electrochemical oscillations in the methanol oxidation on pt
TRANSCRIPT

Electrochemical oscillations in the methanol oxidation on Pt
Jaeyoung Lee *, Christian Eickes, Markus Eiswirth, Gerhard Ertl
Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, D-14195 Berlin, Germany
Received 22 June 2001
Abstract
Experimental observations of temporal dynamics in the electrocatalytic oxidation of methanol (CH3OH) on a polycrystalline
platinum electrode are reported. Hidden negative differential resistance (HNDR) and instabilities of the system were investigated by
means of an electrochemical impedance spectrum analysis and potential oscillations under galvanostatic control. This result could
be applied for a direct methanol fuel cell (DMFC) with higher energy efficiency. # 2002 Published by Elsevier Science Ltd.
Keywords: Methanol; Electro-oxidation; Hidden negative differential resistance; Galvanostatic potential oscillations
1. Introduction
There has been a resurgence of interest in the studies
of the oscillatory behaviour in electrochemical systems
like electrocatalysis, electrodeposition, and electro-dis-
solution [1�/15]. In electrocatalysis, the most important
system with regard to dynamic behaviour of small
organic molecules, is formic acid (HCOOH). Current
oscillations under potentiostatic conditions and poten-
tial oscillations under galvanostatic systems of HCOOH
on a Pt electrode were investigated [5�/7]. However, only
a few studies of potential oscillations during anodic
CH3OH oxidation were reported [8�/10]. Pavela re-
ported that diffusion of CH3OH might be the reason
for the potential oscillations [8]. In early 1960s, Buck
and Griffith [9] observed only small potential oscilla-
tions in the CH3OH oxidation. Krausa and Vielstich
studied the experimental conditions of potential oscilla-
tions during CH3OH oxidation on a Pt electrode and
they suggested a reaction mechanism resulting in
different types of oscillations [10]. The investigation of
the oxidation mechanism of CH3OH and HCOOH on a
Pt electrode has attracted attention, since the under-
standing of the oxidation of C1 fuels can be applied for
the development of clean energy techniques (fuel cell).
While the temporal as well as spatiotemporal behaviour
of HCOOH oxidation has been studied in detail bothexperimentally and theoretically, the mechanistic origin
of bistability and oscillations in CH3OH oxidation is not
well understood [7,11�/15].
In this work, we try to categorise the mechanistic
origin of the CH3OH oxidation by electrochemical
impedance spectroscopy (EIS) and the bifurcation
sequence under galvanostatic conditions. This study
could be also considered as a reference to understandthe electro-oxidation of C2 and C3 compounds [16,17]
and can give important clues to get higher power output
under oscillatory conditions.
2. Experimental
The schematic diagram of the principal three-elec-
trode arrangement is shown in Fig. 1. The electroche-
mical cell body consisted of a glass cylinder capped witha Teflon lid holding all electrodes. A smooth polycrys-
talline Pt ring of 34.5 mm inner diameter and 42.5 mm
outer diameter cut from a Pt plate of 0.1 mm thickness,
thus exhibiting a geometric surface area of 10 cm2, was
used as the working electrode (WE). All solutions were
prepared with ultrapure water (Millipore Milli-Q water,
18 MV �/cm). Prior to each experiment, the Pt ring WE
was first subjected to cleaning with C3H6O and purewater in an ultrasonic bath followed by chemical
cleaning in conc. H2SO4 (98%)�/30% H2O2 (1:1). Then,
a preliminary check of the voltammetric curve between* Corresponding author.
E-mail address: [email protected] (J. Lee).
Electrochimica Acta 47 (2002) 2297�/2301
www.elsevier.com/locate/electacta
0013-4686/02/$ - see front matter # 2002 Published by Elsevier Science Ltd.
PII: S 0 0 1 3 - 4 6 8 6 ( 0 2 ) 0 0 0 7 5 - 0
�/600 and �/700 mV of the WE with scan rate of 100
mV s�1 was performed in 0.5 M H2SO4 (suprapur,
MERCK) deaerated by N2 to verify initial cleanliness.
