fda requirements for suppliers - process vision
TRANSCRIPT

FDA
requirements
for suppliers
Willem van den Biggelaar
Quality & Regulatory Consultant
PROCESS VISION DOWN TO EARTH QUALITY SERVICES

2 of 73 © Process Vision – FDA requirements for suppliers
Let op
• Deze sheets geven alleen maar een summiere indruk
• Nadruk op verschil met ISO 13485
• Nadruk op design, engineering en manufacturing
• Zoals altijd: “The devil is in the details”
• De sheets zijn deels engels, deels nederlands

3 of 73 © Process Vision – FDA requirements for suppliers
Content
• Wie is de FDA
• 21 CFR 820 Quality System
• Design controls
• Production and process controls
• Document and record control
• Process Validation
• Training
• CAPA
• Complaint files
• Tenslotte

4 of 73 © Process Vision – FDA requirements for suppliers
WIE IS DE FDA

5 of 73 © Process Vision – FDA requirements for suppliers
Wie is de FDA
• De Amerikaanse overheidsinstantie FDA controleert en reguleert de volgende producten • Cosmetica
• Voeding
• Medicijnen
• Medische apparatuur
• Producten voor dieren.
• Zij mogen • Produkten weren van de Amerikaanse markt
• Fabrieken stilleggen (in Nederland via Voedsel en Waren authoriteit)
• Boetes opleggen
• Juridisch vervolgen

6 of 73 © Process Vision – FDA requirements for suppliers
FDA historie
• Opgericht in 1906 • Aanleiding: slechte hygiëne en wantoestanden in
vleeswerkende industrie in Chicago en omgeving
• 1938 Food, Drug & Cosmetic act • Aanleiding: Sulfanilamide in poedervorm of tabletten werd
gebruikt voor de streptococcus infecties.
• In 1937 bracht een fabrikant dit product zonder te testen in vloeibare vorm op de markt door het op te lossen in diëthyleen glycol (ook wel antivries): 107 mensen (meest kinderen) overleden

7 of 73 © Process Vision – FDA requirements for suppliers
Belangrijkste regelgeving
• Code of Federal Regulations (CFR) 21 Food and Drugs • Part 1-99 : Algemene regels
• Part 100-199 : Voeding en additieven
• Part 200-299 : Humane medicijnen
• Part 300-499 : OTC medicijnen en antibiotica
• Part 500-599 : Dier voeding en medicijnen
• Part 600-799 : Biologische en bloed producten
• Part 800-899 : Medische (diagnose) apparatuur • 801: Labeling
• 803 Medical Device Reporting
• 807 Registration and listing
• 831 Medical Device Tracking
• 820: Quality System Regulation (QSR)
• 1997: aanvullende wetgeving voor opslaan van elektronische data en
gebruik van elektronische handtekeningen: • 21 CFR Part 11: Electronic records, Electronic signatures.
• Electronic record is bv test data (ook als je die uitprint en archiveert), je vertrouwt op / werkt met de elektronische versie niet de papieren

8 of 73 © Process Vision – FDA requirements for suppliers 8
US safety and effectiveness
no SADEs *
EU
safety and performance
no SADEs*
* Serious Adverse Device Effect
Regulatory Framework
Market access
what’s in the manual?
what’s in it for the patient?
Yet, you may need effectiveness
data to be able to sell the device

9 of 73 © Process Vision – FDA requirements for suppliers
Verschil tussen FDA en ISO 13485
• Compliancy • FDA is verplicht (wetgeving) als je op Amerikaanse markt wilt
• ISO 13485 is vrijwillig, voor Europa is MDD verplicht
• Certificering • ISO 13485 is mogelijk
• FDA is niet mogelijk
• Product labeling • CE label op product verplicht
• Geen FDA label op product wel registratie
• Auditing • ISO 13485 Notified Body audits
• FDA inspectors

10 of 73 © Process Vision – FDA requirements for suppliers
Verschil Inspecties CE/ISO13485 - FDA
2011-Marc-03
EU – Notified Body
1. Vertrouwen
2. Openhartig
3. Vooraf vastgestelde agenda
4. Vast aantal audit dagen
5. Auditor geeft belang en consequenties aan
6. Auditor neemt beslissing over non-conformity
7. Vooraf ingepland
8. Betaald door de klant
US – FDA
1. Wantrouwen (guilty unless proved otherwise)
2. Gesloten
3. Agenda niet bekend, spit door
4. Aantal dagen mag verlengd
5. Auditor doet waarnemingen en rapporteert
6. Beslissingen worden door functionaris bij FDA genomen
7. Onaangekondigd (in Amerika)
8. Overheidsinstantie

11 of 73 © Process Vision – FDA requirements for suppliers
Wanneer komt de FDA?
• Zij komen in 1e instantie altijd bij de zgn. “legal manufacturers(*)” = degene die product op de Amerikaans markt brengt.
• Zij kondigen minimaal 3 maanden vooraf (in Europa) hun komst aan bij de legal manufacturer
• Zij moeten minimaal 1 x in de 2 jaar een inspectie uitvoeren bij een klasse II of klasse III medical device.
(*) Term “legal manufacturer” kent de FDA niet, CE wel Legal entity that is the final approval authority on design changes and assumes Quality Systems responsibility for the development, design and manufacture of the product .
The Legal Manufacturer may perform all of the activities required to develop, design, manufacture, package, sterilize and place a product on the market OR the legal manufacturer may contract (or by letter of appointment) all or any of these activities to a third party.

