endothelin synthesis release fromparathyroid cells · were analyzed by the computer model expfit...
TRANSCRIPT

Proc. Natl. Acad. Sci. USAVol. 88, pp. 6496-6500, August 1991Medical Sciences
Natriuretic peptide receptors regulate endothelin synthesis andrelease from parathyroid cells
(atrial natriuretic peptide/brain natriuretic peptide/peptide hormone receptor/hypertension)
MARIA LAURA DE FEO*, OLGA BARTOLINI*, CLAUDIO ORLANDO*, MARIO MAGGI*, MARIO SERIO*,MARK PINESt, SHMUEL HURWITZt, YOSHIO FUJII*, KAZUSHIGE SAKAGUCHI4,GERALD D. AURBACHt, AND MARIA LUISA BRANDI*§*Department of Clinical Physiopathology, University of Florence, Viale Pieraccini 6, 50139 Florence, Italy; tInstitute of Animal Science, Agricultural ResearchOrganization, The Volcani Center, Bet Dagan 50250, Israel; and tMetabolic Diseases Branch, National Institute of Diabetes and Digestive and KidneyDiseases, National Institutes of Health, Bethesda, MD 20892
Contributed by Gerald D. Aurbach, April 4, 1991
ABSTRACT Cloned rat parathyroid cells (PTr cell line)that produce parathyroid hormone-related peptide plus endo-thelin 1 and primary cultures of human parathyroid cells weretested for growth and differentiation responses to atrial natri-uretic peptide (ANP) and brain natriuretic peptide (BNP).High- and low-affinity binding sites for ANPwere found on PTrcells; BNP appeared to bind to the same receptors with similaraffinities. Either ANP or BNP stimulated production ofcGMPand caused a 30% decrease in Na'-K+-Cl cotransport. Eachpeptide increased synthesis and secretion of endothelin 1 byPTr cells in a dose-dependent fashion, but cell growth was notaffected. Human parathyroid cells (normal and pathological)also responded to ANP or BNP with an increase in cGMPproduction. The finding of receptors for natriuretic hormoneson parathyroid cells with consequent effects on release ofendothelin 1 might be of relevance in understanding the clinicalassociation between hyperparathyroidism and hypertension.
Classical studies have established the physiological role ofparathyroid hormone (PTH) in regulating calcium homeosta-sis through effects on its recognized target tissues, bone andkidney (1). However, the biological significance of otheractions of the parathyroid glands, particularly the inhibitionof smooth muscle contraction in blood vessels and othertissues, is not fully understood (2). Recent research hasshown the parathyroids to be the source of ancillary peptidessuch as PTH-related peptide (PTHrP; refs. 3 and 4) andendothelin 1 (ET-1; refs. 5-7). The latter peptides exertopposing biological effects on smooth muscle, PTHrP inhib-iting and ET-1 stimulating contraction (8, 9). Such observa-tions suggest that the parathyroid glands may physiologicallymodulate vascular smooth muscle tone through the release ofaccessory peptides. Clinical findings supporting this hypoth-esis are the hypertension that often accompanies primaryhyperparathyroidism and the pattern of altered calcium me-tabolism frequently observed in experimental and humanhypertension (see ref. 10 for review). Note also the putative"parathyroid hypertensive factor" described in extracts ofparathyroid tissue from the SHR (spontaneously hyperten-sive) rat (11). A functional interaction between PTH andatrial natriuretic peptide (ANP) in modulating cyclic nucle-otide accumulation and cell proliferation has been observedin chondrocytes and osteoblasts (12, 13). These observations,plus the well-known hypotensive effect of ANP (14), led usto evaluate the possible direct actions of the peptide and ofthe recently characterized brain natriuretic peptide (BNP;ref. 15) on parathyroid cell cultures.
MATERIALS AND METHODSCell Culture. Two systems, a parathyroid cell line obtained
from hyperplastic rat parathyroid glands (PTr cells; ref. 16)and primary cultures of cells from human parathyroid tissue,were used to characterize receptor binding and biologicalresponses to the natriuretic hormones. PTr cells were cul-tured in growth medium consisting of a 1:1 mixture ofDulbecco's modified Eagle's minimal essential medium(DMEM) and Coon's modified Ham's F12 with supplementsas described (16). PTr cells were detached by 0.05% trypsinin phosphate-buffered saline (PBS)/2 mM EDTA and platedon 100-mm dishes. At confluence, growth medium wasremoved and replaced with identical medium but withoutserum or serum substitutes (steady-state medium) and con-taining aprotinin (500 kallikrein inhibitor units/ml; Sigma).The medium was collected on ice at the indicated times afterthe stimulus, centrifuged at 40C, and stored at -20'C untilassayed.Human parathyroid tissue was obtained from patients
undergoing surgery for hyperparathyroidism (parathyroidhyperplasia due to multiple endocrine neoplasia type I, n =2; secondary hyperparathyroidism, n = 1; parathyroid ade-noma, n = 1). Parathyroid specimens were minced in smallfragments with a scalpel and digested with a solution ofPBScontaining collagenase (1 mg/ml, type IV; Sigma) at 37TC ina humidified atmosphere containing 5% CO2. After 8-9 hr ofdigestion, tissue fragments were washed twice with PBS andthen mechanically dispersed by pipet. The cell suspensionwas centrifuged and then plated in Petri culture dishes withCoon's modified Ham's F12 medium containing 20% fetalbovine serum. After 4-6 days of primary culture, cells weredetached with trypsin and plated in 16-mm wells (multiwellplates) for experimental procedures.
