electronic structure theory: this presentation will probably fundamentals to frontiers...
TRANSCRIPT

Electronic structure theory: Fundamentals to frontiers.
2. Density functional theory
MARTIN HEAD-GORDON, Department of Chemistry, University of California, and
Chemical Sciences Division, Lawrence Berkeley National Laboratory
Berkeley CA 94720, USA
This presentation will probably
involve audience discussion, which will create action items. Use PowerPoint to keep track of these action items during your presentation
• In Slide Show, click on the
right mouse button • Select “Meeting Minder” • Select the “Action Items” tab • Type in action items as they
come up • Click OK to dismiss this box This will automatically create
an Action Item slide at the end of your presentation with your points entered.

Outline
1. Basics
2. Limitations of standard functionals 3. Range-separated functionals

Branches of the family tree
• Wavefunction-based electronic structure theory: • Minimize the energy by varying the wavefunction • Tremendously complicated unknown function:
• Modeling the wavefunction yields “model chemistries”
• Density functional theory • The unknown is very simple: • Hohenberg-Kohn theorem guarantees that: • True functional is unknown and probably unknowable • Modeling the functional gives DFT model chemistries.
! = !
!r( )
! = !
!r1, !
r2 ,...,!rn( )
E = E !
!r( ){ }

A brief overview of density functional theory
• First Hohenberg-Kohn theorem (1965): • 1:1 mapping between ground state electron densities and Hamiltonians.
• Proof by contradiction: let H1 and H2 have the same ρ(r) • Use Ψ2 as trial function in H1 problem • Use Ψ1 as trial function in H2 problem
• Contradiction:
E1 !2{ } = !2 T̂ + V̂ ee !2 + v1"# dr > !1 T̂ + V̂ ee !1 + v1"# dr
!1 T̂ + V̂ ee !1 > !2 T̂ + V̂ ee !2
!2 T̂ + V̂ ee !2 > !1 T̂ + V̂ ee !1
E2 !1{ } = !1 T̂ + V̂ ee !1 + v2"# dr > !2 T̂ + V̂ ee !2 + v2"# dr
E1 !2{ } > E1
E2 !1{ } > E2

A brief overview of density functional theory
• First Hohenberg-Kohn theorem (1965): • 1:1 mapping between ground state electron densities and Hamiltonians. • Ground state energy E is determined directly from the Hamiltonian • Hence E is given in terms of the density, ρ(r).
• A formal construction exists for the exact functional, • Constrained search over all wavefunctions yielding ρ(r) (!!!)
• So, in practice the functional must be modeled. • Given a functional, and an external potential (nuclear field) ρ(r) is
found by minimizing over allowed densities.
E = E !
!r( ){ }

Construction of model density functionals
• To model kinetic, exchange, correlation functionals…
• 1A) Find a model problem where the functional can be obtained…
• H atom? Uniform electron gas?
• 1B) Assume a form for the functional and fix the parameters by…
• Known exact conditions (e.g. get model problems right) • Minimizing the errors on known data

• 2) Transfer the functional to problems of interest • Test, test, test…. • If validation is encouraging enough, predict…
• In one of your problems, you will extract the kinetic energy functional that solves the uniform electron gas problem. It is not much more difficult to extract the corresponding exchange functional.
• These are the main ingredients of the Thomas-Fermi model which is a Hohenberg-Kohn density functional.

Kohn-Sham density functional theory
• Largest (unknown) energy contribution is the kinetic energy. • No satisfactory kinetic energy functional yet exists.
• Kohn-Sham framework (a beautiful sidestep): • Use the kinetic energy of a non-interacting system with the
same electron density (a Hartree-Fock type wavefunction). • Leaves exchange and electron correlation (XC) to specify. • Kohn-Sham computational cost: similar to Hartree-Fock. • Still cheap enough to apply to large systems.