After this preliminary check of the Pt electrode, WE was
mounted in the Teflon lid of the cell which already held
both by Hg�/Hg2SO4, K2SO4 (saturated) referenceelectrode and by the platinized Pt counter electrode.
CH3OH (0.03 M, p.a. MERCK) in 0.1 M HClO4
(suprapur, MERCK) was used for CH3OH oxidation
and the electrolyte was extensively bubbled with N2
before each experiment. A nitrogen atmosphere was
maintained over the unstirred solutions during sweep
experiments.
Cyclic voltammogram, galvanostatic scan, and con-stant current method were executed with an in-house-
built potentiostat (Electronic Laboratory of Fritz-Ha-
ber-Institut) and a bi-potentiostat (EG&G, Model 366).
The impedance spectrum was measured by a frequency
response analyzer (S-5720, NF Electronic Instruments,
Yokohama). The frequency range from 10 kHz to 0.1
Hz was digitally recorded with 25 points per decade.
3. Results
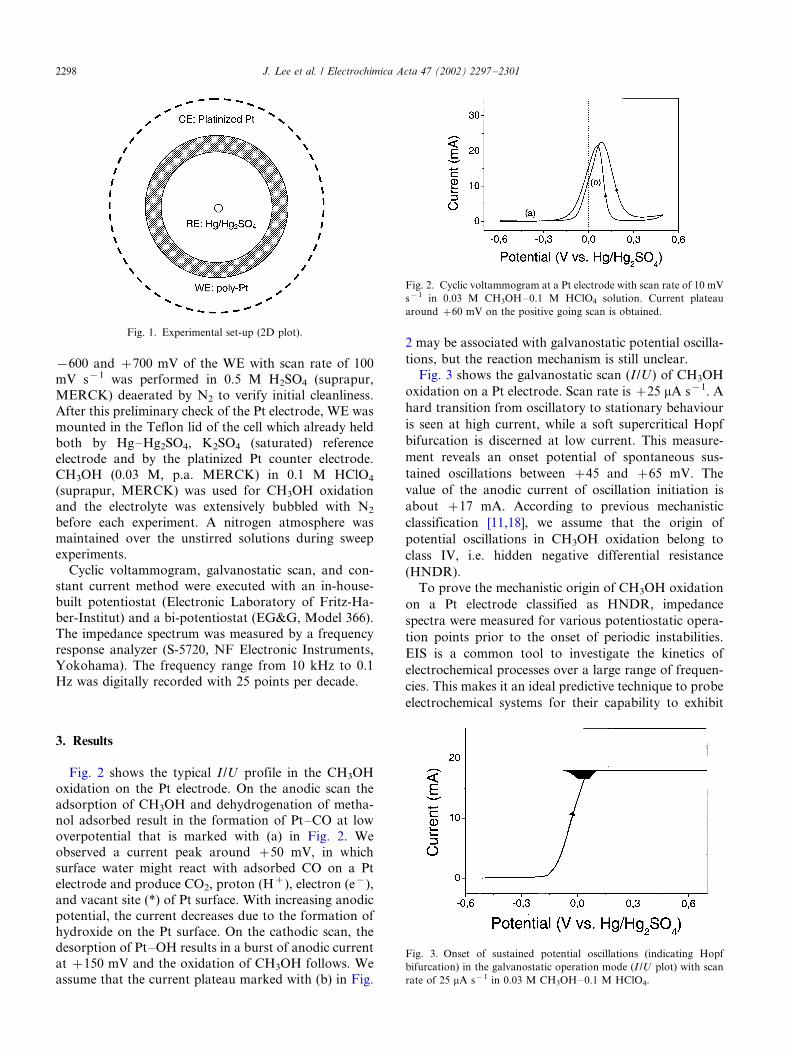
Fig. 2 shows the typical I /U profile in the CH3OH
oxidation on the Pt electrode. On the anodic scan the
adsorption of CH3OH and dehydrogenation of metha-nol adsorbed result in the formation of Pt�/CO at low
overpotential that is marked with (a) in Fig. 2. We
observed a current peak around �/50 mV, in which
surface water might react with adsorbed CO on a Pt
electrode and produce CO2, proton (H�), electron (e�),
and vacant site (*) of Pt surface. With increasing anodic
potential, the current decreases due to the formation of
hydroxide on the Pt surface. On the cathodic scan, thedesorption of Pt�/OH results in a burst of anodic current
at �/150 mV and the oxidation of CH3OH follows. We
assume that the current plateau marked with (b) in Fig.