Binding Studies. PTr cells were grown to subconfluency in16-mm-well plates and then incubated in 20 mM Hepes/DMEM, pH 7.8/0.2% bovine serum albumin at 220C for 30min with 1251I-labeled rat ANP (125I-rANP; specific activity,2000 Ci/mmol; Amersham; 1 Ci = 37 GBq) plus unlabeledrANP or porcine BNP (pBNP) (Peninsula Laboratories) atconcentrations shown. The cells were washed with ice-coldPBS/0.2% bovine serum albumin and solubilized in 1 MNaOH, and extracts were analyzed for radioactivity bycrystal scintillation detection. Association kinetics were de-termined from the rate of specific binding of 0.1 nM (200,000cpm) 125I-rANP. The dissociation rate was determined after
Abbreviations: ANP, atrial natriuretic peptide; hANP and rANP,human and rat ANP; BNP, brain natriuretic peptide; pBNP, porcineBNP; ET-1, endothelin 1; PTH, parathyroid hormone.§To whom reprint requests should be addressed.
6496
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.

Proc. Natl. Acad. Sci. USA 88 (1991) 6497
addition of unlabeled rANP or pBNP (2 ,M) to the reactionmixture at equilibrium.
Cyclic Nucleotide Production. PTr or human parathyroidcells were incubated for the indicated times at 370C withvarious concentrations of rANP, human ANP (hANP), orpBNP in steady-state medium containing 1 mM 3-isobutyl-1-methylxanthine (Sigma). Total (cells and medium) cGMPcontent was assayed by RIA (cGMP RIA kit; DuPont/NEN)after acetylation of samples and standards.
Total cAMP was determined by RIA after ethanol extrac-tion (17) under basal conditions or upon addition of forskolin(0.1 ,uM; Sigma) or pertussis toxin (100 ng/ml; List BiologicalLaboratories, Campbell, CA) with or without hANP or pBNPat 1-1000 nM.
Ion Transport. Rubidium-86 (DuPont/NEN) uptake wasdetermined as a measure of Na'-K+-Cl- cotransport andNa+/K' pump activity in PTr cells as described by O'Don-nell (18). Na'-K+-Cl- cotransport was determined as oua-bain-insensitive, furosemide-sensitive K+ flux [i.e., (K+ fluxin incubation with ouabain) - (K+ flux with ouabain plusfurosemide)]. Na+/K+-ATPase-mediated K+ influx was cal-culated as ouabain-sensitive flux [i.e., (total flux) - (flux inmedium with ouabain)]. Protein content was determinedusing the Pierce protein assay reagent based on the methodof Bradford (19).
Cell Proliferation. PTr cells were incubated on microplatesfor 24 hr at 37°C in serum- and serum substitute-free mediumwithout phenol red. Various concentrations of hANP orpBNP (10 pM to 1 ,uM) were added (five wells for eachexperimental point) and incubations continued for another 24hr at 37°C. Cells were then washed with PBS and incubatedfor 45 min in the same steady-state medium containingHoechst 33342 dye (10lg/ml; Polysciences). Chromatin-associated fluorescence was evaluated in a fluorescencemicrotiter plate reader (Titertek Fluoroscan 11; Flow Labo-ratories) as described (20).Northern Blot Analysis. PTr cells were grown in 100-mm
plates. Subconfluent cultures were incubated with mediumcontaining 0.1 AM ANP or 0.1 ,uM pBNP for 15, 30, 60, 120,or 360 min. Cells were lysed and total RNA was extracted atthe end of incubation as described (7). Samples of total RNA(20 ,ug per lane) were fractionated by electrophoresis in a 1%agarose/formaldehyde gel and transferred to a nylon mem-brane (Nytran; Schleicher & Schuell). 32P-labeled antisenseRNA probe for preproendothelin 1 (preproET-1) was pre-pared as described (7). Rat cyclophilin cDNA (21) wasradiolabeled with 32P by nick-translation and used as acontrol probe. Hybridization with RNA or cDNA probes andautoradiography were carried out as described (7). Densito-metric scans of the autoradiograms were performed on anLKB 2202 Ultroscan laser densitometer.
