enhancement of uvb radiation–mediated apoptosis by...
TRANSCRIPT

Enhancement of UVB radiation–mediated apoptosisby sanguinarine in HaCaT humanimmortalized keratinocytes
Shannon Reagan-Shaw,1 Jorien Breur,1
and Nihal Ahmad1,2,3
1Department of Dermatology, 2University of WisconsinComprehensive Cancer Center, and 3Molecular and EnvironmentalToxicology Center, University of Wisconsin, Madison, Wisconsin
AbstractIn this article, we studied the chemopreventive effects ofsanguinarine on UVB-mediated responses in human HaCaTimmortalized keratinocytes. For our studies, HaCaT cellswere treated with a low dose (50 nmol/L) of sanguinarinefor 24 hours followed by irradiation with UVB (15 or 30mJ/cm2). Our data showed that UVB exposure, at both doses,resulted in decreased cell viability and increased apoptosis.Interestingly, pretreatment of the cells with sanguinarinecaused a significant enhancement in the antiproliferativeresponse of UVB. These responses on UVB and/or sangui-narine treatments were associated with (a) decrease in Bcl-2 and Bcl-XL and (b) increase in Bax, Bid, and Bak proteinlevels. Bax knockdown and Bcl-2 overexpression resultedin a rescue of HaCaT cells from sanguinarine-mediatedapoptosis. DNA cell cycle analysis revealed that UVBtreatment resulted in an accumulation of cells in the G2-Mphase of the cell cycle, whereas pretreatment of sangui-narine resulted in a significant shift of cells in the S phase ata low UVB dose and a further accumulation of cells in theG2-M phase at a higher UVB dose. These effects on cellcycle were accompanied with modulations in the proteinlevels of cyclin (B1, E, and A) and cdc2 and cyclin-dependent kinase 1. Furthermore, sanguinarine treatmentwas found to result in significant modulations in p53,p66Shc, MsrA, and superoxide dismutase levels. Based onour data, we suggest the sanguinarine may protect skincells from UVB-mediated damages via apoptotic elimina-tion of damaged cells that escape programmed cell deathand therefore possess a potential of clonal expansion.[Mol Cancer Ther 2006;5(2):418–29]
IntroductionMore than one million new cases of nonmelanoma skincancers are expected to be newly diagnosed in 2005 inthe United States (1). According to the ‘‘World CancerReport,’’ skin cancer constitutes f30% of all newlydiagnosed cancers in the world (2), and solar UVradiation, particularly its UVB component, is an estab-lished cause of about 90% of skin cancers (3). UVBradiation causes tumor initiation by DNA damage, andits promoting activity includes transcriptional modulationof genes involved in tumor promotion as well asactivation of several signal transduction pathways (4, 5).UVB also indirectly damages DNA by increasing levelsof reactive oxygen species (ROS), which facilitate DNAoxidation (6). Whereas low doses of UVB cause DNAmutation leading to tumor initiation, high dosesresult in irreparable DNA damage causing apoptosis(sunburn) and eventually cell deletion (4, 5). Theavailable options have proven to be inadequate for themanagement of UV damages, including skin cancers.Therefore, there is an urgent need to develop mecha-nism-based novel approaches for the prevention ortreatment of skin cancer. Chemoprevention by naturallyoccurring plant-based agents is being investigated as apotential approach for prevention as well as treatment ofearly treatment of several cancers, including skin cancer(7–12).The solar UV radiation inflict damages to skin cells that
results in the formation of ‘‘initiated’’ cells. The initiated cellsmay ultimately grow into tumors. The initiated cellsgenerally divide much faster (hyperproliferation) thannormal cells and, via the processes of clonal expansion andapoptosis evasion, are transformed into cancerous cells.Therefore, two types of chemopreventive agents could beuseful for the management of skin cancer. First, the agentsthat could inhibit the damages caused by UV may preventthe formation of initiated cells. Second, the agents that couldeliminate the initiated cells (with an ability to becomecancerous) may reduce the risk of cancer development.In this study, we evaluated the chemoprotective
properties of sanguinarine against UVB exposure–medi-ated damages in skin cells. Sanguinarine (13-methyl[1,3]-benzodioxolo[5,6-c]-1,3-dioxolo[4,5-i ]phenanthridinium;Fig. 1A) is derived from the root of Sanguinaria canadensisand is also found in poppy and Fumaria species (13).Sanguinarine has been shown to have antioxidantproperties (14) and to exhibit antimicrobial (15) andanti-inflammatory activities (16). It is used in dentalproducts, such as toothpaste and mouthwashes, to reducegingival inflammation and supragingival plaque forma-tion (17–19). We have shown previously that sanguinar-
Received 7/18/05; revised 11/3/05; accepted 12/8/05.
Grant support: NIH grants CA099076 and CA098368 and Department ofDefense grant W81XWH-04-1-0220.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.
Requests for reprints: Nihal Ahmad, Department of Dermatology,University of Wisconsin, 25B Medical Science Center, 1300 UniversityAvenue, Madison, WI 53706. Phone: 608-263-5359;Fax: 608-263-5223. E-mail: [email protected]
Copyright C 2006 American Association for Cancer Research.
doi:10.1158/1535-7163.MCT-05-0250
418
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

ine induces apoptosis in human epidermoid carcinomacells but not in normal human epidermal keratinocytes atsimilar concentrations (20). We also showed that sangui-narine treatment to immortalized human HaCaT kerati-nocytes resulted in an induction of apoptosis viaactivation of Bcl-2 family proteins (21).In the present study, to assess the photochemopreventive
potential of sanguinarine, we evaluated its effects on UVB-mediated damages inHaCaT keratinocytes in vitro . Our datasuggested that sanguinarine may protect skin cells fromUVB-mediated damages via apoptotic elimination of UV-damaged cells. Our findings may have implications for themanagement of skin cancer and other hyperproliferativeskin conditions.
Materials andMethodsMaterialsSanguinarine chloride (>99.0% pure) was purchased
from Sigma Chemical Co. (St. Louis, MO). The followingantibodies were used: anti-cyclin B1 and anti–cyclin-dependent kinase 2 (cdk2) from Santa Cruz Biotechnol-ogy (Santa Cruz, CA); anti-cdc2, anti-cyclin D1, anti-cyclin D2, anti-cyclin E2, anti–phospho-p53 (Ser15), andanti–phospho-Mdm2 (Ser166) from Cell Signaling Tech-nology (Beverly, MA); anti-cdk4 from BD PharMingen(San Jose, CA); anti-cyclin D3 and anti-Mdm2 fromAbcam (Cambridge, MA); anti-cyclin A and anti-cyclinE1 from NeoMarkers (Fremont, CA); anti-Bcl-2, anti-Bax,anti-Bak, anti-Bid, anti–p66-SHC, anti–phospho p66-SHC(Tyr239), anti-Cu/Zn superoxide dismutase (SOD), anti-
MnSOD, and anti-MsrA from Upstate Biotechnology(Waltham, MA); anti-Bcl-X, anti–p21-WAF1, and anti-p53 from Biosource International (Camarillo, CA); andanti-h-actin from Sigma Chemical Co. (St. Louis, MO).The Bax short hairpin RNA and the full-length Bcl-2plasmids were purchased from Open Biosystems (Hunts-ville, AL).
