epidithiodiketopiperazinesblocktheinteractionbetween ... ·...
TRANSCRIPT

Epidithiodiketopiperazines Block the Interaction betweenHypoxia-inducible Factor-1� (HIF-1�) and p300 by a ZincEjection Mechanism*□S
Received for publication, April 17, 2009, and in revised form, July 1, 2009 Published, JBC Papers in Press, July 9, 2009, DOI 10.1074/jbc.M109.009498
Kristina M. Cook‡§, Stephen T. Hilton¶, Jasmin Mecinovic§, William B. Motherwell�, William D. Figg‡1,and Christopher J. Schofield§
From ‡NCI, National Institutes of Health, Bethesda, Maryland 20814, the §Chemistry Research Laboratory, Department of Chemistry, andthe Oxford Centre for Integrative Systems Biology, University of Oxford, Oxford OX1 3TA, United Kingdom, the ¶Department ofPharmaceutical and Biological Chemistry, School of Pharmacy, University of London, London WC1N 1AX, United Kingdom, and the�Department of Chemistry, University College London, London WC1H 0AJ, United Kingdom
The hypoxic response in humans is regulated by the hypoxia-inducible transcription factor system; inhibition of hypoxia-in-ducible factor (HIF) activity has potential for the treatment ofcancer. Chetomin, a member of the epidithiodiketopiperazine(ETP) family of natural products, inhibits the interactionbetween HIF-� and the transcriptional coactivator p300. Struc-ture-activity studies employing both natural and synthetic ETPderivatives reveal that only the structurally unique ETP core isrequired and sufficient to block the interaction of HIF-1� andp300. In support of both cell-based and animal work showingthat the cytotoxic effect of ETPs is reduced by the addition ofZn2� through an unknownmechanism, ourmechanistic studiesreveal that ETPs react with p300, causing zinc ion ejection. Cellstudies with both natural and synthetic ETPs demonstrated adecrease in vascular endothelial growth factor and antiprolif-erative effects that were abrogated by zinc supplementation.The results have implications for the design of selective ETPsand for the interaction of ETPs with other zinc ion-binding pro-tein targets involved in gene expression.
A major transcriptional activation system, involving HIF2and the p300/CREB-binding protein (CBP) coactivators,
coordinates cellular and physiological responses to hypoxiain animals (for review, see Ref. 1). The oxygen-sensingcomponent of the HIF system is provided by oxygenasesthat catalyze the post-translational hydroxylation of theHIF �-subunit. HIF-� prolyl-hydroxylation signals for pro-teasomal degradation, whereas asparaginyl hydroxylationreduces HIF activity by blocking interaction of the HIF-1�C-terminal activation domain (C-TAD) with the cysteinehistidine-rich domain 1 (CH1) of p300/TAZ1 (transcriptionadaptor zinc-binding domain) domain of CBP (2) (Fig. 1A).Because hypoxia is a general characteristic of solid tumorsand HIF-regulated genes are linked to cancer progression,inhibition of HIF activity has therapeutic potential (forreview, see Ref. 3).Although the C-TAD-CH1 interaction is tight, the observa-
tion that the addition of a single oxygen atom at Asn803 ofHIF-1� (4) significantly reduces binding suggests that it is ame-nable to smallmolecule intervention. Evidence for this proposalcame from the demonstration that a C-TAD-derived peptidereduces HIF activity in cells and attenuated tumor growth in amouse xenograftmodel (5). Subsequently, Kung et al. (6) devel-oped a C-TAD-CH1 interaction assay and screened �600,000compounds that identified a single inhibitor, chetomin (1) (Fig.1B), with a submicromolar IC50 in cellular assays, that pos-sessed significant antitumor activity in mice. In addition toeffectively reducing HIF-dependent transcription, chetominalso radiosensitizes hypoxicHT1080human fibrosarcoma cells(7).Chetomin is one of the epidithiodiketopiperazine (ETP) fam-
ily of fungal secondary metabolites and was initially identifiedas an antibiotic. ETPs contain a unique diketopiperazine scaf-fold with a bridge formed by two or more sulfur atoms; thedisulfide link exists in an unusual conformation that inducessignificant torsional strain (8) (Fig. 1C). Chetomin is an inter-esting, promising lead structure, not only because of its unusualstructure but because it is a rare example of a small moleculethat disrupts a specific protein-protein interaction within a sig-naling pathway (6); there is also evidence that reduced forms ofETPs are selectively trapped in cells, a property that may beused to target hypoxic tumor cells (9). However, the molecularmechanism(s) of action of chetomin, and other ETPs, areunclear.
* This work was supported, in whole or in part, by the National Institutes ofHealth Grant Z01SC 006538. This work was also supported by grants fromCancer Research UK, The Wellcome Trust, and the Biochemical and Bio-technology Research Council. The structure-activity studies of the ETPsand HIF-1�/p300 inhibition were presented in a proffered abstract and oralpresentation at the 99th AACR Annual Meeting, April 12–16, 2008, SanDiego, CA (Cook, K. M., Hilton, S. T., Schofield, C. J., and Figg, W. F. (2008)Drug Design and Delivery: Oral Presentations: Proffered Abstracts, Abstr.4149), available online at the AACR Meeting Abstractions Online web site.
The nucleotide sequence(s) reported in this paper has been submitted to the Gen-BankTM/EBI Data Bank with accession number(s) GQ340758.
□S The on-line version of this article (available at http://www.jbc.org) containssupplemental Figs. 1– 6 and supplemental Schemes 1–3.
1 To whom correspondence should be addressed: 10 Center Dr., 9000 Rock-ville Pike, Bldg. 10, Rm. 5A01, Bethesda, MD 20892. Fax: 301-402-8606;E-mail: [email protected].
2 The abbreviations used are: HIF, hypoxia-inducible factor; HIF-1�, �-sub-unit of hypoxia-inducible factor-1; CBP, CREB-binding protein; CREB,cAMP-response element-binding protein; ETP, epidithiodiketopiperazine;C-TAD, C-terminal activation domain of HIF-1�; CH1, cysteine/histidine-rich domain 1 of p300; GST, glutathione S-transferase; TBST, Tris-bufferedsaline, with Tween 20; ESI-MS, electrospray ionization mass spectrometry;VEGF, vascular endothelial growth factor; DTT, dithiothreitol; ANOVA, anal-ysis of variance.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 284, NO. 39, pp. 26831–26838, September 25, 2009Printed in the U.S.A.
