early clinical trials’ designs and their challenges pamela n munster, md ticr 4-14-2011
TRANSCRIPT

Early Clinical Trials’ Designs
and their Challenges
Pamela N Munster, MD
TICR4-14-2011.

Phase ISafety/Phase
II dose
Phase IIICompare new vs. standard
Phase IVlong term safety
FDA Approval
IND granted
Cell culture, signaling studies, combinations, xenografts and animal models
Animal Tox
Molecular Screens
2554 Phase I trials currently accruing
Historical Drug Development Snapshot
3908 NCI listed
1872 NCI listed
Phase 0
Phase IIEfficacy/Tox

Testing of a novel strategy/compound– New single agent
• First in Man, Proof of Concept • New formulations, schedules or doses
– Combinations of novel agents with existing agents• X combined with chemotherapy• X combined with hormonal therapies
– Novel agent as biological response modifier• Inhibition of Histone deacetylase, • Inhibition of DNA repair (e.g. PARP, Chk1, wee1) • Inhibition of chaperone proteins, (e.g. HSP90)
Types of Treatments in Phase I Clinical Trials (I)

Testing of a novel strategy/compound– Different Modalities
• Electroporation of inhibitory genes, • siRNA, • anti-sense• Immune modifiers
Types of Treatments in Phase I Clinical Trials (II)

Terms and Definitions
– Safety, Toxicity and Tolerability
– Dose limiting toxicities (DLT)
– Maximally Administered Dose (MAD)
– Maximally Tolerated Dose (MTD)
– Recommended Phase II dose (RPTD)
– Dose Escalation Rules
– Pharmacokinetics and Pharmacodynamics
Early Phase Clinical Trials’ Design

Terms and Definitions– Safety, Toxicity and Tolerability
• Standard toxicity assessment by Common Terminology Criteria for Adverse Events (CTCAE) v. 3.0 or more recently 4.0
– Grading» 0-5
– Causality determination: » treatment-emergent vs treatment-related
Early Phase Clinical Trials’ Design

Example: Toxicity Assessment
Presentation:A 47 year old Caucasian male starts an investigational agent on 4-1-11,Four days later he presents with a new rash over his entire right arm with itching. He continues to have mild fatigue, but now has an unquenchable thirst. He is able to do his daily chores. His fasting blood glucose is 271 mg/dL.
Baseline criteria included Grade I fatigue, but otherwise no symptoms.
Assessment: Grading and Relatedness (CTCAE IPhone App, or (ctep.cancer.gov/protocoldevelopment/electronic.../ctcaev3.pdf)Fatigue: Rash:Fasting Blood Glucose:

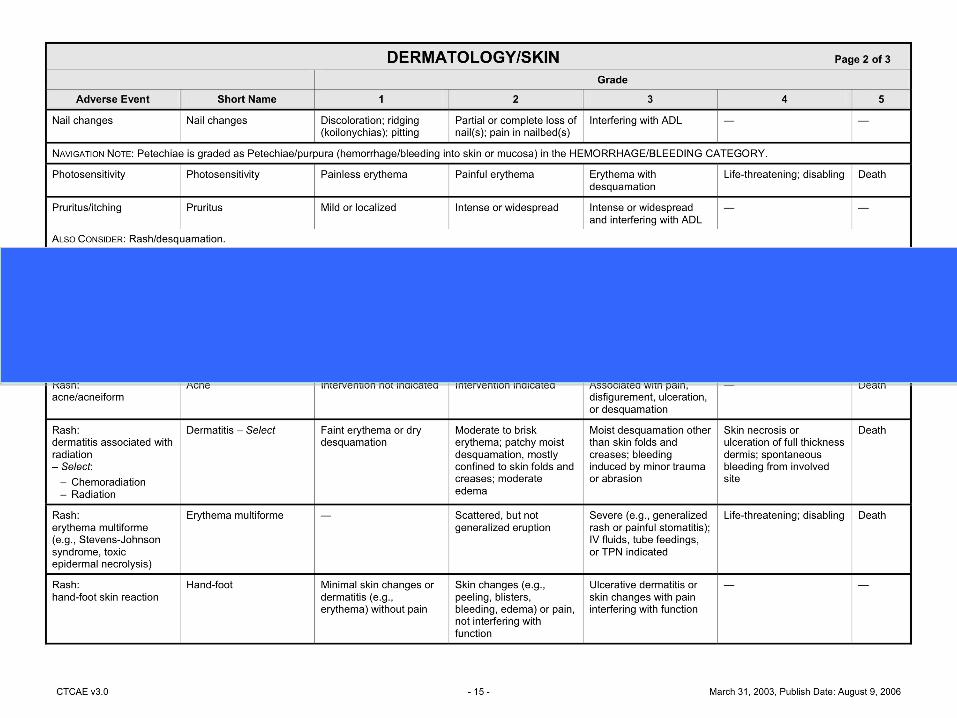

Example: Toxicity Assessment
Presentation:A 47 year old Caucasian male starts an investigational agent on 4-1-11,Four days later he presents with lightheadedness, fatigue, unquenchable thirst. He is able to do his daily chores and has otherwise no symptoms.His fasting blood glucose is 271.Baseline criteria included Grade I fatigue, but otherwise no symptoms.
Assessment:Fatigue: Grade 1, not relatedRash: Grade 2, relatedFasting Blood Glucose: Grade 3, related

Terms and Definitions
Dose limiting toxicities (DLT) within window (usually 3-4 weeks)– Grade 3 non-hematological toxicities
– Grade 4 hematological
– Protocol-specific definitions• E.g. allow for grade 3
– Diarrhea despite optimal care, – Nausea despite optimal care– Hyperglycemia (with treatment for more than 14 days) (PI3k, mTOR) – Hypertension (VEGF inhibitors, VEGFR inhibitors)
Classical Phase I Clinical Trials’ Design

