effective targeting of triple negative breast cancer cells...
TRANSCRIPT

Effective Targeting of Triple Negative Breast Cancer Cells by PF-4942847,
a Novel Oral Inhibitor of Heat Shock Protein 90
Pramod P. Mehta1, Pamela Whalen1, Sangita M. Baxi1, Pei-Pei Kung2, Shinji Yamazaki3,
and Min-Jean Yin1
1Oncology Research, 2Medicinal Chemistry, 3Pharmacokinetics, Dynamics and
Metabolism, Pfizer Worldwide Research and Development,
10724 Science Center Drive, San Diego, CA 92121
Corresponding author: Min-Jean Yin
Email: [email protected]
Tel: (858) 622-7438
Fax: (858) 526-4121
Running title: Targeting TNBCs by a Hsp90 inhibitor PF-4942847
Keywords: Hsp90, AKT, oral cancer drug, triple negative breast cancer
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

2
Abstract
Purpose: Triple negative breast cancer (TNBC) patients have poor prognoses and
survival outcomes such that the development of new targeted therapies is in strong
demand. Mechanisms associated with high proliferation and aggressive tumor
progression, such as PI3K/PTEN aberration, EGFR overexpression, and cell cycle up-
regulation, play important roles in TNBC. The molecular chaperone heat shock protein
90 kDa (Hsp90) is required for the conformational maturation and stability of a variety of
proteins in multiple pathways, such as EGFR, AKT, Raf, cdk4, etc. Therefore, an Hsp90
inhibitor may demonstrate therapeutic benefit in TNBC by targeting multiple pathways.
Experimental Design: The novel oral Hsp90 inhibitor PF-4942847 was characterized in
multiple in vitro and in vivo assays to determine its antitumor activity in TNBC cell lines.
In addition, the correlation of AKT degradation and Hsp70 induction in host peripheral
blood lymphocytes (PBLs) and xenograft tumors was determined.
Results: PF-4942847 induces degradation of multiple client proteins, cell cycle block,
apoptosis, and inhibits cell proliferation in TNBC lines, subsequently leading to tumor
growth inhibition in mouse xenograft models. The correlation of AKT degradation and
Hsp70 induction between PBLs and xenograft tumors reveals a differential modulation of
Hsp90 activity between host and tumor tissues, and suggests that AKT degradation in
PBLs may serve as a pharmacodynamic biomarker in future clinical development.
Conclusions: The novel oral Hsp90 inhibitor, PF-4942847, is a candidate for clinical
development in TNBC by collaboratively targeting multiple signaling pathways.
Additionally, AKT degradation in PBLs may serve as a biomarker in clinical
development.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

3
Translational Relevance
There is a strong need to develop new targeted therapies for triple negative breast cancer
(TNBC) patients. The work presented here shows that a novel oral Hsp90 inhibitor, PF-
4942847, exhibits in vitro and in vivo efficacy in multiple TNBC cell lines; these results
further suggest that PF-4942847 may be of clinical benefit in TNBC patients. The
correlation of AKT degradation between mouse PBLs and xenograft tumors was
determined in response to drug treatment and indicates that AKT degradation in patient
PBLs may have the potential to serve as a biomarker to predict drug modulation in
tumors.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

4
Introduction
Heat shock protein 90 kDa (Hsp90) is a molecular chaperone that regulates the
conformation, stability and activity of numerous key signaling proteins, including protein
kinases (e.g., C-Raf, AKT, ErbB family, Cdk4), steroid receptors (e.g., androgen receptor
and estrogen receptor), and transcription factors (e.g., HIF1α). These Hsp90 client
proteins are involved in multiple pathways of cell transformation and tumor progression;
therefore, targeting Hsp90 offers an opportunity to inhibit multiple pathways in cancer (1,
2). Several natural products, such as geldanamycin and radicicol, bind to Hsp90 at the
NH2-terminal ATP pocket and inhibit the ATPase activity of Hsp90 subsequently leading
to client protein degradation through ubiquitin ligase machinery (3). The geldanamycin
derivative 17-allylamino-17-demethoxy-geldanamycin (17-AAG), 17-DMAG, and IPI-
504 have been developed as potential therapeutics in a variety of clinical trials (4-6).
However, 17-AAG is poorly soluble and has low oral bioavailability, metabolism issues
and hepatotoxicity (7, 8). Because of the potential toxicity of geldanamycin derivatives,
specific small molecular weight Hsp90 inhibitors may be more effective clinical agents.
Several small molecular weight Hsp90 inhibitors, including SNX-5422, CNF2024, STA
9090, and AUY 922, are currently in clinical trials in various tumor types; it is too early
to differentiate these inhibitors from the geldanamycin derivatives in the clinical setting
(9-12).
Breast cancer is a heterogeneous disease for which there are a variety of
biological features, natural history, and treatment options. Gene expression profiling has
allowed us to classify breast cancers into five subtypes based on distinctive gene
expression signatures (13). These five subtypes are luminal A, luminal B, HER2
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

5
positive, basal-like, and normal-like breast cancers. Basal-like tumors are characterized
by the expression of genes that are specific for basal epithelial cells and involved in
cellular proliferation, suppression of apoptosis, cell migration, and cell invasion (13-15).
Basal-like breast cancers (BLBC) are often stained negative by immunochemistry for
estrogen receptor (ER), progesterone receptor (PR), and HER2; this BLBC subpopulation
are thus called triple negative breast cancers (TNBC). Although BLBC and TNBC share
numerous clinical and pathologic features, they are not identical (16, 17). In the majority
of cases, however, these two categories share similar clinical features, prognoses and
treatment options. For convenience, the term “TNBCs” will be used in this study to
collectively describe BLBC and TNBC cell lines and patient populations. Clinical
studies have shown that TNBCs are more aggressive and patients have poorer prognoses
than the other breast cancer subtypes (18, 19). Therefore, the development of targeted
therapies for TNBCs is clearly needed to help this patient population. More than 60% of
TNBCs express EGFR and this could serve as a prognostic marker for TNBC outcomes
(20, 21). Activation of the PI3K pathway either by frequent PTEN alteration, elevated
PIK3CA expression or oncogenic mutation has been identified in TNBCs and is also
associated with a poor prognosis (22-24). Hsp90 regulates multiple signal transduction
pathways by maintaining stability and activity of client proteins such as EGFR, AKT,
Raf, and Cdk4; therefore, targeting Hsp90 function may provide an opportunity to inhibit
tumor progression of TNBCs through EGFR, PI3K, and other proteins (25).
A series of 2-amino-5,7-dihydro-pyrrolo[3,4-d]pyrimidine-6-carboxylic acid
amide compounds were previously described as potent and specific Hsp90 inhibitors; the
detailed structure activity relationship (SAR) has also been reported (26). One of the
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

