electronic structures and transport properties in quasicrystals
TRANSCRIPT

Materials Science and Engineering, B19 (1993) 77-81 77
Electronic structures and transport properties in quasicrystals
T a k e o F u j i w a r a Department of Applied Physics, University of Tokyo, Bunkyo-ku, Tokyo 113 (Japan)
Abstract
The electronic structures in realistic quasicrystals are discussed theoretically. The density of states consists of a set of spiky peaks with widths of order 0.02 eV and has pseudogaps at the Fermi energy with widths of order 1 eV. The Boltzmann theory based on the detailed band structures would predict very high values of resistivity. The temperature dependence of the observed thermoelectric power may be due to the spiky peak structures of the conductivity as a function of the Fermi energy.
I. Introduction
Many kinds of quasicrystals have been found. The compounds AIMnSi and A1LiCu are two typical types of icosahedral quasicrystal. AIMnSi is called the Mackay icosahedron type (MI) because the constituent unit for this quasicrystal is an icosahedral cluster of atoms called the Mackay icosahedron. This class is also called A1- TM type (TM is transition metal), because A1 and TM are essential constituents in compounds of this class. The Mackay icosahedron is an icosahedral cluster of 54 atoms. Between MI clusters, there are several atoms called glue atoms because they are believed to play the role of glue for MI clusters. In AIMnSi, the Mn atoms are on the vertices of the Mackay icosahedron. The AI atoms are at the midpoints between the central hole and the Mn vertices (a sites), and at the midpoints of edges, shared by two Mn atoms on vertices (/3 sites), [1]. This cluster structure is crucial to understanding the structural stability and growth mechanism.
The second class of icosahedral quasicrystals is called the Frank-Kasper type (FK) or triacontahedron type. This name originates from the fact that quasicrystals of this type intimately relate to the famous Frank-Kasper phase of crystalline compounds. A typical example of the second class is A1LiCu. The constituent unit of the FK type quasicrystal is quite different from that of the MI type quasicrystal. In MI type quasicrystals, the atomic decoration of rhombohedra is not uniquely determined. In other words, the way of decorating rhombohedra is determined according to their environment. In FK type quasicrystals, the atomic decoration of prolate and oblate rhombohedra is determined almost uniquely, except that some disorder should be introduced to avoid shorter atomic distances [2]. Furthermore, the structure can
also be understood as duster packing and a constituent unit is a shell-structured triacontahedron of 44 atoms with a vacant center [2].
The above classification, MI or FK, is also made according to different ratios of atomic diameter (d) to quasilattice constant aR
d/aR=0.61 (MI) d/aR=0.57 (FK) (1)
and different electron-to-atom ratios
e/a = 1.6-1.8 (MI) e/a =2.0-2.2 (FK) (2)
Equations (1) show the difference in structures and eqns. (2) imply a possibility that the two are stabilized by different mechanisms [3].
The diffraction pattern of icosahedral quasicrystals can be indexed by six-dimensional indices and, therefore, the structure of icosahedral quasicrystals is specified with the help of a corresponding six-dimensional lattice. From the group theoretical viewpoint, there can be three different Bravais lattices in the six-dimensional space, i.e. primitive, face-center and body-center [4]. Both the AIMnSi and A1LiCu are primitive-type quasi- crystals, which means that the diffraction pattern of the atomic structure is determined by the six-dimen- sional primitive reciprocal lattice. Recently discovered stable quasicrystals AICuFe, AICuRu, A1PdMn etc. are face-center icosahedral quasicrystals, showing the ex- tinction rule of the f.c.c, icosahedral quasicrystals and e/a = 1.75. One other important discovery is the stable and high qualified decagonal quasicrystal A1CoNi, which is quasiperiodic on a plane and periodic along the remaining one direction. The constituent units are observed to be of two types by high resolution electron microscopy. The cluster constituents may be very im-
0921-5107/93/$6.00 © 1993-Elsevier Sequoia. All rights reserved
78 T. Fujiwara / Electronic structures in quasicrystals
portant for understanding the cohesive mechanism of quasicrystals and several physical properties.
