environmental exposures and potential health effects of ...€¦ · (who) meeting on pesticide...
TRANSCRIPT

E Risk Sciences, LLP
March 2012
Environmental Exposures and Potential Health Effects of Bifenthrin A Systematic Review Katherine von Stackelberg, ScD

2 | P a g e
Systematic Review of Environmental Exposures and Potential Health Effects of Bifenthrin
Katherine von Stackelberg, ScD E Risk Sciences, LLP
Keywords: Bifenthrin, weight-of-evidence, review, human health, synthetic pyrethroid, neurotoxicity, carcinogenicity

3 | P a g e
Abstract: This paper explores the association between exposure to bifenthrin, a synthetic pyrethroid, and adverse human health outcomes through a systematic review of the literature. Bifenthrin is a known neurotoxicant to target organisms (e.g., insects) and the toxicological data show the potential for acute effects in mammalian cell cultures, but only at the highest doses tested with no observable dose-response relationships and without cell death. There is no evidence for carcinogenicity, either DNA-reactive or by some other mechanism. There are no documented reproductive or developmental effects in humans. Estimated equivalent in vivo exposures calculated from assay results in human cell cultures are significantly higher than predicted exposure estimates from regulatory agencies or observations. The available biomonitoring data are summarized, and a comparison of predicted exposure estimates from regulatory risk assessments and potential effect levels from the literature are compared to show that expected exposures are less than no adverse observed effect levels from toxicological studies. Given the utility and efficacy of bifenthrin with respect to its termiticidal properties, the analysis qualitatively suggests a net positive risk-benefit tradeoff.

4 | P a g e
Table of Contents Abstract ........................................................................................................................................... 3 Introduction ..................................................................................................................................... 5 Methods........................................................................................................................................... 6
Literature Search and Review ..................................................................................................... 6 Results ............................................................................................................................................. 7
Pharmacokinetics ........................................................................................................................ 7 Oral Exposures ........................................................................................................................ 7 Inhalation ................................................................................................................................ 9 Dermal Absorption.................................................................................................................. 9 Metabolism ............................................................................................................................. 9
Toxicity Studies .......................................................................................................................... 9 Carcinogenicity ..................................................................................................................... 16 Chronic Toxicity ................................................................................................................... 16 Subclinical Effects ................................................................................................................ 18 Regulatory Toxicity Values and Equivalent in vivo Doses Associated with in vitro Effects 24
Comparison of Toxicity Values to Predicted Exposures .............................................................. 25 Conclusions ................................................................................................................................... 26 References and Bibliography ........................................................................................................ 27

5 | P a g e
Introduction Bifenthrin is a synthetic pyrethroid insecticide with biochemical origins in the natural insecticide pyrethrum, an extract of the flower Chrysanthemum cinerariaefolium. It is used primarily as an insecticide on turf, in homes, and for agricultural applications. Synthetic pyrethroids as a class are in the top ten for usage in the home and garden market (Grube et al. 2011), although have fallen in rank since 2004. Synthetic pyrethroids are known neurotoxicants and that is their mode-of-action in target organisms. In mammals, the mechanism of action for neurotoxicity of pyrethroids results from interference with the sodium gate in the nerve membrane by prolonging the open phase of the sodium channel gate when a nerve cell is excited (Soderlund et al. 2002; US EPA 2011). Synthetic pyrethroids are identified as Type I or Type II based on differences in basic structure (the presence or absence of a cyano group in the alpha position) and the overt symptoms of poisoning at high doses in laboratory rodents. Type I pyrethroid acute toxicity is characterized by aggressive behavior, fine tremors, prostration, and high body temperature, referred to as T syndrome, while Type II acute toxicity is characterized by involuntary and irregular movements, choreoathetosis, and generally includes excessive salivation, referred to as CS syndrome (Breckenridge et al. 2009; Burr and Fry 2004). Early published studies (as reported in Ray and Fry 2006) showed a toxicological picture for the synthetic pyrethroids dominated by purely functional or pharmacological neurotoxicity (hyperexcitation) mediated by action on the voltage-gated sodium channel. Subsequent work
refined this picture by showing the additional contribution of actions upon other ion channels, and the heterogeneity of sodium channel actions, but the pyrethroids remained primarily functional neurotoxicants (Soderlund et al. 2002). Pyrethroids without an alpha-cyano group generally show the weakest physiological effect (Wilks 2000). Type II commercial pesticides such as deltamethrin and cypermethrin are generally more acutely toxic than the type I pyrethroids such as permethrin (Ray and Fry 2006). Figure 1 shows the chemical structure of the primary bifenthrin isomer1 as well as the two primary
metabolites (discussed under pharmacokinetics). Bifenthrin is unique among the pyrethroids in that it contains a non-cyano alcohol moiety and yet
demonstrates more of a CS-type acute intoxication (Soderlund et al. 2002). A study by Choi and Soderlund (2006) found that the activation and deactivation kinetics of bifenthrin clustered closely with those of the three established Type I compounds, suggesting a greater similarity with Type I rather than Type II pyrethroids with respect to mechanism of action. Studies show
Figure 1: Structure of Bifenthrin 1(R)cis Acid Isomer (I) and Metabolites (II and III)

6 | P a g e
that acute effects of synthetic pyrethroids are reversible following acute exposures, and that these compounds cause limited cumulative toxicity, if any, following sustained exposure (Soderlund et al. 2002; US EPA 2011). The US EPA recently completed a cumulative risk assessment for the synthetic pyrethroids (US EPA 2011). Both the US EPA and the European Union have developed risk assessments for bifenthrin including predictions of short-term and long-term exposures (US EPA 2011; EC 2010). In addition to a number in vivo studies in rodents and dogs submitted under various regulatory programs, the results of 500 human cell culture assays are now available through the US EPA ToxCast program (Wetmore et al. 2012) and the results for bifenthrin are presented in this paper in the context of the other toxicological and exposure studies. This paper evaluates the weight-of-evidence for adverse effects in humans and explores the relationship between predicted exposures and effect levels from the toxicological data.
Methods Relevant citations are identified by conducting a search of the peer-reviewed literature for toxicological and epidemiologic studies that have evaluated effects of exposure to bifenthrin. With respect to the toxicological studies, we evaluate the stated significance of observed responses, the adequacy of study design and statistical analyses, the presence of dose response relationships, the evidence for and against modes of action appropriate for environmental exposures in the general public, and the consistency of outcomes within and across studies. For the epidemiological studies, we evaluate study design, how exposure was quantified and the potential for exposure misclassification and what implications that might have for presented conclusions, and the potential for concurrent exposures.
Literature Search and Review The literature search included the following terms: effect* [and] bifenthrin (or) pyrethroid; pyrethroid [and] effect* [and] review. All major databases, search engines, and websites, were used, including: • Citation Index/ISI Web of Science • JSTOR • National Library of Medicine/PUBMED/TOXNET • Hazardous Substances DataBase (HSDB) • MEDLINE (OvidSP) • Google Scholar • Google Books • TRIP database • EMBASE (OvidSP) • Scirus • Environmental Research • US EPA Office of Pesticide Programs Public Docket • International Programme on Chemical Safety

7 | P a g e
• FAO Panel of Experts on Pesticide Residues in Food and the Environment and the WHO Expert Group on Pesticide Residues
Studies primarily focused on pharmacokinetics and pharmacodynamics are shown in Table 1. Table 2 presents the in vivo and in vitro animal studies except for the ToxCast data, which are shown in Table 3. Specific ToxCast assays were explored with respect to relevance of a mode of action, and for in vivo exposure levels at which in vitro effects have been observed. Finally, predicted exposure levels from regulatory evaluations of bifenthrin are compared to these toxicological values.
Results The literature search revealed numerous secondary sources for unpublished studies that, in most cases, were submitted to regulatory agencies as part of pesticide evaluation and/or registration in either the United States or Europe. As such, these studies were reviewed by expert panels, these reviews were evaluated to ensure concordance (e.g., were the descriptions and key aspects of the studies the same across the reviews). Table 1 summarizes the studies focused on pharmacokinetics of bifenthrin. Table 2 summarizes the toxicological studies except for the ToxCast assay results, which are presented in Table 3.
Pharmacokinetics Many of the studies identified in Table 1 were only available as summaries from the Joint Food and Agriculture Organization (FAO) of the United Nations and the World Health Organization (WHO) Meeting on Pesticide Residues summary report on bifenthrin (JMPR 1992) which made reference to numerous unpublished pharmacokinetic and toxicological studies submitted to WHO in support of the peer-reviewed evaluation of the potential for adverse effects in humans. These same studies were used by the US EPA Office of Pesticide Programs in their regulatory review of bifenthrin, and these form the basis of the pharmacokinetic summary presented here.
Oral Exposures JMPR (1992) cites an unpublished study by El Naggar et al. (1983) in which rats were treated with a single oral dose of 5 mg/kg bw alcohol (phenyl)-14C-labelled bifenthrin. Approximately 76%-79% of the administered radioactivity was eliminated via the feces and 6-7% via urine within the first 48 hours. Approximately 90% was recovered in excreta after seven days. Radiocarbon residues in most tissues were < 0.1 ppm, except for liver (up to 0.1 ppm), skin (up to 0.4 ppm) and fat (up to 1.7 ppm). A significant portion of the parent chemical was excreted unchanged in the feces. JMPR (1992) cites another unpublished study by El Naggar et al. (1991) in which the authors investigated excretion of bifenthrin following oral administration of a single dose of 2.7 mg/kg bw to female or 5.2 mg/kg bw to male rats. Female rats averaged 30% excretion of radioactivity in bile, approximately 15% in the urine and the remaining 49% via feces. In male rats, the excreted radioactivity averaged 19%, 11% and 25% of the 14C-dose in bile, urine and feces, respectively. In this case, over 90% of the excreted 14C-residue in the bile was in form of polar

