galaxy workshop at the winter school ‐2016
TRANSCRIPT


Plan
Overview of the Genomics Virtual Lab
Introduce Galaxy, a web based platform for analysis nextGen sequencing data
Workshop: RNA‐Seq analysis in Galaxy
Tea break at 15:30‐16:00
Igor MakuninUQ RCC
Michal LorencQUT
Derek BensonUQ RCC

Genomics Virtual LaboratoryAnalysis of nextGen sequencing data is a bottleneck (infrastructure, skills)
Genomics Virtual Lab: take the IT out of Bioinformatics‐ web‐based resources (biologists‐friendly)‐ DIY bioinformatics environment (for geeks)
GVL advantages: ‐ public resources (no charges to users)‐ available immediately

GVL products and servicesGenomics Virtual Lab: genome.edu.au
The main aim: facilitate the genomics research in Australia
Galaxy:• Tutorials and protocols (nextGen sequencing)• Galaxy for tutorials: galaxy‐tut.genome.edu.au• Galaxy for full‐scale analysis: galaxy‐qld.genome.edu.au• “roll your own” Galaxy on the Australian government
funded computer infrastructure (NeCTAR cloud)+ ipython Notebook+ RStudio
Deploy your own computer cluster (NeCTAR cloud)RStudioGenomeSpace
LearnUseGet
Info

Galaxy: how does it look like
Tools
Working window
HistoryTop menu
Upload

Galaxy: possibilitiesYou can:‐ analyze genome‐scale nextGen sequencing data without bash scripting‐ work with big datasets, genomic regions, sequences etc.‐ create and use Galaxy workflows (record steps of your analysis)‐ share results and workflows with a user or make it available to anyone
Private data: ‐ upload through the web interface ‐ ftp (for big datasets)‐ transfer data between different Galaxy servers‐ GenomeSpace
Public data: ‐ UCSC Genome Browser‐ EBA SRA
Over 2,000 tools available through the Galaxy tool shed

Use: GVL GenomeSpace
Data‐centric environment.
Export or import data from different depositories

Use: local Galaxy‐qld server
GVL Galaxy in Queensland: galaxy‐qld.genome.edu.au/galaxyTools:‐ BWA, bowtie2‐ Velvet (microbial genome assembly)‐ Trinity (de novo transcript assembly)‐ tophat2, RNA_STAR (RNA‐Seq)‐ DESeq, edgeR, Cufflinks (differential gene expression)‐ GATK2, variant detection tools‐ Metagenomics tools‐ MACS2, SPP (ChIP‐Seq)‐ SAMtools‐ Picard‐ Local Blast+ search
100s users1000s jobs per month
600 GB for Australian users
Datasets:genome indicesgene annotations

Bad user practice

Very bad user practice
Do not delete jobs in fast succession!

Good user practice for Galaxy‐qld Read GVL FAQ page at genome.edu.au/help/faq
Register with your institutional email and get a bigger disk allocation.
Save the results. The server does not have an external backup.
Use ftp for big datasets – it is faster. Galaxy recognises .gz compression.
Do not store unneeded datasets. Delete temporary files such as SAM. Purge deleted datasets.
Do not start many big jobs in parallel (BWA, bowtie, bowtie2, tophat2, velvet, trinity, rna_star).
Create and use workflows for multi‐step analysis. Use small datasets to build a workflow.
Specify the quality score encoding for FASTQ files.

