enhanced stability of tristetraprolin mrna protects … stability of tristetraprolin mrna protects...
TRANSCRIPT

Enhanced stability of tristetraprolin mRNA protectsmice against immune-mediatedinflammatory pathologiesSonika Patiala, Alan D. Curtis IIb, Wi S. Laia, Deborah J. Stumpoa, Georgette D. Hillc, Gordon P. Flaked, Mark D. Mannieb,and Perry J. Blacksheara,e,1
aSignal Transduction Laboratory, National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709; bDepartment of Microbiology andImmunology, East Carolina University Brody School of Medicine, Greenville, NC 27858; cIntegrated Laboratory Systems, Research Triangle Park, NC 27709;dCellular and Molecular Pathology Branch, National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709; and eDepartments ofMedicine and Biochemistry, Duke University Medical Center, Durham, NC 27710
Edited by Dennis A. Carson, University of California at San Diego, La Jolla, CA, and approved January 4, 2016 (received for review October 7, 2015)
Tristetraprolin (TTP) is an inducible, tandem zinc-finger mRNA bindingprotein that binds to adenylate-uridylate–rich elements (AREs) in the3′-untranslated regions (3′UTRs) of specific mRNAs, such as that encod-ing TNF, and increases their rates of deadenylation and turnover. Sta-bilization of Tnf mRNA and other cytokine transcripts in TTP-deficientmice results in the development of a profound, chronic inflammatorysyndrome characterized by polyarticular arthritis, dermatitis, myeloidhyperplasia, and autoimmunity. To address the hypothesis that in-creasing endogenous levels of TTP in an intact animal might be ben-eficial in the treatment of inflammatory diseases, we generated amouse model (TTPΔARE) in which a 136-base instability motif in the3′UTR of TTP mRNA was deleted in the endogenous genetic locus.These mice appeared normal, but cultured fibroblasts and macro-phages derived from them exhibited increased stability of the other-wise highly labile TTP mRNA. This resulted in increased TTP proteinexpression in LPS-stimulated macrophages and increased levels of TTPprotein in mouse tissues. TTPΔARE mice were protected from collagenantibody-induced arthritis, exhibited significantly reduced inflamma-tion in imiquimod-induced dermatitis, and were resistant to inductionof experimental autoimmune encephalomyelitis, presumably by damp-ening the excessive production of proinflammatory mediators in allcases. These data suggest that increased systemic levels of TTP, sec-ondary to increased stability of its mRNA throughout the body, can beprotective against inflammatory disease in certain models and mightbe viewed as an attractive therapeutic target for the treatment ofhuman inflammatory diseases.
AU-rich elements | mRNA stability | inflammation | deadenylation
Tristetraprolin (TTP) is the prototype of a small family of RNAbinding proteins that can bind to adenylate-uridylate (AU)–rich
elements (AREs) in the 3′-UTR (3′UTR) of its target mRNAs andpromote their rapid turnover (1, 2). TTP-deficient mice developeda chronic systemic inflammatory syndrome (3) that was preventedby interfering with the action of TNF (3–5). Tnf mRNA was thenidentified as a direct target of TTP-mediated destabilization (4, 6);its increase in stability in the TTP KO mice leads to the com-mensurate overproduction of TNF protein (4, 5).TTP mRNA expression exhibits a pattern characteristic of im-
mediate-early response genes in several cell types, with low-to-undetectable levels of expression under basal conditions, and arapid and transient induction upon stimulation (4, 7, 8). Thetransient nature of this induction is largely due to the instability ofthe TTP mRNA itself, part of which is thought to be due to AREslocated within the 3′UTR of TTP mRNA (4, 8, 9). Indeed, TTPhas been suggested to bind to its own AREs and autoregulate itsexpression through a negative feedback loop (9). Although ex-pression of TTP protein in these systems is also rapidly inducible,the protein is more stable than the mRNA after induction, oftenpersisting at high levels for several hours (7, 10).The severe systemic inflammatory phenotype of the TTP KO
mice identified TTP as an endogenous antiinflammatory protein.
We have long wondered whether increasing endogenous levels ofTTP in an intact animal might protect against the development ofimmune and inflammatory diseases. Early attempts to accomplishthis using transgenic delivery of TTP resulted in embryonic lethality,presumably due to unregulated overexpression. To test our hypoth-esis in a different way, we generated a novel knock-in mouse model inwhich an instability motif in the 3′UTR of TTP mRNA was deletedin the mouse genome. We anticipated that, by deleting these in-stability elements, TTP mRNA would be stabilized under physio-logical conditions, and this would result in modest increases in TTPmRNA and protein levels that were still under the control of theendogenous genetic locus.These mice, termed TTPΔARE mice, appear normal but exhibit
increased TTP mRNA stability, as well as increased levels of TTPprotein in their tissues. They were strikingly resistant to the devel-opment of three models of experimental immune-mediated in-flammatory diseases. These data suggest that treatments leading toincreased endogenous TTP levels could be a promising therapeuticapproach in immune and inflammatory human diseases.
ResultsGeneration of TTPΔARE Mice. TTPΔARE mice were generated asdescribed in SI Materials and Methods. Briefly, 136 bases (bases1564–1699 of GenBank accession no. NM_011756) (depicted in
Significance
Inflammation is involved in the pathogenesis of many chronicdiseases. Many deleterious effects of inflammation are mediatedthrough increased production of proinflammatory mediators,known as cytokines and chemokines. Many current therapiesfor these diseases involve blocking single proinflammatorymediators, such as TNF, using parenteral administration ofrecombinant binding proteins. We demonstrate here that agenetic modification in the mouse that increases the expression ofan endogenous antiinflammatory protein, tristetraprolin (TTP),results in protection against mouse models for several humaninflammatory diseases, including rheumatoid arthritis, psoriasis,and multiple sclerosis, presumably by decreasing the productionof proinflammatory cytokines. Our results suggest that increasingTTP expression may be an effective therapeutic strategy in thetreatment of certain inflammatory diseases.