Cell Culture and TreatmentHaCaT cells, an immortalized, nontumorigenic human
keratinocyte cell line, were maintained in DMEM(Invitrogen, Carlsbad, CA) supplemented with 10% fetalbovine serum and 1% antibiotics at standard cell cultureconditions (37jC, 5% CO2 in a humidified incubator). Forour studies, we employed two different experimentalprotocols. In the first protocol, the cells were pretreatedwith sanguinarine followed by UVB exposure; whereas inthe second protocol, the cells were subjected to UVBirradiation first followed by sanguinarine treatment. Forpretreatment studies, the cells (50–60% confluent) weretreated with a low concentration of sanguinarine (50nmol/L) for 24 hours in culture medium. The mediumwas then removed, and PBS was added, and the cellswere exposed to UVB (15 or 30 mJ/cm2). The PBSwas aspirated, culture medium was added, and the cellswere incubated for another 24 hours. Alternatively, forpost-treatment studies, the cells (70% confluent) weretreated with UVB (15 or 30 mJ/cm2) in PBS followedimmediately by 50 nmol/L sanguinarine treatment for 24hours.For UVB irradiation, a ‘‘Daavlin Research Irradiators’’
obtained from Daavlin Co. (Bryan, OH) was used. Thisequipment consists of a fixture mounted on fixed legs
Figure 1. Sanguinarine treatmentsignificantly enhances UVB-mediatedinhibition of cell growth. A, the chem-ical structure of sanguinarine. B, Fol-l ow i n g t r e a tm e n t o f H aC a Tkeratinocytes with sanguinarine (Sang )and/or UVB, the effect on cell viabilitywas measured using trypan blue exclu-sion analysis. Cell viability data isexpressed as the percent viable cellsout of the total number of cells.Columns, mean of three experiments;bars, SD. *, P < 0.01 for untreatedversus treated; ##, P < 0.01 for UVBversus UVB + sanguinarine. C, clonalkeratinocyte growth was assessed us-ing the clonogenic assay. Details of theexperiments are given in Materials andMethods.
Molecular Cancer Therapeutics 419
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

and contains four UVA and four UVB lamps. The exposuresystem is controlled using two Daavlin Flex ControlIntegrating Dosimeters. The dose units, in this equipment,could be entered as mJ/cm2 (for UVB) or Joules (for UVA).For accuracy, the machine is periodically calibrated usingInternational Light IL 1400, digital light meter (Daavlin).
Trypan Blue Exclusion AssayTrypan blue exclusion assay was used to assess the effect
of treatments on the growth and viability of HaCaT cells.Briefly, following treatment of cells with sanguinarine and/or UVB in a six-well plate, as described above, the culturemedium was collected in a 1.5-mL Eppendorf tube. Thecells were trypsinized and collected in the same Eppendorftube. The cells were pelleted by centrifugation, and the cellpellet was resuspended in 300 AL PBS (10 mmol/L, pH 7.4).Trypan blue (0.4% in PBS, 10 AL) was added to a smalleraliquot (10 AL) of cell suspension, and the number of cells(viable unstained and nonviable blue) were counted usinga hemacytometer in duplicate for each sample, with theexperiment repeated at least thrice.
Clonogenic AssayThe reproductive potential of treated HaCaT cells was
assessed using the clonogenic assay, which is consideredto be the optimal assay method for determining survivalafter radiation in vitro . The colony formation assay fornormal keratinocytes treated with UVB has been de-scribed previously (22). Cells were treated with 50 nmol/Lsanguinarine for 24 hours and were collected by trypsi-nization. A trypan blue assay was done, and cells werereplated in triplicate on a six-well tissue culture plate with3,000 cells per well. The cells were allowed to adhereovernight and were then exposed to UVB as describedearlier. The cells were cultured for 14 days with growthmedia being replaced every 3 days. The cells were thenstained with 0.5% crystal violet (in methanol/H2O, 1:1;Sigma), and the colonies were counted.
Apoptosis and Cell Cycle Analysis by FlowCytometryThe extent of apoptosis and cell cycle distribution was
assessed with the APO-BrdUrd TUNEL Apoptosis Assaykit (Molecular Probes, Eugene, OR) as per the manufac-turer’s protocol. Cells were treated as described above in asix-well plate. At 24 hours after UVB exposure, the culturemedium was collected. The cells were gently trypsinizedand added to the culture media and pelleted bycentrifugation. The pellet was washed with PBS, the cellswere counted and (1 � 106) fixed overnight in ethanol(90%). The cells were washed and labeled with UTP-bromodeoxyuridine (BrdUrd) overnight, washed againwith PBS, and incubated with an Alexa 488 anti-BrdUrdantibody followed by counterstaining with propidiumiodide. Cells were analyzed using a FACScan benchtopcytometer (BD Biosciences, San Jose, CA) at the FlowCytometry Facility in the University of Wisconsin Com-prehensive Cancer Center. The analyses were done usingCell Quest software (BD Biosciences) for apoptosis andModFit LT software (Verity Software House, Topsham,ME) for cell cycle analysis, and the data are expressed asthe mean of three experiments showing the same trend.
Western Blot AnalysisFollowing the treatment of the cells with sanguinarine and
UVB in 10-cm dishes, as described above, the medium wasaspirated, and the cells were washed with ice-cold PBS. Ice-cold radioimmunoprecipitation assay buffer [150 mmol/LNaCl, 50 mmol/L Tris-HCl (pH 7.4), 1 mmol/L EDTA, 1%NP40] with freshly added 1 mmol/L phenylmethylsulfonylfluoride and 10 Ag/mL protease inhibitors (ProteaseInhibitor Cocktail Set III, Pierce, Rockford, IL) was addedto the plates. The cells were then scraped, and the cellsuspension was transferred into a microfuge tube on icefor 15 minutes with occasional vortexing, ensuring acomplete cell lysis. The cell suspension was cleared bycentrifugation at 14,000 � g for 15 minutes at 4jC, and thesupernatant (total cell lysate) was either used immediatelyor stored at �70jC. The protein concentration wasdetermined using the Bicinchoninic Acid Protein Assay(Bio-Rad Laboratories, Hercules, CA) as per the manufac-turer’s protocol.For immunoblot analysis, 30 Ag protein was subjected to
SDS-PAGE using 10% to 15% Tris-HCl gel. The protein wastransferred onto a nitrocellulose membrane and blockedwith TBS-Tween (0.1%) plus 5% dry milk. The membranewas probed with an appropriate primary antibody fol-lowed by a secondary horseradish peroxidase–conjugatedantibody. The protein levels were detected by freshlyprepared chemiluminescent solution [100 mmol/L Tris-HCl (pH 8.5), 0.018% H2O2 (v/v), 1.25 mmol/L Luminol,225 nmol/L coumaric acid]. The quantification of proteinwas done by a digital analyses of protein bands (TIFFimages) using UN-SCAN-IT software (Silk Scientific,Orem, UT). The data are expressed as the relative densityof the protein normalized to h-actin.BivariateAnalysis by Flow CytometryFollowing the treatment of the cells with sanguinarine
and UVB in six-well plates, as described above, the culturemedium was collected. The cells were gently trypsinizedand added to the culture media and pelleted by centrifu-gation. The pellet was washed with PBS and counted, andthe cells (1 � 106) were fixed overnight in ethanol (90%).Cells were washed with PBS and pelleted by centrifugation.The pellet was resuspended in 0.25% (v/v) Triton X-100 inPBS on ice and incubated for 5 minutes before washing withPBS and centrifugation. The cells were probed withantibodies diluted in 1% bovine serum albumin (in PBS)for 1 hour at room temperature. The cells were washed with1% bovine serum albumin (in PBS) and centrifuged. The cellpellet was probed with a FITC-conjugated goat anti-mouseIgG antibody for 30 minutes. The cells were diluted in PBSand were analyzed using a FACScan benchtop cytometer(BD Biosciences) at the Flow Cytometry Facility in theUniversity of Wisconsin Comprehensive Cancer Center.The analyses, repeated at least thrice, were done using CellQuest software (BD Biosciences).