Terms and Definitions– Maximally Administered Dose (MAD)
• Highest administered dose (>2/6 pts with DLT)
– Maximally Tolerated Dose (MTD)• Highest tested dose where 0/6 or 1/6 patients
experienced DLT
– Recommended Phase II dose (RPTD)• The dose to be tested in phase II
Classical Phase I Clinical Trials’ Design
Dose escalation designs
Leonardo di Pisa (ca.1202)(Fibonacci)First mentioned in Pingala (200 BC)

Dose escalation designs
– Fibonacci (0, 1, 1, 2, 3, 5, 8, 13, 21, …)
– Modified Fibonacci
– Dose doubling Designs
– Adaptive Designs• real time pharmacokinetics, • statistical adaptations
– Combinations of designs
Early Phase Clinical Trials’ Design

Dose escalation
Cohorts Fib Modified
Fib
Percent increase
Example
(mg)
1 D D ------ 1
2 2 x D 2 x D 100% 2
3 3 x D 3.3 x D 66% 3.3
4 5 x D 5 x D 50% 5
5 8 x D 7 x D 40 % 7
6 13 x D 9 x D 29% 9
7 21 x D 12 x D 33 % 12
8 34 x D 16 x D 33 % 16
Fibonacci and modified Fibonacci Dose escalations
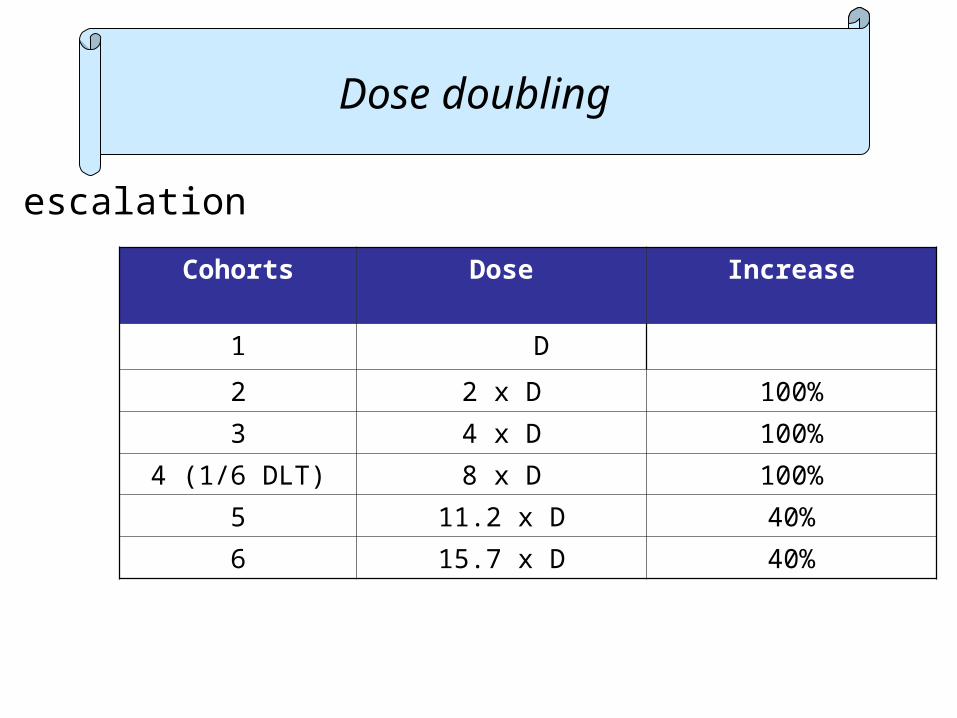
Dose escalation
Cohorts Dose Increase
1 D
2 2 x D 100%
3 4 x D 100%
4 (1/6 DLT) 8 x D 100%
5 11.2 x D 40%
6 15.7 x D 40%
Dose doubling
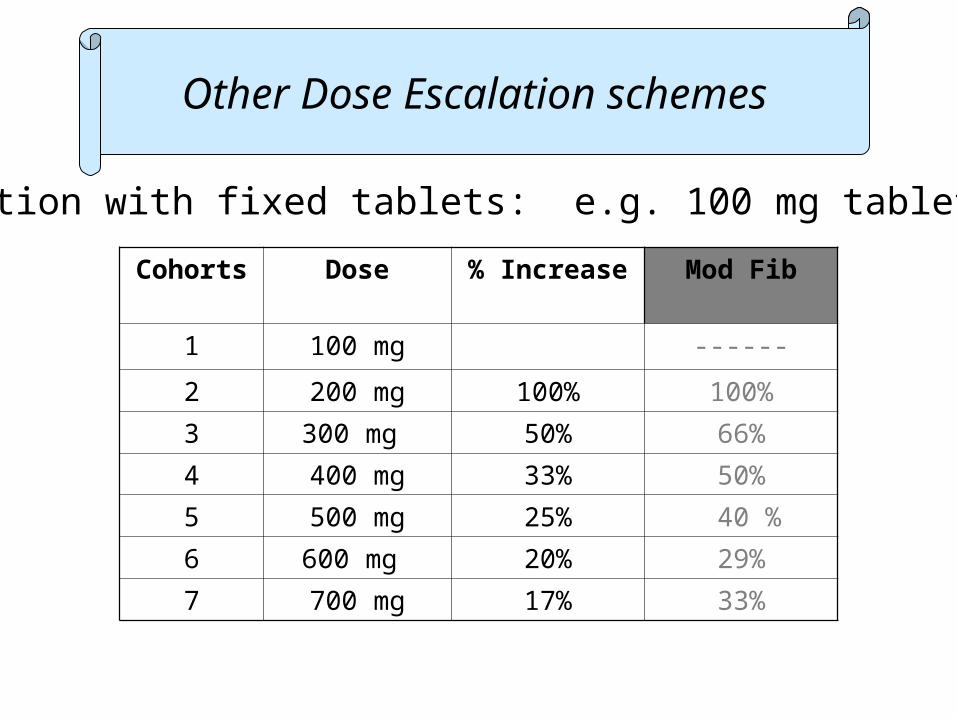
Dose escalation with fixed tablets: e.g. 100 mg tablets
Cohorts Dose % Increase Mod Fib
1 100 mg ------
2 200 mg 100% 100%
3 300 mg 50% 66%
4 400 mg 33% 50%
5 500 mg 25% 40 %
6 600 mg 20% 29%
7 700 mg 17% 33%
Other Dose Escalation schemes