Author contributions: S.P., A.D.C., D.J.S., M.D.M., and P.J.B. designed research; S.P., A.D.C., W.S.L.,D.J.S., G.D.H., G.P.F., and M.D.M. performed research; W.S.L., D.J.S., G.D.H., and M.D.M. contrib-uted new reagents/analytic tools; S.P., A.D.C., W.S.L., D.J.S., G.D.H., G.P.F., M.D.M., and P.J.B.analyzed data; and S.P. and P.J.B. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1519906113/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1519906113 PNAS Early Edition | 1 of 6
IMMUNOLO
GYAND
INFLAMMATION

red in Fig. S1A), comprising an AU-rich region of the TTP mRNA3′UTR, were deleted in the mouse genome. The WT allele, thetargeting construct, and the mutant allele are shown schematicallyin Fig. S1B. The WT, heterozygous, and homozygous TTPΔAREmice could be identified readily by PCR genotyping of tail DNA(Fig. S1C).The body weights of adult (12-wk-old) TTPΔARE mice were
similar to those of sex-matched WT littermates (Fig. S1D). Serumlipid levels were comparable betweenWT and TTPΔAREmice (Fig.S1E), as were the serum activities of several liver enzymes (Fig. S1F).Histopathological (Fig. S2) and immunohistochemical (Fig. S3) ex-amination of major tissues revealed no obvious anatomical or cellularabnormalities in adult TTPΔARE mice. Similarly, complete bloodcounts (Table S1) and bone marrow cell counts (Table S2) did notreveal any evident abnormalities in adult TTPΔARE mice. Thesedata suggest that genetic deletion of 136 bases in the TTP transcriptdid not result in any apparent physiological or developmental defectsin the mice used in this study.
Effect of the TTPΔARE Mutation on TTP mRNA Expression, TTP mRNAStability, and TTP Protein Expression in Cells and Mouse Tissues. Theeffect of the homozygous TTPΔARE mutation on TTP mRNAexpression was examined in cultured primary bone marrow-derived macrophages (BMDM). Under unstimulated conditions,TTP mRNA levels were increased approximately threefold in the
TTPΔARE cells (Fig. 1A). They increased dramatically in cells ofboth genotypes after LPS stimulation, but TTP mRNA levels weresignificantly elevated in the TTPΔARE cells compared with WTcells at 3 h (Fig. 1B). To test TTP mRNA stability, we stimulatedTTP gene transcription in BMDM with LPS, or mouse embryonicfibroblasts (MEF) with 10% (vol/vol) FBS, for 1 h in each case,followed by treatment with actinomycin D to inhibit transcription,and quantitated the remaining RNA at various times. Under theseconditions, TTP mRNA degraded significantly more slowly in boththe TTPΔARE BMDM (Fig. 1C) and the MEF (Fig. 1D). Thesedata demonstrate the increased stability of the mutant TTP mRNAin these cell types.TTP protein was readily detectable in unstimulated conditions in
the TTPΔARE BMDM but not in the WT BMDM (Fig. 1E).Furthermore, TTP protein levels were elevated compared with WTat all times after LPS stimulation in the TTPΔARE cells (Fig. 1E).Most importantly, TTP protein levels were increased in liver,spleen, and thymus from TTPΔARE mice (Fig. 1F).
Effect of the TTPΔARE Mutation on Potential TTP Targets in TTPΔAREBMDM and Intact Mice.We next analyzed the expression of knownor suspected TTP targets in LPS-stimulated BMDM. For Tnf,Il1b, and Cxcl2 mRNAs, the levels in the TTPΔARE cells weresignificantly lower than WT at 6 h, although there was a generalpattern of decreased expression at 3 h as well (Fig. S4A). There
0 1 3 6 240.0
0.3
0.6
0.9
1.2***
Time (hrs), LPS
Rel
ativ
eTT
Pex
pres
sion
(%1h
rW
T)
WTTTP∆ARE
0 30 60 90 1200
50
100
* ****
Actinomycin D (min)
TTP
mR
NA
abun
danc
e(R
elat
ive
toA
ctD
attim
e0)
0 30 60 90 1200
50
100*
Actinomycin D (min)
A B
C D
0
1
2
3
4
Rel
ativ
eTT
Pex
pres
sion
***
W
TTP∆ART E
E
F
55
TTP
0 0.5 1 3 6 24 0 0.5 1 3 6 24
WT TTP∆ARE LPSTime (hrs)
Tubulin
35
M.W.(kDa)
WTTTP ARE∆
WT TTP∆ARE
Live
rSp
leen
Thym
us
TTP
Actin
TTP
Actin
TTP
Actin
1 2 3 1 2 3TTP KO
49
37
49
37
49
37
M.W.(kDa)
0 30 60 90 1201
10
100
Actinomycin D (min)
50
0 30 60 90 1201
10
100
Actinomycin D (min)
50
Fig. 1. TTP mRNA expression and stability, and TTP protein expression in primary cells and tissues derived from homozygous TTPΔARE mice. (A) Relativelevels of TTP mRNA under basal (i.e., unstimulated) conditions in serum-deprived BMDMs (n = 3–4). (B) Time course of expression of TTP mRNA before andafter stimulation (LPS; 1 μg/mL) in BMDMs. Data are expressed as a percentage of WT at 1 h (n = 3–4). (C) TTP mRNA decay in BMDMs and (D) MEFs. BMDMs orMEFs were stimulated with LPS (1 μg/mL) or 10% FBS (vol/vol), respectively, for 1 h, followed by treatment with actinomycin D. Percent remaining mRNA wasmeasured by real-time RT-PCR (n = 4). The insets show semilogarithmic decay plots of the same data, analyzed by nonlinear regression. The approximate half-lives were: WT (BMDMs), ∼32 min; TTPΔARE (BMDMs), ∼80 min; WT (MEFs), ∼28 min; TTPΔARE (MEFs), ∼54 min. (E) TTP protein levels in BMDMs under basaland LPS- (1 μg/mL) stimulated conditions. Tubulin was used as a loading control. (F) TTP protein expression in mouse tissues. The lane labeled TTP KO containsan equal amount of protein from the respective TTP KO mouse tissue, included as a negative control. Actin was used as a loading control. Statistical analysiswas performed by two-tailed Student’s t test for A, C, and D and by two-way ANOVA for B. Error bars represent SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
2 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1519906113 Patial et al.