SEPTEMBER 25, 2009 • VOLUME 284 • NUMBER 39 JOURNAL OF BIOLOGICAL CHEMISTRY 26831
by guest on August 15, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Recent attention has focused on the anticancer properties ofETPs. Many naturally occurring ETPs exert anticancer andother biological activities (for review, see Ref. 10). Gliotoxin (2)is reported to inhibit farnesyltransferase and geranylgeranyl-transferase (11), and chaetocin (3) was reported recently toinhibit a histone methyl transferase (12). Chaetocin has alsobeen shown to possess antimyeloma activity and is selective inkilling myeloma cells over healthy cells (13). Despite the prom-ising anticancer properties found thus far, there has been alongstanding lack of structure-activity relationship or data onthe mechanism of action between ETPs and specific biologicaltargets, in part because ETPs have historically been difficult tosynthesize. Here we describe the synthesis, via a three-compo-nent procedure, of a set of ETPmolecules that we used to carryout structure-activity analyses with the HIF-1�-C-TAD-p300-CH1 interaction.Unexpectedly, the work with isolated proteins and cells
reveals that the ETP disulfide core of the complex chetominstructure is itself sufficient for activity and that the mechanismof action involves disruption of the zinc-binding sites in theCH1domain of p300.Although the zinc sites in p300 are knownto be important for CH1 structure and binding of HIF-1�, theyhad not previously been recognized as a specific target for chet-omin. To our knowledge, no other drug has been known totarget this zinc interaction.
MATERIALS AND METHODS
Synthesis: General Experimental—For full experimentaldetails, see supplemental information. All reactions requir-ing the use of dry conditions were carried out under an atmo-sphere of nitrogen, and all glassware was predried in an oven(110 °C) or flame-dried and cooled under nitrogen prior touse. Stirring was by internal magnetic follower unless other-wise stated. All reactions were followed by TLC, and organicphases extracted were dried with anhydrous magnesium sul-fate. Melting points were determined on a Gallenkampmelt-ing point apparatus. Infrared spectra were recorded on aPerkinElmer Life Sciences 1605 FT-IR spectrophotometerusing NaCl plates. 1H NMR (J values are reported to thenearest 0.5 Hz) and 13C NMR were recorded either on aBruker AMX300 spectrometer or on a Bruker Avance 500spectrometer. High resolution mass spectra were carried outat University College, London University. Mass spectra were
carried out using either a Kratos MS89MS with Kratos DS90software or a Jeol AX505W with a Jeol complement datasystem. Samples were ionized electronically, with an accelerat-ing voltage of �6 kV or by low resolution fast atom bombard-ment in a thioglycerol matrix. High resolution fast atom bom-bardment was carried out at the University of LondonIntercollegate Research Scheme (ULIRS) mass spectrometryfacility at the School of Pharmacy, University of London. Ele-mental analyses of compounds were carried out at the Chem-istry Department, University College, London University.C-TAD-CH1 Fluorescent Binding Assay and Screen—Inhibi-
tion of HIF-1� binding to p300 was measured, essentially asreported (6), by displacement of GST-p300-CH1323–423 from asynthetic biotinylated HIF-1� C-TAD786–826 (Peptide ProteinResearch Ltd., Fareham, UK) immobilized on 96-well strepta-vidin-coated plates. GST-CH1was detected using a Europium-labeled antibody to GST (PerkinElmer Life Sciences). 48.5 nMHIF-1� C-TADwas used to coat plates for 5 h at room temper-ature. Plates were washed four times with TBST (50 mM Tris,150 mM NaCl, 0.05% Tween 20, pH 8.0) buffer. 7.35 nM GST-CH1 was added with compounds or control (1% DMSO) inTBST with 5% BSA, 0.5 mM DTT, and 10 �M ZnCl2 and incu-bated overnight at 4 °C. Plates were washed four times withTBST, and Europium-labeled anti-GST (450 ng/ml) was addedto plates in the buffer used forGST-CH1 addition, and after 2 h,plates were washed six times in TBST. DELFIA enhancementsolution (PerkinElmer Life Sciences) was added before readingwith a Victor3 plate reader (PerkinElmer Life Sciences), usingthe Europium setting under time-resolved fluorescence. Valueswere corrected for background and expressed as a percentageof controls (DMSO) to provide the percentage of CH1 binding.IC50 values were calculated using the Prism version 5.01(GraphPad) graphing program using the non-linear regressionequation log(inhibitor) versus response � variable slope.Purification ofHis6-HIF-1�-C-TAD—Ahexahistidine (His6)-
tagged version of the HIF-1� C-TAD (His6-HIF-1�-C-TADresidues786–826) was prepared as described (14).Purification of GST-CH1323–423—Recombinant N-terminal
GST-tagged p300-CH1 (p300 residues323–423) was overex-pressed and purified as described for GST-tagged p300302–423(6) except for the following modifications; purification used aglutathione-Sepharose 4B (GE Healthcare) column (20 ml)
FIGURE 1. HIF-1�-C-TAD and p300-CH1 interaction and naturally occurring ETPs. A, views from structures of the C-TAD of HIF-1� (amino acids 786 – 826,in blue) interacting with the CH1 of p300 (amino acids 323– 423, in yellow). Protein Data Bank (PDB) ID code 1L3E (2), showing the location of the threezinc-binding sites. A close-up of one of the zinc-coordinating sites of CH1 showing His402, Cys406, Cys411, and Cys414 is shown. B, structure of chetomin.C, structures of the naturally occurring ETPs gliotoxin (2), chaetocin (3), and deacetylsirodesmin PL (4).