6
compounds from this series, PF-4942847, had a low Ki value (6 nM) and exhibited
selectivity in the Invitrogen Kinase Panel against 35 other measured kinases (<10%
inhibition at 1 μM) (26). PF-4942847 was characterized in this study to demonstrate in
vitro and in vivo antitumor activity in a variety of human TNBC cell lines. PF-4942847
induces AKT protein degradation, cell cycle block, apoptosis, and inhibits cell
proliferation in TNBC lines, subsequently leading to tumor growth inhibition of MDA-
MB-231 and MX-1 in mouse xenograft models. The correlation of AKT degradation and
Hsp70 induction between peripheral blood lymphocytes (PBLs) and xenograft tumor was
also determined and reveals a differential modulation of Hsp90 activity between host and
tumor tissues in response to drug treatment suggesting that AKT degradation in PBLs can
potentially be used to guide clinical studies for TNBC.
Materials and Methods Hsp90 inhibitor. PF-4942847 was synthesized as previously described (26). PF-
4942847 was dissolved in DMSO for in vitro cellular assays and formulated in 40%
PEG-400/60% saline (v/v) for animal studies.
Cell lines. All cell lines were purchased from the American Type Culture Collection and
cultured according to ATCC instructions. The gene mutation status of cell lines was
obtained from the Sanger COSMIC database: http://www.sanger.ac.uk/.
Immunoblotting. Cells and xenograft tumors were lysed in lysis buffer (50 mM Tris-
HCl, 0.5% NP-40, 0.5% Triton X-100, 150 mM NaCl, 1 mM Na3VO4, 1 mM NaF, and
protease inhibitor cocktail). Protein (50 μg) was resolved by SDS-PAGE and western
blots were performed with various antibodies (Actin from Sigma, all others from Cell
Signaling) to detect proteins of interest.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

7
Luminex and MesoScale assays. Cells (10,000/well) were seeded in a 96-well microtiter
plate and cultured overnight. PF-4942847 was added to each well at a top concentration
of 10,000 nM with a 3-fold serial dilution ending at 0.0169 nM. After 24 hours, cell
lysates were prepared and analyzed following the manufacturer’s instructions. The
Luminex 100 system (Upstate) was used for AKT protein measurement, and MesoScale
Discovery technology was used for Hsp70 protein measurement.
Cell cycle profiling. TNBC cells were seeded and cultured overnight prior to compound
treatment. Cells were harvested, fixed, stained with propidium iodide (PI), and analyzed
by flow cytometry. A total of 10,000 events were analyzed for each sample, and the
experiments were repeated at least three times.
Cell proliferation and caspase 3/7 assay. For cell proliferation assays, TNBC cells were
seeded at 3000 cells/well in a 96-well plate; PF-4942847 was added to each well as
described above and incubated for 72 hours, followed by addition of 250 μg/ml of
Resazurin (Sigma). After incubation for an additional 6 hours at 37oC, plates were
analyzed by a florescence reader. For the caspase 3/7 assay, cells were plated and
compound was added as described; the cells were assessed by the Caspase 3/7 Assay
(Promega) following the manufacturer’s instructions.
Animal studies. Six- to eight-week-old nu/nu athymic female mice and SCID female
mice were obtained from Jackson Labs; the mice were maintained in pressurized
ventilated caging at the Pfizer La Jolla animal facility. All studies were done in
compliance with Institutional Animal Care and Use Committees guidelines. Tumors
were established by injecting 2x106 cells suspended 1:1 (v/v) with reconstituted basement
membrane (Matrigel, BD Biosciences). For PK/PD studies, mice with established tumors
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

8
of ~400mm3 were randomized and treated with PF-4942847 QD for two days to establish
steady-state of drug exposure. For tumor growth inhibition studies, mice with established
tumors of ~150 mm3 were randomized and treated with vehicle, PF-4942847, or 17-
DMAG. Tumor dimensions were measured with vernier calipers, and tumor volumes
were calculated using this formula: π/6 x (larger diameter) x (smaller diameter)2. Tumor
growth inhibition percentage (TGI %) was calculated as 100 x (1-∆T/∆C). For the
survival study, dosing was stopped as indicated in the graph for all animals and
measurements were taken three times per week; animals were sacrificed and noted
“dead” when the tumor size reached 1500 mm3. For the tumor regression study, mice
with established tumors of ~300mm3 were randomized and treated with vehicle or PF-
4942847 three times per week.
Determination of PF-4942847 concentration in plasma. Plasma concentrations of PF-
4942847 were determined by liquid chromatography tandem mass spectrometry (LC-
MS/MS) following protein precipitation of plasma samples as previously described (27).
Statistical analysis. Prism 5.0 Software (GraphPad Software, San Diego, CA) was used
for statistical analysis and graph generation. Unless otherwise stated, error bars indicate
SD, and p values of <0.05 are denoted by an asterisk (*), p values of <0.005 are denoted
by two asterisks (**), and p values of <0.0005 are denoted by three asterisks (***). A t-
test was performed in the AKT degradation and Hsp70 induction experiments to compare
results from tumor versus PBLs, and the survival study was evaluated by one-way
ANOVA.
Results
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

9
PF-4942847 induces degradation of multiple Hsp90 client proteins in human triple
negative breast cancer cells
We first examined the cellular activity of PF-4942847 (Fig. S1) in a number of
human TNBC cell lines. PF-4942847 was added to MDA-MB-231 cells for multiple
treatment time points, and total protein and phosphorylation levels of AKT, EGFR, cMet,
B-Raf, C-Raf, ERK, and Hsp70 were determined by western blot. The effect of PF-
4942847 on protein degradation and reduction in phosphorylation of Hsp90 clients was
observed beginning at 6 hours after compound addition and was more definite after the
18 hour time point (Fig. 1A). We also observed a preferential loss of p-AKT versus total
AKT at 6 hr and 18 hr (Fig. 1A) suggesting that there may be other key clients degraded
by PF-4942847 that account for this activity. This is most likely due to PF-4942847
interference of Hsp90 maintenance of the stability and functional conformation of client
proteins. Similar results were observed with HCC-70 and MX-1 cells treated with PF-
4942847 (Fig. 1B). Additionally, PF-4942847 induced B-Raf protein degradation
selectively in the cells harboring a mutant B-Raf allele (MDA-MB-231 and MX-1) over
those with two wild type copies (HCC-70). This is consistent with previous studies that
B-Raf mutant protein, but not wild-type B-Raf, is degraded by Hsp90 inhibitors (28-30).
Additionally, the degradation of RTKs such as EGFR and cMet by PF-4972847 leads to
the suppression of ERK phosphorylation in TNBCs, while Hsp70 induction in treated
cells is due to the feedback mechanism in the heat shock protein machinery (31, 32).
Next, a panel of TNBC cell lines was used to investigate AKT protein degradation and
Hsp70 protein induction by plate-based assays to quantitatively determine the AKT
degradation IC50 and Hsp70 induction EC50 values. As shown in Fig. 1C and 1D and
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