8 | P a g e
conjugates and less than 1% could be attributed to the parent compound. Total absorption via the oral pathway of bifenthrin using the sum of average biliary and urinary excretion and tissue concentrations determined in the El Naggar et al. (1991) study yields a value of approximately 50% in females and 36% in males, respectively. The European Commission (EC 2010; EFSA 2009) use a value of 50% absorption via the oral pathway, while the US EPA assumes bifenthrin is 100% absorbed via oral exposures.
Organism DoseFraction in Urine
Fraction in Feces Metabolites Tissue Residues
Rat5 mg/kg bw alcohol (phenyl)-14C-labelled bifenthrin 6%-7%
76%-79% in 48 hrs; 90% in 7 d Parent compound
Radiocarbon residues in most tissues were < 0.1 ppm, except for liver (up to 0.1 ppm), skin (up to 0.4 ppm) and fat (up to 1.7 ppm).
Rat
14C-bifenthrin in one of the following dose regimens: control (vehicle only), a single low-dose of 4 mg/kg bw, multiple low-doses of 4 mg/kg bw/day of non-radiolabelled test material over a two-week period, followed by a single radiolabelled dose of 4 mg/kg bw or a single high-dose of 35 mg/kg bw. 9%-15%
71%-84% in 36 hrs (low-dose); 72 hrs (high-dose) Parent compound
Fat 1 ppm; Most organs < 0.2 ppm (low-dose); < 1ppm (high-dose)
Rat
acid- and alcohol-14C-labelled bifenthrin at single dose levels of 4 mg/kg bw or 35 mg/kg bw. Also unlabelled bifenthrin for 14 days at 4 mg/kg bw/day followed by a single low dose of radiolabelled bifenthrin.
66%-73% (alcohol); 69-83% (acid)
20-25% (alcohol) and 13-22% (acid)
Fecal: hydroxylated parent compound. Urinary: result of hydrolytic and oxidative-hydrolytic processes. Noticable increase in metabolites in multiple low doses not measured
Rat
single oral low-dose of 5.4 mg/kg bw; single oral high-dose of 36-43 mg/kg bw depending on sex; multiple oral low-doses of 4.9 mg/kg bw. "majority"
Feces: hydroxylated parent compound non-conjugates; urinary: hydrolytic or oxidative degradation in conjugate and non-conjugate forms
Average peak concentrations of radioactivity were 9.6 ppm in fat, 1.7 ppm in skin, 0.4 ppm in liver, 0.3 ppm in kidney, 1.7 ppm in ovaries, 3.2 ppm in sciatic nerve, 0.06 ppm in whole blood and 0.06 ppm in plasma
Female ratradiolabeled oral 0.5 mg/kg bw/d for 70 days NA NA
Parent chemical accounted for a majority (65%-85%) of the 14C-residues in fat; also three metabolites.
Half-lives of 51 days (fat), 50 days (skin), 19 days (liver), 28 days (kidney), and 40 days (ovaries and sciatic nerve) were estimated from 14C-depuration.
Lactating goats
radiolabeled oral 2 mg/kg bw/day for 7 days. NA NA
14C-residues isolated as organosoluble, non-conjugated products. Also hydrolytic and intact ester metabolites.
75%-82% of total 14C-residues (ca. 1 ppm). Fat contained 78%-80% (ca. 1.7 ppm) parent compound, muscle 74%-88% (ca. 6.2 ppm), heart 77% (ca. 0.4 ppm), kidney 16%-22% (0.1 ppm) and liver 19%-44% (0.8 ppm)
Lactating goats 2 mg/kg bw/day for 7 days 40%-52% 8%-17% None reported
liver (3.9 ppm), fat (2.8 ppm), kidneys (1.0 ppm) and heart (0.6 ppm)
Long Evans rat and human hepatic microsomes
added 100% cis-bifenthrin in the presence and absence of NADPH NA NA
Intrinsic clearance (measure of the metabolic rate) of bifenthrin was 5- to 15-fold greater in rat relative to human and solely the result of oxidative processes in both NA
See Table 3 for ToxCast assay results.
in vivo Studies
in vitro Studies
Table 1: Pharmacokinetic Studies

9 | P a g e
Inhalation Absorption via inhalation is assumed to be 100%, but inhalation exposures have generally been shown to be very low as compared to dermal or oral, given the low vapor pressure of bifenthrin (US EPA 2011; EC 2010; JMPR 1992).
Dermal Absorption An in vivo study in rats showed that amount of bifenthrin eliminated in the urine and feces was less than 1% of the dose applied, even after 24 hours exposure. The amount absorbed (including the amount in the skin) was 55.14% at 10 hours and 69.1% at 24 hours. A second in vivo study in shaved rats dermally dosed with an aqueous emulsion showed that after a contact time of 24 h, 19% of the dose was recovered on the skin and about 73% in the skin wash with radioactivity in the residual carcass less than 2% (JMPR 1992; EFSA 2008). US EPA recently used a dermal absorption factor of 5% based on uncited studies in their cumulative risk assessment of synthetic pyrethroids (US EPA 2011).
Metabolism Soderlund et al. (2002) report that for workers exposed to allethrin and volunteers or workers exposed to permethrin, cypermethrin, cyfluthrin, or deltamethrin, the metabolites identified in urine samples were consistent with the metabolic pathways for these compounds identified in rodents. An in vitro study using rat and human hepatic microsomes found that in both cases, the primary metabolic pathway was the result of oxidative processes, and the intrinsic clearance (a measure of the metabolic rate) of bifenthrin was 5- to 15-fold greater in the rat relative to human microsomes (Scollon et al. 2009). These authors note that the parent pyrethroid is generally believed to be the neurotoxic entity as metabolism tends to decreases potency (Scollon et al. 2009; Soderlund et al. 2002), although no formal studies of bifenthrin could be found to directly corroborate this conclusion. The most significant metabolite of bifenthrin is 2-methyl-3-phenylbenzoic acid (MPA), the clearest indicator of bifenthrin exposure (Ciner et al. 2010; Smith et al. 2002). Yang et al. (2009) found that bifenthrin weakly activated human pregnane X receptor (PXR) but efficaciously activated rat PXR, suggesting differences in metabolic rates and outcomes in humans as compared to rodents that could have implications for potential toxicity. US EPA ToxCast in vitro assay results (shown in Table 3) indicate that bifenthrin induced P450 metabolic pathways at equivalent in vivo concentrations greater than 0.12 mg/kg-d.
Toxicity Studies Only one study was identified conducted in vivo in humans. A single, short-term epidemiologic study was developed by Srivistava et al. (2005) based on occupational users of bifenthrin. Ten healthy males wearing protective gear sprayed 25 mg/m2 of bifenthrin on interior walls six hours daily for five consecutive days. Clinical and biochemical tests were conducted prior to exposure and on days four and seven following exposure. Tests included lung function, hepatic function,

Table 2: Toxicological Studies for Bifenthrin Guideline/ Reference
Study Type MRID No. (year)/ Classification /Doses Results
870.3100 90-Day oral toxicity (rat)
00141199 (1984) Acceptable/guideline M: 0, 0.88, 3.8, 7.5, 15 mg/kg/day F: 0, 1.04, 4.3, 8.5, 17.2 mg/kg/day
NOAEL = M/F: 3.8/4.3 mg/kg/day LOAEL = M/F: 7.5/8.5 mg/kg/day based on increased incidence of tremors.
870.3150 90-Day oral toxicity (dog)
00141200 (1984) Acceptable/guideline 0, 2.21, 4.42, 8.84, 17.7 mg/kg/day
NOAEL = M/F: 2.21 mg/kg/day LOAEL = M/F: 4.42 mg/kg/day based on increased incidence of tremors.
870.3200 21/28-Day dermal toxicity (rat)
45280501 (2000) Acceptable/guideline 0, 23, 47, 93, 932 mg/kg/day
NOAEL = 47 mg/kg-d LOAEL = 93 mg/kg/day based on staggered gait and exaggerated hindlimb flexion.
870.3200 21/28-Day dermal toxicity (rabbit)
00141198 (1984) Acceptable/guideline 0, 22, 44, 88 442 mg/kg/day
NOAEL = 88 mg/kg/day LOAEL = 442 mg/kg/day based on loss of muscle coordination and increased incidence of tremors.
870.3700a Prenatal developmental in rat (gavage)
00154482 (1983) Acceptable/non-guideline 0, 0.44, 0.88, 1.77, 2.2 mg/kg/day
Maternal NOAEL = 0.88 mg/kg/day LOAEL = 1.77 mg/kg/day based on tremors during gestation. Developmental NOAEL and LOAEL were not established (fetuses were not examined).
870.3700a Prenatal developmental in rat (gavage)
00141201 (1984) Acceptable/guideline 0, 0.44, 0.88, 1.77 mg/kg/day
Maternal NOAEL = 0.88 mg/kg/day LOAEL = 1.77 mg/kg/day based on tremors. Developmental NOAEL = 0.88 mg/kg/day LOAEL = 1.77 mg/kg/day based on increased fetal and litter incidence of hydroureter without nephrosis.
870.3700a Prenatal developmental in rat (diet)
45352301 (2001) Acceptable/guideline 0, 2.4, 4.8, 7.1, 15.5 mg/kg/day
Maternal NOAEL = 7.1 mg/kg/day LOAEL = 15.5 mg/kg/day based on clinical signs and decreased food consumption, body weight gains, and body weight gains (adjusted for gravid uterine weight). Developmental NOAEL = 15.5 mg/kg/day LOAEL was not established.
870.3700b Prenatal developmental in rabbit (gavage)
00145997 (1984) Acceptable/guideline 0, 2.36, 3.5, 7 mg/kg/day
Maternal NOAEL = 2.36 mg/kg/day, LOAEL = 3.5 mg/kg/day based on treatment- related head and forelimb twitching. Developmental NOAEL =7 mg/kg/day, LOAEL not established.

11 | P a g e
Guideline/ Reference
Study Type MRID No. (year)/ Classification /Doses Results
870.3800 Reproduction and fertility effects (rat) 2-yr
00157225 (1986) Acceptable/guideline 0, 1.5, 3.0, 5.0 mg/kg/day
Parental/Systemic NOAEL = M/F: 5.0/3.0 mg/kg/day,LOAEL was not established in males. In females, LOAEL= 5.0 mg/kg/day based on tremors and decreased body weights. Reproductive/ Offspring NOAEL = 5.0 mg/kg/day, Reproductive/ Offspring LOAEL not established.
870.4100b Chronic toxicity 1-yr (dog)
00163065 (1985) Acceptable/guideline 0, 0.66, 1.3, 2.7, 4.4 mg/kg/day
NOAEL = 1.3 mg/kg/day, LOAEL= 2.7 mg/kg/day based on increased incidence of tremors.
870.4300 Chronic/ Carcinogenicity 1-yr (rat)
00157226 (1986) Acceptable/guideline M: 0, 0.6, 2.3, 4.7, 9.7 mg/kg/day F: 0, 0.7, 3.0, 6.1, 12.7 mg/kg/day
NOAEL = M/F: 4.7/3.0 mg/kg/day, LOAEL =M/F: 9.7/6.1 mg/kg/day based on increased incidence of tremors. No conclusive evidence of carcinogenicity
870.4300 Chronic/ Carcinogenicity (mouse)
00157227 (1986) Acceptable/guideline M: 0, 6.7, 25.6, 65.4, 81.3 mg/kg/day F: 0, 8.8, 32.7, 82.2, 97.2 mg/kg/day
NOAEL =M/F: 6.7/8.8 mg/kg/day, LOAEL = M/F: 25.6/32.7 mg/kg/day based on based on increased incidence of tremors. Carcinogenic potential was evidenced by a dose-related increase in the incidence of leiomyosarcomas in the urinary bladder, a significant dose-related trend for combined hepatocellular adenomas and carcinomas in males, and a significantly higher incidence of combined lung adenomas and carcinomas in females.
870.6200a Acute neurotoxicity-rat (gavage)
44862102(1998) Acceptable/Guideline 0, 9.4, 32.8, 70.3 mg/kg/day
NOAEL = 32.8 mg/kg/day, LOAEL=70.3 mg/kg/day based on clinical signs of toxicity, FOB findings, altered motor activity, and mortality (females only).
870.6200b Subchronic neurotoxicity screening battery (rat)
44862103 (1998) Acceptable/Guideline M: 0, 2.7, 5.6, 11.1 mg/kg/day F: 0, 3.5, 6.7, 13.7 mg/kg/day
NOAEL= M/F: 2.7/3.5 mg/kg/day, LOAEL= M/F: 5.6/6.7 mg/kg/day based on neuromuscular findings (tremors, changes in grip strength and landing foot-splay).