Peptide Secretion by PTr Cells. The effects of hANP andpBNP on ET-1 release by PTr cells were tested in cellsincubated at 37°C for 0.5, 1, 2, 6, or 24 hr with the natriuretichormones. The medium (3 ml) was extracted with Sep-PakC18 cartridges (Waters) and ET-1 concentrations were deter-mined by RIA (22). The sensitivity of the assay was 0.5 pg pertube (7.2% coefficient of variation for intraassay error). PTrcells were also analyzed for ANP- and BNP-like immunore-activity by RIA on concentrated extracts ofPTr medium (23);BNP RIA was performed with a commercial kit (PeninsulaLaboratories). The sensitivity of the assays was 0.7 pg per100 Al and 0.9 pg per 100 ,ul for ANP and BNP, respectively(intraassay coefficient of variation, 7.5 and 9%, respectively).
Analysis of Experimental Results. Families of competitivebinding curves for rANP or pBNP receptor interactions wereanalyzed simultaneously with a nonlinear least-squares fit-ting procedure (LIGAND program; ref. 24). Dissociation datawere analyzed by the computer model EXPFIT (25), using abiexponential decay model. The computer program ALLFIT
(26) was used for the analysis of sigmoidal dose-responsecurves obtained in cGMP production studies. Statisticalsignificance was evaluated by one-way analysis of varianceand by Duncan's new multiple range test (27).
RESULTSBinding Studies. The binding of 125I-rANP to PTr cells at
220C was a linear function of the number of cells addedbetween 2 x 105 and 9 x 105 cells (correlation coefficient r =0.986; n = 2); specific binding became maximal at 30-40 min(Fig. lA). The simultaneous computer analysis of threedissociation curves with the program EXPFIT indicated thatthe introduction of a second exponential function signifi-cantly increased the goodness of fit (P < 0.005; k0fn = 2.1988± 1.5292 min-' and kff2 = 0.0364 ± 0.0072 min-', mean ±SD) (Fig. 1B). Mathematical modeling of competition curvesamong 1251-rANP, the corresponding unlabeled peptide, andBNP strongly indicated heterogeneity ofANP binding sites inPTr cells. Indeed, the introduction of a second independentclass of sites significantly improved the goodness of fit (P <0.001). Homologous and heterologous competition curves forrANP and pBNP, obtained by use of a two-site model, areshown in Fig. 2. Both ligands bind with high affinity (rANPpK = 10.7 + 0.24; pBNP pK = 11.04 + 1.6, mean ± SD) tothe low-capacity site (R1, 0.308 ± 0.178 fmol per 106 cells,mean ± SD), with lower affinity (rANP pK = 6.91 ± 0.13;pBNP pK = 6.48 ± 0.16, mean ± SD) for the high-capacitysite (R2, 2182 ± 1255 fmol per 106 cells, mean ± SD).ANP and BNP Increase cGMP Production. cGMP accumu-
lation in PTr cells incubated with 10 nM rANP or pBNPincreased significantly within 1 min, with a further increaseup to 60 min (Fig. 3A). A sharp increase in cGMP productionwas observed with 1 nM rANP, hANP, or pBNP, with EC50values of 15.7 ± 7.8, 33.5 ± 8.4, and 28 ± 6.5 nM (mean ±SD), respectively (Fig. 3B). cGMP content markedly in-
ll 40-
'820.
0
80-
ZA.
A 0 0
0
30 60 90
B
20-~~~~~~
10 20Time, min
30
FIG. 1. Rate of association (A) and dissociation (B) of 1251-rANPfrom binding sites on PTr cells. Cells were incubated for the indicatedtimes at 220C with 0.1 nM 125I-rANP with or without 1 AuM rANP.Results are expressed as percentage of maximum specific binding(calculated as the difference between total and specific binding). Datarepresent the mean of two (A) or three (B) experiments each usingsamples from triplicate wells.
Medical Sciences: De Feo et al.
Du
40 -
20-
n

6498 Medical Sciences: De Feo et al.
m
U.UU-.