10
summarized in Table I, PF-4942847 exhibits IC50 values ranging from 20 nM to 100 nM
in the AKT degradation assay and EC50 values ranging from 20 nM to 40 nM in the
Hsp70 induction assay in a variety of TNBC cell lines. 17-DMAG was included in the
AKT and Hsp70 assays and results indicate that PF-4942847 and 17-DMAG have
comparable effects on the inhibition of Hsp90 activity (Table I).
PF-4942847 blocks cell cycle progression and induces apoptosis resulting in inhibition
of cellular proliferation in TNBCs
PF-4942847 has the ability to induce protein degradation in the signaling
pathways of RTKs (including cMet, EGFR, Ras-Raf-ERK, and AKT); as such, it was
very likely that PF-4942847 would block cell cycle progression and induce cellular
apoptosis via targeting of multiple important signaling pathways in TNBCs. Therefore,
we next investigated cell cycle block and apoptosis activation by the treatment of PF-
4942847 in TNBCs. Results indicate that PF-4942847 induces cell cycle block at the
G2/M transition in both MDA-MB-231 and MX-1 cells (Fig. 2A and 2B, Fig. S2).
Moreover, high concentrations of PF-4942847 also increase the sub-G1 cell population
indicating an increase in apoptotic cells (Fig. 2A and 2B, Fig. S2). PF-4942847 also
induced cellular apoptosis in MDA-MB-231 and MX-1 cells as determined by the
induction of caspase 3/7 activity (Fig. 2C). We next measured the inhibitory activity of
PF-4942847 on cellular proliferation. Results presented in Fig. 2D and summarized in
Table I show that PF-4942847 effectively inhibits cell proliferation in TNBCs with IC50
values ranging from 20 nM to 60 nM. The inhibitory potency of PF-4942847 and 17-
DMAG is comparable. PF-4942847 was also evaluated in cell lines representative of
other breast cancer subtypes; these results demonstrate that PF-4942847 inhibited cellular
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

11
proliferation in a number of breast cancer cell lines with different backgrounds (Table
SI). This further suggests that the inhibition of multiple pathways by disruption of Hsp90
activity is effective in inducing anti-proliferative activity in a variety of breast cancers.
These results clearly indicate that PF-4942847 is a potent Hsp90 inhibitor that induces
protein degradation of multiple key signaling pathways, blocks cell cycle progression,
and induces apoptosis; these combined activities subsequently inhibit cellular
proliferation in TNBCs.
PF-4942847 induces Hsp90 client protein degradation and Hsp70 elevation in MDA-
MB-231 xenograft tumors.
We next determined the in vivo activity of PF-4942847 in mouse xenograft
studies. A MDA-MB-231 xenograft model was used to perform the in vivo experiments
and investigate the relationship between plasma drug exposure and modulation of Hsp90
activity by PF-4942847. In order to reach the steady-state of drug exposure, PF-4942847
was orally administered QD for two days to MDA-MB-231 tumor bearing mice, and free
unbound PF-4942847 was detected in mouse plasma in a dose- and time-dependent
manner after the second dose (Fig. 3A). The free unbound concentrations of PF-4942847
in mouse plasma at seven hours post-dose were 103 nM and 152 nM at 30 mg/kg and 50
mg/kg, respectively, both of which are above the in vitro AKT degradation IC50 value
(20 nM, Table I). Thus, it was determined that PF-4942847 was likely to inhibit Hsp90
activity in xenograft tumors. At the 30 mg/kg dose, MDA-MB-231 tumors were
dissected at various times to determine time-dependent modulation of Hsp90 activity
(Fig. 3B). Additionally, PF-4942847 was administered at several dose concentrations
QD for two days and tumors were dissected at four hours after the second dose to
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

12
determine dose-dependent modulation of Hsp90 activity. PF-4942847 induces total
protein degradation and reduction in phosphorylation of AKT, cMet, B-Raf, C-Raf,
EGFR, and ERK, and induces Hsp70 elevation in MDA-MB-231 xenograft tumors in a
dose-dependent and time-dependent manner as shown in Fig. 3B. AKT and Hsp70 levels
in tumor lysates were also determined by Luminex and MesoScale Discovery,
respectively, to quantitatively determine the changes (Fig. S3) and these results indicate
that AKT degradation and Hsp70 elevation in xenograft tumors treated by PF-4942847
are dose-dependent and time-dependent. Results from western blot analysis as well as
the Luminex and MesoScale assays indicate that AKT and Hsp70 did not fully recover to
normal levels by 24 hours post-treatment, even though the drug exposure at this time
point is below the detection limit (Fig. 3 and Fig. S3). Therefore, PF-4942847 modulates
Hsp90 activity to induce AKT degradation and Hsp70 elevation in MDA-MB-231
tumors, and the recovery of AKT and Hsp70 is delayed in tumors even when the drug has
been cleared from the plasma.
PF-4942847 triggers differential responses of AKT degradation and Hsp70 induction
between xenograft tumors and mouse peripheral blood lymphocytes.
In order to further aid future clinical development studies, we investigated the
relationship of AKT degradation and Hsp70 induction in response to PF-4942847
between xenograft tumors and mouse peripheral blood lymphocytes (PBLs) in order to
determine whether it was possible to correlate Hsp90 activity changes between tumors
and surrogate tissues. Both MDA-MB-231 tumors and mouse PBLs from the same
animal were collected to measure AKT (by Luminex) and Hsp70 levels (by MesoScale
Discovery technology) to determine Hsp90 activity changes induced by PF-4942847
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

