drug repurposing for antimicrobial development: teaching...
TRANSCRIPT

1
Drug Repurposing for Antimicrobial Development: Teaching Old Drugs New Tricks
Learning Objectives 1. Describe major barriers of the traditional drug discovery/development process2. Identify potential advantages that drug repurposing may have over traditional drug
discovery3. List examples of drugs that have been successfully repurposed4. Examine the antimicrobial properties of sertraline
Natalie Boyd, PharmD, MS PhD Candidate, Translational Sciences
Pharmacotherapy Division, The University of Texas at Austin College of Pharmacy Pharmacotherapy Education and Research Center, UT Health San Antonio
June 2nd, 2017

2
Drug Discovery and Development 1. Review of Process
a. Drug Discovery (3-6 years) i. Target identification
• Finding a target that causes or leads to a condition/disease
• Usually receptors or enzymes ii. Target validation
• Validate to confirm functions and effects of the target
• May involve knockout models, protein/gene expression profiles, etc. iii. Compound screening (~10,000) - screen target with thousands of compounds iv. Lead optimization
• Chemically modify selected compound(s) to optimize pharmacokinetic/pharmacodynamic properties
• Oral drug candidates are often gauged using Lipinski’s “rule of five” (Figure 1) a. A set of drug-design criteria b. Associated with lower attrition rate
b. Preclinical (1-2 years) i. Animal modeling ii. Determine absorption, distribution, metabolism,
excretion, toxicity (ADMET) iii. Investigational new drug application
c. Clinical trials - Phase I through IV (5-6 years) (Table 1) d. Food and Drug Administration (FDA) review
i. Generally 1-2 years, unless expedited FDA review (See Appendix A)
Table 1. Characteristics of Phase I Through Phase IV Clinical Trials
Clinical Trial
Patients & Duration
Purpose Approximate %
Success in Each Phase
Phase I
20–100, several months
Test safety of a single dose (Phase Ia) and multiple doses (Phase Ib); includes maximum tolerated dose
70%
Phase II 100–300,
months to 2 years Assess efficacy and side effects
33%
Phase III 300–3,000, 1-4 years
Assess safety and monitoring of adverse reactions 25-30%
Phase IV Varies Post-marketing surveillance for continued
monitoring of safety and efficacy N/A
2. Current Barriers/Challenges in Drug Development a. Movement of a new compound from initial discovery to market launch is slow and costly:
i. Average duration ≈ 10-15 years ii. Minimum cost of ~ $2 billion
b. High attrition = low probability of success i. Only approximately one in 10,000 compounds screened will make it into the market ii. Only ~10% of drugs entering clinical trials will get FDA approval
• Molecular weight ≤ 500
• Log P ≤ 5
• H-bond donors ≤ 5
• H-bond accepters ≤ 10
Figure 1. Lipinski’s Rule-of-Five1, 2
3
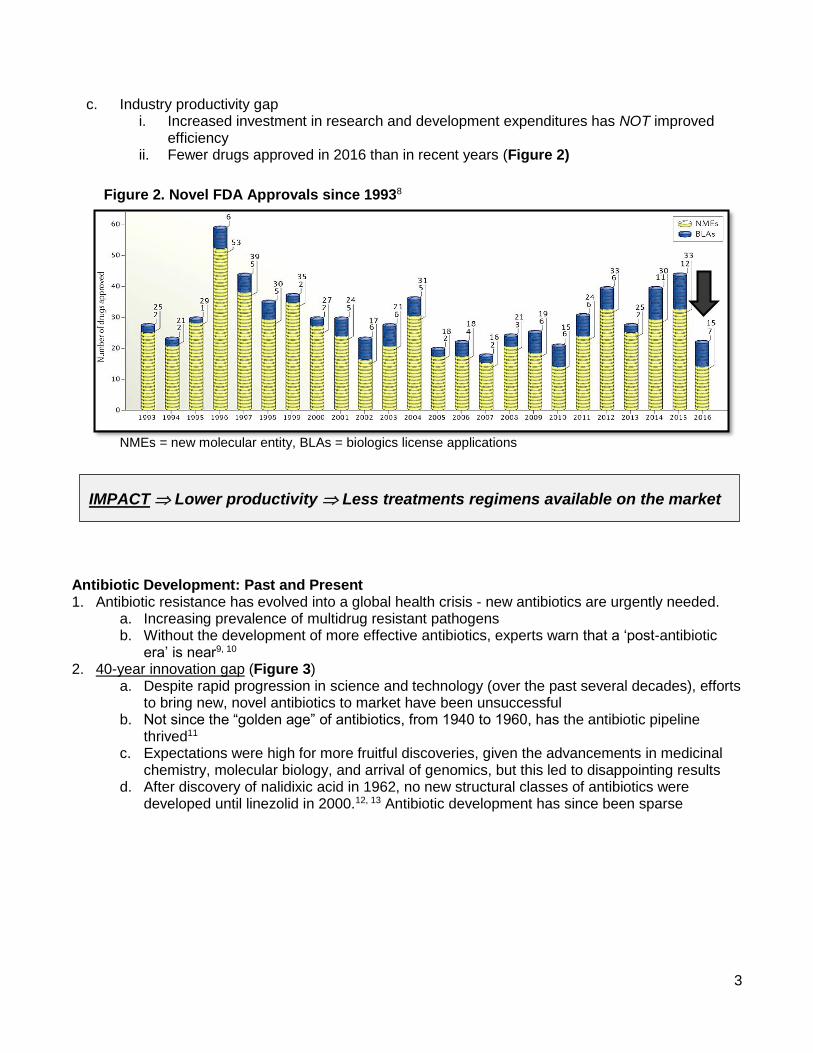
c. Industry productivity gap i. Increased investment in research and development expenditures has NOT improved
efficiency ii. Fewer drugs approved in 2016 than in recent years (Figure 2)
Antibiotic Development: Past and Present 1. Antibiotic resistance has evolved into a global health crisis - new antibiotics are urgently needed.
