gabaa receptor modulation of trigeminovascular nociceptive neurotransmission by midazolam is...
TRANSCRIPT

www.elsevier.com/locate/brainres
Brain Research 1013 (2004) 188–193
Research report
GABAA receptor modulation of trigeminovascular nociceptive
neurotransmission by midazolam is antagonized by flumazenil
Robin James Storer, Simon Akerman, Kevin G. Shields, Peter J. Goadsby*
Headache Group, Institute of Neurology and The National Hospital for Neurology and Neurosurgery, Queen Square, London WC1N 3BG, UK
Accepted 9 March 2004
Abstract
Studies of the pharmacology of trigeminocervical neurons with input from intracranial pain-producing structures have enhanced the
understanding of the basic neurobiology of primary headache, such as migraine. Clinical observations of the treatment of migraine with
medicines acting at the g-aminobutyric acid (GABA) GABAA receptor have lead to studies of their effects on models of trigeminovascular
nociception. Extracellular recordings were made from neurons in the trigeminocervical complex activated by supramaximal electrical
stimulation of superior sagittal sinus (SSS) in the cat. Intravenous administration of the benzodiazepine receptor agonist midazolam, resulted
in a dose-dependent inhibition of superior sagittal sinus evoked trigeminocervical nucleus activity. The inhibition at 50 Ag/kg midazolam was
65F 11% compared to the baseline response (n = 11). Intravenous administration of the benzodiazepine receptor antagonist flumazenil,
resulted in a dose-dependent recovery of superior sagittal sinus evoked trigeminocervical nucleus activity. At a dose of 50 Ag/kg, there was a64F 5% recovery (n = 6). The data demonstrate a potent, reproducible effect of facilitation of GABA transmission at the GABAA receptor
that results in inhibition of trigeminovascular nociceptive transmission. These data are consistent with the useful clinical effects reported with
compounds that can augment GABAergic transmission in the central nervous system (CNS).
D 2004 Elsevier B.V. All rights reserved.
Theme: Sensory systems
Topic: Pain modulation: pharmacology
Keywords: Trigeminal; Dural; Migraine; Headache
1. Introduction
Migraine is an episodic brain disorder that results in
significant morbidity [34] for between 10% and 15% of the
general population [27,38]. Modulation of trigeminal trans-
mission in order to alleviate acute migraine requires an
understanding of the pharmacology of the second order
trigeminal neurons subserving nociception [14]. In recent
years, the pharmacology of this synapse has been studied
with most attention on serotonergic and glutamatergic
transmission [13].
g-Aminobutyric acid (GABA) is well known as an inhib-
itory amino acid neurotransmitter in the central nervous
system (CNS) [39] and may modulate nociceptive response
in the spinal cord [40]. Understanding the role of GABAergic
influences on trigeminovascular nociceptive transmission
0006-8993/$ - see front matter D 2004 Elsevier B.V. All rights reserved.
doi:10.1016/j.brainres.2004.03.068
* Corresponding author. Tel.: +44-207-829-8749; fax: +44-207-813-
0349.
E-mail address: [email protected] (P.J. Goadsby).
within the trigeminal nucleus is important to develop a
comprehensive description of these neurons. It has been
shown that trigeminovascular nociceptive input can be mod-
ulated through GABAergic mechanisms [46]. GABA, when
microiontophoresed onto neurons in the trigeminocervical
complex responding to dural perivascular input, specifically
stimulation of the superior sagittal sinus, inhibits trigeminal
transmission. This effect can be blocked by bicuculline, a
GABAA receptor antagonist, and mimicked by the GABAA
receptor agonist muscimol. In the same series of experiments,
the GABAB receptor agonist baclofen affected only a small
number of neurons with trigeminovascular input and the
GABAB receptor antagonist, 2-hydroxysaclofen, was much
less likely to inhibit trigeminal neurons [46]. More recently it
has been shown that topiramate may inhibit transmission in
the trigeminocervical complex [44], and this compound is
known to modulate GABAergic mechanisms, among other
effects [41]. Midazolam acts at the benzodiazepine modula-
tory site on the GABAA receptor complex to facilitate the
actions of GABA by prolonging channel opening time. We

R.J. Storer et al. / Brain Research 1013 (2004) 188–193 189
sought to determine the effect of benzodiazepine site activa-
tion on the trigeminovascular system in vivo by employing
the specific benzodiazepine site antagonist flumazenil to
reverse the inhibitory effects of midazolam [48].