13
between tumors and normal host tissue. Both Luminex and MesoScale methods had been
previously optimized to ensure that appropriate amounts of tumor and PBL samples were
analyzed and that all data points were within the linear range. Therefore, the Luminex
and MesoScale readout of units per μg of protein tested can be quantitatively compared
from sample to sample. At four hours, PF-4942847 induced similar degrees of AKT
degradation in both tumors and PBLs (Fig. 4A). Similarly, at the 30 mg/kg dose, we
observed comparable AKT degradation in tumors and PBLs (Fig. 4B). Although AKT
degradation might be slightly higher in tumors than PBLs, the differences were not
significant with the exception of the 72 hour time point as shown in Fig. 4B.
Interestingly, we observed a significant difference in Hsp70 induction fold change
between tumors and PBLs (Fig. 4C and D). At four hours, Hsp70 induction is dose-
dependent and significantly higher fold changes were observed in mouse PBLs than
tumors at the 50, 30 and 10 mg/kg doses while the differences between tumors and PBLs
were less pronounced at the 3 and 1 mg/kg doses as indicated in Fig. 4C. At the 30
mg/kg dose, we observed significant differential fold changes of Hsp70 induction in
tumors and PBLs up to the 120 hr time point (Fig. 4D). We further analyzed the data by
comparing the AKT and Hsp70 level per μg of protein (Fig. S4); the analysis revealed
that PF-4942847 induced dose- and time-dependent AKT degradation and Hsp70
induction in both tumors and PBLs. However, while the total amount of AKT per μg of
protein is similar between tumors and PBLs, the total amount of Hsp70 per μg of protein
is much less in PBLs than in tumors. Of note, recovery of AKT and Hsp70 in both the
tumor samples and PBLs was delayed even when the drug concentration was below the
lower limit of detection after 24 hours, and the recovery time appeared to be longer in
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

14
tumors than in PBLs in response to PF-4942847 treatment. Detailed percent inhibition of
AKT and fold increase of Hsp70 in tumors and PBLs are summarized in supplementary
table SII. Taken together, the data demonstrate that PF-4942847 inhibits Hsp90 activity
in both MDA-MB-231 tumors and host PBLs, and furthermore exhibits differential
modulation of AKT and Hsp70 between tumors and PBLs. Additionally, these results
reveal that the relative percent change of AKT in MDA-MB-231 tumors and mouse PBLs
is similar while the relative fold increase of Hsp70 in tumors is much less than those in
PBLs.
PF-4942847 inhibits in vivo tumor growth of human triple negative breast cancer cell
lines MDA-MB-231 and MX-1 in mice.
PF-4942847 has demonstrated in vitro and in vivo inhibition of Hsp90 activity in
a variety of TNBCs and in xenograft MDA-MB-231 tumors. We next determined
whether PF-4942847 was able to induce in vivo tumor growth inhibition (TGI) in mouse
xenograft models. MDA-MB-231 and MX-1 were implanted subcutaneously in nude or
SCID-bg mice, respectively, and treated orally once a day with vehicle or PF-4942847 to
perform the TGI experiments. PF-4942847 was well tolerated at 25 mg/kg in nude mice
and at 20 mg/kg in SCID-bg mice with minimal body weight loss (<5%). PF-4942847
induces 91% and 80% TGI, in MDA-MB-231 and MX-1 xenograft models, respectively
(Fig. 5A and B). Furthermore, the effective concentration (EC50) to induce AKT
degradation and tumor growth inhibition of MDA-MB-231 was calculated using a PK/PD
modeling approach as previously described and those results indicated that AKT
degradation (IC50~19 nM) was well correlated to tumor growth inhibition (EC50~12
nM) in MDA-MB-231 (27). Although 17-DMAG exhibits comparable cellular potency
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

15
as PF-4942847 in TNBC lines, 17-DMAG at the maximal tolerated dose (15 mg/kg, BID,
5 days/wk) only showed 56% tumor growth inhibition in the MDA-MB-231 model. This
is consistent with previous findings where 17-AAG and 17-DMAG have demonstrated
only moderate in vivo tumor growth inhibition activity in breast cancer models (33, 34).
Starting from day 29, MDA-MB-231 tumor-bearing mice were maintained without
further dosing, and tumor volume continued to be measured and animal survival was
assessed (Fig. 6A). Mice treated with PF-4942847 at 25 mg/kg had a longer survival
period compared to the vehicle-treated animals (p<0.005). Two mice in the 25 mg/kg
dose group remained tumor-free at day 120 as shown in Fig. 6A. In the MX-1 model,
PF-4942847 treatment at 20 mg/kg also increased survival of tumor-bearing mice
(p<0.05); however, there were no tumor-free animals at the end of the study, and MX-1
tumors progressed very quickly in the low dose groups such that there was not a
significant survival benefit over the vehicle control group (p>0.05) (Fig. 6B). This
suggests that PF-4942847 is critical in stopping tumor progression but not sufficient to
induce tumor cell death in the MX-1 tumor model. Based on our results where AKT
degradation and Hsp70 induction were sustained beyond PF-4942847 plasma clearance,
we also tested an alternative dosing schedule with PF-4942847. MDA-MB-231 tumor
bearing mice were treated with PF-4942847 orally three times a week (Monday,
Wednesday, and Friday) at 75 mg/kg without significant bodyweight loss; we observed
78% tumor regression in this study. At the end of study day 69, four mice were tumor-
free and the tumor measurements of the remaining mice all had a decreasing trend (Fig.
6C). Therefore, results indicate that PF-4942847 is a potent Hsp90 oral inhibitor that
inhibits tumor growth in triple negative breast cancer models.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

16
Discussion
Preclinical studies have demonstrated the notable sensitivity of HER2-
overexpressing breast tumors to Hsp90 inhibitors (9, 10). 17-AAG (Tanespimycin) is
being developed in the clinic for HER2 positive breast cancer and demonstrates a
moderate clinical response in combination with trastuzumab in patients (35). However,
aside from a study with the IV Hsp90 inhibitor PU-H71 (25), it has not been widely
reported that an Hsp90 inhibitor is effective in inhibition of triple negative breast cancer.
Therefore, we wanted to investigate the potential development opportunities for the oral
Hsp90 inhibitor PF-4942847 in the treatment of triple negative breast cancer. Our results
demonstrate that PF-4942847 inhibited cellular proliferation in a panel of TNBC lines,
eliminated tumor growth in MDA-MB-231 and MX-1 xenograft models, and induced
tumor regression of a MDA-MB-231 xenograft model in mice. In this study, PF-
4942847-induced modulation of AKT and Hsp70 between tumors and host PBLs was
also quantitatively determined and data suggest that AKT degradation in PBLs may be a
better biomarker than Hsp70 induction to predict tumor pharmacodynamic changes in
response to PF-4942847 treatment. Although a number of Hsp90 inhibitors have been
investigated in preclinical and clinical stages, there is not yet an approved drug targeting
Hsp90. PF-4942847 is an orally bioavailable Hsp90 inhibitor with superior physical and
pharmacokinetic properties that differentiate it from other intravenous inhibitors such as
PU-H71 and the geldenamycin derivatives. Additionally, PF-4942847 has demonstrated
excellent anti-tumor activity in preclinical TNBC models as described in this study. Our
results suggest a potential opportunity for PF-4942847 to be developed as an oral anti-
cancer drug in TNBC patients who need better treatment options.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

