and is the solvent
critical in determining polymer properties? In polymer-
izations that terminate by disproportionation, what is
the fate of the unsaturated terminated macromonomer?
Does it copolymerize and form grafts? If so, the struc-
ture of the polymer will change from being linear to a
mixture of linear and grafted chains with conversion.
This was to become a critical study leading to an early
example of what is now called RAFT.21,22 At high
conversions, the shortcomings of both the theory and
the published values for reactivity ratios in copolymer-
ization became apparent, and this also became a ques-
tion that we studied.
Another industrial observation made during my time
in the mineral industry, where I was concerned with
polymer/mineral composites, was to also play a pivotal
role in setting our polymer research program. The ren-
dering of clay (kaolin) particles organophilic for use as
a pigment or filler in polymer composites has been
pursued by many. In my own particular case, we were
using oleic acid to form an organophilic clay in which
the oleic acid, through its double bond, would provide
a reaction point for grafting or reacting into the poly-
mer matrix, the so-called graded seal structure for rein-
forcing composites.23
This was an exciting product to make; some batches
underwent spontaneous combustion! We devised a
process to avoid the spontaneous combustion, but I
needed an explanation for this unusual chemistry.
Some oxidation process seemed to be occurring, but its
exact nature was unclear. Oleic acid is the major fatty
acid of olive oil, and I knew from my experience in
the paint industry that this oil auto-oxidized only
slowly even in the presence of a transition-metal cata-
lyst; the mechanism is reported in my book entitled
The Chemistry of Organic Film Formers.24
We had been studying the effects of clay on poly-
merizations and on peroxide decomposition and had
shown that the strong acid surface that develops when
a clay is dried, even under mild conditions, catalyzes
cumyl peroxide decomposition by an ionic mechanism
(Scheme 2).
However, in the case of oleic acid, I had postulated,
in addition to the ionic decomposition, some acid-
induced homolysis of the aliphatic hydroperoxide
(from oleic acid) and subsequent free-radical chain
reaction to explain the exothermic and spontaneous
combustion.
The clay/oleic acid/air system is difficult to study
experimentally, so I set up a model system to study this
chemistry. Tertiary butyl hydroperoxide was chosen as a
model for the oleyl hydroperoxide postulated as the first
oxidation product of oleic acid, and sulfuric acid was
Scheme 2. Decomposition of cumyl peroxide via clay catalysis.
HIGHLIGHT 5751

used as the equivalent of the clay acidity.25 I chose
methyl methacrylate (MMA) as a solvent because we
expected the radical pathway to be the minor one and
a solvent that polymerized by a radical mechanism
would give a high yield of the polymer for even a few
radicals. Ezio Rizzardo joined the team at this time
and this was to be his introduction to polymer science.
PMMA was formed, and this led to a number of
related studies, including the acid-induced homolysis
of tertiary butyl hydroperoxide.26–28
We carried out traditional organic chemistry on the
PMMA formed. The t-butoxy end groups were cleaved
with boron trichloride to form t-butyl chloride. The
yield, however, was only about 60% of that necessary
for a stoichiometric balance. We then located the
remainder as t-butanol.29
Thus, we knew by 1979 that the initiation of MMA
by t-butoxy radicals was far from that shown in the text
books, in which exclusive addition to the tail of the
monomer was reported. What we did not know was the
extent of head and tail addition or where the hydrogen
abstraction was occurring. I will return to this shortly.
Other groups were also beginning to question the
exclusivity of the steps in radical polymerization. For
example, the ESR study by the Kochi group30 showed
that head addition was possible and that a mixture of
initiating radicals occurred with some monomers. Thus,
the scene was set to revisit radical polymerization.
The quote in the Mayo reference1 was an added
challenge to overcome in convincing CSIRO to fund
our research. They set up a panel of overseas experts
to review both our work on step-growth polymerization
(not reported here) and our free-radical studies. The
panel recommended that I be encouraged to continue
with this work and that extra resources be provided if
possible. They also suggested that given the magnitude
of the discoveries, the universities should be encour-
aged to undertake research in these areas. Thus, the
collaboration began, with funding from CSIRO, of a
group at Griffith University.
Thus, the aims of the polymer group included stud-
ies of each of the steps in radical polymerization. In
particular, because of our interest in defect groups and
their effect on the chemical properties of polymers, we
needed to elucidate all the pathways, not just the major
one in the initiation, propagation, and termination steps.
Additionally, and of critical importance, we aimed to
address the questions posed above for high-conversion
polymerization.
Summaries of these studies can be found in the pub-
lications of our group.19,20,31
In this review, I will briefly mention selected examples
of our work that had significant influence on the future
directions of radical chemistry, particularly early examples
of what was to become know as RAFT21,22 and the focus
on the initiation studies that were to lead to NMP.
COPOLYMERIZATION OFMACROMONOMERS
This study was initiated to address the question that I had
raised earlier, namely, in polymerizations that terminate
by disproportionation, does the unsaturated polymer, a
macromonomer, become incorporated into the structure at
a high conversion. I had asked Geoff Hawthorne to repeat
the work of Enikolopyan et al.32 with cobalt porphyrin,
and so within our laboratories, we were familiar with the
synthesis of low-molecular-weight models for the dispro-
portionation product. Using these models, we were able
to show that copolymerization took place with the PMMA
macromonomer but that the incorporation per polymer
chain was low with MMA and much higher with ethyl
acrylate, styrene, acrylonitrile, and vinyl acetate.33
The study showed that the macromonomer readily
added cyanoisopropyl radicals and did not homopoly-
merize and that the radical formed by addition did not
self-terminate by combination or by reaction with a
cyanoisopropyl radical. Importantly, the molecular
weight of the PMMA formed in the presence of the
methacrylate macromonomer was significantly lower
than that obtained in the absence of the macromono-
mer. This was explained by facile b scission of the
adduct radical, as shown in Scheme 3.