12 of 73 © Process Vision – FDA requirements for suppliers
QSIT (Quality System Inspection Technique)
• Guideline for the FDA inspectors • http://www.fda.gov/ICECI/Inspections/InspectionGuides/uc
m074883.htm
13 of 73 © Process Vision – FDA requirements for suppliers
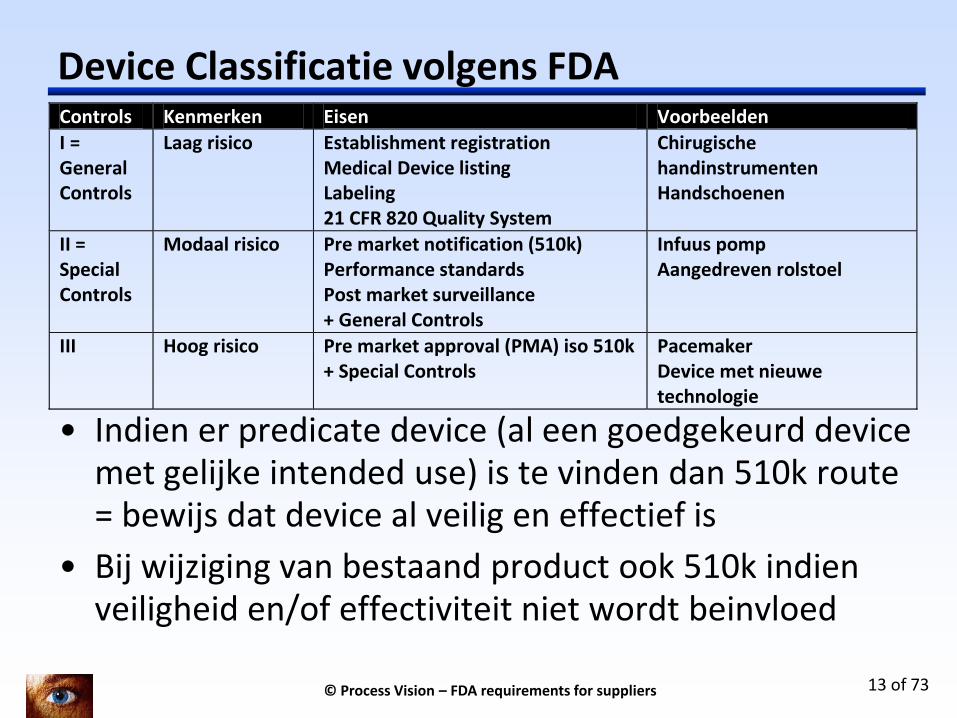
Device Classificatie volgens FDA Controls Kenmerken Eisen Voorbeelden
I = General Controls
Laag risico Establishment registration Medical Device listing Labeling 21 CFR 820 Quality System
Chirugische handinstrumenten Handschoenen
II = Special Controls
Modaal risico Pre market notification (510k) Performance standards Post market surveillance + General Controls
Infuus pomp Aangedreven rolstoel
III Hoog risico Pre market approval (PMA) iso 510k + Special Controls
Pacemaker Device met nieuwe technologie
• Indien er predicate device (al een goedgekeurd device met gelijke intended use) is te vinden dan 510k route = bewijs dat device al veilig en effectief is
• Bij wijziging van bestaand product ook 510k indien veiligheid en/of effectiviteit niet wordt beinvloed

14 of 73 © Process Vision – FDA requirements for suppliers
Plan for 510k clearance (red=also for supplier) 1. Define intended use, indication for use / markets / claims
2. Define strategy on components of the system: seperate FDA routes or combined? Criteria: seperate sale? Future significant changes in one or more components?
3. Define classification (I, II, IIII) conform FDA CDRH database (FDA determines in the end) http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYourDevice/default.htm
4. Conformity assessment route depends on chosen classification (I=general controls, II=+510k, III=+PMA) http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/GeneralandSpecialControls/default.htm
5. Predicate devices 510k database http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm
6. Check if “Accredits persons” may perform 510k review (see CDRH database)
7. Check if “Recognized Consensus Standards” from CDHR database are applicable
8. Select harmonized standards (Design input)
9. Implement QMS that complies with 21 CFR820
10. Plan development & regulatory strategy (including a V&V plan)
11. Appoint US Agent: regulatory representative to FDA located in USA
12. Device Registration and Listing (yearly approx. $ 3000 ) http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/RegistrationandListing/default.htm
13. Comply with medical device requirements (e.g. standards, 510k requirement, labeling )
14. Submit 510k and pay fee (standard $5.000, small business $2500)
15. FDA reviews and issues 510k clearance letter on website

