falsified and sub standard medicines: danger of death · falsified and sub-standard medicines:...
TRANSCRIPT

1
Falsified and Sub-standard Medicines:
Danger of Death
- Unusual in the industrialized countries
- Commonplace in the developing countries
Jacques Pinel

2
FOREWORD
Jacques Pinel was and still is a totemic figure at the medical charity, Médecins Sans Frontières (MSF – Doctors Without Borders).
A pharmacist with a few years of experience in voluntary service overseas in Africa and Asia, Jacques joined MSF in 1979 to work in a refugee camp in Thailand where the charity, still in its infancy, was trying to meet the medical needs of tens of thousands of Cambodian refugees.
Beginning with organizing the camp's pharmacy, Jacques very quickly turned his attention to all the logistics necessary so that the medical team could focus on care-giving, thereby inventing, then developing what would become MSF logistics (medical and non-medical).
From that moment on, he never ceased to think about and share new ideas, launching new projects, creating networks, bringing in potential partners and passing on his experience, always with the goal of improving the aid conveyed to people in need.
Jacques died in 2015, his mind still filled with projects for MSF and its partners.
Starting in the 1990s, Jacques devoted himself more particularly to the issues involving medicines, e.g. the issues of access to medicines required for fighting AIDS, malaria and tuberculosis and the quality of medicines on the international market.
He was the originator of MSF's Campaign for Access to Essential Medicines in 1999 and initiated a series of projects aimed at helping humanitarian organizations and international agencies ensure the quality of their supply.
Throughout his career he witnessed the evolution and growing complexity of the world's medicines markets with the rising risks for the people of developing countries.
Extremely worried about the dangers presented by falsified and poor-quality medicines for these peoples and always believing in the huge importance of education, Jacques spent his later years drafting a document for laymen about these issues.
When he passed away in August, 2015, Jacques had not yet finished his paper.
Jacques' close co-workers and companions (Jean-Michel Caudron, Cécile Macé, Corinne Pouget, Raffaella Ravinetto, Brigitte Renchon, Jean Rigal, Benedetta Schiavetti and Daniel Vandenbergh) therefore decided to finish this document in Jacques' spirit so as to make it available to as many people as possible. The outcome of this process is presented here.
The first chapter is devoted to explaining a few basic notions for a better understanding of the topic of medicines, their quality and their legal and regulatory environment.
The second chapter explains how a medicine can be of poor quality and the distinction to be made between falsified (counterfeit or frankly "fake" medicines) and "sub-standard" medicines.

3
The third chapter examines how the upheavals in the way the world is politically organized and the evolution of the international market have affected the quality of medicines available to the world.
And finally, chapter four explains what measures must be taken to improve the situation for the most deprived countries and the essential role played by the World Health Organization (WHO) with its Prequalification Programme.
Jacques was a fervent supporter of this WHO Prequalification Programme.
He hoped that the responsibility of the Prequalification Programme –initially targeting the antiretrovirals, TB-medicines and the artemisinin-combined therapies–would be widened to include all the medicines on the WHO Model List of Essential Medicines. But he understood perfectly well that this development would be complicated and certainly not attainable in the short-term.
While waiting for this development, one solution for him consisted in improving access to objective and cross-checked information about manufacturers, particularly manufacturers of generics, and pooling resources and means to collect such information as much as possible (between NGOs and international organizations, between governments, etc.).

4
CONTENTS
FOREWORD ........................................................................................................................................................... 2
CONTENTS ............................................................................................................................................................. 4
LIST OF ACRONYMS AND ABBREVIATIONS ..................................................................................................... 5
1 WHAT IS A "TRUE" MEDICINE? ................................................................................................................. 6 1.1 AN ORIGINAL MEDICINE ............................................................................................................................................. 6 1.2 A GENERIC MEDICINE ................................................................................................................................................. 7 1.3 MEDICINE NAMES ...................................................................................................................................................... 8
2 HOW A MEDICINE CAN BE OF POOR QUALITY....................................................................................... 9 2.1 COUNTERFEITS, FALSIFIED OR "FAKE" MEDICINES .................................................................................................... 9 2.2 "SUB-STANDARD" OR "POOR QUALITY" MEDICINES .............................................................................................. 11
3 CHANGES IN INTERNATIONAL CONDITIONS AND THEIR IMPACT ON THE QUALITY OF MEDICINES................................................................................................................................................. 14
3.1 UPHEAVAL IN THE WAY THE WORLD IS ORGANIZED AND ITS CONSEQUENCES FOR THE QUALITY OF MEDICINES . 14 3.1.1 A "preserved" quality in a protectionist and colonialized world .................................................. 14 3.1.2 Questioning quality in a globalized world.............................................................................................. 14
3.2 CONSEQUENCES FOR THE QUALITY OF MEDICINES ................................................................................................ 15 3.2.1 The impact of globalization on the medicines market ...................................................................... 15 3.2.2 The two-speed regulation of a more complicated market .............................................................. 16 3.2.3 Poor quality is unusual in the industrialized countries ..................................................................... 17 3.2.4 The presence of poor-quality products in numerous developing countries is current and
compete with good-quality medicines .................................................................................................... 18
4 WHAT MEASURES COULD CHANGE THE SITUATION FOR THE MOST DEPRIVED COUNTRIES? .. 20 4.1 COUNTERFEIT AND SUB-STANDARD: TWO ISSUES THAT SHOULD NOT BE CONFUSED .......................................... 20
4.1.1 Counterfeits are the work of large- or small-scale criminal organizations: so it is up to the police to fight counterfeiting ....................................................................................................................... 20
4.1.2 Sub-standard medicines are connected to the way National Regulatory Authorities work, which is why it is up to these authorities to fight sub-standard medicines ............................. 20
4.2 THE ROLE OF PHARMACEUTICAL REGULATORY AUTHORITIES ............................................................................... 21 4.2.1 The role of pharmaceutical authorities concerning sub-standard medicines in exporting
countries ............................................................................................................................................................... 21 4.2.2 The role of pharmaceutical authorities of importing countries for sub-standard medicines
................................................................................................................................................................................. 22 4.3 THE WHO'S ROLE FOR SUB-STANDARD MEDICINES .............................................................................................. 23
FOOTNOTES ....................................................................................................................................................... 24
GLOSSARY .................................................................................................................................. 30

5
LIST OF ACRONYMS AND ABBREVIATIONS
AIDS Acquired Immunodeficiency Syndrome
AMA African Medicines Agency
API Active Pharmaceutical Ingredient
ASEAN Association of South East Asian Nations
CPP Certificate of Pharmaceutical Product
DC Developing Countries
DEG Diethylene-glycol
EAC East African Community
EMA European Medicines Agency
FDA Food and Drug Administration
FPP Finished Pharmaceutical Product
GCC Gulf Cooperation Council
GDP Good Distribution Practices
GMP Good Manufacturing Practices
HIV Human Immunodeficiency Virus
ICH International Conference for Harmonization
INN International Non-Proprietary Name
MA Marketing Authorization
MSF Médecins Sans Frontières (Doctors Without Borders)
NGO Non-Governmental Organization
NRA National Regulatory Authority
OOAS Organisation Ouest Africaine de Santé (West African Health Organisation)
PIC/S Pharmaceutical Inspection Convention and Pharmaceutical Inspection Cooperation Scheme
R&D Research & Development
SADC Southern African Development Community
SSFFC Substandard / Spurious / Falsely-labeled / Falsified / Counterfeit (Medical Products)
TB Tuberculosis
UEMOA Union Économique et Monétaire Ouest Africaine (West African Economic and Monetary Union)
USFDA United States (of America) Food and Drug Administration
WHO World Health Organisation
ZAZIBONA Zambia – Zimbabwe – Botswana – Namibia

6
1 WHAT IS A "TRUE" MEDICINE?
Because of their impact on people's health, both individually and collectively speaking, medicines are subject to strict and specific regulations in almost all countries. A pharmaceutical manufacturer wishing to market a medicine in a country must obtain a Marketing Authorization (MA) from the National Regulatory Authority (NRA). To do so the manufacturer must supply the NRA with complete information concerning the following:
- the medicine's efficacy: its beneficial effects enabling cure, relief or protection.
- its safety: the adverse reactions that very generally accompany the beneficial reactions.
- its pharmaceutical quality: the assurance that the medicine manufactured on an industrial scale is equivalent to the "trial" products that have made it possible to determine the medicine's efficacy and safety.
A medicine is a combination of:
- an active ingredient (sometimes several) combined with inactive ingredients (excipients) to obtain a form that a patient can use (tablet, capsule, ampoule, etc.) presented in protective packaging
- information printed on the packaging concerning the conditions of use (instructions, dosage, administration schedule, duration of treatment, adverse reactions, storage conditions, batch number, expiry date)
- Marketing Authorization (therefore use) granted to the manufacturer by the National Regulatory Authority of the country where it is used (particular to each country)
The information supplied to obtain Marketing Authorization must be more or less complete according to whether it is an original medicine (new medicine) or a generic medicine (the copy of an original medicine).
1.1 AN INNOVATOR MEDICINE
An innovator medicinei, also known as an “originator” or “benchmark” or “princeps” medicine, is one the active ingredient of which (or a new dosage or a new presentation) has not yet been used as a medicine for human consumption for its stated purpose. The information that the manufacturer must submit to the National Regulatory Authority is obtained through specific studies carried out with this ingredient:
- Preclinical trials in vitro and in animals, followed by
- Clinical trials on groups of healthy subjects (phase I), then on increasingly larger groups of people suffering from the pathology that the new medicine is intended to treat (phases II and III).

7
These trials make it possible to learn the effect that the active ingredient has on humans concerning:
- its efficacy: indication, dosage(s), administration schedule and duration of treatment, etc.
- its safety: adverse reactions, toxicity, etc.
Other studies determine the way to industrially produce medicines identical to the products that have been used in the efficacy and safety clinical trials:
- the pharmaceutical quality of the industrial medicine (finished pharmaceutical product–FPP)ii: an active ingredient is not usable as such (in the pure, powder or crystal form), for it must often be ingested in very small quantities (to the order of mg). It must be made usable by patients in the form of a medicine (finished product: tablets, capsules, ampoules, etc.), which most often requires a preparation with the addition of inactive ingredients (excipients) in which the active ingredient is dispersed or dissolved so as to obtain a medicine that is stable for a given period and under predefined storage conditions, and this in a reproducible manner (i.e. manufacturing cycle after manufacturing cycle, batch after batch). This manufacturing process is complicated and calls for high-level skills and an advanced technical environment.
The National Regulatory Authority checks and assesses the information provided by the manufacturer, which enables it to determine a ratio between the benefits (treatment or prevention of a disease, improvement in a pathological condition, etc.), and the risks (adverse reactions, etc.) and to assess the conditions of use (cogent instructions corresponding to the clinical trials) as well as the manufacturing quality.
Depending on the outcome of this assessmentiii, the National Regulatory Authority may deliver (or not) an authorization for marketing and use, known as a Marketing Authorization (MA), valid only in the authority's countryiv that granted it and for a finite period of time (in general 3 to 5 years), beyond which the authorization is subject to re-assessment.
1.2 A GENERIC MEDICINE
A generic medicinev is the copy of an original medicine by another pharmaceutical manufacturer.
The preclinical and clinical trials to assess efficacy and safety are not necessary if an MA has already been granted to the original medicine by a stringent National Regulatory Authorityvi. However the generic's technical file submitted for the MA must contain proof that it is equivalent to the original medicine and that it is therefore as effective for patients as the original.
Therefore, the assessment of the technical file is concerned only with pharmaceutical quality, which should have the same specifications as the original (e.g. same active

8
ingredient, same dosage, etc.). Concerning efficacy/safety, it is not necessary to repeat the clinical trials required for the original medicine: if the generic manufacturer can prove that its product's efficacy/safety is equivalent to the original medicine, the therapeutic equivalence has been demonstrated, and the original and generic are interchangeablevii.
The assessment of a generic by a National Regulatory Authority is much less complicated than the assessment of a new medicine. Nonetheless the proof of interchangeability does remain a highly technical activity.
Note: each generic is specific, and its manufacturer must provide proof of the quality of ITS product.
1.3 MEDICINE NAMES
All active ingredients in the composition of medicines have an international name given by the World Health Organization (WHO). This is their International Non-Proprietary Name (INN), also known as the "generic name"viii.
The INN and dosage must be printed on the packaging of all medicines, be they original or generic.
An innovator medicine is generally marketed with a commercial name (also known as a "brand name"ix) chosen by the pharmaceutical manufacturer. This commercial name is generally printed on the packaging in a more visible way than the INN, for the proprietary manufacturer, for obvious commercial reasons, wishes to gain brand recognition for its product.
A generic medicine is usually sold under its INN, but the majority of countries authorize the addition of a new commercial namex. As for the manufacturers of the innovator product, the commercial name is chosen by the generic manufacturer, according to the same commercial objectives.