Thus, this was an early form of RAFT. Later, the
CSIRO group, particularly Graeme Moad, Ezio Riz-
zardo, and San Thang, developed the highly successful
thiocarbonylthio compounds as much more efficient
agents, and they coined the acronym RAFT to describe
such systems.22,34–36
STYRENE–BENZOYL PEROXIDE SYSTEM
Studies on the polymerization of styrene exemplify
clearly a number of the original aims of the research
group: the differences between initiator-derived radi-
cals in reactivity and selectivity, the formation of some
head addition radicals from the monomer, the change
in the initiator efficiency with the conversion, the
effect of a high conversion of the monomer on the
structure of the polymer formed, and the adverse effect
of a small number of unexpected (based on traditional
theories) end groups on the polymer properties.
Head Addition
Although the cyanoisopropyl and the t-butoxy radicals
added exclusively tail to styrene, we were able to show
that benzoyl peroxide derived radicals gave an interest-
ing mixture of products, as shown in Scheme 4.37–39
5752 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 43 (2005)
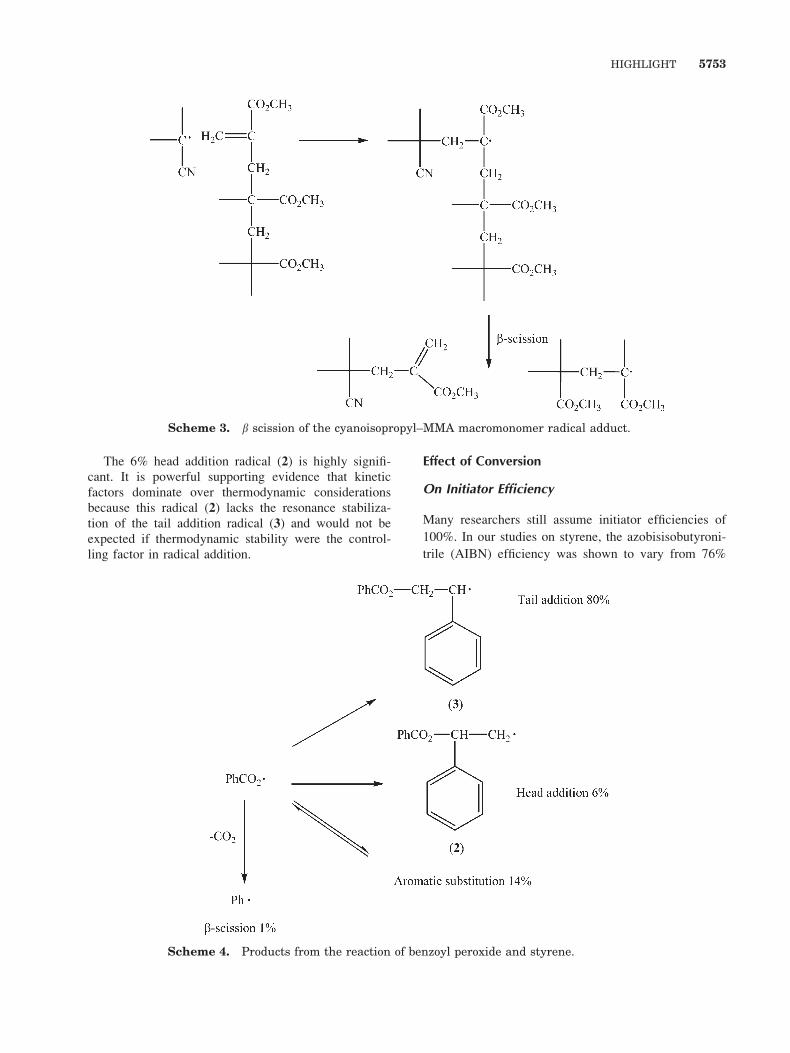
The 6% head addition radical (2) is highly signifi-cant. It is powerful supporting evidence that kinetic
factors dominate over thermodynamic considerationsbecause this radical (2) lacks the resonance stabiliza-
tion of the tail addition radical (3) and would not be
expected if thermodynamic stability were the control-ling factor in radical addition.
Effect of Conversion
On Initiator Efficiency
Many researchers still assume initiator efficiencies of
100%. In our studies on styrene, the azobisisobutyroni-
trile (AIBN) efficiency was shown to vary from 76%
Scheme 4. Products from the reaction of benzoyl peroxide and styrene.
Scheme 3. b scission of the cyanoisopropyl–MMA macromonomer radical adduct.
HIGHLIGHT 5753

at a low conversion down to <20% at a 90–95% con-
version (Fig. 1).31
On Polymer Structure
At high conversions, we were able to show that addi-
tional secondary benzoates, similar to those formed by
head addition in initiation, arise in the termination step
by either primary radical termination or chain transfer
to initiator (Scheme 5).
Thus, the balance between the different end groups
in benzoyl peroxide initiated PS changes with conver-
sion. At a high conversion, PS contains many more
chains terminated by secondary benzoates.37,40
On Properties
We were able to show that secondary benzoate esters
are less thermally stable than the primary ester formed
by tail addition. The primary benzoates persist (>50%
at 300 8C under nitrogen), whereas the secondary ben-
zoates are extremely labile under these conditions
(half-life at 300 8C < 5 min).41,42
These studies highlight the need to specify in detail
the history of polymer samples when properties are
reported. All PSs prepared from benzoyl peroxide are
not the same and will vary with the polymerization
conditions, solvents used, conversion, and so forth.
