from marnett and plastaras, trends genet . 17 , 214 (2001)
DESCRIPTION
Endogenous DNA Damage. from Marnett and Plastaras, Trends Genet . 17 , 214 (2001). Biological Molecules are Labile. RNA is susceptible to hydrolysis. Reduction of ribose to deoxyribose gives DNA greater stability. N-glycosyl bond of DNA is more labile. - PowerPoint PPT PresentationTRANSCRIPT
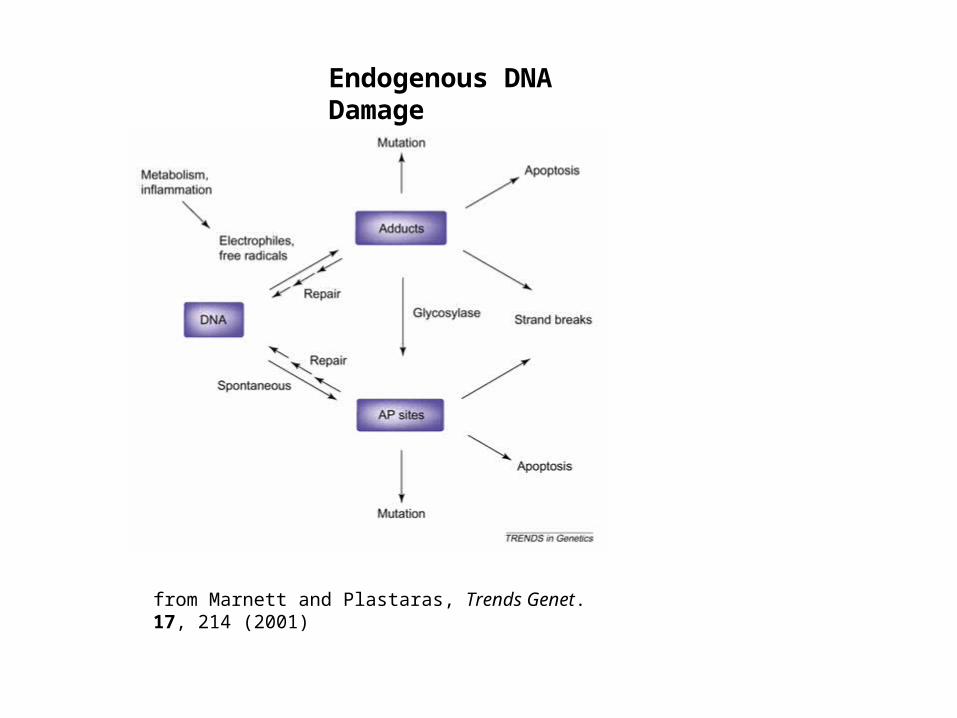
from Marnett and Plastaras, Trends Genet. 17, 214 (2001)
Endogenous DNA Damage

Biological Molecules are Labile
RNA is susceptible to hydrolysis
Reduction of ribose to deoxyribose gives DNA greater stability
N-glycosyl bond of DNA is more labile
DNA damage occurs from normal cellular operations and random interactions with the environment
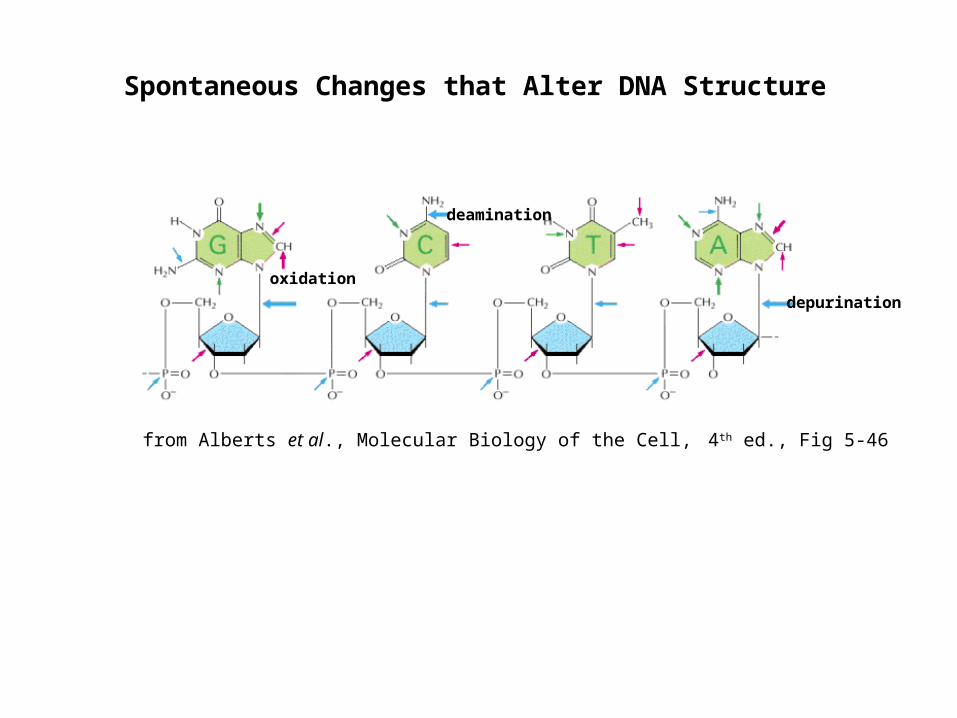
Spontaneous Changes that Alter DNA Structure
from Alberts et al., Molecular Biology of the Cell, 4th ed., Fig 5-46
depurination
deamination
oxidation
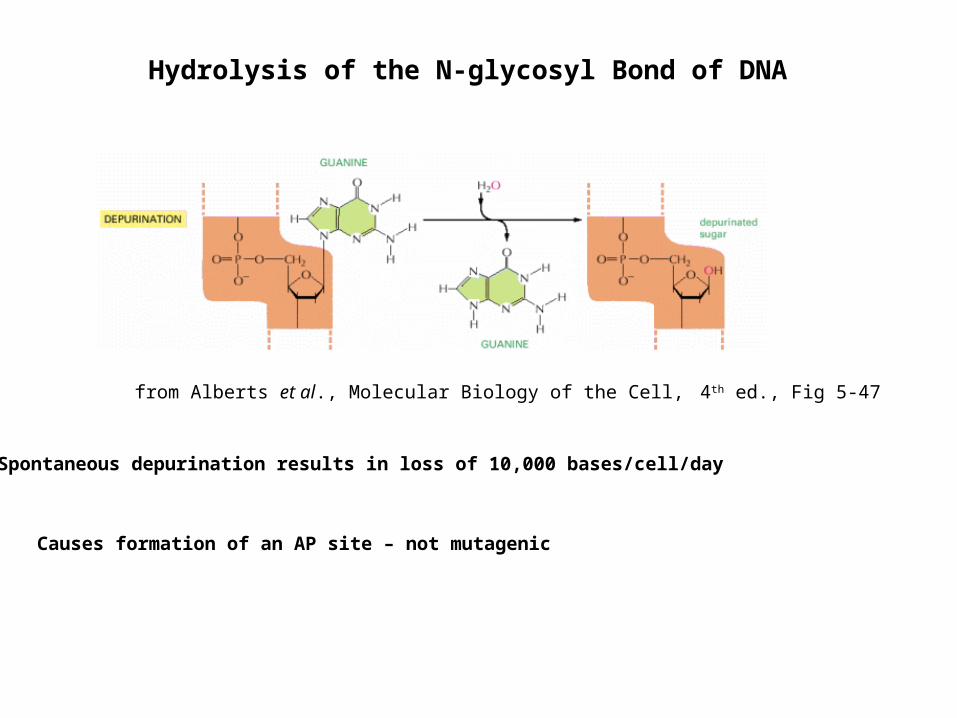
Hydrolysis of the N-glycosyl Bond of DNA
Spontaneous depurination results in loss of 10,000 bases/cell/day