2 may be associated with galvanostatic potential oscilla-
tions, but the reaction mechanism is still unclear.
Fig. 3 shows the galvanostatic scan (I /U ) of CH3OH
oxidation on a Pt electrode. Scan rate is �/25 mA s�1. A
hard transition from oscillatory to stationary behaviour
is seen at high current, while a soft supercritical Hopf
bifurcation is discerned at low current. This measure-
ment reveals an onset potential of spontaneous sus-
tained oscillations between �/45 and �/65 mV. The
value of the anodic current of oscillation initiation is
about �/17 mA. According to previous mechanistic
classification [11,18], we assume that the origin of
potential oscillations in CH3OH oxidation belong to
class IV, i.e. hidden negative differential resistance
(HNDR).
To prove the mechanistic origin of CH3OH oxidation
on a Pt electrode classified as HNDR, impedance
spectra were measured for various potentiostatic opera-
tion points prior to the onset of periodic instabilities.
EIS is a common tool to investigate the kinetics of
electrochemical processes over a large range of frequen-
cies. This makes it an ideal predictive technique to probe
electrochemical systems for their capability to exhibit
Fig. 1. Experimental set-up (2D plot).
Fig. 2. Cyclic voltammogram at a Pt electrode with scan rate of 10 mV
s�1 in 0.03 M CH3OH�/0.1 M HClO4 solution. Current plateau
around �/60 mV on the positive going scan is obtained.
Fig. 3. Onset of sustained potential oscillations (indicating Hopf
bifurcation) in the galvanostatic operation mode (I /U plot) with scan
rate of 25 mA s�1 in 0.03 M CH3OH�/0.1 M HClO4.
J. Lee et al. / Electrochimica Acta 47 (2002) 2297�/23012298

dynamical instabilities. Nyquist plots at three different
applied outer potentials (U ) and the Bode plot at �/65
mV are shown in Fig. 4. At outer potential of �/45 mV
(Fig. 4a), the impedance plot Z (v) exhibits a clockwise
capacitive�/inductive loop indicating dynamically stable
stationary operating points. However, the impedance
plot is drastically changed at higher overpotential �/65
mV, as shown in Fig. 4b. Z (v ) wraps around the origin
anti-clockwise. The intersection of the impedance and
the negative real axis is obtained at a finite frequency,
v0:/1 Hz. Then Z (v ) again exhibits positive real
resistances for very low frequencies. The detailed shape
of the Nyquist plot bears information on the mechan-
istic origin of the oscillatory behaviour, which can be
used to predict the dynamical stability of oscillatory
systems.
Fig. 5 is the Bode plot at �/65 mV and it clearly
demonstrates that the phase angle jumps from �/1808 to
�/1808 at frequency v0:/1 Hz, i.e. this drastic phase
change results in an anti-clockwise impedance profile in
the Nyquist plot (Fig. 4b and c).
Fig. 6a and b show the potential oscillations at
constant anodic current of �/14.5 mA at different times.
It was measured without applying any forced convectionsuch as stirring or nitrogen bubbling. Note that the
value of v0 in impedance spectra (see Fig. 4b and c), the
intersection where Im Z�/0 with Re Z B/0, agrees with
the initial oscillation frequency of 1 Hz obtained in Fig.