9
2 HOW A MEDICINE CAN BE OF POOR QUALITYxi
As with any manufactured product distributed on an industrial scale, a medicine can be of poor quality for numerous reasons. To simplify a complicated topic, the WHO distinguishes between two categories of causes for defects. They can be summarized in the following manner:
- Counterfeits or falsifications when "defects" are due to fraud (intentional acts) in the manufacturing process and/or in distribution.
- Poor quality or sub-standard medicines when the defects have unintentionally or through negligence appeared during the manufacturing process and/or in distribution.
2.1 FALSIFIED, COUNTERFEITS OR "FAKE" MEDICINES
Falsified medicines, "counterfeits", or "fake medicines" are manufactured with the intention to deceive distributors, authorities, prescribers, patients, etc. for financial gain. They are found in both formal and informal/illegal distribution networks.
There is not a systematic link between "falsification" and "poor quality" (a falsified product may potentially contain the right active ingredient, proper dosage, etc.), but most often it proves true. A falsified medicine can therefore not contain the right dosage of the active ingredient, but it can also contain other less expensive active ingredients, inefficacious ingredients or toxic ingredients. There are as many examples of falsifications as there are falsifiers. What's more, the counterfeit is always beyond the control of the National Regulatory Authorities.
Falsifiers manufacture products that resemble as much as possible real medicines without any concern for the consequences on the health of the patients who consume them. Falsifications are manufactured and distributed illegally without the authorization of the country's health authorities. The real origin of a falsified medicine is not the one printed on the packaging (the name of the manufacturer and/or the brand are either usurped or invented).
A falsified medicine should not be called a medicine, however well made it may or may not be (i.e. even if its pharmaceutical quality is acceptable), for it lacks one vital specificity, i.e. traceability and transparency. It is impossible to identify the entire chain between the manufacturer and the patient.

10
The counterfeit issue is illustrated by the following few actual examples among thousands of others:
1) Fake artesunate antimalarial tablets in Southeast Asia:
Ref.: P. Newton et al. Manslaughter by fake artesunate in Asia – Will Africa be next? PloS Med 2006; 3:6
In the early 2000s falsified artesunate tablets fraudulently carrying the name of the genuine Chinese manufacturer, Guilin, were reported in this region (Myanmar, Cambodia, Laos, Vietnam and Thailand). Guilin reacted by printing a hard-to-reproduce hologram on its products. At the same time the regional health facilities procured colorimetric tests enabling them to identify quite simply the presence of artesunate in the tablets.
In 2008 in the same region samples were drawn from 4 medicine-distribution networks (public health facilities, international aid organizations, certified pharmacies and illegal markets). The outcome was serious because whichever the network, nearly half of the collected samples were proven falsifications carrying fake reproductions of the Guilin hologram, and some copies contained much lower quantities of artesunate than that printed but a sufficient quantity to give a positive reaction to the colorimetric tests!
In certain samples advanced chemical analyses detected variable quantities of various ingredients, inactive or active (chloramphenicol, chloroquine, erythromycine, metronidazole, noramidopyrine, mineral powders, etc.) and even safrol, a toxic chemical product used for making Ecstasy. The microscopic examination of the dust on the tablets (in particular pollens) led the Chinese police to an underground manufacturer, which was taken down. It would be naive to think that this police operation put a definitive end to the activities of numerous falsifiers in the region or in the world, as has been shown by more recent studies (http://www.wwarn.org/aqsurveyor/index.html#0; Gaurvika M.L. et al, Poor-quality antimalarial drugs in Southeast Asia and sub-Saharan Africa, Lancet Inf Dis 2012; 12: 488-96).
2) Fake meningitis vaccines in Niger:
Ref.: internal MSF
In 1995 during a meningitis outbreak, Médecins Sans Frontières (MSF) teams observed that the powder from the vials dissolved abnormally in the solvent. The vaccines came from the national stock where the various shipments of vaccines supplied by international aid were centralized.
So the MSF teams switched to a different batch to continue the vaccination campaign. Suspicious samples were sent to the manufacturer whose name was printed on the packaging (Pasteur Vaccins), who concluded that they were falsified and completely inactive.
This kind of problem arises regularly as shown by many warnings issued by the WHO. Among others that can be cited was the warning issued in February, 2015 about the falsification of yellow-fever vaccines circulating in Southeast Asia (http://www.who.int/medicines/publications/drugalerts/Alert2_2016_Fev_Falsified_AMARILyellow-fever-vaccine_searo_fr.pdf?ua=1), or the warning about a falsification of vaccines for

11
meningitis circulating in Niger from May, 2015 (http://www.who.int/medicines/publications/drugalerts/VF_MenomuneAlertFRversion.pdf?ua=1).
3) Cancer medication sold on-line in China:
Ref.: http://www.chinadaily.com.cn/china/2014-05/09/content_17494803.htm (page 7)
Ref.: Report published by the IFPMA: http://www.ifpma.org/uploads/media/UCL-Matrix_Insight-Falsified_Medicines_and_the_Global_Publics_Health.pdf
In 2014 the Chinese authorities warned its population and foreign pharmaceutical authorities about the production and marketing on the Internet in China of falsified cancer medication. These counterfeits deceived Chinese and foreign patients who turned to them because they were experiencing difficulties in buying cancer medication either because of the high price (China, etc.) or stock shortages (Germany and Middle-East countries destabilized by war, etc.).
2.2 "SUB-STANDARD" OR "POOR QUALITY" MEDICINES
The manufacturer of a sub-standard medicine is indeed the one that appears on the packaging and in the documentation, but poor practices have led to defectsxii occurring during the manufacturing phase (poor workmanship) and/or in the distribution phase (degradations). Defects may result from mistakes, negligence and/or insufficiencies in the equipment, facilities, know-how, etc. Sub-standard products are manufactured and distributed in the legal networks.
This term "poor quality" or "sub-standard" generally call to mind a "slight under-dosage but in the end without major consequences for patients". The reality however can be very different, for sub-standard medicines are also the following:
- Overdosed and/or under-dosed medicines: e.g. when weighing is not done correctly or when the mixture between the active ingredient and the excipients is heterogeneous or else when the distribution of the mixture in capsules or tablets is not homogenous, regular or reproducible.
- Unstable medicines: e.g. when active ingredients are degraded in tablets because of faulty packaging that protects them badly from humidity.
- Non-bioavailable medicines: e.g. when the active ingredient is not freed in the digestive tract because the tablet has been over-compressed, which restricts disintegration.

12
- Contaminated medicines: e.g. when highly active substances such as hormonesxiii are handled in the same factory as medicines used in much higher doses (e.g. paracetamol tablets contaminated by hormone powder suspended in air or polluting the machines).
- Non-sterile injectable medicines: e.g. when the time and/or the temperature required for sterilization are not adhered to.
- Labeling mistakes: e.g. when an active ingredient has been poorly labeled or labels are inadvertently switched during manufacturing or when the information on the label and/or the documentation are erroneous or incomplete.
- Degraded medicines becoming inefficacious: e.g. when vaccines are not kept refrigerated during transport or storage.
This not-exhaustive list describes issues encountered in certain countries, especially the low-income countries where National Regulatory Authorities have limited capacities.
The risk of being exposed to sub-standard medicines is greater when the National Regulatory Authorities (in manufacturing or importing countries) are weak and with low technical capacities. They then rarely have the possibility of applying regulations, carrying out controls and inspections or imposing real sanctions.
A few actual examples among thousands of others illustrating the sub-standard issue:
1) Toxic syrups in Haiti:
Ref.: Scalzo A.J. The 1996 Haitian epidemic. J. Toxicol 1996; 34(5): 513-6
In 1996 85 children died after admission to hospital from renal failure. An investigation showed that they had ingested cough medicine. Syrups (cough and analgesics) often contain a sugary liquid excipient (glycerol), but it happens that a chemically similar product frequently used as industrial solvent, diethylene-glycol (DEG), either replaces it or contaminates it (the two products often produced in the same factory). In contrast to glycerol, DEG absorbed into the body is very toxic, especially for the kidneys, leading to either death or serious outcomes (need of life-long dialysis or a kidney transplant). It is obvious that these events do not benefit manufacturers who are also victims of this mix-up and are often treated as criminals.
A similar production mix-up occurred in the USA in 1937, causing the death of 107 people (Ref.: Ballentine V. Taste of raspberries, taste of death. The 1937 Elixir Sulfanilamide Incident. FDA Consumer Magazine, June, 1981). After the accident, regulation was implemented in the USA and all other industrialized countries to supervise the manufacture and marketing of medicines, and since then no other such incident has occurred in these countries. On the other hand this kind of lethal poisoning is still being repeated in the developing countries, e.g. 1969 (South Africa), 1986 (India), 1992 (Nigeria and Bangladesh), 1995 (Haiti), 1998 (India),

13
2006 (Panama), 2008 (Nigeria and 2009 (Bangladesh). This list is certainly incomplete, for it takes into account only the well documented cases.
2) Unsterile infusions in Sudan:
Ref.: internal MSF
In 2004 a medical team working with the displaced populations in Darfur received infusions from a United Nations aid agency. Certain bottles presented mold floating in the solution. A recall of batches was organized, but after six months, out of a total of 15,000 bottles that had entered the country, only 2,200 were identified and withdrawn. It is likely that the remaining bottles were used, as happens frequently in such situations, by simply withdrawing those that were visibly unsterile!
As is often the case, no study either documented or published the consequences of injecting patients with these unsterile solutions.
3) The unintentional mixing of active ingredients in Pakistan:
Ref.: Arie S. Contaminated drugs are held responsible for 120 deaths in Pakistan. BMJ 2012; 344: e951
In 2012 in Lahore, Pakistan, 107 people died after taking tablets of isosorbide to treat heart failure. In fact the medicine contained not just isosorbide, but also pyrimethamine (an antimalarial) with enough strength to block the production of white blood-cells by the bone marrow, leading to the death of 107 people and causing serious adverse reactions in 450 others.
This contamination of one active ingredient by another active ingredient is accidental, the Pakistani manufacturer obviously not gaining any benefit from marketing a sub-standard product with such a production defect and such consequences.

14
3 CHANGES IN INTERNATIONAL CONDITIONS AND THEIR IMPACT ON THE
QUALITY OF MEDICINES
3.1 UPHEAVAL IN THE WAY THE WORLD IS ORGANIZED AND ITS CONSEQUENCES FOR THE QUALITY
OF MEDICINES
3.1.1 A "preserved" quality in a protectionist and colonialized world
Up to the middle of the previous century the world was mainly divided into a dozen major political and economic spheres, i.e. the French, British, Belgian, Portuguese, Spanish, Soviet and Chinese "empires". Each empire was run by an industrialized colonizing nation and included, under its domination, several hundreds of millions of people, and vast geographic areas that were suppliers of raw materials.
- At the trade level, each empire enjoyed a certain economic autonomy with trade being mostly internal. External trade between empires was fairly limited and generally used well known and controlled networks.
- Administratively, numerous public departments were centralized in home countries (defense, courts, police, education, health, etc.). This centralized organizational system enabled savings of scale and a certain operational unity in the entire empire-dominated area. Although colonization had a lot of negative consequences for the colonized people, some consequences were positive, particularly in the field of medicines. Medicines were developed for the diseases predominant in the empires' tropical regions (malaria, sleeping sickness, etc.). In quality terms the medicines used in a hospital in Dakar were the same as in Paris, and those used in Nairobi the same as in London.
3.1.2 Questioning quality in a globalized world
The colonial system gradually fell apart, and by the 1960s and 70s most of the empires had vanished, replaced by a multitude of countries, a majority of which emerged as independent. Previously shared between 10 very centralized spheres, the world now finds itself with nearly 200 countries with on average far fewer inhabitants per country mathematically speaking (even if the world's population has in the meantime more than doubled). Therefore:
Concerning trade, each nation today maintains trade relationships with all other countries, thus weaving a complex web all over the planet, a web well embodied by the Internet's organizational and operational set up. Now, more specifically concerning medicines, each nation maintains commercial relationships with dozens of other nations but must independently ensure the quality of what it imports from sources that can be very distant and diversified.

15
Administratively each newly independent country had to individually ensure services with much more limited human and financial resources than what were once pooled and managed by the dominant country (education, legislation, defense, health, border controls and pharmaceutical manufacturing and regulation), which has had major repercussions on the medicines market.
The former colonizing nations, aware of their weakness due to decolonization, have developed political and economic co-operation and unions to compensate for their reduced size. For example, an agency emerged for medicines, the EMAxiv, within the framework of the European Union, for attributing Marketing Authorizations for 28 countries and 500 million people.
Inversely, initiatives for pooling work and resources of the newly independent nations (e.g. ASEAN, UEMOA, OOAS, SADC, EAC, ZAZIBONA, the Project for the implementation of an "African Medicine Agency"xv), which aim to develop regulation-harmonization projects, are still too recent to be effective, even if they have been under discussion for many years now.
3.2 CONSEQUENCES FOR THE QUALITY OF MEDICINES
3.2.1 The impact of globalization on the medicines market
Competition and the race for the lowest prices
Free trade has led to a concentration of manufacturers in emerging nations with low-cost labor (essentially India and China at present). Mass production intended for the global market has, among other things, caused a dizzying fall in prices, exacerbated by competition. But constant pressure for producing less expensive medicines has necessarily resulted in problems of quality.
The increase of intermediary players on an international scale, making regulatory controls more difficult because of the greater distance from production sources
The distance between production regions (essentially Asia) and the regions of use (the Americas, Europe, Africa, etc.), often with a horde of intermediaries, little binding regulation on the import/export market and the constant pressure for lower prices have led to a perverse system that contributes to supplying the market with poor-quality products.
The former colonizing countries, like all the rich countries, are sufficiently equipped to make informed choices thanks, in particular, to consumers who have acquired a certain heft in regulating the social life of their countries. In these countries, moreover, the facility that puts a product on the domestic market is effectively responsible by law for the consequences of the product's quality on the population.