17
In the current study, mouse peripheral blood lymphocytes (PBLs) were isolated
from MDA-MB-231 tumor bearing mice and then subjected to Luminex and MSD
technology for quantitative detection of AKT and Hsp70, respectively. These new
approaches are superior to conventional western blot analysis as relatively small amounts
of protein are required for the assays and measurements are quantitative compared to
immunoblot analysis. Additionally, these assays can potentially be done with patient
samples that are easily and noninvasively obtained. Our data indicate that PF-4942847
affects Hsp90 function in both tumors and PBLs as AKT reduction and Hsp70 elevation
were observed in both sample sets. We also observed a differential pattern in the changes
of AKT and Hsp70 levels between MDA-MB-231 tumors and PBLs by PF-4942847
treatment. The relative fold change of Hsp70 induction by drug treatment is much higher
in PBLs than tumors, suggesting that the measurement of Hsp70 induction fold changes
in PBLs may over-estimate the changes of Hsp90 activity in tumors. In contrast, the
relative reduction percentages of AKT in tumors and PBLs are similar such that AKT
degradation in PBLs may be a better biomarker than Hsp70 induction to predict the
modulation of Hsp90 activity by PF-4942847 treatment in the clinic.
Our results with 17-DMAG of better in vitro cellular potency than in vivo tumor
growth inhibition are similar to previous studies and implicate that the in vivo toxicity of
geldanamycin derivatives may prevent administration of a dose high enough to reach
sufficient anti-tumor activity in xenograft tumor growth studies, and further suggest that
small molecular weight inhibitors such as PF-4942847 are likely to have optimal anti-
tumor activity due to better drug exposure and safety. Our observation of differential
impact on tumors and normal host tissues with treatment of PF-4942847 indicates that
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

18
there is a preferred therapeutic window to inhibit tumor cell progression while yet
maintaining normal cell function.
In MDA-MB-231 tumors, even when the drug had been cleared from mouse
plasma, the recovery of AKT and Hsp70 was not immediate but relatively slow to reach
basal levels. Therefore, we investigated whether an alternative dosing schedule with
higher dose concentration of PF-4942847 would have better anti-tumor activity by
stronger inhibiting of Hsp90 activity in tumors and allowing recovery of Hsp90 clients in
the normal tissues. Our results indicated that QOD dosing of PF-4942847 in MDA-MB-
231 model (Fig. 6C) induced tumor regression and was superior to QD dosing, even
though a 91% TGI was achieved in the latter. Traditionally, oral daily dosing has been
preferred in the clinic, however, our current data suggest that alternative dosing schedules
should be explored in the clinic to avoid systemic toxicity triggered by daily dosing of
Hsp90 inhibitors while maintaining a low level of client proteins in tumors.
In conclusion, we have characterized a novel oral Hsp90 specific inhibitor, PF-
4942847, and have demonstrated that PF-4942847 is an effective anti-tumor agent to
induce in vitro and in vivo anti-tumor activity in a panel of TNBC cells. Our data suggest
that PF-4942847 is a potential candidate for clinical development to address the unmet
need for therapy targeting triple negative breast cancer. Moreover, the biomarker studies
to determine AKT and Hsp70 changes in tumors and PBLs provide preliminary evidence
that AKT degradation in PBLs is a feasible biomarker approach to evaluate target
modulation in tumors.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

19
Acknowledgements
We thank Gerrit Los, Tod Smeal, Michael R. Gehring, and Martin Wythes for
their support and comments on this study; Leslie Nguyen, Sylvia Vekich, and Andrea
Shen for their help on the determination of PF-4942847 plasma concentration in mice;
and Dawn Nowlin for her insightful comments for quantitative data analysis.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

20
References:
1. Beliakoff J, Whitesell L. Hsp90: an emerging target for breast cancer therapy.
Anticancer Drugs. 2004;15:651-62.
2. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev
Cancer. 2005;5:761-72.
3. Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Structural
basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics
radicicol and geldanamycin. J Med Chem. 1999;42:260-6.
4. Banerji U, O'Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, et al. Phase I
pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-
demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol.
2005;23:4152-61.
5. Kaur G, Belotti D, Burger AM, Fisher-Nielson K, Borsotti P, Riccardi E, et al.
Antiangiogenic properties of 17-(dimethylaminoethylamino)-17-
demethoxygeldanamycin: an orally bioavailable heat shock protein 90 modulator. Clin
Cancer Res. 2004;10:4813-21.
6. Ge J, Normant E, Porter JR, Ali JA, Dembski MS, Gao Y, et al. Design, synthesis,
and biological evaluation of hydroquinone derivatives of 17-amino-17-
demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90. J Med Chem.
2006;49:4606-15.
7. Egorin MJ, Zuhowski EG, Rosen DM, Sentz DL, Covey JM, Eiseman JL. Plasma
pharmacokinetics and tissue distribution of 17-(allylamino)-17-demethoxygeldanamycin
(NSC 330507) in CD2F1 mice1. Cancer Chemother Pharmacol. 2001;47:291-302.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

21
8. Guo W, Reigan P, Siegel D, Zirrolli J, Gustafson D, Ross D. Formation of 17-
allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:quinone
oxidoreductase 1: role of 17-AAG hydroquinone in heat shock protein 90 inhibition.
Cancer Res. 2005;65:10006-15.
9. Chandarlapaty S, Sawai A, Ye Q, Scott A, Silinski M, Huang K, et al. SNX2112,
a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER
kinase-dependent cancers. Clin Cancer Res. 2008;14:240-8.
10. Jensen MR, Schoepfer J, Radimerski T, Massey A, Guy CT, Brueggen J, et al.
NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in
preclinical breast cancer models. Breast Cancer Res. 2008;10:R33.
11. Wang Y, Trepel JB, Neckers LM, Giaccone G. STA-9090, a small-molecule
Hsp90 inhibitor for the potential treatment of cancer. Curr Opin Investig Drugs.
2010;11:1466-76.
12. Lundgren K, Zhang H, Brekken J, Huser N, Powell RE, Timple N, et al. BIIB021,
an orally available, fully synthetic small-molecule inhibitor of the heat shock protein
Hsp90. Mol Cancer Ther. 2009;8:921-9.
13. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al.
Molecular portraits of human breast tumours. Nature. 2000;406:747-52.
14. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene
expression patterns of breast carcinomas distinguish tumor subclasses with clinical
implications. Proc Natl Acad Sci U S A. 2001;98:10869-74.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