Transfection of CellsThe Bax short hairpin RNA and full-length Bcl-2
plasmids were grown up in Luria-Bertani broth andwere purified by a Perfectprep Plasmid Mini kit
UVB Radiation–Mediated Apoptosis by Sanguinarine420
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

(Eppendorf, Hamburg, Germany) according to the man-ufacturer’s instructions. Following pretreatment of thecells with sanguinarine for 24 hours then UVB treatment,the cells were transfected with either of the DNAplasmids. Briefly, each plasmid (0.2 Ag per 25 AL media)and the LipofectAMINE 2000 reagent (Invitrogen; 0.5 ALper 25 AL media) were diluted with serum-free media for5 minutes. Then, the diluted LipofectAMINE was addedto the diluted plasmid and incubated at room tempera-ture for 20 minutes. The PBS was aspirated from the cellsimmediately following UVB treatment, and the plasmid-LipofectAMINE complex was added dropwise to thecells. The cells were incubated at 37jC for 6 hours, atwhich time fetal bovine serum was added to a 10%concentration, and the cells were incubated further foranother 18 hours (for a total incubation of 24 hours afterUVB treatment). At this time, the cells were collected andprocessed for further experiments.
SODActivitySOD activity was assessed employing Bioxytech SOD-
525 kit (Oxis Research, Portland, OR) as per themanufacturer’s protocol. Briefly, following the treatmentof the cells with sanguinarine and UVB in six-well plates,as described above, the culture medium was collected.The cells were gently trypsinized and added to the culturemedia and pelleted by centrifugation. The cells werewashed with PBS and repelleted by centrifugation. Thecells were resuspended in ice-cold PBS and weresonicated for 30 seconds. The cell suspension was clearedby centrifugation at 14,000 � g for 20 minutes at 4jC, andthe supernatant was stored at �70jC. The buffer providedin the kit was warmed, and 40 AL of sample and thesolution R2 were added to a microfuge tube and vortexedbriefly. The sample was incubated at 37jC for 1 minute,and another solution R1 was added and vortexed briefly.The sample was transferred to a spectrophotometriccuvette, and the absorbance was measured over time.The SOD activity is calculated directly from the rate ofabsorbance of the sample versus the average rate of theblank control using the provided ratio table, repeated atleast thrice for each sample.
Statistical AnalysisThe results are expressed as the mean F SD. Statistical
analysis of the data between the untreated versus treatments(*) and UVB-alone treatment versus sanguinarine and UVBtreatment (##) were done by Student’s t test. P < 0.01 wasconsidered statistically significant.
ResultsSanguinarine Enhances UVB-Mediated Inhibition of
Viability and Clonogenic Survival of HaCaT Keratino-cytesHuman HaCaT keratinocytes were either exposed to
sanguinarine alone, UVB alone, or pretreated with sangui-narine followed by UVB exposure. Trypan blue exclusionanalysis was used to assess the effect of treatments on theviability of cells. Our data showed that UVB exposure
(15 or 30 mJ/cm2) resulted in a significant decrease in theviability of HaCaT cells (Fig. 1B).When cells were pretreatedwith 50 nmol/L sanguinarine, this loss of cell viability wassignificantly enhanced at both doses of UVB (Fig. 1B).Sanguinarine alone had no effect on cell viability (Fig. 1B).Interestingly, the after treatment of sanguinarine (followingUVB exposure) did not alter the viability of the cellscompared with UVB-alone treatments, although it wassignificantly different from untreated cells (Fig. 1B).Next, we determined the effects of treatments on the
ability of cells to form cellular colonies and thereby assessthe reproductive potential and the long-term cell survivalafter treatments. As shown by the colony formation assay,sanguinarine treatment alone did not have an effect on theclonogenic survival of the cells, whereas UVB exposureresulted in an inhibitory effect on the colony formationability of HaCaT cells (Fig. 1C). Interestingly, bothsanguinarine pretreatment and post-treatment enhancedthe antiproliferative effects of UVB with a completeinhibition observed with sanguinarine pretreatment at the30 mJ/cm2 dose of UVB (Fig. 1C).
Sanguinarine Enhances UVB-Mediated Induction ofApoptosis via Modulations in Bcl-2 Family Proteins inHaCaTKeratinocytesWe determined whether the loss of cell viability observed
above was mediated via the apoptotic death of HaCaT cells.For this purpose, we employed a terminal deoxynucleo-tidyl transferase–mediated nick-end labeling (TUNEL)assay kit that used BrdUrd incorporation to measureapoptosis by flow cytometry. This assay is based on theprinciple that when DNA strands are cleaved (apoptosis), alarge number of 3V-hydroxyl ends are exposed, which aredetected using an Alexa Fluor 488 dye-labeled anti-BrdUrdmonoclonal antibody. This kit also uses propidium iodidestaining for determining total cellular DNA content. Wefound a significant induction of apoptosis in HaCaT cellstreated with UVB alone for 24 hours (Fig. 2). Similar to thecell viability data, sanguinarine posttreatment did notsignificantly change the number of apoptotic cells com-pared with UVB alone (Fig. 2B). Interestingly, pretreatmentof cells with sanguinarine appreciably increased theapoptotic cell population from 50% (UVB alone) to 67% atthe lower dose of UVB (15 mJ/cm2; Fig. 2). However, at a 30mJ/cm2 UVB dose, apoptosis was significantly increasedfrom 66% (UVB alone) to 89% (sanguinarine pretreatmentplus UVB; Fig. 2). At this point, sanguinarine pretreatmentlooked more promising and was used for further studies.UVB radiation has been shown to activate prodeath Bcl-2
family proteins while also reducing levels of prosurvivalBcl-2 family proteins, thereby inducing apoptosis in theskin (23). Studies by others and from our laboratory haveshown that sanguinarine modulates Bcl-2 family proteinsin favor of apoptosis (21, 24, 25). Immunoblot analysisshowed a significant increase in proapoptotic proteins Bax,Bid, and Bak with a concurrent significant decrease inantiapoptotic proteins Bcl-2 and Bcl-XL when the cells weretreated with UVB (Fig. 3A and B). The pretreatment ofsanguinarine was found to further enhance the modulatory
Molecular Cancer Therapeutics 421
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

effects of UVB towards Bcl-2 family proteins in a way thatfavored apoptosis (Fig. 3A and B). Bcl-2 family proteins canmodulate mitochondrial permeability through oxidativephosphorylation during apoptosis. Changes in the Bax/Bcl-2
ratio suggest a corresponding change in mitochondrialpermeability to release apoptogenic molecules from themitochondria to the cytosol. A bivariate analysis by flowcytometry allows reading of multiple protein levels simul-taneously in the same population of cells. This analysisshowed a significant increase in proapoptotic protein Baxand a significant decrease in antiapoptotic Bcl-2, therebyshifting the Bax/Bcl-2 ratio in favor of apoptosis (Fig. 3C).This shift in the Bax/Bcl-2 ratio toward apoptosis was alsoseen in the Western Blots, further confirming the UVB-caused apoptosis by sanguinarine (Fig. 3).To determine the cause and effect association between
Bax/Bcl-2 and the observed chemopreventive response ofsanguinarine, we did gene overexpression and knock-down experiments. Thus, first, we studied the effect ofknockdown of Bax using vector-based short hairpin RNAand studied its effect on the observed responses ofsanguinarine. Immunoblot analysis showed an efficientknockdown of Bax protein levels in the cells (Fig. 3D).TUNEL analysis confirmed that proapoptotic Bax was acrucial player in sanguinarine pretreatment–mediatedapoptosis as cells were found to be rescued fromapoptosis on transfection with Bax short hairpin RNA(Fig. 3D).In the next experiment, we evaluated the effect of a forced
overexpression of prosurvival Bcl-2 on the observedproapoptotic response of sanguinarine. As shown byWestern Blot analysis (Fig. 3E), the transfection of cells withBcl-2 resulted in an appreciable increase of Bcl-2 proteinlevels. Furthermore, our data also showed that Bcl-2 over-expression resulted in a rescue of HaCaT keratinocytes fromsanguinarine-mediated apoptosis (Fig. 3E). These resultsclearly showed that there is a cause and effect relationshipbetween Bax/Bcl-2 and the induction of apoptosis bysanguinarine.