a. Increasing prevalence of multidrug resistant pathogens b. Without the development of more effective antibiotics, experts warn that a ‘post-antibiotic
era’ is near9, 10 2. 40-year innovation gap (Figure 3)
a. Despite rapid progression in science and technology (over the past several decades), efforts to bring new, novel antibiotics to market have been unsuccessful
b. Not since the “golden age” of antibiotics, from 1940 to 1960, has the antibiotic pipeline thrived11
c. Expectations were high for more fruitful discoveries, given the advancements in medicinal chemistry, molecular biology, and arrival of genomics, but this led to disappointing results
d. After discovery of nalidixic acid in 1962, no new structural classes of antibiotics were developed until linezolid in 2000.12, 13 Antibiotic development has since been sparse
Figure 2. Novel FDA Approvals since 19938
NMEs = new molecular entity, BLAs = biologics license applications
IMPACT Lower productivity Less treatments regimens available on the market

4
3. Major factors that led to the 40-year innovation gap11, 14, 15 a. Perception that the war on antibiotic resistance had been decisively “won.” The concept that
bacterial resistance is inevitable under selective pressure was not appreciated at the time b. Focus shifted from novel discovery to improving pharmacological properties of existing
compounds the Golden Age of medicinal chemistry c. Adherence to “rule-based” drug design (Lipinski) d. Pharma retreat from antibiotic development programs due to low return on investment e. Screening approaches: molecular, target-based versus whole-cell phenotypic16-18
i. Much of the success during the ‘golden age’ was due to empiric whole-cell screening, where they progressed through clinical trials with little or no knowledge of mechanism of action or target.
ii. Following arrival of genomics, bioinformatics, and high-throughput technologies, drug discovery efforts focused on molecular, target-based approaches, but this has not led to more drug approvals
iii. Between 1999 and 2008, 28 of the first-in-class small molecule drugs approved by FDA were discovered by phenotypic screening, compared to 17 discovered by a molecular target-based approach19
Drug Repurposing: An Alternative Pathway 1. Refers to the identification of new uses for existing drugs, candidates under development, or
abandoned compounds/failed compounds20 a. Often used interchangeably with ‘drug repositioning’ b. Other (similar) terms include:
i. reprofiling ii. redirecting iii. redeployment iv. rediscovery v. drug rescue