15 of 73 © Process Vision – FDA requirements for suppliers
21 CFR 820 QUALITY SYSTEM

16 of 73 © Process Vision – FDA requirements for suppliers
7 Key processen in kwaliteitssysteem + 3 regulatory topics (only for legal manufacturers)
Inspectors Focus

17 of 73 © Process Vision – FDA requirements for suppliers
Inhoud 21 CFR 820 in subparts
A. General provision scope,
definitions
B. Quality System Requirements Mngmt resp,
policy, audits, personell
C. Design Controls
D. Document Controls
E. Purchasing Controls
F. Identification and traceability
G. Production and process controls
H. Acceptance activities
I. Non conforming products
J. CAPA
K. Labeling and packaging controls
L. Handling, Storage, distribution and installation
M. Records
N. Servicing
O. Statistical Techniques

18 of 73 © Process Vision – FDA requirements for suppliers
820.20(d) Quality Plan (niet voor ISO 13485)
• Each manufacturer shall establish a quality plan which defines the quality practices, resources, and activities relevant to devices that are designed and manufactured. The manufacturer shall establish how the requirements for quality will be met.
• A quality plan shows how the Quality System requirements are applied to a device or device type to ensure that it meets specifications and quality requirements. The purpose of a quality plan is to communicate the requirements and controls necessary to manufacture, test, and release devices that meet customer needs. The quality plan covers all quality practices, resources, and activities at all stages of manufacturing, including design, procurement, production, and—when applicable—installation and service.
• Kan apart document zijn of referentie naar DMR • Ref DMR, dan aangeven in Quality Manual hoe de DMR
820.20(d) eis afdekt

19 of 73 © Process Vision – FDA requirements for suppliers
DESIGN CONTROLS

20 of 73 © Process Vision – FDA requirements for suppliers
820.30 Design controls
Labeling and packaging is also part of design, so must be conform 820.30

21 of 73 © Process Vision – FDA requirements for suppliers
820.30 (c) Design Input • Ensure requirements are appropriate and address
intended use of device.
• Address incomplete, ambiguous, or conflicting requirements.
• Document, review, and approve input requirements

22 of 73 © Process Vision – FDA requirements for suppliers
820.30 (d) Design Output
• Define and document design output in terms that allow evaluation to design input (measurable)
• Reference acceptance criteria (pass/fail)
• Identify design outputs essential for the proper functioning of device.
• Document, review, and approve design outputs before release.
• Results of design effort at each phase and the end of the total design effort.
• Finished design output is basis for the DMR

23 of 73 © Process Vision – FDA requirements for suppliers
820.30 (e) Design Reviews
• A documented, comprehensive, systematic examination to • Evaluate adequacy of the design requirements
• Evaluate capability of the design to meet requirements
• Identify problems
• Ensure that formal reviews of design results are planned and conducted at appropriate stages.
• Ensure participants include representatives of all functions concerned with design stage being reviewed.
• Document results in the Design History File (DHF).

24 of 73 © Process Vision – FDA requirements for suppliers
820.30 (f) Design Verification
• Verify the device design.
• Confirm that design output meets design input requirements.
• Document results in the DHF

25 of 73 © Process Vision – FDA requirements for suppliers
820.30 (g) Design validation
• Establishing by objective evidence that device specifications conform with user needs and intended use(s).
• Perform under defined operating conditions on initial production units or equivalent.
• Perform process validation in parallel
• Ensure devices conform to defined user needs and intended uses.
• Test of production units under actual or simulated use conditions.
• Perform software validation and risk analysis, where appropriate.
• Document results in the DHF

26 of 73 © Process Vision – FDA requirements for suppliers
820.30 (h) Design Transfer
• Ensure the device design is correctly translated into production specifications

27 of 73 © Process Vision – FDA requirements for suppliers
820.30 (i) Design Changes • Identify, document, validate or verify, review, and
approve design changes before implementation.

28 of 73 © Process Vision – FDA requirements for suppliers
Examples Recognized International Standards
• IEC 60601-1 Basic safety + essential performance
• IEC 60601-1-2 Electromagnetic compatibility
• IEC 60601-1-8 Alarm systems
• IEC 62304 Software life cycle processes
• IEC 62366 Usability engineering
• ISO 14971 Risk management
See http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm

29 of 73 © Process Vision – FDA requirements for suppliers
Risk Management cyclus (ISO 14971) Throughout the life time of the device! Be sure to agree with the legal manufacturer who does what.