16
In the post-colonial phase the newly independent countries have generally not taken into account the impossibility of reproducing and maintaining the kind of administration that had been instituted in each empire. Their population and resources do not make it possible.
Double/multiple standards of manufacturing practices
The focus on prices leads a certain number of manufacturers to produce the same medicine according to two, even several, standards of quality, according to the markets they are intended for, in other words, according to the demand level of the National Regulatory Authorities in the destination countries. A high standard will be achieved for sales in countries with stringent regulatory authorities, demanding the highest quality standard and making it possible to cover the costs of this high standard. Less high standards will be implemented for marketing in countries where requirements are less stringent (and/or lacking the means to enforce themxvi).
3.2.2 The two-speed regulation of a more complicated market
The regulation specifically supervising medicines should make it possible to avoid the negative consequences of the "end of empires" and of globalization, and to ensure the quality of all medicines from the manufacturing phase (including that of active ingredients) wherever it is done, to the administration to the patient.
As products that are manufactured, industrialized, marketed and consumed by everyone, medicines represent an enormous market both internationally and nationally. Like all markets of this kind, a chain of numerous players is necessary between the manufacturing phase (active pharmaceutical ingredients; excipients and packaging materials by other manufacturers; finished products by pharmaceutical manufacturers) and the distribution phase (wholesale distributors whether or not importing, retail pharmacies, etc.) to the patient. The active ingredients and finished products travel from one country to another as imports/exports because in this field, as in many others today, there are practically no countries that carry out all phases on their national soil.
The international market is typified by producer/exporter countries of active ingredients (China and India essentially) and finished products (India, China, South Korea, the Philippines, Russia, etc.). These countries produce for their own domestic market but also export to countries that may be very far away (Africa, Europe, the Americas, etc.).
Often complex networks between these players make traceability difficult and de facto, responsibilities are not always identifiable. The "international zone" (between exporting and importing countries) by definition evades national regulations.
Medicines on domestic markets that can be used, i.e. which possess a Marketing Authorization, are officially only available from legally approved facilities:

17
- Pharmaceutical wholesalers (procurement agencies, manufacturers’ agents, wholesale distributors, etc.) who buy medicines produced locally and/or are imported in bulk, then sell to pharmacies.
- Retail pharmaciesxvii (private pharmacies or hospitals, dispensaries and maternity-ward pharmacies) which dispense medicines to individual patients.
But the truth is quite different because there are illegal networks that coexist with legal networks in all countries.
HOWEVER:
- The illegal networks in the industrialized countries are scarce and generally limited to the increasingly developed illegal sales via the Internet.
- Numerous illegal networks in the developing countries have been created in parallel to the legal network, and in some cases have even become the main distribution network/channel.
- All kinds of situations exist between these two ends of the spectrum.
3.2.3 Poor quality is unusual in the industrialized countries
The medicines market in industrialized countries is strictly regulated and monitored. The existence of poor-quality medicines is extremely limited by regulations, inspections, controls and penalties. All sub-standard and falsified medicines are estimated to account for less than 1% of the market (but these statistics will never be anything more that conjecture).
The illegal networks are extremely limited because of health-insurance systems and social protection and above all by the way these countries are structured (customs, legal system, health, police, etc.). And infractions are severely punished.
Falsified medicines do, however, circulate but mostly in illegal networks to very small, fragmented markets (athletes, bodybuilders, immigrant communities that import medicines from their countries of origin, etc.). These are medicines that are impossible to obtain without a prescription (products known as comfort medicines, i.e. sleeping tablets, antidepressants, sexual stimulants, etc.) or medicines that are misused (opioids, anabolic hormones, etc.) or medicines unavailable in the country in question. The incursion of falsified products is increasing via trade over the Internet, which is hard for the authorities to control. They are sometimes found in the legal distribution networksxviii.
Sub-standard medicines can occasionally be found in the legal network, but warnings, traceability and control systems limit the frequency and the consequencesxix.

18
3.2.4 The presence of poor-quality products in numerous developing countries is current and compete with good-quality medicines
It is impossible to give statistics that show the reality of the presence of poor-quality products in the developing countries, but legal and illegal networks co-exist openly. This situation is in large part the result of the institutional weakness in the countries in question (customs, the courts, legal framework, education, information, etc.), the lack of social-protection systems implemented for disease (health insurance schemes, social security, health/sickness insurance, etc.) and unbalanced trade relations between the Asian manufacturers and the consumers in developing countries.
Legal and illegal networks co-exist, and the illegal network is so common that it often takes on the innocuous name of "informal". All kinds of medicines from all sorts of origins and potentially dangerous are sold on markets and conveyed by peddlers who wander from village to village. The two networks are far from impervious, and in some countries, the legal network (wholesalers, pharmacies) barely offer any more guarantees than the illegal network.
Falsified medicines mostly, but not exclusively, circulate within illegal markets. They are mostly copies that imitate the products of the big international pharmaceutical manufacturers or generic producers. These falsifications concern all types of medicines, not just the comfort medicines but also the vital ones for acute or chronic diseases such as the antimalarials, vaccines and cancer medicines.
The sub-standard medicines are legion in the poorly regulated, most-deprived countries, which are very vulnerable within the framework of international trade relations. A lot of cheap medicines are imported which have often been produced specifically for the developing countries which often do not have enough resources to assess these medicines and guarantee that the Marketing Authorization (MA) granted is a pledge of quality. Generally without an MA in the stringently regulated countries, they have never been subject to stringent assessment.
The sub-standard medicines affect both the illegal and legal markets in both the public and private sectors.
In the public sector (where medicines are "put on the market through national procurement agencies), the pressure placed by buyers, in particular through the call-for-tenders system, often neglects the qualitative aspect and focuses on the cheapest prices. This insistence on price, combined with the globalization of the manufacturing and marketing networks of medicines in the world, has contributed to making the international pharmaceutical market riskier and riskier. This is especially true for the poorest of the importing countries, because of the constant presence of sub-standard medicines connected to this excessive competition, which, in the end, benefits the least scrupulous companies that are the cleverest at understanding how this system works.

19
The reason for the presence of sub-standard medicines in the private sector (whether imported by wholesalers or produced locally) is also this race for the lowest price and the weak resources of the regulatory authorities.

20
4 WHAT MEASURES COULD CHANGE THE SITUATION FOR THE MOST DEPRIVED
COUNTRIES?
4.1 FALSIFIED AND SUB-STANDARD MEDICINES: TWO ISSUES THAT SHOULD NOT BE CONFUSED
Assessing the quality of medicines in a country is usually done from the patient's point of view. But we should be looking upstream from that because the causes of poor quality determine the actions that have to be taken.
4.1.1 Falsified medicines are the work of large- or small-scale criminal organizationsxx: so it is up to the police to fight counterfeiting
Solutions should not be sought from the health authorities or the pharmaceutical industryxxi just as it is neither up to the banks to deal with the problem of fake money nor the chemical industry to tackle the illicit drugs’ issue. To counter this scourge it is mainly up to the national (police, customs, etc.) and international agencies (Interpol, Europol, the World Customs Organization) that are responsible for safety and law enforcement, because criminal networks very often extend beyond national borders.
To date only the WHO, a few regulatory agencies such as the EMA or the USFDA and the Medicrime Convention participate in surveys/investigations that remain under the responsibility of national and international police forces. Economically penalized by counterfeits, the pharmaceutical industry also co-operates in police investigations and is trying to implement systems that attempt to discourage falsifications.
4.1.2 Sub-standard medicines are connected to the way National Regulatory Authorities work, which is why it is up to these authorities to fight sub-standard medicines
The regulations that specifically supervise medicines should make it possible to avoid sub-standards and ensure quality. Such, unfortunately, is more often not the case, and this situation is, to a large extent, the result of the weakness of the institutions in the countries in question (legal framework, regulatory authorities, customs, the courts, education, information, etc.xxii), corruption and the lack of social-protection systems when faced with illness.

21
4.2 THE ROLE OF PHARMACEUTICAL REGULATORY AUTHORITIES
4.2.1 The role of pharmaceutical authorities concerning sub-standard medicines in exporting countries
In most manufacturing countries numerous medicines are neither registered for nor sold on the domestic market. These medicines are known as "for export only" products.
It is an important nuance and can be translated by differences in the Quality Assurance of medicines depending on their destination, for the National Regulatory Authorities of the manufacturing countries do not in fact apply the same level of requirements and control for export-only medicines than those used for products that will be sold on their own market. This difference also exists in the manufacturing countries of multisource generic medicines (in particular India and China) as in the countries with stringent regulation (Europe, the United States, Canada, etc.).
A tacit rule is applied, i.e. the Regulatory Authority considers that it is responsible for ensuring the quality of medicines intended for its own population. As soon as a medicine is exported, it is the responsibility of the Regulatory Authority of the importing country to check that the quality of the imported medicine conforms to its own standards.
This quasi-generalized practice is underpinned by very pragmatic reasoning, i.e. the assessment and control of medicines are extremely costly and high-level, manpower-consuming activities. The Regulatory Authorities devote a maximum of these means to what they consider to be their brief, i.e. the protection of the public health in their own countries.
Unfortunately the impact of this practice can be harmful for patients in the poorest countries. The WHO estimates that in the African region 90% of the National Regulatory Authorities do not have the capacity for effectively controlling their markets and more particularly the quality of the imported productsxxiii.
When medicines are imported from the "stringently-regulated" countries, there might be a misunderstanding, i.e. the importing country (the authorities, medical staff and patients) often have the impression that the quality of these products is somehow guaranteed by its origin, which, in the above-mentioned reasoning, is not necessarily the case.
This state of things and its impact on the developing countries have been known for a long time. The practice is so common that the WHO's Model Certificate of Pharmaceutical Product (CPP) takes it into account, i.e. the Authority that grants the CPP has to indicate if the medicine is indeed marketed in the country of origin or if it solely intended for export.
Several articles have been published on the topic, and civil society in both the developing countries and the manufacturing countries has challenged the political powers on this "double-standard quality".
The European Union reacted with pharmaceutical legislation. Directive 2001/83/EC established a European Community code relating to medicines for human usexxiv. It obliges manufacturers operating in the EU to manufacture all their medicines, both those for the domestic market and those intended for export, in an environment that conforms to the

22
European Production Standards (GMP), which are among the most stringent in the world. All European pharmaceutical manufacturers are inspected by the authorities with the same requirement levels.
Although an undeniable advance, it is still not enough because operating manufacturers and distributors are not obliged to demonstrate the products' pharmaceutical quality for the medicines intended for export, and these products cannot receive authorization for being put on the market in any of the EU member-nations. Poor-quality meals can be prepared even in a modern kitchen; likewise it is entirely possible to manufacture sub-standard medicines in a factory that conforms to the European GMPs. The quality of active ingredients, the formulation of exported medicines, the quality controls during and at the end of production, proof of stability, efficacy and tolerability are among the crucial parameters that are controlled stringently for domestic medicines but not for exported medicines.
It is important that everyone remain aware of these disparities and the potential risks to the most vulnerable patients. It is essential for the "stringent" National Regulatory Authorities to do more to strengthen the controls of medicines manufactured for export.
4.2.2 The role of pharmaceutical authorities of importing countries for sub-standard medicines
Reducing the presence of sub-standards, which are very present in the developing countries, is theoretically within the range of the authorities of the importing countries. But in reality, only a minority of the member-countries of the WHO (fewer than 10% in Africa, according to the WHO itself) have the capacity to "stringently" control their markets.
Medicine quality assurance is a costly activity (funding, human resources, etc.). Even some of the richest countriesxxv think that pharmaceutical quality assurance exceeds their own capacity or is too expensive. More and more countries therefore implement supranational collaboration (EMA, GCC, ICH, PIC/S, etc.xxvi) with the harmonization of standards and technical requirements, mutual recognition of the work done by States or member organizations, information centralization and sharing (databases and other means, etc.). Few have materialized in Africa although the topic has been under discussion for many yearsxxvii).

23
4.3 THE WHO'S ROLE FOR SUB-STANDARD MEDICINES
In 2001 the WHO implemented the Prequalification Programme for medicines (http://apps.who.int/prequal/info_general/documents/advocacy/Advocacy_booklet_2012.pdf) to assess the technical files of recent-medicines (generics as well as innovators) for treating the three diseases defined as priorities (HIV infection, tuberculosis and malaria).
This programme very quickly showed how vital it isxxviii both for international aid bodies (UN agencies, medical NGOs, the Global Fund, etc.) and deprived countries themselves (national procurement agencies, pharmaceutical authorities, etc.) who can use the findings of this programme to select products to be supplied and accelerate their procedures for granting Market Authorization.
This programme includes checking then systematically assessing the information contained in the technical files of each product, and performing on-site audits for the assessment of the manufacturing conditions of active ingredients, the finished product and bioequivalence studies. Its purpose is to provide assurance that the prequalified products are comparable to the standards in the stringently regulated countries.
AIDS, tuberculosis and malaria are obviously not the only pathologies where concerns arise on the quality of medicines, and although the WHO Prequalification Programme has been progressively extended to products for reproductive health and to certain products for mother-child health, infectious hepatitis B and C and the neglected tropical diseases, ideally it should include all the essential medicines on the WHO Essential Medicines Model List.
Although most of the products included in the present Prequalification Programme are manufactured by a relatively small number of producers, the other commonly used medicines in developing countries (antibiotics and other anti-infection medicines, antipyretic and anti-inflammatory medicines and all categories of essential medicines, etc.) are generally manufactured by a large number of producers. Extending the WHO's Prequalification Programme would require considerably more wherewithal and probably pose major organizational and financial problemsxxix.