c. Some investigators have called for more standardized definitions and terminology in order to avoid confusion and misinterpretations of published works in the field21
d. Ideal candidate for repurposing = a drug that has passed safety and tolerability studies
Figure 3. Timeline of Antibiotic Discovery
5
e. Advantages of drug repurposing3, 22-24 i. Reduce development costs ii. Increase efficiency (faster time to approval) iii. Reduce investment risk (for compounds that have cleared Phase-I) iv. Other potential uses: diagnostic tool, biomarker, etc.
f. Repurposing commercially available drugs
i. Example = Gabapentin. Originally indicated for seizure control, but was also found to provide relief for postherpetic neuralgia25
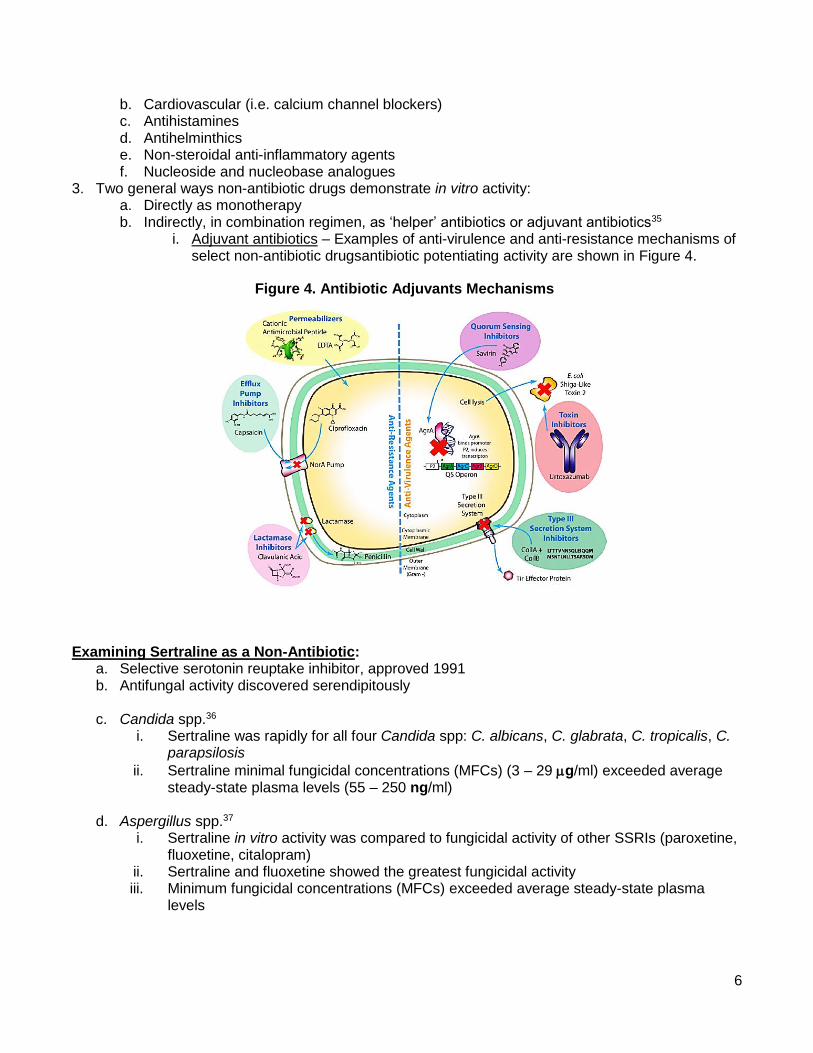
ii. Additional examples provided in Table 2
Drug Initial Approved
Indication Added Indication(s)
Amantadine Influenza Parkinson’s
Amphotericin B Fungal infections Leishmaniosis
Aspirin Inflammation, pain Antiplatelet
Duloxetine Depression
Fibromyalgia
Diabetic neuropathy
Chronic musculoskeletal pain
Everolimus Advanced kidney
cancer Prevention of organ rejection
Finasteride Prostate hyperplasia Hair loss
Gabapentin Epilepsy Postherpetic Neuralgia
Infliximab Crohn’s disease
Rheumatoid arthritis
Severe plaque psoriasis
Ulcerative colitis
Minoxidil Hypertension Hair loss
a. ‘Rescued’ medications (repurposing drugs that had failed and/or been abandoned)26 iii. Thalidomide is a notable example of reviving a discontinued (and once disastrous)
drug. Originally marketed in 1957 to pregnant women for morning sickness, it was found to have devastating teratogenic effects, causing more than 10,000 birth defects. 27, 28
iv. Additional examples provided in Appendix B Non-Antibiotic Drugs and Antibiotic Adjuvants 1. Non-antibiotics = drugs used (or under development) for non-infectious indications that may
possess antimicrobial properties29 2. Several drug classes have been known to demonstrate variable in vitro antibiotic activity:30-34
a. Antipsychotic (i.e. selective serotonin reuptake inhibitors)
Table 2. Successful Repurposing of Existing Drugs3-7
6
b. Cardiovascular (i.e. calcium channel blockers) c. Antihistamines d. Antihelminthics e. Non-steroidal anti-inflammatory agents f. Nucleoside and nucleobase analogues
3. Two general ways non-antibiotic drugs demonstrate in vitro activity: a. Directly as monotherapy b. Indirectly, in combination regimen, as ‘helper’ antibiotics or adjuvant antibiotics35
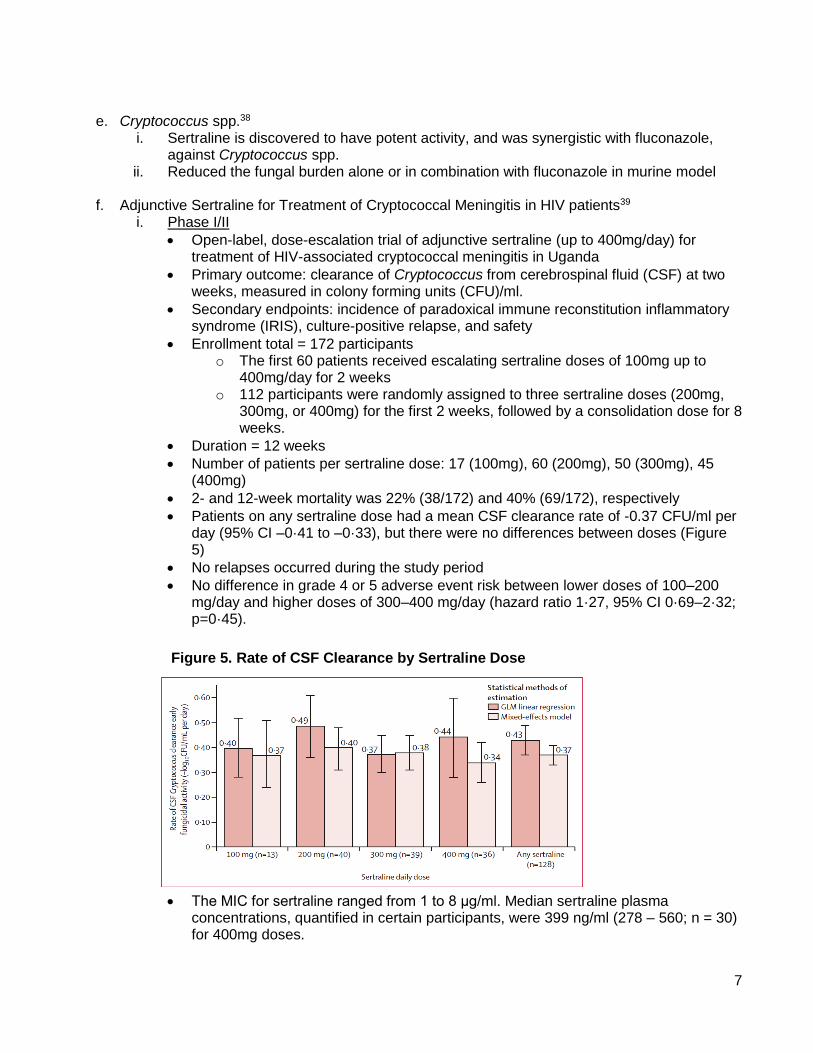
i. Adjuvant antibiotics – Examples of anti-virulence and anti-resistance mechanisms of select non-antibiotic drugsantibiotic potentiating activity are shown in Figure 4.