Adaptive Dose escalation with fixed tablets: e.g. 100 mg tablets
Cohorts Dose Increase
1 100 mg qd
2 100 mg bid 100%
3 100 mg tid 50%
4 200 mg qd 25%
5 200 mg bid 100%
6 200 mg tid 50%
Adaptive Designs (Example)

Cohort Size
Patient Escalation 1+1 (occ. used for lowest doses) 3+3 (most common) 6+6
Cohorts and Sample Size

– Dose Escalation Rules
• PRE-DEFINED CONDITIONS FOR DOSE ESCALATION
– (standard and specific to the individual protocol)
Early Phase Clinical Trials’ DesignTerms and Definitions
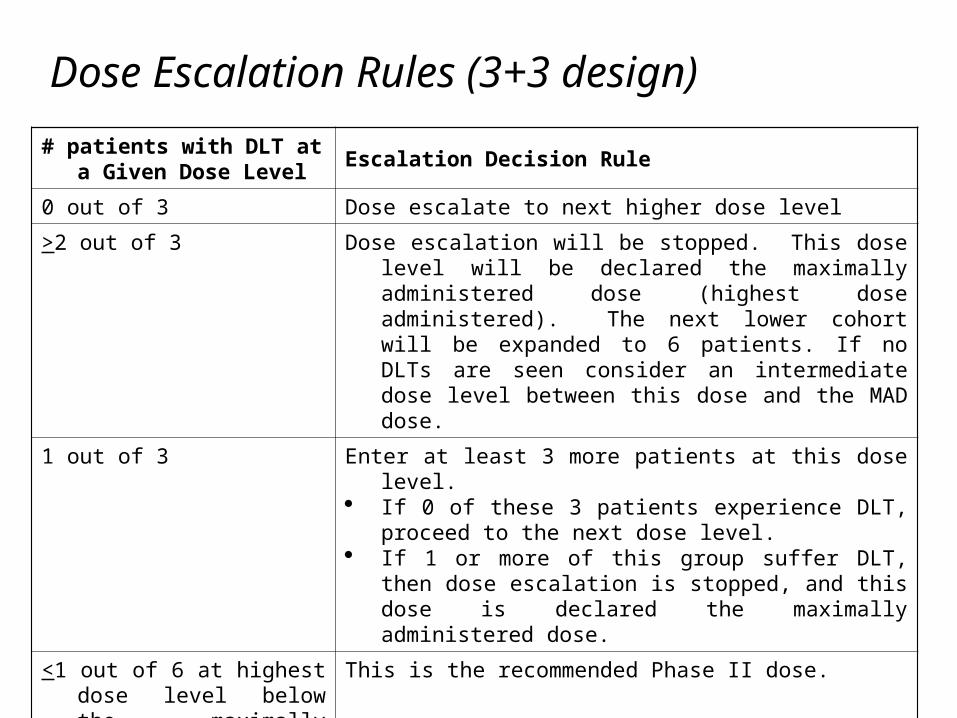
Dose Escalation Rules (3+3 design)
# patients with DLT at a Given Dose Level
Escalation Decision Rule
0 out of 3 Dose escalate to next higher dose level
>2 out of 3 Dose escalation will be stopped. This dose level will be declared the maximally administered dose (highest dose administered). The next lower cohort will be expanded to 6 patients. If no DLTs are seen consider an intermediate dose level between this dose and the MAD dose.
1 out of 3 Enter at least 3 more patients at this dose level. If 0 of these 3 patients experience DLT, proceed to the
next dose level. If 1 or more of this group suffer DLT, then dose
escalation is stopped, and this dose is declared the maximally administered dose.
<1 out of 6 at highest dose level below the maximally administered dose
This is the recommended Phase II dose.
If 0 out of 3 at the highest dose
Consider protocol amendment to dose escalate further

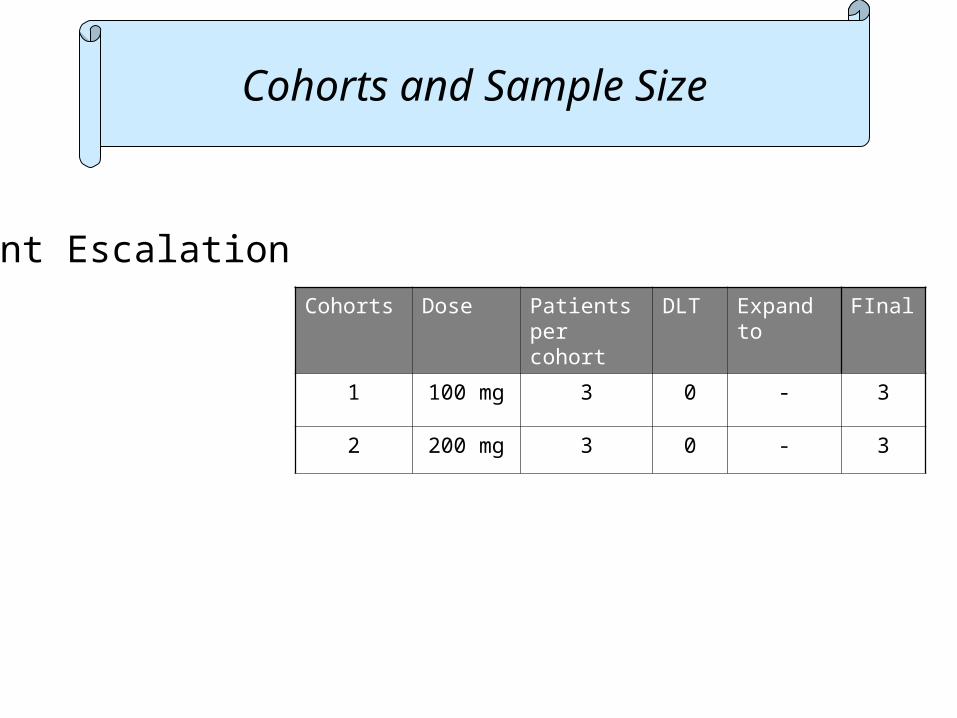
Cohorts and Sample Size
Cohorts Dose Patients per cohort
DLT Expand to
FInal
1 100 mg 3 0 - 3
Patient Escalation 3+3
Cohorts and Sample Size
Cohorts Dose Patients per cohort
DLT Expand to
FInal
1 100 mg 3 0 - 3
2 200 mg 3 0 - 3
Patient Escalation 3+3