were no significant differences at any time point for Il10, Il6, andIl23a mRNAs, although there was a trend toward decreasedexpression at 3 and 6 h in the TTPΔARE cells (Fig. S4A). Incultures of BMDM cells stimulated by LPS, IL-10 concentrationswere significantly lower in the medium of TTPΔARE cells atboth 8 and 24 h, whereas levels of IL-1B and IL-6 were un-changed (Fig. S4B). Surprisingly, levels of TNF and chemokine(C-X-C motif) ligand 2 (CXCL2) were significantly increased at24 h in the medium of the TTPΔARE cells (Fig. S4B).We then injected WT and TTPΔARE mice with three different
doses of LPS and measured the serum levels of TNF, IL10, IL-1B,and CXCL2 in response. In myeloid cell-deficient TTP KO mice,the serum TNF response to LPS was a sensitive indicator of TTPdeficiency, with serum levels increasing by more than 100-fold overWT after low-dose LPS injections (11). In the present study, afterinjection of LPS at 0.5, 3, or 20 mg/kg body weight, the averageserum concentrations of TNF were not significantly different be-tween the WT and the TTPΔARE mice, either during the peakincrease at 1.5 h or at 3 and 6 h (Fig. S5). At the highest dose ofLPS, there was a trend toward lower TNF levels at 1.5 h that didnot achieve statistical significance. Serum levels of IL10 were nodifferent between the two genotypes 1.5 h after 0.5 and 3 mg/kgLPS but were significantly lower at 1.5 h in the TTPΔARE miceinjected with 20 mg/kg and decreased significantly more rapidlyafter that point with all three doses of LPS (Fig. S5). IL-1B levelswere significantly lower in the TTPΔAREmice at 6 h after 20 mg/kgLPS, whereas the levels of CXCL2 did not differ significantly be-tween the two genotypes at any of the three doses of LPS tested.
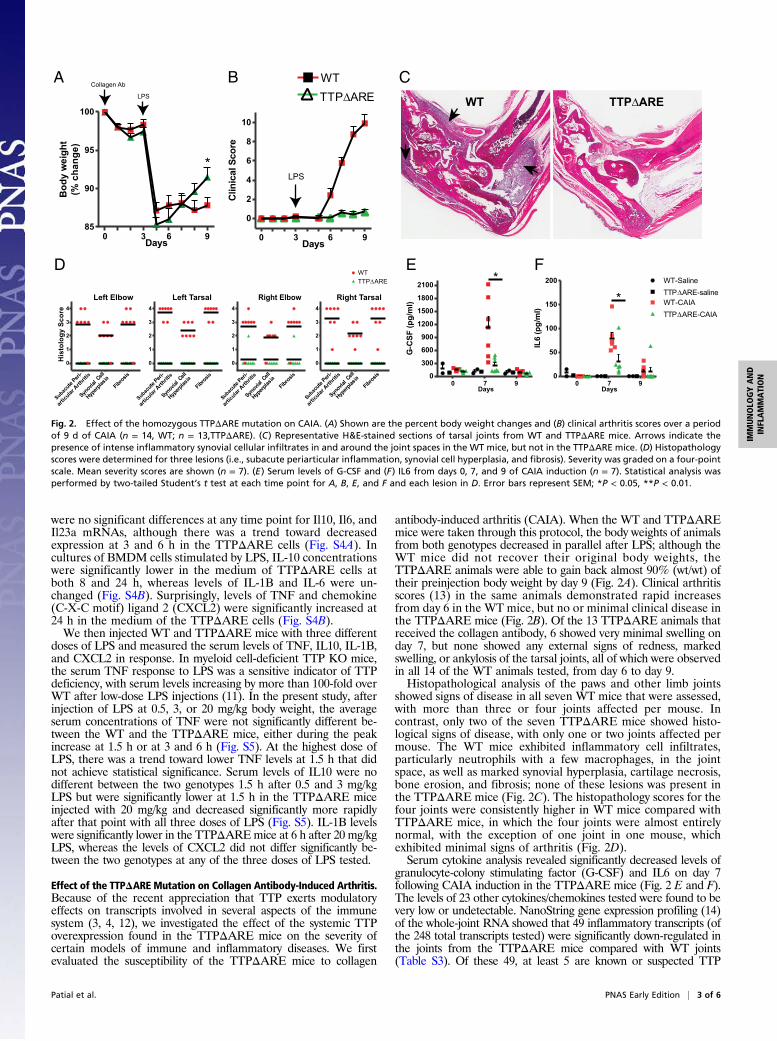
Effect of the TTPΔARE Mutation on Collagen Antibody-Induced Arthritis.Because of the recent appreciation that TTP exerts modulatoryeffects on transcripts involved in several aspects of the immunesystem (3, 4, 12), we investigated the effect of the systemic TTPoverexpression found in the TTPΔARE mice on the severity ofcertain models of immune and inflammatory diseases. We firstevaluated the susceptibility of the TTPΔARE mice to collagen
antibody-induced arthritis (CAIA). When the WT and TTPΔAREmice were taken through this protocol, the body weights of animalsfrom both genotypes decreased in parallel after LPS; although theWT mice did not recover their original body weights, theTTPΔARE animals were able to gain back almost 90% (wt/wt) oftheir preinjection body weight by day 9 (Fig. 2A). Clinical arthritisscores (13) in the same animals demonstrated rapid increasesfrom day 6 in the WT mice, but no or minimal clinical disease inthe TTPΔARE mice (Fig. 2B). Of the 13 TTPΔARE animals thatreceived the collagen antibody, 6 showed very minimal swelling onday 7, but none showed any external signs of redness, markedswelling, or ankylosis of the tarsal joints, all of which were observedin all 14 of the WT animals tested, from day 6 to day 9.Histopathological analysis of the paws and other limb joints
showed signs of disease in all seven WT mice that were assessed,with more than three or four joints affected per mouse. Incontrast, only two of the seven TTPΔARE mice showed histo-logical signs of disease, with only one or two joints affected permouse. The WT mice exhibited inflammatory cell infiltrates,particularly neutrophils with a few macrophages, in the jointspace, as well as marked synovial hyperplasia, cartilage necrosis,bone erosion, and fibrosis; none of these lesions was present inthe TTPΔARE mice (Fig. 2C). The histopathology scores for thefour joints were consistently higher in WT mice compared withTTPΔARE mice, in which the four joints were almost entirelynormal, with the exception of one joint in one mouse, whichexhibited minimal signs of arthritis (Fig. 2D).Serum cytokine analysis revealed significantly decreased levels of