24
FOOTNOTES
i An innovator medicine is generally of recent use (less than fifteen years old). Most often it is international pharmaceutical manufacturers that carry out research and development studies (R&D, leading to receiving an initial MA in a country where the assessment is well regulated and stringent (the United States, the European Union, Japan, Australia, etc.). These countries also grant a commercial monopoly in the form of a patent, intended to provide financial return on the R&D investments. The patent will be obtained in a maximum number of countries where the pharmaceutical manufacturer wishes to market its medicine (with the priority given to countries with big markets, and where the patent can be granted). Although varying from country to country, the duration of the patents is usually 20 years (in reality some fifteen effective years because of the time-lag in going to market). If a pharmaceutical company claims a new use for a medicine that already has an MA, it must supply the clinical trials that correspond to this new use. If it is accepted, this new indication will be incorporated into the MA (strength, treatment duration, etc.).
ii This information about the pharmaceutical quality describes all the manufacturing and control phases, i.e. formulation (quantity of the active ingredient, the list and quantity of inactive excipients for a tablet, an ampule, a vial, etc.), identity and purity control of the active ingredient, content of the active ingredient in the finished product, packaging, stability studies of the finished product, storage conditions, etc. Pharmaceutical quality must ensure that the products manufactured on a large scale for sale are constantly identical to the products that were used for the clinical trials.
iii Here we limit the steps for assessing a medicine to the three criteria of efficacy, safety and pharmaceutical quality. But there can be other non-pharmaceutical criteria that are also taken into account in this assessment, in particular criteria of a pharmacoeconomic nature (e.g. does the offered product provide an economic advantage in relation to the existing market, either for the active ingredient considered or within a specific pharmacological or therapeutic group?).
iv An MA is sometimes valid for a group of countries that have implemented procedures for mutual recognition or centralized registration, as is the case, for example, of the European Union countries. The MA is sometimes called a "registration" or "license" depending on the country. "License" can lead to confusion for it can be synonymous with MA but also be used in the more general sense of authorization (authorization given by an owner to use their name, brand, invention or know-how, but also authorization given by a government, for example, for an import or manufacturing activity, etc.).
v The term generic medicine is very generally applied to the legal copy of an innovator medicine that is no longer patent-protected. The notions of patent and generic vary from one country to another. Because of the different definitions given to the term generic, the WHO prefers using the term multisource medicines (several manufacturers for the same content of active ingredient). The notion of generic medicines is sometimes confused with that of essential medicines. This may have come about in the 1970s when the WHO suggested that countries draw up a list of some hundreds of essential medicines that could treat 90% of their pathologies. On this list, adopted by numerous developing countries, most of the medicines were relatively old even then, so their efficacy and safety well known. Moreover the patent-related issues were no longer of concern to them. These medicines could be freely manufactured by numerous pharmaceutical companies in various countries, and the growing demand made prices fall. However, the two labels of essential medicines and generic medicines are completely different, i.e. the one (generic medicines) is about the notion of

25
innovator-copy and can be linked to intellectual property, while the other (essential medicines) to public health. In other words, the generic of a non-essential original medicine remains a non-essential medicine!
vi The National Regulatory Authorities have differing capacities to carry out their role, most often depending on the developmental conditions in each country, i.e. the pharmaceutical regulation system may be incomplete in some contexts, even completely absent in others, thus leaving space for the presence on the national market of poorly or not-at-all quality-assured medicines. The WHO estimates that the majority of poorest countries' NRAs do not have the capacity to monitor their markets. The WHO defines the "stringent" Regulatory Authorities as those that apply stringent quality standards, similar to those recommended in its technical guidelines about product quality, efficacy and safety and Good Manufacturing Practices (GMPs) (references: (i) WHO Technical Report Series No. 961, 2011. Guide on submission of documentation for prequalification of innovator finished pharmaceutical products approved by stringent regulatory authorities; (ii) WHO Technical Report Series No. 986, 2014. Annex 5 - Guideline on submission of documentation for prequalification of finished pharmaceutical products approved by stringent regulatory authorities). In its definition the WHO circumscribes the "stringent" Regulatory Authorities as those that are members of the ICH (International Council for the Harmonization of Technical Requirements for Pharmaceuticals for Human Use: http://www.ich.org.). To become a member of the ICH a Regulatory Authority must demonstrate it has implemented the ICH's harmonized standards in (i) the GMPs, (ii) the stability of medicines and (iii) the Common Technical Document (CTD) which summarizes the information about the quality, safety and efficacy of a product for Marketing Authorization (MA). An MA granted by a Stringent Regulatory Authority is a reasonable guarantee that the product is not dangerous and is efficacious and of good quality. Several international developmental aid agencies (the Global Fund, for example) have adopted this reference and make it a rule that the priority in selecting supply sources be given to pharmaceutical products (MA) registered with a "stringent" Regulatory Authority identified as member of the ICH.
vii Certain pharmaceutical authorities do not ask for proof of the generic's interchangeability with the original product. They limit themselves to the chemical identity of the active ingredient and the dosage, which does not however guarantee therapeutic equivalence with the original. Which is why the same generic may obtain an MA in one country but not in another.
viii The magazine Prescrire of January, 2016 in an article on the International Non-proprietary Names (INN) gives the following definitions and explanations: INNs correspond to a medicine's active ingredient(s); it is therefore a scientific designation, e.g. "paracetamol" and "morphine" are INNs. An INN can have several commercial names. The INNs are grouped by families to note the similarity of the mechanisms of action, adverse reactions and contraindications and carry a recognition sign of this affiliation in their name. For example the suffix "-zepam" will refer to tranquilizers of the benzodiazepine category; the suffix "-terol" will indicate that the medicine is a bronchus dilator by stimulating the beta-2 adrenergic receptors; etc. INNs must not be confused with generic medicines. Even if it often has the name of the molecule in its name, a generic is one medicine's commercial expression among others. The INNs' purpose is to inform on and give information about a medicine's mechanism of action or its pharmacotherapeutic group. It is very far from the commercial reasoning of brand names. Commercial names are sometimes easy to remember because first of all their goal is to sell, promote and attract customers. The INN objective is entirely different. It is to inform on and give information about a medicine's mechanism of action or its pharmacotherapeutic group. It helps to communicate and recognize the role of a treatment in certain reactions to avoid duplicates.

26
The INN is the real name of the medicine (in the sense of a medicinal ingredient). There are multiple advantages in thinking, speaking and prescribing in INNs rather than a commercial name. INNs provide information about the therapeutic family or the mode of action of the medicines, thus about their adverse effects and their interactions. INNs also make it possible to focus completely independently on what counts in choosing a medicine, i.e. first the choice of ingredient, then the strength and pharmaceutical form.
ix For various reasons the commercial name chosen by a pharmaceutical manufacturer is not always the same depending on the country where the medicine is marketed. Moreover it is sometimes the cause of serious confusion. The same commercial name (or two names that bear a strong resemblance to one another) can potentially be given to two completely different medicines in two different countries, with the obvious risk of confusion and of danger for health that that can carry.
x A country often attributes an MA to several generics of the same innovator (several sources from different pharmaceutical manufacturers) to promote competition not just with the innovator but also among generics. A generic medicine may be marketed as an INN or with a new commercial name depending on each country's regulations. Taxes are moreover levied upon the registration of each MA, which encourages certain pharmaceutical authorities to assign an MA to too many generics of the same innovator. This double naming of a medicine (INN and a commercial name) leads to confusion among patients, managers and even prescribers who sometimes consider them as two different medicines. What's more, when a country authorizes generics to bear a new commercial name, there is even more confusion. Labeling medicines only in INNs largely facilitates management and limits prescription and usage mistakes, but only a few countries enforce this rule.
xi All of these terms –poor-quality medicines, bad medicines, fake medicines, falsifications and counterfeit medicines– do not have universal definitions, So they can designate the following, often each one separately, depending on the country:
- Degraded medicines (tablets that change color, for example). - Inefficacious medicines. - Medicines that cause unintended adverse reactions or toxicity. - Medicines the contents of which are unknown.
This paper considers the terms fake medicines, falsification, counterfeit and counterfeit medicines as synonymous. The very notion of quality for a manufactured product is often a source of confusion. In talking about the quality of a car (its qualities of breaking, handling, comfort, etc.), it must be stated if this quality applies to all the cars of a same model (depending on the model's specifications) or one particular car (the braking condition per wear, for example, or even an occasional assembly-line defect). Thus Médiator® or Vioxx®, qualified as bad medicines, are not medicines of poor pharmaceutical quality, but medicines whose risk-benefit ratio has been poorly assessed (underestimated risks).
xii Some examples of sub-standard medicines demonstrate the unintentional nature of defects, in particular when they financially penalize the manufacturer (some analyzed samples mistakenly show a much higher strength than that shown on the label!). Some defects are visible to the eye, others not so and only become apparent when looked for (which is true for all manufacturing, i.e. a tool, a food item, a building, etc.). Some manufacturers or retailers may take advantage of the insufficient pharmaceutical services in countries where they manufacture and, above all, where they sell their products by deliberately supplying medicines that do not contain all the characteristics promised to or expected by a buyer who will not have the means to check what they are receiving (e.g. using active ingredients that are less pure, thus less expensive, or with poor-quality protective packaging, etc.). These medicines are not included in the WHO definitions of falsifications (the stated manufacturer is indeed the one who

27
legally produced the product), but this kind of sub-standard incorporates a certain amount of duplicity and deception. So, it represents an in-between category between falsification and sub-standard.
xiii A hormone like thyroxine is active at strengths 100,000 times weaker than for paracetamol. (The usual unitary dose for thyroxine is 5 micrograms, while it is 500 mg for paracetamol).
xiv EMA (European Medicine Agency: http://www.ema.europa.eu/ema/).
xv ASEAN (Association of Southeast Asian Nations: www.asean.org); UEMEOA (West African Economic and Monetary Union: www.uemoa.int); WAHO (West African Health Organization: www.wahooas.org); SADC (Southern African Development Community: www.sadc.int); EAC (East-African Community: www.eac.int); ZAZIBONA (Collaboration between national medicines regulatory authorities in Botswana, Namibia, Zambia and Zimbabwe: http://www.mcaz.co.zw/index.php/latest-news/16-zazibona-collaborative-medicines-registration-process).
xvi Beware of the fact that "inferior" quality standards are not necessarily sub-standard medicines. A medicine's quality is linked to a risk factor in terms of public health. It all depends on the defined requirement level (which is then applied in the process of assessing quality), thus of the corresponding risk factor. A reasoned risk level is perfectly acceptable without necessarily endangering the health of the patients who will consume the medicine in question. The difficulty is, of course, in striking a proper balance between this acceptable risk level and the benefit that might be derived on the economic level (by a fall in the price of a product).
xvii In some countries, dangerous medicines are legally sold over the counter by storekeepers. The medicines with the greatest risk for patients can only be prescribed by a health professional (doctor, midwife, etc.).
xviii It does occasionally happen that falsified products enter the legal market. They are expensive and generally patented. Their pharmaceutical quality can be completely comparable to the original so as to go undetected for as long as possible.
xix Mistakes during manufacture in the industrialized nations can occur, but they are much less frequent than in the developing countries for several reasons, i.e. in case of negligence or professional misconduct, the penalties imposed on their author are very dissuasive, and the control, inspection and reaction systems in case of a problem limit the mistakes and the consequences of these mistakes (traceability, warnings by the authorities, batch recalls, etc.).
xx It even happens that a small family or an individual "business" conducts a cottage-industry counterfeiting activity, for example, by filling empty capsules with flour and packaging them in the empty boxes of legitimate medicines.
xxi The pharmaceutical industry is above all composed of commercial enterprises driven by profit. The companies that have gained an international reputation (multinationals, large European, American or Asian manufacturers of generics) are very concerned about their image and devote a lot of money to protecting their products. Their image is tarnished if their medicines are copied in their appearance but not by their effects (nil or toxic).
xxii The WHO has published an assessment of 26 African countries on the capacity of their National Regulatory Authorities: http://www.who.int/healthsystems/Assessment26African_countries.pdf?ua=1
xxiii WHO/AFRO/EDP/04.5, Availability of Drug Regulatory and Quality Assurance Elements in Member States of the WHO African Region, 2004, Brazzaville.

28
xxiv The Official Journal of the European Union. Directive 2001/83/CE of the European Parliament and Council of November 6th, 2001, instituting a community-wide code for medicines for human consumption. The text is available on the website http://ec.europa.eu/health/files/eudralex/vol-1/dir_2001_83/2001_83_ec_fr.pdf. The important entries from the point of view of preventing double standards are articles 40 (by which the member-States take all useful precautions so that manufacturing medicines on their soil be subject to possessing an authorization, which is required even if the medicine is produced for export) and 46 (by which the manufacturing authorization holder is held to respect the guiding principles and lines of the GMPs of the medicines and to use only active ingredients produced in conformity with the GMPs of active ingredients and distributed in conformity with the GDPs of the active ingredients).
xxv The wealthy but lightly-populated States (Singapore, the Gulf States, etc.) rely on the reference regulatory agencies for recognizing (or not) the MAs already attributed without doing their own assessments, which would make no sense given the size of their population. Numerous developing countries, in particular in Africa, have neither the population nor the wherewithal to individually ensure the quality of their medicines. Regional groupings are therefore vital. The new regulation-harmonization projects are working along these lines.
xxvi EMA (European Medicine Agency: http://www.ema.europa.eu/ema/); GCC (Gulf Cooperation Council: https://www.gcc-sg.org/eng/); ICH (International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use: http://www.ich.org/home.html); PIC/S (Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme: http://www.picscheme.org).
xxvii Furthermore there are often questions and debates relating to the responsibility of the manufacturing countries about the quality of the products that are exported (intended for developing countries). The measures implemented by the exporting countries cannot however replace the sovereign responsibility of the importing country's National Regulatory Authority. Indeed it is definitively this authority that will grant the MA and therefore must assess its quality on the legal plain. In comprehending the issue of medicine quality, it is important to distinguish the legislation's context (which steers what must/ can be done) from the ethical considerations (which are not included in the law and which therefore remain on the edge of the decisions that might, for example, be made by a pharmaceutical regulation authority for granting or not an MA). A regulatory authority can only keep to the letter of the applicable law in its own country; it has neither the calling nor the mandate to be a policeman for matters concerning a third country. In other words in legal terms the National Regulatory Authority of the exporting country can only obey the letter of the applicable law in its own country and cannot play the role that must be assumed by the National Regulatory Authority in the importing country.
xxviii The WHO has published a study demonstrating the interest of the Prequalification Programme for the developing countries. In six sub-Saharan African countries 60% of the samples of various antimalarial products from non-prequalified sources were sub-standard, whereas this figure fell to 4% for samples of prequalified sources. Ref.: Survey of the quality of selected antimalarial medicines circulating in six countries of sub-Saharan Africa. WHO (Quality Assurance and Safety of Medicines, Department of Essential Medicines and Pharmaceutical Policies), January, 2011. The same kind of study was done for the anti-tuberculosis medicines: WHO. Survey of the quality of anti-tuberculosis medicines circulating in selected newly independent states of the former Soviet Union. WHO (Quality Assurance and Safety of Medicines, Department of Essential Medicines and Pharmaceutical Policies. WHO Regional Office for Europe), November, 2011.