ETPs Block HIF-1�-p300 Binding by Zinc Ejection Mechanism
26832 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 39 • SEPTEMBER 25, 2009
by guest on August 15, 2019
http://ww
w.jbc.org/
Dow
nloaded from

with further purification by gel filtration chromatography usinga Superdex 200 column (GE Healthcare). Protein was stored inTBST buffer with 10 �M ZnCl2 and 0.5 mM DTT, and proteinconcentration was determined by Bradford assay.Depsipeptide—For crystal structure, see Ref. 15.In Vitro Transcription/Translation and Co-immunoprecipi-
tation of CH1-HIF-1� (6)—In vitro transcription/translationwas performed using the TNT SP6 high-yield protein expres-sion system (Promega) with pCMV HIF-1�-FLAG tag to pro-duce full-length HIF-1� (supplemental information). GST-CH1 was purified as above. HIF-1�-FLAG and GST-CH1 werecombined in the presence of compound or vehicle control(DMSO) inTBSTwith 10�MZnCl2, 0.5mMDTT, and proteaseinhibitors (Complete protease inhibitor tablet, EDTA-free,Roche Applied Science). Mouse monoclonal anti-FLAG anti-body (Sigma) was added with protein G-Sepharose and FastFlow (Sigma) and then incubated for 1 h at room temperature.After 4� TBST washes, the samples were boiled in SDS-PAGEloading buffer, separated on 4–20% Novex Tris-glycine SDS-PAGE gels (Invitrogen), and transferred to polyvinylidenedifluoride membranes, which were blocked in 3% nonfat milk(w/v) and probed with either rabbit anti-FLAG (1:2,000) andgoat anti-rabbit-horseradish peroxidase (HRP) or anti-GST-HRP (1:1,666). Detection was performed using ECL Pluschemiluminescence (Amersham Biosciences). Films werescanned using a regular desktop scanner on the negative filmsetting.Cell Culture—HCT 116 colorectal carcinoma cells were
grown in McCoy’s 5a medium modified supplemented with10% fetal bovine serum, penicillin (50 IU/ml), streptomycin (50�g/ml), and L-glutamine (2 mM), and Hep G2 hepatocellularcarcinoma cells were grown in minimum Eagle’s medium sup-plemented with 10% fetal bovine serum, penicillin (50 IU/ml),streptomycin (50 �g/ml), and L-glutamine (2 mM), at 37 °C in5%CO2 and 95%air. For hypoxic experiments, cells were placedin a modular incubator chamber flushed with 1% O2, 5% CO2,94% N2.Cell Viability Assays—HCT 116 cells were seeded overnight
intowhite 96-well plates in 100�l ofmediumat a concentrationof 5� 104 cells well�1, andHepG2 cells were plated in the samemanner at a concentration of 8 � 104 cells well�1. After over-night incubation at 37 °C, medium was removed and replacedwith 200 �l of medium containing either drug or vehicle con-trol. Plates were placed in either a normoxic incubator or ahypoxic chamber (Billups-Rothenberg) for 18 h. 100 �l ofmediumwere removed for VEGFmeasurement, and cell viabil-ity was determined using the CellTiter-Blue cell viability rea-gent (Promega) according to the manufacturer’s protocol. Forthe viability assessment in the presence of zinc, after the initialovernight incubation of cells at 37 °C, medium was removedand replaced with 100 �l of medium containing 150 �M ZnSO4or ZnCl2. Cells were returned to 37 °C incubator for �90 minbefore adding 100 �l of medium containing 2� final concen-tration of drug or vehicle control. The final concentration ofZn2� for incubation with ETPs was 75 �M. The viability assess-ment was carried out the same as for non-zinc-treated cells.Cell Proliferation Assays—After viability readings were
recorded, 20 �l of culture media containing 10 �M bromode-
oxyuridinewere added to the cells, and theywere placed back inthe 37 °C incubator for 18–24 h. Proliferation was measuredusing the cell proliferation enzyme-linked immunosorbentassay, bromodeoxyuridine (colorimetric) (Roche Applied Sci-ence), according to the manufacturer’s instructions.VEGF Quantification—Tissue culture supernatant levels of
VEGF were determined using a VEGF immunoassay (Calbio-chem) according to the manufacturer’s instructions.Removal of GST Tag from GST-p300-CH1 and Purification
for ESI-MS Studies—After purification of GST-CH1323–423, theGST tag was removed by treatment with PreScission protease(Amersham Biosciences) treatment as specified by the manu-facturer. CH1 was purified by size exclusion chromatographyusing a 300-ml Superdex 75 column (GE Healthcare).Non-denaturing Electrospray Ionization Mass Spectrometric
Assay (16–19)—Aspurified, CH1was amixture of apo and zinccomplexes. To produce the fully tri-metallated form, CH1 wastreated with 3 eq of Zn(II) and an excess of DTT. p300-CH1323–423protein (GST tag-removed CH1, as described above) wasdesalted using a Bio-Spin 6 column (Bio-Rad) into 15 mM
ammonium acetate (pH 7.5) to a final concentration of 20 �M.An aqueous solution of ZnSO4 � 7H2O (100 mM) was thendiluted with MilliQ water to a final working concentrations of100 �M. DTT was dissolved in MilliQ water at a concentrationof 1 mM. ETPs were dissolved in DMSO (10 mM) and thendiluted with ammonium acetate (15 mM, pH 7.5) to a concen-tration of 100 �M. The p300 protein (1.25 �l) was mixed with 3eq of Zn(II) (0.75 �l), 20 eq of DTT (0.5 �l), and 20 eq of ETP (5�l) and incubated for 30 min at room temperature prior toelectrospray ionization mass spectrometry (ESI-MS) analysis.Data were acquired using a quadrupole time-of-flight massspectrometer (Q-TOF micro, Micromass, Altrincham) inter-faced with a Nanomate (Advion Biosciences, Ithaca, NY) with achip voltage of 1.70 kV and a delivery pressure 0.25 p.s.i.(1 p.s.i.� 6.81 kilopascals). The sample cone voltagewas typically200Vwith a source temperature of 40 °C andwith an acquisition/scan time of 10 s/1 s. Calibration and sample acquisition wereperformed in the positive ion mode in the range of 200–5,000m/z. The pressure at the interface between the atmosphericsource and the high vacuum region was fixed at 6.60 mbar.External instrument calibration was achieved by using sodiumiodide. Data were processed with MASSLYNX 4.0 (Waters).Epidithiodiketopiperazines—ETPs were stored frozen in
DMSO. Chetomin and gliotoxin were purchased from Calbio-chem; chaetocin was purchased from Sigma. Deacetyl-sirodesmin PL was a kind gift from Soledade Petras (Universityof Saskatchewan).Software—Graphing was done using Prism version 5.01
(GraphPad) for curves, and IC50 determinations were gener-ated from built-in functions. Molecular modeling was per-formed in PyMOL version 99 (20).Statistical Analysis—Data were analyzed using built-in
GraphPad Prism version 5.01 functions, as specified. All statis-tical tests were two-sided, and the level of significancewas set atp� 0.05. *, p� 0.05, **, p� 0.01, ***, p� 0.001 in the legends forFigs. 2–4, 8, and 9.