Examining Sertraline as a Non-Antibiotic:
a. Selective serotonin reuptake inhibitor, approved 1991 b. Antifungal activity discovered serendipitously
c. Candida spp.36
i. Sertraline was rapidly for all four Candida spp: C. albicans, C. glabrata, C. tropicalis, C. parapsilosis
ii. Sertraline minimal fungicidal concentrations (MFCs) (3 – 29 g/ml) exceeded average steady-state plasma levels (55 – 250 ng/ml)
d. Aspergillus spp.37 i. Sertraline in vitro activity was compared to fungicidal activity of other SSRIs (paroxetine,
fluoxetine, citalopram) ii. Sertraline and fluoxetine showed the greatest fungicidal activity iii. Minimum fungicidal concentrations (MFCs) exceeded average steady-state plasma
levels
Figure 4. Antibiotic Adjuvants Mechanisms
7
e. Cryptococcus spp.38
i. Sertraline is discovered to have potent activity, and was synergistic with fluconazole, against Cryptococcus spp.
ii. Reduced the fungal burden alone or in combination with fluconazole in murine model
f. Adjunctive Sertraline for Treatment of Cryptococcal Meningitis in HIV patients39 i. Phase I/II
• Open-label, dose-escalation trial of adjunctive sertraline (up to 400mg/day) for treatment of HIV-associated cryptococcal meningitis in Uganda
• Primary outcome: clearance of Cryptococcus from cerebrospinal fluid (CSF) at two weeks, measured in colony forming units (CFU)/ml.
• Secondary endpoints: incidence of paradoxical immune reconstitution inflammatory syndrome (IRIS), culture-positive relapse, and safety
• Enrollment total = 172 participants o The first 60 patients received escalating sertraline doses of 100mg up to
400mg/day for 2 weeks o 112 participants were randomly assigned to three sertraline doses (200mg,
300mg, or 400mg) for the first 2 weeks, followed by a consolidation dose for 8 weeks.
• Duration = 12 weeks
• Number of patients per sertraline dose: 17 (100mg), 60 (200mg), 50 (300mg), 45 (400mg)
• 2- and 12-week mortality was 22% (38/172) and 40% (69/172), respectively
• Patients on any sertraline dose had a mean CSF clearance rate of -0.37 CFU/ml per day (95% CI –0·41 to –0·33), but there were no differences between doses (Figure 5)
• No relapses occurred during the study period
• No difference in grade 4 or 5 adverse event risk between lower doses of 100–200 mg/day and higher doses of 300–400 mg/day (hazard ratio 1·27, 95% CI 0·69–2·32; p=0·45).
• The MIC for sertraline ranged from 1 to 8 μg/ml. Median sertraline plasma concentrations, quantified in certain participants, were 399 ng/ml (278 – 560; n = 30) for 400mg doses.