granulocyte-colony stimulating factor (G-CSF) and IL6 on day 7following CAIA induction in the TTPΔARE mice (Fig. 2 E and F).The levels of 23 other cytokines/chemokines tested were found to bevery low or undetectable. NanoString gene expression profiling (14)of the whole-joint RNA showed that 49 inflammatory transcripts (ofthe 248 total transcripts tested) were significantly down-regulated inthe joints from the TTPΔARE mice compared with WT joints(Table S3). Of these 49, at least 5 are known or suspected TTP
A B C
Subacute
Peri-
Synov
ialCe
ll
Hyperp
lasia
Fibros
is
0
1
2
3
4
WTTTP ARE∆
articu
lar A
rthrit
is
0
1
2
3
4
Subacute
Peri-
Synov
ialCe
ll
Hyperp
lasia
Fibros
is
articu
lar A
rthrit
is
ubacute
Peri-
Synov
ialCe
ll
Hyperp
lasia
Fibros
is
articu
lar A
rthrit
is
bacute
Peri-
Synov
ialCe
ll
Hyperp
lasia
Fibros
is
articu
lar A
rthrit
is
S uS
0
1
2
3
4
0
1
2
3
4
His
tolo
gy S
core
Left Elbow Left Tarsal Right Elbow Right Tarsal
D
TTP∆AREWT
LPS
0 3 6 9
0
2
4
6
8
10
DaysC
linic
alSc
ore
Collagen Ab
LPS
0 3 6 985
90
95
100
Days
Bod
yw
eigh
t(%
chan
ge)
*
E F
7
G-C
SF(p
g/m
l)
0
300
600
900
1200
1500
1800
2100*
0 9Days
IL6
(pg/
ml)
0
50
100
150
200 WT-SalineTTP∆ARE-salineWT-CAIATTP ARE-CAIA
*∆
0 7 9Days
WT TTP∆ARE
Fig. 2. Effect of the homozygous TTPΔARE mutation on CAIA. (A) Shown are the percent body weight changes and (B) clinical arthritis scores over a periodof 9 d of CAIA (n = 14, WT; n = 13,TTPΔARE). (C) Representative H&E-stained sections of tarsal joints from WT and TTPΔARE mice. Arrows indicate thepresence of intense inflammatory synovial cellular infiltrates in and around the joint spaces in the WT mice, but not in the TTPΔARE mice. (D) Histopathologyscores were determined for three lesions (i.e., subacute periarticular inflammation, synovial cell hyperplasia, and fibrosis). Severity was graded on a four-pointscale. Mean severity scores are shown (n = 7). (E) Serum levels of G-CSF and (F) IL6 from days 0, 7, and 9 of CAIA induction (n = 7). Statistical analysis wasperformed by two-tailed Student’s t test at each time point for A, B, E, and F and each lesion in D. Error bars represent SEM; *P < 0.05, **P < 0.01.
Patial et al. PNAS Early Edition | 3 of 6
IMMUNOLO
GYAND
INFLAMMATION

targets (2) (i.e., Ccl2, Ccl20, Cxcl1, Il1β, and Il6 mRNAs), and an-other 11 have typical TTP binding sites within their 3′UTRs. Thesedata demonstrate that the homozygous TTPΔARE animals werelargely protected against CAIA, possibly due to the relative down-regulation of inflammatory mediators.
Imiquimod-Induced Dermatitis in WT and TTPΔARE Mice. We nextevaluated the response to imiquimod (IMQ)-induced dermatitis, acommonly used model of human psoriasis (15). IMQ-containingcream was applied daily for five consecutive days to the shavedbacks of 7- to 9-wk-old mice. Clinical signs of psoriasis-like der-matitis, particularly erythema, scaling, and thickening, were ob-served by approximately day 2 or 3. The increase in skinfoldthickness was significantly lower in the TTPΔAREmice than in theWT mice (Fig. 3A).Skin pathology resembling psoriasis, particularly thickening of the
epidermis (acanthosis and hyperkeratosis), was observed in both theWT and the TTPΔARE mice; however, cellular infiltration into thedermis was significantly decreased in the TTPΔARE mice (Fig. 3B,Middle and Right). Moreover, epidermal neutrophilic abscesses/pustules were absent in the TTPΔARE mice (Fig. 3B, Middle). Inaddition, neutrophil infiltration into the dermis was markedly de-creased in the TTPΔARE mice (Fig. 3 C and D). Overall, the WTmice showed more severe epidermal hyperplasia, parakeratosis, anddermal inflammation, whereas the TTPΔARE mice showed morepronounced hyperkeratosis (Fig. 3E). NanoString gene expressionprofiling of day-6 skin RNA demonstrated 11 transcripts that weresignificantly down-regulated in TTPΔARE compared with WT skin(Fig. 3F). Four of the 11 transcripts are known or strongly suspectedTTP targets (i.e., Cxcl1, Il12b, Il1a, and Myc). The remaining seven,
C1qa, Cxcl3, Cxcr1, Cysltr1, Il1r1, Ptgir, and Tnfaip3, althoughnot known to be direct TTP targets, also play critical roles ininflammation. These results suggest that the TTPΔARE mice,although still susceptible to IMQ-induced dermatitis, exhibitedsignificantly decreased inflammation compared with WT mice.