Causes formation of an AP site – not mutagenic
from Alberts et al., Molecular Biology of the Cell, 4th ed., Fig 5-47
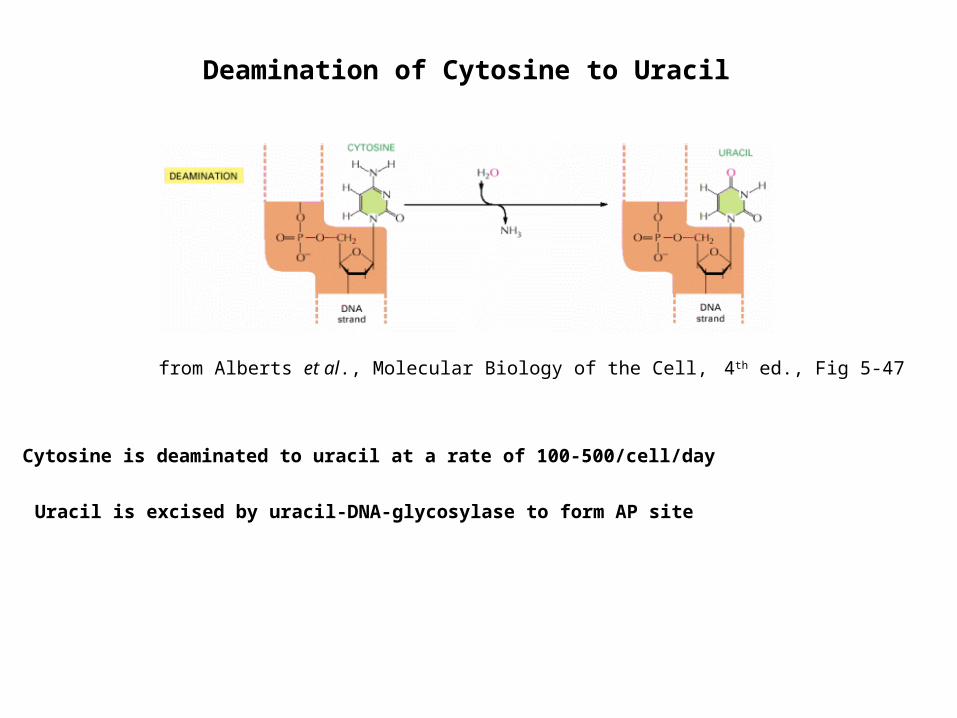
from Alberts et al., Molecular Biology of the Cell, 4th ed., Fig 5-47
Cytosine is deaminated to uracil at a rate of 100-500/cell/day
Uracil is excised by uracil-DNA-glycosylase to form AP site
Deamination of Cytosine to Uracil
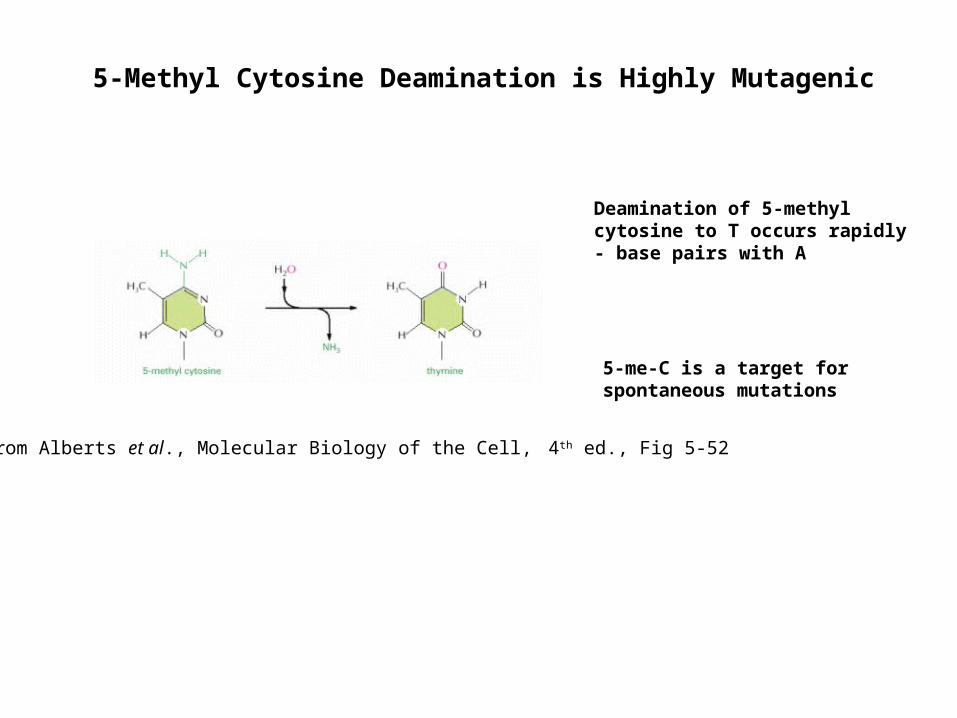
5-Methyl Cytosine Deamination is Highly Mutagenic
from Alberts et al., Molecular Biology of the Cell, 4th ed., Fig 5-52
Deamination of 5-methyl cytosine to T occurs rapidly- base pairs with A
5-me-C is a target for spontaneous mutations
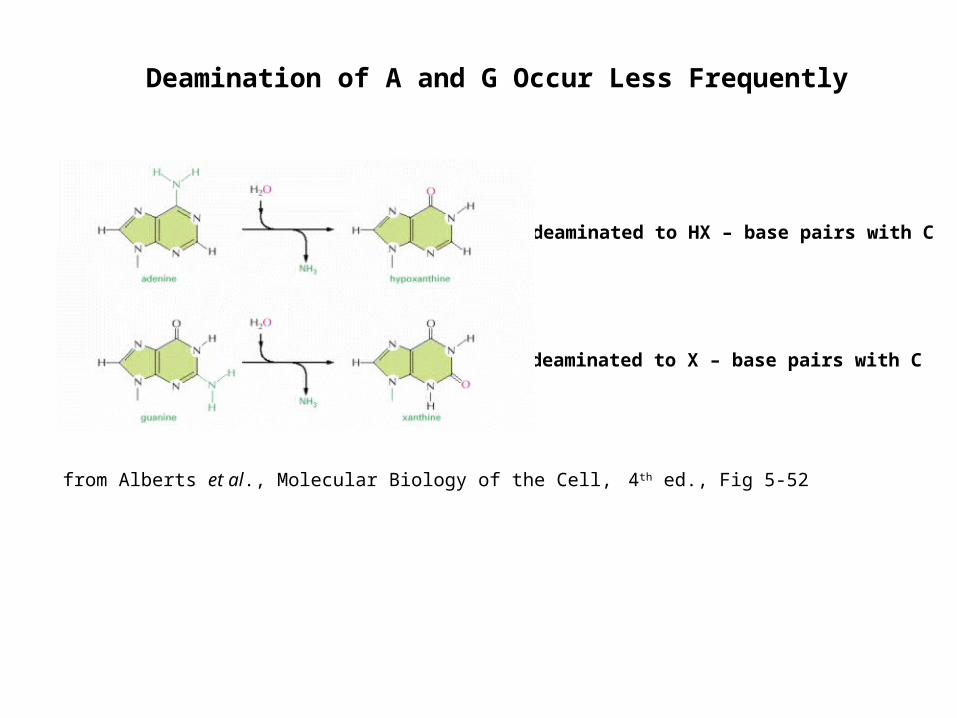
Deamination of A and G Occur Less Frequently
A is deaminated to HX – base pairs with C
G is deaminated to X – base pairs with C
from Alberts et al., Molecular Biology of the Cell, 4th ed., Fig 5-52

Oxidative Damage of DNA
Oxidative damage results from aerobic metabolism, environmental toxins, activated macrophages, and signaling molecules (NO)
Compartmentation limits oxidative DNA damage