30 of 73 © Process Vision – FDA requirements for suppliers
Strenge document control op docs/records
• Consistentie door alle specs, reports, etcera heen • Project namen
• Product namen
• Definities en afkortingen
• Handtekening(en) voor review en goedkeur • Ook na goedkeur bij aanpassingen
• Volgorde goedkeur van belang • Bv test report na test spec, design na requirements
• Werk altijd met approved input documenten • Tussendoor approval is geen probleem
• Correcte document identificatie en versiebeheer (terugvindbaarheid)
• Requirements en test traceability (zie volgende sheet)

31 of 73 © Process Vision – FDA requirements for suppliers
Requirements and test traceability example
Product Validation
Report
System
Verification Report
Detailed Verification
Report
User Needs
Intended Uses Regulatory
standards
System Requirements
Detailed Requirements
Implementation
(Integration) Test
ReportSystem Design
1
2
5
6
7
Detailed Design
3
4
Sa
fety
/ S
ec
uri
ty R
isk
Ma
na
ge
me
nt
File
9
8

32 of 73 © Process Vision – FDA requirements for suppliers
Example design process
Defintion and
Quotation
phaseFEN-PCP01
Concept phaseFEN-PCP02
Design phaseFEN-PCP03
Proto phaseFEN-PCP04
Pilot phaseFEN-PCP05
Series phaseFEN-PCP06
Go Go Go Go Go
CustomerProject Request
Quotation
PO
Require-
mentsQuote
PODesign
Review
Request
Design
Approval
Series Products
Pilot Products
Material flow
Product Safety
Risk
ManagementFEN-PCP13
Product/
Process FMEAFEN-PCP12
Customer TPD
projectFMT-PCP11
Customer
TPD
Customer
Request
for product
Pilot /
Series
Products
Product Creation Process (PCP)
Information flow
Setup Assembly
Process FMT-PCP30
QA / PE in PCP
CM
Glovia
PRP
Glovia
EGMS = Engineering Maintenance Screen
CM = Contract Management
PRP = Project Resource Planning
Project
Leader
Validated
Tools
Safety
risks
Control
Measures
Control
Measures
MSAFEN-PCP16
MSA
needed
MSA
Report
Tool ValidationFEBV-ICT05
TPDAssembly
Binder
Setup Product
Labeling FMT-PCP33
Process
Validation FMT-PCP32

33 of 73 © Process Vision – FDA requirements for suppliers
PRODUCTION & PROCESS CONTROLS

34 of 73 © Process Vision – FDA requirements for suppliers
820.70 (a) Production and process control
• Develop, conduct, control and monitor production processes in order to • Ensure device is conform specifications (defined and approved
during design)
• To prevent non conformance, process controls are to be defined:
• Procedures and standards
• Workmanship
• Manufacturing instructions
• Process validation
• Inspection, testing
• Process Monitoring (e.g. SPC)
• ………..

35 of 73 © Process Vision – FDA requirements for suppliers
820.70 (b) Production and process changes
• Elke verandering in productie proces of product moet • Volgens een procedure lopen
• Gechecked op regulatory impact
• Geverifieerd of gevalideerd indien nodig (820.75)
• Gedocumenteerd volgens doc control (820.40)
• Approved
• Voorbeelden: nieuw product, (interne) verhuizing
• Het beste is als er 1 change proces is voor zowel development en productie

36 of 73 © Process Vision – FDA requirements for suppliers
820.70 (c) Environmental Control
• Degree of environmental control must be consistent with the intended use of the finished device. • Lighting, ventilation, temperature, humidity, air, pressure,
air flow, filtration, airborne contamination, microbial contamination, electrostatic discharge (ESD)
• Requirements for environmental controls must be defined and documented (during design phase)
• Procedures required to ensure control of conditions (monitoring) and periodic inspection (audit) of the control systems.

37 of 73 © Process Vision – FDA requirements for suppliers
820.70(d) Personnel • Define requirements and establish procedures when
unclean employees, inappropriately dressed employees, or employees with medical conditions could adversely affect the quality of the product.
• Hygiene requirements such as hand washing practice
• Storage of food and drink in work areas
• Dress codes and cleanliness

38 of 73 © Process Vision – FDA requirements for suppliers
820.70(e) Contamination Control • Establish and maintain procedures to prevent
contamination of equipment, components, manufacturing materials, in-process devices, finished devices, and returned devices by substances that could adversely affect device safety or effectiveness.
• There must be periodic, documented checks or inspections to verify that the contamination control system is properly functioning.
• Examples pest control, hazardous substances

39 of 73 © Process Vision – FDA requirements for suppliers
820.70(f) Buildings • Buildings shall be of suitable design and contain
sufficient space to perform necessary operations, prevent mixups, and assure orderly handling.
• Floor plan: flow of product and designates: • receiving areas;
• areas for acceptable and rejected products;
• manufacturing areas;
• storage areas for components and finished goods;
• rework, reprocessing, and repair areas;
• office space; and
• non-manufacturing areas (e.g., cafeterias and restrooms).

40 of 73 © Process Vision – FDA requirements for suppliers
820.70(g) Equipment • Ensure that all equipment (e.g., fabrication, molding,
extrusion, assembly, packaging, and sterilization equipment) is appropriately designed and installed to facilitate maintenance, adjustment, cleaning and use.
• Equipment must also meet requirements (made during
design phase) that ensure its proper functioning in the manufacture of the device.
• Provide maintenance procedure, schedule and records
• Some adjustments (defined in DMR or a procedure) are allowed by operator

41 of 73 © Process Vision – FDA requirements for suppliers
820.70(h) Manufacturing Material • Establish procedures for the use and removal of any
manufacturing material and document the removal or reduction of the material (in DHR)
• Examples: cleaning agents, mold-release agents, lubricating oils, latex proteins, sterilant residues
• Only when the manufacturing material could potentially have an adverse effect on product.
• The requirement also applies to processing, reprocessing, repair, and rework.
• Define for third parties allowable manufacturing materials

42 of 73 © Process Vision – FDA requirements for suppliers
820.70(i) Automated Processes • All software used in production or in the Quality
System, whether for design, manufacture, distribution, or traceability, is validated for its intended use.