Effect of the TTPΔARE Mutation on Experimental AutoimmuneEncephalomyelitis. Finally, we assessed the effect of the TTPΔAREmutation on one type of experimental autoimmune encephalomy-elitis (EAE), a mouse model for human multiple sclerosis (16). Weused the C57BL/6-MOG35-55 version of EAE, which has beendescribed recently as the “gold standard” animal model for multiplesclerosis (17). In this experiment, the entire population of WT (n =11) and homozygous TTPΔARE (n = 10) mice was given the en-cephalitogenic stimulus without knowledge of their genotype, withthe genotype code broken only at the end of the experiment. WTmice exhibited progressive weight loss beginning on approximatelyday 12, whereas the TTPΔARE mice did not lose body weight, onaverage (Fig. 4A). The WT mice, but not the TTPΔARE mice,began exhibiting clinical signs of EAE on approximately day 10. Byday 14, 8 of the 11 WT mice exhibited clinical signs of EAE, and anadditional WTmouse was killed because of a maximal clinical scoreof 5.0 on day 13. In contrast, none of the 10 TTPΔARE miceexhibited clinical signs of EAE by day 14. By day 20, another WTmouse was killed because it reached a near-maximal clinical score,and the remaining nine of nine mice exhibited severe clinical signs(clinical scores between 2.0 and 4.0) of EAE. Four of the 10TTPΔARE mice also exhibited clinical signs of EAE by day20; however, the signs exhibited by TTPΔARE mice were of mildseverity (clinical score between 0.5 and 2.0) (Fig. 4B). Another WT
A B
WT
TTPΔARE
0.00
0.07
0.14
0.21
0.28
0.35
*
Incr
ease
in s
kin
fold
thic
knes
s(m
m)
C D
WT TTP∆ARE 0
25
50
75
100
125
Num
bero
fcel
ls
**
E
-1
0
1
2
3
4
His
tolo
gySc
ore
WT
TP∆ARE WT
TTP∆ARE WT
TTP∆AREWT
TTP∆AREWT
TTP∆ARE
Epidermal Hyperplasia
Hyperkeratosis Parakeratosis ParakeratoticInflammation
DermalInflammation
**
**
**
*
WT
TTP∆
AR
E
Control IMQ
H&E
a
pu
d
a
d
IMQ
Transcript WT(mean ± SEM)
TTPΔARE(mean ± SEM)
P value
Known or suspected TTP target or
transcripts with potential TTP binding sites
C1qa 3533±334 2672±189 0.049Cxcl1 74.4±21.1 12.8±5.7 0.018 Known or strongly suspected TTP targetCxcl3 31.2±10.1 6.0±4.8 0.049Cxcr1 10.6±3.6 1.1±0.05 0.027Cysltr1 275.2±30.5 197.5±9.0 0.034Il12b 7.3±1.4 1.1±0.05 0.001 Known or strongly suspected TTP targetIl1a 924.0±112.7 645.6±41.4 0.042 Known or strongly suspected TTP targetIl1r1 1356±48.8 1117±38.2 0.003Myc 1511±105.8 1209±53.8 0.029 Known or strongly suspected TTP targetPtgir 60.4±3.6 42.9±6.5 0.04
Tnfaip3 339.5±22.8 267.0±18.8 0.034
F
Control IMQ
Ly6g
WT
TTP∆
AR
EFig. 3. Inflammation in IMQ-induced dermatitis. (A) Increase in skinfold thickness, measured as the difference between skin fold thickness on day 0 and day 6(n = 7–8). (B) Representative H&E staining of untreated (Left, control) or IMQ-treated (Middle and Right, IMQ) skin from mice of the indicated genotypes. a,acanthosis; d, dermal infiltration; pu, pustule. The right panel shows higher-power magnifications of the inset in the middle panel, demonstrating moreinfiltrating cells in the WT compared with the TTPΔARE section. (C) Shown are Ly6G immunostained sections from control (Left) and IMQ-treated (Right) miceof the indicated genotypes (n = 4–5). (D) Ly6G positive cells in Fig. 4C were quantitated as the number of cells in one high-power field (400×) in an area ofmaximal infiltration from each animal (n = 4–5). (E) Histopathology scores were determined for five lesions, namely, epidermal hyperplasia, hyperkeratosis,parakeratosis, parakeratotic inflammation, and dermal inflammation. Severity was graded on a four-point scale. Each mean score is depicted by a black lineand each point represents data from one animal (n = 7–8). (F) NanoString gene expression analysis was performed on RNA isolated from affected skin. Shownare the normalized counts for the 11 transcripts that were significantly down-regulated in the TTPΔARE group compared with the WT group (n = 6). Statisticalanalysis was performed by two-tailed unpaired Student’s t test for A, D, E, and F. Error bars represent SEM; *P < 0.05, **P < 0.01.
4 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1519906113 Patial et al.

mouse was killed on day 27. By day 30, 8 of the original 11 WTmice remained, all of which exhibited clinical signs of severe EAE,but 10 of the 10 original TTPΔARE mice remained, of which only5 mice exhibited clinical signs of mild EAE. The mean cumulativedisease scores and the mean maximal disease scores for theTTPΔARE mice were significantly lower than those of the WTmice (Fig. 4C). These data demonstrate that the TTPΔARE micewere markedly resistant to the induction of this model of EAEcompared with the WT mice.