Thus, the styrene studies clearly showed the need
for careful selection of the initiator, the change in the
thermal stability with conversion in the benzoyl perox-
ide system, and the effect of the conversion on the ini-
tiator efficiency.
MMA–t-BUTOXY SYSTEM
Effect of the Solvent on End Groups
The variety of products formed in the reactions of
t-butoxy radicals with MMA in the absence of a solvent
Figure 1. AIBN initiator efficiency versus the conversion percentage of styrenemonomer with toluene as the polymerization solvent.
Scheme 5. Termination by primary radical termination or chain transfer to initia-tor (BPO ¼ benzoyl peroxide).
5754 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 43 (2005)

(monomer as the solvent) is discussed in Scheme 9. In
commercial polymerization systems, a solvent is a ne-
cessity, and toluene and 2-butanone, commonly called
MEK, are often used. Residues of these solvents can
be incorporated as the end groups, and this is conver-
sion-dependent; at high conversions, more solvent-
derived groups are found. However, by the careful
selection of the solvent and initiator, it is possible to
form the majority of end groups from solvent-derived
radicals formed by abstraction. For example, when tet-
rahydrofuran (THF) is used as solvent in a t-butoxy-initiated MMA polymerization, virtually no end groups
result from the direct interaction of the t-butoxy radi-
cals with the monomer.43
The use of solvents that compete with the monomer
in a reaction with t-butoxy radicals (e.g., cyclohexane
and THF) is a way of minimizing initiator-derived end
groups, and the use of functional solvents offers the
possibility of end-functional polymers (Scheme 6).43
NITROXIDES AS TRAPPING AGENTS
Our aim in studying initiation was to determine all
pathways, not just the major one, which in most cases
we expected to be tail addition. So the challenge was
to find a technique that would establish all pathways in
the initiation step.
Thus, we considered that 13C NMR and ESR tech-
niques being developed by ourselves and others lacked
the sensitivity necessary to detect these minor pathways.
The stable free radical diphenyl picrylhydrazyl (DPPH)
had been used as a diagnostic test for free-radical poly-
merizations. It was claimed to form DPPH–H by the
abstraction of a hydrogen from the initiating or propa-
gating radical species; that is, the interaction was one
of disproportionation between the two radicals. How-
ever, with Geoff Hawthorne, I had previously used
DPPH in mechanistic studies and shown that the chem-
istry was not straightforward, and a complex mixture
of products could form.44 Scheme 7 shows our results
for its reaction with AIBN.
Moreover, DPPH reacts with minerals and with
acids.45,46
Thus, we needed a trap that would preferably add to
the initiating radicals (i.e., the first radical formed from
the monomer) to form stable products that we could
isolate, separate, and characterize and thereby delineate
the initiation pathways.
A trap that abstracted hydrogen from the initiating
radical would, for example, give another monomer that
could undergo further reaction and so complicate prod-
uct analysis (Scheme 8).
Alkoxyamines were, and still are, used as stabil-
izers in polymer composites, and nitroxides were
known to be inhibitors of radical polymerization. So
we eventually chose as our trapping agent 2,2,6,6-tet-
ramethylpiperidinyl-1-oxy (TEMPO or 4), a nitroxide
that combines with carbon-centered radicals at or near
diffusion-controlled rates to yield stable alkoxyamines.
Later, we greatly extended the range of nitroxides, includ-
ing the new isoindoline nitroxide [1,1,3,3-tetramethylisoin-
doline-2-oxyl (5)]; this proved invaluable because the
aromatic ring allowed for easy detection and quantifi-
cation of the separated alkoxyamine by ultraviolet
spectroscopy (Scheme 9):47–49
Scheme 6. Solvent-derived end groups arising from t-butoxy-initiated radicals inMMA polymerization.
HIGHLIGHT 5755

Because the nitroxides did not react with hetero-
atom-centered radicals, they were ideal for studying the
reactivities of monomers with the common initiators
formed from oxygen-centered radicals, that is, benzoyl
peroxide or t-butoxy sources such as t-butyl hydroper-oxide and di-t-butyl peroxide. In these systems, we
trapped the first monomer-derived radical (Scheme 9).
Some radicals form by unimolecular rearrangement
or fragmentation of the primary radicals to form second-
ary radicals. In this case (Scheme 9), the b scission of
the t-butoxy radicals to methyl radicals that form 10 or
11 and acetone had been studied previously, and the
absolute rate constant was known.48 Thus, the extent of
these b-scission reactions can be used as a radical clock
to calibrate the absolute rate constant of the bimolecular
reactions between the primary radicals (i.e., t-butoxy)and monomer.
Our extensive studies on the initiation processes
using nitroxides as trapping agents are reviewed in a
number of references from our group.19,20,50
I was fortunate to have recruited scientists who
were not only excellent theorists but also gifted experi-
Scheme 8. Generation of monomer via hydrogen ab-straction from an initiating radical.
Scheme 7. Some of the products from the reaction between DPPH and AIBN.
5756 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 43 (2005)

mentalists. They isolated compounds present in minor
amounts, often less than 1%, and noted properties of
the alkoxyamines that collectively led to NMP. Fol-
lowing is a summary of these observations:
1. Compounds with more than one monomer resi-
due were isolated. Thus, with MMA, compound
949 could be theoretically formed from the first
alkoxyamine (8) by thermal dissociation, the
addition of the monomer, and then trapping
(Scheme 10). Similarly, styrene compounds 12and 1338 could be formed by the thermolysis of
14 and 15, respectively, and the insertion of a
monomer unit (Scheme 11). (We did not favor
these mechanisms, and our publications give alter-
native pathways.)