2. Materials and methods
All studies were conducted and terminated under general
anaesthesia in accordance with a project licence issued by the
Home Office of the United Kingdom under the Animals
(Scientific Procedures) Act, 1986. Cats (n = 11) weighing
3.50F 0.53 kg (meanF S.D.) were anaesthetised with a-
chloralose (60 mg/kg i.p., Sigma, St. Louis, MO) and
prepared for physiological monitoring. Halothane (Rhone
Merieux, Essex, UK; 0.5–3% in a 40% oxygen/air mixture)
was administered from an anaesthetic machine during surgi-
cal procedures and then discontinued during experimental
protocols. Catheters were placed in femoral arteries for
arterial blood sampling and continuous measurement of
blood pressure (DTXplus transducer, Ohmeda, Madison,
WI; PM-1000 amplifier, CWE Instruments, Ardmore, PA).
Catheters were also placed in femoral veins allowing fluid
(saline for intravenous infusion BP (0.9% w/v), Baxter
Healthcare, Norfolk, UK) and drug administration. Supple-
mentary doses of a-chloralose in 2-hydroxypropyl-h-cyclo-dextrin (RBI, Natick, MA) were given i.v. at a rate of 5–10
mg/kg per h [45]. The cats were intubated after local
anaesthesia with lidocaine hydrochloride (Intubeaze,
Arnolds, Shrewsbury, UK) and fixed in a stereotaxic frame
(Kopf Instruments, Tujunga, CA).
Jackson/Foley urethral catheters were inserted to drain the
cat bladders, providing more even temperature regulation,
more stable control of blood pressure through control of
bladder distension, and monitoring of urine output. Core
temperature was monitored and maintained between 37 and
39 jC using a rectal thermistor probe and a low-radio-noise
emitting homeothermic heater blanket system (Harvard Ap-
paratus, Holliston, MA). Cats were ventilated with a 40%
oxygen/air mixture (Harvard Apparatus), end-tidal CO2 was
maintained between 2% and 4% and expired oxygen contin-
uously monitored (Datex-Ohmeda, Helsinki, Finland). Heart
rate was monitored by electrocardiogram (CT-1000; CWE
Instruments) or derived from blood pressure changes. The
depth of anaesthesia was monitored periodically throughout
the experiment by testing for sympathetic (pupillary and
cardiovascular) responses to noxious stimulation and with-
drawal reflexes in the absence of neuromuscular blockade.
2.1. Surgery
Midline craniotomies (approximately 20 mm diameter)
and C1/C2 laminectomies were performed allowing access to
the superior sagittal sinus and the area for recording neuronal
activity in the spinal cord. The superior sagittal sinus (SSS)
was isolated by dissecting the dura and falx cerebri adjacent
to them over approximately 15 mm. Small polyethylene
sheets were inserted under the isolated sinuses and laid over
the outlying dura mater then tucked under the edges of the
craniotomy to secure them. To prevent dehydration and to
provide electrical insulation to the cortex, a cylindrical
polypropylene dam was sealed to the bone around the
craniotomy with dental acrylic (Vertex, Zeist, Netherlands)
and filled with liquid paraffin (BDH Laboratory Supplies,
Poole, UK). Possible artefacts from arterial pulsation and
respiratory movement were reduced by bilateral pneumo-
thoraces, held patent with polypropylene tubes; immobiliza-
tion of the spine by clamping a thoracic spinous process to the
stereotaxic frame; clamping the C1 transverse processes to
auxiliary ear bar holders on the frame, and clamping the
remaining caudal portion of the dorsal C2 spinous process to
the frame.
2.2. Stimulation and recording
Isolated SSSs were gently lifted onto a pair of bipolar
platinum hook electrodes connected to a stimulus isolation
unit (SIU5A; Grass Instruments, West Warwick, RI). To
activate primary trigeminal afferents, the SSSs were supra-
maximally stimulated with stimulus-isolated (Grass SIU)
square wave pulses from a Grass S88 stimulator (250 As,110–150 V, 0.3–0.5 Hz) after neuromuscular blockade with
gallamine triethiodide (Concord, Essex, UK), initially 5–10
mg/kg i.v. and maintained with 5–10 mg/kg per h. The dura
mater above the recording regions on the surface of the spinal
cord was reflected after a midline incision and held to the
edges of the laminectomy with N-butyl-cyanoacrylate, fur-
ther stabilizing movement of the cord. Extracellular record-
ings were made using tungsten electrodes. Recording
electrode impedances were typically 0.4–2.1 MV when
measured at 1 kHz in 0.9% saline. After local removal of
the pia mater, the electrodes were lowered into the cord
substance caudal to the C2 roots in the area of the dorsal root
entry zone. The electrodes were advanced or retracted in the
cord substance in 5 Am steps using a microelectrode posi-
tioner consisting of a piezoelectric motor (IW-711, Burleigh
Instruments, Harpenden, UK) and ultra-low-noise controller
(6000ULN) attached to a micromanipulator (Kopf 1760-61).