Recently, several crystalline compounds have been discovered relating to quasicrystals. It is very common that many crystalline structures are stabilized at stoi- chiometry near the quasicrystalline stoichiometry. The local atomic environments of these crystalline structures may be very close to those of the corresponding quasi- crystalline structures, because the diffraction patterns are quite similar. In fact, the crystalline A1MnSi (a- AIMnSi) is constructed by a b.c.c, packing of MI clusters and glue atoms, and the crystalline A1LiCu (R-A1LiCu) is constructed by a b.c.c, packing of triacontahedron clusters. Crystalline Ala3Fen [5], AlaMn [6] are crystalline correspondences of decagonal quasicrystals and they have the cluster-packing structure. In a specific range of atomic concentration of A1CuFc, a phase transfor- mation from a high temperature random-tiling icosa- hedral phase to a low temperature crystalline phase has been observed. The crystalline compounds are called crystalline approximants, whose lattices can be specified generally by approximant rational numbers of the ir- rational number defining the quasiperiodicity of the quasiperiodic lattice. Two-dimensional decagonal and three-dimensional icosahedral lattices are specified by the golden ratio ~-= (v~+ 1)/2 = 1.6180... The golden ratio is expressed by a continued fraction as
~-= 1 + 1/~-= [1; 1, 1, 1, ...] (3)
The optimal approximant of the golden ratio ~" is given as a series
1/1, 2/1, 3/2, 5/3 . . . . , (4)
which is a truncation of the infinite continued fraction (3) at a certain finite step. These rational approximants specify the crystalline approximants.
The high qualified quasicrystalline samples now allow us to observe physical properties. Some physical prop- erties of crystalline compounds of close stoichiometry are very similar to those of quasicrystalline compounds, e& atomic density or electronic specific heats. High qualified samples of quasicrystals show an anomalous electric resistivity, much higher than those in crystalline and amorphous phases. For example, the electric re- sistivity in icosahedral A1LiCu (approximately 870/z12 cm) is six times larger than in crystals, though the electron density at the Fermi energy is almost the same [7]. The electric resistivity of A1CuRu has been reported to be 30 000 /zfl cm [8]. In MgGaZn or AILiCu, the resistivity changes according to the phase transformation under pressure or thermal treatment and that of the quasicrystalline phase shows the highest values [9]. The temperature dependence of the resistivity in normal metals is usually due to scattering by phonons and the resistivity increases with increasing temperature. The
resistivity in several quasicrystals decreases with in- creasing temperature. Magnetoresistance, the Hall coef- ficient and the thermoelectric power also show some anomaly [8]. Some crystalline approximants in the A1- TM class show just the same behavior of the transport properties: high resistivity, temperature dependence of resistivity, etc.
Fujiwara [10], and Fujiwara and Yokokawa [11] first calculated the electronic structure of several crystalline approximants, e.g. a-A1Mn, R-A1LiCu and AI13Fe 4. Re- cently, Hafner and Krajci calculated the density of states in approximants of A1MgZn [12].
In the present paper, we give a short review of our theoretical treatment of the electronic structures of icosahedral (both MI and FK types) and decagonal quasicrystals. The linear muffin-tin orbital method, with atomic sphere approximation (LMTO-ASA), was used throughout the calculations. Our recent calculation of the electronic transport properties is explained shortly.
2. Electronic structures
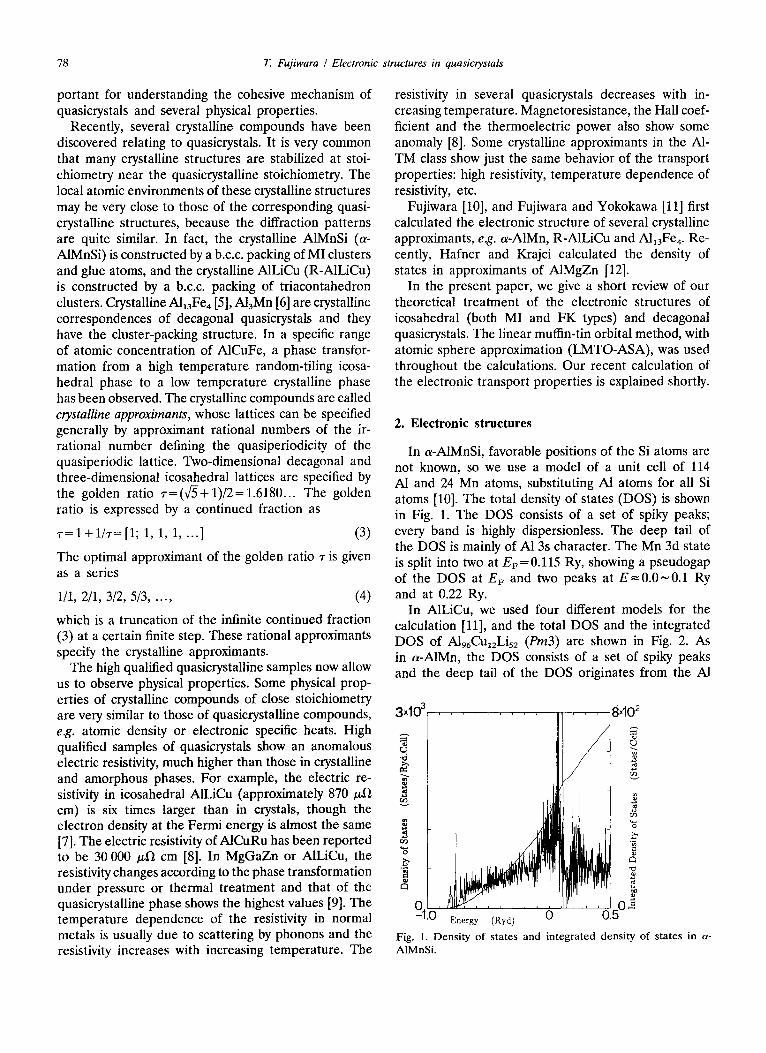
In a-A1MnSi, favorable positions of the Si atoms are not known, so we use a model of a unit cell of 114 A1 and 24 Mn atoms, substituting A1 atoms for all Si atoms [10]. The total density of states (DOS) is shown in Fig. 1. The DOS consists of a set of spiky peaks; every band is highly dispersionless. The deep tail of the DOS is mainly of A1 3s character. The Mn 3d state is split into two at EF=0.115 Ry, showing a pseudogap of the DOS at EF and two peaks at E=0.0~0 .1 Ry and at 0.22 Ry.