Sanguinarine Enhances UVB-Mediated Cell CycleArrest via Modulations in Cell Cycle RegulatoryMolecules in HaCaT KeratinocytesThe DNA damage response culminates in activation of
cell cycle checkpoints and the appropriate DNA repairpathways or, in certain contexts, initiation of apoptoticprograms. The basic purpose of cell cycle regulation is toensure that DNA is faithfully replicated only once duringS phase, and those identical copies of chromosomes areformed and distributed equally to the daughter cellsduring M phase. Lack of fidelity in DNA replication andmaintenance can result in deleterious mutations, leadingto cell death or, in multicellular organisms, cancer. Asingle, high UVB exposure (200 mJ/cm2) to HaCaTkeratinocytes is known to induce G2-M phase cell cyclearrest (26). In this study, the DNA cell cycle analysisrevealed that UVB treatment resulted in a significantaccumulation of cells in the G2-M phase of the cell cycle at24 hours after exposure (Fig. 4). Treatment of sanguinarine(before UVB) resulted in a significant shift of cellaccumulation in the S phase at a 15 mJ/cm2 UVB doseand a further accumulation of cells in G2-M phase at a30 mJ/cm2 UVB dose (Fig. 4).
Figure 2. Sanguinarine treatment significantly enhances UVB-mediatedinduction of apoptosis. Following treatment of HaCaT keratinocytes withsanguinarine (Sang ) and/or UVB, the extent of apoptosis was assessedwith the APO-BrdUrd TUNEL Assay kit. The fragmentation of DNA inapoptotic cells is measured by BrdUrd incorporation, which is visualized byconjugation to an Alexa Fluor 488 dye-labeled anti-BrdUrd antibody. BrdUrdincorporation was analyzed with a flow cytometer (A) followed by acomputational analysis (B) of cells staining positive for BrdUrd. Columns,mean of three experiments; bars, SD. *, P < 0.01 for untreated versustreated; #, P < 0.01 for UVB versus UVB + sanguinarine. A, fromrepresentative experiment repeated thricewith similar results. Details of theexperiments are given in Materials and Methods.
UVB Radiation–Mediated Apoptosis by Sanguinarine422
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

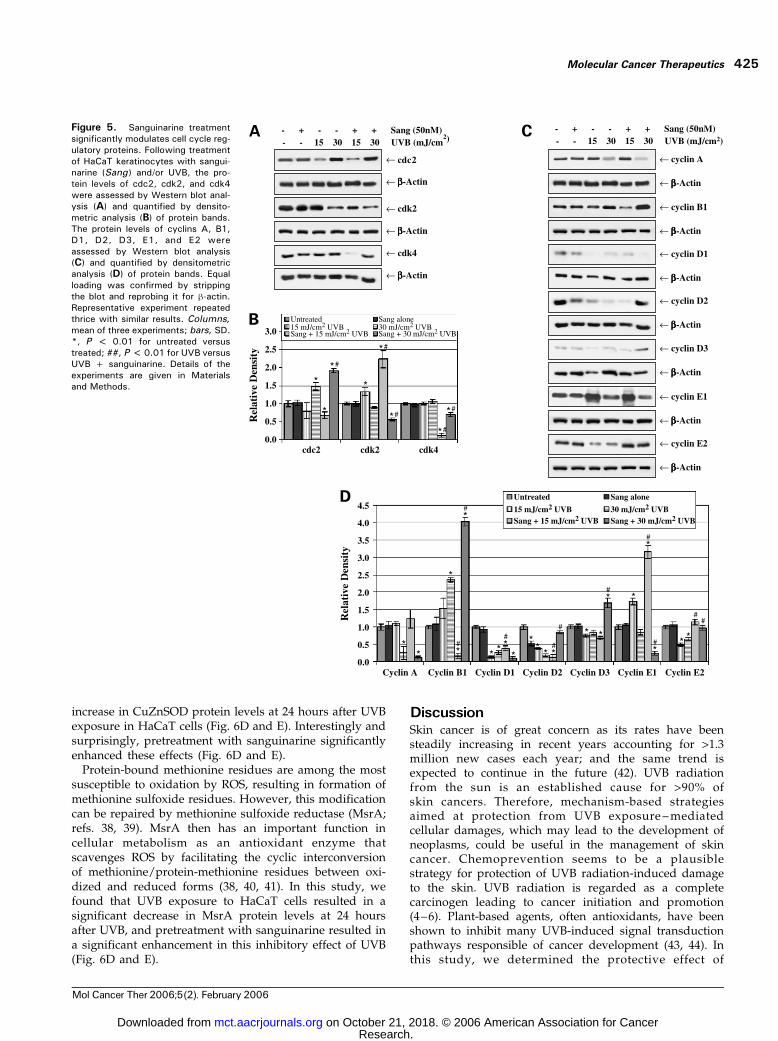
Progression of normal cell division depends on cyclininteraction with cdks and the degradation of cyclins beforechromosomal segregation through ubiquitination. TheG2-Mtransition is regulated by the cyclin B1-cdc2 complex.Binding of cdc2 to cyclin B1 is required for its activity, andrepression of the cyclin B1 gene by p53 also contributes toblocking entry into mitosis. Our data showed that theprotein levels of both cyclin B1 and cdc2 were significantlyelevated in HaCaT cells treated with 30 mJ/cm2 UVB aloneor pretreated with sanguinarine, consistent with the ob-served G2-M phase arrest in these cells (Fig. 5). In addition,protein levels of cyclin B1 and cdc2 were significantlydecreased in cells pretreated with sanguinarine followedby a 15 mJ/cm2 UVB dose (Fig. 5). The cyclin A/E-cdk2complexes play a role in driving replication, regulating the Sphase, and in keeping replication to only once per cell cycle.