Modern Kohn-Sham density functionals
• Local density approximation (LDA): 1960’s, 1970’s • Functional depends only on the density at each point, ρ(r) • LDA overbinds as much as Hartree-Fock (mean field) underbinds!
• Generalized gradient approximations (GGA’s): 1988 • Functional depends on density ρ(r) and its gradients at each r • Greatly improved results! 4-6 kcal/mol error for BLYP, PBE etc.
• Exact exchange mixing (adiabatic connection): 1992 • Mix some Hartree-Fock exchange with GGA’s (Becke) • Best yet! 2-3 kcal/mol error for B3LYP

Classes of Kohn-Sham density functionals
• Local spin density approx
• Example: SVWN
• Generalized gradient approx
• Example: BLYP
• Hybrid density functionals • Wave function exchange • Example: B3LYP
223 atomization energies Mean abs errors (kcal/mol)
G3/99 test set
EXC = dr ! XC" # r( ){ }
EXC = dr ! XC" # r( ),$# r( ){ }
1966
1985
1993

Multiple choice questions….
• In Kohn-Sham DFT, which energy contribution is not strictly a functional of the electron density? – (a) electron-nuclear attraction – (b) exchange-correlation – (c) kinetic energy
• Which of the following properties is obeyed by B3LYP? – (a) variationality – (b) exact for the uniform electron gas – (c) exact for 1-electron systems – (d) size-consistency

Outline
1. Basics
2. Limitations of standard functionals 3. Range-separated functionals

Challenges for density functionals
• Accuracy: lack of systematic improvability confronts…
• (1) Limitations of the exchange functional
• Self-interaction
• (2) Limitations of the correlation functional
• London forces
• Strong correlations

B3LYP dissociation of H2+ (0.65Å to 3Å)
-80
-70
-60
-50
-40
-30
-20
-10
0
rela
tive
ener
gy (
kca
l/m
ol)
HF
B3LYP
3Å 0.65Å

B3LYP dissociation of H2+ (3Å to 13Å)
-60
-50
-40
-30
-20
-10
0
rela
tive
ener
gy (
kca
l/m
ol)
HF
B3LYP
3Å 13Å

Alkali halide dissociation curves
B3LYP
products have fractional charges -- due to electronegativity difference
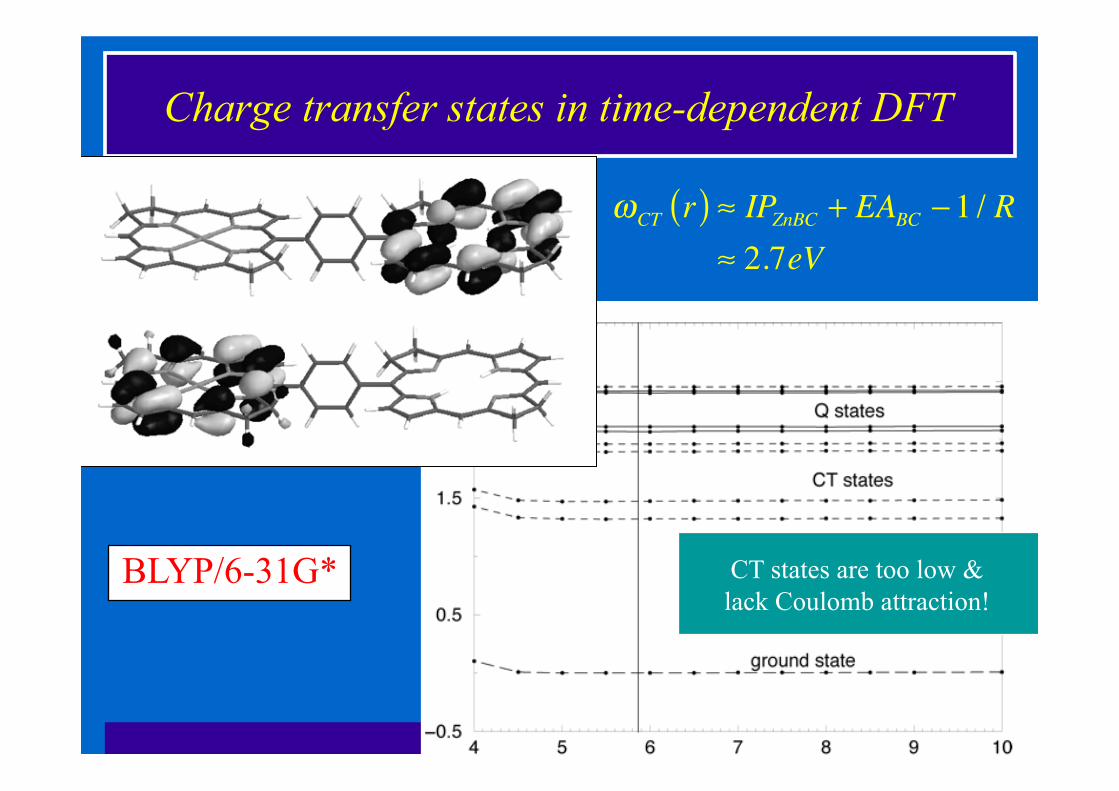
Charge transfer states in time-dependent DFT
CT states are too low & lack Coulomb attraction!
!CT r( ) " IPZnBC + EABC #1 / R" 2.7eV
BLYP/6-31G*