43 of 73 © Process Vision – FDA requirements for suppliers
Example manufacturing process
CleanroomFM-MAP52
ProductionFMT-MAP06
Order PickingFMT-MAP05
Incoming
Inspection
Critical Comp.FMT-MAP04
Release for
ProductionFMT-MAP01
Goods ReceiptFMT-MAP02
ShippingFMT-MAP07
Calibration
EquipmentFMT-MAP09
Work Order
Planning
Pick
list
Work Order
with picked material
Packed
device
Shipped
Device
Material
Purchased items
Non critical
Components
QA status 3
Critical
Components
QA status 1
Approved
Critical
Components
Calibrated
Equipment
Calibraiton
Reports
Device
DHR
LOG
Stock
NC
received
Component
NC
Received
Component
NC
Product before
End test
Non
Conforming
Internal GoodsFMT-MAP10
8D
Scrap
Retour / Repair
to supplier
LOG
Material
On the floor
Returned
Goods
Customer Material
Material flow
CleaningFMT-MAP51
Material
Cleaned
Material
LOG
LOG
Manufacturing Process (MAP)
Logistics (LOG)
QA / PE in Manufacturing (MAP)
Information flow
ESDFMT-MAP50
Use
As is
Stock
DHR = Device History Reocrd
DMR = Device Master Record
NC = Non Conforming
Quality
Managment
Glovia
DMR
Teamcentre
Stock
Supplier
Non
Conforming
ProductsFMT-MAP14
NC
Product after
End test
8D
Customer
Approve
Deliver
with DN or Rework

44 of 73 © Process Vision – FDA requirements for suppliers
DOCUMENT & RECORD CONTROL

45 of 73 © Process Vision – FDA requirements for suppliers
Documentatie: DHF/DHR/DMR
product docs
Order
confirmed
End of
production
Start Design
Phase
Design &
Engineering
Production
EngineeringSales Production
Customer
(Service)
product docs order data service data
DHF index
Design
History
File
(DHF)
DMR index
Device
Master
Record
(DMR=TPD)
DHR Index
Device
History
Record
(DHR)
Device
History
Record
(DHR)
As
delivered
DHR Index
Device
History
Record
(DHR)
End of
Service
Start Series
Phase
Start Proto
Phase
Design
Transfer
production data
Supplier responsibility Customer responsibility
Abbr. Record Type Description
DHF 820.30 (j)
Design History
File
Compilation of records which described the design history of a finished device. There is a history file per
designed device.
It provides proof that product is designed according project plan.
E.g. System Requirements, System Design, test specifications, test reports, project plan.
DMR
(TPD)
820.81
Device Master
Record
Compilation of records containing procedures and specifications for a finished device. There is a master record
per designed device.
This information needed by manufacturing, end users and service.
E.g. assembly instructions, instructions for use, service manual,
DHR 820.184
Device History
Record
Compilation of records containing the production history of a finished device. There is a history record per
produced device (particular unit or batch of devices). It is the order, production and service history of a device
E.g. confirmed sales order, acceptance records (to DMR) and calibration records.

46 of 73 © Process Vision – FDA requirements for suppliers
DHF/DMR/DHR: traceability
• Van elke geproduceerde DHR moet traceerbaar terug te vinden zijn op welke DMR deze gebaseerd is
• Van elke gebruikte DMR moet traceerbaar terug te vinden zijn op welke DHF deze gebaseerd is
Design
History
File
(DHF)
Device
Master
Record
(DMR)
Device
History
Record
(DHR)

47 of 73 © Process Vision – FDA requirements for suppliers
820.184 Inhoud DHR • Aantal geproduceerd inclusief productie datum
• Aantal gedistribueerd
• SN# systeem
• BOM inclusief eventuele SN# van componenten
• Referentie naar gebruikte DMR (Doc Id’s + versies)
• Gebruikte (gecalibreerde) meetapparatuur
• Gebruikte labels (identificatie)
• Identificatie + software versies test programmatuur
• Meetresultaten
• 820.80 Acceptatie resultaten/conclusie/review/approval
• 820.90 Non conforming records

48 of 73 © Process Vision – FDA requirements for suppliers
En nog wat records
• Alle procedures (820.186 Quality System Record)
• Alle niet device gerelateerde records • Voorbeelden
• 820.20 (c) Management review records
• 820.22 Audit records
• 820.25 Training records
• 820.72 Calibration records
• 820.100 CAPA records
• 820.198 Complaint records
• Zijn ook allemaal terug te vinden binnen ISO 13485