We found that the protein levels of cyclin E1were elevated incells treated with 15 mJ/cm2 UVB alone or pretreated withsanguinarine and were significantly decreased in cellspretreated with sanguinarine followed by a 30 mJ/cm2
UVB dose (Fig. 5C and D). On the other hand, cyclinE2 protein levels were found to be significantly down-modulated by UVB treatment and were returned to normallevels with sanguinarine pretreatment (Fig. 5C and D).Our data also showed that the protein levels of cyclin Awere significantly decreased in HaCaT cells treated with30 mJ/cm2 UVB alone or pretreated with sanguinarine(Fig. 5C and D). Furthermore, the protein levels of cdk2were significantly elevated in cells pretreated with sangui-narine followed by a 15mJ/cm2 dose of UVB butwere foundto be significantly decreased in cells pretreated withsanguinarine followed by a 30 mJ/cm2 UVB dose (Fig. 5A
Figure 3. Sanguinarine treatmentsignificantly modulates Bcl-2 familyproteins in favor of apoptosis. Fol-lowing treatment of HaCaT keratino-cytes with sanguinarine (Sang) and/or UVB, the protein levels of Bcl-2,Bax, Bcl-XL, Bid, and Bak wereassessed by Western blot analysis(A) and quantified by densitometricanalysis (B) of protein bands. Equalloading was confirmed by strippingthe blot and reprobing it for h-actin.Representative experiment repeatedthrice with similar results. The effectof treatment on the Bax/Bcl-2 ratio(C) was determined by both densi-tometric analysis of the immunoblotsrelative to h-actin and by a bivariateanalysis by flow cytometry. Col-umns, mean of three experiments;bars, SD. *, P < 0.01 for untreatedversus treated; ##, P < 0.01 forUVB versus UVB + sanguinarine.Following sanguinarine and UVBtreatment, the cells were transfectedwith Bax short hairpin RNA (shRNA ),and Bax protein levels were deter-mined by Western blot analysis. Theeffect of treatments on apoptosiswas determined by TUNEL assay(D). Following sanguinarine andUVB treatment, the cells were trans-fected with Bcl-2, and Bcl-2 proteinlevels were determined by Westernblot analysis. The effect of treatmentson apoptosis was determined byTUNEL assay (E). Columns, mean ofthree experiments; bars, SD. *, P <0.01 for plasmid alone versus plasmid+ treatment; ##, P < 0.01 forplasmid + UVB versus plasmid +UVB + sanguinarine. Details of theexperiments are given in Materialsand Methods.
Molecular Cancer Therapeutics 423
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

and B). Our data also showed a down-regulation of D-typecyclins following UVB treatment, whereas sanguinarinepretreatment resulted in a reversal of levels of both cyclinsD2 and D3 (Fig. 5C and D).
Sanguinarine Enhances UVB-Mediated Activation ofp53 and p66Shc Proteins in HaCaTKeratinocytesThe p53 protein acts as a sensor of UVB damage and as
an initiator of a program of expression of genes involvedin growth arrest, repair, or apoptosis. Chouinard et al.showed that UVB irradiation to HaCaT keratinocytesincreased expression of phospho-p53 (Ser15) protein buthad no appreciable effect on the levels of total p53 proteinat up to 10 hours after UVB exposure (27). Our datashowed a significant increase in phospho-p53 (Ser15)protein levels at 24 hours after UVB exposure, whichwas further enhanced significantly when the cells werepretreated with sanguinarine (Fig. 6A and B). Expressionof total p53 protein was slightly down-modulated inresponse to UVB alone or sanguinarine plus UVBtreatment; however, the effect was not significant (Fig.6A and B). It is known that phosphorylation of p53 onSer15 impairs the binding of Mdm2, which correlates withp53 accumulation following genotoxic stress (28, 29).Mdm2 protein expression was found to be significantlydecreased, whereas phospho-Mdm2 levels were signifi-cantly increased with sanguinarine pretreatment (Fig. 6Aand B) in HaCaT keratinocytes.The p66Shc protein regulates intracellular levels of ROS
and mediates ROS up-regulation during p53-inducedapoptosis (30). p53 induces p66Shc protein up-regulationby increasing its stability (30). Shc proteins are phos-phorylated by all receptor tyrosine kinases and, uponphosphorylation, Shc proteins form stable complexes withcellular tyrosine-phosphorylated polypeptides (31–33).A model has been proposed whereby the p53-p66Shcpathway is a sensor of the levels of intracellular oxidativesignals and regulates intracellular levels of oxidants andof oxidative damage (30). Western blot analysis showeda small, but significant decrease in p66Shc proteinlevels after UVB treatment alone or with sanguinarinepretreatment (Fig. 6A and B). Expression of phospho-p66Shc (Tyr239) was significantly increased at 24 hoursafter UVB treatment, and this effect was found to besignificantly enhanced with sanguinarine pretreatment(Fig. 6A and B).
Sanguinarine Treatment Modulates UVB-MediatedOxidative Stress in HaCaTKeratinocytesThe most important enzymatic antioxidant to protect
cells from UVB damage is SOD (34). Two types of SOD,copper-zinc SOD (Cu/Zn SOD) and manganese-SOD(MnSOD), have been identified in mammalian cells, andkeratinocytes have been reported to contain both enzymesof SOD (35, 36). Examination of total SOD activity showeda significant dose-dependent increase in total SOD activityat 24 hours after UVB exposure, and to our surprise,sanguinarine was found to significantly decrease SODactivity only at the 30 mJ/cm2 UVB dose (Fig. 6C). Earlierstudies have shown that a single UVB exposure to humankeratinocytes decreased MnSOD protein levels and in-creased Cu/Zn SOD protein levels at 24 after UVB (37).Our data showed that UVB exposure resulted in asignificant dose-dependent decrease in MnSOD and
Figure 4. Sanguinarine treatment significantly enhances aUVB-mediatedcell cycle arrest. Following treatment of HaCaT keratinocytes withsanguinarine (Sang ) and/or UVB, cell cycle distribution was assessed usingthe APO-BrdUrd TUNEL Assay kit. The FAC profiles (A), indicating thepositions of G0-G1, S, and G2-M are shown along with computationalanalysis of (B) the cell population in S and G2-M phases. Columns, mean ofthree experiments; bars, SD. *, P < 0.01 for untreated versus treated;##, P < 0.01 for UVB versus UVB+ sanguinarine. A, from representativeexperiment repeated thrice with similar results. Details of the experimentsare given in Materials and Methods.
UVB Radiation–Mediated Apoptosis by Sanguinarine424
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from
increase in CuZnSOD protein levels at 24 hours after UVBexposure in HaCaT cells (Fig. 6D and E). Interestingly andsurprisingly, pretreatment with sanguinarine significantlyenhanced these effects (Fig. 6D and E).Protein-bound methionine residues are among the most
susceptible to oxidation by ROS, resulting in formation ofmethionine sulfoxide residues. However, this modificationcan be repaired by methionine sulfoxide reductase (MsrA;refs. 38, 39). MsrA then has an important function incellular metabolism as an antioxidant enzyme thatscavenges ROS by facilitating the cyclic interconversionof methionine/protein-methionine residues between oxi-dized and reduced forms (38, 40, 41). In this study, wefound that UVB exposure to HaCaT cells resulted in asignificant decrease in MsrA protein levels at 24 hoursafter UVB, and pretreatment with sanguinarine resulted ina significant enhancement in this inhibitory effect of UVB(Fig. 6D and E).
DiscussionSkin cancer is of great concern as its rates have beensteadily increasing in recent years accounting for >1.3million new cases each year; and the same trend isexpected to continue in the future (42). UVB radiationfrom the sun is an established cause for >90% ofskin cancers. Therefore, mechanism-based strategiesaimed at protection from UVB exposure –mediatedcellular damages, which may lead to the development ofneoplasms, could be useful in the management of skincancer. Chemoprevention seems to be a plausiblestrategy for protection of UVB radiation-induced damageto the skin. UVB radiation is regarded as a completecarcinogen leading to cancer initiation and promotion(4–6). Plant-based agents, often antioxidants, have beenshown to inhibit many UVB-induced signal transductionpathways responsible of cancer development (43, 44). Inthis study, we determined the protective effect of
Figure 5. Sanguinarine treatmentsignificantly modulates cell cycle reg-ulatory proteins. Following treatmentof HaCaT keratinocytes with sangui-narine (Sang) and/or UVB, the pro-tein levels of cdc2, cdk2, and cdk4were assessed by Western blot anal-ysis (A) and quantified by densito-metric analysis (B) of protein bands.The protein levels of cyclins A, B1,D1, D2, D3, E1, and E2 wereassessed by Western blot analysis(C) and quantified by densitometricanalysis (D) of protein bands. Equalloading was confirmed by strippingthe blot and reprobing it for h-actin.Representative experiment repeatedthrice with similar results. Columns,mean of three experiments; bars, SD.*, P < 0.01 for untreated versustreated; ##, P < 0.01 for UVB versusUVB + sanguinarine. Details of theexperiments are given in Materialsand Methods.