Tissue culture grade agar (3% (w/v) in saline; Sigma) was set
over the exposed cord after electrode insertion to further
reduce cardiovascularly related movements. Signal from the
recording electrode attached to a high impedance headstage
preamplifier (NL100AK; Neurolog, Digitimer, Herts, UK)
was fed via an AC preamplifier (Neurolog NL104,
gain� 1000) through filters (Neurolog NL125; bandwidth
approximately 300 Hz to 20 kHz) and a 50 Hz noise
eliminator (Hum Bug, Quest Scientific, North Vancouver,
BC) to a second stage amplifier (Neurolog NL106) providing
variable gain (� 20– � 90). This signal (total gain approx-
imately � 20,000– � 90,000) was fed to a gated amplitude
discriminator (Neurolog NL201) and analogue-to-digital
converter (Cambridge Electronic Design, Cambridge, UK)
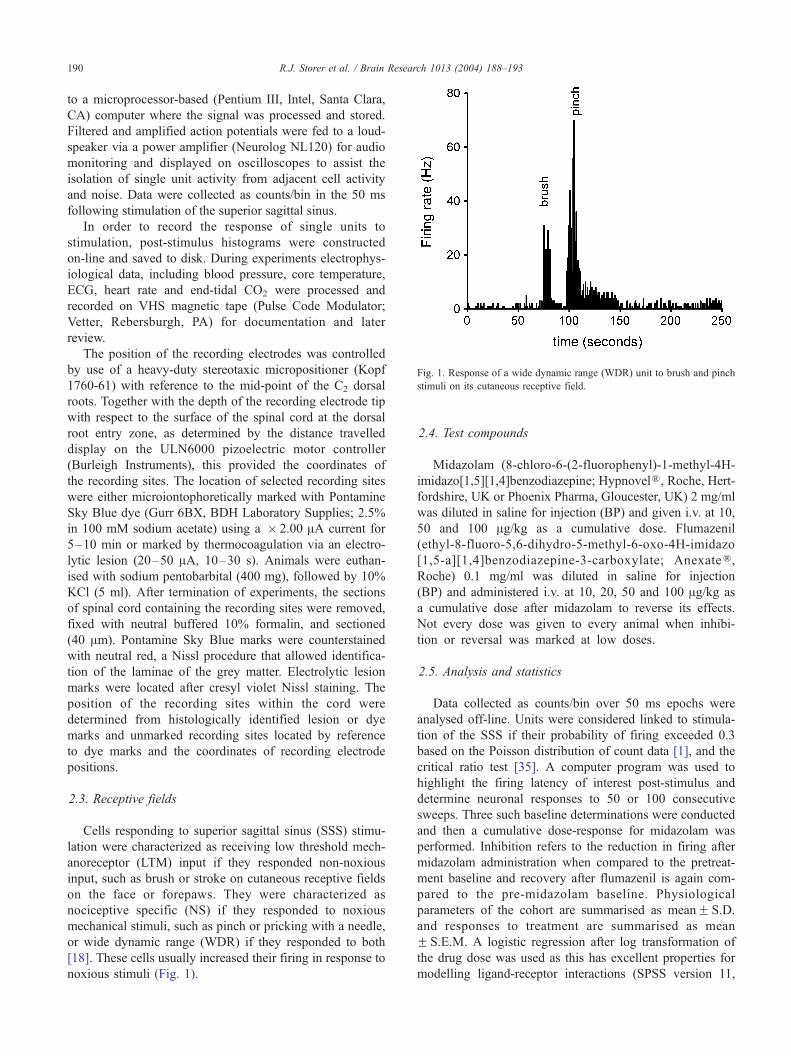
Fig. 1. Response of a wide dynamic range (WDR) unit to brush and pinch
stimuli on its cutaneous receptive field.
R.J. Storer et al. / Brain Research 1013 (2004) 188–193190
to a microprocessor-based (Pentium III, Intel, Santa Clara,
CA) computer where the signal was processed and stored.
Filtered and amplified action potentials were fed to a loud-
speaker via a power amplifier (Neurolog NL120) for audio
monitoring and displayed on oscilloscopes to assist the
isolation of single unit activity from adjacent cell activity
and noise. Data were collected as counts/bin in the 50 ms
following stimulation of the superior sagittal sinus.