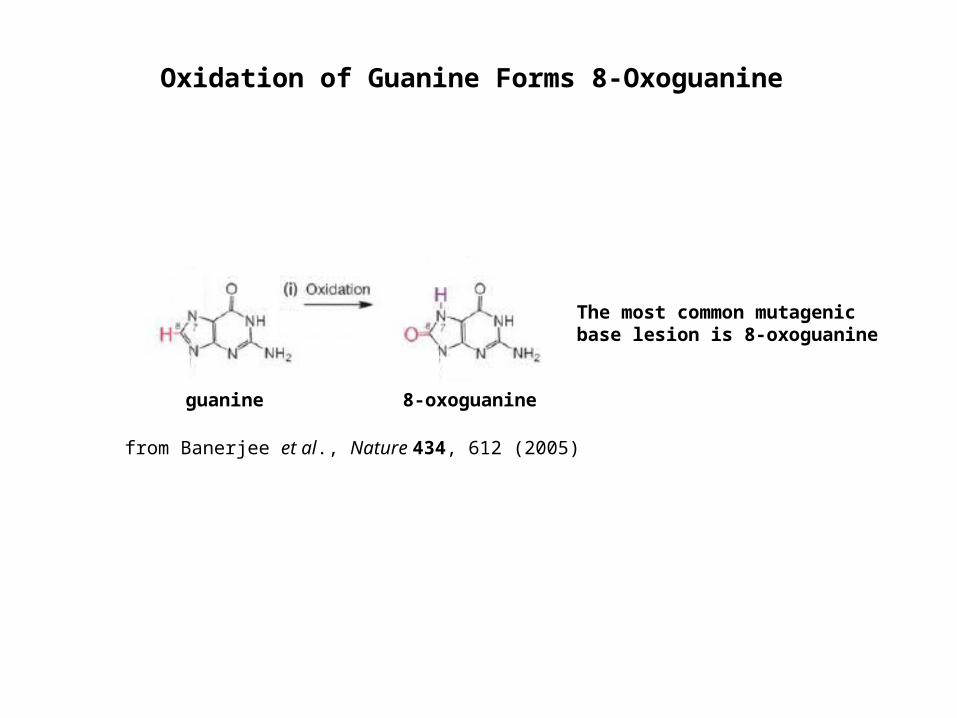
guanine 8-oxoguanine
The most common mutagenic base lesion is 8-oxoguanine
from Banerjee et al., Nature 434, 612 (2005)
Oxidation of Guanine Forms 8-Oxoguanine
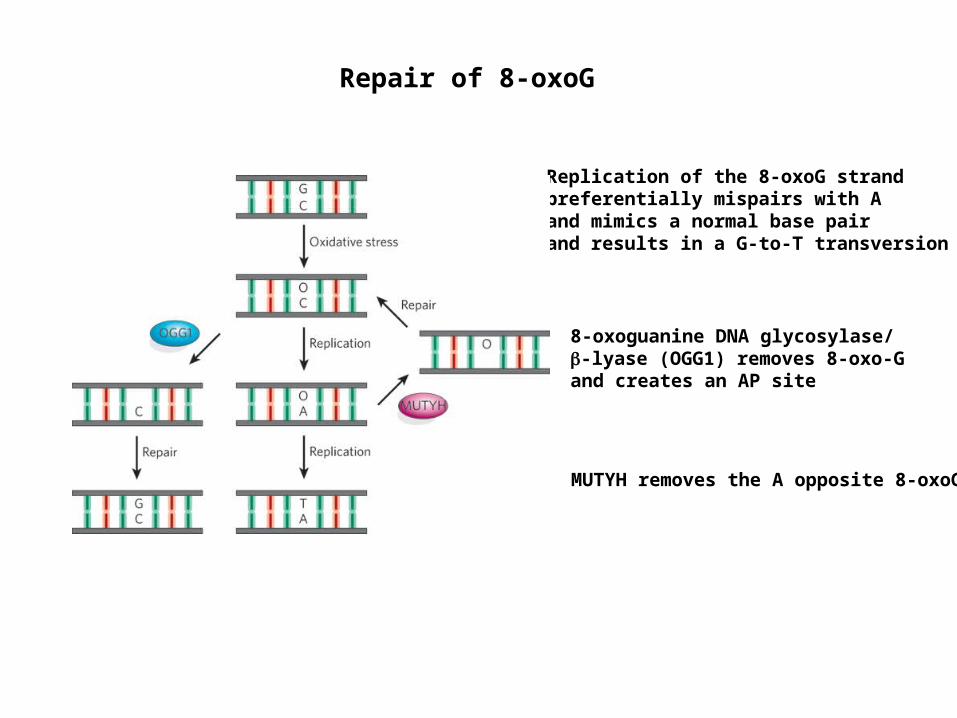
Repair of 8-oxoG
8-oxoguanine DNA glycosylase/-lyase (OGG1) removes 8-oxo-G and creates an AP site
Replication of the 8-oxoG strand preferentially mispairs with A and mimics a normal base pair and results in a G-to-T transversion
MUTYH removes the A opposite 8-oxoG

Oxidation of dNTPs are Mutagenic
cGTP is oxidized to 8-OH-dGTP and is misincorporated opposite A
MutT converts 8-OH-dGTP to 8-OH-dGMP
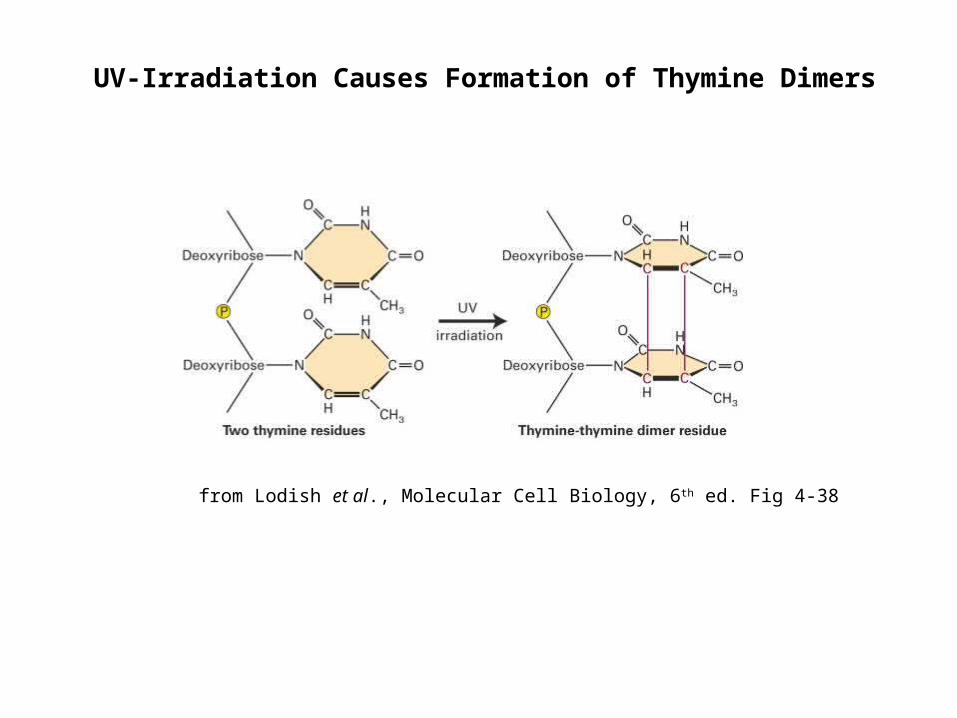
UV-Irradiation Causes Formation of Thymine Dimers
from Lodish et al., Molecular Cell Biology, 6th ed. Fig 4-38

Nonenzymatic Methylation of DNA
Formation of 600 3-me-A residues/cell/day are caused by S-adnosylmethionine
3-me-A is cytotoxic and is repaired by 3-me-A-DNA glycosylase
7-me-G is the main aberrant base present in DNA and is repaired by nonenzymatic cleavage of the glycosyl bond

Effect of Chemical Mutagens