Molecular Cancer Therapeutics 425
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

sanguinarine, a plant alkaloid with documented anti-inflammatory and anticancer effects, against the UVBexposure–mediated damages to human HaCaT keratino-cytes.UVB radiation imparts damage to skin cells, and the cells
respond to this damage in three ways: by tolerating thedamage, by repairing the damage via delays in the cellcycle, or by undergoing apoptosis (a programmed celldeath). The latter two responses represent defense mech-anisms of the system to either repair the damage oreliminate the defective cells (containing damages). How-ever, the unrepaired cells (with genetic abnormalities) mayundergo a clonal expansion to acquire a hyperproliferativephenotype that could ultimately result in a neoplasticcondition.In this study, we found that sanguinarine enhances the
ability of HaCaT cells to undergo cell cycle arrest andapoptosis as a result of UVB-caused damages. This isclearly an important observation because apoptosis is amechanism of defense and acts by opposing the creation ofa damaged (preneoplastic) cell and expansion of this cellinto a clone. Once mutations arise, apoptosis also removesdamaged (preneoplastic) cells that are aberrantly prolifer-ating due to genetic defects.
Similarly, cell cycle arrest increases the time availablefor DNA repair before DNA replication and mutationfixation. The checks and balances governing the cell cycleprevent inappropriate DNA replication, and a breakdownin these checkpoints can lead to genomic instability andcancer. It is known that UVB exposure causes a G2-Mphase cell cycle arrest in the keratinocytes, and our dataverified this finding. However, we showed that sangui-narine pretreatment had a differential effect on thedistribution of cells in different phases of cell cycle. Thus,sanguinarine pretreatment resulted in a significant arrestof cells in the S phase when exposed to 15 mJ/cm2 dose ofUVB and in G2-M phase when exposed to 30 mJ/cm2 doseof UVB. The reason for this differential effect, however,remains unknown at present.Our data also showed the involvement of p53, Bcl-2
family proteins, and cell cycle regulatory proteins duringthe chemoprotective effects of sanguinarine. The mecha-nism of G2-M arrest in mammalian cells is controlled bythe cyclin B1/cdc2 kinase. Activation of this kinase issuppressed by DNA damage, and this may result fromthe imposition of inhibitory phosphorylation on the cdc2kinase as well as down-regulation of cyclin B1 levels. p53represses the cdc2 gene, to help ensure that cells do not
Figure 6. Sanguinarine treatment significantly enhanced UVB-mediated activation of p53 and p66Shc and modulates an oxidative stress response.Following treatment of HaCaT keratinocytes with sanguinarine (Sang ) and/or UVB, the protein levels of p53, phospho-p53, Mdm2, phospho-Mdm2,p66Shc, and phospho-p66Shc were assessed by Western blot analysis (A) and quantitated by densitometric analysis (B) of protein bands. Followingtreatment of HaCaT keratinocytes with sanguinarine and/or UVB, SOD activity was assessed with the Bioxytech SOD-525 kit (C). Following treatment ofHaCaT keratinocytes with sanguinarine and/or UVB, the protein levels of CuZnSOD, MnSOD, and MsrA were assessed by Western blot analysis (D) andquantified by densitometric analysis (E) of protein bands. Equal loading was confirmed by stripping the blot and reprobing it for h-actin. Columns, mean ofthree experiments; bars, SD. *, P < 0.01 for untreated versus treated; ##, P < 0.01 for UVB versus UVB + sanguinarine. Details of the experiments aregiven in Materials and Methods.
UVB Radiation–Mediated Apoptosis by Sanguinarine426
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

escape the initial block. In our study, the cells that werearrested in G2-M phase showed an accumulation inboth cdc2 and cyclin B1 protein levels (Figs. 4 and 5). Onthe other hand, formation and activation of the pre-replication complex requires coordinate actions of cyclinE-cdk2 and cyclin A-cdk2 complexes. And our datashowed that the cells that S-phase arrest as a result ofsanguinarine pretreatment was associated with an accu-mulation of cyclin E, cdk2, and/or cyclin A proteins(Figs. 4 and 5).DNA damage elicited by UVB is thought to be an
important trigger for p53 accumulation and transcrip-tional activation, leading to cell cycle arrest and allowingmore time for DNA repair or elimination of damagedcells through apoptosis. Most nonmelanoma skin cancershave mutations in p53 (45–47). The HaCaT cell line bearsmutations in both alleles of the p53 gene, rendering anonfunctional, transcriptionally inactive protein with anincreased half-life (48). The mutations in p53 present inHaCaT cells are characteristic of the UV signature (48).We found a significant increase in phospho-p53 (Ser15)protein levels that was significantly enhanced withsanguinarine pretreatment along with no appreciablechanges in total p53 protein levels in HaCaT cells (Fig.6). p53 protects mammals from neoplasia by inducingapoptosis, DNA repair, and cell cycle arrest in responseto a variety of stresses. The regulation of tumorsuppressor p53 depends not only on the level ofexpression of this molecule but also on its activationthat regulates its stability and capacity to bind to DNAand trigger transcription (49–51). It is known thatphosphorylation of p53 on Ser15 impairs the binding ofMdm2, which correlates with p53 accumulation followinggenotoxic stress (28, 29). The phosphorylation of p53 isaffected by its conformation, which, in turn, is modifiedby mutation of the protein (52).Another interesting finding of our study was the
observed significant accumulation of phospho-p66Shc(Tyr239) in HaCaT cells exposed to UVB. This responseof UVB was found to be further enhanced by sangui-narine pretreatment (Fig. 6). P66Shc is a splice variant ofp52Shc/p46Shc, a cytoplasmic signal transducer involvedin the transmission of mitogenic signals from tyrosinekinases to Ras (53). P66Shc is not involved in Rasregulation but rather functions in the intracellularpathway that converts oxidative signals into apoptosis(54, 55). P66Shc is serine phosphorylated in cells treatedwith UV or other inducers of oxidative stress, andp66Shc�/� fibroblasts are resistant to UV-induced apo-ptosis, a finding mirrored by the increased UV sensitivityconferred by overexpression of p66Shc (54). p53 inducesp66Shc protein up-regulation by increasing its stability(30). It has been shown that p53 and p66Shc regulatesteady-state levels of intracellular ROS, and the p66Shcgene increases intracellular ROS, thereby affecting therate of oxidative damage to the nucleic acids (30). Thep53/p66Shc pathway could be a sensor for the levels ofintracellular oxidative signals and regulate intracellular
levels of oxidants and of oxidative damage (30). High-intensity oxidative signals would result in high-levelactivation of the p53/p66Shc pathway and apoptosis (30).Low-intensity oxidative signals could result in chronic,low-level activation of the p53/p66Shc signaling path-way, thus allowing moderate ROS increases and accu-mulation of the oxidative damage (30).Thus, because of the documented association of the