29
Lastly, a new study with the same purpose has recently been carried out, dealing with the quality of products for mother/child health: http://www.who.int/medicines/publications/druginformation/issues/WHO_DI_29-3_Medicines.pdf?ua=1
xxix It doesn't in fact seem realistic to consider a programme for all essential medicines because the funds needed for that cannot be found. It would, on the other hand, be realistic to strengthen and accelerate the regulation-harmonization process. From this point of view, the WHO Prequalification Programme also supports capacity-building in the developing countries by holding training sessions and by offering rotational positions to colleagues of these countries in the programme in Geneva.

30
GLOSSARY OF PHARMACEUTICAL TERMS
This glossary gives the lay reader an explanation of the most common technical terms so as to foster
a better understanding of the pharmaceutical issues discussed in the main document.
The glossary gives the following for each term:
1. The definition in English, taken from the English version of the relevant WHO technical
guidelines or, when clarity of the definition is called for, translated from a definition taken
from a technical document in another language (i.e. the French version of a WHO technical
guideline, the Dictionary of the French National Academy of Pharmacy, the Short Manual of
Pharmacovigilance or the Clinical Pharmacology of the revue "Préscrire").
2. The reference to the document with its Internet link
3. A less technical definition and some commentary enabling an understanding of the term's
actual scope for non-specialists.
The glossary contains only terms which are not widely explained in the main document.

31
Term Definition Reference Document Simplified lay definition and comment
Quality Assurance (QA)
Quality Control (QC)
Quality assurance is a wide-ranging concept covering
all matters that individually or collectively influence
the quality of a product. It is the totality of the
arrangements made with the object of ensuring that
pharmaceutical products are of the quality required
for their intended use.
Quality control is concerned with sampling,
specifications and testing, and with the procurement
agency’s documentation and acceptance/rejection
procedures which ensure that the necessary and
relevant tests are actually carried out and that starting
materials, intermediates and finished products are not
accepted for use, sale or supply until their quality has
been judged to be satisfactory.
WHO. Model quality assurance
system for procurement agencies
(MQAS). Annex 3. Technical Report
Series No. 986, 2014.
http://www.who.int/medicines/are
as/quality_safety/quality_assuranc
e/TRS986annex3.pdf
Quality assurance is the set of written procedures that describe
how an organization ensures that its activity (e.g. manufacturing
medicines) is properly carried out.
Quality control is one aspect of quality assurance. It consists in
checking (by analysis at the physicochemical/microbiological
laboratory) that the quality of a medicine (and the materials
used for manufacturing it) complies with the required
specifications.
NB: quality control is one of the steps in quality assurance and
cannot alone be substituted for it.
National Medicine
Regulatory Authority
(NRA)
A national body that administers the full spectrum of
medicine regulatory activities, including at least all of
the following functions in conformity with national
medicine legislation:
■ Marketing authorization of new products and
variations of existing products;
■ Quality control laboratory testing;
■ Monitoring of adverse drug reactions;
■ Provision of information on medicines and
promotion of rational use of medicines;
■ Good manufacturing practice (GMP) inspections and
licensing of manufacturers, wholesalers and
distribution channels;
■ Enforcement operations;
■ Monitoring of drug utilization.
WHO. Model quality assurance
system for procurement agencies
(MQAS). Annex 3. Technical Report
Series No. 986, 2014.
http://www.who.int/medicines/are
as/quality_safety/quality_assuranc
e/TRS986annex3.pdf
A nation's NRA is the public authority responsible for regulating
pharmaceutical activities; among other things it ensures that the
manufacturers and distributors work according to regulatory
requirements and grants the Marketing Authorizations (MAs).
The NRA is a technical agency, independent of the Ministry of
Health, according to WHO recommendations.
In some countries, however, it is a department in the Ministry of
Health (e.g. Pharmacy and Medicine Department , PMD).

32
Term Definition Reference Document Simplified lay definition and comment
Marketing Authorization
(MA)
Marketing License (ML)
An official document issued by the competent
medicines regulatory authority for the purpose of
marketing or free distribution of a product after
evaluation for safety, efficacy and quality. It must set
out, inter alia, the name of the product, the
pharmaceutical dosage form, the quantitative formula
(including excipients) per unit dose (using INNs or
national generic names where they exist), the shelf-life
and storage conditions, and packaging characteristics.
It specifies the information on which authorization is
based (e.g. “The product(s) must conform with all the
details provided in your application and as modified in
subsequent correspondence”). It also contains the
product information approved for health professionals
and the public, the sales category, the name and
address of the holder of the authorization, and the
period of validity of the authorization.
Once a product has been given marketing
authorization, it is included on a list of authorized
products – the register – and is often said to be
“registered” or to “have registration”. Market
authorization may occasionally also be referred to as a
license or product license.
WHO. Marketing Authorization of
Pharmaceutical Products with
Special Reference to Multisource
(Generic) Products - A manual for
National Medicines Regulatory
Authorities (NMRAs) - ‘Blue book’ –
2nd edition - 2011.
http://apps.who.int/iris/bitstream/
10665/44576/1/9789241501453_e
ng.pdf
WHO. Model quality assurance
system for procurement agencies
(MQAS). Annex 3. Technical Report
Series No. 986, 2014.
http://www.who.int/medicines/are
as/quality_safety/quality_assuranc
e/TRS986annex3.pdf
An MA (also known as a registration or license, depending on the
country) is issued by the NRA. It is a compulsory pre-requisite for
distribution, marketing and use of a medicine throughout the
country.
An MA is only granted at the national level (or for a group of
countries, e.g. the EU, which have signed agreements for
centralizing the MAs). Thus there is no worldwide MA.
The MA contains information intended, on the one hand, for
health professionals in the form of a summary of product
characteristics (SPC) and on the other, the general public in the
form of a package leaflet for the medicinal product.
In countries with no stringent regulatory supervision, it may
happen that numerous medicines are de facto distributed
without MAs.

33
Term Definition Reference Document Simplified lay definition and comment
Bioavailability The rate and extent to which the active
pharmaceutical ingredient or active moiety is
absorbed from a pharmaceutical dosage form and
becomes available at the site(s) of action.
WHO. Model quality assurance
system for procurement agencies
(MQAS). Annex 3. Technical Report
Series No. 986, 2014.
http://www.who.int/medicines/are
as/quality_safety/quality_assuranc
e/TRS986annex3.pdf
The Dictionary of the French National Academy of Pharmacy
defines Bioavailability as follows :
“The quantity of and rate with which an active ingredient in a
pharmaceutical form is absorbed and becomes available to the
intended site(s) of action. With sites being hard of access or
sometimes unknown, and the effect most often being systemic,
bioavailability is usually defined as the quantity and rate with
which the active ingredient reaches general circulation or, less
commonly, with which it is excreted via the urine."
(http://dictionnaire.acadpharm.org/w/Acadpharm:Accueil)
Bioavailability corresponds to the capacity of a medicine's
pharmaceutical form to release its active ingredient in the
patient's body. It is represented by a curve that indicates the
amount of active ingredient released and present in the general
circulation over time (or the amount of active ingredient
excreted via the urine over time).
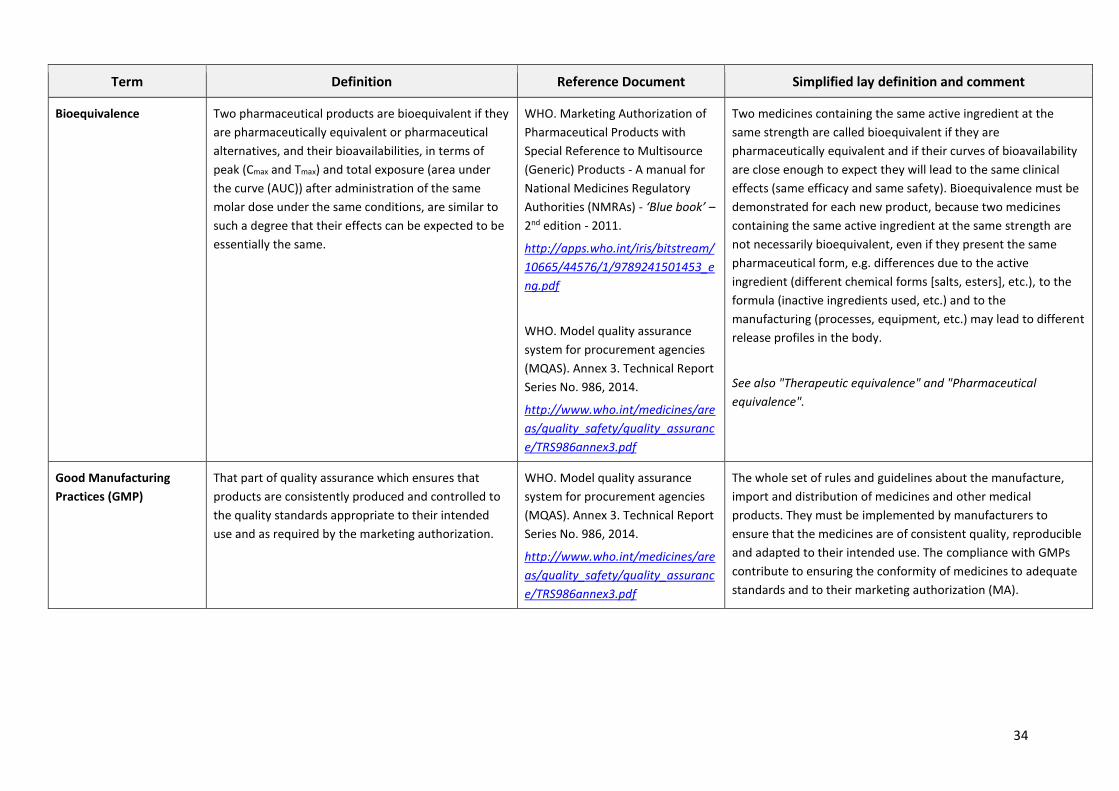
34
Term Definition Reference Document Simplified lay definition and comment
Bioequivalence Two pharmaceutical products are bioequivalent if they
are pharmaceutically equivalent or pharmaceutical
alternatives, and their bioavailabilities, in terms of
peak (Cmax and Tmax) and total exposure (area under
the curve (AUC)) after administration of the same
molar dose under the same conditions, are similar to
such a degree that their effects can be expected to be
essentially the same.
WHO. Marketing Authorization of
Pharmaceutical Products with
Special Reference to Multisource
(Generic) Products - A manual for
National Medicines Regulatory
Authorities (NMRAs) - ‘Blue book’ –
2nd edition - 2011.
http://apps.who.int/iris/bitstream/
10665/44576/1/9789241501453_e
ng.pdf
WHO. Model quality assurance
system for procurement agencies
(MQAS). Annex 3. Technical Report
Series No. 986, 2014.
http://www.who.int/medicines/are
as/quality_safety/quality_assuranc
e/TRS986annex3.pdf
Two medicines containing the same active ingredient at the
same strength are called bioequivalent if they are
pharmaceutically equivalent and if their curves of bioavailability
are close enough to expect they will lead to the same clinical
effects (same efficacy and same safety). Bioequivalence must be
demonstrated for each new product, because two medicines
containing the same active ingredient at the same strength are
not necessarily bioequivalent, even if they present the same
pharmaceutical form, e.g. differences due to the active
ingredient (different chemical forms [salts, esters], etc.), to the
formula (inactive ingredients used, etc.) and to the
manufacturing (processes, equipment, etc.) may lead to different
release profiles in the body.
See also "Therapeutic equivalence" and "Pharmaceutical
equivalence".
Good Manufacturing
Practices (GMP)
That part of quality assurance which ensures that
products are consistently produced and controlled to
the quality standards appropriate to their intended
use and as required by the marketing authorization.
WHO. Model quality assurance
system for procurement agencies
(MQAS). Annex 3. Technical Report
Series No. 986, 2014.
http://www.who.int/medicines/are
as/quality_safety/quality_assuranc
e/TRS986annex3.pdf
The whole set of rules and guidelines about the manufacture,
import and distribution of medicines and other medical
products. They must be implemented by manufacturers to
ensure that the medicines are of consistent quality, reproducible
and adapted to their intended use. The compliance with GMPs
contribute to ensuring the conformity of medicines to adequate
standards and to their marketing authorization (MA).