Figure 5. Rate of CSF Clearance by Sertraline Dose
8
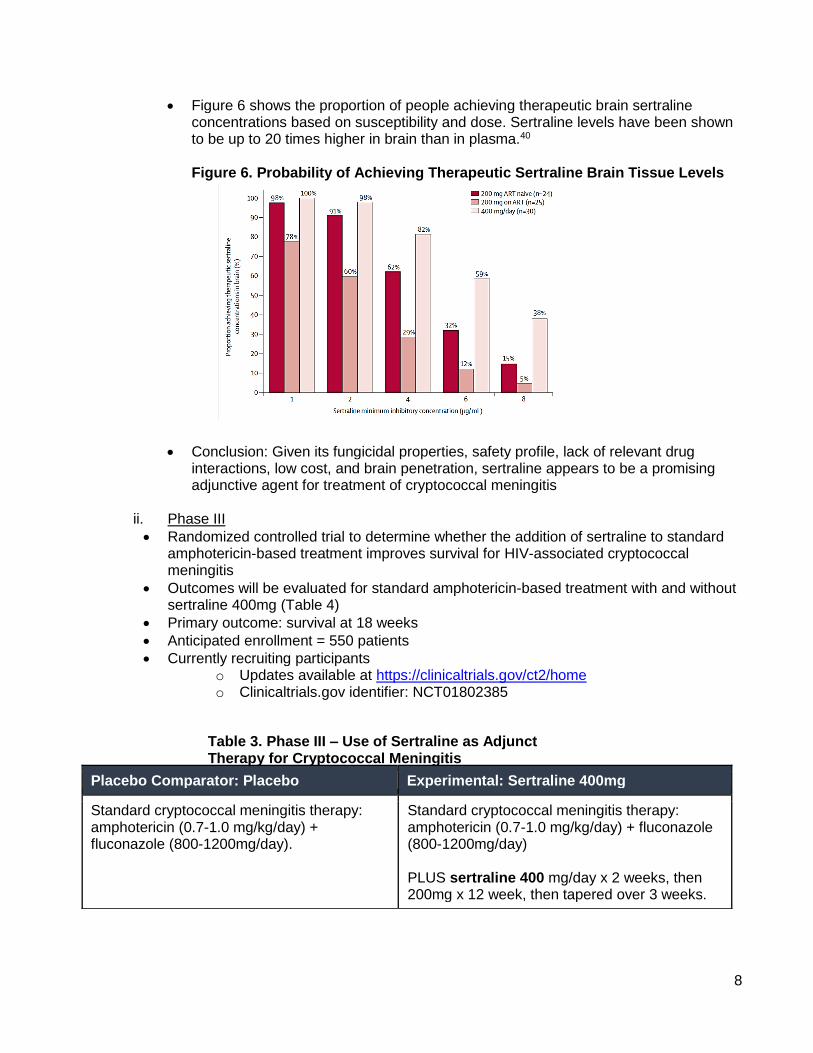
• Figure 6 shows the proportion of people achieving therapeutic brain sertraline concentrations based on susceptibility and dose. Sertraline levels have been shown to be up to 20 times higher in brain than in plasma.40 Figure 6. Probability of Achieving Therapeutic Sertraline Brain Tissue Levels
• Conclusion: Given its fungicidal properties, safety profile, lack of relevant drug interactions, low cost, and brain penetration, sertraline appears to be a promising adjunctive agent for treatment of cryptococcal meningitis
ii. Phase III
• Randomized controlled trial to determine whether the addition of sertraline to standard amphotericin-based treatment improves survival for HIV-associated cryptococcal meningitis
• Outcomes will be evaluated for standard amphotericin-based treatment with and without sertraline 400mg (Table 4)
• Primary outcome: survival at 18 weeks
• Anticipated enrollment = 550 patients
• Currently recruiting participants o Updates available at https://clinicaltrials.gov/ct2/home o Clinicaltrials.gov identifier: NCT01802385
Repurposed s)
Placebo Comparator: Placebo Experimental: Sertraline 400mg
Standard cryptococcal meningitis therapy: amphotericin (0.7-1.0 mg/kg/day) + fluconazole (800-1200mg/day).
Standard cryptococcal meningitis therapy: amphotericin (0.7-1.0 mg/kg/day) + fluconazole (800-1200mg/day) PLUS sertraline 400 mg/day x 2 weeks, then 200mg x 12 week, then tapered over 3 weeks.
Table 3. Phase III – Use of Sertraline as Adjunct Therapy for Cryptococcal Meningitis

9
Examination of Non-Antibiotic Drugs against Staphylococcus aureus Using Novel Screening Assays
Natalie K. Boyd, PharmD, MS1,2 Raymond Benavides, BS2,
Christopher R. Frei, PharmD, MSc1,2
1Pharmacotherapy Division, College of Pharmacy, The University of Texas at Austin;
2Pharmacotherapy Education and Research Center, School of Medicine, UT Health San Antonio, TX
I. Study Objective: The objective of the proposed research is to utilize novel target-based whole cell screening tools to characterize antibiotic effects of select non-antibiotic drugs against Staphylococcus aureus clinical isolates.
II. Central hypothesis: One or more of the selected non-antibiotic drugs will demonstrate antibiotic effects against S. aureus either directly (as monotherapy) or indirectly (in combination with antibiotic drugs).
III. Specific Aims a. Specific Aim 1: Evaluate the in vitro antibiotic activity of non-antibiotic drugs against S.
aureus clinical strains with known resistance genes.
Objective: Determine MICs and MBCs for selected non-antibiotic drugs (amlodipine, azelastine, ebselen, and sertraline) against S. aureus using microplate alamar blue assays.
b. Specific Aim 2: Evaluate the antibiotic-potentiating activity of non-antibiotic drugs against
S. aureus clinical strains with known resistance genes. Objective 2.1: Determine if non-antibiotic drugs (amlodipine, azelastine, ebselen, and sertraline) potentiate ciprofloxacin or tetracycline antibiotic activity using checkerboard titration assays. Objective 2.2: Screen antibiotic-potentiating combinations using an efflux modulation assay and ethidium bromide cartwheel method.
IV. Aim 1 Methods & Rationale: a. Non-antibiotic drugs (Table 4)
Amlodipine, azelastine, ebselen, and sertraline were selected as our test compounds based on several criteria.
• Proven safety and tolerability in humans. Amlodipine, azelastine, and sertraline have been shown to be safe, tolerable, and each have FDA-approved indications. Ebselen is not currently FDA-approved, but has been shown to be safe in humans.41
• Drugs for which antibiotic activity has been demonstrated previously, but with limited data.
10
• Drugs that represented various therapeutic classes.