Cohorts and Sample Size
Cohorts Dose Patients per cohort
DLT Expand to
FInal
1 100 mg 3 0 - 3
2 200 mg 3 0 - 3
3 300 mg 3 0 - 3
Patient Escalation 3+3

Cohorts and Sample Size
Cohorts Dose Patients per cohort
DLT Expand to
FInal
1 100 mg 3 0 - 3
2 200 mg 3 0 - 3
3 300 mg 3 0 - 3
4 400 mg 3 0 - 3
Patient Escalation 3+3

Cohorts and Sample Size
Cohorts Dose Patients per cohort
DLT Expand to
FInal
1 100 mg 3 0 - 3
2 200 mg 3 0 - 3
3 300 mg 3 0 - 3
4 400 mg 3 0 - 3
5 500 mg 3 2 0 3 MAD
Patient Escalation 3+3

Patient Escalation 3+3
Cohorts and Sample Size
Cohorts Dose Patients per cohort
DLT Expand to FInal
1 100 mg 3 0 - 3
2 200 mg 3 0 - 3
3 300 mg 3 0 - 3
4 400 mg 3 1 3 6 (MTD)
5 500 mg 3 2 0 3 MAD

Dose expansion
Purpose:• Estimation of toxicities in larger sample set• Estimation of PK and PD markers• Preliminary Efficacy
Sample size • Minimum 6• General 12-15

Dose expansion
Purpose:• Estimation of toxicities in larger sample set• Estimation of PK and PD markers• Preliminary Efficacy
Sample size • Minimum 6• General 12-15

Pharmacokinetics
and Pharmacodynamics

“Drug Behavior in the Subject”
Pharmacokinetics
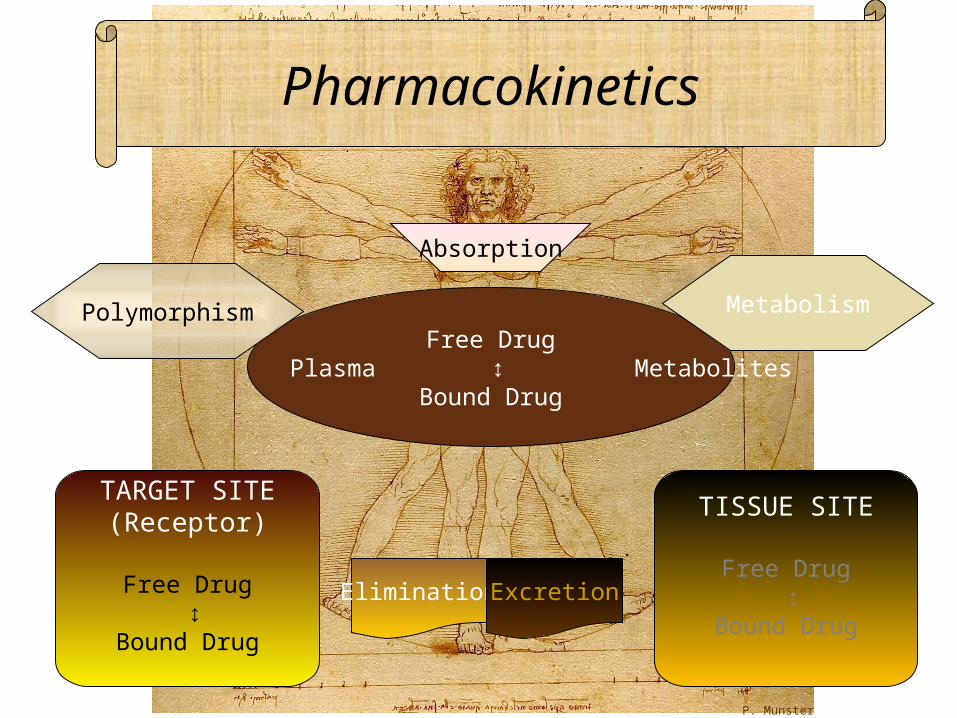
Pharmacokinetics
Free Drug Plasma ↕ Metabolites
Bound Drug
TARGET SITE(Receptor)
Free Drug ↕ Bound Drug
TISSUE SITE
Free Drug ↕ Bound Drug
Metabolism
Absorption
Pharmacokinetics
Elimination Excretion
Polymorphism
P. Munster

MeasurablesRoutinely done: albeit often limited
•Half-life (t1/2) •Distribution volume•Peak concentrations (Cmax)•Time to peak concentration (Tmax)•Area under the curve•Food effects
Not routinely done•Polymorphisms •Drug-drug interactions•PK-PD interactions
Pharmacokinetics

Example of PK sampling in Phase I trial
Heparinized blood samples (10 mL) will be collected on Day1 at 0, 0.5, 1, 2, 4, 6, 8, 24, 48, and 72 hours after the first dose of LY335562 Day 15 at 0, 0.5, 1, 2, 4, 6, 8, and 24 hours after that day’s doseDay 57 at 0, 0.5, 1, 2, 4, 6, 8, and 24 hours after that day’s dose.
Single blood samples for pharmacokinetic studies for the desmethyl metabolite were also collected on days 8, 15, 22, 29, 43, 71, and 85.