Importance of long-range exact exchange
• Ground state potential energy surfaces • Diatomic cation dissociation problem (H2
+, Ar2+, etc)
• Barrier height problems: generally too low • Electrons tend to be too delocalized
• Charge-transfer excited states • D-A Coulomb attraction is missing! • Magnitude of CT states is greatly underestimated • Contaminates the TDDFT spectrum of large molecules

Reducing self-interaction: Range-separation long-range exchange via erf(ωr)
• erf(ωr): long-range. Do exactly. • erfc(ωr): short-range. Do GGA.
• Key contributions: • Savin (1996): concept • Gill et al (1996): solved short-range LSDA exchange • Hirao et al (2001): long-range corrected (LC) functional • Handy, Gerber & Angyan, Scuseria, Perdew, Yang, …
• One can view this as justified within a generalized Kohn-Sham framework, or via adiabatic connection.
1r12
=erfc(! r12 )
r12
+erf (! r12 )
r12

Dispersive effects: e.g. supramolecular interactions
• fullerene-porphyrin dimer
• binding is 31 kcal/mol
• GGA’s give little or no binding energy
Y. Jung, MHG, Phys. Chem. Chem. Phys. 8, 2831 (2006)

Recovering Van der Waals interactions: Empirical dispersion (-D) corrections
• Additional non-local correlation energy contribution:
• C6i are atomic C6 factors; f damps at short-range
• Greatly improves dispersion-dominated interactions: • R. Ahlrichs, R. Penco, G. Scoles, Chem. Phys. 19, 119 (1977) • Q. Wu and W.T. Yang, J. Chem. Phys. 116, 515 (2002) • S. Grimme, J. Comput. Chem. 25, 1463 (2004); 27, 1787 (2006) •
• Not actually a density functional, but.... • Computationally free • Physically reasonable (but double counting problem)
Edisp = !C6
ij
Rij6
i< j
atoms
" fdamp Rij( ) C6ij = C6
iC6j fdamp = 1+ a(Rij / Rr )
!12"# $%!1

Recovering Van der Waals interactions: Double hybrid functionals (assigned paper)
• Gorling-Levy perturbation theory motivates mixing 2nd order perturbation theory (for correlation) with semilocal correlation functionals....
• Physically, PT2 includes non-local long-range correlation that is missing in semilocal functionals...
• But, there is again a double counting problem...