In A1LiCu, we used four different models for the calculation [11], and the total DOS and the integrated DOS of A]96Cu12Li52 (Pro3) are shown in Fig. 2. As in a-AIMn, the DOS consists of a set of spiky peaks and the deep tail of the DOS originates from the A1
3,,10 3 8x10 2
z
n ~
0-1.0 Energy (l~yd) 0 0.5 ON
Fig. 1. Density of states and integrated density of states in a- AIMnSi.

T. Fujiwara / Electronic structures in quasicrystals 79
3.5 . . . . . . . . . . . . . . . 10 3
i . . i x103
i _01 Energy ( R y ) 0 ' O ' ~ 0 1
Fig. 2. Density of states and integrated density of states in R- AILiCu, A196Cu~zLisz (sc Pro3).
i, oo 4oo
if i
Energy (Ry)
Fig. 3. Density of states and integrated density of states in AI~3Fe4.
and Li s-states. The Cu 3d state shows a narrow peak at E = -0.3 to E = -0.1 Ry. The DOS shows a clear opening of a pseudogap at the Fermi energy (EF = 0.051 Ry). Substituting AI atoms for Cu atoms, the position of the pseudogap does not change at all, but the Fermi energy shifts from the true minimum of the pseudogap according to the Cu concentration, showing that the Cu d-states play the role of donors of valence electrons.
The total DOS and the integrated DOS of All3Fe4 are shown in Fig. 3. The DOS consists of a set of spiky peaks and the deep tail of the DOS originates from AI s-states. The main peak at E--- -0.1 to E=0.0 Ry and extending to higher energies is due to Fe 3d states. The pseudogap opens at EF though the density is rather high. The remaining states in the pseudogap are mainly the Fe 3d states.
The common feature in these three DOS curves is that the Fermi wave number kF satisfies the relation
2kv=ap (5)
where Qp is the wavenumber of the strongest peaks in the structure factor or the diffraction pattern. This implies strong electron-quasilattice interaction and the Hume-Rothery mechanism for the stabilization of quasicrystals and crystalline approxdmants. Several ex- perimental observations of photoemission spectra show pseudogaps which are more pronounced in quasicrystals than in crystalline approximants.
3. Transport properties
High qualified A1-TM quasicrystals and their crys- talline approximants have high resistivities. They also show an anomalous temperature and magnetic field dependence of transport properties. The resistivity in normal metals increases with increasing temperatures because of phonon scattering, and the resistivity in semiconductors decreases. Because we have much in- formation on the electronic band structures, it may be interesting to calculate the transport properties based on the band structures and the Boltzmann theory.
The electric current j in the presence of external E and B fields can be written as
ja -- %t3Et3 + tr~rEoB~ + u~t3 Vo T
and the transport coefficients
2 n e27" o'.~=e T(--) = ~o ,,~.kv""(k)v'~(k)[ - - -
(6)
&] o'~t3 v
e3T 2
x[ (7)
e , ?--÷
where e.(k), 1-~o, ~-, T and f are the energy of the band n at k, the unit cell volume, the relaxation time, the temperature and the Fermi-Dirac function respectively, and v.(k)= Vke.(k)/h. The Hall coefficient and the ther- moelectric power are given by equations
(8) It must be mentioned that the expressions in eqn. (7) only include intraband scattering effects and not in- terband scattering effects.