0
0.010 - 0 0
0.005-12 -10 -8
T. log M
-6
FIG. 2. Binding of rANP and pBNP to PTr cells. Analyses oftracer (125I-rANP, 1-100 pM) binding and homologous (rANP, *) orheterologous (pBNP, A) competitive inhibition experiments with 0.1nM tracer were derived from the simultaneous fit of 14 curveswherein each point represented triplicate analyses. B, bound; T,total.
creased in response to natriuretic hormones (1-100 nM) inprimary cultures of human parathyroid tissue (Fig. 4).cAMP production by PTr cells was not affected by hANP
or pBNP up to micromolar concentrations under basal con-ditions or after incubation with forskolin or with pertussistoxin. Neither was there an effect on cultured human cells(data not shown).hANP and pBNP Decrease Na+-K+-CI- Cotransport But
Not Na+/K+ Pump Activity of PTr Cells. Na'-K-Cl-cotransport was assessed in PTr cells as furosemide-sensitive'Rb+ influx with ouabain. A Na'-K-Cl- cotransport sys-tem was found in PTr cells (Table 1). Treatment of cells withhANP or pBNP (100 nM) significantly reduced the cotrans-port (down 30% with respect to baseline; P < 0.001 and P <0.002, respectively); no effect was evident on Na+/K+ pumpactivity.hANP and pBNP Do Not Affect PTr Cell Growth. Prolifer-
ation of PTr cells was not affected by the addition of hANPor pBNP over a wide range of concentrations (1 pM to 1 ,uM)for 24 hr (10.2 0.8 and 9.8 ± 2.7 fluorescence units afterstimulus with 1 ,M hANP or pBNP vs. 11.7 ± 3.6 and 11.8
20 40Time, min
C -12 -8 -4log M
0
3 '
2 'oE
12.CO1
FIG. 3. (A) Rate of cGMP production by PTr cells with ANP orBNP. Cells were incubated at 37°C with no additions (o) or with 10nM rANP (-) or pBNP (-). cGMP concentrations was determined asindicated in Materials and Methods. Data represent the mean oftwoexperiments carried out with triplicate samples. (B) Dose-responseanalyses for ANP- or BNP-stimulated cGMP production with PTrcells in 20-min incubations: A, rANP; e, hANP; *, pBNP. C, control(zero concentration). Data represent the mean of two experimentseach carried out with quadruplicate samples.
10 X
-9 -8 -7 -9 -8 -7hANP,0g M PBNP, M
0~~~ ~ ~ ~ ~ ~~~~0
FIG. 4. Effects of hANP (A) and pBNP (B) on cGMP productionby primary cultures of cells derived from human parathyroid ade-nomas (n), secondary hyperparathyroidism (A), or multiple endo-crine neoplasia, type I (A, o). Cells were treated as described for PI rcells. Results shown are the averages of triplicate analyses and areexpressed as percent of control.
3.7 fluorescence units in controls, respectively; mean +SD, n = 5).hANP and pBNP Increase ET-1Synthesls and Release.
Northern blot hybridization withyihesmP-labeled antisenseRNA probe for preproET-1 showed a considerable increasein message for ET-1 with 0.1a,uM ANP or BNP (Fig. 5). Theresponse was evident within 15 min and became maximal at2 hr.A significant increase in ET-1 content in the culture
medium of PTr cells was detectable after 24-hr incubationswith ANP orBNP (control, 23.7 ± 4.2 pg per 106 cells; 0.1 ,uMhANP and pBNP, 33.4 ± 5.7 and 32.5 + 3.2 pg per 106 cells,respectively, P < 0.05 vs. control; 1 ,uM hANP and pBNP,39.0 ± 6.8 and 40.2 ± 4.8 pg per 106 cells, respectively, P <0.001 vs. control) (Fig. 6). No effect was detectable at shorterincubation times.ANP- and BNP-Like Material in Medium from PTr Cells.
There was no detectable immunoreactivity for ANP or BNPin culture medium derived from PTr cells (3-5 x 106 cells perdish) kept at 37°C for 1, 2, 6, 24, or 48 hr (n = 5; data notshown).
DISCUSSIONThe potential influence of the parathyroids on the cardiovas-cular system was first recognized in 1925 (28). Much later itwas found that PTH causes vasodilatation and increases
Table 1. Effects of ANP and BNP on Na'-K-CPl cotransportand Na+/K+ pump activity in PTr cells
86Rb+ influx, itmol/min per mg of proteinNa+-K+-Cl
Agent cotransport Na+/K+ pump K+ leakNone 0.202 ± 0.010 0.319 ± 0.047 0.043 ± 0.004hANP 0.137 ± 0.006* 0.334 ± 0.136 0.039 ± 0.009None 0.179 ± 0.012 0.563 ± 0.049 0.101 ± 0.005pBNP 0.122 ± 0.005t 0.484 ± 0.092 0.094 ± 0.036
Cells were kept for 10 min in 10mM Hepes/DMEM (Na+, 150mM;K+, 6mM; Cl-, 150 mM) at 37rC, then incubated in the same mediumcontaining 1 mM ouabain plus 1 mM furosemide (Sigma) and/or 0.1A&M hANP or pBNP. After aspiration of the medium, the cells wereincubated for 5 min in identical fresh medium containing I*RbCl (1,uCi/ml). 86Rb uptake was terminated after aspiration of the mediumand extensive washing. Intracellular 86Rb was determined by liquidscintillation spectrometry. Data are the mean ± SD of quadruplicateexperimental points from one of three similar experiments.*P < 0.001 with respect to basal.tp < 0.002 with respect to basal.