p53/p66Shc pathway with the oxidative stress, our dataimplicated that the observed effects of sanguinarinemight be mediated via modulations in the oxidativestress within the cells. To further confirm the involve-ment of oxidative stress, we determined the effect oftreatments on the levels of antioxidant enzyme SOD thatis known to be modulated by the ROS and oxidativestress in the cells.It is important to mention here that ROS, at normal
physiologic levels, play a role in regulating signalingpathways and gene expression and are also involved incancer development (56). It is likely that in skin cancers, adiminished antioxidant defense caused by chronic UVexposure may lead, indirectly, to a clonal expansion ofinitiated, promotable cells that are resistant to excessiveoxidative damage. SOD has been shown to protect humankeratinocytes against UVB-induced injury (37, 57). Inhuman skin, single exposures of solar-simulated UVresulted in a transient reduction of SOD activity (58);however, chronic UV exposure induced epidermal SODactivity (59). An immunohistochemical investigation ofSCC and BCC revealed a decreased CuZnSOD and MnSODexpression within the tumors, indicating a UV-dependentimpairment of the antioxidant defense (35). A single doseof UVB irradiation dose-dependently regulated expressionof MnSOD in HeLa cells, although it had no effect of itsenzymatic activity (60). In contrast, UVB irradiationreduced both the enzymatic activity and the expressionof CuZnSOD in HeLa cells (60). Overexpression of MnSODhas been shown to block or delay apoptosis (34). We founda significant increase in SOD activity at 24 hours after UVBexposure along with a significant increase in CuZnSODand decrease in MnSOD protein levels, and pretreatmentwith sanguinarine significantly enhanced these effects inHaCaT cells (Fig. 6).We also determined effect of treatments on the protein
levels of MsrA, which plays an important role in cellularmetabolism as an antioxidant enzyme that scavenges ROSby facilitating the cyclic interconversion of methionine/protein-methionine residues between oxidized and re-duced forms (38, 40, 41). Mice lacking the MsrA geneexhibit heightened sensitivity to oxidative stress (61). Ourdata showed that UVB exposure to HaCaT cells resulted ina significant decrease in MsrA protein levels, which wasfurther down-regulated with sanguinarine pretreatment(Fig. 6). Thus, our data showed that sanguinarine, oftenreferred as an antioxidant, in fact, enhances the oxidativestress in the cells that are damaged by UVB. Interestingly,sanguinarine did not induce oxidative stress in the cellsthat were not exposed to UVB.
Molecular Cancer Therapeutics 427
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

Taken together, based on our data, we suggest thatsanguinarine may protect skin cells from UVB-mediateddamages via apoptotic elimination of damaged cells thatescape programmed cell death and therefore possess apotential of clonal expansion.
Acknowledgments
We thank undergraduate student Stefanie Jones for her technicalassistance.
References
1. Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA CancerJ Clin 2005;55:10–30.
2. Stewart BW, Kleihues P, editors. World cancer report. Washington(DC): IARC Press; 2003.
3. Gailani MR, Leffell DJ, Ziegler A, et al. Relationship between sunlightexposure and a key genetic alteration in basal cell carcinoma. J NatlCancer Inst 1996;20:349–54.
4. Cleaver JE, Crowley E. UV damage, DNA repair and skin carcinogen-esis. Front Biosci 2002;7:1024–43.
5. Sarasin A. The molecular pathways of ultraviolet-induced carcinogen-esis. Mutat Res 1999;428:5–10.
6. de Gruijl FR. Photocarcinogenesis: UVA vs. UVB radiation. SkinPharmacol Appl Skin Physiol 2002;15:316–20.
7. Hawk ET, Umar A, Viner JL. Colorectal cancer chemoprevention-anoverview of the science. Gastroenterology 2004;126:1423–47.
8. Klein EA, Thompson IM. Update on chemoprevention of prostatecancer. Curr Opin Urol 2004;14:143–9.
9. Mehta RG, Pezzuto JM. Discovery of cancer preventive agents fromnatural products: fromplants to prevention.CurrOncol Rep2002;4:478–86.
10. Park EJ, Pezzuto JM. Botanicals in cancer chemoprevention. CancerMetastasis Rev 2002;21:231–55.
11. Scheer M, Kuebler AC, Zoller JE. Chemoprevention of oral squamouscell carcinomas. Onkologie 2004;27:187–93.
12. Surh YJ. Cancer chemoprevention with dietary phytochemicals. NatRev Cancer 2003;3:768–80.
13. Shamma M, Guinaudeau H. Aporphinoid alkaloids. Nat Prod Rep1986;3:345–51.
14. Vavreckova C, Ulrichova J, Hajduch M, et al. Effect of quaternarybenzo[c]phenanthridine alkaloids sanguinarine, chelerythrine and fagar-onine on some mammalian cells. Acta Univ Palacki Olomuc Fac Med 1994;138:7–10.
15. Mitscher LA, Park YH, Clark D, et al. Antimicrobial agents from higherplants. An investigation of Hunnemannia fumariaefolia pseudoalcoho-lates of sanguinarine and chelerythrine. Lloydia 1978;41:145–50.
16. Lenfeld J, Kroutil M, Marsalek E, et al. Antiinflammatory activity ofquaternary benzophenanthridine alkaloids from Chelidonium majus .Planta Med 1981;43:161–5.
17. Mandel ID. Chemotherapeutic agents for controlling plaque andgingivitis. J Clin Periodontol 1988;15:488–98.
18. Mandel ID. Antimicrobial mouthrinses: overview and update. J AmDent Assoc 1994;125 Suppl 2:2–10S.
19. Walterova D, Ulrichova J, Valka I, et al. Benzo[c]phenanthridinealkaloids sanguinarine and chelerythrine: biological activities and dentalcare applications. Acta Univ Palacki Olomuc Fac Med 1995;139:7–16.
20. Ahmad N, Gupta S, Husain MM, Heiskanen KM, Mukhtar H.Differential antiproliferative and apoptotic response of sanguinarine forcancer cells versus normal cells. Clin Cancer Res 2000;6:1524–8.
21. Adhami VM, Aziz MH, Mukhtar H, Ahmad N. Activation of prodeathBcl-2 family proteins and mitochondrial apoptosis pathway by sanguinar-ine in immortalized human HaCaT keratinocytes. Clin Cancer Res 2003;9:3176–82.
22. Gniadecki R, Hansen M, Wulf HC. Two pathways for induction ofapoptosis by ultraviolet radiation in cultured human keratinocytes. J InvestDermatol 1997;109:163–9.
23. Lee JK, Kim JH, Nam KT, Lee SH. Molecular events associated with
apoptosis and proliferation induced by ultraviolet-B radiation in the skin ofhairless mice. J Dermatol Sci 2003;32:171–9.
24. Weerasinghe P, Hallock S, Tang SC, Liepins A. Role of Bcl-2 familyproteins and caspase-3 in sanguinarine-induced bimodal cell death. CellBiol Toxicol 2001;17:371–81.
25. Weerasinghe P, Hallock S, Liepins A. Bax, Bcl-2, and NF-kappaBexpression in sanguinarine induced bimodal cell death. Exp Mol Pathol2001;71:89–98.
26. Athar M, Kim AL, Ahmad N, et al. Mechanism of ultraviolet B-inducedcell cycle arrest in G2/M phase in immortalized skin keratinocytes withdefective p53. Biochem Biophys Res Commun 2000;277:107–11.
27. Chouinard N, Valerie K, Rouabhia M, Huot J. UVB-mediated activationof p38 mitogen-activated protein kinase enhances resistance of normalhuman keratinocytes to apoptosis by stabilizing cytoplasmic p53. BiochemJ 2002;365:133–45.