DiscussionThe severe inflammatory syndrome seen in the TTP KOmice led tothe identification of Tnf mRNA as a major target of TTP (4, 6). Asa result, the emphasis in most follow-up studies has been on theinvolvement of TTP in innate immunity. More recently, severalstudies have highlighted the importance of other aspects of theimmune response in the pathogenesis of the TTP deficiency syn-drome. For example, mice in which TTP was deleted only frommyeloid cells did not exhibit the TTP deficiency syndrome, althoughthey were hypersensitive to small amounts of injected LPS (11),suggesting the involvement of other cell types in the development ofthe syndrome. In another study, genetic ablation of genes in theIL17/IL23 axis protected mice against the development of the TTPdeficiency syndrome, even in the setting of normal TNF pathwaysand responses (12). These recent studies highlight the concept thatTTP deficiency in an intact animal affects many arms of the im-mune system and is a true systemic disease.Since the earliest discovery of TTP as an endogenous antiin-
flammatory protein, we have been interested in the concept ofattempting to increase levels or activities of TTP in animals, andpotentially in humans, as a possible treatment for inflammatoryconditions. Our initial attempts to accomplish this involved trans-genic mice in which TTP was overexpressed using strong generalpromoters; these invariably led to embryonic lethality. In a differentapproach to increasing TTP expression without loss of its normalphysiological regulation, we developed the knock-in TTPΔAREmouse described in this study, in which an ARE instability elementwas genetically removed from TTP mRNA. Our hope was that thisgerm-line mutation would lead to stabilization of TTP mRNA andoverexpression of the protein throughout the body, without pre-conceived biases about which cell types should be preferentiallytargeted. Similar genetic approaches to the stabilization of ARE-
containing mRNAs have been used in the cases of TNF (18) andIFN-γ (19), among others.Remarkably, the TTPΔARE mice appeared phenotypically
normal, despite elevated and stabilized TTP mRNA in cells derivedfrom them, and increased levels of TTP protein in several tissues.We then tested the hypothesis that the increased “whole-body”levels of TTP might protect the mice against inflammation in fourdistinct experimental models that involved different aspects of theimmune system. First, we tested the response of the TTPΔAREmice to LPS endotoxemia. Somewhat surprisingly, the TTPΔAREmice did not exhibit decreased levels of TNF in the serum after thei.p. injection of three different doses of LPS. However, serum levelsof IL10, whose mRNA is also a known TTP target (20), were lowerin the mutant mice than in the WT mice at the highest LPS doseand returned more rapidly to baseline levels in the TTPΔAREmicethan in the WT mice at all LPS doses. These results suggest that theacute TNF response to LPS was not affected by further increasingendogenous levels of TTP, which are already massively induced inmyeloid cells by LPS in parallel with TNF (11).The second disease model tested was CAIA, often used as an
experimental model of human rheumatoid arthritis (21). We foundthat TTPΔAREmice were essentially completely protected againstthe development of CAIA. The mechanism of this protection islikely to involve the decreased stability of proinflammatory cyto-kine mRNAs in many cells and tissues. For example, we found thatserum levels of G-CSF and IL-6 were significantly reduced inTTPΔARE mice 7 d after the start of the CAIA protocol. Il-6mRNA has been suggested previously to be a target of TTP (22),whereas, to our knowledge, Csf1 mRNA, encoding G-CSF, has notbeen identified as a direct target. However, the 3′UTR of Csf1mRNA in the mouse contains two sequence elements that formthe core of ideal TTP binding sites, UAUUUAU, and several otherclosely related sequences; many of these, including the two core7-mers, are widely conserved among mammals, including humans.G-CSF levels have been shown to be elevated in TTP KO mouseplasma (23), and G-CSF (24) KO mice are protected from colla-gen-induced arthritis. G-CSF is known for its role in maintaininghomeostatic levels of granulocytes (25), and G-CSF administrationresults in neutrophil production (26). G-CSF also enhances neu-trophil trafficking in the synovium, and neutrophil depletion arreststhe progression of arthritis (27), suggesting that G-CSF plays acritical role in mediating inflammatory arthritis by promoting thetrafficking of neutrophils into the synovium. It seems reasonable tosuggest that, among other proinflammatory pathways affected bythe increased levels of TTP, the decreased levels of G-CSF mayhave resulted in decreased manifestations of CAIA.Gene expression profiling of whole-joint RNA revealed decreases
in the levels of at least five known or suspected TTP targets in theTTPΔARE mice, whereas at least 11 other significantly down-reg-ulated transcripts contained potential TTP binding sites and encodeproteins that play critical roles in inflammation. The remaining 33down-regulated transcripts encode components of the complementpathway, toll-like receptor signaling, cytokines/chemokines and theirreceptors, and enzymes that break down extracellular matrix, sup-porting the idea that increased levels of TTP can directly and per-haps indirectly regulate immune response transcripts and protectagainst inflammation.We also explored the response of the TTPΔARE mice to IMQ-
induced dermatitis. We found that the TTPΔARE mice hadless severe epidermal hyperplasia and strikingly reduced neutrophilinfiltration. A previous study demonstrated that neutrophil in-filtration was considerably reduced in IMQ-induced dermatitis inIl1r1 KO mice (28). Interestingly, neutrophil depletion has beenshown to ameliorate the severity of disease in this model of psoriasis(29), and clinical studies have demonstrated disease remission bydrug-induced agranulocytosis, supporting a critical pathogenic roleof neutrophils in psoriasis (30). We found that Il1r1 mRNA ex-pression was significantly reduced in skin from the TTPΔAREmice, suggesting that decreased IL-1R1 signaling may have resultedin reduced dermal neutrophilic infiltration. Four other known orsuspected TTP targets were also decreased in the TTPΔARE mice
A B
C0 5 10 15 20 25 30
80
90
100
110
120
Mea
n%
Initi
alW
eigh
t
TTP∆AREWT
0 5 10 15 20 25 300
1
2
3
4
Clin
ical
Seve
rity
ofEA
E
Days Since Encephalitogenic Challenge
Experimental Group
Prevalence of EAE
Mean Cumulative
Score
Median Cumulative
Score
Mean Max Score
Median Max Score
TTPΔARE 7 of 10 11.7 ± 3.6 11.0 1.7 ± 0.4 2.0Wild Type 11 of 11 51.6 ± 6.7 59.5 3.9 ± 0.2 4.0
Fig. 4. Effect of the TTPΔARE mutation on EAE. (A) Shown are means ± SEMof the percent body weight loss and (B) clinical disease scores observed over aperiod of 30 d during the course of EAE (n = 11,WT; n = 10,TTPΔARE).(C) Shown are data on the prevalence, the cumulative disease scores (meanand median), and the maximal disease scores (mean and median) for the WTand TTPΔARE groups. Cumulative EAE scores were calculated by summingdaily scores for each mouse across the designated time course of disease.Maximal scores were calculated as the most severe EAE score for eachmouse. Mice that did not exhibit EAE had a score of zero for the cumulativeand maximal scores, and these scores were included in the group average.Pairwise comparisons were analyzed by two-tailed t tests. Cumulative scores,P = 0.003; maximal scores, P = 0.0018.