Proc. Natl. Acad Sci. USA 88 (1991)
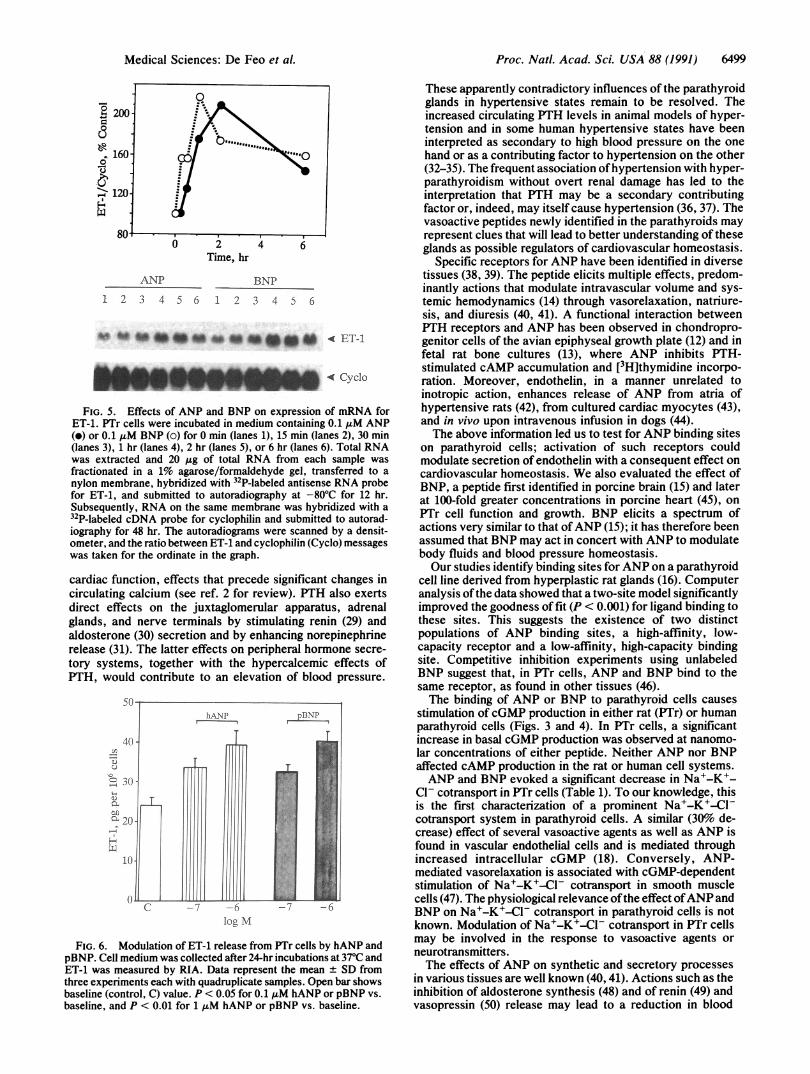
Proc. Natl. Acad. Sci. USA 88 (1991) 6499
0 2 4 6
Time, hr
ANP BNP
1 2 3 4 5 6 1 2 3 4 D 6
ves to"n#af 0 * a n t04 < ET-1
ANUPdAL Cvclo
FIG. 5. Effects of ANP and BNP on expression of mRNA forET-1. PTr cells were incubated in medium containing 0.1 t±M ANP(e) or 0.1 tuM BNP (o) for 0 min (lanes 1), 15 min (lanes 2), 30 min(lanes 3), 1 hr (lanes 4), 2 hr (lanes 5), or 6 hr (lanes 6). Total RNAwas extracted and 20 ,ug of total RNA from each sample wasfractionated in a 1% agarose/formaldehyde gel, transferred to a
nylon membrane, hybridized with 32P-labeled antisense RNA probefor ET-1, and submitted to autoradiography at -80'C for 12 hr.Subsequently, RNA on the same membrane was hybridized with a32P-labeled cDNA probe for cyclophilin and submitted to autorad-iography for 48 hr. The autoradiograms were scanned by a densit-ometer, and the ratio between ET-1 and cyclophilin (Cyclo) messageswas taken for the ordinate in the graph.
cardiac function, effects that precede significant changes incirculating calcium (see ref. 2 for review). PTH also exertsdirect effects on the juxtaglomerular apparatus, adrenalglands, and nerve terminals by stimulating renin (29) andaldosterone (30) secretion and by enhancing norepinephrinerelease (31). The latter effects on peripheral hormone secre-
tory systems, together with the hypercalcemic effects ofPTH, would contribute to an elevation of blood pressure.
hAN-P pB\P
10
C -7 -6 -6
log M
FIG. 6. Modulation of ET-1 release from PTr cells by hANP andpBNP. Cell medium was collected after 24-hr incubations at 370C andET-1 was measured by RIA. Data represent the mean + SD fromthree experiments each with quadruplicate samples. Open bar showsbaseline (control, C) value. P < 0.05 for 0.1 jxM hANP or pBNP vs.
baseline, and P < 0.01 for 1 uM hANP or pBNP vs. baseline.