22
15. Kang SP, Martel M, Harris LN. Triple negative breast cancer: current
understanding of biology and treatment options. Curr Opin Obstet Gynecol. 2008;20:40-
6.
16. Bidard FC, Conforti R, Boulet T, Michiels S, Delaloge S, Andre F. Does triple-
negative phenotype accurately identify basal-like tumour? An immunohistochemical
analysis based on 143 'triple-negative' breast cancers. Ann Oncol. 2007;18:1285-6.
17. Rakha EA, Ellis IO. Triple-negative/basal-like breast cancer: review. Pathology.
2009;41:40-7.
18. Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race,
breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA.
2006;295:2492-502.
19. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al.
Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer
Res. 2007;13:4429-34.
20. Arnes JB, Begin LR, Stefansson I, Brunet JS, Nielsen TO, Foulkes WD, et al.
Expression of epidermal growth factor receptor in relation to BRCA1 status, basal-like
markers and prognosis in breast cancer. J Clin Pathol. 2009;62:139-46.
21. Nalwoga H, Arnes JB, Wabinga H, Akslen LA. Expression of EGFR and c-kit is
associated with the basal-like phenotype in breast carcinomas of African women.
APMIS. 2008;116:515-25.
22. Aleskandarany MA, Rakha EA, Ahmed MA, Powe DG, Paish EC, Macmillan
RD, et al. PIK3CA expression in invasive breast cancer: a biomarker of poor prognosis.
Breast Cancer Res Treat. 2009.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

23
23. Lopez-Knowles E, O'Toole SA, McNeil CM, Millar EK, Qiu MR, Crea P, et al.
PI3K pathway activation in breast cancer is associated with the basal-like phenotype and
cancer-specific mortality. Int J Cancer. 2010;126:1121-31.
24. Marty B, Maire V, Gravier E, Rigaill G, Vincent-Salomon A, Kappler M, et al.
Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway
in basal-like breast cancer cells. Breast Cancer Res. 2008;10:R101.
25. Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, et al.
Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete
responses in triple-negative breast cancer models. Proc Natl Acad Sci U S A.
2009;106:8368-73.
26. Zehnder L, Bennett M, Meng J, Huang B, Ninkovic S, Wang F, et al.
Optimization of Potent, Selective, and Orally Bioavailable Pyrrolodinopyrimidine-
Containing Inhibitors of Heat Shock Protein 90. Identification of Development Candidate
2-Amino-4-{4-chloro-2-[2-(4-fluoro-1H-pyrazol-1-yl)ethoxy]-6-methylphenyl} -N-(2,2-
difluoropropyl)-5,7-dihydro-6H-pyrrolo[3,4-d]pyrimidine-6-carboxam ide. J Med Chem.
2011;54:3368-85.
27. Yamazaki S, Nguyen L, Vekich S, Shen A, Yin MJ, Mehta PP, et al.
Pharmacokinetic-Pharmacodynamic Modeling of Biomarker Response and Tumor
Growth Inhibition to an Orally Available Heat Shock Protein 90 Inhibitor in Human
Tumor Xenograft Mouse Models. J Pharmacol Exp Ther. 2011;published ahead of proint
June 16.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

24
28. Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, et al. V600E B-
Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90
inhibitors. Proc Natl Acad Sci U S A. 2006;103:57-62.
29. da Rocha Dias S, Friedlos F, Light Y, Springer C, Workman P, Marais R.
Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-
allylamino-17-demethoxygeldanamycin. Cancer Res. 2005;65:10686-91.
30. Mehta PP, Kung PP, Yamazaki S, Walls M, Shen A, Nguyen L, et al. A novel
class of specific Hsp90 small molecule inhibitors demonstrate in vitro and in vivo anti-
tumor activity in human melanoma cells. Cancer Lett. 2011;300:30-9.
31. Wegele H, Muller L, Buchner J. Hsp70 and Hsp90--a relay team for protein
folding. Rev Physiol Biochem Pharmacol. 2004;151:1-44.
32. Wegele H, Wandinger SK, Schmid AB, Reinstein J, Buchner J. Substrate transfer
from the chaperone Hsp70 to Hsp90. J Mol Biol. 2006;356:802-11.
33. Beliakoff J, Bagatell R, Paine-Murrieta G, Taylor CW, Lykkesfeldt AE, Whitesell
L. Hormone-refractory breast cancer remains sensitive to the antitumor activity of heat
shock protein 90 inhibitors. Clin Cancer Res. 2003;9:4961-71.
34. Hollingshead M, Alley M, Burger AM, Borgel S, Pacula-Cox C, Fiebig HH, et al.
In vivo antitumor efficacy of 17-DMAG (17-dimethylaminoethylamino-17-
demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative.
Cancer Chemother Pharmacol. 2005;56:115-25.
35. Modi S, Stopeck AT, Linden HM, Solit DB, Chandarlapaty S, Rosen N, et al.
HSP90 Inhibition is Effective in Breast Cancer: A Phase 2 Trial of Tanespimycin
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

25
(17AAG) plus Trastuzumab in Patients with HER2-Positive Metastatic Breast Cancer
Progressing on Trastuzumab. Clin Cancer Res. 2011.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

26
Figure Legends
Figure 1: PF-4942847 induces Hsp90 client protein degradation. A, MDA-MB-231
cells were treated with DMSO (0), 0.1 μM or 1 μM PF-4942847 for 2, 6, 18, 24, and 48
hours. Cell lysates were subject to western blot analysis to detect changes in total protein
or phosphorylation levels of EGFR, p-EGFR, cMet, p-cMet, AKT, p-AKT, B-Raf, C-Raf,
p-ERK and Hsp70. B, MDA-MB-231, HCC-70, and MX-1 were treated with DMSO (0),
0.1 μM or 1 μM PF-4942847 for 24 hours. Western blot analysis was performed to
detect changes in Hsp90 client proteins. Cells were cultured in 96-well microtiter plates
and treated with increasing concentrations of PF-4942847 for 24 hours; C, AKT
degradation was measured by Luminex, and D, Hsp70 induction was determined by
MesoScale Discovery.
Figure 2: PF-4942847 blocks cell cycle progression and induces apoptosis resulting
in inhibition of proliferation of TNBC cell lines. MDA-MB-231 (A) or MX-1 (B) cells
were treated with compounds for 24, 48, or 72 hours, fixed and stained with PI, and
analyzed by flow cytometry. The percentages of the cell population in each cell cycle
phase were as indicated. C, cells were treated with compound for 24 or 48 hours, then
analyzed by caspase 3/7 assay to determine caspase activity. D, cells were cultured and
treated with compound for 72 hours prior to utilizing the resazurin to determine inhibition
of cell proliferation.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