Strongly correlated molecules
Cope rearrangement Oxygen-evolving complex: Mn4O4

No easy answers for strong correlations....
• Either requires a tremendously powerful correlation functional, or, ...
• lies beyond generalized Kohn-Sham theory. For instance using a multi-configuration reference wave-function....
• While this is an important challenge, it is one that is not yet satisfactorily answered today...

Outline
1. Basics
2. Limitations of standard functionals 3. Range-separated functionals

Functional ingredients.... and parameters...
• B97 XC density functional: 12 linear parameters (M=4)
• Long-range exact exchange: 1 non-linear parameter (ω)
• Short-range exact exchange: 1 linear parameter (cX)
s! = "#! / #!4 /3EB97 = dr! "#
LSDA cj# f s#
2( )$% &'j
j=0
M
(
EXLR!HF = !
12
dr1" #i r1( )# j r1( ) dr2
erf $r12( )r12
" #i r2( )# j r2( )ij%
EXSR!HF = !
cX2
dr1" #i r1( )# j r1( ) dr2
erfc $r12( )r12
" #i r2( )# j r2( )ij%

2 types of non-local correlation corrections
• Empirical atom-atom dispersion (-D): 1 parameter (a)
• Similar to R. Ahlrichs, W.T. Yang, S. Grimme... • Computational cost is zero, but not a density functional
• Or: Double hybrid perturbation theory: 2 parameters
• Includes effect of unoccupied orbitals • Significantly more computational expense
Edisp = !C6
ij
Rij6
i< j
atoms
" fdamp Rij( ) C6ij = C6
iC6j fdamp = 1+ a(Rij / Rr )
!12"# $%!1
EPT 2 = cOSEOS(2) + cSSESS
(2)
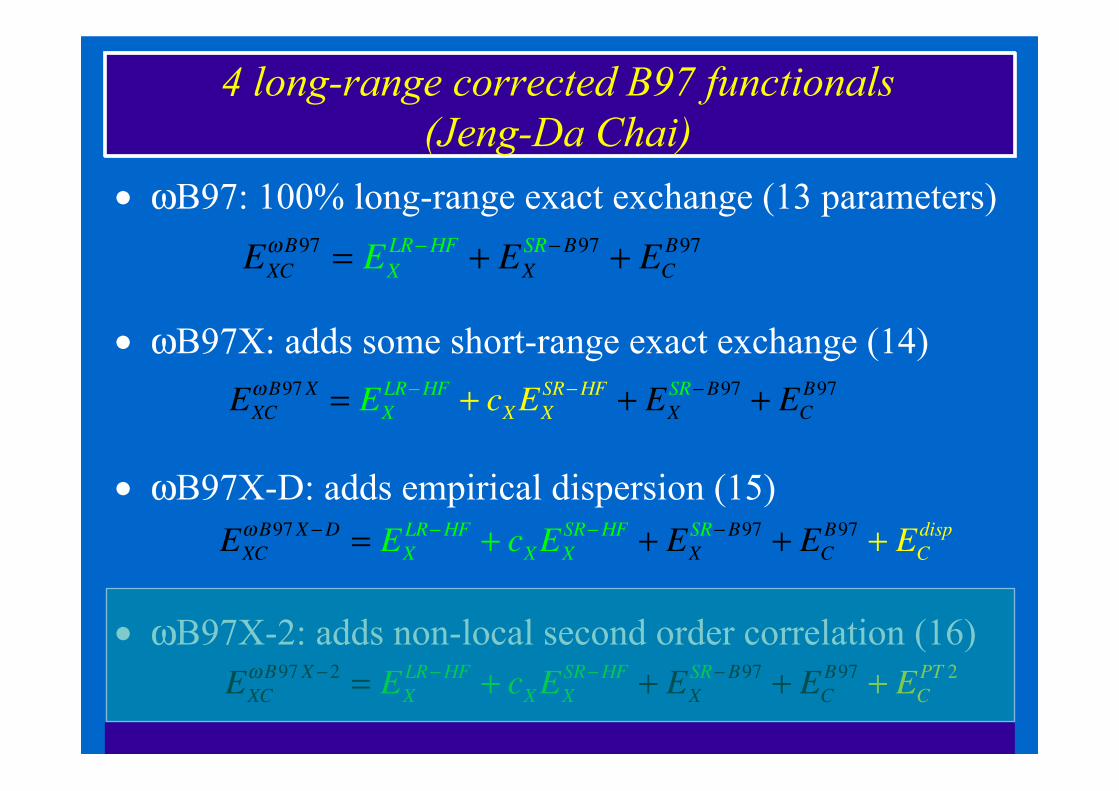
4 long-range corrected B97 functionals (Jeng-Da Chai)
• ωB97: 100% long-range exact exchange (13 parameters)
• ωB97X: adds some short-range exact exchange (14)
• ωB97X-D: adds empirical dispersion (15)
• ωB97X-2: adds non-local second order correlation (16)
EXC!B97 = EX
LR"HF + EXSR"B97 + EC
B97
EXC!B97X = EX
LR"HF + cXEXSR"HF + EX
SR"B97 + ECB97
EXC!B97X"2 = EX
LR"HF + cXEXSR"HF + EX
SR"B97 + ECB97 + EC
PT 2
EXC!B97X"D = EX
LR"HF + cXEXSR"HF + EX
SR"B97 + ECB97 + EC
disp