Patial et al. PNAS Early Edition | 5 of 6
IMMUNOLO
GYAND
INFLAMMATION

skin: Cxcl1, Il12a, Il1a, and Myc mRNAs, and seven additionalsignificantly reduced transcripts, including C1qa, Cxcl3, Cxcr1,Cysltr1, Il1r1, Ptgir, and Tnfaip3 mRNAs, encode important pro-inflammatory proteins. These data support the idea that increasedTTP levels exert a beneficial effect on the course of this diseasemodel by targeting elevated levels of certain cytokines, some ofwhich are direct TTP targets and others that may represent indirectresponders.The fourth disease model tested was EAE (16). Many types of
observation have suggested critical roles for cytokines and chemo-kines, many of whose transcripts are TTP targets, in the pathogenesisof both EAE and human multiple sclerosis (31, 32). Similar to theresults obtained with CAIA, the homozygous TTPΔARE mice werestrikingly resistant to active EAE induction, presumably due to de-creased expression of inflammatory mediators in the blood and CNSfollowing immunization.Taken together, our results support the hypothesis that moder-
ately elevated levels of TTP throughout the body can have beneficialeffects in some mouse models of immune and inflammatory disease.Current therapy of several human diseases of this type involves an-tibody-based molecules that bind directly to the relevant cytokine,such as anti-TNF compounds (33); these are produced by expensiverecombinant DNA techniques and, because of their protein nature,require parenteral administration. Our data support the concept thattherapies aimed at increasing levels or activities of TTP in cellsthroughout the body might benefit patients with these conditions.One possible approach to this is gene therapy; indeed, a previousstudy demonstrated protective effects of adenovirus-delivered TTPin an experimental periodontitis model (34). Other approachesmight involve cell-based therapies, such as hematopoietic stem cell
transplantation of genetically modified cells (35). Ideally, smallmolecules could be identified that, when administered orally, couldcause elevations of TTP throughout the body. If such compoundscould be identified that had minimal toxicity, it seems reasonable tosuggest that they might be useful therapeutically in the treatment ofcertain chronic inflammatory diseases.
Materials and MethodsTTPΔARE knock-in mice with a 136-bp deletion of an AU-rich region of theZfp36 3′UTR were generated at Ozgene using C57BL/6 embryonic stem cellsand standard targeting techniques. Adult male homozygous mutant micewere injected intraperitoneally with three different doses of LPS as acutemodels of inflammation. They were also subjected to the following modelsof inflammatory disease: CAIA (13), IMQ-induced dermatitis (36), and themyelin oligodendrocyte glycoprotein MOG35-55 peptide-induced model ofEAE (37). Detailed materials and methods are contained in SI Materialsand Methods.
All animal procedures were conducted according to US Public Health Servicepolicy on the humane care and use of laboratory animals. The National Instituteof Environmental Health Sciences Institutional Animal Care and Use Committeeapproved all animal procedures used in this study. The East Carolina UniversityInstitutional Animal Care and Use Committee approved the animal protocolused to induce EAE.
ACKNOWLEDGMENTS. We thank Dr. Dave Brar for performing the multiplexcytokine assays, Debra King and Page Myers for technical assistance, Drs. DoriGermolec and Michael Fessler for useful comments on the manuscript, and theHistology and Immunohistochemistry Core and the Molecular Genomics Corefor help with histology and NanoString assays, respectively. This research wassupported by the Intramural Research Program of the NIH, National Institute ofEnvironmental Health Sciences.
1. Blackshear PJ (2002) Tristetraprolin and other CCCH tandem zinc-finger proteins inthe regulation of mRNA turnover. Biochem Soc Trans 30(Pt 6):945–952.
2. Brooks SA, Blackshear PJ (2013) Tristetraprolin (TTP): Interactions with mRNA andproteins, and current thoughts on mechanisms of action. Biochim Biophys Acta1829(6-7):666–679.
3. Taylor GA, et al. (1996) A pathogenetic role for TNF alpha in the syndrome of ca-chexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency.Immunity 4(5):445–454.
4. Carballo E, Lai WS, Blackshear PJ (1998) Feedback inhibition of macrophage tumornecrosis factor-alpha production by tristetraprolin. Science 281(5379):1001–1005.
5. Carballo E, Blackshear PJ (2001) Roles of tumor necrosis factor-alpha receptor subtypes inthe pathogenesis of the tristetraprolin-deficiency syndrome. Blood 98(8):2389–2395.
6. Lai WS, et al. (1999) Evidence that tristetraprolin binds to AU-rich elements andpromotes the deadenylation and destabilization of tumor necrosis factor alphamRNA. Mol Cell Biol 19(6):4311–4323.
7. Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ (2006) Novel mRNA targets fortristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol Cell Biol 26(24):9196–9208.