These apparently contradictory influences of the parathyroidglands in hypertensive states remain to be resolved. Theincreased circulating PTH levels in animal models of hyper-tension and in some human hypertensive states have beeninterpreted as secondary to high blood pressure on the onehand or as a contributing factor to hypertension on the other(32-35). The frequent association ofhypertension with hyper-parathyroidism without overt renal damage has led to theinterpretation that PTH may be a secondary contributingfactor or, indeed, may itself cause hypertension (36, 37). Thevasoactive peptides newly identified in the parathyroids mayrepresent clues that will lead to better understanding of theseglands as possible regulators of cardiovascular homeostasis.
Specific receptors for ANP have been identified in diversetissues (38, 39). The peptide elicits multiple effects, predom-inantly actions that modulate intravascular volume and sys-temic hemodynamics (14) through vasorelaxation, natriure-sis, and diuresis (40, 41). A functional interaction betweenPTH receptors and ANP has been observed in chondropro-genitor cells of the avian epiphyseal growth plate (12) and infetal rat bone cultures (13), where ANP inhibits PTH-stimulated cAMP accumulation and [3H]thymidine incorpo-ration. Moreover, endothelin, in a manner unrelated toinotropic action, enhances release of ANP from atria ofhypertensive rats (42), from cultured cardiac myocytes (43),and in vivo upon intravenous infusion in dogs (44).The above information led us to test for ANP binding sites
on parathyroid cells; activation of such receptors couldmodulate secretion of endothelin with a consequent effect oncardiovascular homeostasis. We also evaluated the effect ofBNP, a peptide first identified in porcine brain (15) and laterat 100-fold greater concentrations in porcine heart (45), onPTr cell function and growth. BNP elicits a spectrum ofactions very similar to that ofANP (15); it has therefore beenassumed that BNP may act in concert with ANP to modulatebody fluids and blood pressure homeostasis.Our studies identify binding sites forANP on a parathyroid
cell line derived from hyperplastic rat glands (16). Computeranalysis of the data showed that a two-site model significantlyimproved the goodness of fit (P < 0.001) for ligand binding tothese sites. This suggests the existence of two distinctpopulations of ANP binding sites, a high-affinity, low-capacity receptor and a low-affinity, high-capacity bindingsite. Competitive inhibition experiments using unlabeledBNP suggest that, in PTr cells, ANP and BNP bind to thesame receptor, as found in other tissues (46).The binding of ANP or BNP to parathyroid cells causes
stimulation of cGMP production in either rat (PTr) or humanparathyroid cells (Figs. 3 and 4). In PTr cells, a significantincrease in basal cGMP production was observed at nanomo-lar concentrations of either peptide. Neither ANP nor BNPaffected cAMP production in the rat or human cell systems.ANP and BNP evoked a significant decrease in Na+-K+-
Cl- cotransport in PTr cells (Table 1). To our knowledge, thisis the first characterization of a prominent Na+-K+-Cl-cotransport system in parathyroid cells. A similar (30o de-crease) effect of several vasoactive agents as well as ANP isfound in vascular endothelial cells and is mediated throughincreased intracellular cGMP (18). Conversely, ANP-mediated vasorelaxation is associated with cGMP-dependentstimulation of Na+-K+-CI- cotransport in smooth musclecells (47). The physiological relevance ofthe effect ofANP andBNP on Na+-K+-Cl- cotransport in parathyroid cells is notknown. Modulation of Na'-K-Cl- cotransport in PTr cellsmay be involved in the response to vasoactive agents orneurotransmitters.The effects of ANP on synthetic and secretory processes
in various tissues are well known (40, 41). Actions such as theinhibition of aldosterone synthesis (48) and of renin (49) andvasopressin (50) release may lead to a reduction in blood
Medical Sciences: De Feo et al.