12 | P a g e
Guideline/ Reference
Study Type MRID No. (year)/ Classification /Doses Results
870.6300 Developmental Neurotoxicity (rat)
46750501 (2006) Acceptable/non-guideline 0, 3.6, 7.2 and 9.0 mg/kg/day (gestation) 0, 8.3, 16.2 and 20.7 mg/kg/day (lactation)
Maternal NOAEL = 3.6 mg/kg/day during gestation and 8.3 mg/kg/day during lactation, LOAEL = 7.2 mg/kg/day during gestation and 16.2 mg/kg/day during lactation based on clinical signs of neurotoxicity (tremors, clonic convulsions, and increased grooming counts). Developmental NOAEL =3.6 mg/kg/day during gestation and 8.3 mg/kg/day during lactation. Developmental LOAEL = 7.2 mg/kg/day during gestation and 16.2 mg/kg/day during lactation based on clinical signs of neurotoxicity (increased grooming counts).
Akhtar et al. (1999)
5 Albino rats oral gavage 0.5 mg/d Talstar over 21 days Serum concentrations of T3 and T4 (P < 0.01) suppressed; serum TSH concentrations increased (P < 0.01)
Holton et al. (1997)
3 F344 rats 20 mg/kg at 6 h and 10 mg/kg at 24 h No histological changes in any part of the brain. Fine tremors only (not enough to interfere with gross motor activities such as eating etc.)
in vitro Studies
JMPR 1992 citing: Haworth (1983); Kennelly et al. (1988)
Mutagenicity; Ames assay; S. typhimurium
75 - 7500 µg/plate; Negative with and without activation.
JMPR 1992 citing: Kirby (1983)
Mouse lymphoma nonactivated 0.018 - 0.24 µl/ml; activated 0.0075 - 0.1 µl/ml
Positive at highest dose with activation; no dose-response.
JMPR 1992 citing: Kennelly (1986)
Mouse lymphoma 15.8 - 500 µg/ml Negative with and without activation.

13 | P a g e
Guideline/ Reference
Study Type MRID No. (year)/ Classification /Doses Results
JMPR 1992 citing: Heidemann (1989)
Chinese hamster ovary cells
10 - 100 µg/ml with and without activation Negative with and without activation.
JMPR 1992 citing: Thilagar (1984b)
Chinese hamster ovary cells chromosome aberration
1000 - 10 000 µg/ml with and without activation Negative with and without activation.
JMPR 1992 citing: Thilagar (1983a)
DNA repair (UDS) rat primary hepatocytes
0.01 - 2.0 µl/ml Positive at highest dose; no dose-response.
JMPR 1992 citing: Thilagar (1983b)
DNA repair (UDS) rat primary hepatocytes
0.5 - 2.5 µl/ml Negative
JMPR 1992 citing: Fautz et al. (1989)
DNA repair (UDS) rat primary hepatocytes
1 - 100 µg/ml Negative
JMPR 1992 citing: Heidemann (1989)
Chinese hamster ovary sister chromatid exchange
1 - 60 µg/ml with and without activation Negative
Goto et al. (2004)
Bhas 42 cells from BALB/c 353 mouse embryo cells transfected with v-Ha-ras oncogene
0 - 5 ppm At least twice the foci of controls at approximately 2ppm; zero foci at 5 ppm. Authors conclude evidence of a good dose-response relationship; however, not clear on what basis determination made.
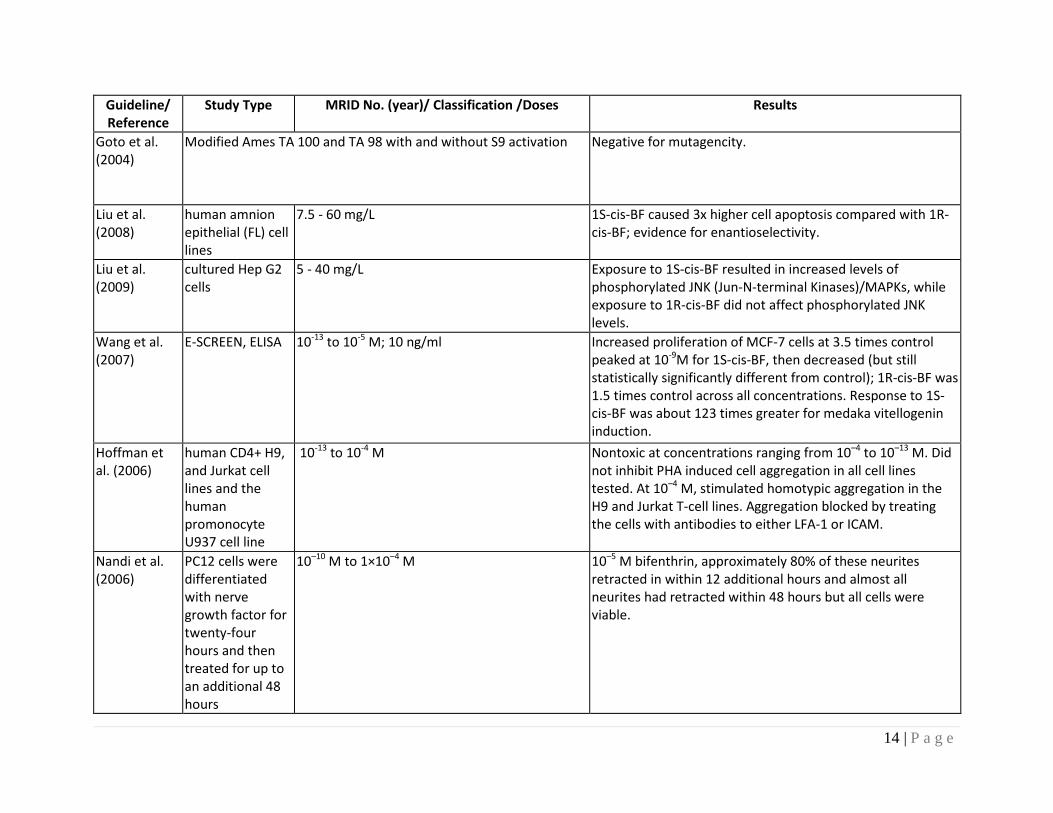
14 | P a g e
Guideline/ Reference
Study Type MRID No. (year)/ Classification /Doses Results
Goto et al. (2004)
Modified Ames TA 100 and TA 98 with and without S9 activation Negative for mutagencity.
Liu et al. (2008)
human amnion epithelial (FL) cell lines
7.5 - 60 mg/L 1S-cis-BF caused 3x higher cell apoptosis compared with 1R-cis-BF; evidence for enantioselectivity.
Liu et al. (2009)
cultured Hep G2 cells
5 - 40 mg/L Exposure to 1S-cis-BF resulted in increased levels of phosphorylated JNK (Jun-N-terminal Kinases)/MAPKs, while exposure to 1R-cis-BF did not affect phosphorylated JNK levels.
Wang et al. (2007)
E-SCREEN, ELISA 10-13 to 10-5 M; 10 ng/ml Increased proliferation of MCF-7 cells at 3.5 times control peaked at 10-9M for 1S-cis-BF, then decreased (but still statistically significantly different from control); 1R-cis-BF was 1.5 times control across all concentrations. Response to 1S-cis-BF was about 123 times greater for medaka vitellogenin induction.
Hoffman et al. (2006)
human CD4+ H9, and Jurkat cell lines and the human promonocyte U937 cell line
10-13 to 10-4 M Nontoxic at concentrations ranging from 10–4 to 10–13 M. Did not inhibit PHA induced cell aggregation in all cell lines tested. At 10–4 M, stimulated homotypic aggregation in the H9 and Jurkat T-cell lines. Aggregation blocked by treating the cells with antibodies to either LFA-1 or ICAM.
Nandi et al. (2006)
PC12 cells were differentiated with nerve growth factor for twenty-four hours and then treated for up to an additional 48 hours
10–10 M to 1×10–4 M 10–5 M bifenthrin, approximately 80% of these neurites retracted in within 12 additional hours and almost all neurites had retracted within 48 hours but all cells were viable.

15 | P a g e
Guideline/ Reference
Study Type MRID No. (year)/ Classification /Doses Results
Tran et al. (2006)
PC12 cells were differentiated with nerve growth factor for 48 hours
10-13 to 10-4 M 10–7 M and 10–5 M bifenthrin, no reduction in cell viability but neurite outgrowth reduced 30% and 55%.
Lu et al. (2010)
PC12 cells 10-9 M to 10-5 M Significant reduction in cell survival and superoxide dimutase, increased production of lactate dehydrogenase, intracellular reactive oxygen species and malondialdehyde, was observed in 1S-cis-BF; less so in 1R-cis-BF (only at 10-5 and no oxidative damage)
ToxCast in vitro assays (see Table 3)

nerve conduction, and electromyogram and no statistically significant differences were found pre- and post-exposure. However, this study was strictly a short-term study, and did not address potential impacts of longer term, chronic exposures.
Carcinogenicity As shown in Table 2, two unpublished rodent studies evaluated the potential carcinogenicity of bifenthrin. Both studies were two-year chronic feeding studies, one in Swiss Webster mice and the other in Sprague-Dawley rats. The mouse study found no significant differences in survival between the groups of either sex. However, tumors were observed in the bladders of two and three male mice at doses of 500 and 600 ppm respectively, and one female mouse at a dose of 200 ppm. No dose-response relationship was observed in either case, although the male cancers were marginally statistically significantly different from controls at the highest dose when considering all doses (Butler et al. 1997). A reevaluation by Butler et al. (1997) found that the histological features of the tumors based on smooth muscle and vascular differentiation suggested a vascular origin. In addition, the authors observed a chronic inflammatory infiltrate and hemosiderin associated with the tumors. There were no metastases observed across either sex. The particular tumor type has since been identified as a submucosal mesenchymal lesion of the urinary bladder of the mouse (Butler et al. 1997; Halliwell 1998) with no relevance to potential human health risks. The other study in rats found no significant treatment-related effects across all endpoints (Unpublished study, McCarty et al. 1986, as reported in JMPR 1992). The US EPA conducted three separate peer review processes to determine the potential carcinogenicity of bifenthrin. All three peer reviews determined that bifenthrin was a Category C, possible human carcinogen, on the basis of two unpublished rodent studies. A joint international meeting of the Food and Agricultural Organization (FAO) Panel of Experts on Pesticide Residues in Food and the Environment and the World Health Organization (WHO) Expert Group on Pesticide Residues, with the cooperation of the International Programme on Chemical Safety (IPCS) met in Rome in 1992 and concluded that although the tumor incidence in the mouse study was of marginal statistical significance, tumorigenic potential for bifenthrin in mice could not be excluded. However, they stopped short of considering bifenthrin a carcinogen in humans (JMPR 1992).
Chronic Toxicity A number of unpublished studies have formed the basis for several different regulatory reviews with respect to bifenthrin as shown in Table 2. DeProspo et al. (1986) as reported in JMPR (1992) administered bifenthrin in the diet at concentrations of 0, 30, 60 or 100 ppm (0, 1.5, 3 or 5 mg/kg/day) to groups of rats (25/sex/group) over two consecutive generations. No mortality was observed. At 100 ppm tremors were observed in first-generation lactating dams. First-generation females showed reduced body-weight gain on days 7 and 14 of the lactation period. Food consumption was depressed in the second generation group at 100 ppm in the males during a single week of exposure. The treatment did not have any effects on the reproductive performance or litter size, litter weight or survival of the progeny. Changes in organ weights at 100 ppm consisted of an elevation of the brain weights of first-generation females. No histomorphologic alterations were observed in tissues from parental or weanling animals. Based on the incidence

