emery and rimoin's principles and practice of medical genetics || bile pigment metabolism and...
TRANSCRIPT

C H A P T E R
69Bile Pigment Metabolism
and Its DisordersNamita Roy Chowdhury and Jayanta Roy Chowdhury
Departments of Medicine and Genetics, Albert Einstein College of Medicine, Bronx, NY, USA
Yesim Avsar
Department of Medicine, Division of Gastroenterology and Liver Diseases, Albert Einstein College of Medicine, Bronx, NY, USA
This article is a revision of the previous edition article by Namita Roy Chowdury, Xia Wang, Jayanta Roy Chowdhury, volume 2, pp 1583–1612, © 2007, Elsevier Ltd.
© 2013, Elsevier Ltd
69.1 INTRODUCTION
Bilirubin, the major yellow pigment in plasma is the deg-radation product of heme, the iron-containing tetrapyr-role chromophore of hemoglobin and several important enzymes. Owing to its internal hydrogen bonding, biliru-bin is very sparsely soluble in water and is toxic to neural and other tissues. It is rendered harmless by a series of physiologic processes, including binding to plasma pro-teins, rapid uptake by hepatocytes, conjugation with sug-ars to form polar derivatives, and efficient bile canalicular excretion. Perhaps because of its distinctive color, biliru-bin has attracted the attention of physicians, chemists, and biologists since antiquity. Excessive accumulation of bilirubin in the serum is an indicator of liver dysfunction, and serum bilirubin analysis is used as a routine “liver function test.” Bilirubin throughput by the liver has also been studied as a model for hepatic disposal of other biologically important organic anions of limited aque-ous solubility. Investigations into the molecular mecha-nisms of inherited disorders of bilirubin metabolism in humans and animals have provided important insights into bilirubin’s metabolic pathways. Newer aspects of bilirubin disposal and its relationship with biliary excre-tion of other substances are continuing to be unveiled by the efforts of numerous laboratories. Definitive treat-ment of some of these disorders remains a therapeutic challenge and continues to stimulate research. Although physicians have been mainly concerned with the toxic effect of bilirubin, the antioxidant property of the pig-ment may impart it a cytoprotective role.
. All rights reserved. 1
This chapter provides a brief description of the chem-istry, toxicity, and disposition of bilirubin, followed by a description of clinical situations in which various aspects of bilirubin throughput are disturbed.
69.2 FORMATION OF BILIRUBIN
Approximately 250–400 mg of bilirubin is produced daily in humans from heme catabolism. Normally, hemoglobin of senescent erythrocytes account for 80% of bilirubin production (1), and the remainder is derived from other heme-containing proteins and enzymes, and free heme. Intravenously administered radiolabeled heme precursors, glycine and δ-aminolevulinic acid, are incorporated into bile pigments in two phases (1,2,4). The “early-labeled peak” of bilirubin (ELB) is excreted in bile during the first 3 days, and contains 20% of the radiolabel. The ELB consists of an initial “fast” com-ponent, comprising two-thirds of the peak in humans, and is largely derived from hepatic hemoproteins such as cytochromes, catalase, peroxidase, and tryptophan pyr-rolase (4), and a rapidly turning over pool of free heme in the cytosol of hepatocytes (5), a fraction of which may be degraded without incorporation into heme proteins (6). Induction of hepatic cytochrome P-450 enhances the ELB (7). As δ-aminolevulinic acid is preferentially incorporated into hepatic hemoproteins, when labeled δ-aminolevulinic acid is used as a precursor, only the ini-tial component of the ELB incorporates radioactivity (4). This is followed by the relatively “slower” phase of the ELB, which normally contains one-third of the peak and

2 CHAPTER 69 Bile Pigment Metabolism and Its D
is derived from erythroid and nonerythroid sources. This phase is enhanced during “ineffective erythropoiesis,” as in congenital dyserythropoietic anemias, megaloblastic anemias, iron-deficiency anemia, and lead toxicity and erythropoietic porphyria (9). The slower component of the ELB is also enhanced during accelerated erythropoie-sis from any cause because of intramedullary destruction of some normoblasts, destruction of reticulocytes in the peripheral circulation (10), and trimming of segments of reticulocytes during maturation (11). Normally, 80% of the radiolabel is excreted as the “late-labeled peak,” which is derived from the hemoglobin of senescent eryth-rocytes and appears approximately 50 days and 100 days after intravenous injection of the precursors in rats and humans, respectively (4). In hemolytic syndromes or acute intravascular or extravascular hemolysis, the life span of erythrocytes is shortened and the late-labeled peak appears earlier.
69.2.1 Enzyme-Mediated Opening of the Heme Ring
Heme (ferroprotoporphyrin IX) consists of four pyrrole rings connected by methane bridges and a single iron in the center (Figure 69-1). The ring is opened by oxidation of the α-methene-bridge carbon, catalyzed by heme oxy-genase, a group of enzymes located in the endoplasmic reticulum. This reaction requires NADPH and oxygen, and results in the elimination of the α-methene-bridge car-bon as CO (12); release of iron, which is reutilized; and formation of biliverdin, a green pigment (Figure 69-1). High levels of heme oxygenase activity are found in organs involved in hemoprotein breakdown, such as the spleen, where senescent erythrocytes are sequestered. In the liver, both hepatocytes and Kupffer cells have heme
FIGURE 69-1 Mechanism of heme ring opening and subsequent reduction of biliverdin to bilirubin. Oxidation of the α-carbon bridge catalyzed by microsomal heme oxygenase results in the elimination of the carbon as CO, resulting in opening of the heme ring.
isorders
oxygenase activity; the activity in the Kupffer cells is as high as in the spleen (13). Three forms of heme oxygenase have been identified (14). Heme oxygenase 1 (HO-1), a 32-kDa protein, is a stress-response protein. It is induced by stress-related agents, such as endotoxins (e.g. lipopoly-saccharide), several cytokines, heavy metals, hypoxia, and reactive oxygen species. HO-1 is also induced by proto-heme IX, oxidized and low-density lipoprotein, and prob-ably also by the shear stress on endothelial cells in the cirrhotic liver (15–17). Nuclear factor-κB and p38 mito-gen-activated protein kinase signaling pathways mediate the lipopolysaccharide-dependent induction of HO-1 via the proximal promoter region of the gene. The lipid per-oxidation end product 4-hydroxy-nonenal (HNE) and phospholipids are natural components of oxidized LDL and mediate the stabilization and nuclear translocation of the anti-inflammatory nuclear factor E2-related factor 2 (Nrf2), thereby enhancing the transcription of HO-1 (18). By converting the pro-oxidant heme to antioxidant biliver-din and bilirubin, it serves as a cellular defense mechanism against oxidative injury. HO-1 expression in response to inflammatory mediators may contribute to resolving inflammatory responses, thereby protecting several organs against oxidative injury. Both CO and biliverdin/bilirubin, which are the products of HO-1, have been implicated in this response. In the gastrointestinal tract, HO-1 induc-tion is associated with protection against injury caused by ischemia-reperfusion, indomethacin, lipopolysaccha-ride-associated sepsis, trinitrobenzene sulfonic acid, and dextran sulfate sodium (19). In the presence of hypercho-lesterolemia, HO-1-deficient mice develop early athero-sclerosis. In humans, HO-1 deficiency is associated with growth retardation, hyperlipidemia, and endothelial dam-age, which may cause consumption coagulopathy and microangiopathic hemolytic anemia. Inducible HO-1 is an important cytoprotectant in the renal vascular endo-thelium and tubular epithelium. It is likely that HO-1 has additional effects that may not be mediated by CO or bilirubin. For example, human mesenchymal stem cells induce subsets of regulatory T-lymphocytes in an HO-1-dependent manner (20). In certain conditions, induction of HO-1 may result in increased iron deposition in tissues, with deleterious effects. In Alzheimer’s disease, HO-1 induction in astroglial cells by β-amyloid and hydrogen peroxide is thought to cause mitochondrial nontransfer-rin iron sequestration, thereby contributing to bioenergy failure (21). Finally, the cytoprotective effect of HO-1 may have a deleterious effect as seen in the case of prostate can-cer progression (22).
Heme oxygenase 2 is a constitutive protein, expressed primarily in the brain and the testis. The catalytic activ-ity of heme oxygenase 3 is very low; this isoform may function mainly as a heme-binding protein. CO released by heme-oxygenase-mediated oxidation of the α-carbon bridge is a potent vasodilator that regulates the vascular tone in the liver and in other organs, such as the heart, under conditions of stress.

ER 69 Bile Pigment Metabolism and Its Disorders 3
CHAPT69.2.1.1 Heme Oxygenase Inhibitors. Non iron metalloprotoporphyrins, such as tin- and zinc proto-porphyrin, bind to heme oxygenase with greater affinity than heme (23), but are not degraded. These dead-end inhibitors of heme degradation (23,24) strongly suppress heme oxygenase activity, thereby suppressing bilirubin formation.
69.2.2 Reduction of Biliverdin to Bilirubin
In most mammals, biliverdin is converted to the orange pigment bilirubin by the action of biliverdin reductase, which utilizes NADH at pH 6.7 and NADPH at pH 8.5 as cofactors (25). Variants of cytosolic biliverdin reduc-tase, isolated from rat liver and spleen (27), are post-translational derivatives of a single-gene product (25,28).
Because bilirubin is almost quantitatively excreted in bile, bilirubin production can be measured by determi-nation of biliary excretion in experimental animals. In humans, it can be estimated by quantifying fecal and uri-nary urobilinogen and stercobilinogen, the bacterial deg-radation products of bilirubin (29). The rate of bilirubin formation can also be determined from the turnover of intravenously administered radioisotope-labeled biliru-bin (30). More conveniently, bilirubin formation can be estimated by measuring CO production using a closed rebreathing system. Oxidation of the α-carbon bridge of heme is the main source of endogenous CO production although a small fraction may be contributed by other sources. Therefore, CO production can be calculated from the CO concentration in the breathing chamber or from an increment in blood carboxyhemoglobin satura-tion (31).
69.3 STRUCTURE OF BILIRUBIN
The planar chemical structure of bilirubin (Figure 69-2) was determined by Fischer and Plieninger (32). Despite the presence of two propionic acid side chains and four amino groups, bilirubin IXα is nearly insoluble in water at physiologic pH. An explanation for this phenomenon was suggested by Fog and Jellum (34) and by Kuenzle et al. (35), who proposed that bilirubin IXα may be inter-nally stabilized by hydrogen bonding between the pro-pionic acid carboxyls and the two external pyrrolenone rings. Such intramolecular hydrogen bonding has been confirmed by X-ray diffraction studies (i). These hydro-gen bonds constrain the molecule into a “ridge tile” con-formation and engage all polar groups of the molecule, rendering it insoluble in water (Figure 69-2). For steric reasons, the hydrogen-bonded structure requires that the interpyrrolic bridges at the 4 and 15 positions of biliru-bin should be in a trans- or Z configuration (37). Addi-tion of methanol, ethanol or 6-M urea interferes with hydrogen-bonded structure and makes bilirubin more labile, water soluble, and rapidly reactive with diazo reagents. Conjugation of the propionic acid carboxyls
with sugar moieties disrupts the hydrogen bonds, result-ing in the formation of water-soluble metabolites that are readily excreted in bile.
69.3.1 Absorption Spectra
Bilirubin IXα has a main absorption band at 450–474 nm in most organic solvents and an extinction coefficient of 48.0–63.4/mmol at its absorption maximum at 1-cm path length.
69.3.2 Photoisomerization of Bilirubin
Exposure of circulating bilirubin to light changes the con-figuration of one or both of the interpyrrolic bridges at the 5 and 15 positions to E or cis-configuration, which ste-rically hinders hydrogen bonding. The resulting 4Z-15E or 4E-15Z isomers lack hydrogen bonds in one half of the molecule, whereas bilirubin IXα-EE lacks hydrogen
O
O
O
O
O
O
15
10NH
NH
NH
NH
NH
NH
NH
NH
H4
H
HO
HO HO
COOH
COOH OH
OHOH
O
O
OO
OO
O
O
10
4
15
A B
C D
FIGURE 69-2 Upper Panel. Hydrogen-bonded structure of biliru-bin. The bilirubin molecule is contorted into a ridge-tile-like configu-ration caused by internal hydrogen bonding (interrupted line) of the propionic acid carboxyls to the amino groups and the lactam oxygen of the pyrrolenone rings of the opposite half of the molecule. The carbon bridges connecting pyrrolenone rings A and B (C-4) and C and D (C-15) are in the Z (trans-) configuration. By engaging the polar groups (propionic acid carboxyls and the amino and lactam groups), hydrogen bonding renders the bilirubin molecule very sparsely soluble in water. Because the central carbon bridge (C-10) is buried deep within the molecule, conjugated bilirubin reacts very slowly with diazo reagents, unless the hydrogen bonds are disrupted by adding “accelerator” reagents (“indirect diazo reaction”). Lower Panel. Glucuronidation of both propionic acid carboxyls results in the formation of bilirubin diglucuronide, the predominant pigment excreted in normal human bile. Glucuronidation disrupts the inter-nal hydrogen bonds, making the molecule water soluble and expos-ing the central carbon bridge (C-10) to diazo reagents, resulting in “direct diazo reaction.”

4 CHAPTER 69 Bile Pigment Metabolism and Its
bonds on both halves of the molecule. Blue light is more efficient in mediating the conformational changes. Fol-lowing absorption of two photons (38), the vinyl substit-uent at position C3 of bilirubin IXα-4E-15Z is cyclized with the methyl substituent on the internal pyrrole ring, forming the structural isomer E-cyclobilirubin or lumi-rubin (39,40). Although the rate of cyclization is slower than that of the formation of configurational isomers, because of the greater stability of lumirubin, this form may be quantitatively more important in phototherapy of neonatal jaundice (39). The photoisomers are more polar than is bilirubin IXα-ZZ and can be excreted in bile without conjugation (42).
69.4 POSSIBLE PHYSIOLOGIC BENEFITS OF BILIVERDIN AND BILIRUBIN
Generation of biliverdin and bilirubin, and the subsequent glucuronidation and canalicular transport of bilirubin, are all energetically expensive mechanisms. The evolutionary conservation of these pathways suggests a physiologic benefit of bilirubin. The antioxidant effect of bilirubin may be important during the neonatal period, when the body concentration of other natural antioxidants is low. In adults, a weak but statistically significant inverse rela-tionship between serum bilirubin levels and the risk of coronary artery disease has been reported (43). Analysis of the Third National Health and Nutrition Examination Survey (NHANES III) database revealed that, in over 176 million individuals in the United States, the odds ratios of a history of colorectal cancer were reduced to 0.295 in men and 0.186 in women per 1-mg/dL increment of serum bilirubin levels (44). Similarly, an inverse relation-ship between serum bilirubin levels and cancer mortal-ity was seen in a large study in Belgian population (45). These impressive statistical data, however, do not estab-lish conclusively a cause-and-effect relationship, because of the possible existence of confounding variables.
69.5 TOXIC EFFECTS OF BILIRUBIN
The toxic effect of bilirubin on the neonatal brain has been known for many centuries. Bilirubin deposition at specific areas of the brain accompanied by struc-tural damage is termed kernicterus. At elevated con-centrations, unconjugated bilirubin, particularly its non-albumin-bound fraction, is toxic to astrocytes and neurons. Mitochondrial damage results in impaired energy metabolism and apoptosis, while injury to plasma membranes may disrupt the transport of neurotrans-mitters (46). The study of mutant Gunn rats that lack hepatic bilirubin glucuronidating activity (see later) has been pivotal in the understanding of the neurotoxicity of bilirubin. This is the only naturally mutant animal model, in which bilirubin-induced brain damage has been known to occur. Structural and functional damages of the cochlear nuclei, resulting in hearing deficiency, are
Disorders
a common complication of several hyperbilirubinemia in human neonates. Cells of the auditory system that receive synaptic input from end-bulbs or calyces appear to be early targets (47). In Gunn rat pups, these morpho-logic changes correlate with brainstem auditory evoked potentials (48). Tight binding of unconjugated bilirubin to plasma albumin inhibits deposition of bilirubin in the brain. Displacement of bilirubin from albumin binding enhances the net transfer of bilirubin into neural tis-sues (50). Administration of sulfonamides in Gunn rats results in reversible brainstem auditory evoked potential abnormalities (51).
Purkinje cells of the cerebellum, hippocampus, and basal ganglia are also common targets of bilirubin depo-sition in Gunn rats and human infants with overt ker-nicterus. Purkinje cells of the cerebellum are affected in Gunn rats at the age of 7 days. Subsequent degeneration of these cells results in cerebellar hypoplasia (52). Pur-kinje cells that recover and persist into adult life may have abnormalities of synapse formation with other Pur-kinje cells or with neural cells of other types. There is enlargement and distortion of cerebellar mitochondria (54) and increased activities of the lysosomal enzymes in Gunn rats by the eighth day of life (55). Cerebellar cyclic GMP concentrations decrease progressively from day 15 to day 30, but cyclic AMP (cAMP) levels remain normal (56). Inhibition of cytochrome c oxidase activity and ascorbate-driven oxygen consumption in neonatal rat cortical neuronal cells by unconjugated bilirubin may lead to neuronal apoptosis (57).
69.5.1 Clinical Features of Bilirubin Neurotoxicity
In most cases, kernicterus occurs during the first few months of life. In patients with inherited absence of bili-rubin glucuronidation, who may survive through infancy, kernicterus may occur during adolescence or early adult-hood. Overt kernicterus usually presents between the third and sixth day of life with loss of the Moro reflex, hypotonia, athetoid movements, and reflex opisthoto-nus in response to startling stimuli. Left untreated, this progresses to atonia and death. In some cases, bilirubin encephalopathy may present late, with cerebellar symp-toms as the presenting feature (58). Chronic effects of bilirubin encephalopathy may be observed in patients who survive acute kernicterus. These include chronic hearing abnormalities, athetoid movements, paralysis of upward gaze, and mental retardation.
Autopsy of infants dying during the acute phase of kernicterus shows bilirubin staining of the hippocam-pus, basal ganglia, and nuclei of the cerebellum and the brainstem (59). However, yellow staining is not found in children dying in the chronic stage of bilirubin enceph-alopathy. In these cases, focal necrosis of neurons and glia are observed. Gliosis of the affected area is seen in chronic cases (60).

69 Bile Pigment Metabolism and Its Disorders 5
CHAPTERBecause histologic lesions are not present from the onset of clinical kernicterus (60), these may not be the initiating pathophysiologic events. Focal bilirubin stain-ing of the brain may also occur in other forms of brain injury, such as cerebral hemorrhage (61), and does not, by itself, establish the diagnosis of kernicterus.
Serum unconjugated bilirubin levels above 20.0 mg/dL are usually considered dangerous (62). However, in some studies, serum bilirubin levels that are not high enough to cause kernicterus have been reported to result in an increased incidence of neurologic abnormalities or decreased intellectual performance later in life (63,64).
69.5.2 Pathophysiology of Kernicterus
Immaturity of the blood–brain barrier in neonates has been implicated in the high incidence of kernicterus in neonates. Tight junctions between capillary endothe-lial cells and foot processes of astroglial cells provide a structural barrier to equilibration of hydrophilic water-soluble substances and proteins in blood with those in the brain (65). The functional counterpart of this bar-rier consists of specific transport mechanisms that are involved in the translocation of ions, water, and nutri-ents from plasma to brain. Despite a long-standing interest in the blood–brain barrier in the newborn, it has been difficult to confirm an enhanced passage of labeled markers (66) into the immature brain, and the evidence for an immature blood–brain barrier in the neonate is not convincing.
Infusion of hypertonic urea (68) or arabinose in the carotid artery of newborn rats results in osmotic shrink-age of capillary endothelial cells, thereby opening the blood–brain barrier transiently. While the barrier remains open, intravenously administered albumin-bound biliru-bin rapidly enters the brain. After discontinuation of the hyperosmolar infusion, the blood–brain barrier becomes reconstituted, and bilirubin is rapidly cleared from the brain (69). Accumulation of unconjugated bilirubin in the cerebrospinal fluid and the brain tissue is limited by its active export, probably mediated by multidrug-resistance-related protein-1 (MRP1/ABCB1), the ATP-dependent pump, present in choroid plexus epithelia, capillary endothelia, astrocytes, and neurons (46). Unconjugated bilirubin may downregulate MRP1/ABCb1 in cells con-stituting the blood–cerebrospinal fluid barrier, thereby potentiating bilirubin encephalopathy (70). Clearance of bilirubin may be inefficient in damaged and edematous brains, which may bind bilirubin (71). Therefore, biliru-bin may be more toxic to brains that are already dam-aged from other causes.
69.5.3 Biochemical Mechanism of Bilirubin Toxicity
Bilirubin inhibits a vast array of metabolic, enzymatic, and synthetic functions of the brain. However, it is not
clear whether these toxic effects, observed in tissue cul-ture or in vitro, are relevant in the clinical manifestation of bilirubin encephalopathy. Bilirubin may uncouple oxidative phosphorylation and inhibit ATPase activity of brain mitochondria (73). Bilirubin reduces local cere-bral glucose utilization in immature rats, particularly in the auditory, visual, hypothalamic, and thalamic regions (74). Bilirubin inhibits non-cAMP-dependent protein kinase activity in vivo (75). Irreversible inhibition of Ca+-activated, phospholipid-dependent protein kinase C activity and cAMP-dependent protein kinase activity has been reported (76,77), suggesting that an abnormality of protein phosphorylation may play a role in bilirubin encephalopathy in the newborn.
69.5.4 Bilirubin Nephrotoxicity
In Gunn rats (78) and hyperbilirubinemic infants (79), deposition of unconjugated bilirubin in the renal medulla results in medullary necrosis and formation of visible bilirubin crystals on the papillae. Impairment of urinary concentration resulting from an abnormal-ity of the ascending loop of Henle occurs in Gunn rats (78,80), but not in mature neonates with hyperbiliru-binemia (81) or adult patients with Crigler–Najjar syn-drome type I (82).
69.6 DISPOSITION OF BILIRUBIN
Similar to many other water-insoluble substances, bil-irubin remains tightly bound to plasma proteins, par-ticularly albumin. In the liver, the albumin–bilirubin complex dissociates and bilirubin, but not albumin, is internalized. Within the cytosol, bilirubin is stored bound to cytosolic proteins, particularly to glutathione-S-transferases (GSTs). Bilirubin is rendered water solu-ble by conjugation with glucuronic acid, catalyzed by a microsomal bilirubin uridine diphosphoglucuronate glucuronosyltransferase (UGT1A1). Bilirubin glucuro-nides are transported across the bile canalicular mem-brane and are carried to the duodenum by the bile duct. Other substances that are sparingly water soluble, or are organic anions in nature, and are destined to be excreted in the bile, share some of these steps with bilirubin. The steps of bilirubin throughput are briefly described in this section and are schematically shown in Figure 69-3.
69.6.1 The Role of Albumin
Owing to its structural peculiarity (see earlier), biliru-bin is only sparingly water soluble at physiologic pH. Binding to albumin keeps the pigment in solution and prevents its deposition into tissues, including the brain. Thus, albumin facilitates the transport of bilirubin to the liver and prevents its toxic effects. Although inher-ited analbuminemia is compatible with life, the biliary recovery of amphipathic compounds such as bilirubin

Disorders
6 CHAPTER 69 Bile Pigment Metabolism and Itsand bromosulfophthalein (BSP) is reduced in genetically analbuminemic rats (83). In this situation, other plasma proteins take over some of the functions of albumin. The neural toxic effect of bilirubin is markedly enhanced in the analbuminemic rat.
There are a primary and a secondary binding site on albumin for bilirubin (84), and some weaker binding sites may exist (86,87). The lysine 240 in human albumin and lysine 238 in bovine serum albumin appear to be primary bilirubin-binding sites (88). Other ligands that bind at the same site as bilirubin, such as sulfonamides, anti-inflammatory drugs, and cholecystographic contrast media, may displace bilirubin competitively from albu-min (86), thereby precipitating bilirubin encephalopa-thy in newborns (89), without changing serum bilirubin levels.
Normally, albumin is present in molar excess to bili-rubin. The reserve bilirubin-binding capacity of albumin acts as a buffer for rapid fluctuations of serum biliru-bin levels as in acute hemolysis. Owing to the influence of many metabolites and drugs on albumin binding of bilirubin, measurement of unbound plasma bilirubin and the reserve bilirubin-binding capacity provides a more accurate estimate of the risk of brain damage from unconjugated bilirubin than does the measurement of total bilirubin concentrations alone. Unbound bilirubin in serum can be quantified by the gel chromatography
B + Alb Sinusoidalmembrane
Contiguousmembrane
Canalicularmembrane
MRP2
UDPGA
UDP
B-glucuronides
BUGT
BGSTs
FIGURE 69-3 A schematic diagram summarizing the metabolism of bilirubin by hepatocytes. In plasma, bilirubin is tightly but reversibly bound to albumin. In liver sinusoids, the albumin–bilirubin complex comes in direct contact with the basolateral domain of the hepato-cycle plasma membrane (sinusoidal membrane) through fenestrae of the specialized hepatic endothelial cells, where the albumin–bilirubin complex dissociates. Bilirubin is taken up by the facilitated diffusion, the molecular basis of which is not fully understood. Within the hepatocyte, bilirubin is stored bound to glutathione-S-transferases (GSRs), which inhibit its efflux from the cell, thereby increasing the net uptake. Glucuronidation of bilirubin in the endoplasmic reticu-lum is catalyzed by uridine diphosphoglucuronate glucuronosyl-transferase-1 (UGT1A1), forming bilirubin monoglucuronide and diglucuronide. Conjugated bilirubin is secreted across the bile cana-liculus by an energy-requiring transporter, MRP2, and to a lesser extent by electrogenic transport. Canalicular transport of bilirubin is shared by other many organic anions, including glucoronides and glutathione conjugates, but not by most bile salts.
(90), peroxidase treatment (52), electrophoretic analysis (91), and fluorometry (92). In conditions associated with the presence of conjugated bilirubin in plasma for a long duration, bilirubin becomes irreversibly bound to albu-min (93). Owing to the long half-life of albumin, this fraction, which is not cleared by the liver or kidney, lin-gers for a long time in serum.
69.6.2 Bilirubin Uptake by Hepatocytes
Bilirubin is delivered to the liver bound to plasma albu-min. Because the endothelial lining of hepatic sinusoids is fenestrated, the albumin–bilirubin complex comes in direct contact with the sinusoidal and basolateral plasma membrane domains of the hepatocyte. Bilirubin dissoci-ates from albumin at or close to the hepatocyte surface before it is taken up into the hepatocyte. Whether this dissociation is facilitated by an albumin receptor at the hepatocyte surface remains speculative. In the presence of spontaneous portosystemic shunts, as in cirrhosis of the liver, or surgically induced diversion of portal blood from the liver, bilirubin produced in the spleen does not come in contact with hepatocytes in the first pass. This results in a mild unconjugated hyperbilirubinemia. An open ductus venosus in the newborn may exacerbate “physiologic” jaundice in infants by a similar mechanism.
Bilirubin is taken up by the hepatocyte by facilitated diffusion (94), which is bidirectional and does not con-sume energy, but needs the presence of inorganic anions, such as Cl− (95,96). The mechanism of bilirubin uptake at the sinusoidal surface is not yet understood precisely (97,98). Unconjugated bilirubin is relatively nonpolar, and some investigators have suggested that it may dif-fuse through the sinusoidal membrane simply by dis-sociating from albumin, without requiring transporter proteins (99). However, concentrative bilirubin uptake is a hepatocyte-specific function (100). Therefore, hepa-tocyte-specific carrier proteins have been sought. OATP2 (also known as OATP-C or SLC21A6) in human liver and OATP4 (S1c21a10) in rat liver are high-affinity transporters of organic anions such as BSP, taurocho-late, estradiol-17β glucuronide, LTC4, estrone-3-sulfate and estrone-1-sulfate, dehydroepiandrosterone sulfate, triiodothyronine, and thyroxin (101,102). Some studies have suggested that OATP-C also transports unconju-gated bilirubin (101), but other studies did not support this conclusion (103). NTCP (SLC10A1) is a specialized carrier for the Na+-dependent hepatic uptake of bile salts (104–107), which does not appear to be directly related to bilirubin uptake. Thus, the physiologically important bilirubin transporter remains to be identified at this time.
69.6.3 Storage of Bilirubin within the Hepatocyte
Within the hepatocyte, bilirubin binds to cytosolic proteins that keep it in solution predominantly. The