Example: Plasma concentration: dose escalations
ASCO 2008, J Infante

Copyright © American Society of Clinical Oncology
Munster, P. N. et al. J Clin Oncol; 19:2002-2009 2001
Fig 1. Mean plasma concentrations of LY353381.HCl versus time following a single dose at visit 1 and multiple doses of LY353381.HCl at visit 3
Example: Plasma concentrations: Drug accumulations
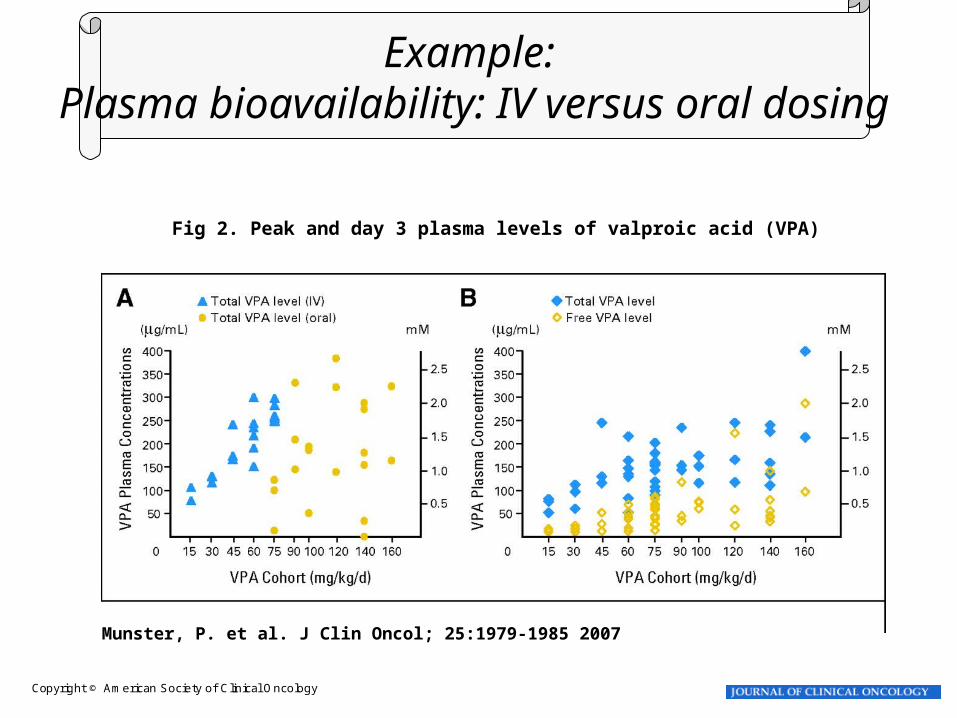
Copyright © American Society of Clinical Oncology
Munster, P. et al. J Clin Oncol; 25:1979-1985 2007
Fig 2. Peak and day 3 plasma levels of valproic acid (VPA)
Example: Plasma bioavailability: IV versus oral dosing

Munster et al CCR 2009
VPA dose level (mg/kg/day)
0
0 15 30 45 60 75 90 100 120 140 160
50
100
150
200
250
300
350
Fre
e V
PA
pla
sma
leve
l (μ
g/m
l)
21%
20%
19%
39%31%
35%43%
45%38%
70%
VPA dose level (mg/kg/day)
To
tal V
PA
pla
sma
leve
l (µ
g/m
l)
0
50
100
150
200
250
300
350
400
450
0 15 30 45 60 75 90 100 120 140 160
Dose Escalation: n=41Dose Expansion: n=15
Example: Plasma Concentrations (Free and Total Drug)
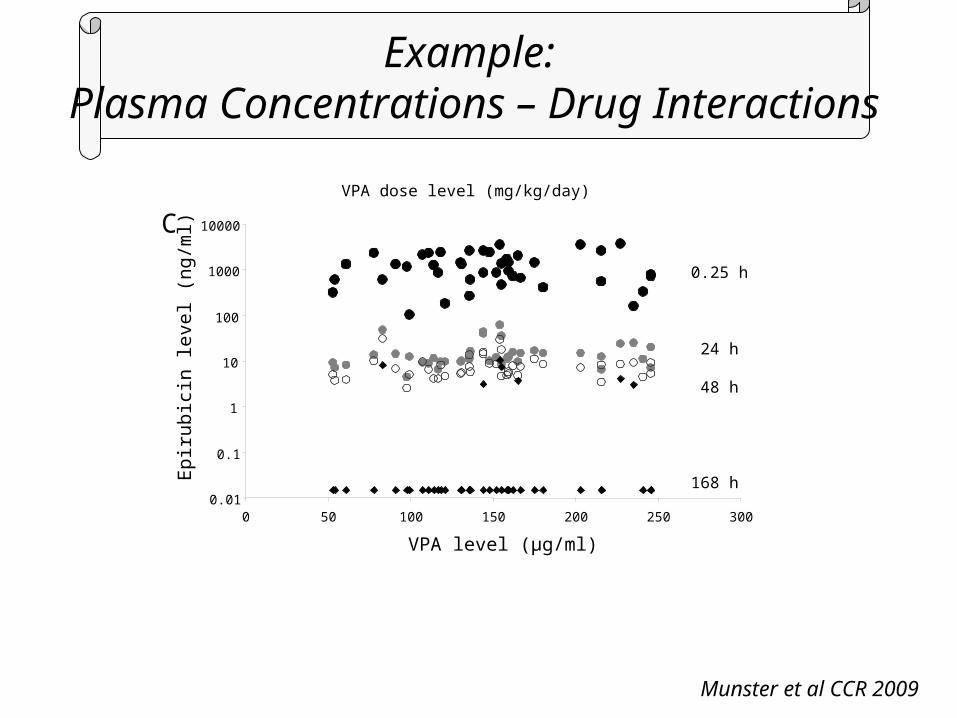
Munster et al CCR 2009
VPA dose level (mg/kg/day)
VPA level (μg/ml)
0.25 h
24 h
48 h
168 h
Epi
rubi
cin
leve
l (ng
/ml)
C
0.01
0.1
1
10
100
1000
10000
0 50 100 150 200 250 300
Example: Plasma Concentrations – Drug Interactions

Metabolism and Polymorphism

[Kirchheiner, 2003]
Polymorphisms in the CYP2D6 gene changes Metabolizers status
PM: Poor metabolizersIM: Intermediate metabolizerEM: Extensive metabolizerUM: Uebermetabolizer