17 | P a g e
of tremors and marginally lower body weights resulted in a no observed effect level (NOEL) of 3 mg/kg-day. A lowest observed effect level was not observed; therefore, the reproductive NOEL was determined to be 5 mg/kg-day (62 FR 62961). Freeman et al. (1984b) as reported in JMPR (1991) and summarized in 62FR69261 orally gavaged groups of rats (25 females/dose/group) on days 6 through 15 of gestation with doses of 0, 0.5, 1 or 2 mg/kg bw/day. Estimation of dose levels were based on a previous pilot developmental study in rats in which bifenthrin was administered in the diet at dose levels of 0, 0.5, 1.0, 2.0, or 2.5 mg/kg-day during days 6 to 15 of gestation. Three of 10 rats at 2.5 mg/kg/day died on days 14-15. Across both studies, tremors were noted at 2.0 mg/kg-day. There were no differences in mean body weight gains or food consumption with respect to the controls. There were no treatment-related differences from controls for reproductive parameters, including pregnancy, number of corpora lutea, implantations, resorptions or litter size. Fetal malformations occurred only sporadically in all groups and without any observable dose-response relationship. The maternal NOEL in this study was 1.0 mg/kg-day, and the maternal LOEL is 2.0 mg/kg/day based on sporadic tremors (gestation days 7 18). The fetal NOEL is >2 mg/kg-day for embryo fetotoxicity. A study by Freeman et al. (1984a) as reported in JMPR (1991) and summarized in 62FR62961 gavaged groups of rabbits (20 females/dose/group) at 0, 2.7, 4 or 8 mg/kg-bw on days 7 through 19 of gestation, based on an earlier dose range finding study. Tremors were observed for most of the animals receiving 8 mg/kg bw/day and head and fore limb twitching were observed during the second half of the dosing period among most of the animals receiving 4 or 8 mg/kg-bw. The test material did not affect the body-weight of the dams, the reproduction parameters, viability or body-weight of the pups, nor the incidence of external and visceral anomalies. The maternal NOEL was estimated at 2.7 mg/kg-day, based on head and forelimb twitching at a LOEL of 4.0 mg/kg/day. The developmental NOEL is 8.0 mg/kg/day, the highest dose tested. JMPR 1992 reports on a study by Algate et al. (1985) in which groups of rats (COBS/Wistar; 3 males/dose group) were orally treated with doses of 0, 1.0, 3.0, 10.0 or 30.0 mg/kg bw/day for consecutive days. Parameters investigated included alertness, locomotor activity, apathy, tremor and abnormal gait. Rats at 30 mg/kg bw/day showed tremor, abnormal gait, respiratory depression and signs of CNS depression (apathy, paralysis). Deaths occurred after developing convulsions. No effects were recorded during the 7-day period after termination of dosing with 1, 3 and 10 mg/kg-d. The minimum effective dose of 30 mg/kg bw/day which caused neurological signs such as paralysis as determined in the Irwin dose-range test (Algate et al. 1985) was used in a tilting-plane test. The test compound was administered orally to groups of rats (5/sex) on two consecutive days. The tilting-plane test (parameter: angle of inclination at which the animals began to slide down a tilted platform) was performed every second day from days 2-16 of the study. The results did not reveal impairment of performance by the treatment and this gave no indication of a delayed neurotoxic effect (Algate et al. 1985). Aldridge (1990) provided an assessment of the toxicological properties of pyrethroids generally with respect to their neurotoxicity and found that exposure of rats to pyrethroids at high doses

18 | P a g e
(often doses that are also associated with death of the animal) led to minor lesions in nerve cells that were not observed at lower doses and were reversible given survival of the animal. Aldridge (1990) concluded that there were no observable neurotoxic actions of the pyrethroids other than those originating from their primary mechanism of action on the sodium channel, and that pyrethroid exposure in “working practice” would not lead to chronic neurotoxicity.
Subclinical Effects Akhtar et al. (1996) conducted a study to evaluate the effect of Talstar, (a commercial formulation of bifenthrin) and a number of other insecticides on the thyroid secretory function in rats. Five rats (ranging in weight from 240 – 320 g) were adminstered 0.5 mg Talstar via oral gavage for 21 days. Serum concentrations of triiodothyronine (T3), thyroxine (T4,) and thyrotrophin (TSH) were determined by using specific radioimmunoassays. Body weight was significantly reduced in Talstar-treated rats (P < 0.01) and serum T3 and T4 levels were significantly suppressed (P < 0.01), while TSH concentrations were stimulated (P < 0.01). Hoffman et al. (2006) exposed human CD4+ H9, and Jurkat cell lines and the human promonocyte U937 cell line to bifenthrin and found that bifenthrin was nontoxic at concentrations ranging from 10–4 to 10–13 M. Bifenthrin did not inhibit PHA induced cell aggregation in all cell lines tested. However, at 10–4 M, bifenthrin activated T-cell function by stimulating ICAM/LFA-1 mediated homotypic aggregation, suggesting that exposure to bifenthrin can increase the risk for and frequency of inflammatory responses. However, that molar concentration approximately corresponds to an in vivo dose of 2.5 mg/kg-d, much higher than predicted exposures (see below). Given that bifenthrin is a known neurotoxicant, studies that have explored subclinical neurotoxicological effects may be most relevant with respect to endpoints in humans. However, the link between alterations in neuronal firing and downstream neurobehavioral syndromes is correlative and not causative (Wolansky et al. 2009). Van Tran et al. (2006) found that bifenthrin inhibited neurite formation by 30% at concentrations of 10-7 M (roughly equivalent to 0.002 mg/kg-d). These authors argue that bifenthrin exposure in utero could lead to neurodevelopmental defects and further argue the possibility that chronic exposure to bifenthrin may lead to neurodegenerative disease (Tran et al. 2006; Nandi et al. 2006). However, both these studies relied on the widely used PC12 cell line, which while exhibiting extensive neurite outgrowth upon differentiation, do not actually give rise to definitive axons or dendrites (Radio and Mundy 2008). The mechanistic basis of induction of differentiation in cell lines is not fully understood, and neurite outgrowth may differ from that occurring in primary neurons. For example, neurites elaborated by the PC12 cell line do not exhibit the properties of either axons or dendrites. Near-lethal doses of pyrethroids cause sparse axonal damage that is reversed in surviving animals. After prolonged exposure to lower doses of pyrethroids axonal damage has not been observed (Vijverberg et al. 1990) although this observation was not specific to bifenthrin. The effects of pyrethroids on the CNS are complex and may also involve antagonism of γ-aminobutyric acid (GABA), modulation of nicotinic cholinergic transmission, enhancement of noradrenalin release, and direct actions on calcium or chloride ion channels. Still, because

19 | P a g e
neurotransmitter-specific pharmacological agents do not protect very well against pyrethroid poisoning, it is unlikely that any one of these effects represents a primary toxic mechanism of action of pyrethroids. More likely, they are secondary to the effects on sodium channels since most neurotransmitters are released secondary to increased sodium entry (Bjorling et al. 2008). There is some concern that some synthetic pyrethroids may possess estrogenic properties (Go et al. 1999). However, with respect to potential effects on thyroid function, the effects are not consistent. In an oral gavage study with Talstar (a commercial bifenthrin formulation), Akhtar et al. (1999) found reduced serum concentrations of thyroid hormones after one week of continuous exposure, while Kaul et al. (1996) found exactly the opposite effect . A key concern with thyroid inhibitors is that impaired thyroid function may alter hormone-mediated events during development, leading to permanent alterations in brain morphology and function. The implication for human effects may be revealed through studies such as Hauser et al. (1993), who demonstrated a possible relationship between thyroid hormone dysfunctions and ADHD by showing that a majority of children with resistance to thyroid hormone (RTh) exhibited ADHD-like symptoms (note: unrelated to bifenthrin in any way). Stein and Weiss (2003) have shown that thyroid hormone concentrations, in children referred to a clinic specializing in learning and behavioral problems, were associated with attentional deficits and hyperactivity and that the ADHD subtype, primarily inattentive, was associated with reductions in free thyroid hormone concentrations. However, there is only one positive study (Akhtar et al. 1999), and that study had very little power (five rats total), and the dose mechanism was oral gavage (as opposed to in the diet, which would represent a more relevant exposure route for humans), the effect was not consistent across studies involving individual pyrethroids, and the statistical significance was borderline (P < 0.01). Finally, as pointed out by McClain (1992), species-specific differences in thyroid gland biochemistry and physiology, notably the lack of thyroid binding globulin in the rodent, urges caution in evaluating hormonally-mediated responses involving thyroid function. US EPA ToxCast Assay Results Table 3 shows the ToxCast assays conducted by the US EPA (Wetmore et al. 2012; Judson et al. 2010; Rotroff et al. 2010). Approximately 400 chemicals, including bifenthrin, were subjected to nearly 500 high-throughput in vitro human cell culture screens for assessing potential effects across multiple cellular pathways. The authors experimentally measured metabolic clearance and plasma protein binding to parameterize a population-based in vitro-to-in vivo extrapolation model for estimating the human oral equivalent dose necessary to produce a steady-state in vivo concentration equivalent to in vitro AC50 (concentration at 50% of maximum activity) and LEC (lowest effective concentration) values from the ToxCast data. The assays range from enzyme induction in specific kinds of cells to up- and down-regulation of different genes. The assay at which effects were noted at the lowest equivalent in vivo dose was for observable changes in cell growth kinetics, followed by unspecified protein binding. It is not clear what relevance these subclinical effects have with respect to in vivo effects, but a number of the assay results at higher concentrations would seem to support some of the immunotoxic effects noted in Tran et al. (2006) and elsewhere. However, these effects may be occurring at doses greater than would typically be experienced by the general population (see next section).