In order to record the response of single units to
stimulation, post-stimulus histograms were constructed
on-line and saved to disk. During experiments electrophys-
iological data, including blood pressure, core temperature,
ECG, heart rate and end-tidal CO2 were processed and
recorded on VHS magnetic tape (Pulse Code Modulator;
Vetter, Rebersburgh, PA) for documentation and later
review.
The position of the recording electrodes was controlled
by use of a heavy-duty stereotaxic micropositioner (Kopf
1760-61) with reference to the mid-point of the C2 dorsal
roots. Together with the depth of the recording electrode tip
with respect to the surface of the spinal cord at the dorsal
root entry zone, as determined by the distance travelled
display on the ULN6000 pizoelectric motor controller
(Burleigh Instruments), this provided the coordinates of
the recording sites. The location of selected recording sites
were either microiontophoretically marked with Pontamine
Sky Blue dye (Gurr 6BX, BDH Laboratory Supplies; 2.5%
in 100 mM sodium acetate) using a � 2.00 AA current for
5–10 min or marked by thermocoagulation via an electro-
lytic lesion (20–50 AA, 10–30 s). Animals were euthan-
ised with sodium pentobarbital (400 mg), followed by 10%
KCl (5 ml). After termination of experiments, the sections
of spinal cord containing the recording sites were removed,
fixed with neutral buffered 10% formalin, and sectioned
(40 Am). Pontamine Sky Blue marks were counterstained
with neutral red, a Nissl procedure that allowed identifica-
tion of the laminae of the grey matter. Electrolytic lesion
marks were located after cresyl violet Nissl staining. The
position of the recording sites within the cord were
determined from histologically identified lesion or dye
marks and unmarked recording sites located by reference
to dye marks and the coordinates of recording electrode
positions.
2.3. Receptive fields
Cells responding to superior sagittal sinus (SSS) stimu-
lation were characterized as receiving low threshold mech-
anoreceptor (LTM) input if they responded non-noxious
input, such as brush or stroke on cutaneous receptive fields
on the face or forepaws. They were characterized as
nociceptive specific (NS) if they responded to noxious
mechanical stimuli, such as pinch or pricking with a needle,
or wide dynamic range (WDR) if they responded to both
[18]. These cells usually increased their firing in response to
noxious stimuli (Fig. 1).
2.4. Test compounds
Midazolam (8-chloro-6-(2-fluorophenyl)-1-methyl-4H-
imidazo[1,5][1,4]benzodiazepine; HypnovelR, Roche, Hert-fordshire, UK or Phoenix Pharma, Gloucester, UK) 2 mg/ml
was diluted in saline for injection (BP) and given i.v. at 10,
50 and 100 Ag/kg as a cumulative dose. Flumazenil
(ethyl-8-fluoro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo
[1,5-a][1,4]benzodiazepine-3-carboxylate; AnexateR,Roche) 0.1 mg/ml was diluted in saline for injection
(BP) and administered i.v. at 10, 20, 50 and 100 Ag/kg as
a cumulative dose after midazolam to reverse its effects.
Not every dose was given to every animal when inhibi-
tion or reversal was marked at low doses.
2.5. Analysis and statistics
Data collected as counts/bin over 50 ms epochs were
analysed off-line. Units were considered linked to stimula-
tion of the SSS if their probability of firing exceeded 0.3
based on the Poisson distribution of count data [1], and the
critical ratio test [35]. A computer program was used to
highlight the firing latency of interest post-stimulus and
determine neuronal responses to 50 or 100 consecutive
sweeps. Three such baseline determinations were conducted
and then a cumulative dose-response for midazolam was
performed. Inhibition refers to the reduction in firing after
midazolam administration when compared to the pretreat-
ment baseline and recovery after flumazenil is again com-
pared to the pre-midazolam baseline. Physiological
parameters of the cohort are summarised as meanF S.D.
and responses to treatment are summarised as mean
F S.E.M. A logistic regression after log transformation of
the drug dose was used as this has excellent properties for
modelling ligand-receptor interactions (SPSS version 11,

R.J. Storer et al. / Brain Research 1013 (2004) 188–193 191
Chicago IL). The effect of midazolam was examined using
regression with dose versus percentage reduction in counts/
bin after superior sagittal sinus stimulation. Similarly, the
effect of flumazenil was tested using a regression of dose
with percentage recovery to the baseline response. P < 0.05
was considered significant.