CHAPTER 69 Bile Pigment Metabolism and Its Disorders 7
bilirubin-binding proteins were originally termed the Y (ligandins) and Z (fatty-acid-binding) proteins (108). Later, the Y proteins or ligandins were found to consist of the α class of GSTs (109). Binding to GSTs does not affect the influx of bilirubin directly, but increases the net input by inhibiting its diffusion out of the hepatocytes (110).
69.6.4 Bilirubin Conjugation
69.6.4.1 Conversion of Bilirubin to Polar Derivatives. Conjugation of the propionic acid carboxyls with sugar moieties, particularly glucuronic acid (111), is the major mechanism by which the internal hydrogen bonds of bilirubin are disrupted and the molecule becomes water soluble. Depending on whether one or both propionic acid carboxyls are glucuronidated, bilirubin monoglucuronide or diglucuronide is formed (113), both of which are efficiently excreted in bile. Bilirubin diglucuronide is the major pigment in normal human and dog bile (111). In addition, smaller amounts of glucosyl and xylosyl conjugates have been described in human T-tube bile (113,115) and bile from other species (116).69.6.4.2 Enzyme-Catalyzed Glucuronidation of Bil-irubin. A family of enzymes termed uridine diphos-phoglucuronate glucuronosyltransferases (UGTs; EC 2.4.1.17), located in the endoplasmic reticulum and nuclear envelope of a variety of cells (117), catalyzes the transfer of the glucuronosyl moiety of UDPglucuro-nate to aglycone substrates. Substrates of this group of enzymes comprise a wide spectrum of substances, includ-ing hormones (e.g. steroid hormones, thyroid hormones, and catecholamines), endogenous metabolites (e.g. bile salts and bilirubin), numerous drugs and their intermedi-ate metabolites, toxins (e.g. carcinogens), and laboratory xenobiotics (118). The products of the transferase reac-tion are ether, ester, thiol, and N-glucuronides. Glucuro-nides are more polar than the aglycone substrates and usually less biologically active. Because of the wide array of substrates that are conjugated via UGTs, this group of enzymes constitutes a major detoxification system of the body.
The catalytic activity of UGTs is partly latent in micro-somal vesicles. Full enzyme activity is obtained by per-turbing the microsomal membrane by detergent (119) or enzymatic (120) treatment. UDP-N-acetylglucosamine, which activates hepatic microsomal UGT activity at low concentrations, may be a physiologic activator of the UGTs. According to one postulated model, the catalytic site of UGT is located inside the lumen of the endoplasmic reticulum, and the donor substrate, UDP-glucuronic acid, must be transported through the mem-brane barrier to the catalytic site (121). In this model, UDP-N-acetylglucosamine is thought to increase UDP-glucuronate transport by activating a putative “perme-ase.” Detergents and other membrane-perturbing agents
may enhance UGT activity in vitro by physically increasing the permeability of the membranes to UDP-glucuronic acid (122). An alternative model proposes that the physical or chemical activators of UGT act by releasing the membrane constraint of UGT (123), and UDP-N-acetylglucosamine acts as an allosteric activator of the enzyme. It should be noted that the two models may not be exclusive of each other.
The carboxy-terminal domain of all UGT isoforms has a high level of sequence homology, and is thought to bind the common substrate, UDPglucuronic acid (124). The amino-terminal domain is less homologous in struc-ture and imparts aglycone substrate specificity to the enzymes (125).69.6.4.3 Families and Subfamilies of UGT. The UGT system consists of a large number of structurally related enzymes, all of which accept UDPglucuronic acid as a donor substrate, but differ in aglycone substrate speci-ficity (126–128,130,131). UGT isoforms differ in onto-genic development (132) and effect of enzyme inducing agents (133). On the basis of the extent of structural homology of the respective messenger RNAs (mRNAs), UGT isoforms have been classified into several families and subfamilies (134). The UGT1A locus expresses one group of UGT isoforms (135). The enzymes have iden-tical carboxy-terminal domains (136–138), but variable amino-terminal regions. The UGT1A locus consists of four consecutive exons (exons 2–5) at the 3′ end, which are used in the mRNAs of all isoforms expressed from this locus and encode the identical carboxy-terminal domain (Figure 69-4). Upstream to these are a series of 13 unique exons, each preceded by a promoter. Only one of these exons is used in a given UGT isoform. The pro-cess of transcription can start at any of the promoters, giving rise to transcripts of different lengths. The pres-ence of a separate promoter upstream to each of these variable-region exons explains independent regulation of enzymes expressed from this gene and their differ-ent organ distribution and expression during ontogeny, enzyme induction, or carcinogenesis. Enzyme activity toward 4-nitrophenol and other simple phenolic sub-strates develops in late fetal life in rats, whereas activity toward bilirubin develops after birth (132). UGT1A6, a 3-methylcholanthrene-inducible isoform, is permanently overexpressed in carcinogen-induced preneoplastic nod-ules in rat liver (137). Triiodothyronine treatment results in a threefold increase in rat liver phenol–UGT activity, whereas bilirubin glucuronidation is reduced by 80% (139).
The unique exon that is located at the 5′ end of the transcript is spliced to exon 2, and the intervening sequences are spliced out. In this manner, the UGT1A locus expresses nine UGT isoforms (the other four unique exons represent pseudogenes). Thus, the UGT1A locus is considered to comprise multiple overlapping genes, each of which is named according to the unique exon that is used in the processed mRNA. For example,

8 CHAPTER 69 Bile Pigment Metabolism and Its Disorders
UGT1A locus
UGT1A6 Primary transcript
UGT1A1 Primary transcript
1A12 1A6 1A5 1A4 1A3 1A2 1A12 3 4 5
1A6 1A5 1A4 1A3 1A2 1A12 3 4 5
1A12 3 4 5
1A1UGT1A1 mRNA2 3 4 5
1A6UGT1A6 mRNA2 3 4 5
FIGURE 69-4 A schematic representation of the human UGT1A locus. This locus is located in chromosome 2 at 2q37 (373). Exons 2–5 are shared by all UGT isoforms expressed from this locus. Upstream to these common-region exons, there is a series of variable-region exons, designated exons UGT1A1 through UGT1A12, only one of which is used in any given UGT isoform. Each of these exons encodes the variable amino-terminal region of one UGT isoform. Each variable-region exon has an upstream promoter element, and is differentially regulated. Depending on the promoter selection, primary transcripts of various lengths are produced. The variable exon 1 at the 5′ end of the transcript is spliced to the common exons 2–5, resulting in the formation of a mature messenger RNA (mRNA) with a unique exon 1. Several species of UGT mRNAs are produced in this manner, all of which have identical 3′ domains. Therefore, a genetic lesion in any of the common-region exons affects all mRNAs expressed from this locus.
if transcription starts at the 3′-most unique exon, the gene is termed UGT1A1. On the other hand, if the transcription starts at the sixth unique exon, the gene is termed UGT1A6. Initially, UGT1A1 and UGT1A4 were reported to have activity toward bilirubin in humans (140), but later, only UGT1A1 was shown to contrib-ute physiologically significant bilirubin glucuronidating activity (53). The UGT1 gene in the rat has a similar exon organization (136), indicating that this gene is highly conserved during evolution.
69.6.5 Excretion of Conjugated Bilirubin across the Bile Canaliculus
In contrast to the transport across the sinusoidal sur-face plasma membranes, canalicular secretion of conju-gated bilirubin and other cholephilic compounds occurs against a high concentration gradient, whereby the liver:bile concentration ratio for organic anions, such as dibromosulfophthalein, may reach 1:1000 (143). Such large gradient cannot be accounted for by the −35 mV potential difference across the canalicular membrane, and suggest the presence of active transport systems (144–147). MRP2 (also termed ABCC2), a member of the ATP-binding cassette (ABC) protein family, serves as the canalicular efflux pump for non-bile-acid organic anions, including bilirubin and other glucuronide and glutathione conjugates (148–150). Genetic deficiency of MRP2/ABCC2 in humans leads to the Dubin–Johnson syndrome, characterized by retention of conjugated bil-irubin in plasma (152,153). The TR− rat, which lacks mrp2/ABCC2 function because of a single-nucleotide deletion of the mrp2/abcc2 gene, is an animal model of Dubin–Johnson syndrome (154,155). Sequence analysis
of the human MRP2/ABCC2 promoter showed a num-ber of putative consensus binding sites for both ubiqui-tous and liver-enriched transcription factors, including AP1, SP1, HNF1, and HNF3β (156,157), as well as the nuclear receptors CAR, PXR, and FXR.
Two other ABC family proteins, mrp1 and mrp3, are expressed at low levels in rat liver, but are upregulated during cholestasis (159,160). Both mrp1 and mrp3 are located in the basolateral membrane of the hepatocytes and pump accumulating bilirubin glucuronides out of the hepatocytes into the plasma (156,161). Mrp1 also pumps glutathione S-conjugates (162), as well as uncon-jugated bilirubin. Mrp3 expression is upregulated in the liver of mrp2-deficient rats and in UGT1A1-deficient Gunn rats (156). Thus, MRP3 (mrp3 in rodents) func-tions as a reverse transporter, which pumps substrates back to the blood when biliary excretion of glucuronides is reduced.
Several other ATP-dependent canalicular transport-ers have been identified and cloned. FICI is a gene mapped to chromosome 18q21 (163), the product of which may couple the hydrolysis of ATP to the trans-location of acidic phospholipids (e.g. phosphatidyl-serine and phosphatidylethanolamine) from the outer to the inner layer of the plasma membrane. The gene for another transport protein termed bile salt export pump, also known as sister of P-glycoprotein, is located on chromosome 2q24 (165). MDR3/ABCB4 in humans (mdr2 in mice) mediates ATP-dependent trans-location of phosphatidylcholine from the inner to the outer leaflet of the bile canalicular plasma membrane (166). Inherited defects of any of these genes result in disordered bilirubin excretion by direct or indirect mechanisms.

69 Bile Pigment Metabolism and Its Disorders 9
CHAPTER69.6.6 Nuclear Receptors
Nuclear receptors may orchestrate the various steps involved in bilirubin throughput: maximum capacities for bilirubin uptake, storage, conjugation, and canalic-ular excretion appear to be similar in vivo. Therefore, reduction of any of these steps can lead to hyperbiliru-binemia. On the other hand, enhancement of bilirubin excretion, which may be needed when bilirubin produc-tion is increased, would require coordinated increase in the capacity of each. It has been proposed that the nuclear receptor, CAR, serves as the co-ordinating mech-anism for physiologic modulation of each of these steps (167,168).
69.6.7 Degradation of Bilirubin in the Gastrointestinal Tract
The small amount of unconjugated bilirubin that reaches the intestine is partly reabsorbed. This reabsorption may be greater when the infant is fed maternal milk than when a baby formula is used. Therefore, breast-feeding may contribute to neonatal hyperbilirubinemia (169). Con-jugated bilirubin is not substantially absorbed from the intestine (170). Intestinal bacteria deconjugate bilirubin (171) and degrade it to urobilinogens and related prod-ucts (172). Urobilinogens absorbed from the intestine are re-excreted in the bile and, to a smaller extent, in the urine. In liver disease and increased bilirubin production, urinary urobilinogen excretion is increased. However, because the extent of reabsorption of urobilinogen by renal tubules varies, and the pigment is unstable in acid urine, quantification of urobilinogen excretion in urine is not of clinical benefit. However, complete absence of urobilinogen in stool and urine indicates complete bile duct obstruction. Urobilinogen is colorless. Its yellow oxidation product, urobilin, contributes to the charac-teristic color of urine and stool.
69.6.8 Extrahepatic Disposition of Bilirubin
During biliary obstruction, urinary excretion becomes the major excretory pathway for bilirubin (173). In chil-dren with biliary atresia, 50–90% of bilirubin excretion may occur through the kidney (174). Unconjugated bil-irubin is tightly bound to albumin and is not filtered by normal renal glomeruli, and therefore does not appear in the urine. A fraction of conjugated bilirubin that is not bound to albumin is filtered by renal glomeruli (173) and is excreted in urine. Therefore, excretion of biliru-bin in the urine, in the absence of albuminuria, indicates the presence of conjugated bilirubin in plasma. Unconju-gated bilirubin entering the renal tubules is reabsorbed, but is not secreted by the tubules (175). Although UGT1A1 activity is present in the proximal small intes-tinal villi (117), the relative contribution of small intes-tine in bilirubin disposition is not known. Small amounts
of unconjugated bilirubin pass to the intestinal lumen across the intestinal epithelium or by exfoliation of the epithelial cells (176).
69.6.9 Alternative Pathways of Bilirubin Disposition
As discussed in the preceding sections of this chapter, exposure to light results in the formation of configura-tional (EZ, ZE, or EE forms) and cyclic (e.g. lumirubin) isomers of bilirubin formed in the presence of ambient light or during phototherapy. These isomers are more polar than bilirubin IXα-ZZ, and are excreted in bile in the unconjugated form (42). A significant fraction of bil-irubin undergoes photodegradation to polar diazo-nega-tive compounds that are excreted in bile and urine (38).
69.7 BILIRUBIN MEASUREMENT
Total serum bilirubin level and the conjugated bilirubin fraction are routinely determined as markers of liver function. In the newborn period, the fractional concen-tration of non-protein-bound bilirubin is a more reli-able predictor of potential neurotoxicity, and helps in determining the need for institution of therapy to reduce serum bilirubin levels. Under some circumstances, the measurement of, covalently, albumin-bound bilirubin may add clinical insight to bilirubin disposition.
Serum bilirubin is usually measured after conver-sion to stable azo-derivatives, analysis of intact tetra-pyrroles being mainly used for research on bilirubin metabolism (177).
69.7.1 Bilirubin Measurement Following Reaction with Diazo Reagents
Reaction with a diazonium ion cleaves the bilirubin mol-ecule at the central carbon bridge, and derivatizes the two resulting dipyrroles at the C9 and C11 positions of bilirubin (178). Because conjugated bilirubin lacks internal hydrogen bonds, the central methane bridge is readily accessible to the diazo reagents. Therefore, conjugated bilirubin reacts rapidly with diazo reagents (“direct” fraction) (179). On addition of “accelerator” substances, such as methanol or caffeine, both conju-gated and unconjugated bilirubin react rapidly (total bilirubin). The “indirect” fraction of bilirubin is calcu-lated by subtracting the “direct” fraction from total bil-irubin. In order to characterize the azodipyrroles, they can be separated by thin-layer chromatography (180) or high-performance liquid chromatography (HPLC) (181). Because up to 15% of unconjugated bilirubin in solution may exhibit direct diazo reaction, this method slightly overestimates conjugated bilirubin. Therefore, a direct-reacting bilirubin concentration of <15% of total bili-rubin is considered normal although normally only up to 4% of serum bilirubin is conjugated. The fraction of

10 CHAPTER 69 Bile Pigment Metabolism and Its Disorders
bilirubin that is covalently bound to albumin also gives a direct reaction (182). As the irreversibly protein-bound bilirubin is cleared slowly from serum after relief of bil-iary obstruction, the finding of direct-reacting bilirubin during this period may give a false impression of contin-ued biliary obstruction.
69.7.2 Chromatographic Analysis of Bilirubin as Intact Tetrapyrrole
For accurate identification of bilirubin and its conjugates for research purposes, methods based on thin-layer chro-matography (180) or HPLC (116,183–185) have been developed. When the identification of specific sugar con-jugates is not required, bilirubin mono- and diconjugates can be converted to mono- and dimethyl esters, respec-tively, by alkaline methanolysis before analysis (186). To measure the covalently albumin-bound fraction of bili-rubin (δ-bilirubin), HPLC is performed on incompletely deproteinated serum (182). The use of chromatographic methods of bilirubin analysis is generally limited to research laboratories.
69.7.3 Slide Test
The Ektachem slide test measures conjugated, uncon-jugated, and irreversibly protein-bound bilirubin using a diazo technique. One slide measures total bilirubin, and another specially coated slide allows only the free and reversibly protein-bound bilirubins to react with the diazo reagent; irreversibly protein-bound bilirubin can be estimated from the difference (187).
69.7.4 Transcutaneous Bilirubinometry
To assess the risk of severe neonatal hyperbilirubinemia, it is useful to evaluate the rate of increase of serum bili-rubin levels during the first 24–48 h of life. Repeated esti-mation of serum bilirubin levels can be accomplished in a noninvasive manner by measuring the yellow color of the skin in reflected light, using computer analysis to circum-vent the interference by underlying skin color (188,189).
69.8 BILIRUBIN IN BODY FLUIDS
69.8.1 Bilirubin in plasma
In the plasma, 96% of total bilirubin is unconjugated although the direct-reacting fraction, as determined by using diazo reagents, may slightly overestimate the conjugated fraction. During bilirubin overproduction, both unconjugated and conjugated fractions increase and their proportion remains unchanged. In contrast, reduced levels of bilirubin glucuronidating activity result in a lower proportion of conjugated bilirubin. In cases of intrahepatic cholestasis, biliary obstruction, or hepato-cellular diseases, as well as in Dubin–Johnson and Rotor
syndromes, both conjugated and unconjugated bilirubin accumulate in plasma, and the proportion of conjugated bilirubin increases. Under these conditions, MRP2 is downregulated, reducing biliary excretion of conjugated bilirubin. Bile pigments accumulating in hepatocytes may be pumped out into plasma by alternative pumps (e.g. MRP1 and MRP3), which are upregulated during chole-stasis. Following prolonged accumulation of conjugated bilirubin, a fraction of the pigment binds to albumin irre-versibly. This fraction, termed δ-bilirubin, gives a direct diazo reaction and can be identified by chromatographic analysis. After resolution of biliary obstruction or intra-hepatic cholestasis, δ-bilirubin may linger in plasma for several weeks because it is not taken up by hepatocytes or excreted in the urine (190).
69.8.2 Bilirubin in Bile
In human bile, over 80% of bilirubin is diglucuronide, with only approximately 4% being unconjugated bili-rubin. When UGT1A1 activity is absent, as in the case of Crigler–Najjar syndrome type I, little or no bilirubin glucuronides are excreted in bile. In Crigler–Najjar syn-drome type II or Gilbert syndromes, in which hepatic UGT1A1 activity is lower than normal, proportions of bilirubin monoglucuronide and unconjugated bilirubin increase in bile. The presence of a significant amount of conjugated bilirubin in bile reliably differentiates Crigler–Najjar syndrome type I from Crigler–Najjar syn-drome type II (see later).
69.9 DISORDERS OF BILIRUBIN METABOLISM
Hyperbilirubinemia may result from increased bilirubin production, reduced uptake from the circulation, abnor-mal intracellular storage, deficiency of UGT1A1 activ-ity, or biliary excretion of bilirubin. In many acquired clinical disorders, such as hepatitis or cirrhosis, several steps of this process are affected. In contrast, in inherited disorders of bilirubin metabolism, a specific step of bili-rubin throughput may be involved. From the standpoint of bilirubin metabolism, these disorders may be classified into those that cause unconjugated hyperbilirubinemia and those associated with significant retention of conju-gated bilirubin in serum.
69.9.1 Metabolic Disorders Causing Unconjugated Hyperbilirubinemia
69.9.1.1 Neonatal Hyperbilirubinemia. Serum bilirubin levels are higher in newborns compared to those in normal adults, with obvious jaundice occurring in about 50% during the first 5 days of life. Normally, serum bilirubin concentrations increase from 1–2 mg/dL at birth to a peak of 5–6 mg/dL in about 72 h, and subsequently decline to <1 mg/dL in 7–10 days (129).

R 69 Bile Pigment Metabolism and Its Disorders 11
CHAPTEIn normal newborns, serum bilirubin is predominantly unconjugated. Exaggeration of this physiologic jaundice raises the concern of bilirubin-induced neuropathy. Serum bilirubin concentrations reach 10 mg/dL in about 16% of newborns, and the level exceeds 15 mg/dL in 5% (192). Jaundice of the newborn results from an increased bilirubin load and a lower capacity of the liver to dispose of bilirubin. Exaggeration of these factors and/or superimposition of additional mechanisms may result in a pathologic level of hyperbilirubinemia. The mechanisms of neonatal jaundice were briefly considered here.
69.9.1.1.1 Increased Bilirubin Load. Bilirubin pro-duction, as measured by carbon monoxide production, is increased in the newborn period (193). The excess bilirubin is derived from erythroid and nonerythroid sources and from shortened erythrocyte half-life (194). Rh incompatibility between mother and fetus used to be a common cause of severe neonatal jaundice before treatment of the mother with anti-Rh immunoglobulins became available (72). ABO blood group incompatibility continues to be a common cause of exaggerated neonatal hyperbilirubinemia (196). Sickle cell disease, hereditary spherocytosis, and toxic or allergic drug reactions are common causes of hemolytic jaundice in the newborn period. Excessive bilirubin production can result from ineffective erythropoiesis, as in thalassemia, vitamin B12 deficiency, and congenital dyserythropoietic anemias. In cases in which the rate of bilirubin production exceeds the bile canalicular excretory capacity, conjugated bili-rubin may accumulate in the serum (197).
69.9.1.1.2 Low Hepatic Bilirubin Uptake. During the first few days of life, the rate of hepatic uptake of bilirubin is lower than that in adults. The low uptake rate may be related to low levels of cytosolic GSTs (199), which increase net bilirubin uptake by binding biliru-bin and thereby reducing its efflux. Delayed closure of the ductus venosus may permit portal blood, which is enriched in conjugated bilirubin from the intestine, to bypass the liver.
69.9.1.1.3 Reduced Bilirubin Glucuronida-tion. Only 1% of the normal adult level of hepatic UGT1A1 activity is present at birth (200). Postnatal maturation of UGT1A1 is birth related, and increases rapidly to adult levels by 14 weeks, regardless of the ges-tational age at birth (201). In some disorders, UGT1A1 activity is inhibited by some inherited factor(s).
69.9.1.1.4 Maternal-Milk Jaundice. Serum biliru-bin levels in breast-fed infants are, in general, higher than in formula-fed babies (202), and, in some cases, may increase to 15–24 mg/dL by the age of 10–19 days (203). The unconjugated hyperbilirubinemia may promptly disappear when breast-feeding is discontinued. If breast-feeding is continued, the hyperbilirubinemia may last for weeks. Maternal-milk jaundice is usually benign (203), but kernicterus has been reported in rare cases (204). The maternal milk contains an inhibitor of UGT1A1
activity in this syndrome (203). It has been postulated that polyunsaturated free fatty acids produced by the action of lipolytic enzymes present in some maternal milk samples may be responsible for the inhibition of the transferase (205). Consistent with this notion, the inhibi-tory effect of maternal milk on bilirubin glucuronidation increases on storage and is destroyed by heating at 56 C (205). Mild maternal-milk jaundice may resolve despite continuation of breast-feeding.
69.9.1.1.5 Maternal-Serum Jaundice. This syn-drome, associated with moderate to severe unconjugated hyperbilirubinemia (8.9–65 mg/dL) within the first 4 days of life, was described by Lucey, Arias, and associates (207,208). The syndrome is thought to be caused by an unidentified inhibitor of UGT1A1 present in the serum of mothers of these infants. The jaundice may persist several weeks and is occasionally associated with kernic-terus.
69.9.1.1.6 Decreased Canalicular Bilirubin Excre-tory Capacity. Maturation of canalicular excretion processes may take longer than does the maturation of uptake and conjugation. Therefore, in the late newborn period, canalicular excretion becomes rate limiting in hepatic bilirubin disposition. In cases in which the bil-irubin load is higher than normal, conjugated bilirubin accumulates in serum (209).
69.9.1.1.7 Increased Intestinal Reabsorp-tion. Intestinal β-glucuronidase-mediated deconjuga-tion releases unconjugated bilirubin in the intestine (210). Because of the lack of an established intestinal flora in the newborn, there is reduced bacterial degra-dation of bilirubin, resulting in an increased absorption of unconjugated bilirubin (210). A greater fraction of unconjugated bilirubin is absorbed from the intestine in infants fed with maternal milk.69.9.1.2 Hyperbilirubinemia Due to Bilirubin Over-production. Where liver function is normal, overpro-duction of bilirubin rarely increases serum bilirubin levels to >3–4 mg/dL. Common causes of overproduction of bilirubin include hematologic conditions associated with hemolysis, such as hereditary spherocytosis, and toxic or idiosyncratic drug reactions. Ineffective erythropoiesis that occurs in thalassemia and vitamin B12 deficiency, and a rare group of disorders called dyserythropoietic anemias, result in unconjugated hyperbilirubinemia of various degrees (211). In patients with hemolytic jaun-dice but normal liver function, a small amount of con-jugated bilirubin produced in the liver may appear in the circulation (212), but the unconjugated:conjugated bilirubin ratio remains normal. In sickle cell anemia, a combination of hemolysis and liver abnormalities may lead to the accumulation of both unconjugated and con-jugated bilirubin.69.9.1.3 Inherited Disorders of Bilirubin Glucuro-nidation. Three inherited syndromes associated with a deficiency of bilirubin glucuronidation have been described. A near-complete deficiency of UGT1A1

12 CHAPTER 69 Bile Pigment Metabolism and Its Disorders
activity results in Crigler–Najjar syndrome type 1. Severe but incomplete deficiency of UGT1A1 activity results in Crigler–Najjar syndrome type 2 also known as Arias syn-drome. A mild reduction of UGT1A1 activity results in a common benign disorder, Gilbert syndrome, in which a mild and fluctuating unconjugated hyperbilirubinemia is the only significant clinical feature. Clinical and bio-chemical features of these syndromes are summarized in Table 69-1 and are described in this section.
69.9.1.3.1 Crigler–Najjar Syndrome Type 1. Cri-gler and Najjar (213) described this rare, recessively inherited syndrome in 1952 in six infants from three unrelated families. After the discovery of UGT, this syn-drome was found to result from an absence of UGT1A1 activity (214). All patients had lifelong severe nonhe-molytic unconjugated hyperbilirubinemia. Five of the six infants in the initial report died of bilirubin-induced encephalopathy within 15 months. The sixth patient remained free of brain damage until the age of 15 years, when kernicterus developed and death followed in 6 months (214). Another related patient remained without brain damage until 18 years of age, but then developed bilirubin encephalopathy and died at the age of 24 (215). The original family described by Crigler and Najjar had a high degree of consanguinity. Several members of this family had other recessively inherited disorders, such as Morquio syndrome, homocystinuria, metachromatic leukodystrophy, and bird-headed dwarfism. In subse-quently studied families, the other recessively inherited disorders were not observed. Since 1952, several hundred other patients with Crigler–Najjar syndrome type 1 have been described in all races, and an autosomal-recessive inheritance has been established (214). Icterus is often the only finding on physical examination. However, in many patients, residual neurologic abnormalities, a
sequel to a previous episode of bilirubin encephalopathy, may be detected. With routine use of phototherapy and plasmapheresis for reversing acute bilirubin encephalop-athy, many patients with Crigler–Najjar syndrome type 1 now survive through childhood. However, many survi-vors develop kernicterus around puberty or in early adult life (215). Use of phototherapy usually prolongs sur-vival without encephalopathy to adolescence, and liver transplantation results in long-term survival (see later). Because of a relatively high concentration of unconju-gated bilirubin in bile, pigment stones are common.
69.9.1.3.1.1 Laboratory Tests. Serum bilirubin lev-els usually range from 20 to 25 mg/dL, but may reach 50 mg/dL (213–215). Serum bilirubin is unconjugated, and there is no bilirubinuria. The concentration of serum bilirubin increases during intercurrent illnesses, and decreases on exposure to the sun or bright artificial light (41). The bile is paler than normal (217) and contains only small amounts of unconjugated bilirubin (218). The stool color is normal despite reduced fecal urobilinogen excretion (213). There is no evidence of hemolysis (217), and bilirubin is produced at a normal rate (219). Because bile canalicular transport is normal, BSP (213) and indo-cyanin green (220) and cleared normally from serum, and the biliary tree is normally visualized by cholecysto-graphic agents.
69.9.1.3.1.2 Liver Histology. Liver histology is nor-mal, except that, in several patients, bilirubin plugs were found to be deposited in bile canaliculi and bile ducts (213,218), probably resulting from biliary excretion of unconjugated bilirubin or its photoisomers.
69.9.1.3.1.3 Abnormalities of Hepatic UGTs. Hepatic UGT activity toward bilirubin is virtually absent in all patients with Crigler–Najjar syndrome type 1. In addition, many of these patients have reduction of the
TABLE 69-1 Inherited Disorders Causing Unconjugated Hyperbilirubinemia
Crigler–Najjar Syndrome Type 1
Crigler–Najjar Syndrome Type 2 Gilbert Syndrome
Serum bilirubin concentration 340–850 Mm <340 μM Usually <50 μMRoutine liver function tests Normal Normal NormalSerum bile acid levels Normal Normal NormalOral cholecystography Normal Normal NormalLiver histology Normal Normal NormalBile proportion of monoglucuronide Usually pale: contains small
amounts of unconjugated bilirubin
Increased proportion of bilirubin monoglucuronide
Increased bilirubin
Hepatic BUGT activity None 10% of normal or less 25–40% of normalEffect of phenobarbital None Reduction by 25% or more Reduction on serum bilirubinMode of inheritance Autosomal recessive Autosomal recessive Autosomal recessivePrevalence Rare Uncommon Common, ~5% in general
populationPrognosis Kernicterus is the rule Usually benign; kernicterus is rare BenignAnimal model monkey, Southdown
sheepGunn rat – Bolivian squirrel mutant
BUGT, bilirubin uridine diphosphoglucuronate glucuronosyltransferase.