Nitrous acid causes deamination of C to U and A to HX
U base pairs with AHX base pairs with C
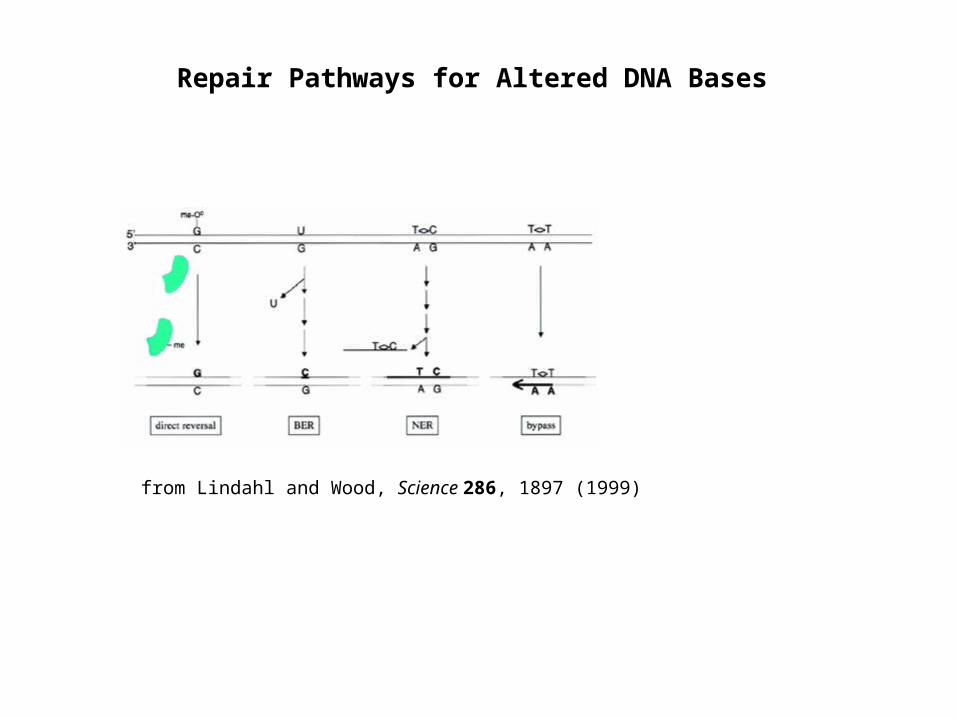
Repair Pathways for Altered DNA Bases
from Lindahl and Wood, Science 286, 1897 (1999)

Direct Repair of DNA
Photoreactivation of pyrimidine dimers by photolyase restores the original DNA structure
O6-methylguanine is repaired by removal of methyl group by MGMT
1-methyladenine and 3-methylcytosine are repaired by oxidative demethylation
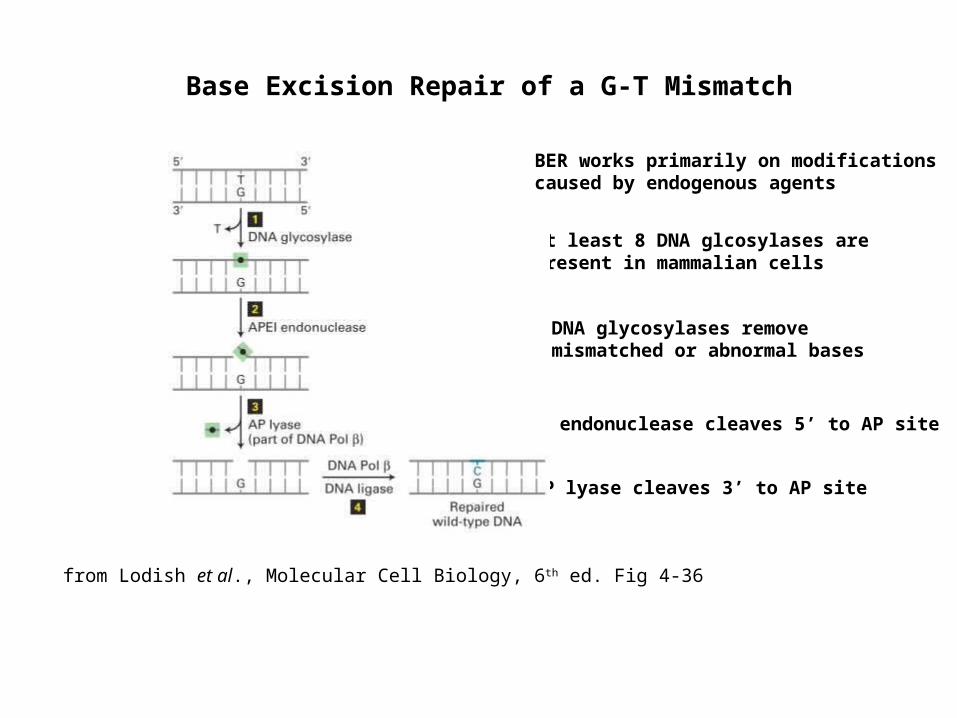
Base Excision Repair of a G-T Mismatch
At least 8 DNA glcosylases are present in mammalian cells
DNA glycosylases remove mismatched or abnormal bases
AP endonuclease cleaves 5’ to AP site
AP lyase cleaves 3’ to AP site
from Lodish et al., Molecular Cell Biology, 6th ed. Fig 4-36
BER works primarily on modifications caused by endogenous agents

Mechanism of hOGG1 Action
hOGG1 binds nonspecifically to DNA
Contacts with C results in the extrusion of corresponding base in the opposite strand
G is extruded into the G-specific pocket, but is denied access to the oxoG pocket