ETPs Block HIF-1�-p300 Binding by Zinc Ejection Mechanism
SEPTEMBER 25, 2009 • VOLUME 284 • NUMBER 39 JOURNAL OF BIOLOGICAL CHEMISTRY 26833
by guest on August 15, 2019
http://ww
w.jbc.org/
Dow
nloaded from
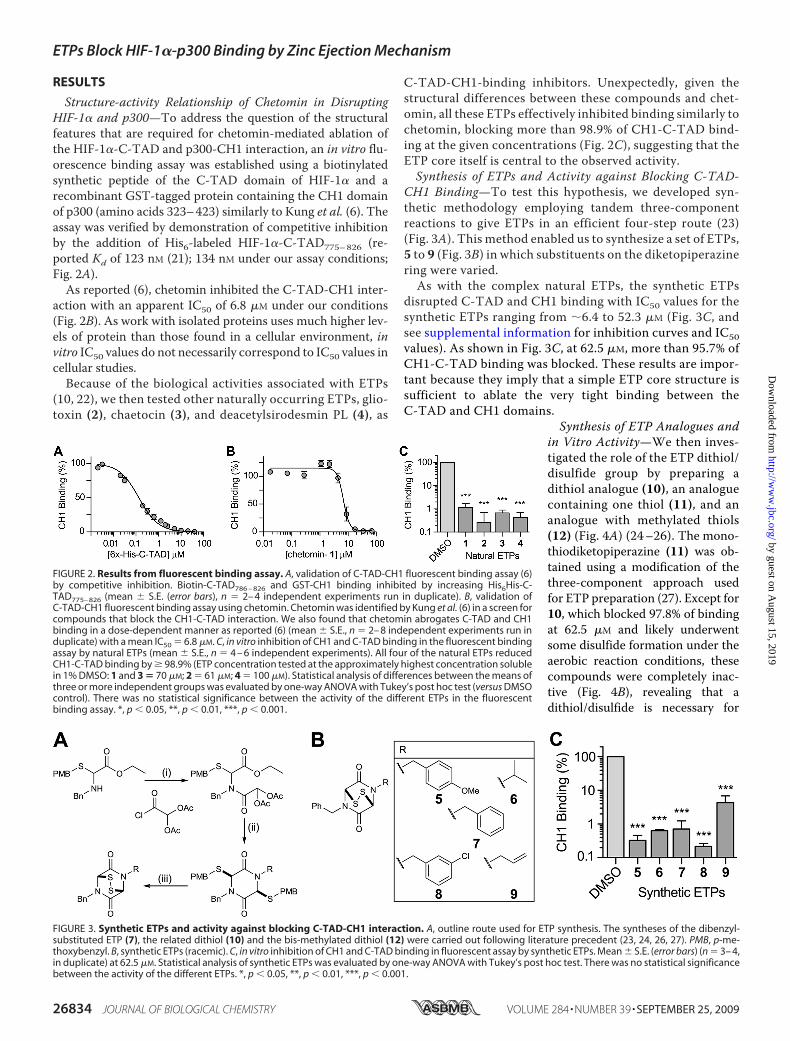
RESULTS
Structure-activity Relationship of Chetomin in DisruptingHIF-1� and p300—To address the question of the structuralfeatures that are required for chetomin-mediated ablation ofthe HIF-1�-C-TAD and p300-CH1 interaction, an in vitro flu-orescence binding assay was established using a biotinylatedsynthetic peptide of the C-TAD domain of HIF-1� and arecombinant GST-tagged protein containing the CH1 domainof p300 (amino acids 323–423) similarly to Kung et al. (6). Theassay was verified by demonstration of competitive inhibitionby the addition of His6-labeled HIF-1�-C-TAD775–826 (re-ported Kd of 123 nM (21); 134 nM under our assay conditions;Fig. 2A).As reported (6), chetomin inhibited the C-TAD-CH1 inter-
action with an apparent IC50 of 6.8 �M under our conditions(Fig. 2B). As work with isolated proteins uses much higher lev-els of protein than those found in a cellular environment, invitro IC50 values do not necessarily correspond to IC50 values incellular studies.Because of the biological activities associated with ETPs
(10, 22), we then tested other naturally occurring ETPs, glio-toxin (2), chaetocin (3), and deacetylsirodesmin PL (4), as
C-TAD-CH1-binding inhibitors. Unexpectedly, given thestructural differences between these compounds and chet-omin, all these ETPs effectively inhibited binding similarly tochetomin, blocking more than 98.9% of CH1-C-TAD bind-ing at the given concentrations (Fig. 2C), suggesting that theETP core itself is central to the observed activity.Synthesis of ETPs and Activity against Blocking C-TAD-
CH1 Binding—To test this hypothesis, we developed syn-thetic methodology employing tandem three-componentreactions to give ETPs in an efficient four-step route (23)(Fig. 3A). This method enabled us to synthesize a set of ETPs,5 to 9 (Fig. 3B) in which substituents on the diketopiperazinering were varied.As with the complex natural ETPs, the synthetic ETPs
disrupted C-TAD and CH1 binding with IC50 values for thesynthetic ETPs ranging from �6.4 to 52.3 �M (Fig. 3C, andsee supplemental information for inhibition curves and IC50values). As shown in Fig. 3C, at 62.5 �M, more than 95.7% ofCH1-C-TAD binding was blocked. These results are impor-tant because they imply that a simple ETP core structure issufficient to ablate the very tight binding between theC-TAD and CH1 domains.
Synthesis of ETP Analogues andin Vitro Activity—We then inves-tigated the role of the ETP dithiol/disulfide group by preparing adithiol analogue (10), an analoguecontaining one thiol (11), and ananalogue with methylated thiols(12) (Fig. 4A) (24–26). The mono-thiodiketopiperazine (11) was ob-tained using a modification of thethree-component approach usedfor ETP preparation (27). Except for10, which blocked 97.8% of bindingat 62.5 �M and likely underwentsome disulfide formation under theaerobic reaction conditions, thesecompounds were completely inac-tive (Fig. 4B), revealing that adithiol/disulfide is necessary for
FIGURE 2. Results from fluorescent binding assay. A, validation of C-TAD-CH1 fluorescent binding assay (6)by competitive inhibition. Biotin-C-TAD786 – 826 and GST-CH1 binding inhibited by increasing His6His-C-TAD775– 826 (mean � S.E. (error bars), n � 2– 4 independent experiments run in duplicate). B, validation ofC-TAD-CH1 fluorescent binding assay using chetomin. Chetomin was identified by Kung et al. (6) in a screen forcompounds that block the CH1-C-TAD interaction. We also found that chetomin abrogates C-TAD and CH1binding in a dose-dependent manner as reported (6) (mean � S.E., n � 2– 8 independent experiments run induplicate) with a mean IC50 � 6.8 �M. C, in vitro inhibition of CH1 and C-TAD binding in the fluorescent bindingassay by natural ETPs (mean � S.E., n � 4 – 6 independent experiments). All four of the natural ETPs reducedCH1-C-TAD binding by � 98.9% (ETP concentration tested at the approximately highest concentration solublein 1% DMSO: 1 and 3 � 70 �M; 2 � 61 �M; 4 � 100 �M). Statistical analysis of differences between the means ofthree or more independent groups was evaluated by one-way ANOVA with Tukey’s post hoc test (versus DMSOcontrol). There was no statistical significance between the activity of the different ETPs in the fluorescentbinding assay. *, p � 0.05, **, p � 0.01, ***, p � 0.001.
FIGURE 3. Synthetic ETPs and activity against blocking C-TAD-CH1 interaction. A, outline route used for ETP synthesis. The syntheses of the dibenzyl-substituted ETP (7), the related dithiol (10) and the bis-methylated dithiol (12) were carried out following literature precedent (23, 24, 26, 27). PMB, p-me-thoxybenzyl. B, synthetic ETPs (racemic). C, in vitro inhibition of CH1 and C-TAD binding in fluorescent assay by synthetic ETPs. Mean � S.E. (error bars) (n � 3– 4,in duplicate) at 62.5 �M. Statistical analysis of synthetic ETPs was evaluated by one-way ANOVA with Tukey’s post hoc test. There was no statistical significancebetween the activity of the different ETPs. *, p � 0.05, **, p � 0.01, ***, p � 0.001.