Scheme 10. Possible mechanism for the formation of MMA dimer from the ther-molysis of alkoxyamine.
Scheme 9. Products from the reaction of t-butoxy radicals with MMA in the pres-ence of 5.
HIGHLIGHT 5757

2. In the separation of alkoxyamines, their instabil-
ity in a gas–liquid chromatography column was
noted.51
3. In the recrystallization of some alkoxyamines, the
use of solvents with high boiling points resulted in
the formation of the characteristic color of the ni-
troxide. We therefore routinely used solvents with
low boiling points or solvent/nonsolvent mixtures
to recrystallize alkoxyamines; this provided com-
pelling evidence for the reversibility of alkoxy-
amine formation.52
4. Other observations on the reversibility of the
alkoxyamines were as follows:
a. Graeme Moad and I noted that the cis–trans
ratio of styrene dimers changed on heating
(Scheme 12).53
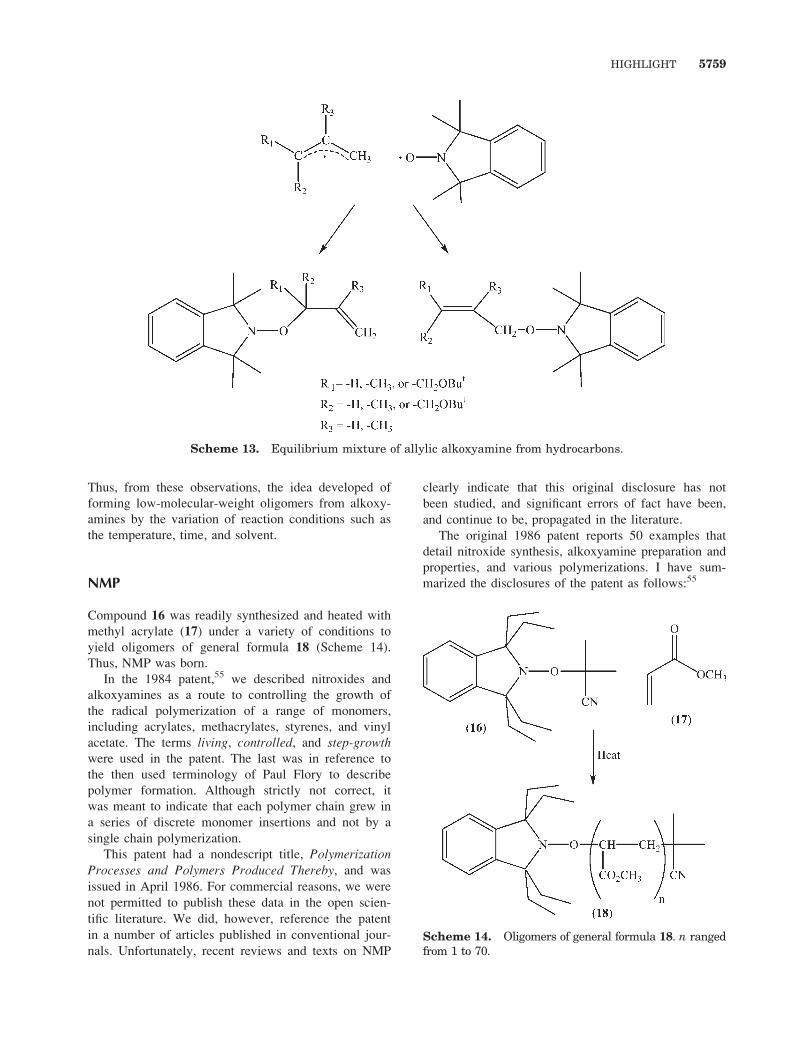
b. The allylic alkoxyamine from hydrocarbons
reverted to an equilibrium mixture of products
on heating (Scheme 13).54
5. Solvent effects showed that highly polar solvents
(methanol and acetic acid) caused alkoxyamine
mixtures to rearrange.43
Scheme 12. Interconversion of cis–trans isomers of styrene dimers.
Scheme 11. Possible mechanism for the formation of styrene dimer from the ther-molysis of alkoxyamine.
5758 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 43 (2005)
Thus, from these observations, the idea developed of
forming low-molecular-weight oligomers from alkoxy-
amines by the variation of reaction conditions such as
the temperature, time, and solvent.
NMP
Compound 16 was readily synthesized and heated with
methyl acrylate (17) under a variety of conditions to
yield oligomers of general formula 18 (Scheme 14).
Thus, NMP was born.
In the 1984 patent,55 we described nitroxides and
alkoxyamines as a route to controlling the growth of
the radical polymerization of a range of monomers,
including acrylates, methacrylates, styrenes, and vinyl
acetate. The terms living, controlled, and step-growthwere used in the patent. The last was in reference to
the then used terminology of Paul Flory to describe
polymer formation. Although strictly not correct, it
was meant to indicate that each polymer chain grew in
a series of discrete monomer insertions and not by a
single chain polymerization.
This patent had a nondescript title, PolymerizationProcesses and Polymers Produced Thereby, and was
issued in April 1986. For commercial reasons, we were
not permitted to publish these data in the open scien-
tific literature. We did, however, reference the patent
in a number of articles published in conventional jour-
nals. Unfortunately, recent reviews and texts on NMP
clearly indicate that this original disclosure has not
been studied, and significant errors of fact have been,
and continue to be, propagated in the literature.