oxoG moves out of the G-specific pocket, enters the oxoG-specific pocket, and excised from the DNA
from David, Nature 434, 569 (2005)
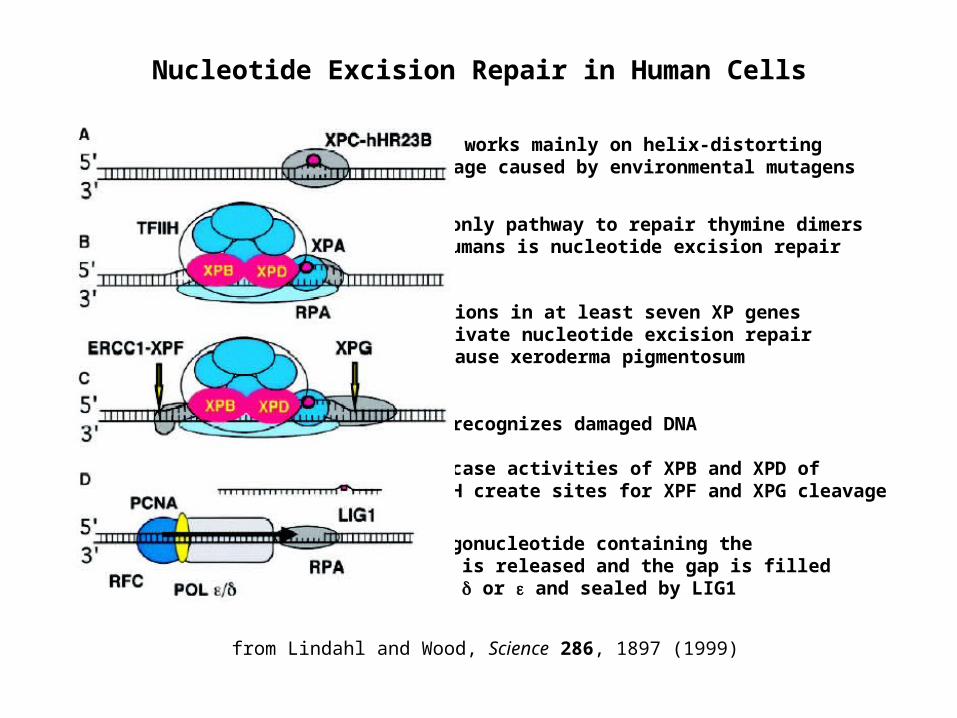
Nucleotide Excision Repair in Human Cells
Mutations in at least seven XP genes inactivate nucleotide excision repair and cause xeroderma pigmentosum
The only pathway to repair thymine dimers in humans is nucleotide excision repair
XPC recognizes damaged DNA Helicase activities of XPB and XPD of TFIIH create sites for XPF and XPG cleavage
NER works mainly on helix-distorting damage caused by environmental mutagens
An oligonucleotide containing the lesion is released and the gap is filled by POL or and sealed by LIG1
from Lindahl and Wood, Science 286, 1897 (1999)

Transcription-coupled Repair
Repair of the transcribed strand of active genes is corrected 5-10-fold as fast as the nontranscribed strand
All the factors required for NER are required for transcription-coupled repair except XPC
The arrest of POL II progression at a lesion served as a damage recognition signal
Recruitment of NER factors also involves CS-A and CS-B