8. Lai WS, Stumpo DJ, Blackshear PJ (1990) Rapid insulin-stimulated accumulation of anmRNA encoding a proline-rich protein. J Biol Chem 265(27):16556–16563.
9. Tchen CR, Brook M, Saklatvala J, Clark AR (2004) The stability of tristetraprolin mRNAis regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself.J Biol Chem 279(31):32393–32400.
10. Cao H, Tuttle JS, Blackshear PJ (2004) Immunological characterization of triste-traprolin as a low abundance, inducible, stable cytosolic protein. J Biol Chem 279(20):21489–21499.
11. Qiu LQ, Stumpo DJ, Blackshear PJ (2012) Myeloid-specific tristetraprolin deficiency inmice results in extreme lipopolysaccharide sensitivity in an otherwise minimal phe-notype. J Immunol 188(10):5150–5159.
12. Molle C, et al. (2013) Tristetraprolin regulation of interleukin 23 mRNA stabilityprevents a spontaneous inflammatory disease. J Exp Med 210(9):1675–1684.
13. Khachigian LM (2006) Collagen antibody-induced arthritis. Nat Protoc 1(5):2512–2516.
14. Geiss GK, et al. (2008) Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 26(3):317–325.
15. Flutter B, Nestle FO (2013) TLRs to cytokines: Mechanistic insights from the imiquimodmouse model of psoriasis. Eur J Immunol 43(12):3138–3146.
16. Miller SD, Karpus WJ (2007) Experimental autoimmune encephalomyelitis in themouse. Curr Protoc Immunol 78(15.1):15.1.1–15.1.18.
17. Ben-Nun A, et al. (2014) From classic to spontaneous and humanized models ofmultiple sclerosis: Impact on understanding pathogenesis and drug development.J Autoimmun 54:33–50.
18. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G (1999) Impaired on/offregulation of TNF biosynthesis in mice lacking TNF AU-rich elements: Implications forjoint and gut-associated immunopathologies. Immunity 10(3):387–398.
19. Hodge DL, et al. (2014) IFN-gamma AU-rich element removal promotes chronic IFN-gamma expression and autoimmunity in mice. J Autoimmun 53:33–45.
20. Stoecklin G, et al. (2008) Genome-wide analysis identifies interleukin-10 mRNA astarget of tristetraprolin. J Biol Chem 283(17):11689–11699.
21. Nandakumar KS, Holmdahl R (2005) Efficient promotion of collagen antibody in-duced arthritis (CAIA) using four monoclonal antibodies specific for the major epi-topes recognized in both collagen induced arthritis and rheumatoid arthritis.J Immunol Methods 304(1-2):126–136.
22. Zhao W, Liu M, D’Silva NJ, Kirkwood KL (2011) Tristetraprolin regulates interleukin-6expression through p38 MAPK-dependent affinity changes with mRNA 3′ un-translated region. J Interferon Cytokine Res 31(8):629–637.
23. Kaplan IM, et al. (2011) Deletion of tristetraprolin caused spontaneous reactivegranulopoiesis by a non-cell-autonomous mechanism without disturbing long-termhematopoietic stem cell quiescence. J Immunol 186(5):2826–2834.
24. Lawlor KE, et al. (2004) Critical role for granulocyte colony-stimulating factor in in-flammatory arthritis. Proc Natl Acad Sci USA 101(31):11398–11403.
25. Lieschke GJ, et al. (1994) Mice lacking granulocyte colony-stimulating factor havechronic neutropenia, granulocyte and macrophage progenitor cell deficiency, andimpaired neutrophil mobilization. Blood 84(6):1737–1746.
26. Roberts AW (2005) G-CSF: A key regulator of neutrophil production, but that’s not all!Growth Factors 23(1):33–41.
27. Eyles JL, et al. (2008) A key role for G-CSF-induced neutrophil production and traf-ficking during inflammatory arthritis. Blood 112(13):5193–5201.
28. Tortola L, et al. (2012) Psoriasiform dermatitis is driven by IL-36-mediated DC-kera-tinocyte crosstalk. J Clin Invest 122(11):3965–3976.
29. Sumida H, et al. (2014) Interplay between CXCR2 and BLT1 facilitates neutrophil in-filtration and resultant keratinocyte activation in a murine model of imiquimod-induced psoriasis. J Immunol 192(9):4361–4369.
30. Toichi E, Tachibana T, Furukawa F (2000) Rapid improvement of psoriasis vulgarisduring drug-induced agranulocytosis. J Am Acad Dermatol 43(2 Pt 2):391–395.
31. Axtell RC, Steinman L (2009) Gaining entry to an uninflamed brain. Nat Immunol10(5):453–455.
32. Reboldi A, et al. (2009) C-C chemokine receptor 6-regulated entry of TH-17 cells intothe CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol10(5):514–523.
33. Taylor PC, Feldmann M (2009) Anti-TNF biologic agents: Still the therapy of choice forrheumatoid arthritis. Nat Rev Rheumatol 5(10):578–582.
34. Patil CS, et al. (2008) Targeting mRNA stability arrests inflammatory bone loss. MolTher 16(10):1657–1664.
35. Cieri N, et al. (2014) Adoptive immunotherapy with genetically modified lymphocytesin allogeneic stem cell transplantation. Immunol Rev 257(1):165–180.
36. van der Fits L, et al. (2009) Imiquimod-induced psoriasis-like skin inflammation in miceis mediated via the IL-23/IL-17 axis. J Immunol 182(9):5836–5845.
37. Islam SM, Curtis AD, 2nd, Taslim N, Wilkinson DS, Mannie MD (2014) GM-CSF-neuroantigen fusion proteins reverse experimental autoimmune encephalomyelitisand mediate tolerogenic activity in adjuvant-primed environments: Association withinflammation-dependent, inhibitory antigen presentation. J Immunol 193(5):2317–2329.
38. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4):402–408.
6 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1519906113 Patial et al.