Table 3: ToxCast Assay Results ToxCast Assay (Judson et al.
2011) Lowest Oral Equivalent
Dose (mg/kg-d)
Median Oral Equivalent
Dose (mg/kg-d)
Upper Oral Equivalent
Dose (mg/kg-d)
Assay Description
ACEA_LOCinc 0.01 0.02 0.04 Change in cell growth kinetics NVS_TR_hNET 0.01 0.03 0.06 Protein binding; biochemical BSK_3C_Vis_down 0.04 0.09 0.16 Unknown BSK_BE3C_uPA_down 0.04 0.09 0.16 This gene encodes a serine protease involved in
degradation of the extracellular matrix; possibly tumor cell migration and proliferation. Polymorphism associated with late-onset Alzheimer's; decreased affinity for fibrin-binding.
BSK_hDFCGF_MMP1_up 0.04 0.09 0.16 Upregulation of protein involved in the breakdown of extracellular matrix in normal physiological processes.
BSK_KF3CT_MMP9_down 0.04 0.09 0.16 Downregulation of protein involved in the breakdown of extracellular matrix in normal physiological processes.
BSK_SAg_CD38_up 0.04 0.09 0.16 Upregulation of CD38; novel multifunctional ectoenzyme widely expressed in cells and tissues especially in leukocytes. Cell adhesion, signal transduction and calcium signaling.
ATG_PXR_TRANS 0.11 0.24 0.44 This gene product belongs to the nuclear receptor superfamily, members of which are transcription factors characterized by a ligand-binding domain and a DNA-binding domain. The encoded protein is a transcriptional regulator of the cytochrome P450 gene CYP3A4, binding to the response element of the CYP3A4 promoter as a heterodimer with the 9-cis retinoic acid receptor RXR. It is activated by a range of compounds that induce CYP3A4.
CLZD_CYP1A1_48 0.12 0.25 0.44 Encodes a member of the cytochrome P450 superfamily of enzymes.
BSK_hDFCGF_MCSF_down 0.12 0.26 0.47 Downregulation of a gene encoding a cytokine protein that controls the production, differentiation, and function of macrophages.

21 | P a g e
ToxCast Assay (Judson et al. 2011)
Lowest Oral Equivalent
Dose (mg/kg-d)
Median Oral Equivalent
Dose (mg/kg-d)
Upper Oral Equivalent
Dose (mg/kg-d)
Assay Description
BSK_hDFCGF_Proliferation_down
0.12 0.26 0.47 Downregulation; dermal fibroblast.
BSK_hDFCGF_VCAM1_up 0.12 0.26 0.47 Upregulation of a member of the Ig superfamily that encodes a cell surface sialoglycoprotein expressed by cytokine-activated endothelium. This type I membrane protein mediates leukocyte-endothelial cell adhesion and signal transduction, and may play a role in the development of artherosclerosis and rheumatoid arthritis.
BSK_SAg_CD69_down 0.12 0.26 0.47 Downregulation of calcium dependent lectin superfamily of type II transmembrane receptors. Expression of the encoded protein is induced upon activation of T lymphocytes, and may play a role in proliferation.
CLZD_CYP2B6_48 0.12 0.26 0.48 Encodes a member of the cytochrome P450 superfamily of enzymes.
CLZD_CYP3A4_48 0.13 0.27 0.49 Encodes a member of the cytochrome P450 superfamily of enzymes.
CLZD_GSTA2_48 0.14 0.30 0.55 Enzymes that function in the detoxification of electrophilic compounds.
CLZD_CYP2B6_24 0.19 0.39 0.70 Encodes a member of the cytochrome P450 superfamily of enzymes.
CLZD_CYP3A4_24 0.25 0.52 0.92 Encodes a member of the cytochrome P450 superfamily of enzymes.
BSK_3C_hLADR_down 0.28 0.58 1.03 Downregulation of a gene in the immune system by presenting peptides derived from extracellular proteins.
BSK_3C_Proliferation_down 0.28 0.58 1.03 Downregulation. BSK_3C_Thrombomodulin_up 0.28 0.58 1.03 Upregulation of endothelial-specific type I membrane
receptor that binds thrombin. This binding results in the activation of protein C, which degrades clotting factors Va and VIIIa and reduces the amount of thrombin generated.

22 | P a g e
ToxCast Assay (Judson et al. 2011)
Lowest Oral Equivalent
Dose (mg/kg-d)
Median Oral Equivalent
Dose (mg/kg-d)
Upper Oral Equivalent
Dose (mg/kg-d)
Assay Description
BSK_4H_Pselectin_down 0.28 0.58 1.03 Downregulation of selectin P (granule membrane protein 140kDa, antigen CD62).
BSK_4H_VCAM1_down 0.28 0.58 1.03 Downregulation of a member of the IG superfamily. BSK_hDFCGF_CollagenIII_up 0.28 0.58 1.03 Upregulation of pro-alpha1 chains of type III collagen, a
fibrillar collagen that is found in extensible connective tissues such as skin, lung, uterus, intestine and the vascular system, frequently in association with type I collagen.
BSK_LPS_IL8_down 0.28 0.58 1.03 Downregulation of a member of the CXC chemokine family. This chemokine is one of the major mediators of the inflammatory response.
BSK_LPS_MCSF_down 0.28 0.58 1.03 Downregulation of a cytokine that controls the production, differentiation, and function of macrophages.
BSK_LPS_PGE2_down 0.28 0.58 1.03 Downregulation of prostaglandin E receptor 2 (subtype EP2).
BSK_LPS_VCAM1_down 0.28 0.58 1.03 Downregulation of a member of the IG superfamily. BSK_SAg_Eselectin_down 0.28 0.58 1.03 Downregulation of cytokine-stimulated endothelial cells;
thought to be responsible for the accumulation of blood leukocytes at sites of inflammation by mediating the adhesion of cells to the vascular lining.
BSK_SAg_IL8_down 0.28 0.58 1.03 Downregulation of CXC chemokine family; major mediators of the inflammatory response.
BSK_SAg_PBMCCytotoxicity_up 0.28 0.58 1.03 Increased cytotoxicity. BSK_SAg_Proliferation_down 0.28 0.58 1.03 Decreased proliferation. ATG_PXRE_CIS 0.29 0.60 1.08 Transcriptional regulator of cytochrome P450 gene. CLZD_ABCB1_24 0.31 0.64 1.15 ABC proteins transport various molecules across extra-
and intra-cellular membranes. ATG_VDRE_CIS 0.43 0.91 1.62 Cytochrome P450. CLZD_CYP1A1_24 0.57 1.19 2.13 Cytochrome P450.

23 | P a g e
ToxCast Assay (Judson et al. 2011)
Lowest Oral Equivalent
Dose (mg/kg-d)
Median Oral Equivalent
Dose (mg/kg-d)
Upper Oral Equivalent
Dose (mg/kg-d)
Assay Description
NCGC_PXR_Agonist_human 0.59 1.23 2.20 Transcriptional regulator of the cytochrome P450 gene CYP3A4.
ACEA_LOC2 0.68 1.43 2.56 Change in cell growth kinetics ACEA_LOCdec 0.68 1.43 2.56 Change in cell growth kinetics ACEA_IC50 0.71 1.48 2.65 Change in cell growth kinetics ATG_ERa_TRANS 0.76 1.60 2.86 Estrogen receptor, a ligand-activated transcription factor
composed of several domains important for hormone binding, DNA binding, and activation of transcription.
BSK_3C_MCP1_down 0.83 1.73 3.09 Family of secreted proteins involved in immunoregulatory and inflammatory processes.
BSK_4H_Eotaxin3_down 0.83 1.73 3.09 Downregulation of gene coding proteins involved in immunoregulatory and inflammatory processes.
BSK_4H_MCP1_down 0.83 1.73 3.09 Downregulation of gene coding proteins involved in immunoregulatory and inflammatory processes.
BSK_hDFCGF_PAI1_down 0.83 1.73 3.09 Unknown BSK_SAg_CD40_down 0.83 1.73 3.09 Downregulation of a gene mediating a broad variety of
immune and inflammatory responses including T cell-dependent immunoglobulin class switching.
BSK_SM3C_Proliferation_down 0.83 1.73 3.09 Human vascular cells downregulation of proliferation. CLM_Hepat_DNADamage_48hr 1.24 2.60 4.65 DNA damage in rat hepatocytes. CLM_CellLoss_72hr 2.67 5.58 10.00 Cell loss in rat hepatocytes. CLM_MicrotubuleCSK_72hr 3.02 6.31 11.31 Microtubule damage in rat hepatocytes. CLM_Hepat_Steatosis_48hr 3.86 8.06 14.44 Steatosis in rat hepatocytes. CLM_MitoMembPot_1hr 4.08 8.51 15.26 Mitochondrial function in HepG2 rat hepatocytes.

Regulatory Toxicity Values and Equivalent in vivo Doses Associated with in vitro Effects Table 4 presents the range of regulatory values used to develop risk assessments in the United States and Europe as part of pesticide registration activities. In general, these values are all based on a NOAEL, e.g., a dose at which no effects were observed in the original study, of 1.5 mg/kg-d from the one-year feeding study in dogs (EPA 1988; Serota et al. 1985 as cited in JMPR 1992) but incorporating different safety factors (typically 100 - 300). These doses, designed to be protective of any potential health effects of exposure to bifenthrin given that they are based on a no effect level, are in the 0.004 to 0.03 mg/kg-d range. The duration column shows over what time period these exposures can occur -- long-term (e.g., lifetime), medium-term (e.g., a year or two), or short-term (e.g., days).
We estimated equivalent in vivo doses from toxicological studies to explore whether subclinical effects noted through in vivo and in vitro toxicological studies might occur at relevant exposure
concentrations. The methodology is based on the extensive physiological based pharmacokinetic (PBPK) modeling performed as part of the US EPA ToxCast program (Wetmore et al. 2012; Rotroff et al. 2010; Judson et al. 2010). These authors conducted modeling to be able to convert in vitro ToxCast assay results to equivalent in vivo exposure concentrations. Table 3 presents those values for bifenthrin and the various ToxCast assays. The lowest concentration associated with any assay is 0.01 mg/kg-d for potential effects on cell growth. This value is at the high end of the regulatory range shown in Table 4. Going back to Table 2, there are a number of in vitro studies, particularly for neurotoxic endpoints, that noted effects, but typically at concentrations in the 10-4 to 10-5 M range, which translate to equivalent in vivo concentrations of 1-10 mg/kg-d, significantly higher than the regulatory values. However, Tran et al. (2006) in their study involving the PC12 neuronal cell line found bifenthrin inhibited nerve growth factor-mediated neurite outgrowth by 30% and 55% at concentrations of 10-7 and 10-6, respectively, and that concentrations of technical grade bifenthrin of 10–6 M and 10–3 M inhibited neurite outgrowth by approximately 35% and 75% respectively. Following the approach of Wetmore et al. (2012), the equivalent in vivo dose at the lowest 10-7 M concentration would be 0.002 mg/kg-d, lower than the lowest regulatory value.
Source Value (mg/kg-d) DurationUS EPA IRIS 0.015 long-term, lifetime exposuresUS EPA 2003 0.004 long-term, lifetime exposuresUS EPA 2003 0.033 short-term exposuresEC 2010 0.015 short-term exposuresEC 2010 0.0075 medium-term exposuresEC 2010 0.0075 long-term, lifetime exposures
Table 4: Regulatory Values Used in Risk Assessments