• Apart from ebselen, we also chose drugs that have been shown to inhibit P-glycoprotein efflux pumps, as this characteristic has been correlated with intrinsic antibiotic activity.42-44
• Note that our range of concentrations for susceptibility testing exceeds average plasma concentrations. These were chosen, in part, based on previous in vitro studies.38, 39, 45-49 Additional factors such as route of administration, site of infection, and drug concentration at tissue sites were also considered.
Table 4. Selected Non-Antibiotic Drugs
SSRI=selective serotonin reuptake inhibitor
b. Bacterial isolates
Non-antibiotic MICs and MBCs were determined for five clinical isolates (Table 5) and one quality control strain (S. aureus 29213). Each isolate is genetically and phenotypically distinct from one another.
• Clinical isolates were selected from previous clinical and epidemiological studies by our research group.50-52
• Isolates were obtained from wound swab cultures of patients with skin and soft tissue infections (SSTIs) who were seen at primary care clinics in the South Texas Ambulatory Research Network (STARNet) from 2010 to 2013.
• Our group has already identified resistance genes and determined susceptibilities in these isolates using whole genome sequencing and Vitek 2 automated testing system (bioMerieux, Durham, NC), respectively.52
Table 5. Whole Genomic Sequencing and Antibiotic Susceptibilities† for Clinical Isolates
†From VITEK® 2 automated system; shaded areas indicate resistance; OX = oxacillin, CLIN = clindamycin, ERY = erythromycin, GENT = gentamicin, TMP-SMX = trimethoprim-sulfamethoxazole, DOX = doxycycline, TET = tetracycline, VAN = vancomycin, CIP = ciprofloxacin
Generic drug name
Drug class or primary
indication
FDA approved
Mean peak plasma
concentrations
Concentrations for MIC
determination (µg/ml)
Amlodipine Ca+ channel
blocker Yes 6-14 ng/ml 12.5 - 256
Azelastine Antihistamine Yes 200 pg/ml 12.5 - 400
Ebselen Antioxidant No N/A 0.25 - 8
Sertraline SSRI Yes 100 - 500 ng/ml 2.5 - 160
Isolate Resistance Genes OX CLIN ERY GENT TMP-SMX
DOX TET VAN CIP
A1019 mecA ≥ 4, R
≤ 0.25 ≤ 0.5 ≤ 0.5 ≤ 10 ≤ 2 ≤ 4 ≤ 0.5 ≤ 0.5
A32 mecA, ermC, tetK,
gyrA(S84L) ≥ 4, R
≥ 4, R ≥ 8, R
≤ 0.5 20 ≤ 2 ≥ 16,
R 1
≥ 8, R
B60 gyrA(S84L), norA ≤ 0.5 ≤ 0.25 ≤ 0.5 ≤ 0.5 ≤ 10 ≤ 2 ≤ 4 ≤ 0.5 ≥ 8, R
B72 ermC, tetK ≤ 0.5 ≥ 4, R ≥ 8, R
≤ 0.5 > 320,
R 4
≥ 16, R
1 ≤ 0.5
J1019 norA ≤ 0.5 ≤ 0.25 ≤ 0.5 ≤ 0.5 ≤ 10 ≤ 2 ≤ 4 ≤ 0.5 ≤ 0.5

11
c. Susceptibility testing
• MICs. Standard broth microdilution procedures were followed according to Clinical Laboratory and Standards Institute (CLSI) methods,53 but with the addition of alamar blue dye. This method follows standard broth microdilution procedures, but with the addition of alamar blue dye, which indicates bacterial growth by change in color (blue to pink) and fluorescence intensity. Rationale for alamar blue. The MIC measure itself, as a single inhibitory concentration, provides little insight about the antibiotic properties of a drug. Both rate and extent of bacterial killing (or inhibition) are defining characteristics of an antibiotic and are therefore critical to understand during early drug discovery.54, 55 These additional endpoints can be captured by simply adding a growth indicator, such as alamar blue. Resazurin, the active ingredient in alamar blue, is a cell permeable non-toxic blue dye that undergoes an oxidation-reduction reaction after entering the cell, changing from non-fluorescent blue (oxidized form) to highly fluorescent, pink-colored resorufin (reduced form). The extent of this color conversion represents cell viability and can be assessed qualitatively, by visual inspection of color change, and quantitatively, with fluorometric readings
• MBCs. Minimum bactericidal concentrations were determined according to CLSI procedures.56 After MICs were recorded, contents from wells for which there is no growth will be plated onto Mueller-Hinton agar plates and incubated 18-24 hours at 35 ± 2°C.
MBCs were defined as the minimum drug concentration that results in a ≥ 99.9% reduction (≥ 3-log10) in CFU/ml from initial inoculum.
d. Fluorometric analysis.
• Fluorescence monitoring was included as an additional measure of antibiotic activity and was measured with Synergy HT Microplate Reader (excitation, 530 nm; emission 590 nm).
• Growth control wells represent 100% fluorescence, while Mueller-Hinton broth with alamar blue represents negative controls. The percent of bacterial growth inhibited by non-antibiotic drugs will be calculated with respect to the growth control. Percent (%) inhibition for a given drug concentration will be calculated
as: (1 −Fluorescencetest drug
Fluorescencegrowth control ) X 100.