6500 Medical Sciences: De Feo et al.
pressure. We found that both ANP and BNP stimulate thesynthesis of ET-1 in PTr cells. Characteristically ET-1 isrecognized as a product of endothelial cells (8). It exhibitsvasoconstricting activity in vitro and pressor action in vivo(51); hence it could be a factor in the pathophysiology ofhypertension. On the other hand it has yet to be establishedwhether endothelin acts as a circulating hormone or merelyas a local factor (52).As shown here, the parathyroid glands are a newly recog-
nized target tissue for actions of the natriuretic and hypoten-sive peptides ANP and BNP. The parathyroids represent oneof the tissues most replete with mRNA for ET-1, which mayact as an autocrine mediator of the effects of calcium onparathyroid function (7). Natriuretic hormones could influ-ence secretory processes in parathyroid cells by increasingET-1 synthesis. One might speculate that beyond local ef-fects, stimulation of ET-1 synthesis and release from para-thyroid cells by natriuretic hormones could be a regulatoryinfluence on intravascular volume and vascular smooth mus-cle tone. On the other hand, however, in our in vitroexperiments the ANP-related peptides have produced rathermodest increases in ET-1 release. It is uncertain whethercomparable effects in vivo would be sufficient to affectperipheral vascular tone. In any event, further studies areneeded to establish the physiological importance of natri-uretic hormones on'parathyroid cell function, the significanceof ET-1 eleaboration by parathyroid cells, and the relation-ship of these findings, if any, to the pathogenesis of hyper-tension in hyperparathyroidism.
We thank Dr. C. Tosti-Guerra for BNP measurement and Dr. T.Masaki for kindly supplying the cDNA for preproET-1.
1. Aurbach, G. D., Marx, S. J. & Spiegel, A. M. (1991) in Wil-liams Textbook ofEndocrinology, eds. Wilson, J. D. & Foster,D. V. (Saunders, Philadelphia), 8th Ed., in press.
2. Mok, L. L. S., Nickols, G. A., Thompson, J. C. & Cooper,C. W. (1989) Endocrine Rev. 10, 420-436.
3. Ikeda, K., Weir, E. C., Sakaguchi, K., Burtis, W. J., Zimering,M., Mangin, M., Dreyer, B. E., Brandi, M. L., Aurbach, G. D.& Broadus, A. E. (1989) Biochem. Biophys. Res. Commun.162, 108-115.
4. Zajac, J. D., Callaghan, J., Elridge, C., Diefenbach-Jagger, H.,Suva, L. J., Hudson, P., Moseley, J. M., Michelangeli, V. P.& Pasquini, G. (1989) Mol. Cell. Endocrinol. 67, 107-112.
5. De Feo, M. L., Bartolini, O., Benvenuti, S., Franceschelli, F.,Maggi, M., Modigliani, U., Tanini, A., Serio, M. & Brandi,M. L. (1990) J. Endocrinol. Invest. 13 (Suppl. 1), 8 (abstr.).
6. Fujii, Y., Moreira, J. E., Maggi, M., Orlando, C., Aurbach,G. D., Brandi, M. L. & Sakaguchi, K. (1989) J. Bone Miner.Res. 5 (Suppl. 2), 269 (abstr.).
7. Fujii, Y., Moreira, J. E., Orlando, C., Maggi, M., Aurbach,G. D., Brandi, M. L. & Sakaguchi, K. (1991) Proc. Natl. Acad.Sci. USA 88, 4235-4239.
8. Nickols, G. A., Nana, A. D., Nickols, M. A., DiPette, D. J. &Asimakis, G. K. (1989) Endocrinology 125, 834-841.
9. Yanagisawa, M., Kurihara, H., Kimura, S., Tomobe, Y.,Kobayashi, M., Mitsui, Y., Yazaki, Y., Katsutoshi, G. &Masaki, T. (1988) Nature (London) 332, 411-415.
10. Resnick, L. M. (1989) in Endocrine Mechanisms in Hyperten-sion, eds. Laragh, J. H., Brenner, B. M. & Kaplan, N. M.(Raven, New York), pp. 265-286.
11. Pang, P. K. T. & Lewanczuk, R. Z. (1989) Am. J. Hypertens.2, 898-902.
12. Pines, M. & Hurwitz, S. (1988) Endocrinology 123, 360-365.13. Vargas, J. J., Holden, S. N., Fall, P. M. & Raisz, L. G. (1989)
Endocrinology 125, 2527-2531.14. Goetz, K. L. (1988) Am. J. Physiol. 254, El-E15.15. Sudoh, T., Kangawa, K., Minamino, N. & Matsuo, H. (1988)
Nature (London) 332, 78-81.16. Sakaguchi, K., Santora, A., Zimering, M., Curcio, F., Aur-
bach, G. D. & Brandi, M. L. (1987) Proc. Natl. Acad. Sci. USA84, 3269-3273.
17. Brooker, G., Harper, J. F., Terasaki, W. L. & Moylon, R. D.(1979) Adv. Cyclic Nucleotide Res. 10, 1-33.
18. O'Donnell, M. E. (1989) Am. J. Physiol. 257, C36-C44.19. Bradford, M. M. (1976) Anal. Biochem. 72, 248-254.20. Richards, W. L., Song, M. K., Krutzch, H., Evarts, R. P.,
Maiden, E. & Thorgeirsson, S. S. (1985) Exp. Cell Res. 159,235-246.