Fig. 3. Dose-dependent inhibition of superior sagittal sinus evoked
trigeminocervical nucleus activity by intravenous midazolam. The ordinate
represents the percentage reduction in neuronal firing to stimulation of the
superior sagittal sinus with increasing doses of midazolam when compared
to the baseline pre-midazolam firing responses. Data are plotted as mean
with standard error bars.
3. Results
Animals from which data is reported had cardiorespira-
tory parameters that were normal for the anaesthetised cat.
Blood gas levels were measured at intervals throughout the
experiment and were within normal limits: arterial blood pH
7.37F 0.03 and pCO2 3.62F 0.28 kPa.
3.1. Localization and neuronal characteristics
Extracellular recordings were made and data collected
from 11 neurons in the trigeminocervical complex of cats
[20]. Cells were located 4 mm rostral to 4 mm caudal to the
midpoint of the C2 rootlets, 0–150 Am lateral to the dorsal
root entry zone at a depth of approximately � 1400 Am to
around � 3000 Am below the (dorsal) cord surface (Fig. 2).
Cells responded to electrical sagittal sinus stimulation with
latencies consistent with A-y fibres (fibre input/afferents;
typically 8–10 ms). Cells received wide dynamic range
(WDR) or nociceptive specific (NS) mechanoreceptor input
Fig. 2. Localization of recording sites. A transverse section through the
spinal cord at the level of C2 is represented. Dye or lesion marked recording
sites (.) were identified histologically. The positions of unmarked or
unrecovered sites were identified by reference to the surface of the cord or
the position of dye marks made in electrode tracts and electrode tip
coordinates at the recording site (o). Although the positions of the recorded
units are mapped to only one side of the cord in the figure, they represent
results obtained from both the left-hand side and right-hand side of the
spinal cord. The scale bar represents a distance of 1 mm in both directions.
from cutaneous VI or VII receptive fields on the face and
forepaws.
3.2. Benzodiazepine receptor agonist
Intravenous administration of midazolam resulted in a
dose-dependent inhibition of superior sagittal sinus evoked
trigeminocervical nucleus activity (Fig. 3). The inhibition at
50 Ag/kg was 65F 11% compared to the baseline response
(n = 11). The overall dose effect was significant (F1,24 =
13.1, P < 0.002, R2 = 0.84).
3.3. Benzodiazepine receptor antagonist
Intravenous administration of flumazenil resulted in a
dose-dependent recovery of superior sagittal sinus evoked
Fig. 4. Dose-dependent recovery of superior sagittal sinus evoked
trigeminocervical nucleus activity by intravenous administration of
flumazenil. The ordinate is the response after flumazenil as a percentage
of the pre-midazolam treatment baseline data, plotted as the mean with
standard error bars.

R.J. Storer et al. / Brain Research 1013 (2004) 188–193192
trigeminocervical nucleus activity (Fig. 4). At a dose of 50
Ag/kg, there was a 64F 5% recovery (n = 6). The dose effect
of flumazenil was significant (F1,20 = 30.5, P < 0.001,
R2 = 0.94).
4. Discussion
This study demonstrates that the benzodiazepine receptor
agonist midazolam can dose-dependently inhibit nociceptive
neurons in the trigeminocervical complex with input from
the dura mater. Furthermore, to demonstrate the receptor
specificity of this effect, the inhibition by midazolam is
substantially and dose-dependently reversed by the benzo-
diazepine receptor antagonist flumazenil. Given that mid-
azolam has its effect on GABAergic transmission through
allosteric modulation of the GABAA receptor, these new
data are consistent with our previous characterization of the
importance of GABAA transmission in the trigeminocervi-
cal complex [46], and reinforce that adequate study of that
mechanism is necessary to fully understand trigeminovas-
cular nociceptive transmission.
Disordered central metabolism of the inhibitory neuro-
transmitter GABA has been implicated in migraine patho-
genesis [47]. Increases in the plasma concentrations of the
excitatory amino acids L-glutamate and L-aspartate during
migraine episodes have been reported [11]. Butalbital,
which is commonly used for migraine and tension-type
headache [10,37], may act through allosteric barbiturate
binding sites on GABAA receptors. Progesterone and related
steroids have been reported to be effective in a limited way
in migraine treatment [4,29,43]; although these approaches
are not used widely in clinical practice [26]. These com-
pounds could exert their effects through GABAA receptor
neurosteroid binding sites [25]. Moreover, clinical studies in
migraine have also indicated the efficacy of potentially
GABAergic drugs, including valproate [16,19,21,32], top-
iramate [5,33], propofol [24] and baclofen [17].