80 T. Fujiwara / Electronic structures in quasicrystals
The preliminary calculations based on the details of the band structure of a-A1MnSi show that (n/m)~ = 0.5, approximately 0.1 per unit cell (defined in eqn. (7)). The value (n/m),~ does not synchronize with that of the DOS but has similar spiky structures. The value of the conductivity is not determined simply by the DOS but by various factors such as v,(k) on the Fermi surface. An essential source of a small value of (n/ m)~,,, is the existence of multiple electron and hole pockets on the Fermi surface. By assuming (n/m),~ = 0.1 per unit cell and ~- = 3.0 × 10- ~5 s with a unit cell volume 1"~ o = 2.0387 x 10 - ~ cm -3 of ~-AIMnSi, the conductivity is estimated as o~=41.4 12 -~ cm -~ or the resistivity p = 24 000 izl~ cm.
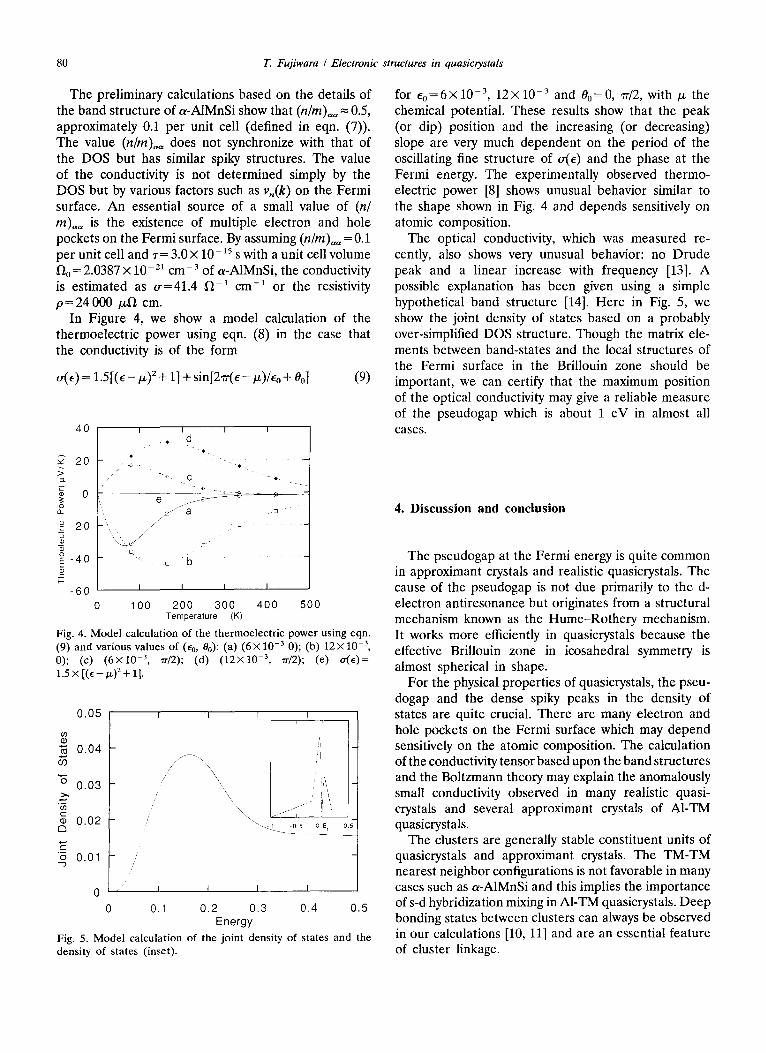
In Figure 4, we show a model calculation of the thermoelectric power using eqn. (8) in the case that the conductivity is of the form
o(e) --- 1.5[(e - /x) 2 + 1] + sin[2~r(e - t~)/Eo + O4 (9)
40
~ 20 :=L
%-- o
0 a .
-r_ -20
~ - 4 0 ,xz F--
-60
I I I I _. d
e- , - , @
. f
J 3
•4 c "b
J
I I I I
O0 200 300 400 Temperature (K)
500
Fig. 4. Model calculation of the thermoelectric power using eqn. (9) and various values of (E0, 00): (a) (6× 10 -3 0); (b) 12x 10 -3, 0); (c) (6x lO -3, 'rr/2); (d) (12X10 -3, rr/2); (e) ~r(E)= 1 . 5 X [ ( e - p . )2 + 1 ] .