CHAPT
level of glucuronidation of phenolic substrates (221). Immunologic analysis using monoclonal and polyclonal antibodies revealed variability of expression of UGT pro-teins in the liver of different patients with Crigler–Najjar syndrome type 1 (221).
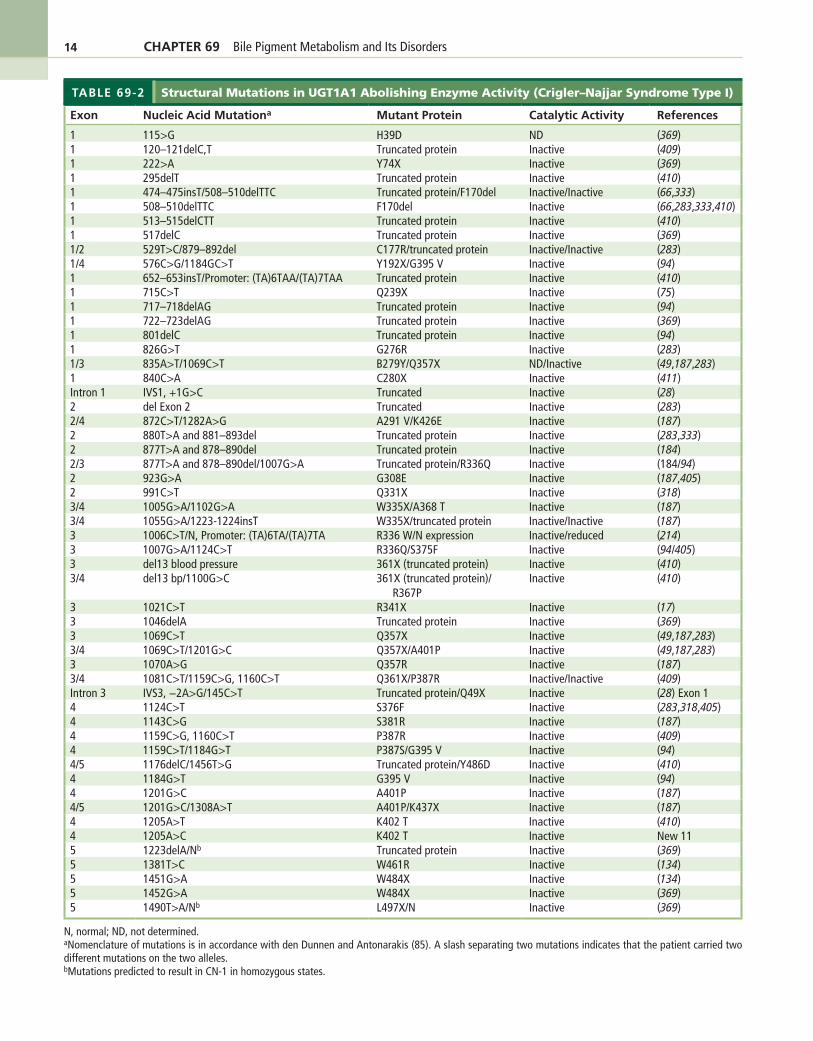
Cloning and characterization of the human UGT1A locus in 1992 (135,140) has permitted the elucidation of the genetic basis of Crigler–Najjar syndrome type 1 (48,49,223). Because UGT1A1 is the only UGT isoform that contributes significantly to bilirubin glucuronida-tion, genetic lesions within any of the five exons com-prising the UGT1A1 gene may lead to complete or near-complete loss of hepatic bilirubin glucuronidation. Such genetic lesions may consist of point mutations, deletions, or insertions within the coding region. Intronic mutations at splice donor or acceptor sites may lead to inappropriate splicing, resulting in loss of bilirubin–UGT activity (224). The genetic lesions may result in mutation of a single critical amino acid or deletion of segments of the enzyme. The various genetic lesions described in the literature (Table 69-2) have been reviewed recently (226–230). In cases where the genetic lesions are pre-sent in exons 2–5, all UGT isoforms expressed from the UGT1A locus are affected. However, when the mutation is located in the unique first exon of UGT1A1, only bili-rubin glucuronidation is affected.
A large number of mutations can result in Crigler–Najjar syndrome type 1, and, in most cases, no particular mutation is common in a given community. An exception to this is seen in the Amish-Mennonite community, in which Crigler–Najjar syndrome type I is relatively com-mon and all patients carry the same mutation (N. Roy Chowdhury et al., unpublished observation), indicating transmission of this mutation from a common ancestor through intermarriage within the small community.
Molecular diagnosis of Crigler–Najjar syndrome is achieved by amplification of the five exons of UGT1A1 and their flanking sequences by polymerase chain reac-tion (PCR) and sequence determination of the ampli-cons. The same strategy can be applied to perform prenatal diagnosis using chorionic villus samples as the starting material (232). Although in a great majority of cases, inheritance of genetic lesions from both parents is required for the manifestation of Crigler–Najjar syn-drome, recently an instance of uniparental isodisomy has been reported in which both mutant alleles were inher-ited from the father (233). The mother’s UGT1A1 geno-type was normal. This case highlights the desirability of analyzing the genotype of both parents to determine the mode of inheritance of Crigler–Najjar syndrome.
The Gunn rat, and animal model of Crigler–Najjar syndrome type 1. Gunn rats are a mutant strain of Wistar rats that exhibit lifelong nonhemolytic unconjugated hyperbilirubinemia inherited as an autosomal-recessive characteristic (234) because of the deficiency of UGT activity toward bilirubin (235). The Gunn rat is the only animal model that develops bilirubin encephalopathy
ER 69 Bile Pigment Metabolism and Its Disorders 13
spontaneously (236). Studies performed in Gunn rats have provided important information on bilirubin tox-icity and have helped in developing new therapeutic modalities for hyperbilirubinemia, including cell trans-plantation and gene therapies (224,237–239). Gunn rats have a single guanosine base deletion in the common-region exon 4, which results in a frameshift, leading to a premature termination codon. As a consequence, 150 amino acid residues at the carboxy-terminal end of all isoforms encoded by the UGT1A locus are deleted (240) and their activities affected (241). However, UGT iso-forms expressed from other UGT genes are unaffected (242).
69.9.1.3.1.4 Treatment of Crigler–Najjar Syndrome Type 1. Temporizing measures are directed at reducing serum bilirubin levels. Definitive treatment consists of partial- or whole-liver transplantation. New experimen-tal therapies based on liver cell transplantation and gene therapy are currently being developed. These treatment modalities are briefly discussed here.
69.9.1.3.1.4.1 Phototherapy. Phototherapy is the most commonly used medical therapy for severe uncon-jugated hyperbilirubinemia (218). Banks of fluorescent lamps with devices for shielding the eyes, or “light blan-kets,” lower serum bilirubin levels by converting bil-irubin IXα-ZZ to its photoisomers (see the section on bilirubin chemistry). Thickening of skin, pigmentation, and decreased surface area compared to body mass result in a diminution of effect of phototherapy beyond the age of 3 or 4 years (37,218).
69.9.1.3.1.4.2 Plasmapheresis. During neurologic emergencies, serum bilirubin concentration can be acutely reduced by plasmapheresis (218). Following removal of bilirubin that is tightly bound to plasma albumin, bilirubin is mobilized from tissue stores to the plasma. Attempts to remove plasma bilirubin by affin-ity chromatography on albumin-conjugated agarose gel columns were hindered in human subjects because of the removal of formed elements of blood (243).
69.9.1.3.1.4.3 Orthotopic Liver Transplantation. Presently, the transplantation of whole liver or a segment of the liver is the only available definitive treatment for Crigler–Najjar syndrome type 1 (244,245). Although this procedure is associated with some risk in these patients, it has been curative in several cases and has dramatically improved the outlook for these patients.
69.9.1.3.1.4.4 Experimental Methods for Reduction of Serum Bilirubin Levels.
69.9.1.3.1.4.4.1 Inhibition of Heme Oxygenase Activ-ity. Noniron metalloporphyrins are strong inhibitors of microsomal heme oxygenase activity (23). Adminis-tration of tin-protoporphyrin has been shown to sup-press neonatal hyperbilirubinemia in rhesus monkeys (246,247). Injection of tin-mesoporphyrin at 0.5 μmol/kg three times a week for 13–23 weeks in two 17-year-old boys with Crigler–Najjar syndrome type 1 resulted in a modest reduction in serum bilirubin concentrations
14 CHAPTER 69 Bile Pigment Metabolism and Its Disorders
TABLE 69-2 Structural Mutations in UGT1A1 Abolishing Enzyme Activity (Crigler–Najjar Syndrome Type I)
Exon Nucleic Acid Mutationa Mutant Protein Catalytic Activity References
1 115>G H39D ND (369)1 120–121delC,T Truncated protein Inactive (409)1 222>A Y74X Inactive (369)1 295delT Truncated protein Inactive (410)1 474–475insT/508–510delTTC Truncated protein/F170del Inactive/Inactive (66,333)1 508–510delTTC F170del Inactive (66,283,333,410)1 513–515delCTT Truncated protein Inactive (410)1 517delC Truncated protein Inactive (369)1/2 529T>C/879–892del C177R/truncated protein Inactive/Inactive (283)1/4 576C>G/1184GC>T Y192X/G395 V Inactive (94)1 652–653insT/Promoter: (TA)6TAA/(TA)7TAA Truncated protein Inactive (410)1 715C>T Q239X Inactive (75)1 717–718delAG Truncated protein Inactive (94)1 722–723delAG Truncated protein Inactive (369)1 801delC Truncated protein Inactive (94)1 826G>T G276R Inactive (283)1/3 835A>T/1069C>T B279Y/Q357X ND/Inactive (49,187,283)1 840C>A C280X Inactive (411)Intron 1 IVS1, +1G>C Truncated Inactive (28)2 del Exon 2 Truncated Inactive (283)2/4 872C>T/1282A>G A291 V/K426E Inactive (187)2 880T>A and 881–893del Truncated protein Inactive (283,333)2 877T>A and 878–890del Truncated protein Inactive (184)2/3 877T>A and 878–890del/1007G>A Truncated protein/R336Q Inactive (184/94)2 923G>A G308E Inactive (187,405)2 991C>T Q331X Inactive (318)3/4 1005G>A/1102G>A W335X/A368 T Inactive (187)3/4 1055G>A/1223-1224insT W335X/truncated protein Inactive/Inactive (187)3 1006C>T/N, Promoter: (TA)6TA/(TA)7TA R336 W/N expression Inactive/reduced (214)3 1007G>A/1124C>T R336Q/S375F Inactive (94/405)3 del13 blood pressure 361X (truncated protein) Inactive (410)3/4 del13 bp/1100G>C 361X (truncated protein)/
R367PInactive (410)
3 1021C>T R341X Inactive (17)3 1046delA Truncated protein Inactive (369)3 1069C>T Q357X Inactive (49,187,283)3/4 1069C>T/1201G>C Q357X/A401P Inactive (49,187,283)3 1070A>G Q357R Inactive (187)3/4 1081C>T/1159C>G, 1160C>T Q361X/P387R Inactive/Inactive (409)Intron 3 IVS3, −2A>G/145C>T Truncated protein/Q49X Inactive (28) Exon 14 1124C>T S376F Inactive (283,318,405)4 1143C>G S381R Inactive (187)4 1159C>G, 1160C>T P387R Inactive (409)4 1159C>T/1184G>T P387S/G395 V Inactive (94)4/5 1176delC/1456T>G Truncated protein/Y486D Inactive (410)4 1184G>T G395 V Inactive (94)4 1201G>C A401P Inactive (187)4/5 1201G>C/1308A>T A401P/K437X Inactive (187)4 1205A>T K402 T Inactive (410)4 1205A>C K402 T Inactive New 115 1223delA/Nb Truncated protein Inactive (369)5 1381T>C W461R Inactive (134)5 1451G>A W484X Inactive (134)5 1452G>A W484X Inactive (369)5 1490T>A/Nb L497X/N Inactive (369)
N, normal; ND, not determined.aNomenclature of mutations is in accordance with den Dunnen and Antonarakis (85). A slash separating two mutations indicates that the patient carried two different mutations on the two alleles.bMutations predicted to result in CN-1 in homozygous states.

CHAPTER 69 Bile Pigment Metabolism and Its Disorders 15
(248). The place of this agent in the treatment of Crigler–Najjar syndrome type 1 is yet to be determined.
69.9.1.3.1.4.4.2 Oxidative Degradation of Bilirubin. Bilirubin oxidase from Myrothecium verrucaria (249) catalyzes the oxidation of bilirubin to a colorless prod-uct. Perfusion of human blood containing bilirubin through filters packed with immobilized bilirubin oxi-dase resulted in the degradation of 90% of the bilirubin per pass (249). When such columns were connected to the circulation of a Gunn rat, serum bilirubin levels are decreased by 50% in 30 min. There are some concerns, however, regarding the removal of formed elements of blood by these columns. Intravenous injection of biliru-bin oxidase, linked to polyethylene glycol to increase its half-life in circulation, has resulted in significant reduc-tion of serum bilirubin levels in Gunn rats, but only for a few hours (249).
69.9.1.3.1.4.4.3 Induction of P-450c. Induction of cytochrome P-450c activity results in increased oxi-dative degradation of bilirubin in Gunn rat liver, lead-ing to the reduction of serum bilirubin levels. Several indoles present in cruciferous vegetables, such as cab-bage, cauliflower, and Brussels sprouts, induce P4501A1 and P4501A2 in rat liver and intestine (250). Indole-3-carbinol, an inducer of P4501A2, is being studied for a potential therapeutic effect in Crigler–Najjar syndrome type 1 (250).
69.9.1.3.1.4.4.4 Replacement of UGT1A1 Activity. UGT1A1 activity is present in excess in normal liver. Therefore, partial replacement of UGT1A1 should reduce serum bilirubin levels in Crigler–Najjar syndrome type 1 to a nontoxic level. Transplantation of hepato-cytes into the Gunn rat liver by portal venous infusion, injection into the splenic pulp (251), intraperitoneal injection of microcarrier-bound hepatocytes (85,252), or intraperitoneal implantation of alginate–polylysine-encapsulated hepatocytes (254) resulted in reduction of serum bilirubin levels in Gunn rats. After intrasplenic injection, a great majority of the hepatocytes rapidly translocate to the liver, where, in the absence of immune rejection, they survive and function throughout the life span of the recipient (255). On the basis of the expe-rience gained from these preclinical studies, isolated allogenic human hepatocytes were transplanted into the liver of a patient with Crigler–Najjar syndrome type 1 through a catheter placed percutaneously into the portal vein (258,259). Transplantation of 7.5 billion hepato-cytes resulted in the lowering of plasma bilirubin con-centration by about 50% and permitted reduction of the duration of phototherapy. Two and half years later, bili-rubin glucuronide excretion in bile continued, but serum bilirubin level gradually increased, probably because of increased bilirubin production or reduced effectiveness of phototherapy. The patient received an auxiliary liver transplantation, which has kept her serum bilirubin within normal limits (J. Roy Chowdhury, personal com-munication).
The clinical course of this case, as well as the experi-ence with other patients who received hepatocyte trans-plantation (260), indicates that the number of adult hepatocytes that can be transplanted at a single pro-cedure is not likely to be sufficient for curing inherited liver-based metabolic disorders (261). Moreover, there is an increasing shortage of good-quality donor livers for hepatocyte isolation (259,261). For these reasons, strategies are being explored to induce preferential pro-liferation of the transplanted normal hepatocytes over the mutant host cells. Since adult hepatocytes have a remarkable capacity to proliferate, massive repopulation of the liver by transplanted hepatocytes requires not only a proliferate stimulus to the engrafted cells, but also pre-parative manipulations of the host liver that prevent the replication of the host liver cells. Controlled hepatic irra-diation in combination with a variety of mitotic stimuli is being evaluated for extensive repopulation of the liver by engrafted wild-type or genetically modified hepatocytes (141,264–266). Recent success in obtaining hepatocyte-like cells by differentiating human embryonic stem cells or induced pluripotent cells derived by reprogramming somatic cells, such as skin fibroblasts offer hopes for a renewable source of functional transplantable hepato-cytes (24,268).
69.9.1.3.1.4.4.5 Gene Therapy. Supplementation with a normal UGT1A1 gene is an attractive potential therapeutic modality. Methods for gene introduction into the liver using recombinant viruses or ligands that mediate receptor-directed endocytosis are being devel-oped for this purpose. These approaches have been reviewed (224,269). In the ex vivo approach, liver cells isolated from a resected liver lobe of a mutant subject are established in primary culture and transduced with normal genes using recombinant retroviruses. The trans-duced cells are then transplanted into the subject, from whom the cells were obtained, thereby circumventing the need for immunosuppression. This approach resulted in modest reduction of serum low-density lipoprotein (LDL) cholesterol levels in LDL receptor–deficient rab-bits (Watanabe Heritable Hyperlipidemic strain) (270) and in patients with familial hypercholesterolemia (271). However, advances in techniques of gene transfer (196) to the liver and ability to conditionally immortalize hepa-tocytes (272) should improve the possibility of success of ex vivo gene therapy. Recombinant adenoviruses are highly efficient in transferring genes to quiescent hepato-cytes in vivo. Adenoviruses remain episomal and express transgenes very efficiently. Administration of these vec-tors to transfer human UGT1A1 gene complementary DNA (cDNA) into Gunn rats resulted in rapid reduction in serum bilirubin levels.
However, these episomal vectors are eventually lost following cell division, and, because they are highly immunogenic, they cannot be administrated repeatedly. The use of viral gene-deleted helper-dependent adeno-vectors can prolong the duration of transgene expression

16 CHAPTER 69 Bile Pigment Metabolism and Its D
and result in lifelong amelioration of jaundice in Gunn rats (273), but, for clinical gene therapy, the expression is not expected to be long enough to span the life of a human subject without repeated administration of the vectors. Repeated gene transfer using adenoviral vec-tors is possible by using generalized immunosuppression around the time of virus administration (274) or spe-cific host tolerization to adenoviral proteins (275–278). However, this approach is difficult to translate into clin-ical application. In another approach, an immunomod-ulatory gene, such as adenoviral E3 or CTLA4-Ig, has been expressed along with the therapeutic transgene to prevent immune response against the cells infected with adenovirus (279,280). Coexpression of CTLA4-Ig, an inhibitor of costimulation of T lymphocytes by antigen-presenting cells, permits repeated administration of the adenovector, resulting in lifelong correction of jaundice in Gunn rats. However, the safety of abrogating host immunity toward adenoviruses, which are potential human pathogens, remains doubtful.
To avoid the need for readministration of gene-ther-apy vectors, we and others have explored the use of vec-tors that integrate into the host genome. Simian virus 40 (SV40) is a DNA virus of the papova family. Recombi-nant SV40 viruses have been developed by replacing the coding region for the T antigens with a target gene. These vectors can infect quiescent hepatocytes, are nonimmu-nogenic, and integrate into the host genome (281,282), permitting long-term expression of the transgene. Gunn rats treated with a recombinant SV40 virus expressing UGT1A1 showed significant long-term reduction in serum bilirubin levels (283). Because the recombinant virus does not evoke an immune response, the vector can be administered repeatedly.
Vectors based in nonrecombinant lentiviruses can integrate into the genome of both dividing and quiescent cells, such as hepatocytes. Administration of recombi-nant lentiviruses in utero on the nineteenth gestational day in Gunn rat embryos resulted in UGT1A1 expres-sion in the liver and reduction of serum bilirubin levels in Gunn rats (284). Both recombinant SV40 and lenti-viral vectors have a broad range of cellular targets and transduce many tissue types after systemic administra-tion (283,284).
Nonviral, plasmid-based vectors are also being explored for gene therapy on the Gunn rat model. Both naked DNA and carriers containing ligands that are endocytosed via liver-specific receptors (e.g. the asialo-glycoprotein receptor) are being evaluated (285–287). To enhance integration of the therapeutic transgene, some investigators are examining the feasibility of using the Sleeping Beauty transposon system, which results in the excision of the transgene at specific flanking sequences and splicing into the host chromosomal DNA (288). In a very different approach, oligonucleotides are used to cor-rect single-base mutations or deletions utilizing the cell’s intrinsic mismatch repair system (289). The nonviral
isorders
gene transfer or gene repair strategies hold great prom-ise for safe gene therapy of inherited jaundice although, at their current state, these approaches are not efficient enough for immediate clinical application.
69.9.1.3.2 Crigler–Najjar Syndrome Type 2. Crigler–Najjar syndrome type 2 is also known as Arias Syn-drome. Arias (8) reported a milder variant of the clas-sical Crigler–Najjar syndrome, which is now termed Crigler–Najjar syndrome type 2, in which serum uncon-jugated bilirubin concentrations usually ranged from 8 to 18 mg/dL. During intercurrent illness, general anes-thesia, or prolonged fasting, bilirubin levels could be as high as 40 mg/dL (291,292). Kernicterus is unusual in this group, but can be precipitated during episodes of exacerbated hyperbilirubinemia (8,217,291,292). As in Crigler–Najjar syndrome type 1, there is no evidence of hemolysis or other liver dysfunction. Crigler–Najjar syndrome type 2 is clinically differentiated from type 1 by >25% reduction of serum bilirubin after administra-tion of enzyme-inducing agents (e.g. phenobarbital) and the presence of significant amounts of bilirubin glucuro-nides in bile although the proportion of bilirubin mono-glucuronide exceeds 30% of total conjugated bilirubin (normal, ~10%) (292,293), reflecting a reduced hepatic UGT1A1 activity. The liver histology is normal, and UGT1A1 activity is usually reduced to approximately 10% of normal (292,293).
69.9.1.3.2.1 Genetic Lesions. Molecular genetic studies that have performed so far support an autoso-mal-recessive mode of inheritance (294). As in Crigler–Najjar syndrome type 1, the genetic lesions are found in the coding region of UGT1A1. However, in Crigler–Najjar syndrome type 2, these mutations always con-sists of a single amino acid transition that significantly reduces the UGT1A1 activity, without completely abol-ishing it. Table 69-3 lists the reported genetic lesions found in Crigler–Najjar syndrome type 2. In some cases, the mutation may increase the Km for bilirubin (295). As the in vitro UGT1A1 assays are performed at high bilirubin concentrations, their measurements may over-estimate the in vitro activity of the enzyme in these cases. The various lesions causing Crigler–Najjar syndrome type 2 have been reviewed (228).
69.9.1.3.3 Gilbert Syndrome. This disorder, asso-ciated with a mild, chronic, fluctuating unconjugated hyperbilirubinemia was described by Gilbert and Lere-boullet (296). Although most patients present as isolated cases, familial incidence also has been noted (297).
69.9.1.3.3.1 Clinical Features. Gilbert syndrome is usually diagnosed in young adults who are incidentally found to have mild, predominantly unconjugated hyper-bilirubinemia. Bilirubin levels may be elevated only inter-mittently and are usually <3 mg/dL, but may increase during intercurrent illness, stress, fasting, or menstrual periods (297). Mild icterus is the only positive finding on physical examination. Vague symptoms, such as fatigue and abdominal discomfort, reported by some patients