49 of 73 © Process Vision – FDA requirements for suppliers
Good Documentation Practices (GDP) • Gebruik permanente blauwe inkt
• Eenduidige datum notatie: bv JJJJ-MMM-DD
• Eenduidige naam notaitie: bv initialen + achternaam
• Leesbaar, bv in blokletters
• Geen lege cellen, NA alleen als is toegestaan
• Correcties? Doorhalen met enkele lijn, naam + handtekening + datum + reden
• Gebruik geen “aanhangsels” zonder identificatie
• Afronden getallen alleen bij uitkomst, niet bij bewerking
• Handtekening = verklaring dat je iets hebt gedaan, reden moet helder zijn voor de persoon die tekent
• Handtekening bij afwezigheid alleen door bevoegde personen
• Maak goede templates en train de mensen op GDP

50 of 73 © Process Vision – FDA requirements for suppliers
Template voorbeeld - 1

51 of 73 © Process Vision – FDA requirements for suppliers
Template voorbeeld - 2

52 of 73 © Process Vision – FDA requirements for suppliers
Digitale handtekeningen? CFR 11
ELECTRONIC RECORDS AND ELECTRONIC SIGNATURES
• Part 11 will not apply when: • computers are used to generate paper printouts of
electronic records and
• these paper records meet all predicate rule requirements and
• persons rely on the paper records to perform regulated activities.

53 of 73 © Process Vision – FDA requirements for suppliers
PROCESS VALIDATIE

54 of 73 © Process Vision – FDA requirements for suppliers
820.75 Process Validatie
• 820.3(z)(1) Process validation • Establishing by objective evidence that a process
consistently produces a result or product meeting its predetermined specifications.
Different from design validation
• 820.3(z)(2) Design Validation • Establishing by objective evidence that device specifications
conform with user needs and intended use(s).

55 of 73 © Process Vision – FDA requirements for suppliers
Typical Process Validation examples
• Solderen
• Schoonmaken
• Lijmen
• Sterilisatie
• Spuitgieten
• Sealen
• Lassen
• Software controlled systemen
• Vertinnen
• Vriesdrogen
• Warmtebehandeling
If requirements of
product can only be
assured by destructive
testing then validation
is needed

56 of 73 © Process Vision – FDA requirements for suppliers
Software tool validatie niet alleen in productie zoals bij ISO 13485
• Maar ook in development & engineering : • Compilers / linkers
• CAD/CAM systemen
• Calculaties
• DHF/DMR archiving/versioning systems
• …....
• En op het kwaliteitssyteem: • CAPA database
• QMS Change request tool
• Customer complaint database
• QMS procedure version / archive tool
• Performance Indicator generation (Metric/KPI)
• ……..

57 of 73 © Process Vision – FDA requirements for suppliers
Software validation approach

58 of 73 © Process Vision – FDA requirements for suppliers
TRAINING

59 of 73 © Process Vision – FDA requirements for suppliers
Personell
• Hire sufficient personnel with necessary education, background, training, and experience.
• Make personnel aware of device defects that could occur from improper job performance.
• Make personnel aware of defects and errors that could be encountered as part of their job.
• Establish procedures for identifying training needs and to ensure personnel are adequately trained.

60 of 73 © Process Vision – FDA requirements for suppliers
Training
• Kwaliteits systeem: functies/rollen
• Functie omschrijving: taken en verantwoordelijkheden
• Organigram: wie heeft welke functie
• Skills matrix: wie heeft welke taken + training status
• Training records: wie is getrained op welke taak
• Bovenstaand “bouwwerk” moet consistent en compleet zijn