ETPs Block HIF-1�-p300 Binding by Zinc Ejection Mechanism
26834 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 39 • SEPTEMBER 25, 2009
by guest on August 15, 2019
http://ww
w.jbc.org/
Dow
nloaded from

activity. The lack of activity of a disulfide-linked depsipeptideprovides evidence for the requirement of a disulfide bridged/dithiol diketopiperazine for activity.ETPs Block CH1 Binding to Full-length HIF-1� in Vitro—Be-
cause assays with HIF-1� and HIF-binding proteins (the HIFhydroxylases) have shown that binding affinities can vary sig-nificantly with the length of the HIF fragment (28–30), wetested whether the ETP compounds could prevent CH1 frombinding to full-lengthHIF-1�. A co-immunoprecipitation assayusing full-length HIF-1� and p300-CH1 demonstrated dose-dependent inhibition with both the natural and the syntheticETPs (Fig. 4C, and see supplemental information for the entirerange of compounds) as seen in the in vitroC-TAD-CH1 assay.However, compound 9, which was slightly less active than theother ETPs in the in vitro fluorescence assay (although not to
statistical significance), failed toblock CH1 binding to full-lengthHIF-1� at the same concentrationas the other ETPs. This indicatesthat appropriate modifications canlead to a decrease in activity and thatselectivity of ETPs might be ac-hieved (Fig. 4C).ETPs Cause Zinc Ejection from
CH1 as Mechanism of Action—Themolecular mechanism of actionof the ETPs was then investigatedby using non-denaturing ESI-MSto analyze ETP-induced modifica-tions. We did not observe ETP-mediated modification of theC-TAD under our conditions(although it contains a cysteine(Cys800)). In contrast, we observedstriking effects on p300-CH1 uponETP treatment.Upon treatment of CH1 with an
excess of the natural ETPs (chet-omin, chaetocin, gliotoxin, and de-acetylsirodesmin PL), complete zinc
ion release was observed from CH1. The results with chetominand chaetocin were complex, probably because they containtwo ETP cores, so we carried out further investigations withgliotoxin, which contains a single ETP core. When CH1 wastreated with gliotoxin (5 eq), partial zinc ejection was observed(CH1�Zn-CH1�Zn2:CH1�Zn3 in an �1:3:4 ratio, respectively).In the presence of 10 eq of gliotoxin, the two main speciesobserved were apo CH1 and CH1�Zn, in an �1:1 ratio.With 20eq of gliotoxin, complete release of zinc gave apo CH1 as theonly observed species (Fig. 5A).Next we screened the synthetic ETPs for zinc ejection activ-
ity. Interestingly, and consistent with the structure-activityrelationship studies, the results showed that 6, 7, and 10 causedpartial zinc release (yielding apo CH1 and all three metallated
FIGURE 4. Synthetic ETP analogues and activity against blocking C-TAD-CH1 interaction. A, synthetic ETP analogues (racemic). B, in vitro inhibition of CH1and C-TAD binding in fluorescence assay by synthetic ETP analogues. Compounds possessing the ETP core in both oxidized (1–9) and reduced (10) form ablateCH1-C-TAD binding. Analogues possessing a single thiol (11) or methylated thiols (12) are inactive. (Mean � S.E. (error bars), n � 2–3, at 62.5 �M.) Statisticalanalysis of synthetic ETPs was evaluated by one-way ANOVA with Tukey’s post hoc test. There was no statistical significance between the activity of thedifferent ETPs. *, p � 0.05, **, p � 0.01, ***, p � 0.001. C, co-immunoprecipitation (IP) of FLAG-HIF-1� and bound GST-CH1 in the presence of ETPs. Shown is theamount of CH1 pulled down. Compounds with reduced/oxidized ETP core block the CH1-HIF-1� interaction, except for 9, indicating that selectivity of ETPs maybe achievable. The single thiol analogue (11) and methylated thiols (12) analogue did not block the interaction. For the entire range of compounds andcontrols, see supplemental information.
FIGURE 5. ETPs block the C-TAD-CH1 interaction by zinc ejection from CH1, demonstrated by ESI-MS.A, non-denaturing ESI-MS analyses on the effect of increasing amounts of gliotoxin (2) on zinc ion binding byCH1. B, non-denaturing electrospray ionization mass spectrometric analyses on the effect of various syntheticETPs and ETP analogues on zinc ion binding by CH1. Note the variations in efficiency and note that theanalogues 11 and 12, lacking the ETP core, do not cause zinc ion ejection.