CHAPTER 69 Bile Pigment Metabolism and Its Disorders 17
TABLE 69-3 Structural Mutations in UGT1A1 Reducing Enzyme Activity (Crigler–Najjar Syndrome Type 2 and Gilbert Syndrome)
Exon Nucleic Acid Mutationa Mutant Protein Catalytic Activity References
1 44T>G L15R Reduced activity (212)1 44T>G/572C>T L15/R/S191F Reduced activit/inactivet New #11¼ 111C>A/1207C>T P34Q/R403C Reduced activity (94)1 211G>Ab G71R Reduced activity (9,371,412)1 211G>A/Nc G71R/N Reduced activity (371,412,413)1/5 211G>A and 1456T>G G71R and Y486D Reduced activity1 395T>C/Nc L132P/N Reduced activity (412)1 524T>A L175Q Reduced activity (283)½ 524T>A/973delG L175Q/truncated protein Reduced activity/truncated-inactive (283)¼ 576C>G/1130G>T/Promoter: (TA)6TAA/
(TA)7TAAY192X/H377 V Inactive/reduced activity (94/369)
1 625T>C R209 W Reduced activity (36,113,283)¼ 625 T<C/1186delG R209 W/truncated protein Reduced activity/truncated-inactive1 671T>G/722-723delAG V224 G/truncated protein Residual activity/inactive (369)1 686C>Ac P229Q/N Reduced activity (371)1 717–718delAG/671T>G Truncated protein/V224 G Inactive/reduced activity (274)Intron 1/3 865-1G>A/1007G>T Splicing aberration/R336L Inactive/reduced activity (94)2/3 877T>A&878–890del/1060T>C Truncated protein/W354R Inactive/reduced activity (94,184)2 881T>C/N, Promoter: (TA)6TAA1/(TA)7TAA I294 T/N Reduced activity (214)2 992A>G Q331R Reduced activity (193)2 991C>T/Nc Q331X/N Reduced activity (161)3/intron 3 1006C>T/1304>1G>T R336 W/splicing aberration Reduced activity/inactive 214/943/5 1006C>T/1489delG R336 W/A498PfsX3 Reduced activity/inactive New #114 1099C>G/Nc R367 G/N Reduced activity (371,412)4 1130A>G/N H377R/N ND/N (369)4 1133G>T G377 V ND (369)4 1160C>A P387H Reduced activity New #115 1391A>C/N, Promoter: (TA)7TAA/(TA)7TAA Z464A/N Residual activity (174)5 1433C>A A478D Reduced activity (94)5 1456T>Gb Y486D Reduced activity (125,371)
A slash separating two mutations indicates that the patient carried two different mutations on the two alleles.N, normal; ND, not determined.aNomenclature of mutations is in accordance with den Dunnen and Antonarakis (85).bIn these cases, the hyperbilirubinemia was mild and the patients were classified as Gilbert syndrome.cIn all these cases, a heterozygous mutation was found to be the cause of the phenotype and a dominant-negative mechanism has been proposed to explain the hyperbilirubinemia.
may be manifestations of anxiety. Except for the pre-dominantly unconjugated hyperbilirubinemia, routine blood tests are negative. Because canalicular excretion processes are normal, oral cholecystography allows visu-alization of the gallbladder. Liver biopsy is not routinely indicated, but, when performed, shows normal histol-ogy, except for a nonspecific accumulation of lipofuscin pigment in the centrilobular zones (298).
69.9.1.3.3.2 Incidence. Gilbert syndrome is per-haps the most common inherited disorder in man, the reported incidence ranging from 3% to 7% of the pop-ulation (299,300). Distribution of serum bilirubin lev-els in the general population follows a skewed (301) or bimodal (299,300) pattern rather than a Gaussian distri-bution, and it has been proposed that subjects with Gil-bert syndrome should be considered to represent one end of this spectrum. Therefore, the definition of Gilbert syn-drome is somewhat arbitrary, making the determination
of its precise incidence difficult. Although it is possible to distinguish patients with Gilbert syndrome from the general population by determination of biliary biliru-bin diglucuronide:monoglucuronide ratio and UGT1A1 activity in liver biopsy specimens, these tests are imprac-tical for population studies. Because of the higher average serum bilirubin levels in males (299), Gilbert syndrome is diagnosed with much more frequency in males than in females (300). Gilbert syndrome is often recognized around puberty, which may be related to the inhibition of bilirubin glucuronidation by endogenous steroid hor-mones (302).
69.9.1.3.3.3 Bilirubin Glucuronidation. Hepatic UGT1A1 activity, as determined by in vitro assays, is present at a consistently low level (around 30% of nor-mal) in Gilbert syndrome (303,304).
69.9.1.3.3.4 Organic Anion Uptake. In addition to reduced UGT1A1 activity, some patients with Gilbert

18 CHAPTER 69 Bile Pigment Metabolism and Its Di
syndrome exhibit abnormalities of organic anion trans-port (306). However, based on studies of handling to bilirubin loads and the pattern of bilirubin conjugates secreted in bile, other investigators have suggested that the initial uptake of bilirubin is normal for Gilbert syndrome and the reduced plasma clearance may be explained by a decreased rate of glucuronidation only (307). The plasma disappearance of organic anions other than bilirubin (e.g. BSP or indocyanin green) may be abnormal in a minority of cases of Gilbert syndrome (308,309). No mechanistic basis for the association of decreased UGT1A1 activity and reduced organic anion uptake in these cases is known, and the coexistence of the two abnormalities may be merely coincidental.
69.9.1.3.3.5 Diagnosis. Gilbert syndrome is diag-nosed in individuals with mild unconjugated hyperbiliru-binemia without evidence of hemolysis or structural liver disease. Although hemolysis is not a feature of Gilbert syndrome, coexistent compensated hemolysis is found in many patients because increased bilirubin load results in clinically obvious jaundice (310). A presumptive diagno-sis of Gilbert syndrome is made when mild unconjugated hyperbilirubinemia is noted on several occasions, and serum levels of alanine and aspartate aminotransferases, alkaline phosphatase, γ-glutamyl transpeptidase, and fasting and postcibal bile acid are all normal. Sequence determination of the promoter region upstream to exon 1 of UGT1A1 provides a simple means of diagnosis. If confirmation of diagnosis is required, chromatographic determination of the relative content of bilirubin mono-glucuronide and diglucuronide in bile is of potential use in the diagnosis of Gilbert syndrome. Fasting or nicotinic acid tests provide suggestive data, but are not specific for Gilbert syndrome. Needle biopsy of the liver is not necessary for the diagnosis.
69.9.1.3.3.6 Effect of Fasting. Reduction of daily caloric intake to 400 kcal for 48 h results in elevation of serum bilirubin levels in all individuals (26). How-ever, this response may be exaggerated in Gilbert syn-drome (312). Fasting may induce hyperbilirubinemia by multiple physiologic mechanisms. Both increased bilirubin production (313) and reduced hepatic biliru-bin clearance (314) have been reported. Reduction of hepatic UDPglucuronic acid content has been proposed as a mechanism of the exaggerated hyperbilirubinemic response to fasting in rats (315). However, because fast-ing also exacerbates hyperbilirubinemia in homozygous Gunn rats that lack UGT1A1 activity (316), reduction of bilirubin glucuronidation cannot be a complete expla-nation for fasting hyperbilirubinemia. Because of the overlap with the normal response to caloric deprivation (312), the fasting test does not reliably distinguish Gil-bert syndrome from other hyperbilirubinemic states.
69.9.1.3.3.7 Nicotinic Acid Tests. Intravenous administration of nicotinic acid increases unconjugated hyperbilirubinemia, probably by increasing erythrocyte fragility, splenic heme oxygenase activity, and splenic
sorders
bilirubin formation (317). Nicotinic acid administration has been proposed as a provocative test for Gilbert syn-drome; however, similar to fasting, it does not clearly distinguish patients with Gilbert syndrome from normal subjects or those with hepatobiliary disease (317).
69.9.1.3.3.8 Bilirubin Conjugates in Bile. As in Crigler–Najjar syndrome type 2, bile in Gilbert syndrome contains an increased proportion of bilirubin mono-glucuronide (293), reflecting reduced hepatic UGT1A1 activity in these syndromes.
69.9.1.3.3.9 Genetic Basis of Gilbert Syndrome. Gil-bert syndrome is associated with a variant TATAA box in the promoter upstream to exon 1 of UGT1A1. The normal TATAA element has the sequence A[TA]6TAA, whereas subjects with Gilbert syndrome are homozy-gous for elongation of the dinucleotide repeat, so that the sequence of the TATAA element is T[TA]7TAA (318). All subjects with Gilbert syndrome of white, black, or Asian Indian origin who have had genetic analysis were found to be homozygous for this variant promoter. Promoter–reporter studies show that an increased TATAA box length reduces UGT1A1 expres-sion (318). Patients with the Gilbert genotype have been shown to have lower hepatic microsomal UGT1A1 activity (319). UGT1A1 alleles carrying the Gilbert-type TATAA element have been designated as UGT1A1*28 (320). However, all subjects who are homozygous for the variant promoter do not exhibit the clinical pheno-type, indicating that additional variables, particularly the rate of bilirubin production, are important for the manifestation of the phenotype. Notably, although the inheritance is autosomal (chromosome 2q37), women with this genotype usually do not manifest jaundice, probably because of lower daily bilirubin production. The gene frequency of the Gilbert-type promoter in the Western hemisphere is approximately 0.3. Thus, 9% of the general population is homozygous for this genotype. The gene frequency may be lower in Japan. Some muta-tions of the coding region of UGT1A1 may be associated with mild hyperbilirubinemia, compatible with the clini-cal diagnosis of Gilbert syndrome (321,322) (Table 69-3). These mutations have not been observed in populations other than those of Far Eastern origin. Some of these mutations have been reported to be dominant negative, suggesting that they can partially inactivate a normal allele (323).
Because of the high frequency of the Gilbert-type TATAA element in the general population, some het-erozygous carriers of structural mutations may carry the Gilbert-type promoter on the structurally normal allele. This reduces the expression of the only structurally nor-mal allele. Therefore, a combination of these mutations may give rise to intermediate levels of hyperbilirubine-mia (324,325). This explains the long-standing observa-tion that intermediate levels of hyperbilirubinemia are common among relatives of patients with Crigler–Najjar syndrome. This type of combination is a more common

CHAPTER
cause of intermediate levels of hyperbilirubinemia than is homozygous mutation of the coding region in UGT1A1.
69.9.1.3.3.10 Health Implications of Gilbert Syn-drome. Gilbert syndrome is generally considered innoc-uous, and its recognition is considered important mainly for reassuring the patient and the physician that no underlying liver disease is responsible for the mild hyper-bilirubinemia. However, the clinical and epidemiologic significance of Gilbert syndrome is continuing to be unveiled. The UGT1A1*28 genotype has been reported to be associated with accelerated or prolonged neonatal hyperbilirubinemia (326,327). In children with a combi-nation of glucose-6-phosphate dehydrogenase deficiency and the UGT1A1*28 genotype, neonatal serum bilirubin concentrations can rise to dangerously high levels (328). Patients with hereditary spherocytosis who also have the UGT1A1*28 genotype may develop pigment gallstones early in life (329).
Although the mild hyperbilirubinemia associated with Gilbert syndrome should not cause any significant toxic-ity in adults and may indeed be cytoprotective by virtue of the antioxidant effect of bilirubin, the reduced expression of UGT1A1 activity may have some significant conse-quences. The pharmacogenetics of UGT1A1 polymor-phism is receiving increasing attention (320). Oxidative paracetamol metabolism is associated with drug toxic-ity. Conflicting evidence has been reported for increased oxidative metabolism of paracetamol and its decreased glucuronidation in subjects with the UGT1A1*28 allele (330,331). Gilbert syndrome is associated with a high incidence of diarrhea in patients treated with the antican-cer drug, irinotecan (332).
69.9.1.3.3.11 Animal Model. Bolivian squirrel mon-keys have higher serum unconjugated bilirubin concen-trations and a greater hyperbilirubinemic response to fasting than does a closely related Brazilian population
69 Bile Pigment Metabolism and Its Disorders 19
(333,334). Plasma clearance of intravenously adminis-tered bilirubin, hepatic UGT activity toward bilirubin, and bilirubin diglucuronide:monoglucuronide ratio in bile are lower in the Bolivian population. In these respects, Bolivian squirrel monkeys are a model of Gil-bert syndrome. Fasting hyperbilirubinemia is rapidly reversed by oral or intravenous administration of carbo-hydrates, but not by lipid administration (333).
69.9.2 Disorders Associated with Predominantly Conjugated Hyperbilirubinemia
Accumulation of conjugated bilirubin in serum may result from leakage of bilirubin glucuronides from hepatocytes or regurgitation from bile in inflammatory or ischemic diseases of the hepatocyte, and intrahepatic cholestasis or biliary obstruction. However, in specific disorders of organic anion storage or transport (e.g. Rotor and Dubin–Johnson syndromes), conjugated bilirubin may be retained in plasma in the absence of other evidence of liver dysfunction. Table 69-4 provides a comparison of the clinical and pathophysiologic features of these inher-ited disorders. In addition, there are inherited abnormal-ities of bile canalicular transport proteins that cause a group of disorders collectively termed progressive famil-ial intrahepatic cholestasis. In another genetic condition, paucity of bile ducts leads to conjugated hyperbilirubine-mia. A brief description of these disorders follows.69.9.2.1 Dubin–Johnson Syndrome. A syndrome characterized by chronic nonhemolytic jaundice due to accumulation of conjugated bilirubin in serum and grossly pigmented, but otherwise histologically normal, livers was described by Dubin and Johnson (335) and by Sprinz and Nelson (336). Mild icterus is the only signif-icant physical finding. Rarely, hepatosplenomegaly has
TABLE 69-4 Inherited Disorders Causing Retention of Conjugated Bilirubin
Dubin–Johnson Syndrome Rotor Syndrome
Serum bilirubin Predominantly conjugated, usually 50–85 μM, can be as high as 340 μM
Predominantly conjugated, usually 50–100 μM,occasionally as high as 340 μM
Routine liver function tests Normal except for hyperbilirubinemia Normal except for hyperbilirubinemiaSerum bile salt levels Normal NormalPlasma BSP retention Normal at 45 min; secondary rise at 90 min Elevated; but no secondary rise at 90 minBSP infusion studies Tm very low; storage normal Tm and storage both reducedOral cholecystogram Usually does not visualize the gallbladder Usually visualizes the gallbladderUrinary coproporphyrin excretion Total—normal; >80% as coproporphyrin I Total—elevated; ∼50–75%, as coproporphyrin I patternAppearance of liver Grossly black NormalHistology of liver Dark pigments, predominantly in centrilobular Normal, no increase in pigmentation areas; otherwise
normalMode of inheritance Autosomal recessive Autosomal recessivePrevalence Rare (1:1300 in Middle Eastern Jews) RarePrognosis Benign BenignAnimal model Mutant TR− rats/mutant Corriedale sheep/Golden
lion tamarin monkeyNone
BSP, bromosulfophthalein.

20 CHAPTER 69 Bile Pigment Metabolism and Its Disorders
been observed (337,338). Although patients are usually asymptomatic, an occasional patient complains of weak-ness and vague abdominal pain. Serum bile acid levels are normal (339), and pruritis is absent. Serum bilirubin levels are increased by intercurrent illness, oral contra-ceptives, and pregnancy (339). Although the syndrome is occasionally recognized during the neonatal period (340), the diagnosis is most often made after puberty. In some cases, jaundice is first noticed during pregnancy or contraceptive use (339).
69.9.2.1.1 Laboratory Tests. Liver function tests, including serum bile acid levels, are normal (339). Serum bilirubin levels fluctuate. Usual levels range from 2 to 5 mg/dL, but levels as high as 20–25 mg/dL have been observed. Over 50% of total serum bilirubin is direct-reacting, and bilirubin is excreted in urine. Because the canalicular excretion abnormality includes a wide variety of non-bile-acid organic anions, oral cholecystography, even using a “double dose” of contrast material, usually does not visualize the gallbladder. However, visualiza-tion may occur 4–6 h after intravenous administration of meglumine iodipamide (Biligrafin) (341). Grossly, the liver is black, and light microscopy reveals a dense pig-ment (342). Following infusion of 3H-epinephrine into mutant Corriedale sheep (an animal model for Dubin–Johnson syndrome), the isotope is incorporated into the hepatic pigment (343). The pigment differs from authen-tic melanin (344), but could be composed of polymers of epinephrine metabolites (345). Following liver disease, such as acute viral hepatitis, the pigment is cleared from the liver and reaccumulates slowly after recovery (346).
69.9.2.1.2 Organic Anion Transport. The hepatic secretion of bilirubin glucuronides, the leukotriene LTC4, reduced and oxidized glutathione, and numerous glucuronide and glutathione conjugates are disturbed in these patients (347,348). In contrast, bile acid secre-tion is unaffected. Hepatic storage is normal, and the transport abnormality is limited to canalicular excre-tion (349). After intravenous injection of BSP, plasma BSP concentration decreases at near-normal rate for 45 min, indicating a normal sinusoidal uptake process. However, in 90% of patients, plasma BSP concentra-tion increases after this time, so that the concentration at 90 min is greater than that at 45 min because of reflux of glutathione-conjugated BSP from hepatocytes into the circulation (67). A similar secondary rise occurs after intravenous administration of bilirubin (351). Because the secondary rise of plasma BSP occurs in other hepa-tobiliary disorders as well (352), this phenomenon is not pathognomonic of Dubin–Johnson syndrome.
69.9.2.1.3 Urinary Coproporphyrin Excretion. In normal subjects, approximately 75% of the urinary cop-roporphyrin is isomer III, which is the precursor of heme. The total urinary coproporphyrin excretion is normal in Dubin–Johnson syndrome, but over 80% of the copro-porphyrin is isomer I (353). The proportion of urinary coproporphyrin I in the urine of neonates is higher than
in adults, but the levels are not as high as in Dubin–John-son syndrome (354). In unaffected parents or children of subjects with Dubin–Johnson syndrome, total urinary coproporphyrin excretion was reduced by 40% as com-pared to normal, because of a 50% reduction in cop-roporphyrin III excretion (355). The mechanism of the abnormal pattern of urinary porphyrin excretion and its relationship to the organic anion transport defect is not known. When the history and physical examination are consistent, the urinary coproporphyrin excretion pattern is diagnostic of Dubin–Johnson syndrome.
Organic anions other than bile acids are transported into the bile canaliculus from the hepatocyte against a concentration gradient by an ATP-dependent energy-consuming process, mediated by a protein that had been originally termed the canalicular multispecific organic anion transporter (cMOAT), and is now known as MRP2 or ABC-C2 (3,149,150,357,358) (Figure 69-1). These anions include conjugated bilirubin and other gluc-uronide- or glutathione-conjugated substances. MRP2 is one of the ABC transporters (357). Direct evidence for its involvement in canalicular transport came from the discovery of a frameshift mutation in the gene encoding in the TR− rat (154).
Despite the absence of MRP2, the serum bilirubin lev-els are only mildly elevated in Dubin–Johnson syndrome, suggesting alternative pathways for the secretion of bil-irubin conjugates. Currently it is not known whether other canalicular transport proteins mediate the resid-ual canalicular transport of bilirubin glucuronides in Dubin–Johnson syndrome. As discussed earlier, absence of MRP2 leads to accumulation of organic anions within the hepatocytes and upregulation of the expression of MRP1 and MRP3 in the basolateral domain of the hepa-tocyte plasma membrane. These and possibly other ATP-consuming pumps may contribute to the active export of unconjugated bilirubin and bilirubin glucuronides from the hepatocyte to plasma via the space of Disse (159).
69.9.2.1.4 Genetic Basis and Inheritance. Dubin–Johnson syndrome has been reported in all races and both sexes. There is a high incidence (1:1300) in Persian Jews (338), in whom it is associated with clotting factor VII deficiency (359). On the basis of the urinary cop-roporphyrin excretion abnormality, the inheritance pat-tern is clearly recognized as autosomal recessive (355). Dubin–Johnson syndrome is caused by mutations of the MRP2 (ABCC2) gene, causing a deficiency of canalicular MRP2 expression or function (152,154,360).
The human cDNA has been isolated on the basis of homology with rat MRP2 (153). The human MRP2 gene has been localized to chromosome 10q23–q24 (361), and the exon–intron organization has been elucidated (362,363). Several different mutations (Table 69-5) have been identified in this gene in patients with Dubin–Johnson (153,362–365). Mutations most commonly involve the crucial ATP-binding region. Mutation at an intronic splice donor site has also been identified in a patient with

CHAPTER 69 Bile Pigment Metabolism and Its Disorders 21
TABLE 69-5 Mutations Identified in Patients with Dubin–Johnson Syndrome
Exon Mutationa Mutation Protein References
Nucleic Acid 3/28 298C>T/3928C>T R100X/R1310X– (374)Truncated protein 13 1669–1815del Truncated protein (98)Intron 15 IVS15, +2T>C Truncated protein (18)16 2026G>C G676R (181)17 2125T>C W709R (199)18 2302C>T R768 W (98)18 2272–2439del Truncated protein (98)18 splice site/29 2439+2T>C/4145A>G 2272del168/Q1382R (102)23 3196C>T R1066X–truncated protein (150)25 3449G>A R1150H (37)25 3517A>T I1173F (37)30 4175–4180del Truncated protein (266)
aNomenclature of mutations is in accordance with den Dunnen and Antonarakis (85).
Dubin–Johnson syndrome (364). Some mutations may lead to impaired glycosylation of MRP2, leading to pre-mature proteasome-dependent degradation (366). The genetic analyses have confirmed an autosomal-recessive pattern of inheritance.
69.9.2.1.5 Animal Models69.9.2.1.5.1 Mutant Corriedale Sheep. In this
mutant strain, biliary excretion of conjugated bilirubin, glutathione-conjugated BSP, iopanoic acid, and indocya-nine green is decreased, whereas taurocholate transport is normal (367). The secretion of the organic cation pro-caine amide ethobromide is unaffected (3) and, interest-ingly, the secretion of unconjugated BSP is unimpaired (368). There is a mild hyperbilirubinemia, with 60% of the bilirubin being conjugated. The liver is pigmented but otherwise of normal histology (343). Total urinary cop-roporphyrin excretion is normal, with increased excre-tion of coproporphyrin isomer I and decreased isomer III excretion. In all these respects, the mutant Corriedale sheep is a model of Dubin–Johnson syndrome.
69.9.2.1.5.2 TR− Rats. These rats have a metabolic defect strongly resembling that in people with Dubin–Johnson syndrome and the mutant Corriedale sheep. Biliary secretion of conjugated bilirubin and many other organic anions is impaired (369), and coproporphyrin I is the major porphyrin excreted in urine (370). The liver is not normally pigmented, but, when the rats are fed a diet enriched in tryptophan, tyrosine, and phenylalanine, intracellular pigment deposition can be demonstrated. Impaired excretion of anionic metabolites of tyrosine, phenylalanine, and tryptophan may result in their reten-tion, oxidation, polymerization, and subsequent lyso-somal accumulation in the liver (371). As in human Dubin–Johnson syndrome, TR− rats (372) and rats of the Eisai hyperbilirubinemic rats (EHBR) strain, which have a similar defect, absence of MRP2 (mrp2 in rats) in the canalicular membrane leads to impaired canalicular secretion of bilirubin and other glucuronides, the leu-kotriene LTC4, glutathione, and glutathione conjugates (347). Experiments with the rodent models showed that
bilirubin conjugates and bile acids are secreted into the bile canaliculus via different pathways. Bile acids with free 3-OH groups are transported by the bile salt export pump (BSEP), but 3-OH-conjugated bile acids are trans-ported by mrp2, as evidenced by impaired secretion of bile acid conjugates in TR− and EHBR rats (373).
69.9.2.1.5.3 A Non-Human Primate Model. The Golden Lion tamarin monkey, which manifests conju-gated hyperbilirubinemia, is another animal model for Dubin–Johnson syndrome (374).69.9.2.2 Rotor Syndrome. Rotor, Manahan, and Flo-rentin described several patients with chronic predomi-nantly conjugated hyperbilirubinemia from two families (93). Hematologic tests and blood biochemistries were normal. Liver histology was normal and, in contrast to Dubin–Johnson syndrome, there was no pigmentation. The basic abnormality of this disorder is different from that of Dubin–Johnson syndrome (375,376). Rotor syndrome is harmless and is characterized by chronic predominantly conjugated hyperbilirubinemia without evidence of hemolysis (93,375,376). It is rare, but has been described in several races.
69.9.2.2.1 Organic Anion Excretion. After intrave-nous injection of 5 mg/kg BSP, over 25% of injected BSP is retained in serum at 45 min, but there is no secondary rise of plasma BSP level and conjugated BSP does not appear in the plasma (377). There is also marked plasma retention of intravenously administrated unconjugated bilirubin (378) and ICG (379). Phenotypically normal heterozygotes have mild abnormal BSP retention at 45 min (377). In contrast to the findings in Dubin–Johnson syndrome, oral cholecystographic agents visualize the gallbladder in Rotor syndrome (380).
69.9.2.2.2 Hepatic Storage. Transport maximum and hepatic storage of BSP has been determined by a constant infusion technique (377). In Dubin–Johnson syndrome, the transport is markedly abnormal, but the hepatic storage capacity is normal. In contrast, in Rotor syndrome, the storage capacity was reduced by 75–90%, whereas the transport maximum was reduced by only