Both GABAA and GABAB receptors are located at both
peripheral and central sites. In the CNS, GABA immuno-
reactivity has been demonstrated in the spinal cord [6,31].
Both GABAA and GABAB receptors are present in the
dorsal horn of rats [3,36] and humans [3]. Immunoreactivity
to glutamic acid decarboxylase (GAD), the biosynthetic
enzyme for GABA, has been demonstrated in the trigeminal
nucleus caudalis (TNC) of the cat spinal dorsal horn [2].
Ultrastructural localisation has indicated that inhibitory
GABAergic controls in the trigeminal nucleus caudalis
involve both pre- and post-synaptic mechanisms, and are
probably mediated via direct contacts onto ascending pro-
jection neurons, as well as via synaptic contacts onto
nociceptive primary afferent fibres [2]. GABAA receptors
are hetero-oligomeric transmembrane proteins that act as
ligand-gated chloride ion channels. They contain distinct
binding sites for benzodiazepines, barbituates, and other
allosteric modulators of chloride ion flux [30,42]. The
action of valproate may be mediated through GABAA
receptors [7]. Expression of the proto-oncogene c-fos as a
marker of nociceptive neuronal activity within the trigem-
inal nucleus caudalis is reduced by valproate [8] and
allopregnanolone, a neurosteroid progesterone metabolite,
which modulates GABAA receptor activity through an
allosteric binding site [7].
It seems likely that GABAergic modulation of trigemi-
novascular nociceptive transmission forms part of the brain-
stem modulatory systems that normally gate head pain. It is
known that activation of neurons in the periaqueductal grey
(PAG) matter can inhibit trigeminovascular nociceptive
transmission [22]. At the level of the PAG, GABAA receptor
activation is important [23]. In the trigeminocervical com-
plex locally ejected muscimol, a GABAA receptor agonist,
inhibits neurons responding to trigeminovascular input and
to L-glutamate microiontophoresis [46]. These observations
are consistent with a post-synaptic GABAA receptor and
that GABAA receptors are usually not found in pre-synaptic
locations [28]. It has been shown that GABAA receptors
located in the spinal cord have an antinociceptive effect
when activated [9,12,15]. The new data seem consistent
with a general principle effect of GABAA modulation on
nociceptive afferent traffic.
In conclusion, we show that midazolam, a benzodiaze-
pine receptor agonist that facilitates GABAA receptor mod-
ulation, can dose-dependently inhibit trigeminal neurons
activated by stimulation of normally pain-producing trige-
minovascular afferents. Furthermore, the benzodiazepine
receptor antagonist flumazenil dose-dependently inhibits
the effect of midazolam. Taken together, the data provide
further support for a potent and important GABAA receptor
mediated modulation of trigeminovascular nociceptive
transmission that may explain some of the clinical effects
of preventive anti-migraine treatments.
Acknowledgements
The authors thank Paul Hammond and Michele Lasa-
landara for their excellent technical assistance. This work
has been supported by the Wellcome Trust, The Guarantors
of Brain (KGS) and the Patrick Bertould Trust (KGS). PJG
is a Wellcome Senior Research Fellow.
References
[1] P. Armitage, G. Berry, Statistical Methods in Medical Research, 3rd
edn., Blackwell, Oxford, 1994.
[2] A.I. Basbaum, E.J. Glazer, W. Oertel, Immunoreactive glutamic
acid decarboxylase in the trigeminal nucleus caudalis of the cat:
a light- and electron-microscopic analysis, Somatosens. Res. 4
(1986) 77–94.
[3] N.G. Bowery, A.L. Hudson, G.W. Price, GABAA and GABAB recep-
tor site distribution in the rat central nervous system, Neuroscience 20
(1987) 365–383.

R.J. Storer et al. / Brain Research 1013 (2004) 188–193 193
[4] W.G. Bradley, P. Hudgson, J.B. Foster, D.J. Newell, Double-blind
controlled trial of a micronized preparation of flumedroxone (Demi-
gran) in prophylaxis of migraine, BMJ 3 (1968) 531–533.
[5] J.L. Brandes, D. Jacobs, W. Neto, S. Bhattacharaya, Topiramate in the
prevention of migraine headache: a randomized, double-blind, place-
bo-controlled, parallel study (MIGR-002), Neurology 60 (Suppl. 1)
(2003) A238.