ETPs Block HIF-1�-p300 Binding by Zinc Ejection Mechanism
SEPTEMBER 25, 2009 • VOLUME 284 • NUMBER 39 JOURNAL OF BIOLOGICAL CHEMISTRY 26835
by guest on August 15, 2019
http://ww
w.jbc.org/
Dow
nloaded from
species). In contrast, the monothiol (11) and thioether (12)analogues, all of which were inactive in the binding assays, didnot cause zinc release, demonstrating that a dithiol/disulfideETP core is essential for the activity (Fig. 5B).These results reveal that the ETP disulfide core structure is
both sufficient and, within the set of tested compounds, neces-sary for inhibition of the HIF-1�-CH1 interaction. The mech-anism of action involvingmodification of the zinc-binding siteson CH1 domain of p300 is supported by observations, employ-ingNMR studies, that CH1 becomes less structured in the pres-ence of chetomin (6). Consistent with the previous study ofchetomin (6), we did not acquire evidence for irreversible cova-lentmodification of CH1.We did, however, observe complexesbetween metallated, but not apo, forms of CH1, on treatment
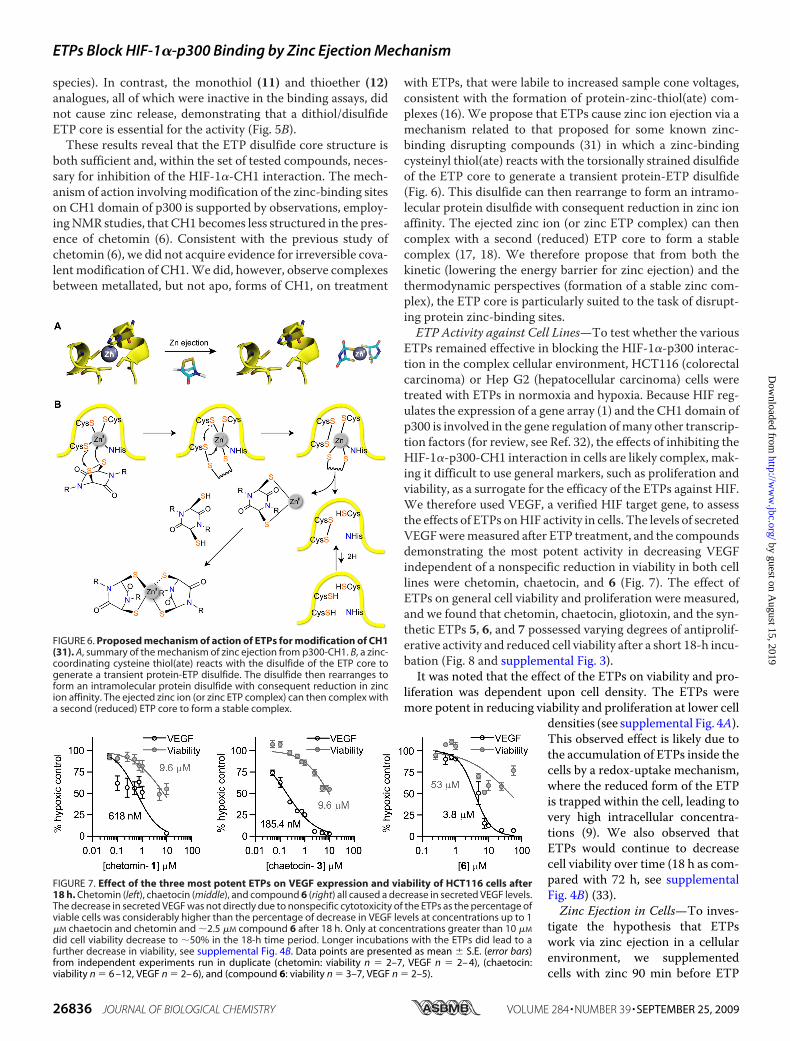
with ETPs, that were labile to increased sample cone voltages,consistent with the formation of protein-zinc-thiol(ate) com-plexes (16). We propose that ETPs cause zinc ion ejection via amechanism related to that proposed for some known zinc-binding disrupting compounds (31) in which a zinc-bindingcysteinyl thiol(ate) reacts with the torsionally strained disulfideof the ETP core to generate a transient protein-ETP disulfide(Fig. 6). This disulfide can then rearrange to form an intramo-lecular protein disulfide with consequent reduction in zinc ionaffinity. The ejected zinc ion (or zinc ETP complex) can thencomplex with a second (reduced) ETP core to form a stablecomplex (17, 18). We therefore propose that from both thekinetic (lowering the energy barrier for zinc ejection) and thethermodynamic perspectives (formation of a stable zinc com-plex), the ETP core is particularly suited to the task of disrupt-ing protein zinc-binding sites.ETP Activity against Cell Lines—To test whether the various
ETPs remained effective in blocking the HIF-1�-p300 interac-tion in the complex cellular environment, HCT116 (colorectalcarcinoma) or Hep G2 (hepatocellular carcinoma) cells weretreated with ETPs in normoxia and hypoxia. Because HIF reg-ulates the expression of a gene array (1) and the CH1 domain ofp300 is involved in the gene regulation ofmany other transcrip-tion factors (for review, see Ref. 32), the effects of inhibiting theHIF-1�-p300-CH1 interaction in cells are likely complex, mak-ing it difficult to use general markers, such as proliferation andviability, as a surrogate for the efficacy of the ETPs against HIF.We therefore used VEGF, a verified HIF target gene, to assessthe effects of ETPs onHIF activity in cells. The levels of secretedVEGFweremeasured after ETP treatment, and the compoundsdemonstrating the most potent activity in decreasing VEGFindependent of a nonspecific reduction in viability in both celllines were chetomin, chaetocin, and 6 (Fig. 7). The effect ofETPs on general cell viability and proliferation were measured,and we found that chetomin, chaetocin, gliotoxin, and the syn-thetic ETPs 5, 6, and 7 possessed varying degrees of antiprolif-erative activity and reduced cell viability after a short 18-h incu-bation (Fig. 8 and supplemental Fig. 3).It was noted that the effect of the ETPs on viability and pro-
liferation was dependent upon cell density. The ETPs weremore potent in reducing viability and proliferation at lower cell
densities (see supplemental Fig. 4A).This observed effect is likely due tothe accumulation of ETPs inside thecells by a redox-uptake mechanism,where the reduced form of the ETPis trapped within the cell, leading tovery high intracellular concentra-tions (9). We also observed thatETPs would continue to decreasecell viability over time (18 h as com-pared with 72 h, see supplementalFig. 4B) (33).Zinc Ejection in Cells—To inves-
tigate the hypothesis that ETPswork via zinc ejection in a cellularenvironment, we supplementedcells with zinc 90 min before ETP
FIGURE 6. Proposed mechanism of action of ETPs for modification of CH1(31). A, summary of the mechanism of zinc ejection from p300-CH1. B, a zinc-coordinating cysteine thiol(ate) reacts with the disulfide of the ETP core togenerate a transient protein-ETP disulfide. The disulfide then rearranges toform an intramolecular protein disulfide with consequent reduction in zincion affinity. The ejected zinc ion (or zinc ETP complex) can then complex witha second (reduced) ETP core to form a stable complex.
FIGURE 7. Effect of the three most potent ETPs on VEGF expression and viability of HCT116 cells after18 h. Chetomin (left), chaetocin (middle), and compound 6 (right) all caused a decrease in secreted VEGF levels.The decrease in secreted VEGF was not directly due to nonspecific cytotoxicity of the ETPs as the percentage ofviable cells was considerably higher than the percentage of decrease in VEGF levels at concentrations up to 1�M chaetocin and chetomin and �2.5 �M compound 6 after 18 h. Only at concentrations greater than 10 �M
did cell viability decrease to �50% in the 18-h time period. Longer incubations with the ETPs did lead to afurther decrease in viability, see supplemental Fig. 4B. Data points are presented as mean � S.E. (error bars)from independent experiments run in duplicate (chetomin: viability n � 2–7, VEGF n � 2– 4), (chaetocin:viability n � 6 –12, VEGF n � 2– 6), and (compound 6: viability n � 3–7, VEGF n � 2–5).
ETPs Block HIF-1�-p300 Binding by Zinc Ejection Mechanism
26836 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 39 • SEPTEMBER 25, 2009
by guest on August 15, 2019
http://ww
w.jbc.org/
Dow
nloaded from

treatment. We found that as compared with growth mediumwithout zinc, therewas substantially increased cell viability (34)and proliferation, as well as restored production of VEGF, inboth Hep G2 cells and HCT116 cells (Fig. 9 and supplementalFigs. 5 and 6).