Disorders
22 CHAPTER 69 Bile Pigment Metabolism and Its50% (377). The findings in Rotor syndrome are similar to those in “familial hepatic storage disease,” which is associated with predominantly conjugated hyperbilirubi-nemia and normal liver histology (381). These two dis-orders may represent the same pathophysiologic entity.
69.9.2.2.3 Urinary Coproporphyrin Excretion. In Rotor syndrome, total urinary coproporphyrin is increased two- to fivefold over normal. The proportion of coproporphyrin I in urine is approximately 65% of total (376). These findings are similar to those seen in many other hepatobiliary disorders (382), and distinguish Rotor syndrome from Dubin–Johnson syndrome. In a recent report, however, two brothers with clinical Rotor syndrome had over 80% of urinary coproporphyrins as isomer I (383). Obligate heterozygotes have a copropor-phyrin excretory pattern that is intermediate between that of control subjects and patients with Rotor syn-drome. By urinary coproporphyrin analysis, Rotor syn-drome appears to be inherited as an autosomal-recessive characteristic (376). The urinary coproporphyrin abnormality may be due to reduced biliary excretion of coproporphyrins, with concomitant increase in renal excretion. The mechanism of the organic anion transport defect in Rotor syndrome is not known.
69.9.3 Hyperbilirubinemia Resulting from Inherited Cholestasis Syndromes
69.9.3.1 Progressive Familial Intrahepatic Choles-tasis. These inherited disorders do not specifically affect bilirubin metabolism, but cause the retention of both unconjugated and conjugated bilirubin in plasma by affecting bile flow. Three life-threatening heterogeneous disorders, characterized by defective secretion of bile acids or other components of bile, may result in cholestasis of various degrees of severity and are collectively termed progressive familial intrahepatic cholestasis (PFIC) (384). Benign recurrent intrahepatic cholestasis (BRIC) is a disorder that is genetically related to PFIC I. These disorders usually present during infancy or childhood, and are often associated with growth failure and progressive liver disease. Several additional genetic cholestatic disorders have been described during the past few years. PFIC syndromes and the other inherited cholestatic diseases have been reviewed recently (385) and are discussed in brief here.
69.9.3.1.1 Progressive Familial Intrahepatic Cho-lestasis Type I. PFIC I was originally described in a family in the Amish-Mennonite community and was named Byler disease, after the family name of the first patient (386). The disease is associated with severe life-threatening cholestasis, and is caused by mutations in the P-type ATPase gene, FIC1 (also termed ATP8B1), which has been localized to chromosome 18q21 (163). FIC-1 couples the hydrolysis of ATP to the translo-cation of acidic phospholipids. How the mutation of FIC-1 causes cholestasis and abnormal bile acid
transport is understood only partially (387). Lack of FIC-1 may lead to diminished nuclear translocation of the nuclear receptor FXR (388). Cholestasis may result from consequent downregulation of the BSEP (Section 76.345.3.361.3).
69.9.3.1.2 Benign Recurrent Intrahepatic Chole-stasis. This disorder was first described in 1959 (389). It presents in adolescence or early adulthood (390,391) with recurrent episodes of conjugated hyperbilirubinemia associated with malaise, anorexia, pruritis, weight loss, and malabsorption. Laboratory tests reveal biochemical evidence of cholestasis without severe hepatocellular injury (392,393). Such episodes last for weeks to months, followed by a complete return to normalcy clinically, biochemically, and histologically. In a given patient, recurrent attacks resemble each other in symptoms, signs, and duration. Liver histology reveals noninflammatory intrahepatic cholestasis without fibrosis regardless of the number and severity of attacks. During remission, liver histology returns to normal whether examined by light or electron microscopy (394).
Family studies suggest a recessive inheritance pattern. Surprisingly, this relatively benign disorder was also found to be associated with mutations in the FIC-1 gene, other mutations of which cause the much more severe disorder PFIC I.
There is no specific treatment for BRIC. Liver trans-plantation has not been considered because of the episodic and nonprogressive nature of the disease.
69.9.3.1.3 Progressive Familial Intrahepatic Cho-lestasis Type II. This disorder resembles Byler disease clinically, but occurs in non-Byler families, mainly in the Middle East and Europe. It is caused by defects in the gene that codes for the sister P-glycoprotein (SPGP), also known as BSEP or ABCB11, which is a canalicu-lar ATP-dependent transporter of bile acids from the hepatocytes into the bile (395). The BSEP gene has been localized to chromosome 2q24 (165). A large number of different point mutations in BSEP have been described in patients with PFIC II.
Although both PFIC I and PFIC II are associated with life-threatening cholestasis, serum γ-glutamyl transpep-tidase levels are normal or nearly so in both disorders, which differentiate them from PFIC III (165,384). BSEP is expressed specifically in the liver and, as expected, liver transplantation ameliorates all manifestations of PFIC II.
69.9.3.1.4 Progressive Familial Intrahepatic Cho-lestasis Type III. PFIC III involves mutations in the multidrug resistance protein-3 P-glycoprotein (PGY3, also known as MDR3/ABCB4) (Figure 69-1). MDR3/ABCB4 is thought to transport phosphatidylcholine from the inner lipid leaflet of the bile canaliculus to the outer leaflet, thereby replenishing the phospholipid, which is continuously removed into the bile because of contact with bile acids. In the absence of phosphatidyl-choline translocation, bile acids damage the canalicular

CHAPTER 69 Bile Pigment Metabolism and Its Disorders 23
membrane, causing progressive destruction of small bile ducts (209,396). In contrast to the findings in PFIC I and II, serum γ-glutamyl transpeptidase activity is increased in PFIC III (384). MDR3/ABCB4 deficiency in humans has been associated with a wide spectrum of hepatobi-liary disorders, including small-duct primary sclerosing cholangitis (397) and cholesterol gallstones (398). Interestingly, heterozygosity for nonsense or missense mutations of the MDR3/ABCB4 gene has been associ-ated with the heretofore enigmatic familial intrahepatic cholestasis of pregnancy (399,400).
69.9.3.1.5 Treatment. The treatment options currently available for PFICs are limited. Experimen-tal transplantation of normal hepatocytes in an mdr2 knockout mouse model of PFIC III has resulted in massive repopulation of the liver with the transplanted cells, resulting in long-term amelioration of the phospho-lipid transport defect in that condition (401,402).
69.9.3.1.6 Other Inherited Cholestatic Disor-ders. The cholangiopathy of North American Indian childhood cirrhosis is caused by the genetic lesions of a gene (CIRH1A) that encodes a mitochondrial scaffold protein. This disorder resembles clinically extrahe-patic biliary atresia, except that there is no biliary tract obstruction (403).
GRACILE syndrome, another form of potentially lethal intrahepatic cholestasis associated with fetal growth retardation, aminoaciduria, iron overload, and lactic acidosis (GRACILE), is caused by genetic lesions of another mitochondrial scaffold gene, BCS1L (404). Inter-estingly, although North American Indian childhood cirrhosis and GRACILE syndrome are both caused by mutations of genes expressing mitochondrial scaffold proteins, neither of these syndromes includes features of usual mitochondriopathy, such as central nervous system lesions or hepatocellular steatosis.
Alagille syndrome is a multisystem development anomaly affecting the hepatobiliary system, eyes, verte-brae (sagittal, cleft), heart, kidneys, and central nervous system (405). Most patients exhibit a characteristic facies. The central nervous system lesions can be sec-ondary to malnutrition, but in patients with carefully corrected nutritional balance, intracranial bleeding is the most common central nervous system complica-tion (406). The syndrome is inherited as an autosomal-dominant condition with markedly variable penetrance. Lesions of the Jagged1 (JAG1) gene, mapped to chro-mosome 20p12 (407), are responsible for this disease. Jagged-1 is a ligand of Notch, and plays a critical role in the Notch signaling pathway that is important in the nor-mal development of various organs. In about 70% of the patients with Alagille syndrome, deletion or mutations of the JAG1 gene can be found, the coding region being normal in the remaining patients. Interestingly, 50–70% of these mutations are de novo, and not found in the parents. Paucity of bile ducts, which was considered sine qua non for Alagille syndrome is found in only 89% of
cases (406). Liver transplantation is required eventually in 21–50% of patients who have hepatic symptoms in infancy.
Villin disease, related to lesions of the VILLIN1 gene, was reported to be the basis of cholestatic disease in several children, who eventually developed cirrhosis, requiring liver transplantation (408).
ACKNOWLEDGMENTS
This work was supported in part by the following grants: NIH DK 079017 (to NRC), Liver Research Core Center (P30-DK 41296), NYSTEM C024346 and C026440 (to JRC), and a research grant from Oxalosis and Hyperox-aluria Foundation (to NRC).
REFERENCES
1. Berk, P. D.; Howe, R. B.; Bloomer, J. R.; Berlin, N. I. Studies on Bilirubin Kinetics in Normal Adults. J. Clin. Invest. 1979, 49, 2176.
2. London, I. M.; West, R.; Shemin, D.; Rittenberg, D. On the Origin of Bile Pigment in Normal Man. J. Biol. Chem. 1950, 184, 351.
3. Alpert, S.; Mosher, M.; Shanske, A.; Arias, I. M. Multiplic-ity of Hepatic Excretory Mechanisms for Organic Anions. J. Gen. Physiol. 1969, 53, 238.
4. Robinson, S. H. Origins of the Early-Labeled Peak. In Bile Pigments: Chemistry and Physiology. U. S; Berk, P. D.; Ber-lin, N. I., Eds.; Government Printing Office: Washington, DC, 1977; pp 175.
5. Yannoni, C. Z.; Robinson, S. H. Early-Labelled Heme Syn-thesis in Normal Rats and Rats with Iron Deficiency Anemia. Biochim. Biophys. Acta 1976, 428, 533.
6. Grandchamp, B.; Bissel, D. M.; Licko, V.; Schmidt, R. For-mation and Disposition of Newly Synthesized Heme in Adult Rat Hepatocytes in Primary Cultures. J. Biol. Chem. 1981, 256, 11677.
7. Levitt, M.; Schacter, B. A.; Zipursky, A.; Israels, L. G. The Nonerythropoietic Component of Early Bilirubin. J. Clin. Invest. 1968, 47, 1281.
8. Arias, I. M. Chronic Unconjugated Hyperbilirubinemia without Overt Signs of Hemolysis in Adolescents and Adults. J. Clin. Invest. 1962, 41, 2233.
9. Schwartz, S.; Johnson, J. A.; Stephenson, B. D., et al. Eryth-ropoietic Defects in Protoporphyria: A Study of Factors Involved in Labeling of Porphyrins and Bile Pigments from ALA3H and Glycine-14C. J. Lab. Clin. Med. 1971, 78, 411.
10. Hamer, J. W.; Fitserrald, P. H. Disturbed Marrow Cell Pro-liferation in Primary Shunt Hyperbilirubinemia. Blood 1973, 41, 38.
11. Come, S. E.; Shohet, S. B.; Robinson, S. H. Surface Remod-eling vs. Whole-Cell Hemolysis of Reticulocytes Produced with Erythroid Stimulation or Iron Deficiency Anemia. Blood 1974, 44, 817.
12. Tenhunen, R.; Marver, H. S.; Schmidt, R. Microsomal Heme Oxygenase: Characterization of the Enzyme. J. Biol. Chem. 1969, 244, 6388.
13. Bissel, Dm; Hammaker, L.; Schmid, R. Liver Sinusoidal Cells: Identification of a Sub-Population for Erythrocyte Catabolism. J. Cell. Biol. 1972, 54, 107.
14. Elbirt, K. K.; Bonkovsky, H. L. Heme Oxygenase: Recent Advances in Understanding its Regulation and Role. Proc. Assoc. Am. Physicians 1999, 111, 438.

24 CHAPTER 69 Bile Pigment Metabolism and Its Disorders
15. Fernandez, M.; Bonkovsky, H. L. Increased Heme Oxy-genase-1 Gene Expression in Liver Cells and Splanchnic Organs from Portal Hypertensive Rats. Hepatology 1999, 29, 1672.
16. Ishizawa, S.; Yoshida, T.; Kikuchi, G. Induction of Heme Oxygenase in Rat Liver. J. Biol. Chem. 1983, 258, 4220.
17. Makino, N.; Suematsu, M.; Sugiura, Y., et al. Altered Expression of Heme Oxygenase-1 in the Livers of Patients with Portal Hypertensive Diseases. Hepatology 2001, 33, 32.
18. Jian, Z.; Li, K.; Liu, L.; Zhang, Y.; Zhou, Z.; Li, C.; Gao, T. Heme Oxygenase-1 Protects Human Melanocytes from H(2)O(2)-Induced Oxidative Stress via the Nrf2-ARE Pathway. J. Invest. Dermatol. 2011 Mar 17, [Epub ahead of print].
19. Naito, Y.; Takagi, T.; Uchiyama, K.; Yoshikawa, T. Heme Oxygenase-1: A Novel Therapeutic Target for Gastrointesti-nal Diseases. J. Clin. Biochem. Nutr. 2011, 48, 126.
20. Mougiakakos, D.; Jitschin, R.; Johansson, C. C.; Okita, R.; Kiessling, R.; Le Blanc, K. The Impact of Inflamma-tory Licensing on Heme Oxygenase-1-Mediated Induction of Regulatory T Cells by Human Mesenchymal Stem Cells. Blood 2011 Mar 9, [Epub ahead of print].
21. Schipper, H. M. Heme Oxygenase-1 in Alzheimer Disease: A Tribute to Moussa Youdim. J. Neural. Transm. 2011, 118, 381.
22. Li, Y.; Su, J.; Dingzhang, X.; Zhang, J.; Yoshimoto, M.; Liu, S.; Bijian, K.; Gupta, A.; Squire, J. A.; Alaoui Jamali, M. A.; Bismar, T. A. PTEN Deletion and Heme Oxygenase-1 Over-expression Cooperate in Prostate Cancer Progression and Are Associated with Adverse Clinical Outcome. J. Pathol. 2011 Jan 10, http://dx.doi.org/10.1002/path. 2855, [Epub ahead of print].
23. Yoshinga, T.; Sassa, S.; Kappas, A. The Occurrence of Molecular Interactions Among NADPH-Cytochrome C Reductase, Heme Oxygenase and Biliverdin Reductase in Heme Degradation. J. Biol. Chem. 1982, 257, 7778.
24. Drummond, G. S.; Kappas, A. Chemoprevention of Neona-tal Jaundice: Potency of Tin-Protoporphyrin in an Animal Model. Science 1982, 217, 6466.
25. Fakhrai, H.; Maines, M. D. Expression and Characterization of a Rat Kidney Biliverdin Reductase: Evidence Suggesting that the Liver and Kidney Enzymes Are the Same Gene Tran-script Product. J. Biol. Chem. 1992, 267, 4023.
26. Bensinger, T. A.; Maisels, M. J.; Marlson, D. E.; Conrad, M. E. Effect of Low Caloric Diet on Endogenous Carbon Mon-oxide Production. Normal Adults and Gilbert’s Syndrome. Proc. Soc. Exp. Biol. Med. 1973, 144, 417.
27. Huang, T. J.; Trakshel, G. M.; Maines, M. D. Detection of Ten Variants of Biliverdin Reductase in Rat Liver by Two-Dimensional Gel Electrophoresis. J. Biol. Chem. 1989, 264, 7844.
28. Frydman, R. B.; Tomaro, M. L.; Awruch, J.; Frydman, B. Interconversion of the Molecular Forms of Biliverdin Reductase from Rat Liver. Biochim. Biophys. Acta 1983, 759, 257.
29. Dhar, G. J. Enterohepatic Circulation and Plasma Transport of Urobilinogen. In Bile Pigments: Chemistry and Physiol-ogy; Berk, P. D.; Berlin, N. I., Eds.; U.S. Government Print-ing Office: Washington, DC, 1977; pp 526.
30. Jones, E. A.; Bloomer, J. R.; Berk, P. D., et al. Quantitation of Hepatic Bilirubin Synthesis in Man. In Bile Pigments: Chemistry and Physiology; Berk, P. D.; Berlin, N. I., Eds.; U. S. Government Printing Office: Washington, DC, 1977; pp 189.
31. Berk, P. D.; Rodkey, F. L.; Blaschke Collison, H. A.; Wag-goner, J. G. Comparison of Plasma Bilirubin Turnover and Carbon Monoxide Production in Man. J. Lab. Clin. Med. 1974, 83, 29.
32. Fischer, H.; Plieninger, H. Synthese des Biliverdins (Uterover-dins) und Bilirubins der Biliverdine XIII, und III, Sowie der Vinulneoxanthosaure. Hoppe–Seylers A Physiol Chem 1942, 274, 231.
33. Berk, P. D.; Rodkey, F. L.; Blaschke Collison, H. A.; Wag-goner, J. G. Comparison of Plasma Bilirubin Turnover and Carbon Monoxide Production in Man. J. Lab. Clin. Med. 1974, 83, 29.
34. Fog, J.; Jellum, E. Structure of Bilirubin. Nature 1963, 198, 88.
35. Kuenzle, C. C.; Weibel, M. H.; Pelloni, R. R. A Proposed Novel Structure for the Metal Chelates of Bilirubin. Bio-chem. J. 1973, 130, 1147.
36. Bonnet, R. J.; Davis, E.; Hursthouse, M. B. Structure of Bili-rubin. Nature 1976, 262, 326.
37. Manitto, P.; Monti, D.; Garbagnati, E. pH Dependence of the Water Solubility of Bilirubin Photoderivatives and its Relevance to Phototherapy. Pediatr. Res. 1984, 18, 378.
38. Moan, J. Light Therapy of Newborns with Hyperbilirubine-mia. [Translated from Norwegian] Tidsskrift for Den Nor-ske Laegeforening 1992, 112, 1459.
39. Ennever, J. F.; Dreising, T. J. Quantum Yields for the Cycli-zation and Configurational Isomerization of 4E, 15Z Biliru-bin. Photochem. Photobiol. 1991, 53, 25.
40. Iho, S.; Onishi, S. Kinetic Study of the Photochemical Changes of (ZZ)-Bilirubin IXα Bound to Human Serum Albumin: Demonstration of (EZ)-Bilirubin IXα as an Inter-mediate in Photochemical Changes From (ZZ)-Bilirubin IXα to (EZ)-Cyclobilirubin IXα. Biochem. J. 1985, 226, 251.
41. Bloomer, J. R.; Berk, P. D.; Howe, R. B.; Berlin, N. I. Biliru-bin Metabolism in Congenital Nonhemolytic Jaundice. Pedi-atr. Res. 1971, 5, 256.
42. Stoll, M. S. Phototherapy of Jaundice. In Bile Pigments and Jaundice; Ostrow, J. D., Ed.; Marcel Dekker: New York, 1986; pp 551.
43. Breimer, L. H.; Wannamethee, G.; Ebrahim, S.; Shaper, A. G. Serum Bilirubin and Risk of Ischemic Heart Disease in Middle-Aged British Men. Clin. Chem. 1995, 41, 1504.
44. Zucker, S. D.; Horn, P. S.; Serman, K. E. Serum Bilirubin Levels in the U.S. Population: Gender Effect and Inverse Cor-relation with Colorectal Cancer. Hepatology 2004, 40, 827.
45. Temme, E. H. M.; Zhang, J.; Schouten, E. G.; Kesteloot, H. Serum Bilirubin and 10-Year Mortality Risk in a Belgian Population. Cancer Causes Control 2001, 12, 887.
46. Ostrow, J. D.; Pascolo, L.; Brites, D.; Tiribelli, C. Molecular Basis of Bilirubin-Induced Neurotoxicity. Trends Mol. Med. 2004, 10, 65.
47. Shapiro, S. M. Binaural Effects in Brainstem Auditory Evoked Potentials of Jaundiced Gunn Rats. Hear. Res. 1991, 53, 41.
48. Shapiro, S. W.; Conlee, J. W. Brainstem Auditory Evoked Potentials Correlate with Morphological Changes in Gunn Rat Pups. Hear. Res. 1991, 57, 16.
49. Bosma, P. J.; Roy Chowdhury, J.; Huang, T. J., et al. Mechanism of Inherited Deficiencies of Multiple UDP- Glucuronosyltransferase Isoforms in Two Patients with Cri-gler–Najjar Syndrome, Type I. FASEB J. 1992, 6, 2859.
50. Shapiro, S. M. Bilirubin Toxicity in the Developing Nervous System. Pediatr. Neurol. 2003, 29, 410.
51. Shapiro, S. M. Reversible Brainstem Auditory Evoked Poten-tial Abnormalities in Jaundiced Gunn Rats Given Sulfon-amide. Pediatr. Res. 1993, 34, 629.
52. Takagishi, Y.; Yamamura, H. Purkinje Cell Abnormalities and Synaptogenesis in Genetically Jaundiced Rats (Gunn Rats). Brain Res. 1989, 492, 116.
53. Bosma, P. J.; Seppen, J.; Goldhoorn, B., et al. Bilirubin UDP-Glucuronosyltransferase 1 Is the Only Relevant Bilirubin Glucuronidating Isoform in Man. J. Biol. Chem. 1994, 269, 17960.

CHAPTER 69 Bile Pigment Metabolism and Its Disorders 25
54. Schutta, H. S.; Johnson, L.; Neville, H. S. Mitochondrial Abnormalities in Bilirubin Encephalopathy. J. Neuropathol. Exp. Neurol. 1970, 29, 296.
55. Sato, H.; Keino, H.; Aono, S., et al. Different Behaviours Among Lysosomal Enzymes in the Cerebellum of Jaundiced Gunn Rats with Cerebellar Hypoplasia. J. Neurochem. 1987, 48, 1823.
56. Aono, S.; Kato, T.; Tanaka, R., et al. Cyclic Nucleotides and the Activity of Glia Maturation Factor in the Hypoplastic Cerebellum of Developing Jaundiced Gunn Rats. J. Neuro-chem. 1988, 50, 700.
57. Vaz, A. R.; Delgado-Esteban, M.; Brito, M. A.; Bolaños, J. P.; Brites, D.; Almeida, A. Bilirubin Selectively Inhibits Cytochrome C Oxidase Activity and Induces Apoptosis in Immature Cortical Neurons: Assessment of the Protective Effects of Glycoursodeoxycholic Acid. J. Neurochem. 2010, 112, 56.
58. Labrune, P. H.; Myara, A.; Francoual, J., et al. Cerebella Symptoms as the Presenting Manifestations of Bilirubin Encephalopathy in Children with Crigler–Najjar Type I Dis-ease. Pediatrics 1992, 89, 768.
59. Zuelzer, W. W.; Mudgett, R. T. Kernicterus: Etiologic Study Based on an Analysis of 55 Cases. Pediatrics 1950, 6, 452.
60. Vaughan, V. C.; Allen, F. C.; Diamond, L. K. Erythroblasto-sis Fetalis. IV. Further Observations on Kernicterus. Pediat-rics 1950, 6, 706.
61. Lucey, J. F. Bilirubin and Brain Damage—A Real Mess. Pediatrics 1982, 69, 381.
62. Meador, C. K. The Art and Science of Nondisease. N. Engl. J. Med. 1965, 272, 92.
63. Naeye, R. L. Amniotic Fluid Infections, Neonatal Hyperbili-rubinemia and Psychomotor Impairment. Pediatrics 1978, 62, 497.
64. Van de Bor, M.; Ens-Dokkum, M.; Schreuder, A. M., et al. Hyperbilirubinemia in Low Birth Weight Infants and Out-come at 5 Years of Age. Pediatrics 1992, 89, 359.
65. Rappaport, S. I. Sites and Functions of Blood–Brain Barrier. In Blood–Brain Barrier in Physiology and Medicine; Raven Press: New York, 1976; pp 43.
66. Purpura, D. P.; Carmichael, M. W. Characteristics of Blood-Brain Barrier to Gamma-Aminobutyric Acid in Neonatal Cat. Science 1960, 131, 410.
67. Charbonnier, A.; Brisbois, P. Etude Chromatographique de la BSP au cours de l’Epreuve Clinique d’Epuration Plasma-tique de Ce Colorant. Rev. Intern. Hepatol. 1960, 10, 1163.
68. Laas, R.; Helmke, K. Regional Cerebral Blood Flow Fol-lowing Unilateral Blood-Brain Barrier Alteration Induced by Hyperosmolar Perfusion in the Albino Rat. In Cerebral Cir-culation and Metabolism; Cervos-Navarro, J.; Fritschka, E., Eds.; Raven Press: New York, 1981; pp 317.
69. Levine, R. L.; Fredericks, W. R.; Rappaport, S. I. Clearance of Bilirubin from Rat Brain after Reversible Osmotic Open-ing of the Blood–Brain Barrier. Pediatr. Res. 1985, 19, 1040.
70. Gazzin, S.; Berengeno, A. L.; Strazielle, N.; Fazzari, F.; Raseni, A.; Ostrow, J. D.; Wennberg, R.; Ghersi-Egea, J. F.; Tiribelli, C. Modulation of Mrp1 (ABCc1) and Pgp (ABCb1) by Bilirubin at the Blood-CSF and Blood-Brain Barriers in the Gunn Rat. PLoS One 2011, 6, e16165.
71. Lee, K.-S.; Gartner, L. M. Management of Unconjugated Hyperbilirubinemia in the Newborn. Semin. Liver Dis. 1983, 3, 52.
72. Clarke, C. A.; Donohoe, W. T.A.; Finn, R., et al. Prevention of Rh Hemolytic Disease: Final Results of the “High Risk” Clinical Trial. A Combined Study From Centers in England and Baltimore. Br. Med. J. 1971, 2, 607.
73. Mustafa, M. G.; Cowger, M. L.; Kind, T. E. Effects of Biliru-bin on Mitochondrial Reactions. J. Biol. Chem. 1969, 244, 6403.
74. Roger, C.; Koziel, V.; Vert, P.; Nehlig, A. Effects of Biliru-bin Infusion on Cerebral Glucose Utilization in the Immature Rat. Dev. Brain Res. 1993, 76, 115.
75. Morphis, I.; Constantopoulos, A.; Matsaniotis, N.; Papaphi-lis, A. Bilirubin-Induced Modulation of Cerebral Protein Phosphorylation in Neonate Rabbits In Vivo. Science 1982, 218, 156.
76. Constantopoulos, A.; Matsaniotis, N. Bilirubin Inhibition of Protein Kinase: Its Prevention by Cyclic AMP. Cytobios 1976, 17, 17.
77. Sano, K.; Nakamura, H.; Tamotsu, M. Mode of Inhibitory Action of Bilirubin on Protein Kinase C. Pediatr. Res. 1985, 19, 587.
78. Martinez-Maldonado, M.; Suki, W. N.; Schenker, S. Nature of the Urinary Concentrating Defect in the Gunn Strain of Rat. Am. J. Physiol. 1969, 216, 1386.
79. Haymaker, W.; Margoles, C.; Pentschew, A., et al. Kernic-terus and Its Importance in Cerebral Palsy. In Proceedings of the 11th Annual Meeting of the American Academy of Cerebral Palsy; Charles C Thomas: Springfield, IL, 1961; pp 21.
80. Odell, G. B.; Bolen, J. L.; Poland, R. L., et al. Protection from Bilirubin Nephropathy in Jaundiced Gunn Rats. Gastroen-terology 1974, 66, 1218.
81. Engle, W. D.; Avant, B. S., Jr. Neonatal Hyperbilirubinemia and Renal Function. J. Pediatr. 1982, 100, 113.
82. Axelson, R. A. Spontaneous Renal Papillary Necrosis in the Gunn Rat. Pathology 1973, 5, 43.
83. Inoue, M.; Hirata, E.; Morino, Y., et al. The Role of Albu-min in the Hepatic Transport of Bilirubin: Studies in Mutant Analbuminemic Rats. J. Biochem. 1985, 97, 737.
84. Jacobsen, J. Binding of Bilirubin to Human Serum Albumin: Determination of the Dissociation Constants. FEBS Lett. 1969, 5, 112.
85. Demetriou, A. A.; Levenson, S. M.; Whiting, J., et al. Replacement of Hepatic Functions in Rats by Transplanta-tion of Microcarrier-Attached Hepatocytes. Science 1986, 233, 1190.
86. Brodersen, R. Bilirubin Solubility and Interaction with Albu-min and Phospholipid. J. Biol. Chem. 1979, 254, 2364.
87. Brodersen, R.; Funding, L.; Pederson, A. O.; Rojgaard- Petersen, H. Binding of Bilirubin to Low-Affinity Sites of Human Serum Albumin In Vitro Followed by Co-Crystallization. Scand. J. Clin. Lab. Invest. 1972, 29, 433.
88. Jacobsen, C. Lysine Residue 240 of Human Serum Albumin is Involved in High-Affinity Binding of Bilirubin. Biochem. J. 1978, 171, 453.
89. Odell, G. B. The Dissociation of Bilirubin from Albumin and Its Clinical Implications. J. Pediatr. 1959, 55, 268.
90. Kapitulnik, J.; Valaes, T.; Kaufmann, N. A.; Blondheim, S. H. Clinical Evaluation of Sephadex Gel Filtration in Esti-mation of Bilirubin Binding in Serum in Neonatal Jaundice. Arch. Dis. Child 1974, 49, 886.
91. Athanassiadis, S.; Chopra, D. R.; Fisher, M.; McKenna, J. An Electrophoretic Method for Detection of Unbound Bili-rubin and Reserve Bilirubin Binding Capacity in Serum of Newborns. J. Lab. Clin. Med. 1974, 83, 968.
92. Lamolla, A. A.; Eisinger, J.; Blumberg, W. E., et al. Fluo-rometric Study of the Partition of Bilirubin Among Blood Components: Basis for Rapid Microassays of Bilirubin and Bilirubin Binding Capacity in Whole Blood. Anal. Biochem. 1979, 15, 25.
93. Rotor, A. B.; Manahan, L.; Florentin, A. Familial Nonhe-molytic Jaundice with Direct van den Bergh Reaction. Acta Med. Philos. 1948, 5, 37.
94. Scharschmidt, B. F.; Waggoner, J. G.; Berk, P. D. Hepatic Organic Anion Uptake in the Rat. J. Clin. Invest. 1975, 56, 1280.