0.05
03
0.04 co
"6 0.03
C
0.02 £3
C 0.01 /
/
j / /
I I I I J
,// ~ / ',\
;/' \ \ \ / \ \
/ i i\,
j . / I "/
-05 O=E 0.5
I I I I
0.1 0.2 0.3 0.4 0.5 Energy
Fig. 5. Model calculation of the joint density of states and the density of states (inset).
for Co=6×10 -3, 12x10 -3 and 00=0, 7r/2, with p~ the chemical potential. These results show that the peak (or dip) position and the increasing (or decreasing) slope are very much dependent on the period of the oscillating fine structure of a(e) and the phase at the Fermi energy. The experimentally observed thermo- electric power [8] shows unusual behavior similar to the shape shown in Fig. 4 and depends sensitively on atomic composition.
The optical conductivity, which was measured re- cently, also shows very unusual behavior: no Drude peak and a linear increase with frequency [13]. A possible explanation has been given using a simple hypothetical band structure [14]. Here in Fig. 5, we show the joint density of states based on a probably over-simplified DOS structure. Though the matrix ele- ments between band-states and the local structures of the Fermi surface in the Brillouin zone should be important, we can certify that the maximum position of the optical conductivity may give a reliable measure of the pseudogap which is about 1 eV in almost all cases.
4. Discussion and conclusion
The pseudogap at the Fermi energy is quite common in approximant crystals and realistic quasicrystals. The cause of the pseudogap is not due primarily to the d- electron antiresonance but originates from a structural mechanism known as the Hume-Rothery mechanism. It works more efficiently in quasicrystals because the effective Brillouin zone in icosahedral symmetry is almost spherical in shape.
For the physical properties of quasicrystals, the pseu- dogap and the dense spiky peaks in the density of states are quite crucial. There are many electron and hole pockets on the Fermi surface which may depend sensitively on the atomic composition. The calculation of the conductivity tensor based upon the band structures and the Boltzmann theory may explain the anomalously small conductivity observed in many realistic quasi- crystals and several approximant crystals of A1-TM quasicrystals.
The clusters are generally stable constituent units of quasicrystals and approximant crystals. The TM-TM nearest neighbor configurations is not favorable in many cases such as ~-A1MnSi and this implies the importance of s-d hybridization mixing in A1-TM quasicrystals. Deep bonding states between clusters can always be observed in our calculations [10, 11] and are an essential feature of cluster linkage.

T. Fujiwara / Electronic structures in quasicrystals 81
Acknowledgment
The author is thankful for useful discussions with J. Poon, J. Hafner, S. Burkov, F. Cyrot-Lackmann, C. Berger and D. Mayou.
References
1 V. Elser and C. L. Henley, Phys. Rev. Lett., 55 (1985) 2883. 2 C. L. Henley and V. Elser, Philos. Mag. B, 53 (1986) L59.
M. Audier, J. Pannetier, M. Leblanc, C. Janot, J.-M. Lang and B. Dubost, Physica B, 153 (1988) 136.
3 A. Inoue, in T. Fujiwara and T. Ogawa (eds.), Quasicrystals, Springer, Heidelberg, 1990.
4 D. S. Rokhsar, N. D. Mermin and D. C. Wright, Phys. Rev. B, 35 (1987) 5487.
5 C. L. Henley, J. Non-Cryst. Solids, 75 (1988) 91.
6 K. Hiraga, to be published in Philos. Mag. 7 K. Kimura, H. Iwahashi, T. Hashimoto, S. Takeuchi, U.
Mizutani, S. Ohashi and G. Itoh, J. Phys. Soc. Jpn., 58 (1989) 2472.
8 B. D. Biggs, S. J. Poon and N. D. Munirathnam, Phys. Rev. Lett., 65 (1990) 2700. T. Klein, C. Berger, D. Mayou and F. Cyrot-Lackmann, Phys. Rev. Lett., 66 (1991) 2907.
9 Y. Asayama, Y. Mori, M. Kobayashi, H. Kawamura, K. Kimura and S. Takeuchi, J. Phys. Soc. Jpn., 58 (1989) 2231. U. Mizutani, Y. Sakabe and T. Matsuda, J. Phys. Condens. Matter, 2 (1989) 6153.
10 T. Fujiwara, Phys. Rev. B, 40 (1989) 942. 11 T. Fujiwara and T. Yokokawa, Phys. Rev. Lea., 66 (1991)
333. 12 J. Hafner and M. Kxajci, Phys. Rev. Leg., 68 (1992) 2321. 13 C. C. Homes, T. Timusk, X. Wu, Z. Altounian, A. Sahnoune
and J. O. Str6m-Olsen, Phys. Rev. Lett., 67 (1991) 2694. 14 S. E. Burkov, T. Timusk and N. W. Ashcroft, private com-
munications.