The original 1986 patent reports 50 examples that
detail nitroxide synthesis, alkoxyamine preparation and
properties, and various polymerizations. I have sum-
marized the disclosures of the patent as follows:55
Scheme 14. Oligomers of general formula 18. n rangedfrom 1 to 70.
Scheme 13. Equilibrium mixture of allylic alkoxyamine from hydrocarbons.
HIGHLIGHT 5759
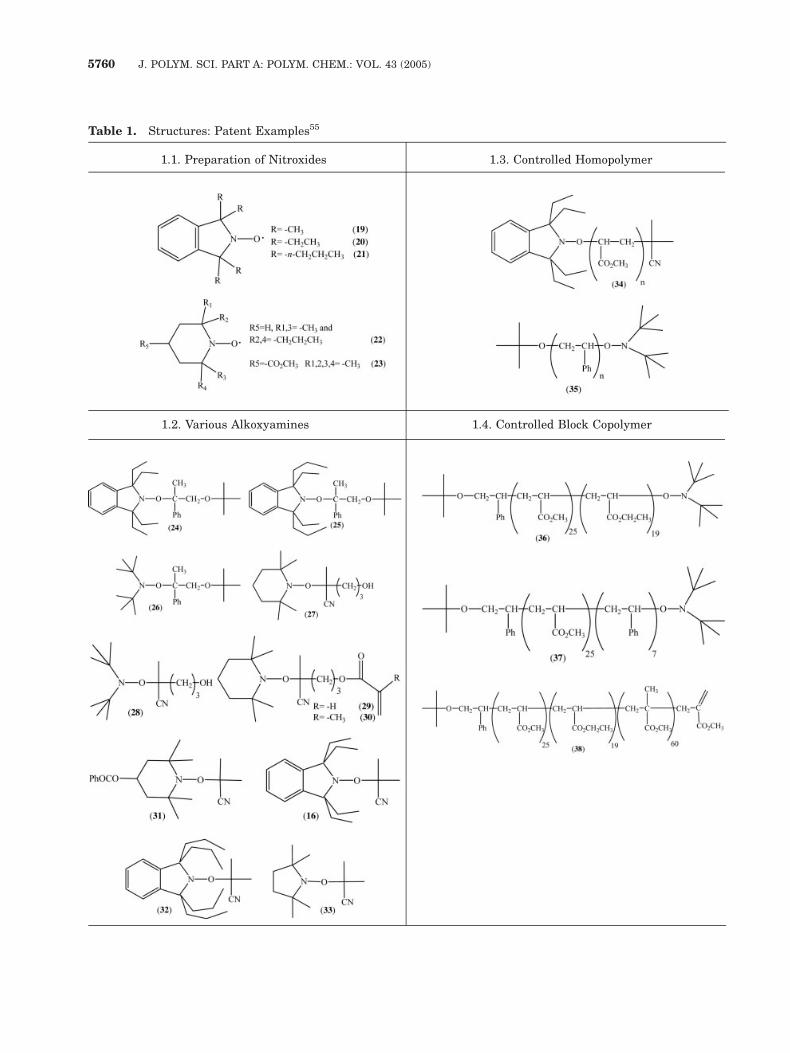
Table 1. Structures: Patent Examples55
1.1. Preparation of Nitroxides 1.3. Controlled Homopolymer
1.2. Various Alkoxyamines 1.4. Controlled Block Copolymer
5760 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 43 (2005)
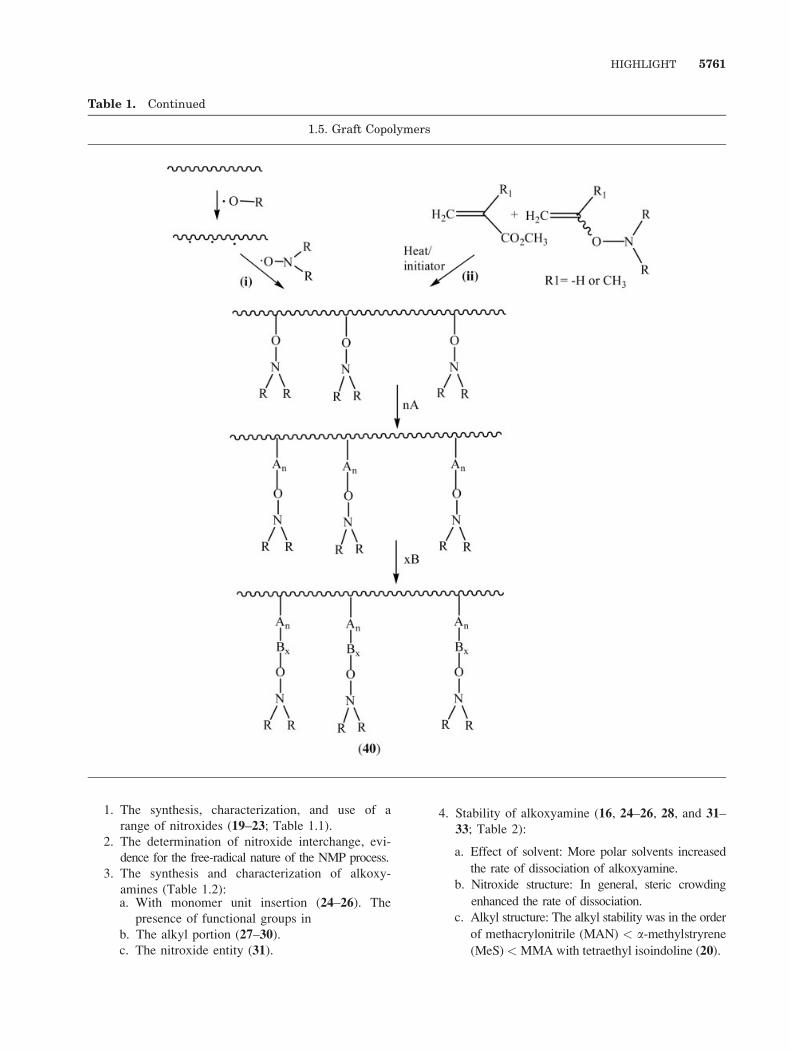
1. The synthesis, characterization, and use of a
range of nitroxides (19–23; Table 1.1).