35
Term Definition Reference Document Simplified lay definition and comment
Patent Industrial title of ownership accorded by governments
to creators of an invention so that they can be granted
the protection of and exclusive rights to use for a
given amount of time.
Translated from:
Dictionary of the French National
Academy of Pharmacy (in French).
http://dictionnaire.acadpharm.org
/w/Acadpharm:Accueil
The title of ownership attributed to the inventor so as to protect
the intellectual property rights for an invention. Patents are
regulated by the TRIPS agreement of the World Trade
Organization (WTO). Under TRIPS the patent gives its title holder
a monopoly of 20 years for manufacturing and using their
invention commercially.
In practice, the monopoly for medicines is shorter (generally
some fifteen years or even less because of the development time
between the granting of the patent and obtaining an MA.
If a medicine is under patent in a given country, generics can only
be marketed after the patent's expiry. Some exceptions are
possible, especially for public-health reasons (see below:
Compulsory License).
Fixed dose combination
(FDC)
A combination of two or more active pharmaceutical
ingredients in a fixed ratio of doses. This term is used
generically to mean a particular combination of active
pharmaceutical ingredients irrespective of the
formulation or brand. It may be administered as
single-entity products given concurrently or as a
finished pharmaceutical product (FPP).
WHO. Multisource (generic)
pharmaceutical products:
guidelines on registration
requirements to establish
interchangeability. Annex 7.
Technical Report Series, No. 937,
2006.
http://apps.who.int/prequal/info_g
eneral/documents/trs937/who_trs
_937__annex7_eng.pdf
Fixed dose combinations (FDCs) are used to promote compliance
and limit the emergence of resistances. This is, for instance, the
case for the artemisinin-derivative-based medicines for malaria,
as well as for the antiretrovirals and the tuberculostatic
medicines.

36
Term Definition Reference Document Simplified lay definition and comment
Packaging All operations, including filling and labeling, that a bulk
product has to undergo in order to become a finished
product.
Remark: Filling of a sterile product under aseptic
conditions or a product intended to be terminally
sterilized, would not normally be regarded as part of
packaging.
WHO. WHO Good manufacturing
practices for pharmaceutical
products: main principles. Annex 2.
WHO Technical Report Series, No.
986, 2014.
http://www.who.int/medicines/are
as/quality_safety/quality_assuranc
e/TRS986annex2.pdf
By extending the definition: a generic term used for designating
packaging items.
A medicine's packaging materials should should be of adequate
quality so as to make it possible to conserve its characteristics
during transport, storage and distribution.
A distinction is made between primary packaging in direct
contact with the medicine (e.g. tablets' blister, plastic perfusion
pouch, etc.) and secondary packaging (e.g. cardboard boxes) that
protect the primary packaging but is not in direct contact with
the medicine. The materials used for primary packaging should
protect but not interact with the medicine.
Packaging is also used to provide essential information to
patients and prescribers (e.g. strength, storage conditions, expiry
dates, instructions for use, interactions, etc.).
Manufacturing date Date established for each batch, corresponding to the
final manufacturing completion date. It is usually
expressed by a month and a year. The date of analysis
leading to the batch release can be taken as the
manufacturing date on the condition that the time
between the start of manufacture and the release of
the product does not exceed a twentieth of the shelf
life.
Translated from the French version
of:
WHO. Marketing Authorization of
Pharmaceutical Products with
Special Reference to Multisource
(Generic) Products - A manual for
National Medicines Regulatory
Authorities (NMRAs) - ‘Blue book’ -
Series Pharmaceutical Regulation,
No. 13 - 2008.
http://apps.who.int/medicinedocs/
documents/s16166f/s16166f.pdf
Printing the manufacturing date on the primary and secondary
packaging is not always made compulsory by national
regulations, although the WHO recommends it.
This date MUST always appear on the certificates of analysis.

37
Term Definition Reference Document Simplified lay definition and comment
Expiry date The date given on the individual container (usually on
the label) of a pharmaceutical product up to and
including the date on which the product is expected to
remain within specifications, if stored correctly. It is
established for each batch by adding the shelf-life to
the date of manufacture.
WHO. WHO Good distribution
practices for pharmaceutical
products. Annex 5. WHO Technical
Report Series, No. 957, 2010.
http://www.who.int/medicines/are
as/quality_safety/quality_assuranc
e/GoodDistributionPracticesTRS957
Annex5.pdf
This date must be clearly expressed by a month and a year and
printed on the primary and secondary packaging, along with
storage conditions. It corresponds to the product's shelf life,
derived from the outcomes of stability studies.
See below: Shelf life.
Shelf life The period of time during which an API or FPP, if
stored correctly, is expected to comply with the
specification as determined by stability studies on a
number of batches of the API or FPP. The shelf-life is
used to establish the expiry date of each batch.
WHO. Stability testing of active
pharmaceutical ingredients and
finished pharmaceutical products.
Annex 2. WHO Technical Report
Series, No. 953, 2009.
http://apps.who.int/medicinedocs/
documents/s19133en/s19133en.pd
f
A medicine must conserve its chemical, physical, microbiological
and biopharmaceutical attributes for its entire shelf life. The
shelf life is determined by stability studies carried out on a
certain number of commercial batches (i.e. the packages for
sale). The study conditions depend on the climate conditions in
which the medicines will be stored and used. The WHO defines
four major climate zones: temperate (I), sub-tropical (II), hot and
dry (III) and hot and humid (IV).
The shelf life and the expiry date must be assessed for any new
product. If a product is exposed to more aggressive conditions
than those indicated for its storage (for instance in case a vaccine
undergoes a break in the cold chain during storage), the declared
shelf life and expiry date are no longer reliable.
International Non
Proprietary Name (INN)
International Non proprietary Names (INN) identify
pharmaceutical substances or active pharmaceutical
ingredients.
WHO. Programs (webpage).
Essential medicines and health
products. Guidance on
International Non-proprietary
Names (INN).
http://www.who.int/medicines/ser
vices/inn/innguidance/en/
Each INN is a unique name that is globally recognized and is
public property. The WHO is responsible for attributing INNs for
pharmaceutical ingredients.
The purpose of the INNs is to clearly identify a medicine's action
mechanism and/or its pharmacotherapeutic group. The INNs
help in the management and prescription of medicines, because
they indicate the medicine's therapeutic family (essential
information for caregivers), and it avoids confusion that may be
caused by the commercial names.
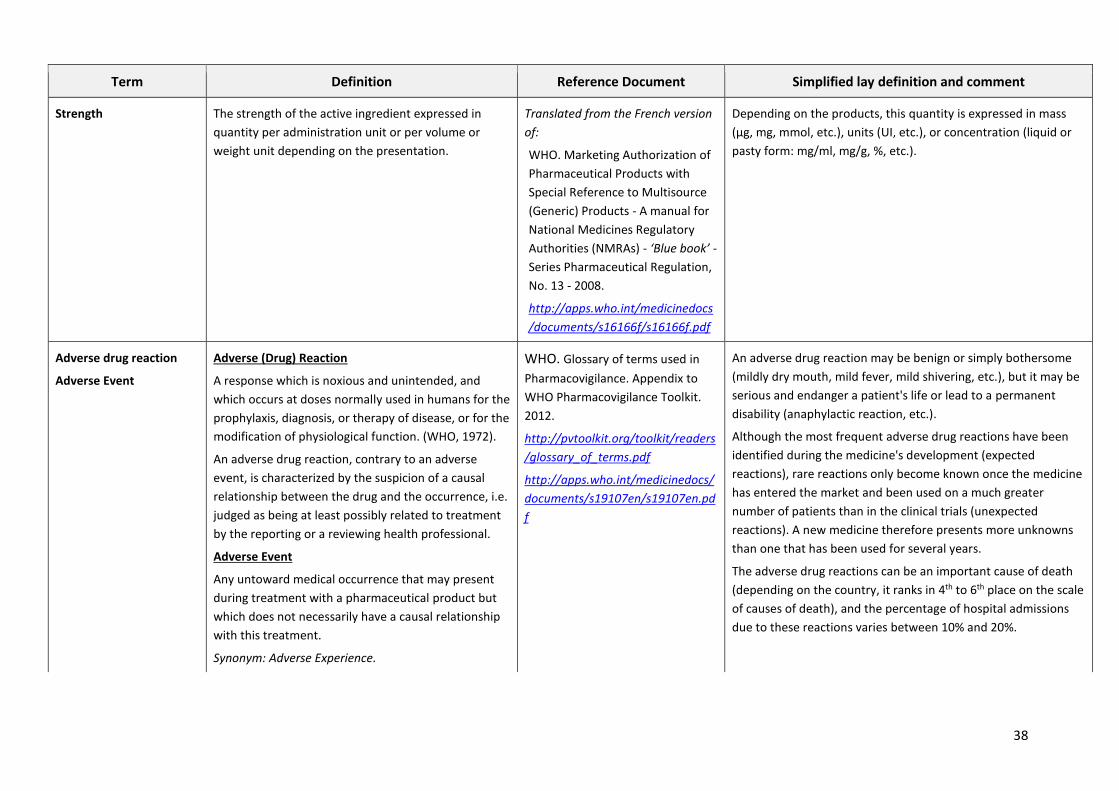
38
Term Definition Reference Document Simplified lay definition and comment
Strength The strength of the active ingredient expressed in
quantity per administration unit or per volume or
weight unit depending on the presentation.
Translated from the French version
of:
WHO. Marketing Authorization of
Pharmaceutical Products with
Special Reference to Multisource
(Generic) Products - A manual for
National Medicines Regulatory
Authorities (NMRAs) - ‘Blue book’ -
Series Pharmaceutical Regulation,
No. 13 - 2008.
http://apps.who.int/medicinedocs
/documents/s16166f/s16166f.pdf
Depending on the products, this quantity is expressed in mass
(µg, mg, mmol, etc.), units (UI, etc.), or concentration (liquid or
pasty form: mg/ml, mg/g, %, etc.).
Adverse drug reaction
Adverse Event
Adverse (Drug) Reaction
A response which is noxious and unintended, and
which occurs at doses normally used in humans for the
prophylaxis, diagnosis, or therapy of disease, or for the
modification of physiological function. (WHO, 1972).
An adverse drug reaction, contrary to an adverse
event, is characterized by the suspicion of a causal
relationship between the drug and the occurrence, i.e.
judged as being at least possibly related to treatment
by the reporting or a reviewing health professional.
Adverse Event
Any untoward medical occurrence that may present
during treatment with a pharmaceutical product but
which does not necessarily have a causal relationship
with this treatment.
Synonym: Adverse Experience.
WHO. Glossary of terms used in
Pharmacovigilance. Appendix to
WHO Pharmacovigilance Toolkit.
2012.
http://pvtoolkit.org/toolkit/readers
/glossary_of_terms.pdf
http://apps.who.int/medicinedocs/
documents/s19107en/s19107en.pd
f
An adverse drug reaction may be benign or simply bothersome
(mildly dry mouth, mild fever, mild shivering, etc.), but it may be
serious and endanger a patient's life or lead to a permanent
disability (anaphylactic reaction, etc.).
Although the most frequent adverse drug reactions have been
identified during the medicine's development (expected
reactions), rare reactions only become known once the medicine
has entered the market and been used on a much greater
number of patients than in the clinical trials (unexpected
reactions). A new medicine therefore presents more unknowns
than one that has been used for several years.
The adverse drug reactions can be an important cause of death
(depending on the country, it ranks in 4th to 6th place on the scale
of causes of death), and the percentage of hospital admissions
due to these reactions varies between 10% and 20%.