27
Figure 3: PF-4942847 triggers Hsp90 client protein degradation in MDA-MB-231
xenograft tumors. A, PF-4942847 was administered orally QD for 2 days to MDA-MB-
231 tumor bearing mice at 10, 30, and 50 mg/kg; plasma concentration of PF-4942847
was determined and plotted at select time points. B, MDA-MB-231 tumor bearing mice
were treated QD for 2 days with PF-4942847 at 30 mg/kg, and tumors were harvested at
4, 7, and 24 hours after the second dose to determine time-dependent protein changes.
Or, mice were treated QD for 2 days with PF-4942847 at 10, 30, and 50 mg/kg, and
tumors were harvested at four hours after the second dose to determine dose-dependent
protein changes. Tumor lysates were subject to western blot analysis for various Hsp90
client proteins as indicated.
Figure 4: PF-4942847 induces a differential modulation of AKT and Hsp70 between
MDA-MB-231 tumors and mouse peripheral blood leukocytes (PBLs). Mouse PBLs
from MDA-MB-231 bearing animals were collected and analyzed for AKT degradation
(A and B) and Hsp70 induction (C and D) after treatment with PF-4942847. A t-test was
performed to compare each tumor versus PBLs paired sample, p<0.05 is denoted by one
asterisk (*), p<0.005 by two asterisks (**), and p<0.0005 by three asterisks (***).
Figure 5: PF-4942847 demonstrates anti-tumor activity in MDA-MB-231 and MX-1
xenograft models. MDA-MB-231 (A) or MX-1 (B) cells were implanted into nude and
SCID-bg mice, respectively. When the tumors reached ~150 mm3, tumor-bearing mice
were randomized to various groups and treated with vehicle or various doses of PF-
4942847 as indicated. Tumors were measured three times/week, and tumor sizes were
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

28
recorded and plotted against study days. Tumor growth inhibition by PF-4942847 was
calculated as described. Tumor growth inhibition by 17-DMAG in the MDA-MB-231
xenograft model was also performed (C).
Figure 6: PF-4942847 induces tumor regression in the MDA-MB-231 xenograft
model. MDA-MB-231 (A) or MX-1 (B) tumor bearing mice were treated with various
dose concentrations of PF-4942847. When the average tumor size in the vehicle group
was above 1500mm3, mice were taken down from the study, and PF-4942847 treatment
was stopped for all other groups to perform the survival study. The last dosing days are
as indicated in each graph. One-way ANOVA analysis was performed to compare each
group to the vehicle group, p values<0.05 are indicated by one asterisk (*) and p values
<0.005 are indicated by two asterisks in the graph (**). MDA-MB-231 (C) cells were
implanted into nude mice and allowed to reach ~300 mm3. Once tumors reached
sufficient size, mice were dosed with PF-4942847 three times per week at 75 mg/kg PO
in order to examine whether PF-4942847 would induce tumor regression.
Table I: Summary of IC50 values for AKT degradation, Hsp70 induction, and cell
proliferation inhibition by PF-4942847 and 17-DMAG treatment in a panel of TNBCs.
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

Figure 1A.
C. AKT degradation
10-1 100 101 102 103 1040
20406080
100120
MDA-MB-231
MX-1HCC-70
PF-4942847 Conc. (nM)
% o
f con
trol
D.
PF-4942847 (µM) 0 0.1 1MDA-MB-231 HCC-70 MX-1
EGFR
cMet
Hsp70
B-Raf
AKT
C-RafpERK
Actin
B.0 0.1 1 0 0.1 1
p-EGFR
p-cMet
p-AKT
PF-4942847 (µM)0 0.1 1
Hsp70 induction
10-1 100 101 102 103 1040
2
4
6
8
MDA-MB-231HCC-70MX-1
PF-4942847 Conc. (nM)
Indu
ctio
n fo
ld
MDA-MB-231 2 hr0 0.1 1
6 hr0 0.1 1
18 hr0 0.1 1
24 hr 48 hr0 0.1 1
EGFR
cMet
Hsp70
B-Raf
AKT
C-RafpERK
Actin
p-EGFR
p-cMet
p-AKT
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

Figure 2
C
D Cell proliferation inhibition
10-1 100 101 102 103 1040
20
40
60
80
100
120MDA-MB-231HCC-70MX-1
PF-4942847 conc. (nM)
% o
f con
trol
BA
DMSO
PF-847 0.1uM
PF-847 1uMDMSO
PF-847 0.1uM
PF-847 1uMDMSO
PF-847 0.1uM
PF-847 1uM0
25
50
75
100 subG1G1/G0SG2/M
24hr 48hr 72hrMDA-MB-231
% o
f Cel
ls
DMSO
PF-847 0.1uM
PF-847 1uMDMSO
PF-847 0.1uM
PF-847 1uMDMSO
PF-847 0.1uM
PF-847 1uM0
25
50
75
10024hr 48hr 72hr
subG1G1/G0SG2/M
MX-1
% o
f Cel
ls
Caspase 3/7 activity
24hrs 48hrs 24hrs 48hrs0
1
2
3
4PF-847 0.1uMPF-847 1uMPaclitaxol 1uM
MX-1 MDA-MB-231
Act
ivat
ion
fold
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

Hsp70
EGFR
B-raf
AKT
cMet
C-Raf
pERK
Figure 3.A
B ________ ________ ________Vehicle________ 4hr 7hr 24hr
Actin
Vehicle ________ ______ ________________ 50mg/kg 30mg/kg 10mg/kg
p-EGFR
p-cMet
p-AKT
PF-4942847 conc. in plasma
01 4 7 240
100
200
300
400
10mg/kg30mg/kg50mg/kg
post dose time (hr)
Unb
ound
dru
g co
nc. (
nM)
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

Figure 4
A. B.
C. D.
*
*** **
AKT degradation at 4hr
50mg/k
g
30mg/k
g
10mg/k
g
3mg/k
g
1mg/k
g0
20
40
60
80
100
120 TumorPBL
% o
f con
trol
AKT degradation at 30 mg/kg
4hr 7hr24hr
48hr72hr
96hr120hr
144hr0
20
40
60
80
100
120 TumorPBL
% of
contr
olHsp70 induction at 4hr
50mg/k
g
30mg/k
g
10mg/k
g
3mg/k
g
1mg/k
g0
10
20
30
40TumorPBL
Indu
ctio
n fo
ld
Hsp70 induction at 30 mg/kg
4hr
7hr
24hr
48hr
72hr
96hr
120h
r14
4hr
048
12162024 Tumor
PBL
Indu
ctio
n fo
ld****** *** ***
***
*** ***
***
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