DISCUSSION
Our results have isolated the active pharmacophore in chet-omin by revealing that the ETP core in itself was both necessaryand sufficient for ablation of the HIF-p300 interaction. Using
non-denaturing mass spectrometry, we investigated the mech-anism of disruption and found that both the natural and thesynthetic ETPs cause zinc ion ejection from the CH1 domain ofp300. This is important from a medicinal perspective becausemost of the identified targets for ETPs are actually zinc-bindingproteins (or have zinc-binding partners).In a cancer cell-based model, the addition of zinc eliminated
the antiproliferative effects of both natural and synthetic ETPsand drastically increased the percentage of viable cells. Mostimportantly, the addition of zinc restored the cell’s productionof VEGF in hypoxia, a protein that is under the direct transcrip-tional control of HIF.The potentially lethal poisoning of grazing animals by the
fungus Pithomyces chartarum and its treatment provide evi-dence that ETPs directly target zinc binding in animals (10).This fungus poses a serious agricultural problem in parts of theworld that have ryegrass pastures and hot and/or humidweather (10). As a source of the sporidesmin family of ETPs, thefungus can cause liver toxicity and facial/udder eczema, a phe-nomenon that can be reversed by adding zinc ions to the diet,which is consistent with our proposed mechanism.It is very likely that ETPs operate by more than one mecha-
nism in cells (including redox cycling/producing reactive oxi-dizing species (10). However, it is striking that some, but not all(e.g. thioredoxin (35)), proposed ETP targets either are knownzinc-binding proteins (e.g. farnesyltransferase/geranylgeranyl-transferase-gliotoxin (11)) or are associated with zinc-bindingproteins (histone methyl transferase-chaetocin (12)), suggest-ing that the disruption of zinc bindingmay be one of the poten-tial generic mechanisms of ETP action and account, at least inpart, for their toxicity (10).It is notable that ETP biosynthesis itself inmicroorganisms is
positively regulated by a zinc-containing transcription factor(36); it is possible that inhibition of this transcription factor byETPs could provide a feedback loop. Further, nuclear factor-�B(NF-�B), which is known to bind the p300-CH1 domain fortranscription (37), is a proposed target of gliotoxin (38).Because the p65 subunit of NF-�B subunit binds the CH1domain of p300, disruption of the CH1 domain could accountfor all or part of the gliotoxin-mediated loss in activity ofNF-�B.
Further work is needed to determine whether ETP-mediateddisruption of the CH1 domain affects all CH1 interactions andwhether ETPs disrupt interactions with other domains of p300,such as thatwith PGC-1� (39), andwhether the stability of p300is affected. Because of the multiple transcription factors thatbind the CH1 domain, loss of CH1 structure and functionalitycould account for the extensive antitumor effects seen withchetomin in vivo (6), as seen previously with the overexpressionof a CH1 polypeptide that decreased HIF transcription and ledto a significant decrease in tumor size (5).In addition to the potential consequences of zinc binding in
vivo, other proposed targets of ETPs are likely to contribute tothe anticancer effects: for example, the effects of chaetocin onthe thioredoxin system (40), which itself is a proposed target forcancer drug development. Finally, the identification of themechanismbywhich ETPs inhibit theHIF-1�-p300 interactionvia zinc ejection now opens the way to the generation of ETP
FIGURE 8. Effect of both natural and synthetic ETPs on HCT116 cell viabil-ity (A) and proliferation (B) in normoxia (mean � S.E. (error bars); viabil-ity: n > 4; proliferation: n > 3). Statistical analysis of differences betweenthe means of three or more independent groups was evaluated by one-wayANOVA with Tukey’s post hoc test (versus DMSO control). See supplementalinformation for more supporting data. *, p � 0.05, **, p � 0.01, ***, p � 0.001.
FIGURE 9. Effect of representative synthetic ETP, compound 6, on HCT116cell viability, proliferation, and VEGF secretion in the presence orabsence of zinc (mean � S.E. (error bars); viability: n > 4; proliferation: n> 3; VEGF n > 3). HCT116 and Hep G2 cells were treated with 25 �M com-pound 6, with or without zinc supplementation for 18 h. A, viability of HCT116and Hep G2 cells under normoxic and hypoxic (1% O2) conditions. The per-centage of viable cells was considerably higher in zinc-treated cells versusnon-zinc-treated. B, zinc supplementation increases proliferation to levelssimilar to the control, in both cell lines. C, enzyme-linked immunosorbentassay quantification of secreted VEGF normalized to DMSO control in hypoxicHCT116 and Hep G2 cells. Zinc supplementation restores VEGF production inhypoxic HCT116 and Hep G2 cells. For data on other ETPs, see supplementalinformation. Statistical significance of differences between the means of ETP-treated samples and ETP-treated samples with zinc supplementation wasevaluated by unpaired Student’s t test. *, p � 0.05, **, p � 0.01, ***, p � 0.001.
ETPs Block HIF-1�-p300 Binding by Zinc Ejection Mechanism
SEPTEMBER 25, 2009 • VOLUME 284 • NUMBER 39 JOURNAL OF BIOLOGICAL CHEMISTRY 26837
by guest on August 15, 2019
http://ww
w.jbc.org/
Dow
nloaded from

derivatives that are rationally targeted to specific proteintargets.
Acknowledgments—We thank Dr. Soledade Petras (University of Sas-katchewan) for generously providing deacetylsirodesmin PL. Wethank the National Cancer Institute, National Institutes of Health,Cancer ResearchUK, TheWellcome Trust, and Biochemical and Bio-technology Research Council, for funding, Shandiz Shahbazi forassistance with cell culture and Dr. Douglas Price for advice andencouragement.
REFERENCES1. Hirota, K., and Semenza, G. L. (2006)Crit. Rev. Oncol. Hematol. 59, 15–262. Freedman, S. J., Sun, Z. Y., Poy, F., Kung, A. L., Livingston, D.M.,Wagner,
G., and Eck, M. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 5367–53723. Kaelin, W. G., Jr., and Ratcliffe, P. J. (2008)Mol. Cell 30, 393–4024. Lando, D., Peet, D. J., Whelan, D. A., Gorman, J. J., and Whitelaw, M. L.
(2002) Science 295, 858–8615. Kung, A. L., Wang, S., Klco, J. M., Kaelin, W. G., and Livingston, D. M.