39
Term Definition Reference Document Simplified lay definition and comment
Pharmaceutical
equivalence
Products are pharmaceutical equivalents if they
contain the same amount of the same active
substance(s) in the same dosage form; if they meet
the same or comparable standards; and if they are
intended to be administered by the same route.
Pharmaceutical equivalence does not necessarily
imply therapeutic equivalence, as differences in the
excipients and/or the manufacturing process can lead
to differences in product performance.
WHO. Marketing Authorization of
Pharmaceutical Products with
Special Reference to Multisource
(Generic) Products - A manual for
National Medicines Regulatory
Authorities (NMRAs) - ‘Blue book’ –
2nd edition - 2011.
http://apps.who.int/iris/bitstream/
10665/44576/1/9789241501453_e
ng.pdf
See below: Therapeutic equivalence.
Therapeutic equivalence Two pharmaceutical products are considered to be
therapeutically equivalent if they are pharmaceutically
equivalent or pharmaceutical alternatives and after
administration in the same molar dose, their effects,
with respect to both efficacy and safety, are
essentially the same when administered to patients by
the same route under the conditions specified in the
labeling.
WHO. Marketing Authorization of
Pharmaceutical Products with
Special Reference to Multisource
(Generic) Products - A manual for
National Medicines Regulatory
Authorities (NMRAs) - ‘Blue book’ –
2nd edition - 2011.
http://apps.who.int/iris/bitstream/
10665/44576/1/9789241501453_e
ng.pdf
Therapeutic equivalence between two medicines containing the
same active ingredient at the same strength is generally
demonstrated in vivo (by bioequivalence studies or clinical trials)
and in some cases by in vitro studies (comparative dissolving
profiles).
In a bioequivalence study, the same strength of the generic
medicine to be tested and the innovator medicine (reference) is
administered to a certain number of healthy subjects, and the
concentration profiles of active ingredients are measured in the
blood and compared.
Bioequivalence studies are needed to demonstrate that a generic
medicine (a pharmaceutical equivalent) is a therapeutic
equivalent with the innovator medicine, thus interchangeable
with it.
See also Bioequivalence and Bioavailability.
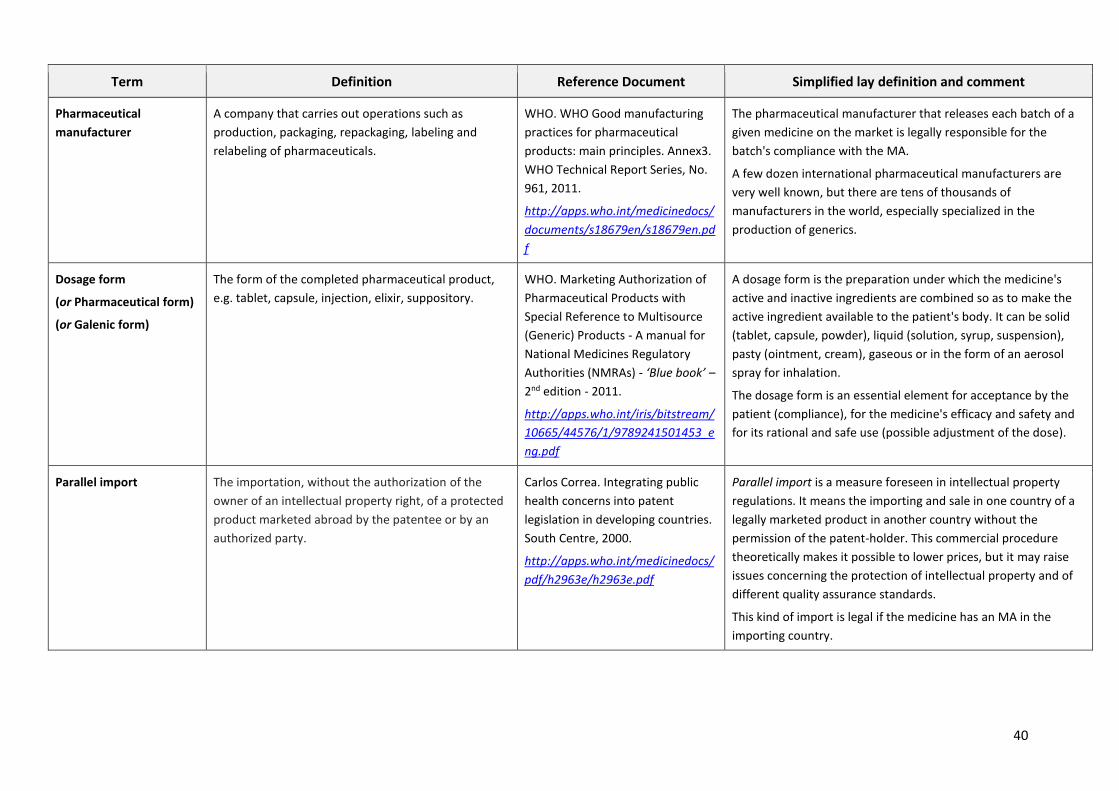
40
Term Definition Reference Document Simplified lay definition and comment
Pharmaceutical
manufacturer
A company that carries out operations such as
production, packaging, repackaging, labeling and
relabeling of pharmaceuticals.
WHO. WHO Good manufacturing
practices for pharmaceutical
products: main principles. Annex3.
WHO Technical Report Series, No.
961, 2011.
http://apps.who.int/medicinedocs/
documents/s18679en/s18679en.pd
f
The pharmaceutical manufacturer that releases each batch of a
given medicine on the market is legally responsible for the
batch's compliance with the MA.
A few dozen international pharmaceutical manufacturers are
very well known, but there are tens of thousands of
manufacturers in the world, especially specialized in the
production of generics.
Dosage form
(or Pharmaceutical form)
(or Galenic form)
The form of the completed pharmaceutical product,
e.g. tablet, capsule, injection, elixir, suppository.
WHO. Marketing Authorization of
Pharmaceutical Products with
Special Reference to Multisource
(Generic) Products - A manual for
National Medicines Regulatory
Authorities (NMRAs) - ‘Blue book’ –
2nd edition - 2011.
http://apps.who.int/iris/bitstream/
10665/44576/1/9789241501453_e
ng.pdf
A dosage form is the preparation under which the medicine's
active and inactive ingredients are combined so as to make the
active ingredient available to the patient's body. It can be solid
(tablet, capsule, powder), liquid (solution, syrup, suspension),
pasty (ointment, cream), gaseous or in the form of an aerosol
spray for inhalation.
The dosage form is an essential element for acceptance by the
patient (compliance), for the medicine's efficacy and safety and
for its rational and safe use (possible adjustment of the dose).
Parallel import The importation, without the authorization of the
owner of an intellectual property right, of a protected
product marketed abroad by the patentee or by an
authorized party.
Carlos Correa. Integrating public
health concerns into patent
legislation in developing countries.
South Centre, 2000.
http://apps.who.int/medicinedocs/
pdf/h2963e/h2963e.pdf
Parallel import is a measure foreseen in intellectual property
regulations. It means the importing and sale in one country of a
legally marketed product in another country without the
permission of the patent-holder. This commercial procedure
theoretically makes it possible to lower prices, but it may raise
issues concerning the protection of intellectual property and of
different quality assurance standards.
This kind of import is legal if the medicine has an MA in the
importing country.

41
Term Definition Reference Document Simplified lay definition and comment
Drug interaction We speak of a drug interaction when the simultaneous
administration of two or more medicines leads to
enhancing the desired or counteracting the adverse
effects of at least one of these medicines.
Translated from:
Short manual of
Pharmacovigilance and clinical
pharmacology. Prescrire, 2011 (in
French).
http://www.prescrire.org/fr/101/3
24/47316/0/PositionDetails.aspx
A drug interaction occurs when two or more medicines,
prescribed at the same time or close to one another, react
together.
This reaction might cause either an increase or a decrease in the
action of one or more medicines, but it can also determine
changes in the efficacy/safety profile, e.g. causing new
therapeutic or adverse reactions.
Drug interactions can be beneficial and sought if there is an
additive-synergy or enhancing end (e.g. L-dopa and peripheral
dopadecarboxylase inhibitors; penicillin and the betalactamase
inhibitor) or, on the contrary, create adverse incompatibilities
(e.g. simultaneously using a medicine for sleep–a sedative–and
an anti-allergy product–an antihistamine– can reduce alertness
and reactions, making driving a car or using machines
dangerous).
Licensing:
Voluntary license
Authorization given by a right-holder (licensor) to
someone (licensee) to exercise acts that only the
licensor can legally do.
Carlos Correa. Integrating public
health concerns into patent
legislation in developing countries.
South Centre, 2000.
http://apps.who.int/medicinedocs/
pdf/h2963e/h2963e.pdf
A voluntary license is a measure foreseen in intellectual-property
regulations. The holder of a pharmaceutical patent has the
discretion to grant other parties exclusive or non-exclusive
licenses to manufacture, import and/or distribute the
pharmaceutical product for which he holds the patent. Voluntary
licenses can enable considerable containment of costs.
At the discretion of the patent-holder, these agreements are
generally concluded for strategic reasons (e.g. entry into a
market) rather than as actions to make the product more
financially affordable, and in some cases they may not even lead
to lowering the price at all.

42
Term Definition Reference Document Simplified lay definition and comment
Licensing:
Compulsory license
Authorization given by a judicial or administrative
authority to a third party for the use of a patented
invention, without the consent of the patentee, on
various grounds of general interest (absence of
working, public health, anticompetitive practices,
emergency, national defense).
Carlos Correa. Integrating public
health concerns into patent
legislation in developing countries.
South Centre, 2000.
http://apps.who.int/medicinedocs/
pdf/h2963e/h2963e.pdf
The compulsory licensing system is a measure foreseen in
intellectual-property regulations. In the case of medicines it
enables a government to grant a license to a company, a
governmental body or any other party for reasons of public
health to manufacture/use a generic version of a patented
medicine, even without the permission of the patent-holder.
In public-health terms compulsory licenses may be an important
instrument for promoting competition and making the new
essential medicines more affordable. There are certain
prerequisites to be met before a compulsory license can be
granted, e.g. a voluntary license must be requested of the
patent-holder beforehand, and the patent-holder must receive
adequate payment for use of the invention.
The WHO has recommended the use of compulsory licenses in
cases of patent-rights abuse or to meet a national emergency so
as to ensure that the medicines' prices correspond to the local
government's/population's buying power.
Batch A defined quantity of starting material, packaging
material, or product processed in a single process or
series of processes so that it is expected to be
homogeneous.
It may sometimes be necessary to divide a batch into a
number of sub-batches, which are later brought
together to form a final homogeneous batch.
In the case of terminal sterilization, the batch size is
determined by the capacity of the autoclave.
In continuous manufacture, the batch must
correspond to a defined fraction of the production,
characterized by its intended homogeneity.
The batch size can be defined either as a fixed quantity
or as the amount produced in a fixed time interval.
WHO. WHO Good manufacturing
practices for pharmaceutical
products: main principles. Annex 2.
WHO Technical Report Series, No.
986, 2014.
http://www.who.int/medicines/are
as/quality_safety/quality_assuranc
e/TRS986annex2.pdf
In pharmaceutical manufacturing, every batch must be released
by a person authorized by the manufacturer. By releasing the
batch, the authorized person certifies that the batch has been
manufactured and monitored according to the characteristics
described in the Marketing Authorization.
A batch is identified by a unique code (batch number),
reproduced on the finished product's primary and secondary
packaging as well as on the documents associated with its
manufacture and guaranteeing its traceability (batch record, the
batch's certificate of analysis, etc.).

43
Term Definition Reference Document Simplified lay definition and comment
Generic product
Interchangeable
pharmaceutical product
Multisource
pharmaceutical product
Generic product
The term generic product has somewhat different
meanings in different jurisdictions. Use of this term is
therefore avoided as much as possible, and the term
“multisource pharmaceutical product” (see below) is
used instead.
Generic products may be marketed either under the
approved non proprietary name (INN) or under a
brand (proprietary) name. They may be marketed in
dosage forms and/or strengths different from those of
the innovator products.
Where the term “generic product” is used, it means a
pharmaceutical product, usually intended to be
interchangeable with the innovator product, which is
usually manufactured without a license from the
innovator company and marketed after expiry of the
patent or other exclusivity rights.
The term should not be confused with generic names
for APIs.
Interchangeable pharmaceutical product
An interchangeable pharmaceutical product is one
which is therapeutically equivalent to a comparator
product and can be interchanged with the comparator
in clinical practice.
Multisource pharmaceutical product
Multisource pharmaceutical products are
pharmaceutically equivalent products that may or may
not be therapeutically equivalent. Multisource
pharmaceutical products that are therapeutically
equivalent are interchangeable.
WHO. Marketing Authorization of
Pharmaceutical Products with
Special Reference to Multisource
(Generic) Products - A manual for
National Medicines Regulatory
Authorities (NMRAs) - ‘Blue book’ –
2nd edition - 2011.
http://apps.who.int/iris/bitstream/
10665/44576/1/9789241501453_e
ng.pdf
A generic product is a copy of an innovator medicine (also known
as an originator medicine, a benchmark medicine or a princeps
medicine). The definition of a generic product may change
depending on the country. However, generics are always meant
to be pharmaceutically equivalent products (same quantity of
the same active ingredients(s) in the same dosage form but not
necessarily the same excipients – see the definition of
Pharmaceutical Equivalent), but not systematically
therapeutically equivalent.
For a medicine to be interchangeable, thus known as "generic",
its bioequivalence with the original medicine must be proven. In
strictly regulated countries the bioequivalence between a
generic medicine and the original medicine must be
demonstrated prior to its being put on the market. But such is
not the case in all countries. For this reason the WHO talks rather
of "multisource" medicines.
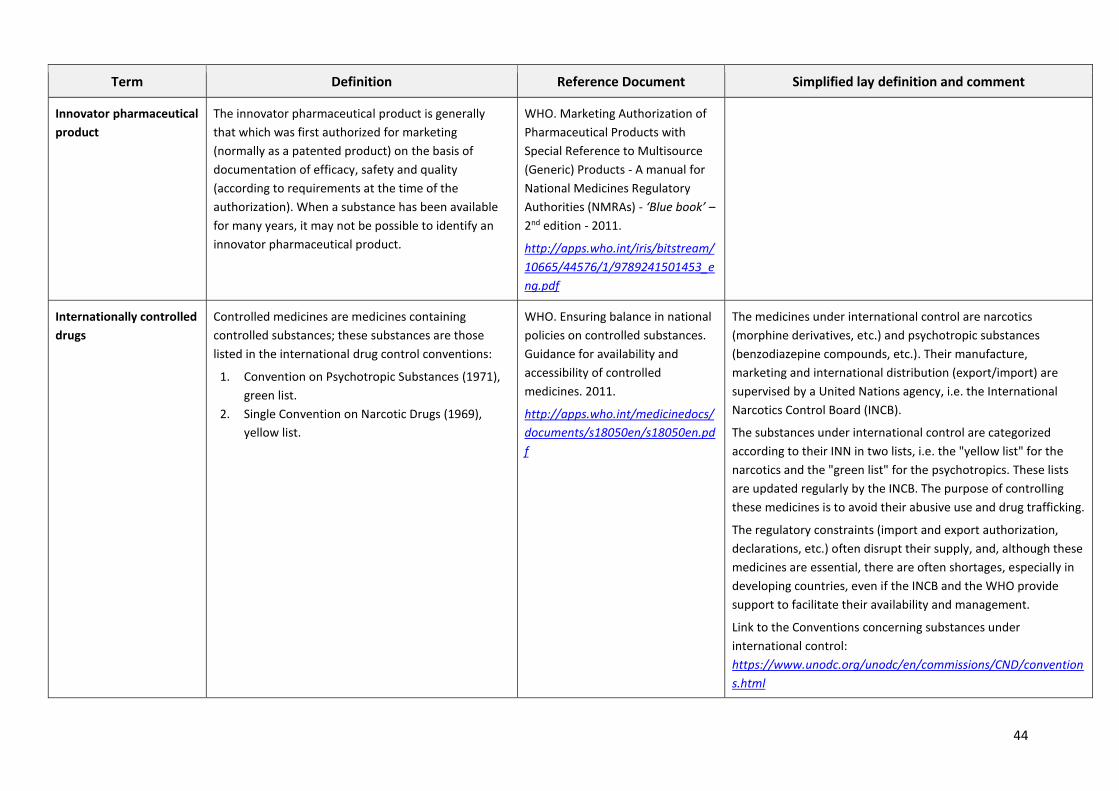
44
Term Definition Reference Document Simplified lay definition and comment
Innovator pharmaceutical
product
The innovator pharmaceutical product is generally
that which was first authorized for marketing
(normally as a patented product) on the basis of
documentation of efficacy, safety and quality
(according to requirements at the time of the
authorization). When a substance has been available
for many years, it may not be possible to identify an
innovator pharmaceutical product.
WHO. Marketing Authorization of
Pharmaceutical Products with
Special Reference to Multisource
(Generic) Products - A manual for
National Medicines Regulatory
Authorities (NMRAs) - ‘Blue book’ –
2nd edition - 2011.
http://apps.who.int/iris/bitstream/
10665/44576/1/9789241501453_e
ng.pdf
Internationally controlled
drugs
Controlled medicines are medicines containing
controlled substances; these substances are those
listed in the international drug control conventions:
1. Convention on Psychotropic Substances (1971),
green list.
2. Single Convention on Narcotic Drugs (1969),
yellow list.
WHO. Ensuring balance in national
policies on controlled substances.
Guidance for availability and
accessibility of controlled
medicines. 2011.
http://apps.who.int/medicinedocs/
documents/s18050en/s18050en.pd
f
The medicines under international control are narcotics
(morphine derivatives, etc.) and psychotropic substances
(benzodiazepine compounds, etc.). Their manufacture,
marketing and international distribution (export/import) are
supervised by a United Nations agency, i.e. the International
Narcotics Control Board (INCB).
The substances under international control are categorized
according to their INN in two lists, i.e. the "yellow list" for the
narcotics and the "green list" for the psychotropics. These lists
are updated regularly by the INCB. The purpose of controlling
these medicines is to avoid their abusive use and drug trafficking.
The regulatory constraints (import and export authorization,
declarations, etc.) often disrupt their supply, and, although these
medicines are essential, there are often shortages, especially in
developing countries, even if the INCB and the WHO provide
support to facilitate their availability and management.
Link to the Conventions concerning substances under
international control:
https://www.unodc.org/unodc/en/commissions/CND/convention
s.html