Pharmacodynamics

Pharmacodynamics
“Drug Effects on the subjects and tumors”
• Symptoms• Signs• Changes in lab values• Molecular and Biological effects

Pharmacodynamics: signs and symptoms
Munster et al JCO 2007
Munster P et al. Clin Cancer Res 2009;15:2488-2496©2009 by American Association for Cancer Research
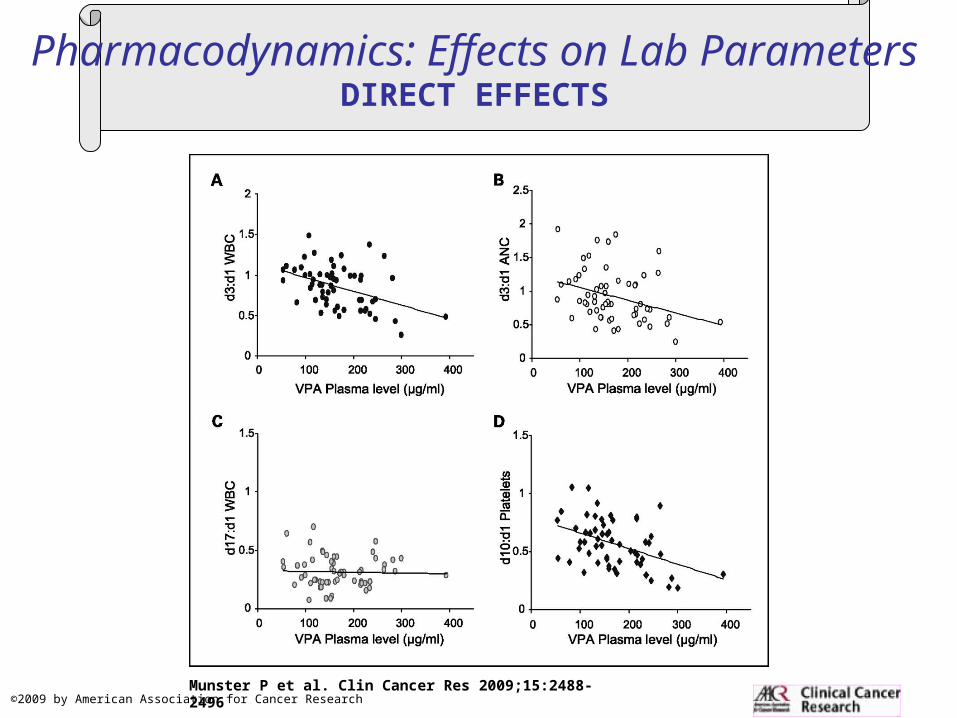
Pharmacodynamics: Effects on Lab ParametersDIRECT EFFECTS

Copyright © American Society of Clinical OncologyMunster, P. et al. J Clin Oncol; 25:1979-1985 2007
Fig 1. Valproic acid (VPA) effects on epirubicin-associated toxicities
Indirect effects
Pharmacodynamics: Effects on Lab ParametersINDIRECT EFFECTS

HORMONERECEPTORSTATUS
HER2STATUS
VISCERADISEASE
PRIOR AROMATASEINHIBITORS
PRIORTAMOXIFEN
ACET-H4
DOSEMOD
Patients with Partial Response: 8/43 (19%)Pt 1Pt 2Pt 3Pt 4Pt 5Pt 6 Pt 7Pt 8
ER+ / PR+ER+ / PR+ER+ / PR+ER+ / PR-ER+ / PR+ER+ / PR+ER+ / PR+ER+ / PR+
Not amplNot amplNot amplNot amplNot amplNot amplNot amplNot ampl
YesYesYesYesNoYesYesNo
LetrozoleAnastrozole, ExemestaneAnastrozole, ExemestaneLetrozole, Exemestane AnastrozoleLetrozole, ExemestaneAnastrozole, ExemestaneLetrozole
TamoxifenTamoxifen Tamoxifen
YesYesNoYesYesYes-Yes
300200300300
Patients with Stable Disease > 24 weeks: 9/43 (21%)Pt 1Pt 2Pt 3Pt 4Pt 5Pt 6Pt 7Pt 8Pt 9
ER+ / PR-ER+ / PR+ER+ / PR-ER+ / PR+ER+ / PR-ER+ / PR-ER+ / PR+ER+ / PR+ER+ / PR+
Not amplNot amplNot amplNot amplNot amplNot amplAmplNot amplNot ampl
NoYesYesNoYesYesNoYesNo
Letrozole, AnastrozoleLetrozole, Exemestane,Anastrozole, LetrozoleLetrozoleAnastrozoleAnastrozole, LetrozoleLetrozole, ExemestaneLetrozoleLetrozole
TamoxifenTamoxifen
Tamoxifen TamoxifenTamoxifen
YesYes-YesYesYesYesYesYes
200200300300
Pharmacodynamics: PD effects (histone acetylation) and Responses