2. The determination of nitroxide interchange, evi-
dence for the free-radical nature of the NMP process.
3. The synthesis and characterization of alkoxy-
amines (Table 1.2):a. With monomer unit insertion (24–26). The
presence of functional groups in
b. The alkyl portion (27–30).c. The nitroxide entity (31).
4. Stability of alkoxyamine (16, 24–26, 28, and 31–33; Table 2):
a. Effect of solvent: More polar solvents increased
the rate of dissociation of alkoxyamine.
b. Nitroxide structure: In general, steric crowding
enhanced the rate of dissociation.
c. Alkyl structure: The alkyl stability was in the order
of methacrylonitrile (MAN) < a-methylstryrene
(MeS)<MMA with tetraethyl isoindoline (20).
Table 1. Continued
1.5. Graft Copolymers
HIGHLIGHT 5761

5. Control of polymer architectures:
a. Homopolymers (34 and 35; Table 1.3).
b. Block copolymers (36–38; Table 1.4).
c. Graft copolymers (40; Table 1.5).
i. By forming alkoxyamine on the backbone
by copolymerizing the unsaturated alkoxy-
amines.
ii. By forming polymeric alkoxyamine by ab-
straction from a preformed polymer.
6. Location of specific end groups. By selection of
monomer mixtures, one terminal alkoxyamine is
stable under polymerization conditions.
7. Formation of macromonomers that are not avail-
able by other means.
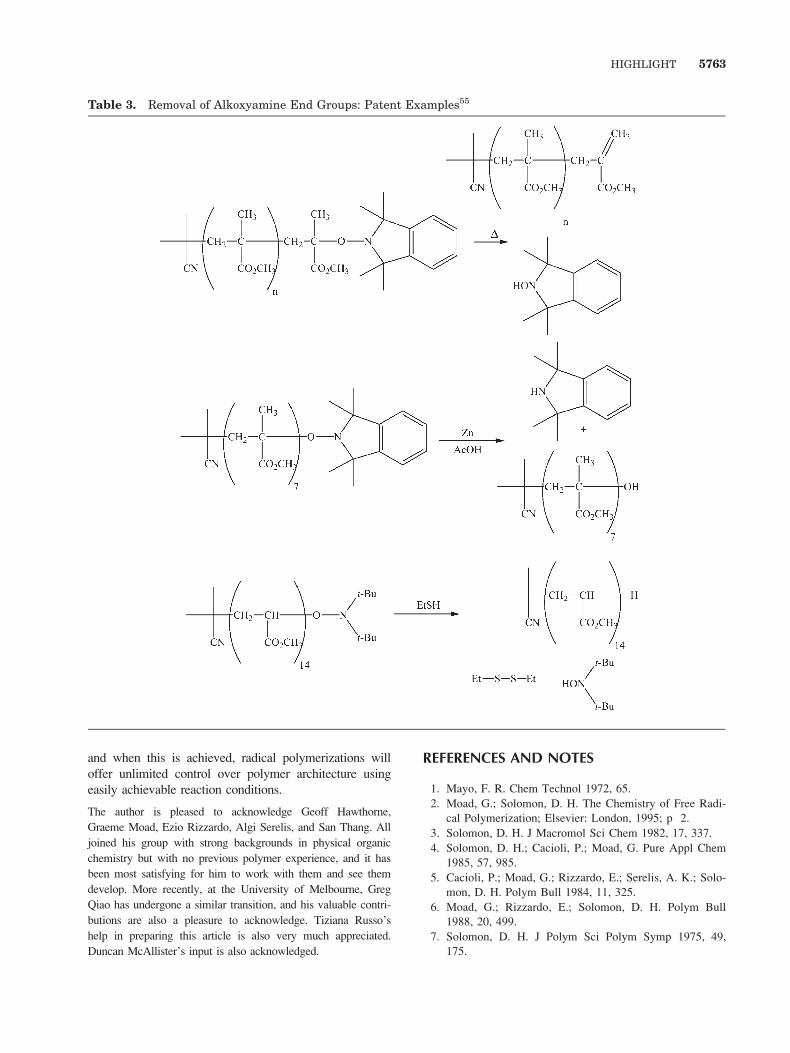
8. Removal of alkoxyamine end groups (Table 3).
9. Formation of low-molecular-weight functional
polymers for further use in, for example conden-
sation polymerization, that is, diol.
NARROW-POLYDISPERSITY POLYMERSAND COPOLYMERS BY NMP
The molecular weight distributions (MWDs) of most of
the examples in this original patent were broad. However,
Graeme Moad recognized the potential of this living sys-
tem for preparing narrow-polydispersity polymers by
NMP. We published the theoretical requirements for nar-
row MWDs,56,57 and these have since been demon-
strated in a variety of systems by many workers.58
NMP received scant attention until Georges et al.58
used NMP to prepare narrow-polydispersity PS. There
followed a period of intense interest in NMP; for
example, the Hawker group59,60 added to the range of
suitable nitroxides and polymer structures. Thus, NMP
enabled the synthesis of a range of polymer structures
not previously attainable by radical chemistry, including
low-molecular-weight, narrow-polydispersity homopoly-
mers and copolymers (block and graft copolymers), as
well as many other intriguing structures not accessible by
other chemistries.