28. Bean LJ, Stark GR. Phosphorylation of serines 15 and 37 is necessaryfor efficient accumulation of p53 following irradiation with UV. Oncogene2001;20:1076–84.
29. Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-inducedphosphorylation of p53 alleviates inhibition by MDM2. Cell 1997;91:325–34.
30. Trinei M, Giorgio M, Cicalese A, et al. A p53–66Shc signallingpathway controls intracellular redox status, levels of oxidation-damagedDNAandoxidative stress-induced apoptosis. Oncogene 2002;21:3872–8.
31. Songyang Z, Margolis B, Chaudhuri M, Shoelson SE, Cantley LC. Thephosphotyrosine interaction domain of SHC recognizes tyrosine-phos-phorylated NPXY motif. J Biol Chem 1995;270:14863–6.
32. van der GP, Wiley S, Lai VK, et al. A conserved amino-terminal Shcdomain binds to phosphotyrosine motifs in activated receptors andphosphopeptides. Curr Biol 1995;5:404–12.
33. McGlade J, Cheng A, Pelicci G, Pelicci PG, Pawson T. Shc proteinsare phosphorylated and regulated by the v-Src and v-Fps protein-tyrosinekinases. Proc Natl Acad Sci U S A 1992;89:8869–73.
34. Shindo Y, Hashimoto T. Ultraviolet B-induced cell death in fourcutaneous cell lines exhibiting different enzymatic antioxidant defences:involvement of apoptosis. J Dermatol Sci 1998;17:140–50.
35. Kobayashi T, Matsumoto M, Iizuka H, Suzuki K, Taniguchi N.Superoxide dismutase in psoriasis, squamous cell carcinoma and basalcell epithelioma: an immunohistochemical study. Br J Dermatol 1991;124:555–9.
36. Kobayashi T, Saito N, Takemori N, et al. Ultrastructural localization ofsuperoxide dismutase in human skin. Acta Derm Venereol 1993;73:41–5.
37. Sasaki H, Akamatsu H, Horio T. Effects of a single exposure to UVBradiation on the activities and protein levels of copper-zinc and manganesesuperoxide dismutase in cultured human keratinocytes. PhotochemPhotobiol 1997;65:707–13.
38. Moskovitz J, Weissbach H, Brot N. Cloning the expression of amammalian gene involved in the reduction of methionine sulfoxideresidues in proteins. Proc Natl Acad Sci U S A 1996;93:2095–9.
39. Moskovitz J, Berlett BS, Poston JM, Stadtman ER. The yeast peptide-methionine sulfoxide reductase functions as an antioxidant in vivo . ProcNatl Acad Sci U S A 1997;94:9585–9.
40. Stadtman ER, Moskovitz J, Berlett BS, Levine RL. Cyclic oxidationand reduction of protein methionine residues is an important antioxidantmechanism. Mol Cell Biochem 2002;234–5:3–9.
41. Levine RL, Berlett BS, Moskovitz J, Mosoni L, Stadtman ER.Methionine residues may protect proteins from critical oxidative damage.Mech Ageing Dev 1999;107:323–32.
42. Jemal A, Tiwari RC, Murray T, et al. Cancer statistics, 2004. CACancer J Clin 2004;54:8–29.
43. Afaq F, Mukhtar H. Photochemoprevention by botanical antioxidants.Skin Pharmacol Appl Skin Physiol 2002;15:297–306.
44. Sander CS, Chang H, Hamm F, Elsner P, Thiele JJ. Role of oxidativestress and the antioxidant network in cutaneous carcinogenesis. Int JDermatol 2004;43:326–35.
45. Kanjilal S, Strom SS, Clayman GL, et al. p53 mutations in non-melanoma skin cancer of the head and neck: molecular evidence for fieldcancerization. Cancer Res 1995;55:3604–9.
46. Oram Y, Orengo I, Baer SC, Ocal T. p53 Protein expression insquamous cell carcinomas from sun-exposed and non-sun-exposed sites.J Am Acad Dermatol 1994;31:417–22.
UVB Radiation–Mediated Apoptosis by Sanguinarine428
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

47. Ziegler A, Leffell DJ, Kunala S, et al. Mutation hotspots due tosunlight in the p53 gene of nonmelanoma skin cancers. Proc Natl Acad SciU S A 1993;90:4216–20.
48. Lehman TA, Modali R, Boukamp P, et al. p53 mutations in humanimmortalized epithelial cell lines. Carcinogenesis 1993;14:833–9.
49. Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphorylationof Ser-20 mediates stabilization of human p53 in response to DNAdamage. Proc Natl Acad Sci U S A 1999;96:13777–82.
50. Giaccia AJ, Kastan MB. The complexity of p53 modulation: emergingpatterns from divergent signals. Genes Dev 1998;12:2973–83.
51. Steegenga WT, van der Eb AJ, Jochemsen AG. How phosphorylationregulates the activity of p53. J Mol Biol 1996;263:103–13.
52. Adler V, Pincus MR, Minamoto T, et al. Conformation-dependentphosphorylation of p53. Proc Natl Acad Sci U S A 1997;94:1686–91.
53. Pelicci G, Lanfrancone L, Grignani F, et al. A novel transformingprotein (SHC) with an SH2 domain is implicated in mitogenic signaltransduction. Cell 1992;70:93–104.
54. Migliaccio E, Giorgio M, Mele S, et al. The p66shc adaptor proteincontrols oxidative stress response and life span in mammals. Nature 1999;402:309–13.
55. Migliaccio E, Mele S, Salcini AE, et al. Opposite effects of the p52shc/
p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fossignalling pathway. EMBO J 1997;16:706–16.
56. Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors ofDNA damage: variations on a theme? Curr Opin Cell Biol 2001;13:225–31.
57. Sasaki H, Akamatsu H, Horio T. Protective role of copper, zincsuperoxide dismutase against UVB-induced injury of the human keratino-cyte cell line HaCaT. J Invest Dermatol 2000;114:502–7.
58. Punnonen K, Autio P, Kiistala U, Ahotupa M. In-vivo effects of solar-simulated ultraviolet irradiation on antioxidant enzymes and lipid perox-idation in human epidermis. Br J Dermatol 1991;125:18–20.
59. Punnonen K, Lehtola K, Autio P, Kiistala U, Ahotupa M. Chronic UVBirradiation induces superoxide dismutase activity in human epidermisin vivo. J Photochem Photobiol B 1995;30:43–8.
60. Isoherranen K, Peltola V, Laurikainen L, et al. Regulation ofcopper/zinc and manganese superoxide dismutase by UVB irradiation,oxidative stress and cytokines. J Photochem Photobiol B 1997;40:288–93.
61. Moskovitz J, Bar-Noy S, Williams WM, et al. Methionine sulfoxidereductase (MsrA) is a regulator of antioxidant defense and lifespan inmammals. Proc Natl Acad Sci U S A 2001;98:12920–5.
Molecular Cancer Therapeutics 429
Mol Cancer Ther 2006;5(2). February 2006
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

2006;5:418-429. Mol Cancer Ther Shannon Reagan-Shaw, Jorien Breur and Nihal Ahmad sanguinarine in HaCaT human immortalized keratinocytes
mediated apoptosis by−Enhancement of UVB radiation
Updated version
http://mct.aacrjournals.org/content/5/2/418
Access the most recent version of this article at:
Cited articles
http://mct.aacrjournals.org/content/5/2/418.full#ref-list-1
This article cites 59 articles, 13 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/5/2/418.full#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://mct.aacrjournals.org/content/5/2/418To request permission to re-use all or part of this article, use this link
Research. on October 21, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from