26 CHAPTER 69 Bile Pigment Metabolism and Its Dis
95. Satlin, L. M.; Amin, V.; Wolkoff, A. W. Organic Anion Transporting Polypeptide Mediates Organic Anion/HCO3
- Exchange. J. Biol. Chem. 1997, 272, 26340.
96. Wolkoff, A. W.; Samuelson, A. C.; Johansen, K. L., et al. Influence of Cl- on Organic Anion Transport in Short-Term Cultured Rat Hepatocytes and Isolated Perfused Rat Liver. J. Clin. Invest. 1987, 79, 1259.
97. Stremmel, W.; Berk, P. D. Hepatocellular Uptake to Sulfo-bromophthalein and Bilirubin is Selectively Inhibited by an Antibody to the Liver Plasma Membrane Sulfobromophtha-lein/Bilirubin Binding Protein. J. Clin. Invest. 1986, 78, 822.
98. Torres, A. M.; Lunazzi, G. C.; Stremmel, W.; Tiribelli, C. Bilitranslocase and Sulfobromophthalein Bilirubin-Binding Protein Are Both Involved in the Hepatic Uptake of Organic Anions. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 8136.
99. Zucker, S. D.; Goessling, W. Mechanism of Hepatocellu-lar Uptake of Albumin-Bound Bilirubin. Biochim. Biophys. Acta 2000, 1463, 197.
100. Laperche, Y.; Preaux, A. M.; Berthelot, P. Two Systems Are Involved in the Sulfobromophthalein Uptake by Rat Liver Cells: One is Shared with Bile Acids. Biochem. Pharmacol. 1981, 30, 1333.
101. Cui, Y.; Konig, J.; Leier, I., et al. Hepatic Uptake of Bilirubin and Its Conjugates by the Human Organic Anion-Transporting Polypeptide 2 (Symbol SLC21A6). J. Biol. Chem. 2000, 276, 9626.
102. Suzuki, H.; Sugiyama, Y. Transport of Drugs Across the Hepatic Sinusoidal Membrane: Sinusoidal Drug Influx and Efflux in the Liver. Semin. Liver Dis. 2000, 20, 251.
103. Wang, P.; Kim, R. B.; Roy-Chowdhury, J.; Wolkoff, A. W. Organic Anion Transport Protein SLC21A6 (OATP2) is Not Sufficient for Bilirubin Transport. J. Biol. Chem. 2003, 278, 20695.
104. Kouzuki, H.; Suzuki, H.; Ito, K., et al. Contribution of Sodium Taurocholate Co-Transporting Polypeptide to the Uptake of its Possible Substrates into Rat Hepatocytes. J. Pharmacol. Exp. Ther. 1998, 286, 1043.
105. Stieger, B.; Hagenbuch, B.; Cornacchia, L., et al. Molecular Properties of the Na+-Dependent Taurocholate Cotransport-ing Polypeptide (NTCP) of Rat River. Hepatology 1993, 18, 143A.
106. Yamazaki, M.; Suzuki, H.; Hanano, M.; Sugiyama, Y. Dif-ferent Relationships between Cellular ATP and Hepatic Uptake Among Taurocholate, Cholate, and Organic Anions. Am. J. Physiol. 1993, 264, G693.
107. Yamazaki, M.; Suzuki, H.; Sugiyama, Y., et al. Uptake of Organic Anions by Isolated Rat Hepatocytes: A Classifica-tion in Terms of ATP-Dependency. J. Hepatol. 1992, 14, 41.
108. Levi, A. J.; Gatmaitan, Z.; Arias, I. M. Two Hepatic Cyto-plasmic Protein Fractions, Y and Z, and Their Possible Role in the Hepatic Uptake of Bilirubin, Sulfobromophthalein, and Other Anions. J. Clin. Invest. 1969, 48, 2156.
109. Rowe, J. D.; Nieves, E.; Listowsky, I. Subunit Diversity and Tissue Distribution of Human Glutathione S-Transferases: Interpretations Based on Electrospray Ionization-MS and Peptide Sequence-Specific Antisera. Biochem. J. 1997, 325, 481.
110. Wolkoff, A. W.; Goresky, C. A.; Sellin, J., et al. Role of Ligandin in Transfer of Bilirubin from Plasma into Liver. Am. J. Physiol. 1979, 236, E638.
111. Gordon, E. R.; Goresky, C. A.; Chan, T. H.; Perlin, A. S. The Isolation and Characterization of Bilirubin Diglucuro-nide, the Major Bilirubin Conjugate in Dog and Human Bile. Biochem. J. 1976, 155, 477.
112. Fakhrai, H.; Maines, M. D. Expression and Characterization of a Rat Kidney Biliverdin Reductase: Evidence Suggesting that the Liver and Kidney Enzymes Are the Same Gene Tran-script Product. J. Biol. Chem. 1992, 267, 4023.
orders
113. Heirwegh, K. P. M.; Van Hees, G. P.; Leroy, P., et al. Het-erogeneity of Bile Pigment Conjugates as Revealed by Chro-matography of Their Ethyl Anthranilate Azopigments. Biochem. J. 1979, 120, 877.
114. Fischer, H.; Plieninger, H. Synthese des Biliverdins (Uterover-dins) und Bilirubins der Biliverdine XIII, und III, Sowie der Vinulneoxanthosaure. Hoppe–Seylers A Physiol. Chem. 1942, 274, 231.
115. Kuenzle, C. C. Bilirubin Conjugates of Human Bile: The Excretion of Bilirubin Conjugates as the Acyl Glycosides of Aldobiouronic Acid, Pseudoaldobiouronic Acid and Hex-uronosylhexuronic Acid, with a Branched-Chain Hexuronic Acid as One of the Components of the Hexuronosylhexuro-nide. Biochem. J. 1970, 119, 411.
116. Spivak, W.; Carey, M. C. Reverse-Phase HPLC Separation, Quantification and Preparation of Bilirubin and Its Conju-gates from Native Bile. Biochem. J. 1985, 225, 787.
117. Roy Chowdhury, J.; Novikoff, P. M.; Roy Chowdhury, N.; Novikoff, A. B. Distribution of UDP-Glucuronosyltransferase in Rat Tissue. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 2990.
118. Dutton, G. J.; Burchell, B. Newer Aspects of Glucuronida-tion. Prog. Drug Metab. 1977, 2, 1.
119. Jansen, P. L. M. Studies on UDP Glucuronosyltransferase. Doctoral Dissertation; University of Nijmegen: The Nether-lands, 1975.
120. Vessey, D. A.; Zakim, D. Regulations of Microsomal Enzymes by Phospholipids. J. Biol. Chem. 1971, 246, 4649.
121. Hauser, S. C.; Ziurys, J. C.; Gollan, J. L. A Membrane Trans-porter Mediates Access of Uridine-5′-Diphosphoglucuronic Acid from the Cytosol into the Endoplasmic Reticulum of Rat Hepatocytes: Implications for Glucuronidation Reac-tions. Biochim. Biophys. Acta 1988, 967, 149.
122. Heirwegh, K. P. M.; Campbell, M.; Meuwissen, J. A. T. P. Compartmentation of Membrane Bound Enzymes: Some Basic Concepts and Consequences for Kinetic Studies. In Conjugation Reactions in Drug Biotransformation; Aitio, A., Ed.; Elsevier: Amsterdam, 1978; pp 191.
123. Hallinan, T. Comparison of Compartmented and of Con-formational Phospholipid-Constraint Models for the Intra-membranous Arrangement of UDP-Glucuronyltransferase. In Conjugation Reactions in Drug Biotransformation; Aitio, A., Ed.; Elsevier: Amsterdam, 1978; pp 257.
124. Drake, R. R.; Igari, Y.; Lester, R., et al. Application of 5-Azido-UDP-Glucose and 5-Azido-UDP-Glucuronic Acid Photoaffinity Probes for the Determination of the Active Site Orientation of Microsomal UDP-Glucosyltransferases and UDP-Glucuronosyltransferases. J. Biol. Chem. 1992, 267, 11360.
125. Mackenzie, P. I. Expression of Chimeric cDNAs in Cell Culture Defines a Region of UDP-Glucuronosyltransferase Involved in Substrate Selection. J. Biol. Chem. 1990, 265, 3432.
126. Bock, K. W.; Josling, D.; Lilenblum, W. M.; Pfeil, H. Puri-fication of Rat Liver Glucuronosyltransferase-Separation of Two Enzyme Forms Inducible by 3-Methyl-Cholanthrene or Phenobarbital. Eur. J. Biochem. 1977, 98, 19.
127. Burchell, B. Purification of UDP Glucuronosyltransferase from Untreated Rat Liver. FEBS Lett. 1977, 78, 101.
128. Falany, C. N.; Tephly, T. R. Separation, Purifica-tion and Characterization of Three Isozymes of UDP- Glucuronosyltransferase from Rat Liver Microsomes. Arch. Biochem. Biophys. 1983, 227, 248.
129. Gartner, L. M.; Lee, K.; Vaisman, S., et al. Development of Bilirubin Transport and Metabolism in the Newborn Rhesus Monkey. J. Pediatr. 1977, 90, 513.
130. Gorski, J. P.; Kasper, C. B. Purification and Properties of Microsomal UDP Glucuronosyl Transferase from Rat Liver. J. Biol. Chem. 1977, 252, 1336.

R 69 Bile Pigment Metabolism and Its Disorders 27
CHAPTE131. Roy Chowdhury, J.; Roy Chowdhury, N.; Falany, C. N., et al. Isolation and Characterization of Multiple Forms of Rat Liver UDP-Glucuronate Glucuronosyltransferase. Bio-chem. J. 1986, 233, 827.
132. Wishart, G. F. Functional Heterogeneity of UDP Glucurono-syl Transferase as Indicated by Its Differential Development and Inducibility by Glucocorticoids. Biochem. J. 1978, 174, 485.
133. Bock, K. W.; Frohling, W.; Remmer, H.; Rexer, B. Effects of Phenobarbital and 3-Methyl Cholanthrene on Substrate Specificity of Rat Liver Microsomal UDP Glucuronosyltrans-ferase. Biochim. Biophys. Acta 1973, 327, 46.
134. Mackenzie, P. I.; Owens, I. S.; Burchell, B., et al. The UDP Glycosyltransferase Gene Superfamily: Recommended Nomenclature Update Based on Evolutionary Divergence. Pharmacogenetics 1997, 7, 255.
135. Ritter, J. K.; Chen, F.; Sheen, Y. Y., et al. A Novel Complex Locus UGT1 Encodes Human Bilirubin, Phenol and other UDP-Glucuronosyltransferase Isozymes with Identical Car-boxy Termini. J. Biol. Chem. 1992, 267, 3257.
136. Iyanagi, T. Molecular Basis of Multiple UDP- Glucuronosyltransferase Isoenzyme Deficiencies in the Hyperbilirubinemic Rat (Gunn Rat). J. Biol. Chem. 1991, 266, 24048.
137. Roy Chowdhury, N.; Saber, M. A.; Lahiri, P., et al. Expres-sion of Specific UDP-Glucuronosyltransferase Isoforms in Carcinogen-Induced Preneoplastic Rat Liver Nodules. Hepa-tology 1991, 13, 31.
138. Sato, H.; Koiwai, O.; Tanabe, K.; Kashiwamata, S. Isolation and Sequencing of Rat Liver Bilirubin-UDP- Glucuronosyltransferase cDNA: Possible Alternate Splicing of a Common Primary Transcript. Biochem. Biophys. Res. Commun. 1990, 169, 260.
139. Roy Chowdhury, J.; Roy Chowdhury, N.; Moscioni, A. D., et al. Differential Regulation by Triiodothyronine of Sub-strate-Specific Uridinediphosphoglucuronate Glucuronosyl Transferases in Rat Liver. Biochim. Biophys. Acta 1983, 761, 58.
140. Ritter, J. K.; Crawford, J. M.; Owens, I. S. Cloning of Two Human Liver Bilirubin-UDP-Glucuronosyltransferase cDNAs with Expression in COS-1 Cells. J. Biol. Chem. 1991, 266, 1043.
141. Guha, C.; Parashar, B.; Deb, N. J., et al. Long-Term Normal-ization of Serum Bilirubin Levels by Massive Repopulation of Gunn Rat Liver by Normal Hepatocytes, Transplanted after Preparative Hepatic Irradiation and Partial Hepatec-tomy. Hepatology 2002, 36, 354.
142. Bosma, P. J.; Seppen, J.; Goldhoorn, B., et al. Bilirubin UDP-Glucuronosyltransferase 1 Is the only Relevant Bilirubin Glucuronidating Isoform in Man. J. Biol. Chem. 1994, 269, 17960.
143. Meijer, D. K. F. Transport and Metabolism in the Hepato-Biliary System. In Schulz, S. G.; Forte, J. G.; Rauner, B. B., Eds. Handbook of the Gastrointestinal System; Oxford Uni-versity Press: New York, 1989; Vol. III, pp 717.
144. Arias, I. M.; Che, M. X.; Gatmaitan, Z., et al. The Biology of the Bile Canaliculus. Hepatology 1993, 17, 318.
145. Kobayashi, K.; Komatsu, S.; Nishi, T., et al. ATP-Dependent Transport for Glucuronides in Canalicular Plasma Membrane Vesicles. Biochem. Biophys. Res. Commun. 1991, 176, 622.
146. Nishida, T.; Gatmaitan, Z.; Roy Chowdhry, J.; Arias, I. M. Two Distinct Mechanisms for Bilirubin Glucuronide Trans-port by Rat Bile Canalicular Membrane Vesicles: Demon-stration of Defective ATP-Dependent Transport in Rats (TR−) with Inherited Conjugated Hyperbilirubinemia. J. Clin. Invest. 1992, 90, 2130.
147. Zimniak, P.; Awasthi, Y. C. ATP-Dependent Transport Sys-tems for Organic Anions. Hepatology 1993, 17, 330.
148. Cole, S. P. C.; Bhardwaj, G.; Gerlach, J. H., et al. Overex-pression of a Transporter Gene in a Multidrug-Resistant Human Lung Cancer Cell Line. Science 1992, 258, 1650.
149. Ishikawa, T.; Muller, M.; Klunemann, C., et al. ATP- Dependent Primary Active Transport of Cysteinyl Leukot-rienes Across Liver Canalicular Membranes. J. Biol. Chem. 1990, 265, 19279.
150. Nishida, T.; Hardenbrook, C.; Gatmaitan, Z.; Arias, I. M. ATP-Dependent Organic Anion Transport System in Normal and TR− Rat Liver Canalicular Membranes. Am. J. Physiol. 1992, 262, G629.
151. Hallinan, T. Comparison of Compartmented and of Con-formational Phospholipid-Constraint Models for the Intra-membranous Arrangement of UDP-Glucuronyltransferase. In Conjugation Reactions in Drug Biotransformation; Aitio, A., Ed.; Elsevier: Amsterdam, 1978; pp 257.
152. Kartenbeck, J.; Leuschner, U.; Mayer, R.; Keppler, D. Absence of the Canalicular Isoform of the MRP Gene-Encoded Conjugate Export Pump from the Hepatocytes in Dubin–Johnson Syndrome. Hepatology 1996, 23, 1061.
153. Paulusma, C. C.; Kool, M.; Bosma, P. J., et al. A Mutation in the Human cMOAT Gene Causes the Dubin–Johnson Syn-drome. Hepatology 1997, 25, 1539.
154. Paulusma, C. C.; Bosma, P. J.; Zaman, G. J. R., et al. Con-genital Jaundice in Rats with a Mutation in a Multidrug Resistance-Associated Protein Gene. Science 1996, 271, 1126.
155. Takikawa, H.; Sano, N.; Narita, T., et al. Biliary Excretion of Bile Acid Conjugates in a Hyperbilirubinemic Mutant Sprague–Dawley Rat. Hepatology 1991, 14, 352.
156. Stockel, B.; Konig, J.; Nies, A. T., et al. Characterization of the 5′-Flanking Region of the Human Multidrug Resistance Protein 2 (MRP2) Gene and its Regulation in Comparison with the Multidrug Resistance Protein 3 (MRP3) Gene. Eur. J. Biochem. 2000, 267, 1347.
157. Tanaka, T.; Uchiumi, T.; Hinoshita, E., et al. The Human Multidrug Resistance Protein 2 Gene: Functional Character-ization of the 5′-Flanking Region and Expression in Hepatic Cells. Hepatology 1999, 30, 1507.
158. Heirwegh, K. P. M.; Van Hees, G. P.; Leroy, P., et al. Het-erogeneity of Bile Pigment Conjugates as Revealed by Chro-matography of Their Ethyl Anthranilate Azopigments. Biochem. J. 1979, 120, 877.
159. Konig, J.; Rost, D.; Cui, Y.; Keppler, D. Characterization of the Human Multidrug Resistance Protein Isoform MRP3 Localized to the Basolateral Hepatocyte Membrane. Hepa-tology 1999, 29, 1156.
160. Vos, T. A.; Ros, J. E.; Havinga, R., et al. Regulation of Hepatic Transport Systems Involved in Bile Secretion During Liver Regeneration in Rats. Hepatology 1999, 29, 1833.
161. Keppler, D.; Konig, J. Hepatic Secretion of Conjugated Drugs and Endogenous Substances. Semin. Liver Dis. 2000, 20, 265.
162. Hirohashi, T.; Suzuki, H.; Sugiyama, Y. Characterization of the Transport Properties of Cloned Rat Multidrug Resis-tance-Associated Protein 3 (MRP3). J. Biol. Chem. 1999, 274, 15181.
163. Bull, L. N.; Van Eijk, M. J.; Pawlikowaska, L., et al. A Gene Encoding a P-Type ATPase Mutated in Two Forms of Hereditary Cholestasis. Nat. Genet. 1998, 18, 219.
164. Huang, T. J.; Trakshel, G. M.; Maines, M. D. Detection of Ten Variants of Biliverdin Reductase in Rat Liver by Two-Dimensional Gel Electrophoresis. J. Biol. Chem. 1989, 264, 7844.
165. Strautnieks, S. S.; Kagalwalla, A. F.; Tanner, M. S., et al. Identification of a Locus for Progressive Familial Intrahe-patic Cholestasis (PFIC2) on Chromosome 2q24. Am. J. Hum. Genet. 1997, 61, 630.

28 CHAPTER 69 Bile Pigment Metabolism and Its
166. de Vree, J.; Jacquemin, E.; Strum, E., et al. Mutations in the MDR3 Gene Cause Progressive Familial Intrahepatic Cho-lestasis. Proc. Acad. Natl. Sci. U.S.A. 1998, 25, 282.
167. Huang, W.; Zhang, J.; Chua, S. S., et al. Induction of Bili-rubin Clearance by the Constitutive Androstane Receptor (CAR). Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 4156.
168. Roy-Chowdhury, J.; Locker, J.; Roy Chowdhury, N. Nuclear Receptors Orchestrate Detoxification Pathways. Dev. Cell 2003, 4, 607.
169. Alonso, E. M.; Whitington, P. F.; Whitington, S. H., et al. Enterohepatic Circulation of Nonconjugated Bilirubin in Rats Fed with Human Milk. J. Pediatr. 1991, 118, 425.
170. Lester, R.; Schmid, R. Intestinal Absorption of Bile Pigments. II. Bilirubin Absorption in Man. N. Engl. J. Med. 1963, 269, 178.
171. Brodersen, R.; Herman, L. S. Intestinal Reabsorption of Unconjugated Bilirubin: A Possible Contributing Factor in Neonatal Jaundice. Lancet 1963, 1, 1242.
172. Stoll, M. S.; Lim, C. D.; Gray, C. H. Chemical Variants of the Urobilins. In Bile Pigments: Chemistry and Physiology; Berk, P. D.; Berlin, N. I., Eds.; U.S. Government Printing Office: Washington, DC, 1977; pp 483.
173. Fulop, M.; Sanson, J.; Brazeau, P. Dialyzability, Protein Binding, and Renal Excretion of Plasma Conjugated Biliru-bin. J. Clin. Invest. 1965, 44, 666.
174. Cameron, J. L.; Filler, R. M.; Iber, E. L., et al. Metabolism and Excretion of C14-Labeled Bilirubin in Children with Bili-ary Atresia. N. Engl. J. Med. 1966, 274, 231.
175. Gollan, J. L.; Dallinger, K. J.C.; Billing, B. H. Excretion of Conjugated Bilirubin in the Isolated Perfused Rat Kidney. Clin. Sci. Mol. Med. 1978, 54, 381.
176. Schmid, R.; Hammaker, L. Metabolism and Disposition of C14-Bilirubin in Congenital Nonhemolytic Jaundice. J. Clin. Invest. 1963, 42, 1720.
177. Doumas, B. T.; Wu, T. W. The Measurement of Bilirubin Fractions in Serum. Crit. Rev. Clin. Lab. Sci. 1991, 28, 415.
178. Hutchinson, D. W.; Johnson, B.; Knell, A. J. The Reaction between Bilirubin and Aromatic Diazo Compounds. Bio-chem. J. 1972, 127, 907.
179. Van den Bergh, A. A. H.; Muller, P. Ueber eine Direkte und eine Indirekte Diazoreaktion auf Bilirubin. Biochem. Z 1916, 77, 90.
180. Heirwegh, K. P. M.; Fevery, J. B.; Meuwissen, J. A. T. P., et al. Recent Advances in the Separation and Analysis of Diazo-Positive Bile Pigments. Methods Biochem. Anal. 1974, 22, 205.
181. Trotman, B. W.; Roy Chowdhury, J.; Wirt, G. D.; Bernstein, S. E. Azodipyrrole Analysis of Unconjugated and Conjugated Bilirubin Using Diazotized Ethylanthranilate in Dimethyl-sulfoxide. Anal. Biochem. 1982, 121, 175.
182. Lauff, J. J.; Kasper, M. E.; Ambros, R. T. Quantitative Liq-uid Chromatographic Estimation of Bilirubin Species in Pathological Serum. Clin. Chem. 1983, 29, 800.
183. Onishi, S.; Itho, S.; Kawade, N., et al. An Accurate and Sen-sitive Analysis by High Pressure Liquid Chromatography of Conjugated and Unconjugated Bilirubin IX in Various Bio-logical Fluids. Biochem. J. 1980, 185, 281.
184. Roy Chowdhury, J.; Roy Chowdhury, N. Quantitation of Bilirubin and Its Conjugates by High Pressure Liquid Chro-matography. Falk Hepatol. 1982, 11, 1649.
185. Roy Chowdhury, J.; Roy Chowdhury, N.; Wu, G., et al. Bilirubin Monoglucuronide and Diglucuronide Formation by Human Liver In Vitro: Assay by High Pressure Liquid Chromatography. Hepatology 1981, 1, 622.
186. Blanckaert, N.; Kabra, P. M.; Farina, F. A., et al. Measure-ment of Bilirubin and Its Mono- and Diconjugates in Human Serum by Alkaline Methanolysis and High Performance Liq-uid Chromatography. J. Lab. Clin. Med. 1980, 96, 198.
Disorders
187. Kubasik, N. P.; Mayer, T. K.; Basker, A. G., et al. The Measurement of Fractionated Bilirubin by Ektachem Film Slides: Method Validation and Comparison of Conjugated Bilirubin Measurements with Direct Bilirubin in Obstruc-tive and Hepatocellular Jaundice. Am. J. Clin. Pathol. 1985, 84, 518.
188. Maisels, M. J.; Kring, E. Transcutaneous Bilirubinometry Decreases the Need for Serum Bilirubin Measurement and Saves Money. Pediatrics 1997, 99, 599.
189. Tayba, R.; Gribetz, D.; Gribetz, I.; Holtzman, I. R. Non-Invasive Estimation of Serum Bilirubin. Pediatrics 1998, 102, 28.
190. Weiss, J. S.; Gautam, J. J.; Lauff, M. W., et al. The Clinical Importance of a Protein-Bound Fraction of Serum Bilirubin in Patients with Hyperbilirubinemia. N. Engl. J. Med. 1983, 309, 147.
191. Gartner, L. M.; Lee, K.; Vaisman, S., et al. Development of Bilirubin Transport and Metabolism in the Newborn Rhesus Monkey. J. Pediatr. 1977, 90, 513.
192. Hardy, J. B.; Peeples, M. O. Serum Bilirubin Levels in New-born Infants: Distributions and Associations with Neuro-logical Abnormalities During the First Year of Life. Johns Hopkins Med J. 1971, 128, 265.
193. Maisels, M. J.; Pathak, A.; Nelson, N. M., et al. Endogenous Production of Carbon Monoxide in Normal and Erythro-blastic Newborn Infants. J. Clin. Invest. 1971, 50, 1.
194. Vest, M.; Strebel, L.; Hauensiein, D. The Extent of “Shunt” Bilirubin and Erythrocyte Survival in the Newborn Infant Measured by the Administration of (15N) Glycine. Biochem. J. 1965, 95, 11c.
195. Clarke, C. A.; Donohoe, W. T.A.; Finn, R., et al. Prevention of Rh Hemolytic Disease: Final Results of the “High Risk” Clinical Trial. A Combined Study From Centers in England and Baltimore. Br. Med. J. 1971, 2, 607.
196. Hsia, D. Y.-Y.; Gellis, S. S. Studies on Erythroblastosis Feta-lis Due to ABO Incompatibility. Pediatrics 1954, 13, 503.
197. Snyder, A. L.; Satterlee, W.; Robinson, S. H.; Schmid, R. Conjugated Plasma Bilirubin in Jaundice Caused by Pigment Overload. Nature 1967, 213, 93.
198. Keppler, D. Multidrug Resistance Proteins (MRPs, ABCCs): Importance for Pathophysiology and Drug Therapy. Handb. Exp. Phar. 2011, 201, 299.
199. Levi, A. J.; Gatmaitan, Z.; Arias, I. M. Deficiency of Hepatic Organic Anion-Binding Protein, Impaired Organic Anion Uptake by Liver and “Physiologic” Jaundice in Newborn Monkeys. N. Engl. J. Med. 1970, 283, 1136.
200. Odell, G. B. “Physiologic” Hyperbilirubinemia in the Neo-natal Period. N. Engl. J. Med. 1967, 277, 193.
201. Kawade, N.; Onishi, S. The Prenatal and Postnatal Devel-opment of UDP-Glucuronosyltransferase Activity Towards Bilirubin and the Effect of Premature Birth on this Activity in Human Liver. Biochem. J. 1981, 196, 257.
202. Arthur, L. J. H.; Bevan, B. R.; Holton, J. B. Neonatal Hyper-bilirubinemia and Breast Feeding. Dev. Med. Child Neurol. 1966, 8, 279.
203. Arias, I. M.; Gartner, L. M.; Seifter, S.; Furman, M. Pro-longed Neonatal Unconjugated Hyperbilirubinemia Asso-ciated with Breast Feeding and a Steroid, Pregnana-3α, 20β-diol, in Maternal Milk that Inhibits Glucuronide For-mation In Vitro. J. Clin. Invest. 1964, 42, 2037.
204. Maisels, M. J.; Newman, T. B. Kernicterus in Otherwise Healthy Breast-Fed Term Newborns. Pediatrics 1995, 96, 730.
205. Foliot, A.; Ploussard, J. P.; Housett, E., et al. Breast Milk Jaundice: In Vitro Inhibition of Rat Liver Bilirubin-Uridine Diphosphate Glucuronosyltransferase Activity and Z Pro-tein-Bromosulfophthalein Binding by Human Breast Milk. Pediatr. Res. 1976, 10, 594.