Nucleotide Excision Repair Pathway in Mammals
Cockayne’s Syndrome and Trichothiodystrophy are multisystem disorders defective in transcription-coupled DNA repair

Mismatch Repair in E. coli
MutS binds to mismatch and recruits MutL
Activates endonuclease activity of MutH and nicks the nearest unmethylated GATC
Recruits MutU (helicase) and exonucleases
DNA pol III fills in the gap
Newly replicated DNA is hemimethylated

Mismatch Repair in Human Cells
Defective mismatch repair is the primary cause of certain types of human cancers
MSH2 and MSH6 bind to mismatch-containing DNA and distinguish between the template and newly synthesized strand
from Lodish et al., Molecular Cell Biology, 6th ed. Fig 4-37
MutL introduces random nicks at distal sites on the same strand
The gap is filled in by DNA polymerase and DNA ligase
EXO1 at 5’-side of the mismatch activates a 5’-3’ exonuclease and removes mismatch
MMR complex identifies newly synthesized strand by the presence of a 3’-terminus
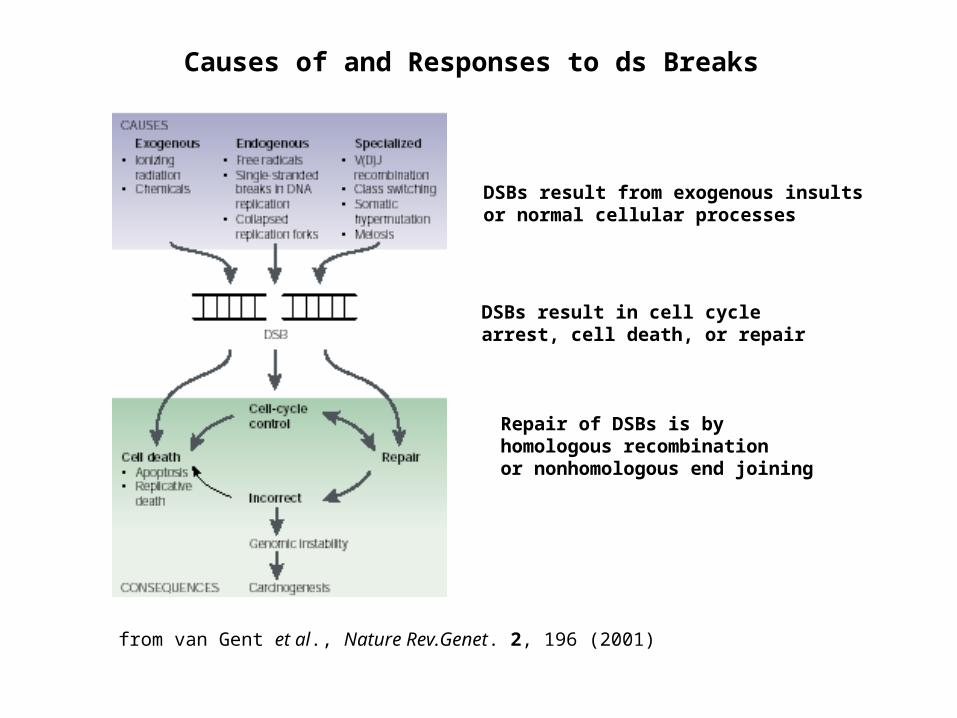
Causes of and Responses to ds Breaks