45
Term Definition Reference Document Simplified lay definition and comment
Misuse
(or Irrational
use/prescribing)
Prescribing that does not conform to good standards
of treatment - for example, extravagant prescribing,
overprescribing, incorrect prescribing, multiple
prescribing, or underprescribing of medication.
In the purposes of the guidelines on controlled
substances, the term “Misuse” is defined as the non-
medical and non-scientific use of substances
controlled under the international drug control
treaties or under national law.
WHO. Drug and Therapeutics
Committees - A Practical Guide
(2003).
http://apps.who.int/medicinedocs/
en/d/Js4882e/
WHO. Ensuring balance in national
policies on controlled substances.
Guidance for availability and
accessibility of controlled
medicines. 2011.
http://apps.who.int/medicinedocs/
documents/s18050en/s18050en.pd
f
The term is used by the WHO to describe the inappropriate
prescription and/or use of a medicine. In addition it can be used
to describe the abuse or misuse of a medicine by users for
recreational, fraudulent or lucrative (resale) purposes, in either
common or criminal law.
Misuse is, for example, common with psychoactive substances
and can lead to abuse by and drug dependency in users.
Pharmacopoeia Originally the art of preparing medicines, then
inventorying the medicines in use at any given time,
often called a Codex; at present an official register,
periodically revised by a scientific commission,
published in a country or a group of countries, the
standards of which are enforced on pharmaceutical
activities.
The pharmacopoeias are, in particular, composed of
monographs relating to raw materials or the
preparations entering into the manufacture of
medicines. They are a collection of specifications that
define the qualitative and quantitative characteristics
of these substances or preparations with a view to
ensuring optimal quality compatible with the
requirements of public health.
Translated from:
Dictionary of the French National
Academy of Pharmacy (in French).
http://dictionnaire.acadpharm.org
/w/Acadpharm:Accueil
An official publication of a country that lists raw materials (active
and inactive substances) and sometimes the finished products
(the given form of a medicine), it describes their characteristics
as well as the methods and standards that enable quality control.
Not all countries have the means to establish and update a
national pharmacopoeia. The pharmacopoeias of certain
countries (Great Britain, the United States, France, Japan, China,
India, etc.) or groups of countries (the European Union) are used
as internationally recognized references.
With the purpose of harmonizing medicinal quality standards
since 1951, the WHO publishes the International Pharmacopoeia
(Ph.Int.), adapted to the environment of the developing
countries (DCs).

46
Term Definition Reference Document Simplified lay definition and comment
Pharmaceutical product 1. Any medicine intended for human use or
administered to food-producing animals, presented
in its finished dosage form or as an active ingredient
for use in such dosage form, that is subject to
control by pharmaceutical legislation in both the
exporting state and the importing state.
2. Any substance or combination of substances which
has a therapeutic, prophylactic or diagnostic use, or
is intended to modify physiological functions, and is
presented in a dosage form suitable for
administration to humans.
1. WHO. Guidelines for
implementation of the WHO
Certification Scheme on the
Quality of Pharmaceutical
Products Moving in International
Commerce. Annex 10. WHO
Technical Report Series, No. 863,
1996.
http://apps.who.int/medicinedoc
s/pdf/s5516e/s5516e.pdf
2. WHO. Guidance for
organizations performing in vivo
bioequivalence studies. Annex 9.
WHO Technical Report Series,
No. 996, 2016.
http://www.who.int/medicines/
publications/pharmprep/WHO_T
RS_996_annex09.pdf
In the current acceptance: pharmaceutical product encompasses
all products subject to pharmaceutical regulation (medicines,
vaccines, medical devices, diagnostic tests, etc.).

47
Term Definition Reference Document Simplified lay definition and comment
Pharmacovigilance Pharmacovigilance is the science and activities relating
to the detection, assessment, understanding and
prevention of adverse effects or any other medicine-
related problem.
WHO. Pharmacovigilance: Ensuring
the Safe Use of Medicines - WHO
Policy Perspectives on Medicines,
No. 009, October 2004.
http://apps.who.int/medicinedocs/
en/d/Js6164e/
Pharmacovigilance is a system of information gathering dealing
with the detection, understanding and assessment of adverse
effects or any other problem associated with medicine use. This
system was widely implemented in Europe and the United States
after the serious adverse effects caused by the use of
thalidomide in 1961.
The clinical trials carried out prior to marketing a medicine to
assess therapeutic efficacy and acceptability are necessarily done
on a limited number of people. After entering the market, the
medicine is used by a much larger number and diversity of
patients who may also use other medicines simultaneously. By
documenting and centrally reporting the observed adverse drug
reactions, it is possible to know the adverse effects better,
especially when they are infrequent.
This monitoring (pharmacovigilance) relies essentially on the
spontaneous reporting of cases by physicians, pharmacists and
patients. At the national level it is most often the National
Medicine Regulatory Authority that is in charge of
pharmacovigilance.
In the developing countries, pharmacovigilance, when it exists,
focuses above all on the follow-up of strategic and recent
medicines (antiretrovirals, malaria medicines, etc.) or of misuses
(to reduce unnecessary expenses). The assessment of the
medicine quality in these countries should also be incorporated
into the notion of pharmacovigilance. It is in fact vital to
determine beforehand if the adverse effects reported are linked
to the medicine under consideration or attributable to the use of
sub-standard or falsified medicines.

48
Term Definition Reference Document Simplified lay definition and comment
Benefit risk ratio The ratio of benefit to risk in the use of a drug; a
means of expressing a judgment concerning the role
of the drug in the practice of medicine, based on
efficacy and safety data along with consideration of
misuse potential, severity and prognosis of the
disease, etc. The concept may be applied to a single
drug or in comparisons between two or more drugs
used for the same condition.
WHO. The Use of Essential Drugs:
Ninth Report of the WHO Expert
Committee (including the Model
List of Essential Drugs). 2000.
http://apps.who.int/medicinedocs/
fr/d/Js2281e/
The ratio between the therapeutic benefit of a medicine and its
adverse effects as part of usage in conformity with the MA. An
MA is only delivered if the beneficial effects are considered
greater than the harmful effects.
After entering the market, with hindsight and usage by a much
larger number of patients than during the clinical trials, the
benefit/risk ratio can sometimes be inverted, leading to the
withdrawal of the MA.
NB: depending on their contexts and their risk-assessment
methods, two countries may arrive at a noticeably different
assessment of the benefit/risk ratio for the same medicine.
Active Pharmaceutical
Ingredient (API)
That portion of a medicine (drug) that has therapeutic
properties.
WHO. Drug and Therapeutics
Committees - A Practical Guide
(2003).
http://apps.who.int/medicinedocs/
en/d/Js4882e/
A natural, synthetic or biological ingredient that gives the
medicine its therapeutic activity. A medicine is composed of
active ingredient(s) and a set of inactive ingredients (excipients:
water, lactose, starch, etc.) required in its preparation to obtain
a form that may be used by a patient or an animal (tablet,
capsule, injectable ampule, vial, etc.).
Inactive Pharmaceutical
Ingredient
(or Excipient)
An inert substance used to give a pharmaceutical
preparation a suitable form or consistency.
WHO. Drug and Therapeutics
Committees - A Practical Guide
(2003).
http://apps.who.int/medicinedocs/
en/d/Js4882e/
The amount of excipients In a medicine is often much greater than the amount of active ingredient(s). A quality defect in one of the excipients can therefore have important impact despite the ingredient's "inactive" nature, and in certain cases potentially deadly (e.g. poisoning by diethylene-glycol, an impurity of glycerin that is frequently used as an excipient in the manufacture of syrups).
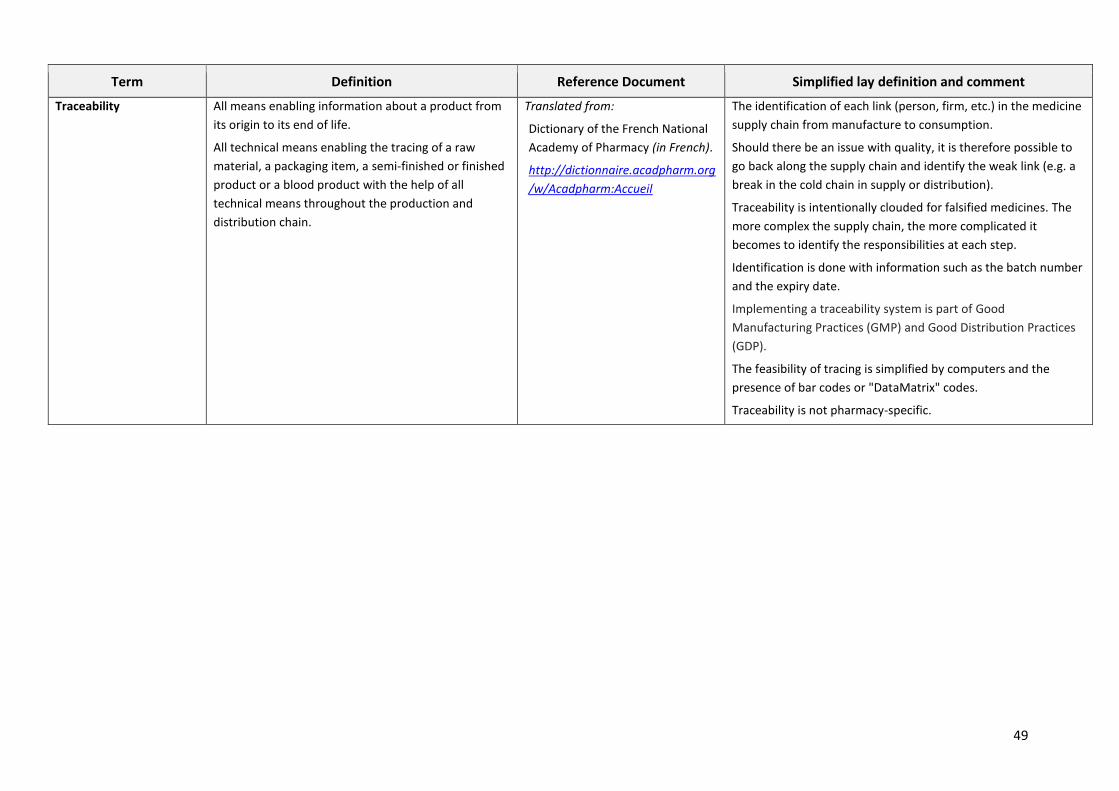
49
Term Definition Reference Document Simplified lay definition and comment
Traceability All means enabling information about a product from
its origin to its end of life.
All technical means enabling the tracing of a raw
material, a packaging item, a semi-finished or finished
product or a blood product with the help of all
technical means throughout the production and
distribution chain.
Translated from:
Dictionary of the French National
Academy of Pharmacy (in French).
http://dictionnaire.acadpharm.org
/w/Acadpharm:Accueil
The identification of each link (person, firm, etc.) in the medicine
supply chain from manufacture to consumption.
Should there be an issue with quality, it is therefore possible to
go back along the supply chain and identify the weak link (e.g. a
break in the cold chain in supply or distribution).
Traceability is intentionally clouded for falsified medicines. The
more complex the supply chain, the more complicated it
becomes to identify the responsibilities at each step.
Identification is done with information such as the batch number
and the expiry date.
Implementing a traceability system is part of Good
Manufacturing Practices (GMP) and Good Distribution Practices
(GDP).
The feasibility of tracing is simplified by computers and the
presence of bar codes or "DataMatrix" codes.
Traceability is not pharmacy-specific.