[6] S.M. Carlton, E.S. Hayes, Light microscopic and ultrastructural anal-
ysis of GABA-immunoreactive profiles in the monkey spinal cord,
J. Comp. Neurol. 300 (1990) 162–182.
[7] F.M. Cutrer, M.A. Moskowitz, The actions of valproate and neuro-
steroids in a model of trigeminal pain, Headache 36 (1996) 579–585.
[8] F.M. Cutrer, V. Limmroth, G. Ayata, M.A. Moskowitz, Attenuation
by valproate of c-fos immunoreactivity in trigeminal nucleus caudalis
induced by intracisternal capsaicin, Br. J. Pharmacol. 116 (1995)
3199–3204.
[9] E.J. Drower, D.L. Hammond, GABAergic modulation of nociceptive
threshold: effects of THIP and bicuculline microinjected in the ventral
medulla of the rat, Brain Res. 450 (1988) 316–324.
[10] A.H. Elkind, Drug abuse and headache, Med. Clin. North Am. 75
(1991) 717–732.
[11] M.D. Ferrari, J. Odink, K.D. Box, M.J.A. Malessy, G.W. Bruyn,
Neuroexcitatory plasma amino acids are elevated in migraine, Neu-
rology 40 (1990) 1582–1586.
[12] H.L. Fields, M.M. Heinricher, P. Mason, Neurotransmitters in nocicep-
tive modulatory circuits, Annu. Rev. Neurosci. 14 (1991) 219–245.
[13] P.J. Goadsby, The pharmacology of headache, Prog. Neurobiol. 62
(2000) 509–525.
[14] P.J. Goadsby, R.B. Lipton, M.D. Ferrari, Migraine—current under-
standing and treatment, N. Engl. J. Med. 346 (2002) 257–270.
[15] M.M. Heinricher, H.J. Kaplan, GABA-mediated inhibition in rostral
ventromedial medulla: role in nociceptive modulation in the lightly
anesthetized rat, Pain 47 (1991) 105–113.
[16] R. Hering, A. Kuritzky, Sodium valproate in the prophylactic treat-
ment of migraine: a double-blind study versus placebo, Cephalalgia 12
(1992) 81–84.
[17] R. Hering-Hanit, Baclofen for prevention of migraine, Cephalalgia 19
(1999) 589–591.
[18] J.W. Hu, J.O. Dostrovsky, B.J. Sessle, Functional properties of neu-
rons in cat trigeminal subnucleus caudalis (medullary dorsal horn). I.
Responses to oral-facial noxious and nonnoxious stimuli and projec-
tions to thalamus and subnucleus oralis, J. Neurophysiol. 45 (1981)
173–192.
[19] R. Jensen, T. Brinck, J. Olesen, Sodium valproate has a prophylactic
effect in migraine without aura: A triple-blind, placebo-controlled
crossover study, Neurology 44 (1994) 647–651.
[20] H. Kaube, K.A. Keay, K.L. Hoskin, R. Bandler, P.J. Goadsby, Ex-
pression of c-Fos-like immunoreactivity in the caudal medulla and
upper cervical cord following stimulation of the superior sagittal sinus
in the cat, Brain Res. 629 (1993) 95–102.
[21] J. Klapper, On behalf of the Divalproex Sodium in Migraine Prophy-
laxis Study Group, Divalproex sodium in migraine prophylaxis: a
dose-controlled study, Cephalalgia 17 (1997) 103–108.
[22] Y.E. Knight, P.J. Goadsby, The periaqueductal gray matter modulates
trigeminovascular input: a role in migraine? Neuroscience 106
(2001) 793–800.
[23] Y.E. Knight, T. Bartsch, P.J. Goadsby, Trigeminal antinociception
induced by bicuculline in the periaqueductal grey (PAG) is not af-
fected by PAG P/Q-type calcium channel blockade in rat, Neurosci.
Lett. 336 (2003) 113–116.
[24] J.C. Krusz, V. Scott, J. Belanger, Intravenous propofol: unique ef-
fectiveness in treating intractable migraine, Headache 40 (2000)
224–230.
[25] J.J. Lambert, D. Belelli, C. Hill-Venning, H. Callachan, J.A. Peters,
Neurosteroid modulation of native and recombinant GABAA recep-
tors, Cell. Mol. Neurobiol. 16 (1996) 155–174.
[26] J.W. Lance, P.J. Goadsby, Mechanism and Management of Headache,
6th edn., Butterworth-Heinemann, London, 1998.
[27] R.B. Lipton, W.F. Stewart, S. Diamond, M.L. Diamond, M. Reed,
Prevalence and burden of migraine in the United States: data from the
American Migraine Study II, Headache 41 (2001) 646–657.