V. Aim 2 Methods & Rationale (for antibiotic-potentiating activity):
a. Antibiotics and reference efflux pump Inhibitor. Two antibiotics, ciprofloxacin and tetracycline, and a reference efflux pump inhibitor, reserpine, will be included in this study.
• Rationale. Ciprofloxacin and tetracycline were chosen for the purposes of determining antibiotic-potentiating activity of non-antibiotics and to screen for efflux-inhibition activity (to identify efflux-related mechanism).
• Ciprofloxacin and tetracycline are known substrates for several multidrug resistant efflux pumps (Appendix C).
• NorA, one of the most widely studied efflux pumps in S. aureus, is frequently evaluated with ciprofloxacin as the antibiotic substrate.

12
• TetK is another important efflux pump in S. aureus, conferring high-level resistance to tetracycline. Reserpine has been well-described in its ability to reduce or reverse efflux pump activity on antibiotic and/or biocide substrates.57-59
• As a known efflux pump inhibitor, reserpine is used as an indicator of NorA and TetK efflux pump activity, but is not used clinically due to toxic concentrations required for efflux inhibition.
b. Susceptibility testing. MICs, MBCs, and fluorometric analysis for ciprofloxacin, tetracycline, and reserpine were performed using Aim 1 methods (See IV).
c. Antibiotic-potentiating activity
• Checkerboard Assay.
We determined if non-antibiotic drugs potentiate ciprofloxacin or tetracycline antibiotic activity using a checkerboard titration assay with alamar blue dye.
Serial dilutions of a non-antibiotic drug (amlodipine, azelastine, ebselen, or sertraline) and ciprofloxacin or tetracycline were added at concentrations below the MIC. An example of checkerboard design is shown in Figure 7, where tetracycline was added vertically in each column and non-antibiotics were added across rows.
Figure 7. Checkerboard Template.
A reduction of MIC ≥ 4-fold (or more than one dilution) was considered significant for antibiotic-potentiation.
• Efflux Modulation & Ethidium Bromide Agar Cartwheel Assay57-59 This methodology assesses the presence of efflux pump activity in clinical bacterial isolates.
The isolates are streaked in solid media containing increasing concentrations of EtBr and the fluorescence emitted—which is inversely proportional to their capacity to extrude the compound—is compared to the fluorescence of control strains (i.e. isolates with known efflux activity). An example is described in Appendix D.
VI. Summary of Results
(Only tetracycline, and not ciprofloxacin, combinations are described here) a. Amlodipine, azelastine, and sertraline MICs were 128 µg/ml, 200 µg/ml, and 20 µg/ml,
respectively, for all S. aureus isolates. b. Ebselen MICs ranged from 0.25 µg/ml (SA 29213) to 1 µg/ml (isolate B72). c. Baseline tetracycline (TET) MICs were 32 µg/ml for resistant isolates A32 and B72
13
d. Combining TET with sub-MIC concentrations of non-antibiotics led to a four- to 16-fold decrease in TET MICs (Table 6).
e. Eight- to 16-fold reductions in TET MICs occurred for seven of the eight TET/non-antibiotic combinations, which effectively restored TET susceptibility to S. aureus (≤ 4 µg/ml). Table 6. TET MICs (µg/ml), Alone and in Combination with Non-Antibiotics
AML = amlodipine, AZE = azelastine, EBS = ebselen, SERT = sertraline
VII. Conclusions
a. Complete reversal of tetracycline resistance occurred in seven of the eight TET/non-antibiotic combinations against two S. aureus clinical isolates.
b. Reversal of tetracycline resistance in isolates with tetK gene is suggestive of efflux pump inhibition. However, additional studies are needed for confirmation.
c. Overall, further studies exploring the antibiotic-potentiating effects of non-antibiotics tested herein.
VIII. Strengths a. This study is novel in that it combines two strategic approaches: drug repurposing and
target-based whole cell screens. b. Another unique aspect of this study is the use of the microplate alamar blue assays. The
extent of this color conversion represents cell viability and can be assessed qualitatively, by visual inspection of color change, and quantitatively, with fluorometric readings,
IX. Limitations a. Static drug concentrations: A disadvantage of MIC and MBC testing is that each drug is
tested at a fixed concentration. Although a range of drug concentrations are evaluated, each drug concentration remains constant throughout the incubation period. This differs from drug concentrations in vivo, which fluctuate with dose administration and elimination.
b. Variability in MBC determinations: We anticipate that some variability may be observed with MBC results. What defines bacteriostatic vs. bactericidal has been controversial, as no standardized definition exists. Instead these values (i.e. MBC/MIC ratios) have been arbitrarily assigned based on concept.60 Furthermore, MBC determinations have been shown to be less reproducible as compared to the MIC by broth microdilution, which maintains a precision of ± 1 dilution.