61 of 73 © Process Vision – FDA requirements for suppliers
CAPA
62 of 73 © Process Vision – FDA requirements for suppliers
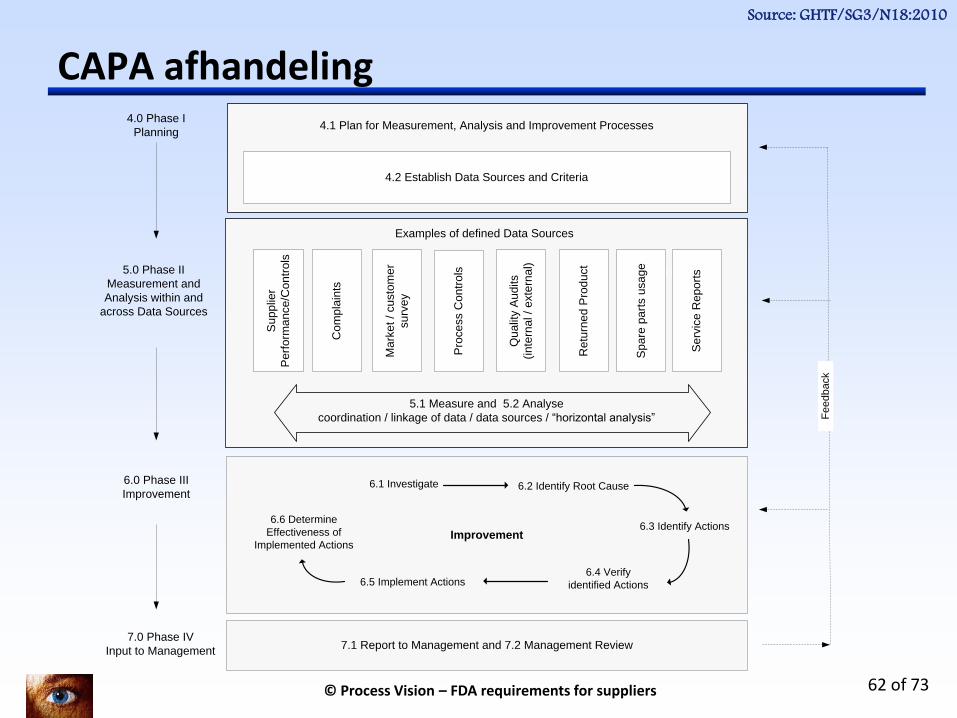
CAPA afhandeling
5.1 Measure and 5.2 Analyse
coordination / linkage of data / data sources / “horizontal analysis”
5.0 Phase II
Measurement and
Analysis within and
across Data Sources
6.0 Phase III
Improvement
4.0 Phase I
Planning4.1 Plan for Measurement, Analysis and Improvement Processes
7.0 Phase IV
Input to Management7.1 Report to Management and 7.2 Management Review
Examples of defined Data Sources
Within each data source
4.2 Establish Data Sources and Criteria
Fe
ed
ba
ck
Co
mp
lain
ts
Se
rvic
e R
ep
ort
s
Sp
are
pa
rts u
sa
ge
Qu
alit
y A
ud
its
(in
tern
al / e
xte
rna
l)
Su
pp
lier
Pe
rfo
rma
nce/C
on
tro
ls
Pro
ce
ss C
on
tro
ls
Re
turn
ed
Pro
du
ct
Ma
rke
t / cu
sto
me
r
su
rve
y
Improvement
6.1 Investigate 6.2 Identify Root Cause
6.3 Identify Actions
6.4 Verify
identified Actions6.5 Implement Actions
6.6 Determine
Effectiveness of
Implemented Actions
Source: GHTF/SG3/N18:2010

63 of 73 © Process Vision – FDA requirements for suppliers
CAPA geintegreerd met risk management Source: GHTF/SG3/N15R8

64 of 73 © Process Vision – FDA requirements for suppliers
COMPLAINT FILES

65 of 73 © Process Vision – FDA requirements for suppliers
820.198 Complaint Files (also FDA focus) • Be sure to involve all disciplines necessary:
• Clinical expert, compliance expert, marketing, …….
• Be sure that root cause is effectively solved (via CAPA)
• Establish and maintain procedures for receiving, reviewing, and evaluating complaints.
• Procedure must ensure that • Oral complaints are documented upon receipt.
• All complaints processed uniform and timely.
• Complaints evaluated to determine whether complaint represents a Medical Device Report (adverse event).
• Review and evaluate all complaints if investigation is necessary.
• Records of investigation are maintained
• No investigation? Justification + name individual responsible for the decision.

66 of 73 © Process Vision – FDA requirements for suppliers
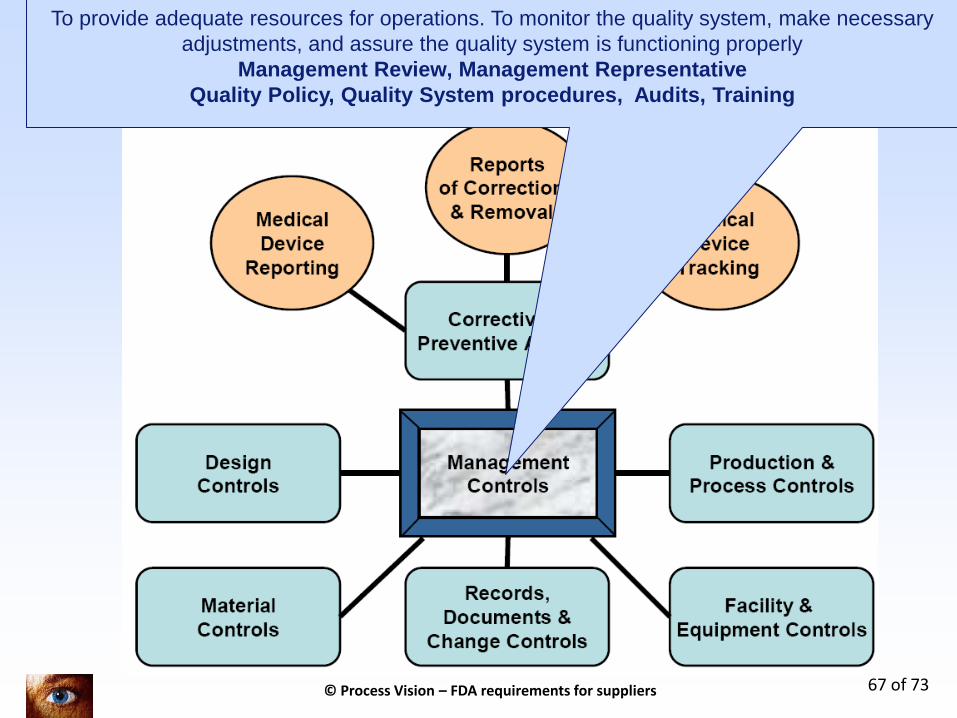
And the other sub systems briefly explained
67 of 73 © Process Vision – FDA requirements for suppliers
7 Key processen in kwaliteitssysteem + 3 regulatory topics (only for legal manufacturers)
To provide adequate resources for operations. To monitor the quality system, make necessary
adjustments, and assure the quality system is functioning properly
Management Review, Management Representative
Quality Policy, Quality System procedures, Audits, Training