[28] H. Liu, H. Wang, M. Sheng, L.Y. Jan, Y.N. Jan, A.I. Basbaum,
Evidence for presynaptic N-methyl-D-aspartate autoreceptors in the
spinal cord dorsal horn, Proc. Natl. Acad. Sci. U. S. A. 91 (1994)
8383–8387.
[29] P.O. Lundberg, Prophylactic treatment of migraine with flumedrox-
one, Acta Neurol. Scand. 45 (1969) 309–326.
[30] R.L. Macdonald, R.W. Olsen, GABAA receptor channels, Annu. Rev.
Neurosci. 17 (1994) 569–602.
[31] R. Magoul, B. Onteniente, M. Geffard, A. Calas, Anatomical distri-
bution and ultrastructural organization of the GABAergic system in
the rat spinal cord. An immunocytochemical study using anti-GABA
antibodies, Neuroscience 20 (1987) 1001–1009.
[32] N.T. Mathew, J.R. Saper, S.D. Silberstein, L. Rankin, H.G. Markley,
S. Solomon, A.M. Rapoport, C.J. Silber, R.L. Deaton, Migraine pro-
phylaxis with divalproex, Arch. Neurol. 52 (1995) 281–286.
[33] N.T. Mathew, J. Schmitt, D. Jacobs, W. Neto, Topiramate in the
migraine prevention (MIGR-001): effect on migraine frequency, Neu-
rology 60 (Suppl. 1) (2003) A336.
[34] M. Menken, T.L. Munsat, J.F. Toole, The global burden of disease
study—implications for neurology, Arch. Neurol. 57 (2000) 418–420.
[35] J. Nagler, N. Conforti, S. Feldman, Alterations produced by cortisol in
the spontaneous activity and responsiveness to sensory stimuli of
single cells in the tuberal hypothalamus of the rat, Neuroendocrino-
logy 12 (1973) 52–66.
[36] J.M. Palacios, J.K. Wamsley, M.J. Kuhar, High affinity GABA recep-
tors—autoradiographic localization, Brain Res. 222 (1981) 285–307.
[37] N.H. Raskin, Headache, Churchill-Livingstone, New York, 1988.
[38] B.K. Rasmussen, J. Olesen, Symptomatic and nonsymptomatic head-
aches in a general population, Neurology 42 (1992) 1225–1231.
[39] E. Roberts, Disinhibition as an organizing principle in the nervous
system—the role of the GABA system. Application to neurologic and
psychiatric disorders, in: E. Roberts, T.N. Chase, D.B. Tower (Eds.),
GABA in Nervous System Function, Raven Press, New York, 1976,
pp. 515–539.
[40] L.A. Roberts, C. Beyer, B.R. Komisaruk, Nociceptive responses to
altered GABAergic activity at the spinal cord, Life Sci. 39 (1986)
1667–1674.
[41] R.P. Shank, J.F. Gardocki, A.J. Streeter, B.E. Maryanoff, An over-
view of the preclinical aspects of topiramate: pharmacology, phar-
macokinetics, and mechanism of action, Epilepsia 41 (Suppl. 1)
(2000) S3–S9.
[42] W. Sieghart, GABAA receptors: ligand-gated Cl� ion channels mod-
ulated by multiple drug-binding sites, Trends Pharmacol. Sci. 13
(1992) 446–450.
[43] I. Singh, I. Singh, D. Singh, Progesterone in the treatment of mi-
graine, Lancet i (1947) 745–747.
[44] R.J. Storer, P.J. Goadsby, Topiramate inhibits trigeminovascular traf-
fic in the cat: a possible locus of action in the prevention of migraine,
Cephalalgia 23 (2003) 726.
[45] R.J. Storer, P. Butler, K.L. Hoskin, P.J. Goadsby, A simple method,
using 2-hydroxypropyl-h-cyclodextrin, of administering a-chloralose
at room temperature, J. Neurosci. Methods 77 (1997) 49–53.
[46] R.J. Storer, S. Akerman, P.J. Goadsby, GABA receptors modulate
trigeminovascular nociceptive neurotransmission in the trigeminocer-
vical complex, Br. J. Pharmacol. 134 (2001) 896–904.
[47] K.M.A. Welch, E. Chabi, K. Bartosh, V.S. Achar, J.S. Meyer, Cere-
brospinal fluid g aminobutyric acid levels in migraine, BMJ 3 (1975)
516–517.
[48] J.G. Whitwam, Flumazenil and midazolam in anaesthesia, Acta
Anaesthesiol. Scand. 39 (Suppl. 108) (1995) 15–22.