Repair of DSBs is by homologous recombination or nonhomologous end joining
DSBs result from exogenous insults or normal cellular processes
DSBs result in cell cycle arrest, cell death, or repair
from van Gent et al., Nature Rev.Genet. 2, 196 (2001)
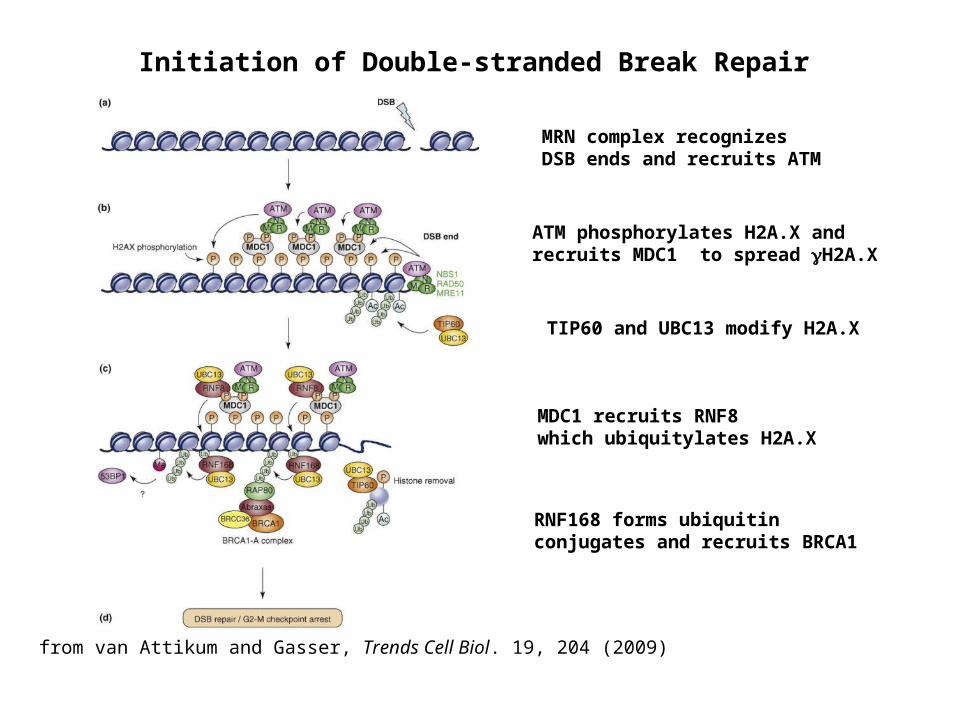
Initiation of Double-stranded Break Repair
from van Attikum and Gasser, Trends Cell Biol. 19, 204 (2009)
MRN complex recognizes DSB ends and recruits ATM
ATM phosphorylates H2A.X and recruits MDC1 to spread H2A.X
TIP60 and UBC13 modify H2A.X
MDC1 recruits RNF8 which ubiquitylates H2A.X
RNF168 forms ubiquitin conjugates and recruits BRCA1
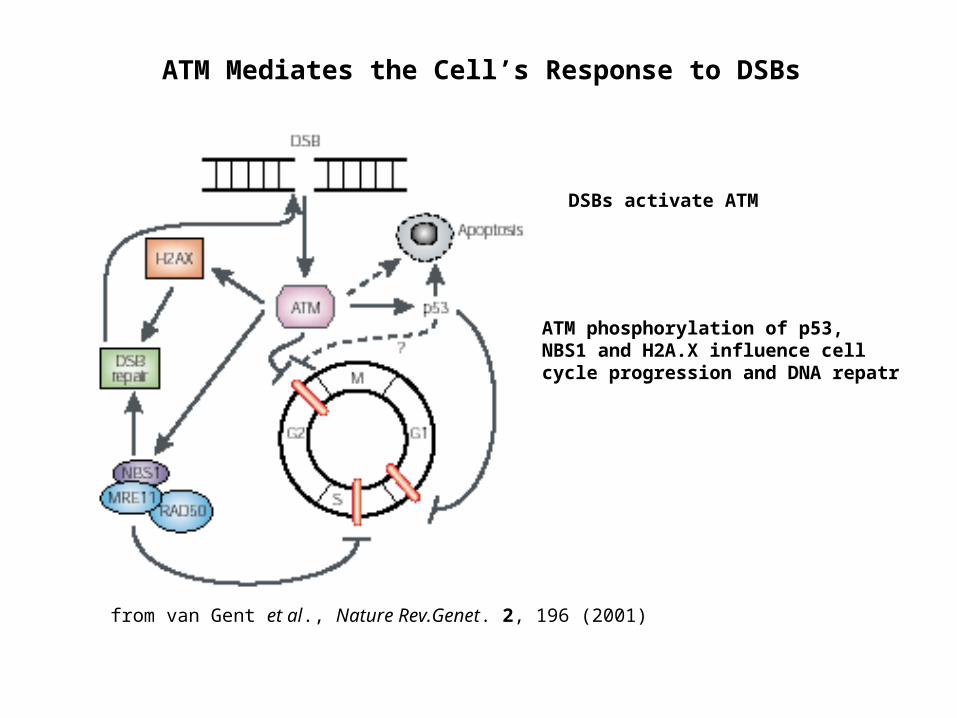
ATM Mediates the Cell’s Response to DSBs
from van Gent et al., Nature Rev.Genet. 2, 196 (2001)
DSBs activate ATM
ATM phosphorylation of p53, NBS1 and H2A.X influence cell cycle progression and DNA repatr

ssDNAs with 3’ends are formed and coated with Rad51, the RecA homolog
Rad51-coated ssDNA invades the homologous dsDNA in the sister chromatid
The 3’-end is elongated by DNA polymerase, and base pairs with ss 3-end of the other broken DNA