H4 acetylation occurred only in 58% of treated patients%
cha
nge
in h
isto
ne a
cety
latio
n
Acetyl-H4
Day 1 Day 8
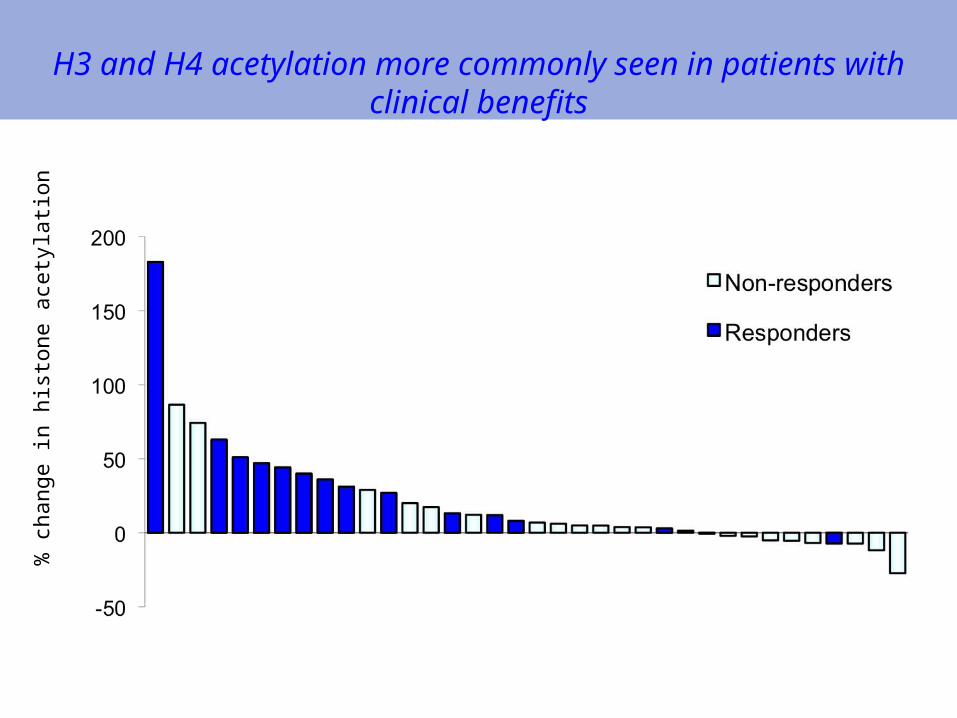
H3 and H4 acetylation more commonly seen in patients with clinical benefits
% c
hang
e in
his
tone
ace
tyla
tion

Correlative studies for the clinical trial (II)H3 and H4 acetylation more commonly seen in patients
with clinical benefits
0
10
20
30
40
50
ALL R NR
% c
hang
e in
ace
tyl-H
4%
cha
nge
in a
cety
l-H4
CALL R NR
*
*
*P=0.022, n=36
Rel
ativ
e H
DA
C2
Exp
ress
ion P=0.04, n=36
NR R

No correlation between H4 acetylation and toxicity%
cha
nge
in h
isto
ne a
cety
latio
n in
PB
MC
s
% p
atie
nts
with
sev
ere
toxi
citie
s
R NR

• What are your goals for your trial?
• The objectives of a trial must be stated in specific terms.
• The endpoints (or outcomes), determined for each study participant, are the quantitative measurements required by the objectives.
Objectives and Endpoints

Phase I trials
• Primary Objectives– Safety and Tolerability– Recommended Phase II dose
• Secondary Objectives– PK and PD– ……….
Examples: Objectives and Endpoints

• Who is the target population in your trial?
• Inclusion criteria– Population criteria– Disease Criteria– Overall health criteria
• Exclusion criteria– Comorbid conditions– Confounding factors
Objectives and Endpoints

• Who is the target population in your trial?
• Inclusion criteria• Exclusion criteria
Objectives and Endpoints

Inclusion
• 18 years of age or older
• Histologically confirmed metastatic or unresectable melanoma
• Glutamic acid-for-valine substitution at amino acid position 600 in the BRAF gene or other activating BRAF mutation, as determined by high-throughput genotyping (VE600)
• Patients may have received any number of prior systemic treatments for their cancer
• At least one measurable site of disease by CT, according to standard RECIST criteria
• ECOG Performance Status 0-1
• Estimated life expectancy > 12 weeks
• Absolute neutrophil count > 1500 per cubic mm
• Platelet Count > 100,000 per cubic mm
• Hemoglobin > 9 g/dl
• Serum bilirubin < 1.5 x upper limit of normal, Serum AST and ALT < 2.5 x upper limit of normal
• Serum Creatinine < 1.5 x upper limit of normal
• For women of childbearing potential, negative serum pregnancy test and use of physician-approved method of birth control throughout the study
Examples: Inclusion CriteriaBRAF inhibitors in patients with melanoma carrying a BRAF Mutations

Exclusion Criteria:
• Have received chemotherapy or radiotherapy within 4 weeks prior to entering the study or a targeted therapy within 2 weeks prior to entering the study
• Have not recovered from adverse events due to agents previously administered (CTCAE v. 3.0 grade 1 or baseline)
• Currently receiving other investigational agents
• Known brain metastases, unless treated and stable off of corticosteroids for at least four weeks
• Prior treatment with a selective inhibitor of RAF or MEK
• Uncontrolled intercurrent illness, including but not limited to, clinically significant active infection; symptomatic congestive heart failure, unstable angina pectoris, and/or cardiac arrhythmia other than atrial fibrillation; psychiatric illness/social situations that would limit compliance with study requirements
• Refractory nausea or vomiting, swallowing disorder, or malabsorption syndrome that would interfere with swallowing or absorbing the study medication
• Pregnant or breast feeding women
• Uncontrolled hypertension
• A mean left ventricular ejection fraction (LVEF) less than 45%
Examples: Exclusion Criteria

• No clear relationship between dose and toxicity • No definite threshold toxicity (DLT)• No clear relationship between efficacy and dose• No clear target modulation• Unrecognized down-target effects
– Activating or inhibitory
• Efficacy not likely
Difficult to choose dose or schedule in these circumstances
Emerging Challenges

• Imaging– PET scanning
• Tissue Perfusion • Blood Volume• Glucose metabolism• Estrogen metabolism
– DCE-MRI• Pharmacodynamic Studies
– Target inhibition in end-organ• Tumor biopsies• Circulating tumor cells
– Target inhibition in surrogate tissue• Peripheral Blood Mononuclear Cells
• Pharmacokinetics– Targeted Drug Levels:
• Serum Markers
Alternative and/or Additional Endpoints

• Traditional Phase I designs are valid for novel chemotherapy drugs and for some novel agents but….
• Many targeted agents require careful assessment and incorporation of correlative endpoints or novel imaging modalities to decide the dose, schedule, and optimal therapeutic partners
• Availability of real-time PK and PD (biomarkers)
• Validation of endpoints in larger studies with enriched populations
Conclusion