6. As discussed in previous work [18,19], the critical
potential, where the Nyquist plot blows up such that
approaches 9/�, marks the galvanostatic transition to
periodic behaviour, i.e. Hopf bifurcation. The value ofv0 represents the intrinsic frequency of the oscillation in
the electrocatalytic oxidation of CH3OH on a pure
polycrystalline Pt.
4. Discussion
According to previous work [4], stepwise dehydro-
genation mechanism for the oxidation of methanol is asfollows:
CH3OH�Pt 0 Pt�CH2OH�H��e� (1)
Pt�CH2OH�Pt 0 Pt2�CHOH�H��e� (2)
Pt2�CHOH�Pt 0 Pt3�COH�H��e� (3)
Pt3�COH 0 Pt(3�x)�CO�xPt�H��e� (4)
H2O�Pt 0 Pt�OHad�H��e� (5)
Pt(3�x)�CO�Pt�OHad0(4�x)Pt�CO2��H�
�e� (6)
After applying an anodic current the surface will be
covered by CH3OH residues with different oxidation
states. If reaction rates of dehydrogenation from Eqs.(1)�/(4) are slow, only few vacant Pt sites remain and
thus further dehydrogenation or oxidation of the
CH3OH residues is slowed down further. Therefore,
Fig. 4. Electrochemical impedance behaviour of methanol oxidation at a Pt electrode measured at three outer potentials of: (a) �/45; (b) �/65; and (c)
�/85 mV.
Fig. 5. The Bode plot at �/65 mV shows the phase shift as a function
of frequency. Experimental condition is same as in Fig. 2. Closed and
open circles indicate log jZ j and phase angle, respectively.
J. Lee et al. / Electrochimica Acta 47 (2002) 2297�/2301 2299

the adsorption of H2O must be considered (Eq. (5)) for
the complete oxidation of CH3OH as shown in Eq. (6),
since H2O is the source of oxygen. However, this
adsorption process also needs free Pt sites. Thus, the
potential rises until greater amounts of Pt�/OH are
formed and more free Pt sites are present by the
autocatalytic reaction (see Eq. (6)) of the oxidation ofPt�/CO and the conversion of the residues can proceed
with a higher reaction rate. During this reaction the
potential will decrease slowly until larger areas of the
surface are empty, then the potential decreases rapidly
for about 200 mV in a negative direction.
Of late, it was [11] suggested in a simple theoretical
calculation that higher power output of direct C1 fuel
cell might be possible by applying autonomous and/orexternally driven potential oscillations. This result is due
to smaller dissipation (energy loss) in the average
dissipation in periodic condition compared to the
stationary system. The detail theoretical explanation is
as follows.
Dissipation (D ) and efficiency (h ) are defined as D�/
dS /dt (S , entropy) and h�/Pout/Pin, respectively and we
normalise the efficiency like h�/1�/D ? according toD�/Pin�/Pout. Dissipation is time-dependent term.
�D��v
2p g2p=v
0
D(t) dt (period�2p=v) (7)
DD��D��Dstationary (8)
We can calculate average value of dissipation (�D�)
when the period is 2p /v . To increase the power
efficiency of direct C1 fuel cell, negative DD is wanted
in time-periodic processes.
In linear thermodynamics, DD is always bigger than
zero. But in nonlinear systems, the phase between fluxand force can vary over a wide range. If it is p (or �/p),
DD has maximum negative value, that is maximum
efficiency can be obtained by applying a periodic
perturbation which results in a phase of 1808 phase
between flux (current) and force (potential).
DD�kDU 2 v
2p g2p=v
0
cos vt cos(vt�8 ) dt
�1
2kDU 2 cos 8 (9)
8:0 0 DD�1
2kDU 2 (10a)
8:p 0 DD��1
2kDU 2 (10b)
where 8 is the phase between I and U ; �D�, averagevalue of dissipation when period is 2p /v . The required
perturbation period can be directly seen from the
impedance spectra.