DNA polymerase and DNA ligase fills in gaps
from Lodish et al., Molecular Cell Biology, 5th ed. Fig 23-31
Repair of ds Breaks by Homologous Recombination
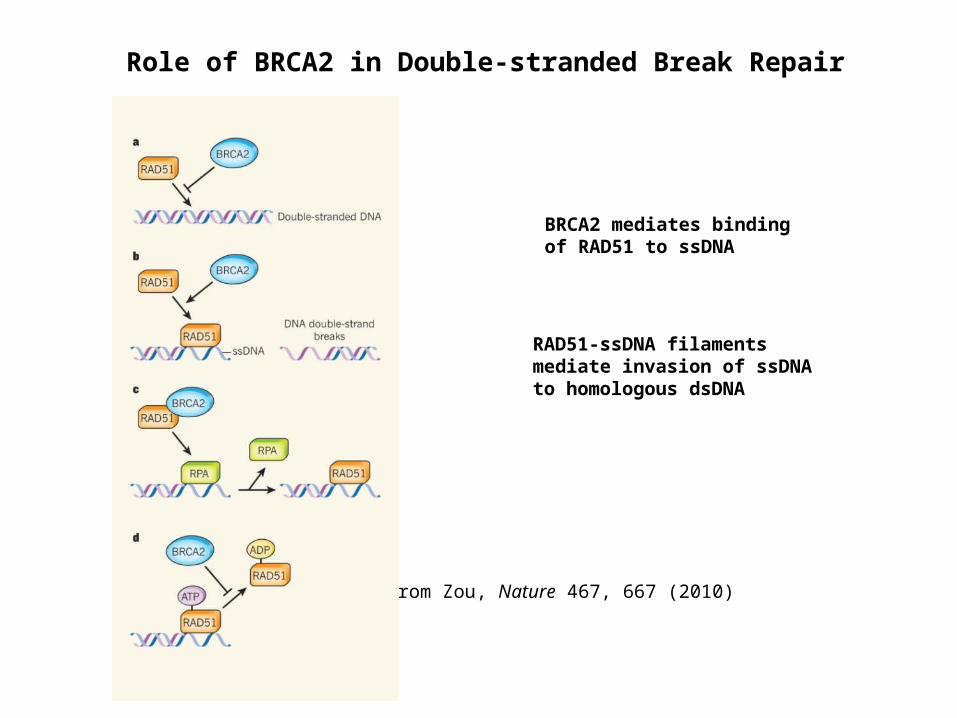
Role of BRCA2 in Double-stranded Break Repair
BRCA2 mediates binding of RAD51 to ssDNA
RAD51-ssDNA filaments mediate invasion of ssDNA to homologous dsDNA
from Zou, Nature 467, 667 (2010)
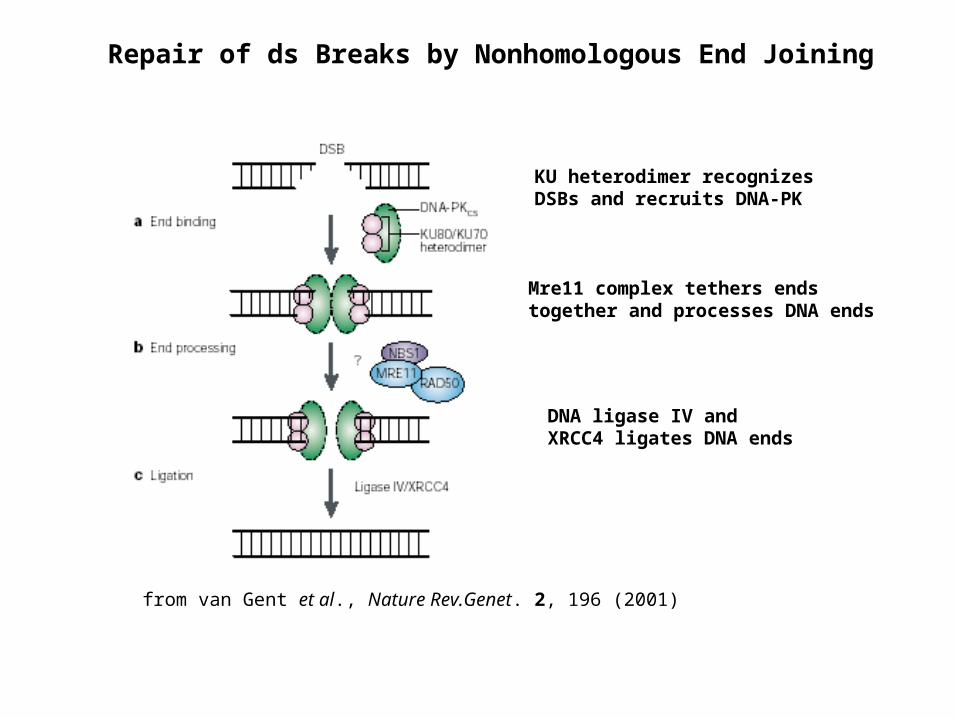
from van Gent et al., Nature Rev.Genet. 2, 196 (2001)
Repair of ds Breaks by Nonhomologous End Joining
KU heterodimer recognizes DSBs and recruits DNA-PK
Mre11 complex tethers ends together and processes DNA ends
DNA ligase IV and XRCC4 ligates DNA ends
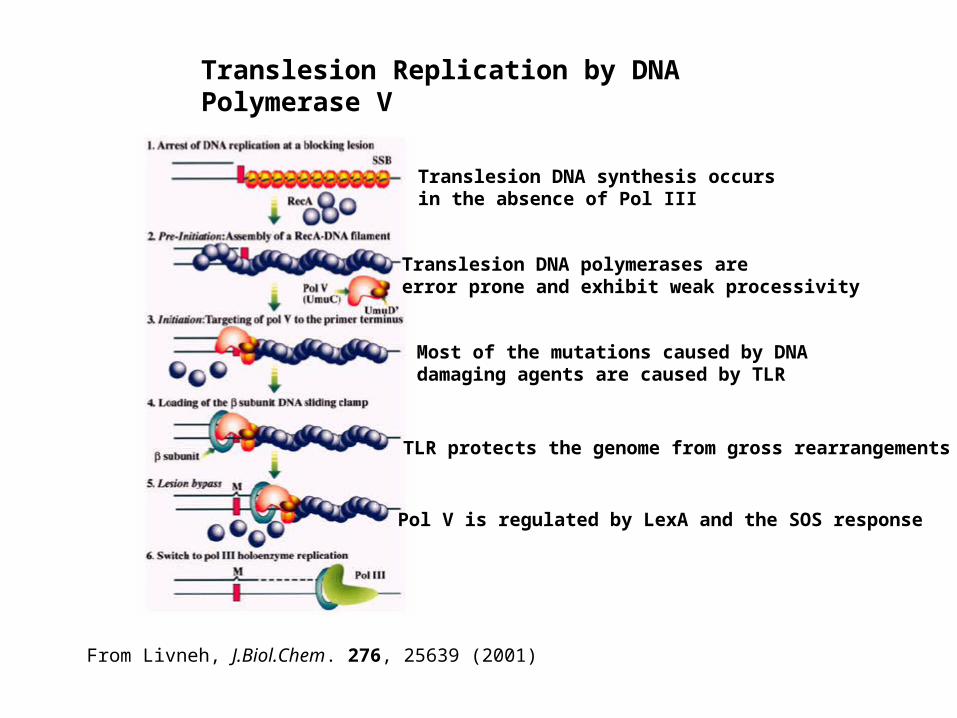
Translesion Replication by DNA Polymerase V
From Livneh, J.Biol.Chem. 276, 25639 (2001)
Translesion DNA synthesis occurs in the absence of Pol III
Translesion DNA polymerases are error prone and exhibit weak processivity
Most of the mutations caused by DNA damaging agents are caused by TLR
TLR protects the genome from gross rearrangements
Pol V is regulated by LexA and the SOS response