25 | P a g e
Comparison of Toxicity Values to Predicted Exposures This section compiles predicted exposure estimates from risk assessment models developed by the US EPA (US EPA 2011) and the European Commission (EC 2010). Specific details of that modeling is presented in those documents and not repeated here, but these are regulatory evaluations designed to predict exposures protective of both applicators and the general public across a wide variety of uses of bifenthrin. The EC estimates are based on a biocidal use of bifenthrin in which applicators are exposed via the inhalation and dermal pathways through professional use of a water-based formulation containing 0.03% bifenthrin. The analysis assumes 11.4% absorption via the dermal pathway (by far the majority of the exposure) and 100% absorption via the inhalation pathway. Oral exposure is assumed to be negligible in this case. For the general public using bifenthrin as a wood preservative, the EC analysis assumes some oral exposure following application of the bifenthrin, in addition to dermal and inhalation exposures. Oral absorption is assumed at 50%. For the US EPA exposure estimates, dermal, oral, and inhalation are included, where oral also includes potential residues on agricultural products (agricultural use is not authorized in Europe). Figure 2 presents the compiled exposure estimates with the regulatory toxicity values in different colors (violet = 0.002 mg/kg-d from Tran et al. 2006; red = 0.004 mg/kg-d from US EPA 2003; blue = 0.0075 mg/kg-d from EC 2010, and green = 0.015 from EC 2010). The short-term value of 0.03 from US EPA 2003 is not shown as it is much higher than any other value on the graph. The top half of the graph shows predicted exposure estimates for "direct" exposures -- including professional applicators (top four scenarios) or home residential use. The bottom part of the graph below the gray line shows predicted exposures for "indirect" uses -- home residential and garden use involving professional applications and residual exposures to individuals in the home. The range of values presented for the EC study in the top half of the graph are based professional applicators using bifenthrin with protective gear (lowest values) to no protective gear (highest values). In general, predicted exposures for the general population fall below any regulatory levels of concern. The violet line is a subclinical effect and is only included because it is the only evidence of any effects occurring at concentrations that might relevant to potential exposures. The red and blue lines are based on a no observed adverse effect level, and the green line, protective of short term exposures, is based on an observation of tremors in rats. Applicators who fail to use protective gear do exceed the regulatory NOAEL, but fall short of actual effect levels. In addition, the use of bifenthrin as a biocide on wood involves an application method likely to be very different from what is experienced in the United States, where the primary use of bifenthrin is as a direct-acting insecticide (e.g., sprayed into crevices, etc.) or on agricultural crops to which the general public would only be exposed through residues.

26 | P a g e
Conclusions The available studies for bifenthrin suggest a low probability of adverse health effects associated with exposure at environmentally-relevant concentrations. For cancer, specifically bladder cancer, as an endpoint, of the two chronic two-year rodent studies, the one in mice showed a marginally statistically significant increase in a type of vascular bladder cancer. Three separate and independent peer reviews convened by EPA concluded that bifenthrin was a class C, possible human carcinogen, on the basis that the particular observed tumor was rare, but considering the ameliorating factors that no dose response relationship was observed, the only incidence of the tumor that was statistically significant (marginally so) was at the highest dose, and that the tumors were only observed in males, the panel could not conclude that bifenthrin was carcinogenic although did not rule it out, either. The rat study showed no increases in tumors of any kind. A joint international meeting of the Food and Agricultural Organization (FAO) Panel of Experts on Pesticide Residues in Food and the Environment and the World Health Organization (WHO) Expert Group on Pesticide Residues, with the cooperation of the International Programme on Chemical Safety (IPCS) which met in Rome in 1992 concluded that bifenthrin is not carcinogenic and shows no evidence of mutagenicity. The results of in vitro and in vivo test results that predict human carcinogenicity support a determination that the evidence for bifenthrin exposure leading to carcinogenic outcomes in humans is marginal to non-existent. A series of studies using the rat PC12 cell line found statistically significant decreases in neurite outgrowth at the highest concentration tested, but with concomitant cell viability, indicating no
Figure 2: Estimated Exposure Levels (mg/kg-d) from US EPA (2003) and EC (2010) Under Regulatory Evaluations of Bifenthrin. Reference Lines are Violet (Tran et al. 2006 Subclinical Effects); Red (US EPA 2003 for Chronic Exposures); Blue (EC 2010 and US EPA IRIS); Green (EC 2010 Short Term Exposures)

27 | P a g e
neuronal cell death, a prerequisite for neurodegenerative effects in humans. Consequently, the evidence for chronic neurotoxicological effects in humans is marginal to non-existent. The US EPA recently published a cumulative risk assessment for the synthetic pyrethroids generally and focused on short-term neurotoxicological impacts related solely to disruption of sodium-gated channels and found that the cumulative impact of synthetic pyrethroids allowed for additional risk in the "risk cup" (US EPA 2011). A number of studies have demonstrated enantiospecific toxicity (attributable to 1R-cis-bifenthrin); however, given that most commercial mixtures will likely contain some proportion of both enantiomers and so the contribution of any one is difficult to predict. Similarly, the ToxCast assay results did not specify which enantiomer was used, and different results could have been observed across one or the other. Both the US EPA (2011) and the European Commission (2010) have estimated potential exposures to bifenthrin as part of regulatory evaluations. Comparing these predicted exposures to regulatory toxicological levels shows that only exposures to applicators who directly handle bifenthrin, and typically assuming no protective gear, are likely to exceed regulatory values. These regulatory values are derived from a study in dogs and are based on a NOAEL with uncertainty and safety factors. The regulatory values are an order of magnitude lower than the lowest value from a series of 500 human cell culture assays (ToxCast). The ToxCast results also showed that bifenthrin induces P450 metabolism, typically a detoxifying mechanism, although at concentrations higher than predicted by regulatory exposure models. However, were exposures to be underestimated or were to increase, these metabolic pathways would serve to detoxify the parent compound as has also been observed in the in vivo pharmacokinetic studies summarized in Table 1. Bifenthrin is a synthetic derivative of a pyrethroid found in crysanthemums. The synthetic derivative is specifically designed such that the mechanism for acute toxicity preferentially selects the target organism, thereby reducing potential toxicity to mammals, and toxicity to mammals is an order of magnitude less than to target organisms (US EPA 2011). The risk of adverse health effects resulting from chronic exposures is judged to be low based on the marginal to non-existent evidence for health effects; nonetheless this low risk must be considered in the larger context of the benefits received from use of the constituent in products to exterminate pests, primarily termites. Given the utility and efficacy of bifenthrin with respect to its termiticidal properties, the analysis qualitatively suggests a net positive risk-benefit tradeoff.
References and Bibliography Akhtar N, Kayani SA, Ahmad MM, et al. 1996. Insecticide-induced changes in secretory activity of the thyroid gland in rats. J Appl Toxicol 16(5):397-400. Aldridge WN. 1990. An assessment of the toxicological properties of pyrethroids and their neurotoxicity. Crit Rev Toxicol 21:89–104. Algate DR, Goor DL, Leach RM and Munt PL. 1985. FMC 54800*: An investigation of the possible delayed neurological effects using the tilting-plane test. Unpublished report No. A85-

28 | P a g e
1795 prepared by Huntingdon Research Centre for FMC Corp., Princeton, NJ, USA. Huntington Study Number FMC 87&88/85657. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Benson ES, Bullock WL, Myhr BC and Ruoff DL. 1984. Mutagenicity evaluation of FMC 54800 technical in the sex-linked recessive lethal test in Drosophila melanogaster. FMC report No. A83-1104. Unpublished report prepared by Litton Bionetics, Inc. for FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Bjørling-Poulsen M, Andersen HR and Grandjean P. 2008. Review. Potential developmental neurotoxicity of pesticides used in Europe. Env Health 7:50-77. doi:10.1186/1476-069X-7-50 Bradbury SP and JR Coats. 1989. Comparative toxicology of the pyrethroid insecticides. Rev Environ Cont Toxicol 108:134-177 Breckenridge CB, Holden L, Sturgess N, Weiner M, Sheets L, Sargent D, Soderlund DM, Choi JS, Symington S, Clark JM, Burr S, Ray D. 2009. Evidence for a separate mechanism of toxicity for the Type I and the Type II pyrethroid insecticides. Neurotoxicology 30 Suppl 1:S17-31. Burr SA and Ray DE. 2004. Structure-Activity and interaction effects of 14 different pyrethroids on voltage-gated chloride ion channels. Toxicol Sci 77:341 - 346. Butler WH, Cohen SH and Squire RA. 1997. Mesenchymal tumors of the mouse urinary bladder with vascular and smooth muscle differentiation. Toxicol Pathol 25:268-274. Cao Z, Shafer TJ, Crofton KM, Gennings C and Murray TF. 2011a. Additivity of pyrethroid actions on sodium influx in cerebrocortical neurons in primary culture." Environ Health Perspect 119(9):1239-1246. Cao Z, Shafer TJ and Murry TF. 2011b. Mechanisms of pyrethroid insecticide-induced stimulation of calcium influx in neocortical neurons. J Pharmacol Exper Ther 336(1):197-205. Cheng T, Bronson DC, Cuirle EM, Robinson RA and Wu J. 1988. Metabolism of 14C-bifenthrin (FMC 54800) in rats. Unpublished report No. PC-0092 from Hazleton Laboratories, Inc., Madison, WI, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Choi J-S and Soderlund DM. 2006. Structure–activity relationships for the action of 11 pyrethroid insecticides on rat Nav1.8 sodium channels expressed in Xenopus oocytes. Toxicol Appl Pharmacol 21(3):233-244. Ciner FL, Plunkett Jr RW, Martin MF, Harris SA and Croley TR. 2010. LC/MS/MS determination of urinary concentrations of insecticides and herbicides in professional applicators. White paper developed by the Division of Consolidated Laboratory Services, State of Virginia. http://www.dgs.state.va.us/DivisionofConsolidatedLaboratoryServices/News/WhitePapers/tabid/524/Default.aspx, accessed March, 2011.