(2000) Nat. Med. 6, 1335–13406. Kung, A. L., Zabludoff, S. D., France, D. S., Freedman, S. J., Tanner, E. A.,
Vieira, A., Cornell-Kennon, S., Lee, J., Wang, B., Wang, J., Memmert, K.,Naegeli, H. U., Petersen, F., Eck, M. J., Bair, K. W., Wood, A. W., andLivingston, D. M. (2004) Cancer Cell 6, 33–43
7. Staab, A., Loeffler, J., Said, H. M., Diehlmann, D., Katzer, A., Beyer, M.,Fleischer, M., Schwab, F., Baier, K., Einsele, H., Flentje, M., and Vorder-mark, D. (2007) BMC Cancer 7, 213
8. Hilton, S. T., Motherwell, W. B., Potier, P., Pradet, C., and Selwood, D. L.(2005) Bioorg. Med. Chem. Lett. 15, 2239–2242
9. Bernardo, P.H., Brasch,N., Chai, C. L., andWaring, P. (2003) J. Biol. Chem.278, 46549–46555
10. Chai, C. L., and Waring, P. (2000) Redox. Rep. 5, 257–26411. Vigushin, D. M., Mirsaidi, N., Brooke, G., Sun, C., Pace, P., Inman, L.,
Moody, C. J., and Coombes, R. C. (2004)Med. Oncol. 21, 21–3012. Greiner, D., Bonaldi, T., Eskeland, R., Roemer, E., and Imhof, A. (2005)
Nat. Chem. Biol. 1, 143–14513. Isham, C. R., Tibodeau, J. D., Jin,W., Xu, R., Timm,M.M., and Bible, K. C.
(2007) Blood 109, 2579–258814. Coleman, M. L., McDonough, M. A., Hewitson, K. S., Coles, C., Meci-
novic, J., Edelmann, M., Cook, K. M., Cockman, M. E., Lancaster, D. E.,Kessler, B. M., Oldham, N. J., Ratcliffe, P. J., and Schofield, C. J. (2007)J. Biol. Chem. 282, 24027–24038
15. Shigematsu,N., Ueda,H., Takase, S., Tanaka,H., Yamamoto, K., andTada,T. (1994) J. Antibiot. 47, 311–314
16. Lienard, B. M., Selevsek, N., Oldham, N. J., and Schofield, C. J. (2007)Chem. Med. Chem. 2, 175–179
17. Woodcock, J. C., Henderson,W., andMiles, C.O. (2001) J. Inorg. Biochem.85, 187–199
18. Woodcock, J. C., Henderson,W.,Miles, C. O., andNicholson, B. K. (2001)J. Inorg. Biochem. 84, 225–232
19. Selevsek, N., Tholey, A., Heinzle, E., Lienard, B. M., Oldham, N. J.,Schofield, C. J., Heinz, U., Adolph,H.W., and Frere, J.M. (2006) J. Am. Soc.Mass Spectrom. 17, 1000–1004
20. DeLano,W. L. (2002) PyMOL version 99, the PyMOLMolecular GraphicsSystem, DeLano Scientific LLC, San Carlos, CA
21. Freedman, S. J., Sun, Z. Y., Kung, A. L., France, D. S., Wagner, G., and Eck,M. J. (2003) Nat. Struct. Biol. 10, 504–512
22. Gardiner, D. M., Waring, P., and Howlett, B. J. (2005) Microbiology 151,1021–1032
23. Aliev, A. E., Hilton, S. T., Motherwell, W. B., and Selwood, D. L. (2006)Tetrahedron Lett. 47, 2387–2390
24. Jiang, H., Newcombe, N., Sutton, P., Lin, Q. H., Mullbacher, A., andWar-ing, P. (1993) Aust. J. Chem. 46, 1743–1754
25. Fukuyama, T., Nakatsuka, S. I., and Kishi, Y. (1981) Tetrahedron 37,2045–2078
26. Seya, H., Nozawa, K., Nakajima, S., Kawai, K., and Udagawa, S. (1986)J. Chem. Soc. Perkin Trans. 1, 109–116
27. Hilton, S., Motherwell, W., and Selwood, D. (2004) Synlett. 2609–261128. Dames, S. A., Martinez-Yamout, M., De Guzman, R. N., Dyson, H. J., and
Wright, P. E. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 5271–527629. Koivunen, P., Hirsila, M., Gunzler, V., Kivirikko, K. I., and Myllyharju, J.
(2004) J. Biol. Chem. 279, 9899–990430. Ehrismann, D., Flashman, E., Genn, D. N., Mathioudakis, N., Hewitson,
K. S., Ratcliffe, P. J., and Schofield, C. J. (2007) Biochem. J. 401, 227–23431. Loo, J. A., Holler, T. P., Sanchez, J., Gogliotti, R., Maloney, L., and Reily,
M. D. (1996) J. Med. Chem. 39, 4313–432032. Goodman, R. H., and Smolik, S. (2000) Genes Dev. 14, 1553–157733. Jordan, T. W., and Pedersen, J. S. (1986) J. Cell Sci. 85, 33–4634. Duncan, E. J., Thompson, M. P., and Phua, S. H. (2005) Toxicol. Lett. 159,
164–17235. Choi, H. S., Shim, J. S., Kim, J. A., Kang, S. W., and Kwon, H. J. (2007)
Biochem. Biophys. Res. Commun. 359, 523–52836. Fox, E. M., Gardiner, D. M., Keller, N. P., and Howlett, B. J. (2008) Fungal.
Genet. Biol. 45, 671–68237. Gerritsen,M. E.,Williams,A. J., Neish,A. S.,Moore, S., Shi, Y., andCollins,
T. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 2927–293238. Pahl, H. L., Krauss, B., Schulze-Osthoff, K., Decker, T., Traenckner, E. B.,
Vogt, M., Myers, C., Parks, T., Waring, P., Muhlbacher, A., Czernilofsky,A. P., and Baeuerle, P. A. (1996) J. Exp. Med. 183, 1829–1840
39. Puigserver, P., Adelmant, G., Wu, Z., Fan, M., Xu, J., O’Malley, B., andSpiegelman, B. M. (1999) Science 286, 1368–1371
40. Tibodeau, J., Benson, L., Isham, C., Owen, W., and Bible, K. (2009) Anti-oxid. Redox. Signal. 11, 1097–1106
ETPs Block HIF-1�-p300 Binding by Zinc Ejection Mechanism
26838 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 39 • SEPTEMBER 25, 2009
by guest on August 15, 2019
http://ww
w.jbc.org/
Dow
nloaded from

William D. Figg and Christopher J. SchofieldKristina M. Cook, Stephen T. Hilton, Jasmin Mecinovic, William B. Motherwell,
) and p300 by a Zinc Ejection Mechanismα (HIF-1αFactor-1Epidithiodiketopiperazines Block the Interaction between Hypoxia-inducible
doi: 10.1074/jbc.M109.009498 originally published online July 9, 20092009, 284:26831-26838.J. Biol. Chem.
10.1074/jbc.M109.009498Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2009/07/09/M109.009498.DC1
http://www.jbc.org/content/284/39/26831.full.html#ref-list-1
This article cites 38 references, 13 of which can be accessed free at
by guest on August 15, 2019
http://ww
w.jbc.org/
Dow
nloaded from