@SRR391845.1639 ILLUMINA‐C32_FC:3:1:80:12/1TAGCAGCACATCATGGTTTACATCGTATGCCGTCTT+IIHIDIIIIIIIIIIIIIHIHIIIIIDGIBGGGGGG
FASTQ quality score encodingQual. = 40Offset = 3339+33 = 73ASCII(73): I

FASTQ quality score in Galaxy
Many old illumina datasets have a proprietary data encoding (offset 64)Currently most NGS datasets use Sanger encoding (offset 33)
Galaxy
By default Galaxy assign ‘fastq’ data type to uploaded FASTQ files.In this case the offset is not specified, and many tools do not recognize the data
fastqillumina – old illumina quality score encoding (offset 64, illumina 1.3+)fastqsanger – new illumina 1.8+ / Sanger quality score encodingNearly all modern NGS data use Sanger encoding (fastqsanger in Galaxy)
Solution:‐ specify a proper format, eg fastqsanger or fastqillumina, during the data upload ‐ change the format via Attributes > Datatype‐ use NGS: QC and manipulation > FASTQ Groomer tool

Troubleshooting GVL FAQ page at genome.edu.au/help/faq
Read the error report!
Search for the error message. https://biostar.usegalaxy.org
Report the error to the local Galaxy administrators.

Thank you!GVL site: www.genome.edu.auGalaxy for tutorials: galaxy‐tut.genome.edu.auGalaxy Queensland: galaxy‐qld.genome.edu.au
Contributors and participants:
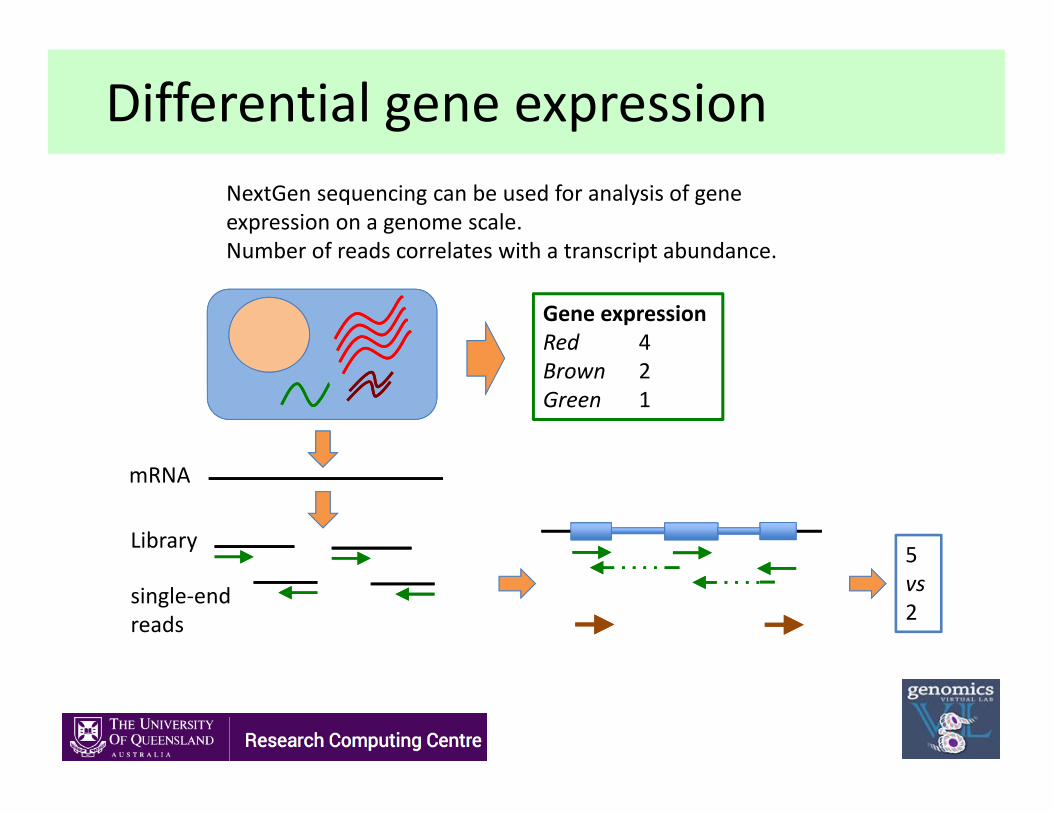
Differential gene expressionNextGen sequencing can be used for analysis of gene expression on a genome scale.Number of reads correlates with a transcript abundance.
Library
single‐endreads
5vs2
mRNA
Gene expressionRed 4Brown 2Green 1
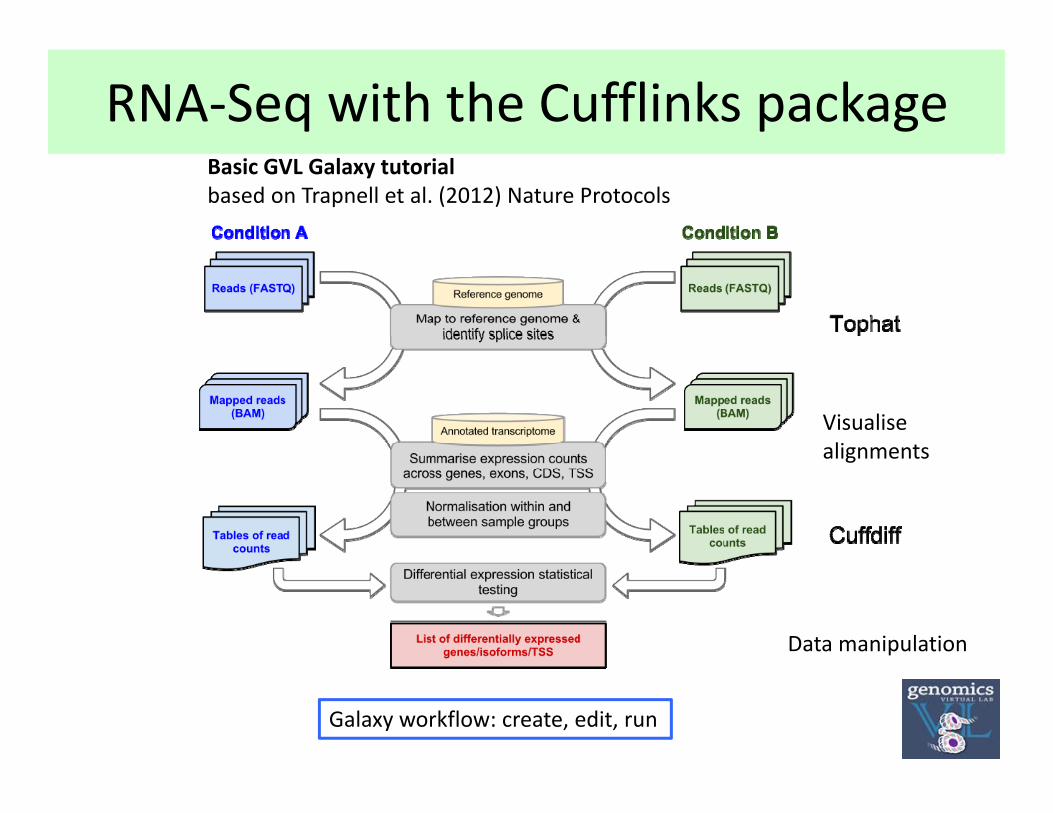
RNA‐Seq with the Cufflinks packageBasic GVL Galaxy tutorialbased on Trapnell et al. (2012) Nature Protocols
Visualisealignments
Galaxy workflow: create, edit, run
Data manipulation

Setup for the workshophttps://genome.edu.au
2. Register on a personal GVL Galaxy
1. Go to the GVL website:
The servers will be available for one week after the workshop.

Galaxy workflow
History menu > Extract Workflow
1. Name the workflow
2. Uncheck all BAM filesKeep the gene annotation fileensembl_dm3.chr4.gtf as input dataset
Extracted workflows do not keep genome assembly for aligner tools.
We will edit the workflow.

Workflow Edit1. Delete one replicate per condition 2. Add ‘Condition 1 input’ (or 2) in
Annotation / Notes for tophat jobs3. Check the assembly in tophat jobs4. Rename the output in Filter
Add Actions > Rename dataset5. Save the workflow
6. Ran the workflow7. Select FASTQ files from C1 and C2
(check the genome assembly)8. Save the results into a new history