29 | P a g e
Coats JR. 1990. Mechanisms of toxic action and structure-activity relationships for organochlorine and synthetic pyrethroid insecticides. Environ Health Perspect 87:255-62. DeProspo JR, Bullock WL, Fletcher MJ, Freeman C, McCarty JD and Nadaskay N. 1983. Pilot teratology study in rabbits with FMC 54800 technical. Unpublished report No. A83-976 prepared by FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. DeProspo JR et al. 1986. Multigeneration reproduction study with FMC 54800 technical in rats. Unpublished report No. A83-977 prepared by FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported by JMPR 1992. El Naggar SF, Dow KD, Robinson RA and Stenzel JI. 1983. Excretion/tissue distribution of alcohol-14C FMC 54800 in rat. Unpublished report No. P-0775 from FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. El Naggar SF, Dow KD, Hogya JP, Newman JE and Robinson RA. 1986. Analysis of tissues and milk from goats administered 14C FMC 54800. Unpublished report No. P-1367 from FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation. El Naggar SF, Gavin DM, Gross EM, Hogya JP, Kooge C, Newman JE, Reynolds JL, Robinson RA and Wu J. 1986b. Metabolism of FMC 54800 in rats - identification of products in excreta. Unpublished report No. P-1439 from FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992 El Naggar SF, Barge MS, Schocken MJ and Tilka MA. 1991. Metabolism study: quantitative estimates of urinary, faecal and biliary excretion of alcohol (phenyl)-14C bifenthrin in the laboratory rat. Unpublished report No. P-2570 from FMC Corp.,Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. European Commission (EC). 2010. Directive 98/8/EC concerning the placing biocidal products on the market. Inclusion of active substances in Annex I or IA to Directive 98/8/EC. Assessment Report Bifenthrin. Fautz R, Jackson A and Völkner W. 1989. Unscheduled DNA synthesis in primary hepatocytes of male rats in vitro with bifenthrin. FMC report No. A90-3153. Unpublished report prepared by Cytotest Cell Research GMBH & Co. KG (CCR Project 175408) for FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Federal Register. 2003. Environmental Protection Agency, 40 CFR Part 180, [OPP–2002–0358; FRL–7304–4] Bifenthrin; Pesticide Tolerance. FR23056 68(83):23056, April 30, 2003. Freeman C, Bullock WL, DeProspo JR, Fletcher MJ, Malloy AV, McConnell RF and Nadaskay N. 1984a. Teratology study in rabbits with FMC 54800 technical. Unpublished report No. A83-1092 prepared by FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported by JMPR 1992.

30 | P a g e
Freeman C, Barta WD, Bullock WL, DeProspo JR, Fletcher MJ and Nadaskay N. 1984b. Teratology study in rats with FMC 54800 technical. Unpublished report No. A83-1091 prepared by FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Geiger LE, Ballester EJ, Barbera J and Malloy AV. 1986. Oncogenicity of FMC 54800: Lifetime feeding study in albino mice. Unpublished report No. A83-974 prepared by FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Go V, Garey J, Wolff MS, Pogo BGT and Golde TE. 1999. Estrogenic potential of certain pyrethroid compounds in the MCF-7 human breast carcinoma cell line. Environ Health Perspect 107(3):173-177. Golde TE. 2009. Review. The therapeutic importance of understanding mechanisms of neuronal cell death in neurodegenerative disease. Molec Neurodegen 4(16):1-17. Goto S, Asada S, Fushiwaki Y, Mori Y, Tanaka N, Umeda M, Nakajima D and Takeda K. 2004. Tumor-promoting activity and mutagenicity of 5 termiticide compounds. J UOEH 26(4):423-430. Gray AJ. 1985. Pyrethroid structure-toxicity relationships in mammals. Neurotoxicology 6:127–137. Grube A, Donaldson D, Kiely T and Wu L. US EPA. 2011. Pesticides industry sales and usage 2006 and 2007 market estimates. Biological and Economic Analysis Division, Office of Pesticide Programs. Halliwell WH. 1998. Submucosal mesenchymal tumors of the mouse urinary bladder. Toxicol Pathol 26(1):128-136. Harris SA and Wells KM. 2007. Dose prediction modeling for epidemiologic assessment of pesticide exposure risks in occupational cohorts. In Assessing Exposures and Reducing Risks to People from the Use of Pesticides, Chapter 13, pp. 187-200. American Chemical Society Symposium Series, Vol. 951. Washington DC: American Chemical Society. Hauser P, Zametkin AJ, Martinez P et al. 1993. Attention deficit-hyperactivity disorder in people with generalized resistance to thyroid hormone. N Engl J Med 328:997–1001. Hawkins DR, Elsom LF, Jackson R, Shillam KWG and Robinson RA. 1986. Bioaccumulation of 14C-FMC 54800 in the rat. Unpublished report No. PC-0045 from Huntingdon Research Centre, Huntingdon, England. Submitted to WHO by FMC Corporation. Haworth S R, Barta WD, Bullock WL, Burke JK, Karten NS, Kott S, Lawlor TE, Olevine SM and Plunkett RJ. 1983. Salmonella/mammalian-microsome plate incorporation mutagenicity assay (Ames Test): FMC 54800. FMC report No. A83-838. Unpublished report prepared by

31 | P a g e
Microbiological Associates for FMC Corp., Princeton, NJ., USA. Submitted to WHO by FMC Corporation by JMPR 1992. Heidemann A, Bonk C and Völkner W. 1989. Gene mutation assay in Chinese hamster ovary (CHO) cells in vitro with bifenthrin. FMC report No. A89-3099. Unpublished report prepared by Cytotest Cell Research GMBH & Co. KG (CCR Project 144022) for FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Heidemann A, Bonk C and Völkner W. 1989. Sister chromatid exchange assay in Chinese hamster ovary (CHO) cells in vitro with bifenthrin. FMC report No. A89-3016. Unpublished report prepared by Cytotest Cell Research GMBH & Co. KG (CCR Project 144011) for FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Hoffman N, Tran V, Daniyan A, Ojugbele O, Pryor SC, Bonventre JA, Flynn K and Weeks BS. 2006. Bifenthrin activates homotypic aggregation in human T-cell lines. Med Sci Monit 12(3):BR87-94. Holton JL, Nolan CC, Burr SA, Ray DE and Cavanagh JB. 1997. Increasing or decreasing nervous activity modulates the severity of the glio-vascular lesions of 1,3-dinitrobenzene in the rat: effects of the tremorgenic pyrethroid, bifenthrin, and of anaesthesia. Acta Neuropathol 93:159–165. Jacobson-Kram D. 2008. Commentary: Regulatory toxicology and the critical path: Predicting long-term outcomes from short-term studies. Veterin Pathol 45:707-709. Joint Meeting on Pesticide Residues (JMPR), International Programme on Chemical Safety. 1992. Joint meeting of the FAO Panel of Experts on Pesticide Residues in Food and the Environment and the WHO Expert Group on Pesticide Residues. Bifenthrin, Part II: Toxicology. http://www.inchem.org/documents/jmpr/jmpmono/v92pr04.htm, accessed January 2010. Judson RS, Houck KA, Kavlock RJ, Knudsen TB, Martin MT, Mortensen HM, Reif DM, Rotroff DM, Shah I, Richard AM and Dix DJ. 2010. In vitro screening of environmental chemicals for targeted testing prioritization: The ToxCast Project. Environ Health Perspect 118(4):485–492. doi:10.1289/ehp.0901392 Kennelly JC, Garner JV and Weiner M. 1986. Study to determine the ability of FMC 54800 to induce mutations to 6-thioguanine resistence in mouse lymphoma L5178Y cells using a fluctuation assay. FMC report No. A86-2059. Unpublished report prepared by Microtest Research Limited (Lab Project ID MFC 1/ML/KF17/M114) for FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Kennelly JC, Cuirle EM, Garner JV and Malloy AV. 1988. Study to determine the ability of FMC 54800 to induce mutation in four histidine-requiring strains of Salmonella typhimurium using liver S-9 from (a) male or (b) female Swiss Webster mice or (c) male Sprague Dawley Rats. FMC report No. A88-2651. Unpublished report prepared by Microtest Research Limited

32 | P a g e
(study number: MFC 1/S) for FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported by JMPR 1992. Kirby PE, Breidenthal G, Bullock WL, Carey EM, Johnson JL, Karten NS, Malloy AV and O'Keefe TR. 1983. L5178Y TK+/- Mouse lymphoma mutagenesis assay: FMC 54800 technical: FMC report No. A83-978. Unpublished report prepared by Microbiological Associates for FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Laskowski DA. 2002. Physical and chemical properties of pyrethroids. Rev Environ Contam Toxicol 174:49-170. Lawrence LJ and Casida JE. 1982. Pyrethroid toxicology: mouse intracerebral structure-toxicity relationships. Pestic Biochem Physiol 18:9–14. Liu H, Zhao M, Zhang C, Ma Y and Liu W. 2008. Enantioselective cytotoxicity of the insecticide bifenthrin on a human amnion epithelial (FL) cell line. Toxicology 253(1-3):89-96. Liu HG, Xu LH, Zhao MR, et al. 2009. Enantiomer-specific, bifenthrin-induced apoptosis mediated by MAPK signalling pathway in Hep G2 Cells. Toxicology 261(3):119-125. Liu J, Yang Y, Yang Y, Zhang Y and Liu W. 2010. Disrupting effects of bifenthrin on ovulatory gene expression and prostaglandin synthesis in rat ovarian granulosa cells. Toxicology 282(1-2):47-55. Losa S, Johnstone A and Shafer T. 2009. Relative potencies of type I and type II pyrethroids for inhibition of spontaneous firing in neuronal networks. Toxicologist 102:Abstract 2126. Lu C, Barr DB, Pearson M, Bartell S and Bravo R. 2006. A longitudinal approach to assessing urban and suburban children’s exposure to pyrethroid pesticides. Environ Health Perspect 114:1419–1423. Lu C, Barr DB, Pearson MA, Walker LA and Bravo R. 2009. The attribution of urban and suburban children’s exposure to synthetic pyrethroid insecticides: a longitudinal assessment. J Expo Sci Environ Epidemiol 19(1):69–78. Lu X, Hu F, Ma Y, Wang C, Zhang Y and Zhao M. 2010. The role of oxidative stress in enantiomer-specific, bifenthrin-induced cytotoxicity in PC12 cells. Environ Toxicol, Early View. McCarty JD, Ballester EJ, Barbera J, Barta W, Geiger LE and McCarty JD. 1986. Combined chronic oral toxicity and oncogenicity of FMC 54800: 2-year feeding study in albino rats. Unpublished report No. A83-952 prepared by FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. McClain R. 1992. Thyroid gland neoplasia: Non-genotoxic mechanisms. Toxicol Letters 64/65:397-408.