69 Bile Pigment Metabolism and Its Disorders 29
CHAPTER206. Koiwai, O.; Aono, S.; Adachi, Y., et al. Crigler–Najjar Syn-drome Type II Is Inherited Both as a Dominant and as a Recessive Trait. Hum. Mol. Genet. 1996, 5, 645.
207. Arias, I. M.; Wolfson, S.; Lucey, J. F.; McKay, R. J., Jr. Tran-sient Familial Neonatal Hyperbilirubinemia. J. Clin. Invest. 1965, 44, 1442.
208. Lucey, J. F.; Driscol, J. J. Physiological Jaundice Re-Exam-ined. In Sass-Kortsak, A., Ed.; Kernicterus, 1961.
209. Nies, A. T.; Gatmaitan, Z.; Arias, I. M. ATP-Dependent Phosphatidylcholine Translocation in Rat Liver Canalicular Plasma Membrane Vesicles. J. Lipid Res. 1996, 37, 1125.
210. Poland, R. L.; Odell, G. B. Physiologic Jaundice: The Enterohepatic Circulation of Bilirubin. N. Engl. J. Med. 1971, 284, 1.
211. Israels, G.; Zipursky, A. Primary Shunt Hyperbilirubinemia due to an Alternate Path of Bilirubin Production. Am. J. Med. 1959, 27, 693.
212. Schalm, L.; Weber, A. P. Jaundice with Conjugated Bilirubin in Hyperhaemolysis. Acta Med. Scand. 1964, 176, 549.
213. Crigler, J. F.; Najjar, V. A. Congenital Familial Non- hemolytic Jaundice with Kernicterus. Pediatrics 1952, 10, 169.
214. Childs, B.; Sidbury, J. B.; Migeon, C. J. Glucuronic Acid Conjugation by Patients with Familial Non-Hemolytic Jaun-dice and Their Relatives. Pediatrics 1959, 23, 903.
215. Berk, P. D.; Martin, F.; Blaschke, T. F., et al. Unconjugated Hyperbilirubinemia: Physiological Evaluation and Experi-mental Approaches to Therapy. Ann. Intern. Med. 1975, 82, 552.
216. Bloomer, J. R.; Berk, P. D.; Howe, R. B.; Berlin, N. I. Biliru-bin Metabolism in Congenital Nonhemolytic Jaundice. Pedi-atr. Res. 1971, 5, 256.
217. Arias, I. M.; Gartner, L. M.; Cohen, M., et al. Chronic Nonhemolytic Unconjugated hyperbilirubinemia with Gluc-uronosyltransferase Deficiency: Clinical, Biochemical, Phar-macologic, and Genetic Evidence for Heterogeneity. Am. J. Med. 1969, 47, 395.
218. Wolkoff, A. W.; Roy Chowdhury, J.; Gartner, L. A., et al. Crigler–Najjar Syndrome (Type I) in an Adult Male. Gastro-enterology 1979, 76, 3380.
219. Billing, G. H.; Gray, C. H.; Kulcycka, A., et al. The Metabo-lism of 14C-Bilirubin in Congenital Nonhaemolytic Hyper-bilirubinaemia. Clin. Sci. 1964, 27, 163.
220. Blaschke, T. F.; Berk, P. D.; Rodkey, F. L., et al. Effects of Glutethimide and Phenobarbital on Hepatic Bilirubin Clearance, Plasma Bilirubin Turnover, and Carbon Mon-oxide Production in Man. Biochem. Pharmacol. 1974, 23, 2795.
221. Van Es, H. H.G.; Goldhoorn, B.; Paul-Abrahamse, M., et al. Immunochemical Characterization of UDP-Glucuronosyl-transferase in Four Patients with the Crigler–Najjar Type I Syndrome. J. Clin. Invest. 1990, 85, 1199.
222. Bosma, P. J.; Roy Chowdhury, J.; Huang, T. J., et al. Mechanism of Inherited Deficiencies of Multiple UDP- Glucuronosyltransferase Isoforms in Two Patients with Crigler–Najjar Syndrome, Type I. FASEB J. 1992, 6, 2859.
223. Ritter, J. K.; Yeatman, M. T.; Ferriera, P.; Owens, I. S. Iden-tification of a Genetic Alteration in the Code for Bilirubin UDP-Glucuronosyltransferase in the UGT1 Gene Complex of a Crigler–Najjar Syndrome, Type I Patient. J. Clin. Invest. 1992, 90, 150.
224. Ghosh, S. S.; Takahashi, M.; Thummala, N. R., et al. Liver-Directed Gene Therapy: Promises and Prospects at the Turn of the Century. J. Hepatol. 2000, 32, 238.
225. Lamolla, A. A.; Eisinger, J.; Blumberg, W. E., et al. Fluo-rometric Study of the Partition of Bilirubin among Blood Components: Basis for Rapid Microassaysof Bilirubin and Bilirubin Binding Capacity in Whole Blood. Anal. Biochem. 1979, 15, 25.
226. Clarke, D. J.; Moghrabi, N.; Monaghan, G., et al. Genetic Defects of UDP-Glucuronosyltransferase-1 (UGT1) Gene That Cause Familial Non-Hemolytic Unconjugated Hyper-bilirubinemias. Clin. Chim. Acta 1997, 266, 63.
227. Jansen, P. L. M.; Oude Elferink, R. P. J. Hereditary Hyper-bilirubinemia: A Molecular and Mechanistic Approach. Semin. Liver. Dis. 1988, 8, 168.
228. Kadakol, A.; Ghosh, S. S.; Sappal, B. S., et al. Genetic Lesions of Bilirubin Uridine Diphosphoglucuronate Glucuronosyl-transferase Causing Crigler–Najjar and Gilbert’s Syndromes: Correlation of Genotype to Phenotype. Hum. Mutat. 2000, 16, 297.
229. Petit, F. M.; Gajdos, V.; Francoual, J., et al. Allelic Hetero-geneity of Crigler–Najjar Type I Syndrome: A Study of 24 Cases. Clin. Genet. 2004, 66, 571.
230. Servedio, V.; d’Apolito, M.; Maiorano, N., et al. Spectrum of UGT1A1 Mutations in Crigler–Najjar (CN) Syndrome Patients: Identification of Twelve Novel Alleles and Geno-type–Phenotype Correlation. Hum. Mutat. 2005, 25, 325.
231. Lester, R.; Schmid, R. Intestinal Absorption of Bile Pigments. II. Bilirubin Absorption in Man. N. Engl. J. Med. 1963, 269, 178.
232. Kadakol, A.; Deocharan, B.; Mukhopadhyay, L., et al. Rapid Prenatal Diagnosis of Crigler–Najjar Syndrome Type 1 by Genetic Analysis of Chorionic Villus Samples. Hepatology 1998, 28, 316A.
233. Petit, F. M.; Gajdos, V.; Parisot, F., et al. Parental Isodisomy for Chromosome 2 as the Cause of Crigler–Najjar Syndrome Type I Syndrome. Eur. J. Hum. Genet. 2005, 13, 278–282.
234. Gunn, C. H. Hereditary Acholuric Jaundice in a New Mutant Strain of Rats. J. Hered. 1938, 29, 137.
235. Schmid, R.; Axelrod, J.; Hammaker, L.; Swarn, R. L. Con-genital Jaundice in Rats due to Defective Glucuronide For-mation. J. Clin. Invest. 1958, 37, 1123.
236. Lathe, G. H.; Walker, M. An Enzymatic Defect in Human Neonatal Jaundice and in Gunn’s Strain of Jaundiced Rats. Biochem. J. 1957, 67, 9.
237. Cohen, A. N.; Kapitulnik, J.; Ostro, J. D., et al. Effects of Phenobarbital on Bilirubin Metabolism and Its Response to Phototherapy in the Jaundiced Gunn Rat. Hepatology 1985, 5, 310.
238. Roy Chowdhury, J.; Hays, R. M.; Wu, C. H., et al. Disruption of Microtubules Results in Prolonged Persistence and Expres-sion of Genes Targeted to the Liver In Vivo by Receptor-Mediated Endocytosis. Gastroenterology 1993, 104, A981.
239. Roy Chowdhury, J.; Van Es, H. H. S.; Roy Chowdhury, N. Gunn Rat: An Animal Model for Deficiency of Bilirubin Con-jugation. In Pathophysiology of Bile Secretion; Berk, P. D.; Tavoloni, N., Eds.; Raven Press: New York, 1992; pp 713.
240. Roy Chowdhury, J.; Huang, T. J.; Kesari, K., et al. Molecular Basis for the Lack of Bilirubin-Specific and 3-Methylcholanthrene-Inducible UDP-Glucuronosyltransferase Activities in Gunn Rats: The Two Isoforms Are Encoded by Distinct mRNA Species that Share an Identical Single Base Deletion. J. Biol. Chem. 1991, 266, 18294.
241. Flock, E. V.; Bollman, J. L.; Owen, C. A.; Zollman, P. E. Conjugation of Thyroid Hormones and Analogues by the Gunn Rat. Endocrinology 1965, 77, 303.
242. Roy Chowdhury, N.; Kondapalli, R.; Roy Chowdhury, J. The Gunn Rat: An Animal Model for Inherited Deficiency of Bilirubin Glucuronidation. In Animal Models in Liver Research; Cornelius, C. E.; Marshak, R. R.; Melby, E. C., Eds.; Academic Press: New York, 1993; pp 150.
243. Scharschmidt, B. F.; Martin, J. F.; Shapiro, L. J., et al. The Use of Calcium Chelating Agents and Prostaglandin El to Eliminate Platelet and White Blood Cell Losses Resulting from Hemoperfusion through Uncoated Charcoal, Albu-min-Agarose Gel, and Neutral and Cation Exchange Resin. J. Lab. Clin. Med. 1977, 90, 110.

Disorders
30 CHAPTER 69 Bile Pigment Metabolism and Its244. Kaufman, S. S.; Wood, R. P.; Shaw, B. W., et al. Orthotopic Liver Transplantation for Type I Crigler–Najjar Syndrome. Hepatology 1986, 6, 1259.
245. Van der Veere, C. N.; Sinaasappel, M.; McDonagh, A. F., et al. Current Therapy for Crigler–Najjar Syndrome Type 1: Report of a World Registry. Hepatology 1996, 24, 311.
246. Cornelius, C. S.; Rodgers, P. A. Prevention of Neonatal Hyperbilirubinemia in Rhesus Monkey by Tin-Protoporphyrin. Pediatr. Res. 1984, 18, 728.
247. Galbraith, R. A.; Drummond, G. S.; Kappas, A. Suppres-sion of Bilirubin Production in the Crigler–Najjar, Type I Syndrome: Studies with Heme Oxygenase Inhibitor Tin-Mesoporphyrin. Pediatrics 1992, 89, 175.
248. Lavin, A.; Sung, C.; Klibanov, A. M.; Langer, R. Enzymatic Removal of Bilirubin from Blood: A Potential Treatment of Neonatal Jaundice. Science 1985, 230, 543.
249. Sugi, K.; Inoue, M.; Morino, Y. Degradation of Plasma Bili-rubin by a Bilirubin Oxidase Derivative which Has a Rela-tively Long Half-Life in the Circulation. Biochim. Biophys. Acta 1989, 991, 405.
250. Kapitulnik, J. The Role of Cytochrome P-450 in the Alter-native Pathways of Bilirubin Metabolism in Congenitally Jaundiced Gunn Rats and Infants with the Crigler–Najjar Syndrome, Type I. In Proceedings of the International Bili-rubin Workshop; 1992; pp 53, Trieste.
251. Vroemen, J. P. A. M.; Blankaert, N.; Buurman, W. A., et al. Treatment of Enzyme Deficiency by Hepatocyte Transplan-tation in Rats. J. Surg. Res. 1985, 39, 267.
252. Demetriou, A. A.; Levenson, S. M.; Novikoff, P. M., et al. Survival, Organization, and Function of Microcarrier-Attached Hepatocytes Transplanted in Rats. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 7475.
253. Demetriou, A. A.; Levenson, S. M.; Whiting, J., et al. Replacement of Hepatic Functions in Rats by Transplanta-tion of Microcarrier-Attached Hepatocytes. Science 1986, 233, 1190.
254. Bruni, S.; Chang, T. M. S. Encapsulated Hepatocytes for Controlling Hyperbilirubinemia in Gunn Rats. Int. J. Artif. Organs 1991, 14, 239.
255. Gupta, S.; Aragona, E.; Vemuru, R. P., et al. Permanent Engraftment and Function of Hepatocytes Delivered to the Liver: Implications for Gene Therapy and Liver Repopula-tion. Hepatology 1991, 14, 144.
256. Moghrabi, N.; Clarke, D. J.; Burchell, B.; Boxer, M. Coseg-regation of Intragenic Markers with a Novel Mutation that Causes Crigler–Najjar Syndrome Type I: Implication in Car-rier Detection and Prenatal Diagnosis. Am. J. Hum. Genet. 1993, 53, 722.
257. Mor-Cohen, R.; Zivelin, A.; Rosenberg, N., et al. Identifi-cation and Functional Analysis of Two Novel Mutations in the Multidrug Resistance Protein 2 Gene in Israeli Patients with Dubin–Johnson Syndrome. J. Biol. Chem. 2001, 276, 36923.
258. Fox, I. J.; Roy Chowdhury, J.; Kaufman, S. S., et al. Treat-ment of Crigler–Najjar Syndrome Type I with Hepatocyte Transplantation. N. Engl. J. Med. 1998, 333, 1422.
259. Roy Chowdhury, J.; Strom, S. C.; Roy Chowdhury, N., et al. Hepatocyte Transplantation in Humans: Gene Therapy and More. Pediatrics 1998, 102, 647.
260. Lysy, P. A.; Najimi, M.; Stephenne, X.; Bourgois, A.; Smets, F.; Sokal, E. M. Liver Cell Transplantation for Crigler–Najjar Syndrome Type I: Update and Perspectives. World J. Gastroenterol. 2008, 14, 3464.
261. Fox, I. J.; Roy-Chowdhury, J. Hepatocyte Transplantation. J. Hepatol. 2004, 40, 878.
262. Nguyen, T. H.; Oberholzer, J.; Birraux, J., et al. Highly Effi-cient Lentiviral Vector-Mediated Transduction of Nondivid-ing, Fully Reimplantable Primary Hepatocytes. Mol. Ther. 2002, 6, 199.
263. Guha, C.; Parashar, B.; Deb, N. J., et al. Long-Term Normal-ization of Serum Bilirubin Levels by Massive Repopulation of Gunn Rat Liver by Normal Hepatocytes, Transplanted after Preparative Hepatic Irradiation and Partial Hepatec-tomy. Hepatology 2002, 36, 354.
264. Guha, C.; Sharma, A.; Gupta, S., et al. Amelioration of Radi-ation-Induced Liver Damage in Partially Hepatectomized Rats by Hepatocyte Transplantation. Cancer Res. 1999, 59, 587.
265. Lee, S. W.; Wang, X.; Roy Chowdhury, N.; Roy Chowdhury, J. Hepatocyte Transplantation: State of the Art and Strate-gies for Overcoming Existing Hurdles. Ann. Hepatol. 2004, 3, 48.
266. Takahashi, M.; Deb, N. J.; Kawashita, Y., et al. A Novel Strategy for In Vivo Expansion of Transplanted Hepatocytes Using Preparative Hepatic Irradiation and FasL-Induced Hepatocellular Apoptosis. Gene Ther. 2003, 10, 304.
267. Nies, A. T.; Gatmaitan, Z.; Arias, I. M. ATP-Dependent Phosphatidylcholine Translocation in Rat Liver Canalicular Plasma Membrane Vesicles. J. Lipid Res. 1996, 37, 1125.
268. Si-Tayeb, K.; Noto, F. K.; Nagaoka, M.; Li, J.; Battle, M. A.; Duris, C.; North, P. E.; Dalton, S.; Duncan, S. E. Highly Efficient Generation of Human Hepatocyte-Like Cells from Induced Pluripotent Stem Cells. Hepatology 2010, 51, 297.
269. Roy Chowdhury, N.; Lu, Y.; Jiang, J., et al. Comparison of Gene Therapy Strategies for Treatment of Liver Diseases. J. Gastroenterol. Hepatol. 2004, 19, S295.
270. Roy Chowdhury, J.; Grossman, M.; Gupta, S., et al. Long-Term Improvement of Hypercholesterolemia After Ex Vivo Gene Therapy in LDL-Receptor Deficient Rabbits. Science 1991, 54, 1802.
271. Grossman, M.; Raper, S. E.; Kozarsky, K., et al. Successful Ex Vivo Gene Therapy Directed to Liver in a Patient with Familial Hypercholesterolemia. Nat. Genet. 1994, 6, 335.
272. Tada, K.; Roy Chowdhury, N.; Prasad, V. R., et al. Long-Term Amelioration of Bilirubin Glucuronidation Defect in Gunn Rats by Transplanting Genetically Modified Immor-talized Autologous Hepatocytes. Cell Transplant. 1998, 7, 607.
273. Toietta, G.; Mane, V. P.; Norona, W. S., et al. Lifelong Elim-ination of Hyperbilirubinemia in the Gunn Rat with a Single Injection of Helper-Dependent Adenoviral Vector. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3930.
274. Ilan, Y.; Jona, V. K.; Sengupta, K., et al. Transient Immuno-suppression with FK506 Permits Long-Term Expression of Therapeutic Genes Introduced into the Liver Using Recom-binant Adenoviruses. Hepatology 1997, 26, 949.
275. Ilan, Y.; Attavar, P.; Takahashi, M., et al. Induction of Cen-tral Tolerance by Intrathymic Inoculation of Adenoviral Antigens into the Host Thymus Permits Long-Term Gene Therapy in Gunn Rats. J. Clin. Invest. 1996, 98, 2640.
276. Ilan, Y.; Prakash, R.; Davidson, A., et al. Oral Tolerization to Adenoviral Antigens Permits Long-Term Gene Expres-sion Using Recombinant Adenoviral Vectors. J. Clin. Invest. 1997, 99, 1098.
277. Ilan, Y.; Sauter, B.; Roy Chowdhury, N., et al. Oral Toleriza-tion to Adenoviral Proteins Permits Repeated Adenovirus-Mediated Gene Therapy in Rats with Preexisting Immunity to Adenovirus. Hepatology 1998, 27, 1368.
278. Takahashi, M.; Ilan, Y.; Roy Chowdhury, N., et al. Long-Term Correction of Bilirubin UDP-Glucuronosyltransferase Deficiency in Gunn Rats by Administration of a Recombi-nant Adenovirus during the Neonatal Period. J. Biol. Chem. 1996, 271, 26536.
279. Ilan, Y.; Droguett, G.; Roy Chowdhury, N., et al. Insertion of the Adenoviral E3 Region into a Recombinant Viral Vector Prevents Antiviral Humoral and Cellular Immune Responses and Permits Long-Term Gene Expression. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 2587.

CHAPTER 69 Bile Pigment Metabolism and Its Disorders 31
280. Thummala, N. R.; Ghosh, S. S.; Lee, S. W., et al. A Non-Immunogenic Adenoviral Vector, Coexpressing CTLA4Ig and Bilirubin-Uridinediphosphoglucuronate Glucurono-syltransferase Permits Long-Term, Repeatable Transgene Expression in the Gunn Rat Model of Crigler–Najjar Syn-drome. Gene Ther. 2002, 9, 981.
281. Strayer, D. S.; Branco, F.; Zern, M. A., et al. Durability of Transgene Expression and Vector Integration: Recombinant SV40-Derived Gene Therapy Vectors. Mol. Ther. 2002, 6, 227.
282. Strayer, D. S.; Zern, M. A.; Roy Chowdhury, J. What Can SV40 Vectors Do for Gene Therapy? Curr. Opin. Mol. Ther. 2002, 4, 313.
283. Sauter, B. V.; Parashar, B.; Roy Chowdhury, N., et al. Long Term Correction of Bilirubin Glucuronidation in the Biliru-bin-UDP-Glucuronosyltranferase Deficient Gunn Rat by In Vivo Gene Therapy with Recombinant SV40. Gastroenterol-ogy 2000, 119, 1348.
284. Seppen, J.; van der Rijt, R.; Looije, N., et al. Long-Term Cor-rection of Bilirubin UDP Glucuronyltransferase Deficiency in Rats by In Utero Lentiviral Gene Transfer. Mol. Ther. 2003, 8, 593.
285. Bommineni, V. R.; Roy Chowdhury, N.; Wu, G. Y., et al. Depolymerization of Hepatocellular Microtubules after Par-tial Hepatectomy. J. Biol. Chem. 1994, 269, 25200.
286. Roy Chowdhury, N.; Nays, R. M.; Bommineni, V. R., et al. Microtubular Distribution Prolongs the Expression of Human Bilirubin-Uridinediphosphoglucuronate Glucuronosyltransfer-ase-1 Gene Transferred into Gunn Rat Livers. J. Biol. Chem. 1996, 271, 2341.
287. Wilson, J. M.; Wu, G. Y.; Wu, C. H., et al. Hepatocyte-Directed Gene Transfer In Vivo Leads to Transient Improve-ment of Hypercholesterolemia in LDL-Receptor-Deficient Rabbits. J. Biol. Chem. 1992, 67, 963.
288. Kren, B. T.; Ghosh, S. S.; Linchen, C. L., et al. Hepatocyte-Targeted Delivery of Sleeping Beauty Mediates Efficient Gene Transfer In Vivo. Gene Ther. Mol. Biol. 2003, 7, 229.
289. Kren, B. T.; Parashar, B.; Bandhopadhyaya, B., et al. Correc-tion of UDP-Glucuronosyltransferase Gene Defect in Gunn Rat Model of Crigler–Najjar Syndrome Type I with a Chime-ric Oligonucleotide. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 10349.
290. Arias, I. M. Chronic Unconjugated Hyperbilirubinemia without Overt Signs of Hemolysis in Adolescents and Adults. J. Clin. Invest. 1962, 41, 2233.
291. Gollan, J. L.; Huang, S. M.; Billing, B.; Sherlock, S. Prolonged Survival in Three Brothers with Severe Type II Crigler–Najjar Syndrome: Ultrastructural and Metabolic Studies. Gastroen-terology 1975, 68, 1543.
292. Gordon, E. R.; Shaffer, E. A.; Sass-Kortsak, A. Bilirubin Secretion and Conjugation in Crigler–Najjar Syndrome Type II. Gastroenterology 1976, 70, 761.
293. Fevery, J.; Blanckaert, N.; Heirwegh, K. P. M., et al. Unconju-gated Bilirubin and an Increased Proportion of Bilirubin Mono-conjugates in the Bile of Patients with Gilbert’s Syndrome and Crigler–Najjar Syndrome. J. Clin. Invest. 1977, 60, 970.
294. Bosma, P. J.; Golhoorn, B.; Oude Elferink, R. P., et al. A Mutation in Bilirubin Uridine 5′-Diphosphate Glucuronos-yltransferase Isoforms 1 Causing Crigler–Najjar Syndrome Type II. Gastroenterology 1993, 105, 216.
295. Seppen, J.; Bosma, P. J.; Goldhoorn, B. G., et al. Discrimina-tion between Crigler–Najjar Type I and II by Expression of Mutant Bilirubin Uridine Diphosphate-Glucuronosyltrans-ferase. J. Clin. Invest. 1994, 94, 2385.
296. Gilbert, A.; Lereboullet, P. La Cholamae Simple Familiale. Sem. Med. 1901, 21, 241.
297. Thompson, R. P. H. Genetic Transmission of Gilbert’s Syn-drome. In Familial Hyperbilirubinemia; Okolicsanyl, L., Ed.; Wiley: New York, 1981; pp 91.
298. Novikoff, A. B.; Essner, E. The Liver Cell. Am. J. Med. 1960, 19, 102.
299. Owens, D.; Evans, J. Population Studies on Gilbert’s Syn-drome. J. Med. Genet. 1975, 12, 152.
300. Powell, L. W.; Hemingway, E.; Billing, B. H.; Sherlock, S. Idiopathic Unconjugated Hyperbilirubinemia (Gilbert’s Syn-drome): A Study of 42 Families. N. Engl. J. Med. 1967, 277, 1108.
301. Bailey, A.; Robinson, D.; Dawson, A. M. Does Gilbert’s Dis-ease Exist? Lancet 1977, 1, 931.
302. Muraca, M.; Fevery, J. Influence of Sex and Sex Steroids on Bilirubin-Uridinediphosphate Glucuronosyltransferase Activity of Rat Liver. Gastroenterology 1984, 87, 308.
303. Arias, I. M.; London, I. M. Bilirubin Glucuronide Forma-tion In Vitro: Demonstration of a Defect in Gilbert’s Disease. Science 1957, 126, 563.
304. Gollan, J. L.; Bateman, C.; Billing, B. H. Effects of Dietary Composition on the Unconjugated Hyperbilirubinemia of Gilbert’s Syndrome. Gut 1976, 17, 335.
305. Roy Chowdhury, J.; Grossman, M.; Gupta, S., et al. Long-Term Improvement of Hypercholesterolemia after Ex Vivo Gene Therapy in LDL-Receptor Deficient Rabbits. Science 1991, 54, 1802.
306. Berk, P. D.; Bloomer, J. R.; Howe, R. B.; Berlin, N. I. Consti-tutional Hepatic Dysfunction (Gilbert’s Syndrome): A 1970.
307. Goresky, C. A.; Gordon, E. R.; Shaffer, E. A., et al. Defini-tion of a Conjugation Dysfunction in Gilbert’s Syndrome: Studies of the Handling of Bilirubin Loads and of the Pattern of Bilirubin Conjugates Secreted in Bile. Clin. Sci. Mol. Med. 1978, 1, 63.
308. Berk, P. D.; Blaschke, T. F.; Waggoner, J. G. Defective BSP Clearance in Patients with Constitutional Hepatic Dysfunc-tion (Gilbert’s Syndrome). Gastroenterology 1972, 63, 472.
309. Cobelli, C.; Ruggeri, A.; Toffolo, G., et al. BSP vs Biliru-bin Kinetics in Gilbert’s Syndrome. In Familial Hyperbili-rubinemia; Okolicsanyl, L., Ed.; Wiley: New York, 1981; pp 121.
310. Berk, P. D.; Blaschke, T. F. Detection of Gilbert Syndrome in Patients with Hemolysis: A Method Using Radioactive Chro-mium. Ann. Intern. Med. 1972, 77, 527.
311. Bensinger, T. A.; Maisels, M. J.; Marlson, D. E.; Conrad, M. E. Effect of Low Caloric Diet on Endogenous Carbon Mon-oxide Production. Normal Adults and Gilbert’s Syndrome. Proc. Soc. Exp. Biol. Med. 1973, 144, 417.
312. Felscher, B. F.; Rickard, D.; Redeker, A. G. The Reciprocal Relation between Caloric Intake and the Degree of Hyper-bilirubinemia in Gilbert’s Syndrome. N. Engl. J. Med. 1970, 283, 170.
313. Bakken, A. F.; Thaler, M. M.; Schmidt, R. Metabolic Regu-lation of Heme Catabolism and Bilirubin Production: Hor-monal Control of Hepatic Heme Oxygenase Activity. J. Clin. Invest. 1972, 51, 530.
314. Bloomer, J. R.; Barrett, P. V.; Rodkey, F. L.; Berlin, N. I. Studies on the Mechanisms of Fasting Hyperbilirubinemia. Gastroenterology 1971, 61, 479.
315. Felscher, B. F.; Carpio, N. M.; Van Couvering, K. Effect of Fasting and Phenobarbital on Hepatic UDP-Glucuronic Acid Formation in the Rat. J. Lab. Clin. Med. 1979, 93, 414.
316. Gollan, J. L.; Hatt, K. J.; Billing, B. H. The Influence of Diet on Unconjugated Hyperbilirubinemia in the Gunn Rat. Clin. Sci. Mol. Med. 1975, 49, 229.
317. Davidson, A. R.; Rojas-Beuno, A.; Thompson, R. P. H.; Wil-liams, R. Reduced Caloric Intake and Nicotinic Acid Provo-cation Tests in Diagnosis of Gilbert’s Syndrome. Br. Med. J. 1975, 2, 480.
318. Bosma, P. J.; Roy Chowdhury, J.; Bakker, C., et al. A Sequence Abnormality in the Promoter Region Results in Reduced Expression of Bilirubin-UDP-Glucuronosyltransferase-1 in Gilbert Syndrome. N. Engl. J. Med. 1995, 333, 1171.