Challenges of PD-PK interaction.
Back Up slides:Example of a Clinical Trial

Example of a clinical trial
xxxxx
- Single Arm Phase I/II Study of vorinostat 400 mg and tamoxifen at 20 Single Arm Phase I/II Study of vorinostat 400 mg and tamoxifen at 20 mgmg
- Patients with estrogen receptor-positive metastatic breast cancer who progressed on prior therapy with an aromatase inhibitors and up to three chemotherapy regimens
- Primary Objective: Overall Response Rate
xxx
xxx
x
x
x
Toxicity visitClinical Assessment CBC, CMP, TMPBMCStaging Studies
Vorinostat (400 mg/day)
Tamoxifen (20 mg/d)
Cycle 1 (4 weeks)
xx
Cycle 2 (4 weeks)

HDAC inhibitors
Histone Deacetylases and Histone AcetylTransferases (HATs) modulate gene expression by the removal or the addition of acetyl groups to lysine residues of histones or other non-histone targets.

Vorinostat Background
• Member of the class of Histone deacetylase inhibitors
• Cmax reported >1 uM (FDA package insert)
• Narrow therapeutic window with wide interpatient variability
• Expected Responses: <10% in breast cancer

C T V VT
Estrogen receptor
0
2000
4000
6000
MCF-7
Control Vor 1uMVPA 2mM
mR
NA
0
50
100
150
200
250 PR
Progesterone receptor
MCF-7
C T V VTm
RN
A
HDAC inhibitor effects on Estrogen (ER) and Progesterone (PgR) receptors
Control Vor 1uMVPA 2mM
prot
ein
Bicaku et al, Canc Res 2008
C T V VT
ER-positive cells (MCF-7 and T47D)
prot
ein
ER

The effects on Estrogen (ER) and Progesterone (PgR) receptors
are mediated through HDAC2
A
ER
HDAC1
PR
HDAC1 siRNA - + - +
ER
HDAC6
PR
- +HDAC2 siRNA
PR
ER
HDAC2
% A
po
pto
sis
UntreatedTamoxifen
Control HDAC2 HDAC1 HDAC6
siRNA scramble
0
5
10
15
20
25
30
HDAC6 siRNA

Munster et BMJ 2009
Vorinostat dose per day (mg)
Vor
inos
tat
leve
ls (
ng/m
l)Vorinostat dose and plasma levels from Phase I studies

Munster et al BMJ 209
Vorinostat effects on down-stream targets: Chromatin remodeling genes
-6
-5-4
-3
-2-1
0
12
3
-4
-2
0
2
4
6
8
10 Acetyl-H4
HP-1Topo IIα
X-f
old
chan
ge
in e
xpre
ssio
nX
-fol
d ch
ang
e in
exp
ress
ion
1 2 3 4 5 6 7 8 9 10 11 12 pt
x-fo
ld in
crea
se in
ace
tyla
tion
Tumor vs PBMC acetylation
0
1
2
3
PBMC tumor PBMC tumor
acetyl-H3acetyl-H4
Tumor
Tumor
A
C
B

Acetyl-H4
Topo IIα
Topo IIβ
SMC2
DNMT-1
HP-1
- +vorinostat
-6
-5-4
-3
-2-1
0
12
3
-4
-2
0
2
4
6
8
10Acetyl-H4
HP-1Topo IIα
X-f
old
chan
ge
in e
xpre
ssio
nX
-fol
d ch
ang
e in
exp
ress
ion
1 2 3 4 5 6 7 8 9 10 11 12

Summary and Conclusion of the clinical trial
Rationale of the trial: combined modification of the estrogen receptor by and anti-estrogen and an HDAC inhibitor may increase the benefits: expected response with either drug <10%
Observed Response: Complete and Partial response: 20%stable disease > 6 months: 20%Accepted clinical benefit’s rate: 40%
SummaryResults are interesting, but unclear whether we should go forward with this combination

Could careful assessment of PD and biomarkers improve
patient selections?

Correlative studies: Histone acetylation
biomarker in responders vs non-responders
Ch
an
ge
in H
4 h
isto
ne
ace
tyla
tion
(%
)
Ch
an
ge
in H
4 h
isto
ne
ace
tyla
tion
(%
)
All R NR
-50
0
50
100
150
200Non-responders
Responders
% c
han
ge
in h
isto
ne a
cety
latio
n

Correlative studies: HDAC2biomarker in responders vs
non-responders

Toxicities: on target or off target effect
% p
atie
nts
with
se
vere
toxi
citie
s
RR NRNR

What did we learn?
1) The drug does not accomplish the desired effects in 50% of the treated patients.2) Toxicities do not correlate with response or acetylation
What do we need to know?
- Ineffective drugs or ineffectively used?- Are the plasma levels insufficient?- Is the dosing adequate?- Normal interpatient variability or predetermined metabolism?- Could we have preselected these patients?
What did we learn?
1) The drug does not accomplish the desired effects in 50% of the treated patients.2) Toxicities do not correlate with response or acetylation
What do we need to know?
- Ineffective drugs or ineffectively used?- Are the plasma levels insufficient?- Is the dosing adequate?- Normal interpatient variability or predetermined metabolism?- Could we have preselected these patients?
Open Questions

•More detailed and expanded pharmacokinetic analysis
•Insightful pharmacokinetic interpretation?
•Rapid turn around of PKs to allow the adaptive designs.
•Correlation of PK with pharmacodynamic endpoints.
•Evaluation of polymorphisms for enzymatic metabolites, gender, ethnic and racial differences
What would help us improve?

Acknowledgements Clinical collaborators
Susan Minton, Mira Lacevic, Roohi Ismail-Khan, Laurie Sullivan, Sadie Aguila, Hope Rugo, Michelle Melisko, Mark Moasser,
Extramural Funding Agencies/Foundations NCI/NHI DOD Merck
Munster Lab Douglas Marchion Elona Bicaku Morgen Schmitt Dawn Morelli Golda Collamat, Soe Maunglay Scott Thomas, Christine Christian Ted Thurn,
With gratitude and admiration for all Patients and Their Families and Advocates

Thank you