FUTURE
The original CSIRO alkoxyamine/nitroxide patent
expired this year and is no longer a restriction in the
commercialization of NMP by others. We can confi-
dently expect increased activity as systems described
in scientific conferences become commercial realities.
Other forms of controlled living radical polymerization
have been developed. In 1995, the groups led by Sawa-
moto61 and Matyjaszewski62 reported atom transfer poly-
merization, and more effective catalysts for these systems
were developed in the same year by the group led by
Percec.63 A major advance in 1998 was the discovery of
RAFT35,36 using thiocarbonylthio compounds by the
CSIRO team now led by Ezio Rizzardo. Although each
technique has its virtues, combinations of these methodol-
ogies and the interconversion of one form to another are
opening up exciting possibilities for the synthesis of
highly controlled polymer structures by radical polymer-
ization. At the time of the discovery of NMP living ionic
systems were the standard we aimed to emulate; the sit-
uation has now changed drastically. Radical polymeriza-
tions now offer the versatility of radical chemistry, a
greater tolerance of impurities, and the ability to form
structures not accessible with ionic systems.
Thus, of the three classical steps in radical polymer-
ization, we now have control over initiation and termina-
tion. Control over propagation remains the holy grail,
Table 2. Solvent Effects: Patent Examples55
Solvent Alkoxyamine Half-Life at 60 8C (min)
Ethyl acetate 16 3324 7525 1026 18a
28 8.5a
31 13032 3133 280
Light petroleum 16 38Acetonitrile 16 22Dimethylformamide 16 20Methanol 16 17Methanol/water (9:1) 16 16Methanol/acetic acid (9:1) 16 15
a Half-life at 40 8C.
5762 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 43 (2005)
and when this is achieved, radical polymerizations will
offer unlimited control over polymer architecture using
easily achievable reaction conditions.
The author is pleased to acknowledge Geoff Hawthorne,
Graeme Moad, Ezio Rizzardo, Algi Serelis, and San Thang. All
joined his group with strong backgrounds in physical organic
chemistry but with no previous polymer experience, and it has
been most satisfying for him to work with them and see them
develop. More recently, at the University of Melbourne, Greg
Qiao has undergone a similar transition, and his valuable contri-
butions are also a pleasure to acknowledge. Tiziana Russo’s
help in preparing this article is also very much appreciated.
Duncan McAllister’s input is also acknowledged.
REFERENCES AND NOTES
1. Mayo, F. R. Chem Technol 1972, 65.
2. Moad, G.; Solomon, D. H. The Chemistry of Free Radi-
cal Polymerization; Elsevier: London, 1995; p 2.
3. Solomon, D. H. J Macromol Sci Chem 1982, 17, 337.
4. Solomon, D. H.; Cacioli, P.; Moad, G. Pure Appl Chem
1985, 57, 985.
5. Cacioli, P.; Moad, G.; Rizzardo, E.; Serelis, A. K.; Solo-
mon, D. H. Polym Bull 1984, 11, 325.
6. Moad, G.; Rizzardo, E.; Solomon, D. H. Polym Bull
1988, 20, 499.
7. Solomon, D. H. J Polym Sci Polym Symp 1975, 49,
175.
Table 3. Removal of Alkoxyamine End Groups: Patent Examples55
HIGHLIGHT 5763

8. Butler, G. B.; Crawshaw, A.; Miller, W. L. J Am Chem
Soc 1958, 80, 3615.
9. Solomon, D. H. J Macromol Sci Chem 1975, 9, 97.
10. Solomon, D. H.; Hawthorne, D. G. J Macromol Sci Rev
Macromol Chem 1976, 15, 143.
11. Johns, S. R.; Rizzardo, E.; Solomon, D. H.; Willing, R. I.
Makromol Chem Rapid Commun 1983, 4, 29.
12. Moad, G.; Rizzardo, E.; Solomon, D. H.; Johns, S. R.;
Willing, R. I. Makromol Chem Rapid Commun 1984, 5, 793.
13. Moad, G.; Rizzardo, E.; Solomon, D. H. Polym Bull
1984, 12, 471.
14. Moad, G.; Solomon, D. H.; Johns, S. R.; Willing, R. I.
Macromolecules 1984, 17, 1094.
15. Cacioli, P.; Hawthorne, D. G.; Johns, S. R.; Solomon,
D. H.; Rizzardo, E.; Willing, R. I. J Chem Soc Chem
Commun 1985, 1355.
16. Hawthorne, D. G.; Johns, S. R.; Solomon, D. H.; Will-
ing, R. I. Aust J Chem 1979, 32, 1155.
17. Hawthorne, D. G.; Solomon, D. H. J Polym Sci Polym
Symp 1976, 55, 211.
18. Moad, G.; Solomon, D. H. Aust J Chem 1990, 43, 215.
19. Moad, G.; Solomon, D. H. The Chemistry of Free Radi-
cal Polymerization; Pergamon: London, 1995.
20. Moad, G.; Solomon, D. H. The Chemistry of Radical
Polymerization; Elsevier: London, 2005.
21. Moad, G.; Solomon, D. H. The Chemistry of Radical
Polymerization; Elsevier: London, 2005; p 501.
22. Chiefari, J.; Rizzardo, E. In Handbook of Radical Poly-
merization; Matyjaszewski, K.; Davis, T. P., Eds.; Wiley-
Interscience: Hoboken, NJ, 2002; p 646.