32 CHAPTER 69 Bile Pigment Metabolism and Its Disorders
319. Raijmakers, M. T.; Jansen, P. L.; Steegers, E. A.; Peters, W. H. Association of Human Liver Bilirubin UDP- Glucuronyltransferase Activity with a Polymorphism in the Promoter Region of the UGT1A1. J. Hepatol. 2000, 33, 348.
320. Wells, P. G.; Mackenzie, P. I.; Roy Chowdhury, J., et al. Glucuronidation and the UDP-Glucuronosyltransferases in Health and Disease. Drug Metab. Dispos. 2004, 32, 281.
321. Maruo, Y.; Sato, H.; Yamano, T., et al. Gilbert’s Syndrome Caused by Homozygous Missense Mutation (Tyr486Asp) of Bilirubin-UDP Glucuronosyl Transferase. J. Pediatr. 1998, 132, 1045.
322. Soeda, Y.; Yamamoto, K.; Adachi, Y., et al. Predicted Homo-zygous Mis-Sense Mutation in Gilbert’s Syndrome. Lancet 1995, 346, 1494.
323. Koiwai, O.; Nishizawa, M.; Hasada, K., et al. Gilbert’s Syn-drome Is Caused by a Heterozygous Missense Mutation in the Gene for Bilirubin UDP-Glucuronosyltransferase. Hum. Mol. Genet. 1995, 4, 1183.
324. Kadakol, A.; Sappal, B. S.; Ghosh, S. S., et al. Interaction of Coding Region Mutations and the Gilbert-Type Pro-moter Abnormality of the UGT1A1 Gene Causes Moderate Degrees of Unconjugated Hyperbilirubinemia and May Lead to Neonatal Kernicterus. J. Med. Genet. 2001, 38, 244.
325. Labrune, P.; Myara, A.; Chalas, J., et al. Association of a Homozygous (TA)8 Promoter Polymorphism and a N400D Mutation of UGT1A1 in a Child with Crigler–Najjar Type II Syndrome. Hum. Mutat. 2002, 20, 399.
326. Bancroft, J. D.; Kreamer, B.; Gourley, G. R. Gilbert Syn-drome Accelerates Development of Neonatal Jaundice. J. Pediatr. 1998, 132, 656.
327. Roy Chowdhury, N.; Deocharan, B.; Bejjanki, H. R., et al. Presence of the Genetic Marker for Gilbert Syndrome is Associated with Increased Level and Duration of Neonatal Jaundice. Acta Paediatr. 2002, 91, 100.
328. Kaplan, M.; Renbaum, P.; Levy-Lahad, E., et al. Gilbert Syn-drome and Glucose-6-Phosphate Dehydrogenase Deficiency: A Dose-Dependent Genetic Interaction Crucial to Neonatal Hyperbilirubinemia. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 12128.
329. del Giudice, E. M.; Perrotta, S.; Nobili, B., et al. Coinheri-tance of Gilbert Syndrome Increases the Risk for Developing Gallstones in Patients with Hereditary Spherocytosis. Blood 1999, 94, 2259.
330. de Morais, S. M.; Uetrecht, J. P.; Wells, P. G. Decreased Gluc-uronidation and Increased Bioactivation of Acetaminophen in Gilbert’s Syndrome. Gastroenterology 1992, 102, 577.
331. Ullrich, D.; Sieg, A.; Blume, R., et al. Normal Pathways for Glucuronidation, Sulphation and Oxidation of Paracetamol in Gilbert’s Syndrome. Eur. J. Clin. Invest. 1987, 17, 237.
332. Iyer, L.; King, C. D.; Whitington, P. F., et al. Genetic Predis-position to the Metabolism of Irinotecan (CPT-11): Role of Uridine Diphosphate Glucuronosyltransferase Isoform 1A1 in the Glucuronidation of Its Active Metabolite (SN-38) in Human Liver Microsomes. J. Clin. Invest. 1998, 101, 847.
333. Portman, O. W.; Alexander, M.; Roy Chowdhury, J., et al. Effects of Nutrition on Hyperbilirubinemia in Bolivian Squirrel Monkeys. Hepatology 1984, 4, 454.
334. Portman, O. W.; Roy Chowdhury, J.; Roy Chowdhury, N., et al. A Non-Human Primate Model for Gilbert’s Syndrome. Hepatology 1984, 4, 175.
335. Dubin, I. N.; Johnson, F. B. Chronic Idiopathic Jaundice with Unidentified Pigment in Liver Cells: A New Clinico-pathologic Entity with a Report of 12 Cases. Med. (Balti-more) 1954, 33, 155.
336. Sprinz, H.; Nelson, R. S. Persistent Nonhemolytic Hyper-bilirubinemia Associated with Lipochrome-Like Pigment in Liver Cells: Report of Four Cases. Ann. Intern. Med. 1954, 41, 952.
337. Dubin, I. N. Chronic Idiopathic Jaundice: A Review of Fifty Cases. Am. J. Med. 1958, 23, 268.
338. Shani, M.; Seligshon, U.; Gilon, E., et al. Dubin–Johnson Syndrome in Israel: Clinical, Laboratory, and Genetic Aspects of 101 Cases. West J. Med. 1970, 39, 549.
339. Cohen, L.; Lewis, C.; Arias, I. M. Pregnancy, Oral Contra-ceptives, and Chronic Familial Jaundice with Predominantly Conjugated Hyperbilirubinemia (Dubin–Johnson Syndrome). Gastroenterology 1972, 62, 1182.
340. Nakata, F.; Oyanagi, K.; Fujiwara, M., et al. Dubin–Johnson Syndrome in a Neonate. Eur. J. Pediatr. 1979, 132, 299.
341. Morita, M.; Kihava, T. Intravenous Cholecystography and Metabolism of Meglumine Iodipamide (Biligrafin) in Dubin–Johnson Syndrome. Radiology 1971, 95, 57.
342. Ehrlick, J. C.; Novikoff, A. B.; Platt, R.; Essner, E. Hepato-cellular Lipofuscin and the Pigment of Chronic Idiopathic Jaundice. Bull N. Y. Acad. Med. 1960, 36, 488.
343. Arias, I. M.; Bernstein, L.; Roffler, R.; Ben Ezzer, J. Black Liver Diseases in Corriedale Sheep: Metabolism of Tritiated Epinephrine and Incorporation of Isotope into the Hepatic Pigment In Vivo. J. Clin. Invest. 1965, 44, 1026.
344. Swartz, H. M.; Sarna, T.; Varma, R. R. On the Nature and Excretion of the Hepatic Pigment in the Dubin–Johnson Syn-drome. Gastroenterology 1979, 76, 958.
345. Arias, I. M.; Blumberg, W. The Pigment in Dubin–Johnson Syndrome. Gastroenterology 1979, 77, 820.
346. Ware, A.; Eigenbrodt, E.; Naftalis, J.; Combes, B. Dubin–Johnson Syndrome and Viral Hepatitis. Gastroenterology 1974, 67, 560.
347. Bohan, A.; Boyer, J. L. Mechanisms of Hepatic Transport of Drugs: Implications for Cholestatic Drug Reactions. Semin. Liver Dis. 2002, 22, 123.
348. Goresky, C. A. The Hepatic Uptake Process: Its Implications for Bilirubin Transport. In Jaundice; Goresky, C. A.; Fisher, M. M., Eds.; Plenum: New York, 1975; pp 159.
349. Goresky, C. A. The Hepatic Uptake and Excretion of Sul-fobromophthalein and Bilirubin. Can. Med. Assoc. J. 1965, 92, 851.
350. Charbonnier, A.; Brisbois, P. Etude Chromatographique de la BSP au cours de l’Epreuve Clinique d’Epuration Plasma-tique de Ce Colorant. Rev. Intern. Hepatol. 1960, 10, 1163.
351. Schoenfield, L. J.; McGill, D. B.; Hunton, D. B., et al. Studies of Chronic Idiopathic Jaundice (Dubin–Johnson Syndrome): Demonstration of Hepatic Excretory Defect. Gastroenterol-ogy 1963, 44, 101.
352. Rodes, J.; Zubizarreta, A.; Bruguera, M. Metabolism of Bromsulphophthalein in Dubin–Johnson Syndrome: Diag-nostic Value of the Paradoxical Increase in Plasma Levels of BSP. Am. J. Dig. Dis. 1972, 17, 545.
353. Kaplowitz, N.; Javitt, N.; Kappas, A. Coproporphyrin I and III Excretion in Bile and Urine. J. Clin. Invest. 1972, 51, 2895.
354. Wolkoff, A. W.; Arias, I. M. Coproporphyrin Excretion in Amniotic Fluid and Urine From Premature Infants: A Pos-sible Maturation Defect. Pediatr. Res. 1974, 8, 591.
355. Wolkoff, A. W.; Cohen, L. E.; Arias, I. M. Inheritance of the Dubin–Johnson Syndrome. N. Engl. J. Med. 1973, 288, 113.
356. Alpert, S.; Mosher, M.; Shanske, A.; Arias, I. M. Multiplic-ity of Hepatic Excretory Mechanisms for Organic Anions. J. Gen. Physiol. 1969, 53, 238.
357. Jedlitschky, G.; Leier, I.; Buchholz, U., et al. ATP-Depen-dent Transport of Glutathione S-Conjugates by the Multi-drug Resistance-Associated Protein. Cancer Res. 1994, 54, 4833.
358. Kobayashi, K.; Sogame, Y.; Hara, H.; Hayashi, K. Mecha-nism of Glutathione S-Conjugate Transport in Canalicular and Basolateral Rat Liver Plasma Membranes. J. Biol. Chem. 1990, 265, 7737.

CHAPT
359. Seligsohn, U.; Shani, M.; Ramot, B., et al. Dubin–Johnson Syndrome in Israel. II. Association with Factor-VII Defi-ciency. Q. J. Med. 1970, 39, 569.
360. Kamisako, T.; Leier, I.; Cui, Y., et al. Transport of Mono-glucuronosyl and Bisglucuronosyl Bilirubin by Recombinant Human and Rat Multidrug Resistance Protein 2. Hepatology 1999, 30, 485.
361. Allikmets, R.; Gerrard, B.; Hutchinson, A.; Dean, M. Char-acterization of the Human ABC Superfamily: Isolation and Mapping of 21 New Genes Using the Expressed Sequence Tags Database. Hum. Mol. Genet. 1996, 5, 1649.
362. Toh, S.; Wada, M.; Uchuimi, T., et al. Genomic Structure of the Canalicular Multispecific Organic Anion Transporter Gene (MRP2/cMOAT) and Mutations in the ATP-Binding Cassette Region in Dubin–Johnson Syndrome. Am. J. Hum. Genet. 1999, 64, 739.
363. Tsujii, H.; Konig, J.; Rost, D., et al. Exon–Intron Organiza-tion of the Human Multidrug Resistance Protein 2 (MRP2) Gene Mutated in Dubin–Johnson Syndrome. Gastroenterol-ogy 1999, 117, 653.
364. Kajihara, S.; Hisatomi, A.; Mizuta, T., et al. A Splice Site Mutation in the Human Canalicular Multispecific Organic Anion Transporter (cMOAT) Gene Causes Dubin–Johnson Syndrome. Biochem. Biophys. Res. Commun. 1998, 253, 454.
365. Wada, M.; Toh, S.; Taniguchi, K., et al. Mutations in the Canalicular Multispecific Organic Anion Transporter Gene, a Novel ABC Transporter, in Patients with Hyperbilirubinemia II/Dubin–Johnson Syndrome. Hum. Mol. Genet. 1998, 7, 203.
366. Keitel, V.; Karenbeck, J.; Nies, A. T., et al. Impaired Protein Maturation of the Conjugate Export Pump Multidrug Resis-tance Protein 2 as a Consequence of a Deletion Mutation in Dubin–Johnson Syndrome. Hepatology 2000, 32, 1317.
367. Cornelius, C. E.; Arias, I. M.; Osburn, B. I. Hepatic Pig-mentation with Photosensitivity: A Syndrome in Corriedale Sheep Resembling Dubin–Johnson Syndrome in Man. J. Am. Vet. Med. Assoc. 1965, 146, 709.
368. Barnhart, J. L.; Gronwall, R. R.; Combes, B. Biliary Excre-tion of Sulfobromophthalein Compounds in Normal and Mutant Corriedale Sheep: Evidence for a Disproportion-ate Transport Defect for Conjugated Sulfobromophthalein. Hepatology 1981, 1, 441.
369. Jansen, P. L. M.; van Klinken, J. W.; van Gelder, M., et al. Preserved Organic Anion Transport in Mutant TR Rats with a Hepatobiliary Secretion Defect. Am. J. Physiol. 1993, 265, G445.
370. Jansen, P. L. M.; Peters, W. H. M.; Lamers, W. H. Heredi-tary Chronic Conjugated Hyperbilirubinemia in Mutant Rats Caused by Defective Hepatic Anion Transport. Hepa-tology 1985, 5, 573.
371. Kitamura, T.; Alroy, J.; Gatmaitan, Z., et al. Defective Bili-ary Excretion of Epinephrine Metabolites in Mutant (TR) Rats: Relation to the Pathogenesis of Black Liver in the Dubin-Johnson Syndrome and Correidale Sheep with an Analogous Excretory Defect. Hepatology 1992, 15, 1154.
372. Jansen, P. L. M.; Groothuis, G. M. M.; Peters, W. H. M.; Meijer, D. K. F. Selective Hepatobiliary Transport Defect for Organic Anions and Neutral Steroids in Mutant Rats with Hereditary Conjugated Hyperbilirubinemia. Hepatology 1987, 7, 71.
373. Takikawa, H.; Nishikawa, K.; Sano, N., et al. Mechanisms of Biliary Excretion of Lithocholate-3-Sulfate in Eisai Hyper-bilirubinemic Rats (EHBR). Dig. Dis. Sci. 1995, 40, 1792.
374. Schulman, F. Y.; Montali, R. J.; Bush, M., et al. Dubin–Johnson-Like Syndrome in Golden Lion Tamarins (Leonto-pithecus rosalia rosalia). Vet. Pathol. 1993, 30, 491.
375. Haverback, B. J.; Wirtschafter, S. K. Familial Nonhemolytic Jaundice with Normal Liver Histology and Conjugated Bili-rubin. N. Engl. J. Med. 1960, 262, 113.
ER 69 Bile Pigment Metabolism and Its Disorders 33
376. Wolkoff, A. W.; Wolpert, E.; Pascasio, F. N.; Arias, I. M. Rotor’s Syndrome: A Distinct Inheritable Pathophysiologic Entity. Am. J. Med. 1976, 60, 173.
377. Wolpert, E.; Pascasio, F. M.; Wolkoff, A. W.; Arias, I. M. Abnormal Sulfobromophthalein Metabolism in Rotor’s Syn-drome and Obligate Heterozygotes. N. Engl. J. Med. 1977, 296, 1099.
378. Schiff, L.; Billing, B. H.; Oikawa, Y. Familial Nonhemolytic Jaundice with Conjugated Bilirubin in the Serum. N. Engl. J. Med. 1959, 260, 1315.
379. Kawasaki, H.; Kinwa, N.; Irisa, T.; Hirayama, C. Dye Clear-ance Studies in Rotor’s Syndrome. Am. J. Gastroenterol. 1979, 71, 380.
380. Porush, J. G.; Delman, A. J.; Feuer, M. M. Chronic Idio-pathic Jaundice with Normal Liver Histology. Arch. Intern. Med. 1962, 109, 102.
381. Dhumeaux, D.; Berthelot, P. Chronic Hyperbilirubinemia Associated with Hepatic Uptake and Storage Impairment: A New Syndrome Resembling that of the Mutant Southdown Sheep. Gastroenterology 1975, 69, 988.
382. Aziz, M. A.; Schwartz, S.; Watson, C. J. Studies of Copro-porphyrin VIII: Reinvestigation of the Isomer Distribution in Jaundice and Liver Diseases. J. Lab. Clin. Med. 1964, 63, 596.
383. Rapacini, G. L.; Topi, G. C.; Anti, M., et al. Porphyrins in Rotor Syndrome: A Study on an Italian Family. Hepatogas-troenterol 1986, 33, 11.
384. Jansen, P. L.; Muller, M. M. Progressive Familial Intrahe-patic Cholestasis Types 1, 2, and 3. Gut 1998, 42, 766.
385. Knisely, A. S. Progressive Familial Intrahepatic Cholestasis: An Update. Pediatr. Dev. Pathol. 2004, 7, 309.
386. Clayton, R. J.; Iber, F. L.; Ruebner, B. H., et al. Byler Dis-ease: Fatal Familial Intrahepatic Cholestasis in an Amish Kindred. Am. J. Dis. Child 1969, 117, 112.
387. Klomp, L. W.; Vargas, J. C.; van Mil, S. W., et al. Character-ization of Mutations in ATP8B1 Associated with Hereditary Cholestasis. Hepatology 2004, 40, 27.
388. Chen, F.; Ananthanarayanan, M.; Emre, S., et al. Progressive Familial Intrahepatic Cholestasis, Type 1, Is Associated with Decreased Farnesoid X Receptor Activity. Gastroenterology 2004, 126, 756.
389. Summerskill, W. H. J.; Walshe, J. M. Benign Recurrent Intrahepatic Obstructive Jaundice. Lancet 1959, 2, 686.
390. de Pagter, A. G. F.; Van Berge Henegouwen, G. P.; Bokkel-Huinnuk, J. A.; Brandt K-, H. Familial Benign Recurrent Intrahepatic Cholestasis. Gastroenterology 1976, 71, 202.
391. Tygstrup, N.; Jensen, B. Intermittent Intrahepatic Cholesta-sis of Unknown Etiology in Five Young Males from the Faroe Islands. Acta Med. Scand. 1969, 185, 523.
392. Summerfield, J. A.; Scott, J.; Berman, M., et al. Benign Recur-rent Intrahepatic Cholestasis: Studies of Bilirubin Kinetics, Bile Acids, and Cholangiography. Gut 1980, 21, 154.
393. Summerskill, W. H. J. The Syndrome of Benign Recurrent Cholestasis. Am. J. Med. 1965, 38, 298.
394. Biempica, L.; Gutstein, S.; Arias, I. M. Morphological and Biochemical Studies of Benign Recurrent Cholestasis. Gas-troenterology 1967, 52, 521.
395. Thompson, R.; Strautnieks, S. BSEP: Function and Role in Progressive Familial Intrahepatic Cholestasis. Semin. Liver Dis. 2001, 21, 545.
396. den Dunnen, J. T.; Antonarakis, S. E. Mutation Nomencla-ture Extensions and Suggestions to Describe Complex Muta-tions: A Discussion. Hum. Mutat. 2000, 15, 7.
397. Strautnieks, S. S.; Knisley, A. S.; Gerred, S., et al. Mutations in MDR3 in Adult Onset Cholangiopathy. In Bile Acids: From Genomics to Disease and Therapy. Proceedings of Falk Symposium 129; Keppler, D.; Leuschner, U.; Paumgart-ner, G.; Stiehl, A., Eds.; Kluwer Dordrecht, 2003; pp 184.

34 CHAPTER 69 Bile Pigment Metabolism and Its
398. Rosmorduc, O.; Hermelin, B.; Poupon, R. MDR3 Gene Defect in Adults with Symptomatic Intrahepatic and Gall-bladder Cholesterol Cholelithiasis. Gastroenterology 2001, 120, 1459.
399. Dixon, P. H.; Weerasekera, N.; Linton, K. J., et al. Hetero-zygous MDR3 Missense Mutation Associated with Intra-hepatic Cholestasis of Pregnancy: Evidence for a Defect in Protein Trafficking. Hum. Mol. Genet. 2000, 9, 1209.
400. Jacquemin, E.; Cresteil, D.; Manouvrier, S., et al. Heterozy-gous Non-Sense Mutation Associated with Intrahepatic Cho-lestasis of Pregnancy. Lancet 1999, 353, 210.
401. de Vree, J. M. L.; Ottenhoff Smith, A. J.; Aten, J.; Oude Elf-erink, R. P. J. Long Term Effects of Hepatocyte Transplanta-tion in an Animal Model of Progressive Familial Intrahepatic Cholestasis. Hepatology 1999, 30, 409A.
402. de Vree, J. M. L.; Ottenhoff Smith, A. J.; Aten, J., et al. Rapid Correction of Mdr2 Deficiency by Transplantation of Mdr3 Transgenic Hepatocytes. Hepatology 1998, 28, 387A.
403. Drouin, E.; Russo, P.; Tuchweber, B., et al. North American Indian Cirrhosis in Children: A Review of 30 Cases. J. Pedi-atr. Gastroenterol. Nutr. 2000, 31, 395.
404. Rapola, J.; Heikkila, P.; Fellman, V. Pathology of Lethal Fetal Growth Retardation Syndrome with Aminoaciduria, Iron Overload and Lactric Acidosis (GRACILE). Pediatr. Pathol. Mol. Med. 2002, 21, 183.
405. Emerick, E. M.; Rand, E. B.; Goldmuntz, E., et al. Features of Alagille Syndrome in 92 Patients: Frequency and Relation to Prognosis. Hepatology 1999, 29, 822.
Disorders
406. Piccoli, D. A.; Spinner, N. A. Alagille Syndrome and the Jag-ged1 Gene. Semin. Liver Dis. 2001, 21, 525.
407. Oda, T.; Elkaholoun, A. G.; Meltzer, P. S.; Chandrashek-harappa, S. C. Identification and Cloning of the Human Homolog (Jag1) of the Rat Jagged1 Gene from the Alagille Syndrome Critical Region at 20p12. Genomics 1997, 43, 376.
408. Phillips, M. J.; Azuma, T.; Meredith, S. L., et al. Abnormali-ties in Villin Gene Expression and Canalicular Microvillus Structure in Progressive Cholestatic Liver Disease in Child-hood. Lancet 2003, 362, 1090.
409. Ciotti, M.; Chen, F.; Rubatelli, F. F.; Owens, I. S. Coding and a TATA Box Mutation at the Bilirubin UDP-Glucuronosyltransferase Gene Cause Crigler–Najjar Syndrome Type 1 Disease. Biochim. Biophys. Acta 1998, 1407, 40.
410. Nong, S. H.; Xie, Y. M.; Chen, G. R.; Zhang, B. T. A Case with Type I Crigler–Najjar Syndrome. Zhonghua Er Ke Za Zhi 2003, 41, 382.
411. Aono, S.; Yamada, Y.; Keino, H., et al. A New Type of Defect in the Gene for Bilirubin Uridine 5′-Diphosphate-Glucuronosyltransferase in a Patient with Crigler–Najjar Syndrome Type I. Pediatr. Res. 1994, 35, 629.
412. Aono, S.; Adachi, Y.; Uyama, E., et al. Analysis of Genes for Bilirubin UDP-Glucuronosyltransferase in Gilbert’s Syn-drome. Lancet 1995, 345, 958.
413. Aono, S.; Yamada, Y.; Keino, H., et al. Identification of a Defect in the Gene for Bilirubin UDP-Glucuronosyltransferase in a Patient with Crigler–Najjar Syndrome Type II. Biochem. Biophys. Res. Commun. 1993, 197, 1239.
Biographies
Namita Roy Chowdhury, PhD is a professor in the Departments of Medicine and Genetics at Albert Einstein College of Medicine, New York. Dr N Roy Chowdhury received her PhD in Physiology from Calcutta University, India and her postdoctoral training at the University of Chemical Biology, Calcutta, India. She joined the Marion Bessin Liver Research Center of Albert Einstein College of Medicine in 1977, where she rose through the ranks to full professor. Her research is focused on the molecular mechanisms inherited liver diseases, particularly diseases of bilirubin metabolism.
Jayanta Roy Chowdhury, MD, MRCP, AGAF is a professor in the Department of Medicine (Gastrointestinal and Liver Diseases) and Genetics at Albert Einstein College of Medicine, New York. Dr J Roy Chowdhury received his Medical degree from Medical College, Calcutta, India. He received his postgraduate clinical training at Mount Sinai Hospital Services, New York, State University of New York at Stony Brook, and Royal Infirmary, Edinburgh, Scotland, UK. Dr J Roy Chowdhury joined the Marion Bessin Liver Research Center at Albert Einstein College of Medicine in 1975. He is the Scientific Director of the Cell Culture and Genetic Engineering Core of the Liver Research Center and also of the Gene Therapy Core of Albert Einstein College of Medicine. Dr J Roy Chowdhury’s research focuses on the molecular mechanisms of metabolic liver diseases, and gene-based and cell-based therapies of liver diseases.