Figure 5.A.
PF-4942847 in MDA-MB-231
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 300
200
400
600
800
1000
1200
1400
1600
1800vehicle
25 mg/kg10 mg/kg3 mg/kg1 mg/kg
PO, QDn=10/group
91%
76%
64%
47%
Study Day (post tumor implant)
Tum
or v
olum
e (m
m3 )
Dosing
B.
17-DMAG in MDA-MB231
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 300
100
200
300
400
500
600Vehicle10mg/kg
56%IP BIDx5/wkn=7/group
Dosing
Study Day (post tumor implant)
Tum
or v
olum
e (m
m3 )
C.
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 420
250
500
750
1000
1250
vehicle
20 mg/kg10 mg/kg5 mg/kg
PO, QDn=8/group
PF-4942847 in MX-1
34%
51%
80%Dosing
Study Day (post tumor implant)
Tum
or v
olum
e (m
m3 )
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592
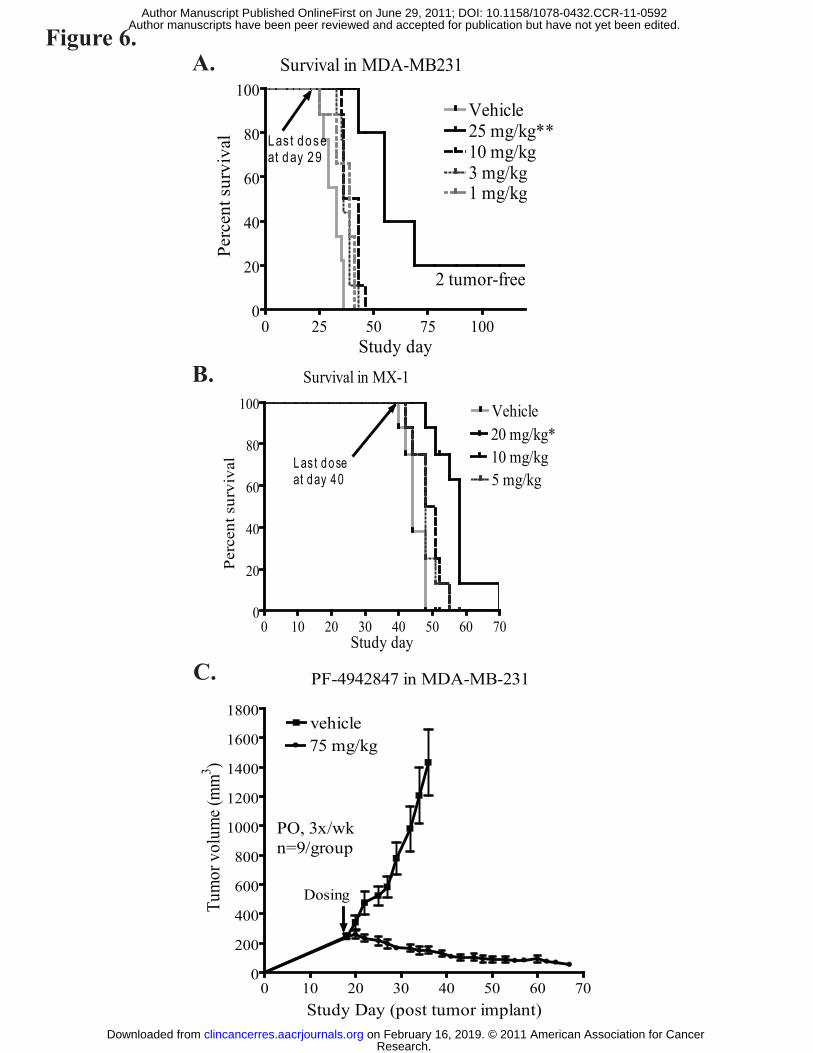
Figure 6.A.
B.
C. PF-4942847 in MDA-MB-231
0 10 20 30 40 50 60 700
200
400
600
800
1000
1200
1400
1600
1800
PO, 3x/wkn=9/group
vehicle75 mg/kg
Study Day (post tumor implant)
Tum
or v
olum
e (m
m3 )
Dosing
0 25 50 75 1000
20
40
60
80
100Vehicle25 mg/kg**10 mg/kg3 mg/kg1 mg/kg
2 tumor-free
Survival in MDA-MB231
Las t doseat day 29
Study day
Perc
ent s
urvi
val
0 10 20 30 40 50 60 700
20
40
60
80
100 Vehicle20 mg/kg*10 mg/kg5 mg/kg
Perc
ent s
urvi
val Las t dose
at day 40
Survival in MX-1
Study day
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

Table I: Summary of IC50 values in AKT degradation, Hsp70 induction, and cell proliferation assays
Mean SD Mean SD Mean SD Mean SD Mean SD Mean SD
MDA-MB-231 19.4 11.8 24.8 11.2 24.1 3.3 3.5 1.4 25.9 6.5 8.6 1.2
HCC-70 61.4 21.3 22.8 6.5 23.4 4.9 7.7 1.9 57.3 18.8 4.9 1.8
MX-1 93.7 15.6 87.4 20.7 17.2 1.6 2.7 0.2 24.7 2.4 5.2 0.2
Hs-578sT 28.5 3.6 9.5 3.2 23.6 3.4 3.4 1.4 18.1 8.3 7.5 0.2
BT-549 74.5 15.4 22.7 10.6 21.4 4.2 4.6 1.3 19.1 6.9 5.5 0.3
MDA-MB-468 86.9 19.5 89.1 20.3 37.8 6.2 12.3 2.9 37.0 4.1 13.5 3.6
HCC-38 51.7 7.3 35.8 6.3 35.6 8.1 13.8 1.5 23.7 2.4 11.1 0.3
EC50 (nM)
Hsp70 induction Cell proliferation inhibiton
IC50 (nM)
PF-4942847 17-DMAGPF-4942847 17-DMAG
IC50 (nM)
AKT degradation
PF-4942847 17-DMAG
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592

Published OnlineFirst June 29, 2011.Clin Cancer Res Pramod P. Mehta, Pamela M. Whalen, Sangita M Baxi, et al. PF-4942847, a Novel Oral Inhibitor of Heat Shock Protein 90Effective Targeting of Triple Negative Breast Cancer Cells by
Updated version
10.1158/1078-0432.CCR-11-0592doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2011/08/10/1078-0432.CCR-11-0592.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/early/2011/06/29/1078-0432.CCR-11-0592To request permission to re-use all or part of this article, use this link
Research. on February 16, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 29, 2011; DOI: 10.1158/1078-0432.CCR-11-0592