33 | P a g e
Nandi A, Chandi D, Lechesa R, Pryor S, McLaughlin A, Bonventre J, Flynn K and Weeks B. 2006. Bifenthrin causes neurite retraction in the absence of cell death: A model for pesticide associated neurodegeneration. Med Sci Monit 12(5):BR169-173. Pessah IN, Seegal RF, Lein PJ, LaSalle J, Yee BK, Van De Water J and Berman RF. 2008. Immunologic and neurodevelopmental susceptibilities of autism. NeuroToxicology 29(3):532–545. Pohl HR, Mumtaz MM, Scinicariello F, Hansen H. 2009. Binary weight-of-evidence evaluations of chemical interactions—15 years of experience. Reg Toxicol Pharmacol 54(3):264–271. Putman DL, Bullock WL, Malloy AV, McCarvill J and Putman DL. 1983a. Activity of FMC 54800 technical in the morphological transformation of BALB/3T3 mouse embryo cells in the absence of exogenous metabolic activation. Unpublished report No. A83-980 prepared by FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Radio NM and Mundy WR. 2008. Review. Developmental neurotoxicity testing in vitro: Models for assessing chemical effects on neurite outgrowth. NeuroToxicology 29:361–376. Ray DE and Fry JR. 2006. A reassessment of the neurotoxicity of pyrethroid insecticides. Pharmacol Ther 111(1):174-93. Rotroff DM, Wetmore BA, Dix DJ, Ferguson SS, Clewell HJ, Houck KA, LeCluyse EL, Andersen ME, Judson RS, Smith CM, Sochaski MA, Kavlock RJ, Boellmann F, Martin MT, Reif DM, Wambaugh JF and Thomas RS. 2010. Incorporating human dosimetry and exposure into high-throughput in vitro toxicity screening. Toxicol Sci 117(2):348–358. Sadowska-Woda I, Popowicz and Karowicz-Bilińska A. 2010. Bifenthrin-induced oxidative stress in human erythrocytes in vitro and protective effect of selected flavonols. Toxicol In Vitro 24(2):460-464. Scollon EJ, Starr JM, Godin SJ, DeVito MJ and Hughes MF. 2009. In vitro metabolism of pyrethroid pesticides by rat and human hepatic microsomes and cytochrome p450 isoforms. Drug Metab Dispos 37(1):221-8. Selim S, El Naggar SF, Isbell D, Mazur P and Patterson R. 1986a. Absorption, distribution and excretion studies of FMC 54800 in the rat. Unpublished report No. PC-0047 from Biological Test Center, Irvine, CA, USA. Submitted to WHO by FMC Corporation as reported by JMPR 1992. Selim S, Chun CL, El Naggar SF, Isbell D, Mazur P and Patterson R. 1986b. The kinetics of FMC 54800 in the blood of rats following a single oral dose. Unpublished report No. PC-0048 from Biological Test Center, Irvine, CA, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992.

34 | P a g e
Sonich-Mullin C, Fielder R, Wiltse J, Baetcke K, Dempsey J, Fenner-Crisp P, Grant D, Hartley M, Knaap A, Kroese D. 2001. IPCS conceptual framework for evaluating a MOA for chemical carcinogenesis. Regul. Toxicol. Pharmacol. 34:146–152. Shafer TJ and Meyer DA. 2004. Effects of pyrethroids on voltage-sensitive calcium channels: a critical evaluation of strengths, weaknesses, data needs, and relationship to assessment of cumulative neurotoxicity. Toxicol Appl Pharmacol 196(2):303-18. Shafer TJ, Meyer DA and Crofton KM. 2005. Developmental neurotoxicity of pyrethroid insecticides: critical review and future research needs. Environ Health Perspect 113(2):123-36. Skandrani D, Gaubin Y, Vincen C, Beau B, Murat JC, Soleilhavoup J-P and Croute F. 2006. Relationship between toxicity of selected insecticides and expression of stress proteins (HSP, GRP) in cultured human cells: Effects of commercial formulations versus pure active molecules. Biochimica et Biophysica Acta (BBA) 1760(1):95-103. Smith PA, Thompson MJ and Edwards JW. 2002. Estimating occupational exposure to the pyrethroid termiticide bifenthrin by measuring metabolites in urine. J Chromatogr B Analyt Technol Biomed Life Sci 778(1-2):113-20. Soderlund DM, Clark JM, Sheets LP, Mullin LS, Piccirillo VJ, Sargent D, Stevens JT and Weiner ML. 2002. Mechanisms of pyrethroid neurotoxicity: implications for cumulative risk assessment. Toxicol 171(1):3-59. Srivastava HC, Kumar GP, Hassan A, Dabhi M, Pant CS and Yadav RS. 2005. Evaluation of possible health effects of pyrethroid insecticides, bifenthrin 10% WP, and deltamethrin 25% WG, on spraymen exposed in a field trial in India. Bull Environ Contam Toxicol 75(3):413-20. Stein MA and Weiss RE. 2003. Thyroid function tests and neurocognitive functioning in children referred for attention deficit/hyperactivity disorder. Psychoneuroendocrinology 28:304–316. Thilagar A, Brauninger RM, Bullock WL, Karten PV, Kott S, Malloy AV and Pant KJ. 1983a. Unscheduled DNA synthesis in rat primary hepatocytes: FMC 54800 technical. FMC Report No. A83-985. Unpublished report prepared by Microbiological Associates for FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Thilagar A, Brauninger RM, Bullock WL, Karten PV and Malloy AV. 1983b. Unscheduled DNA synthesis in rat primary hepatocytes: FMC 54800 technical. FMC Report No. A83-1043. Unpublished report prepared by Microbiological Associates for FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported by JMPR 1992. Thilagar A, Bullock WL, Karten NS, Kott S and Malloy AV. 1984a. CHO/HGPRT Mutation assay in the presence and absence of exogenous metabolic activation: FMC 54800 technical. FMC Report No. A83-1144. Unpublished report prepared by Microbiological Associates for

35 | P a g e
FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported by JMPR 1992. Thilagar A, Bullock WL, Karten NS, Kott S, Kumaroo PV and Malloy AV. 1984b. Chromosome aberrations in Chinese hamster ovary (CHO) cells: FMC 54800 technical. FMC Report No. A83-1105. Unpublished report prepared by Microbiological Associates for FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported by JMPR 1992. Tran V, Hoffman N, Mofunanaya A, Pryor SC, Ojugbele O, McLaughlin A, et al. 2006. Bifenthrin inhibits neurite outgrowth in differentiating PC12 cells. Med Sci Monitor 12:BR57–62. Tullman RH and Robinson RA. 1986. Analysis of FMC 54800 residues in plasma from rats dosed orally with 14C FMC 54800. Unpublished report No. P-1448 from FMC Corp., Princeton, NJ, USA. Submitted to WHO by FMC Corporation. Tulve NS, Egeghy PP, Fortmann RC, Whitaker DA, Nishioka MG, Naeher LP and Hilliard A. 2007. Multimedia measurements and activity patterns in an observational pilot study of nine young children. Jnl Exp Sci Environ Epidem 18:31-44. US Environmental Protection Agency (US EPA). 1988. Whiting, R, Health Effects Division. Biphenthrin Reference Dose for Chronic Oral Exposure (RfD) Acc. 264637. July 20, 1988. Available from http://www.epa.gov/pesticides/chemical/foia/cleared-reviews/reviews/128825/128825.htm, accessed November, 2009. US Environmental Protection Agency (US EPA). 1992. Third carcinogenicity peer review of bifenthrin. Memorandum within the Office of Pesticide Programs. Available from http://www.epa.gov/pesticides/chemical/foia/cleared-reviews/reviews/128825/128825.htm, accessed November, 2009. US Environmental Protection Agency (US EPA). 1997. Risk assessment for extension of tolerances for synthetic pyrethroids. Office of Prevention, Pesticides and Toxic Substances. US Environmental Protection Agency (US EPA). 2003. Bifenthrin, pesticide tolerance. Federal Register 68(83):28056-23068. US Environmental Protection Agency (US EPA). 2007. Bifenthrin: Human-health risk assessment for proposed uses on mayhaw, root vegetables, (except sugar beets, crop subgroup 1B), peanut, pistachio, soybean, and fruiting vegetables (Crop Group 8). PC 128825. US Environmental Protection Agency (US EPA). 2011. Pyrethroid cumulative risk assessment. Available from http://epa.gov/pesticides/cumulative/common_mech_groups.htm; accessed December 2011. Vijverberg HPM and van den Bercken J. 1990. Neurotoxicological effects and the mode of action of pyrethroid insecticides. Crit Rev Tox 21(2):105-126.

36 | P a g e
Wang C, Chen F, Zhang Q, et al. 2009. Chronic toxicity and cytotoxicity of synthetic pyrethroid insecticide cis-bifenthrin. Jnl Env Sci – China 21(12):1710-1715. Wang L, Liu W, Yang C, Pan Z, Gan J, Xu C, Zhao M and Schlenk D. 2007. Enantioselectivity in estrogenic potential and uptake of bifenthrin. Environ Sci Technol 41(17):6124-6128. Weeks BS and Perez PP. 2006. PolicosanolPlus and Neuroprevin ameliorate pesticide-mediated inhibition of neurite outgrowth and neurite degeneration. Med Sci Monit 12(12):BR379-384. Wetmore BA, Wambaugh JF, Ferguson SS, Sochaski MA, Rotroff DM, Freeman K, Clewell III HJ, Dix DJ, Andersen ME, Houck KA, Allen B, Judson RS, Singh R, Kavlock RJ, Richard AM and Thomas RS. 2012. Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol Sci 125(1):157–174. Wilks MF. 2000. Pyrethroid-induced paresthesia—a central or local toxic effect? J Toxicol Clin Toxicol 38:103– 105. Wolansky MJ, Gennings C, DeVito MJ and Crofton KM. 2009. Evidence for dose-additive effects of pyrethroids on motor activity in rats. Environ Health Perspect 117(10):1563-70. Wolansky MJ, Harrill JA. 2008. Neurobehavioral toxicology of pyrethroid insecticides in adult animals: a critical review. Neurotoxicol Teratol 30(2):55-78. Wolansky MJ, McDaniel KL, Moser VC and Crofton KM. 2007. Influence of dosing volume on the neurotoxicity of bifenthrin. Neurotoxicol Teratol 29(3):377-384. Wu J, Barge M, Cuirle EM and Robinson RA. 1988. Metabolism of 14C-bifenthrin (FMC 54800) in rats - analyis and quantitation of metabolites in excreta. Unpublished report No. PC-0093 from Xenobiotic Laboratories, Inc., Princeton, NJ, USA. Submitted to WHO by FMC Corporation as reported in JMPR 1992. Yang D, Wang X, Chen YT, Deng R and Yan B. 2009. Pyrethroid insecticides: isoform-dependent hydrolysis, induction of cytochrome P450 3A4 and evidence on the involvement of the pregnane X receptor. Toxicol Appl Pharmacol 237(1):49-58. Zhao M, Chen F, Wang C, Zhang Q, Gan J, Liu W. 2010. Integrative assessment of enantioselectivity in endocrine disruption and immunotoxicity of synthetic pyrethroids. Environ Pollut 2010 Jan 22. [Epub ahead of print]