21. Danielson, P. E., Forss-Petter, S., Brow, M. A., Calavetta, L.,Douglass, J., Milner, R. J. & Sutcliffe, J. G. (1988) DNA 7,261-267.
22. Orlando, C., Brandi, M. L., Peri, A., Giannini, S., Fantoni, G.,Calabresi, E., Serio, M. & Maggi, M. (1990) Endocrinology 126,1780-1782.
23. Laffi, G., Marra, F., Pinzani, M., Meacci, E., Tosti-Guerra, C.,De Feo, M. L. & Gentilini, P. (1989) Liver 9, 315-321.
24. Munson, P. J. & Rodbard, D. (1980) Anal. Biochem. 107,220-239.
25. Guardabasso, V., Munson, P. J. & Rodbard, D. (1988) Comp.Methods Progr. Biomed. 27, 55-63.
26. De Lean, A., Munson, P. J. & Rodbard, D. (1978) Am. J.Physiol. 235, E97-E102.
27. Duncan, D. B. (1955) Biometrics 11, 1-42.28. Collip, J. B. & Clark, E. P. (1925) J. Biol. Chem. 64, 485-507.29. Davis, J. 0. & Freeman, R. H. (1976) Physiol. Rev. 56, 1-56.30. Olgaard, K., Dangaard, H. & Egfiord, M. (1986) Kidney Int. 29,
168 (abstr.).31. Campese, V. M. (1986) Am. J. Nephrol. 6 (Suppl. 1), 29-32.32. Young, E. V., Bukowski, R. D. & McCarron, D. A. (1988)
Proc. Soc. Exp. Biol. Med. 187, 123-141.33. Doris, P. A., Harvey, S. & Pang, P. K. T. (1987) Life Sci. 41,
1383-1389.34. Resnick, L. M. & Laragh, J. H. (1985) Am. J. Med. 78,
385-398.35. McCarron, D. A., Pingree, P. A., Rubin, R. J., Goucher,
S. M., Molitch, M. & Krutzik, S. (1980) Hypertension 2,162-168.
36. Hellstrm, J., Birke, G. & Edvall, C. A. (1958) Br. J. Urol. 30,13-24.
37. Sangal, A. K. & Beevers, D. G. (1983) Br. Med. J. 286,498-499.
38. Jacobs, J. W., Vlasuk, G. P. & Rosenblatt, M. (1987) in AtrialNatriuretic Factor, eds. Rosenblatt, M. & Jacob, J. W. (Saun-ders, Philadelphia), pp. 63-77.
39. Waldman, S. A. & Murad, F. (1989) Bioessays 10, 16-19.40. Gutkowska, J. & Nemer, M. (1989) Endocrine Rev. 10, 519-
536.41. Inagami, T. (1989) J. Biol. Chem. 264, 3043-3046.42. Winquist, R. J., Scott, A. L. & Vlasuk, G. P. (1989) Hyper-
tension 14, 111-114.43. Fukuda, Y., Hirata, Y., Taketani, S., Takatsugu, K., Oikawa,
S., Nakazato, H. & Kobayashi, Y. (1989) Biochem. Biophys.Res. Commun. 164, 1431-1436.
44. Stasch, J. P., Hirth-Dietrich, C., Kadza, S. & Neuser, D. (1989)Life Sci. 45, 869-875.
45. Minamino, M., Aburaya, M., Ueda, S., Kangawa, K. &Matsuo, H. (1988) Biochem. Biophys. Res. Commun. 155,740-746.
46. Hirata, Y., Shichiri, M., Emori, T., Marumo, F., Kangawa, K.& Matsuo, H. (1988) FEBS Lett. 238, 415-418.
47. O'Donnell, M. E. & Owen, N. E. (1986) Proc. Natl. Acad. Sci.USA 83, 6132-6136.
48. Kudo, T. & Baird, A. (1984) Nature (London) 312, 756-757.49. Obana, K., Naruse, K., Naruse, H., Sakurai, H., Demura, T.,
Inagami, T. & Shizume, K. (1985) Endocrinology 117, 1282-1284.
50. Obana, K., Naruse, M., Inagami, T., Brown, A. B., Naruse,K., Kurimento, F., Sakurai, H., Demura, H. & Shizume, K.(1985) Biochem. Biophys. Res. Commun. 132, 1088-1094.
51. Le Monnier de Gouville, A., Lippton, H. L., Cavero, I.,Summer, W. R. & Hyman, A. L. (1989) Life Sci. 45, 1499-1513.
52. Saito, Y., Nakao, K., Itoh, H., Yamada, T., Mukoyama, M.,Arai, H., Hosoka, K., Shirakami, G., Suga, S. & Jougasaki, M.(1989) Biochem. Biophys. Res. Commun. 161, 320-326.
Proc. Natl. Acad. Sci. USA 88 (1991)