It should be noted that the above thermodynamic
considerations only compare the performance of a given
electrode under stationary and periodic operation.
Certainly a crucial point, not addressed in this work,
is an improvement of the electrode kinetics, such as Ru-modification of a Pt electrode, which can reduce the
amount of CO poisoning [20].
5. Conclusions
EIS and the galvanostatic scan of CH3OH oxidation
on Pt exhibits a HNDR and a Hopf bifurcation at low
overpotential. Therefore, this oscillator belong to class
IV according to an earlier mechanistic classification
[11,18], as did HCOOH oxidation [5,7]. Comparison of
stationary conditions with oscillatory systems, higher
efficiency of direct C1 fuel cell is theoretically possible.Thus, further study will be carried out to get higher
power output by applying an external pulse with correct
frequency of a 1 Hz obtained by EIS technique.
Fig. 6. Potential oscillations at constant current of �/14.5 mA show oscillations frequency of 1 Hz. It is in good agreement with the result obtained in
Fig. 4b and c. (a) At initial oscillations time; and (b) at hard transition time.
J. Lee et al. / Electrochimica Acta 47 (2002) 2297�/23012300

Acknowledgements
Partial financial support by the Deutsche Forschungs-
gemeinschaft (DFG) through Sfb 555 is gratefullyacknowledged.
References
[1] T. Matsuda, Y. MuLouyama, H. Hommura, S. Yae, Y. Nakato,
J. Electrochem. Soc. 144 (1997) 2996.
[2] E.W. Bohannan, L.-Y. Huang, F.S. Miller, M.G. Shumsky, J.A.
Switzer, Langmuir 15 (1999) 813.
[3] D.T. Bowlin, A. Scheeline, A. Pearlstein, Electrochim. Acta 43
(1998) 417.
[4] V.S. Bagotsky, Yu.B. Vassiliev, O.A. Khazova, J. Electroanal.
Chem. 81 (1977) 229.
[5] J. Lee, P. Strasser, M. Eiswirth, G. Ertl, Electrochim. Acta 47
(2001) 501.
[6] W. Vielstich, Fuel Cells, Wiley, New York 1970.
[7] J. Lee, J. Christoph, P. Strasser, M. Eiswirth, G. Ertl, J. Chem.
Phys. 115 (2001) 1485.
[8] T.O. Pavela, Suomen Kemistilehti 30 (1957) 138.
[9] R.P. Buck, L.R. Griffith, J. Electrochem. Soc. 109 (1962) 1005.
[10] M. Krausa, W. Vielstich, J. Electroanal. Chem. 399 (1995) 7.
[11] P. Strasser, Ph.D. thesis, Freie Universitat Berlin, Berlin, 1999.
[12] C. Lamy, J.M. Leger, J. Clavilier, R. Parsons, J. Electroanal.
Chem. 150 (1983) 71.
[13] R.R. Adzic, W.E. O’Grady, S. Srinivasan, J. Electrochem. Soc.
128 (1981) 1913.
[14] J. Clavilier, C. Lamy, J.M. Leger, J. Electroanal. Chem. 125
(1981) 249.
[15] S. Sriramulu, T.D. Jarvi, E.M. Stuve, J. Electroananl. Chem. 467
(1999) 132.
[16] S. Chen, M. Schell, J. Electroanal. Chem. 478 (1999) 108.
[17] J.-W. Kim, S.-M. Park, J. Electrochem. Soc. 146 (1999) 1075.
[18] P. Strasser, M. Eiswirth, M.T.M. Koper, J. Electroanal. Chem.
478 (1999) 50.
[19] M.T.M. Koper, Adv. Chem. Phys. 92 (1996) 161.
[20] J. Lipkowski, P.N. Ross, Electrocatalysis, Wiley-VCH, New York
1998.
J. Lee et al. / Electrochimica Acta 47 (2002) 2297�/2301 2301