68 of 73 © Process Vision – FDA requirements for suppliers
7 Key processen in kwaliteitssysteem + 3 regulatory topics (only for legal manufacturers)
To assure that all products that are accepted, used, and distributed meet specifications
“Products” includes components, manufacturing materials, in-process devices, finished devices,
and returned devices
Purchase,
statistical techniques,
incoming, in-process, final acceptance,
identification & traceability,
labeling, packaging, handling,
storage, distribution
Non conforming products

69 of 73 © Process Vision – FDA requirements for suppliers
7 Key processen in kwaliteitssysteem + 3 regulatory topics (only for legal manufacturers)
To assure:
Specifications and procedures are adequate
Only current documents are used
Changes are reviewed, approved and incorporated into documents
Documents are maintained for the required length of time
Device Master Record (DMR),
Device History Record (DHR),
Document control
Quality System Records (QSR)
70 of 73 © Process Vision – FDA requirements for suppliers
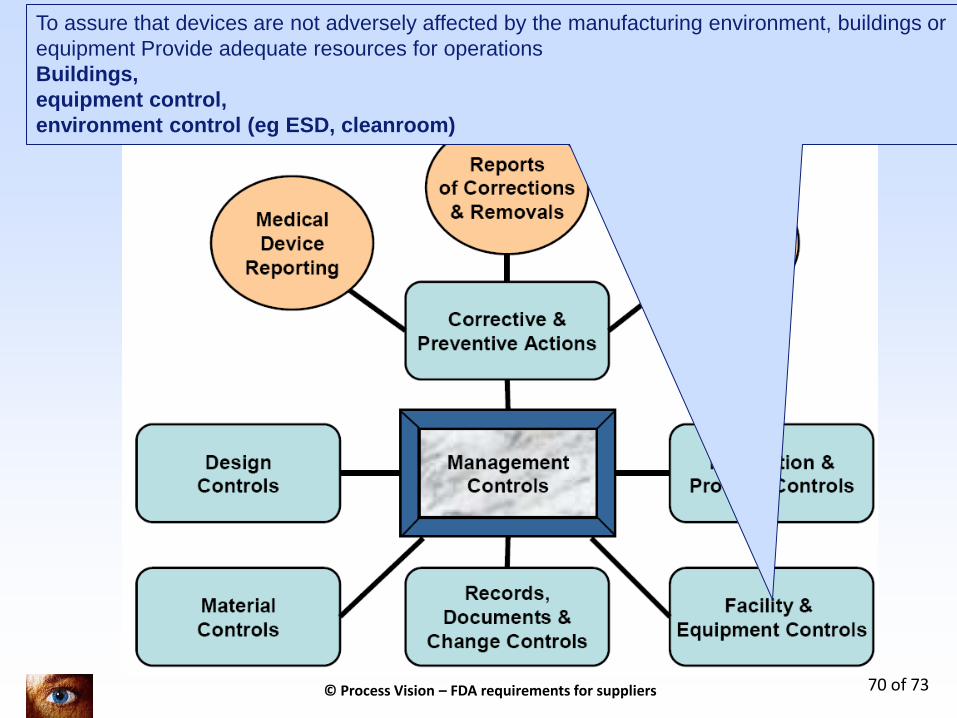
7 Key processen in kwaliteitssysteem + 3 regulatory topics (only for legal manufacturers)
To assure that devices are not adversely affected by the manufacturing environment, buildings or
equipment Provide adequate resources for operations
Buildings,
equipment control,
environment control (eg ESD, cleanroom)

71 of 73 © Process Vision – FDA requirements for suppliers
TENSLOTTE

72 of 73 © Process Vision – FDA requirements for suppliers
FDA references
Home page http://www.fda.gov/MedicalDevices/default.htm
A-Z http://www.fda.gov/SiteIndex/default.htm
510k database search http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm
Product code database http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm
Recognised standards http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm
CFR 21
Device Registration
and Listing
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm
http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYour
Device/RegistrationandListing/default.htm
Training Overview http://www.fda.gov/Training/CDRHLearn/ucm162015.htm
Training QSR 820 http://www.fda.gov/MedicalDevices/ResourcesforYou/Industry/ucm126252.htm

73 of 73 © Process Vision – FDA requirements for suppliers
How to continue.......
• Perform FDA Gap analysis on current Quality Management System (QMS)
• Implement gap analysis in QMS
• Pick a pilot project to validate the QMS changes
• Implement the project with QMS changes
• Evaluate