Why must these functionals be “trained”?
• All parameters should be determined self-consistently... subject to constraints that preserve the LDA limit – hence cannot adopt existing B97 values
• For ωB97 and ωB97X: – GGA parameters: short-range exchange; semi-local correlation – Range separator: compromise across problems of interest
• For ωB97X-D: – Additionally minimize the correlation double-counting error

Training set: 412 data points (Jeng-Da Chai)
• Bond-breaking energies: G3/99 dataset (296) – Curtiss, Raghavachari, Redfern, Pople, JCP 112, 7374 (2000)
• Barrier heights for simple chemical reactions (76) – Zhao, Truhlar et al, JPC A 108, 2715 (2005), 109, 2012 (2006)
• Non-covalent interactions (22) – Jurecka, Sponer, Cerny, Hobza, PCCP 8, 1985 (2006)
• Absolute atomic energies (18) – Chakravorty, Gwaltney, Davidson, Parpia, Fischer, PR A 47, 3649 (1993)

Comparison of optimizable functionals: All trained identically (Jeng-Da Chai)
HCTH*: 12 parameter GGA (like ωB97 with ω=0) B97*: 13 parameter hybrid (like ωB97X with ω=0)
ωB97: 13 parameter range-separated. ωopt=0.4
ωB97X: 14 parameter range-separated hybrid ωopt=0.3, cX=0.16 ωB97X-D: 15 parameter, with dispersion ωopt = 0.2, cX = 0.22
– All are exact for the uniform electron gas (constraints)...
– What is the value of range separation? And dispersion?

223 G3/99 atomization energies (Jeng-Da Chai)
Training set data
GGA hybrid range-separated family

38 non-hydrogen transfer barriers (Jeng-Da Chai)
Training set data
range-separated family hybrid GGA

22 intermolecular interactions (Jeng-Da Chai)
Training set data
range-separated family hybrid GGA

Test set data
Test performance for energies (Jeng-Da Chai)

Alanine tetrapeptide conformational energies
• Compare against basis set limit MP2 • 27 conformations • Calculations by Daniel Lambrecht

Conclusions and open issues
• For molecular problems, particularly where self-interaction is significant, range-separated functionals are a significant improvement over hybrids – ωB97, ωB97X, and ωB97X-D are widely useful – though significant weaknesses remain... – and further testing & comparison is desirable (e.g. vs M06)
• Challenges include – strong correlation (unresolved) – can self-interaction can be further reduced? – increased exact exchange degrades performance for metals*