23. Solomon, D. H. Chem Aust 1982, 49, 192.
24. Solomon, D. H. The Chemistry of Organic Film For-
mers; Wiley: New York, 1967.
25. Solomon, D. H.; Hawthorne, D. G. Chemistry of Pig-
ments and Fillers; Wiley: New York, 1983; p 192.
26. Rizzardo, E.; Solomon, D. H. J Macromol Sci Chem
1977, 11, 1697.
27. Rizzardo, E.; Solomon, D. H. J Macromol Sci Chem
1980, 14, 33.
28. Quint, G.; Rizzardo, E.; Solomon, D. H.; Spurling, T. H.
J Macromol Sci Chem 1980, 15, 527.
29. Rizzardo, E.; Solomon, D. H. J Macromol Sci Chem
1979, 13, 1005.
30. Elson, I. H.; Mao, S. W.; Kochi, J. K. J Am Chem Soc
1974, 97, 335.
31. Solomon, D. H.; Moad, G. Makromol Chem Macromol
Symp 1987, 10, 109.
32. Enikolopyan, N. S.; Smirnov, B. R.; Ponomarev, G. V.;
Belgovskii, I. M. J Polym Sci Polym Chem Ed 1981,
19, 879.
33. Cacioli, P.; Hawthorne, D. G.; Laslett, R. L.; Rizzardo, E.;
Solomon, D. H. J Macromol Sci Chem 1986, 23, 839.
34. Moad, G.; Rizzardo, E.; Thang, S. H. Aust J Chem
2005, 58, 379.
35. Phuong, L.-T.; Moad, G.; Rizzardo, E.; Thang, S. H.
WO 97-US12540, 1997.
36. Chiefari, J.; Chong, Y. K.; Ercole, F.; Kristina, J.; Jeff-
ery, J.; Le, T. P. T.; Mayadunne, R. T. A.; Meijs, G. F.;
Moad, C. L.; Moad, G.; Rizzardo, E.; Thang, S.-H. Mac-
romolecules 1998, 31, 5559.
37. Moad, G.; Rizzardo, E.; Solomon, D. H. J Macromol
Sci Chem 1982, 17, 51.
38. Moad, G.; Rizzardo, E.; Solomon, D. H. Macromole-
cules 1982, 15, 909.
39. Jones, M. J.; Moad, G.; Rizzardo, E.; Solomon, D. H.
J Org Chem 1989, 54, 1607.
40. Moad, G.; Solomon, D. H.; Johns, S. R.; Willing, R. I.
Macromolecules 1982, 15, 1188.
41. Moad, G.; Solomon, D. H.; Willing, R. I. Macromole-
cules 1988, 21, 855.
42. Krstina, J.; Moad, G.; Solomon, D. H. Eur Polym J
1989, 25, 767.
43. Grant, R. D.; Griffiths, P. G.; Moad, G.; Rizzardo, E.;
Solomon, D. H. Aust J Chem 1983, 36, 2447.
44. Hawthorne, D. G.; Solomon, D. H. J Macromol Sci
Chem 1972, 6, 661.
45. Solomon, D. H.; Hawthorne, D. G. J Macromol Sci
Chem 1971, 5, 575.
46. Solomon, D. H.; Swift, J. D. J Polym Sci Part A: Gen
Pap 1965, 3, 3107.
47. Rizzardo, E.; Solomon, D. H. Polym Bull 1979, 1, 529.
48. Griffiths, P. G.; Rizzardo, E.; Solomon, D. H. J Macro-
mol Sci Chem 1982, 17, 45.
49. Griffiths, P. G.; Moad, G.; Rizzardo, E.; Solomon, D. H.
Aust J Chem 1983, 36, 397.
50. Handbook of Radical Polymerization; Matyjaszewski, K.;
Davis, T. P., Eds.; Wiley-Interscience: Hoboken, NJ, 2002;
p 127.
51. Schreck, V. A.; Serelis, A. K.; Solomon, D. H. Aust JChem 1989, 42, 375.
52. Solomon, D. H. University of Melbourne, Melbourne,Australia. Unpublished results, 2005.
53. Moad, G.; Solomon, D. H. University of Melbourne,Melbourne, Australia. Unpublished results, 2005.
54. Cuthbertson, M. J.; Rizzardo, E.; Solomon, D. H. Aust JChem 1983, 36, 1957.
55. Solomon, D. H.; Rizzardo, E.; Cacioli, P. U. S. Patent 4,581, 429, 1986.
56. Johnson, C. H. J.; Moad, G.; Solomon, D. H.; Spurling,T. H.; Vearing, D. J. Aust J Chem 1990, 43, 1215.
57. Johnson, C. H. J.; Moad, G.; Solomon, D. H.; Spurling,T. H. Aust J Chem 1986, 39, 1943.
58. Georges, M. K.; Veregin, R. P. N.; Kazmaier, P. M.;Hamer, G. K. Macromolecules 1993, 26, 2987.
59. Hawker, C. J.; Bosman, A. W.; Harth, E. Chem Rev2001, 101, 3661.
60. Hawker, C. J. In Handbook of Radical Polymerization;Matyjaszewski, K.; Davis, T. P., Eds.; Wiley-Interscience:Hoboken, NJ, 2002; p 463.
61. Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashamura,T. Macromolecules 1995, 28, 1721.
62. Wang, J.-S.; Matyjaszewski, K. Macromolecules 1995, 28,7901.
63. Percec, V.; Barboiu, B. Macromolecules 1995, 28, 7970.
5764 J. POLYM. SCI. PART A: POLYM. CHEM.: VOL. 43 (2005)