Clinical Isolate
TET Baseline MICs
TET MICs (In Combination with Non-Antibiotic Drugs)
AML 64 µg/ml
AZE 100 µg/ml
EBS 0.25 µg/ml
SERT 10 µg/ml
A32 (MRSA)
32 2 4 2 4
B72 (MSSA)
32 2 2 8 4

14
References 1. Lipinski, C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today
Technol 1, 337-341 (2004). 2. Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J
Pharmacol Toxicol Methods 44, 235-249 (2000). 3. Barratt, M.J. & Frail, D. Drug repositioning : bringing new life to shelved assets and existing
drugs. (John Wiley & Sons, Hoboken, N.J.; 2012). 4. Sardana, D. et al. Drug repositioning for orphan diseases. Brief Bioinform 12, 346-356 (2011). 5. Padhy, B.M. & Gupta, Y.K. Drug repositioning: re-investigating existing drugs for new
therapeutic indications. J Postgrad Med 57, 153-160 (2011). 6. DiMasi, J.A. Innovating by developing new uses of already-approved drugs: trends in the
marketing approval of supplemental indications. Clinical therapeutics 35, 808-818 (2013). 7. Rumore, M.M. Medication Repurposing in Pediatric Patients: Teaching Old Drugs New Tricks. J
Pediatr Pharmacol Ther 21, 36-53 (2016). 8. Mullard, A. 2016 FDA drug approvals. Nat Rev Drug Discov 16, 73-76 (2017). 9. Cima, G. WHO warns of 'post-antibiotic era'. J Am Vet Med Assoc 244, 1356-1357 (2014). 10. Fowler, T., Walker, D. & Davies, S.C. The risk/benefit of predicting a post-antibiotic era: is the
alarm working? Ann N Y Acad Sci 1323, 1-10 (2014). 11. Walsh, C.T. & Wencewicz, T.A. Prospects for new antibiotics: a molecule-centered perspective.
The Journal of antibiotics 67, 7-22 (2014). 12. Palumbi, S.R. Humans as the world's greatest evolutionary force. Science 293, 1786-1790
(2001). 13. Ronald, A.R., Turck, M. & Petersdorf, R.G. A critical evaluation of nalidixic acid in urinary-tract
infections. N Engl J Med 275, 1081-1089 (1966). 14. Singh, S.B. Confronting the challenges of discovery of novel antibacterial agents. Bioorg Med
Chem Lett 24, 3683-3689 (2014). 15. Silver, L.L. Challenges of antibacterial discovery. Clin Microbiol Rev 24, 71-109 (2011). 16. Zheng, W., Thorne, N. & McKew, J.C. Phenotypic screens as a renewed approach for drug
discovery. Drug Discov Today 18, 1067-1073 (2013). 17. Matano, L.M., Morris, H.G., Wood, B.M., Meredith, T.C. & Walker, S. Accelerating the discovery
of antibacterial compounds using pathway-directed whole cell screening. Bioorg Med Chem (2016).
18. Farha, M.A. & Brown, E.D. Unconventional screening approaches for antibiotic discovery. Ann N Y Acad Sci 1354, 54-66 (2015).
19. Swinney, D.C. & Anthony, J. How were new medicines discovered? Nat Rev Drug Discov 10, 507-519 (2011).
20. Beachy, S.H., Johnson, S.G., Olson, S. & Berger, A.C. Drug repurposing and repositioning : workshop summary. (The National Academies Press, Washington, D.C.; 2014).
21. Langedijk, J., Mantel-Teeuwisse, A.K., Slijkerman, D.S. & Schutjens, M.H. Drug repositioning and repurposing: terminology and definitions in literature. Drug Discov Today 20, 1027-1034 (2015).
22. Pryor, R. & Cabreiro, F. Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem J 471, 307-322 (2015).
23. Strittmatter, S.M. Overcoming Drug Development Bottlenecks With Repurposing: Old drugs learn new tricks. Nat Med 20, 590-591 (2014).
24. Toumi, M., Murteira, S., Caban, A. & Kornfeld, A. Drug Repurposing as An Efficient Strategy In Drug Development - Example Of Cns Area. Value Health 17, A435 (2014).
25. Carter, G.T. & Galer, B.S. Advances in the management of neuropathic pain. Phys Med Rehabil Clin N Am 12, 447-459 (2001).
26. Sharlow, E.R. Revisiting Repurposing. Assay Drug Dev Technol 14, 554-556 (2016).

15
27. McBride, W.G. Studies of the etiology of thalidomide dysmorphogenesis. Teratology 14, 71-87 (1976).
28. Vargesson, N. Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res C Embryo Today 105, 140-156 (2015).
29. Ejim, L. et al. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat Chem Biol 7, 348-350 (2011).
30. Nzila, A., Ma, Z. & Chibale, K. Drug repositioning in the treatment of malaria and TB. Future Med Chem 3, 1413-1426 (2011).
31. Stewart, P.S. Prospects for Anti-Biofilm Pharmaceuticals. Pharmaceuticals (Basel) 8, 504-511 (2015).
32. Thangamani, S., Mohammad, H., Abushahba, M.F., Sobreira, T.J. & Seleem, M.N. Repurposing auranofin for the treatment of cutaneous staphylococcal infections. International journal of antimicrobial agents 47, 195-201 (2016).
33. Perlmutter, J.I. et al. Repurposing the antihistamine terfenadine for antimicrobial activity against Staphylococcus aureus. Journal of medicinal chemistry 57, 8540-8562 (2014).
34. Carlson-Banning, K.M. et al. Toward repurposing ciclopirox as an antibiotic against drug-resistant Acinetobacter baumannii, Escherichia coli, and Klebsiella pneumoniae. PloS one 8, e69646 (2013).
35. Wright, G.D. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance: (Trends in Microbiology 24, 862-871; October 17, 2016). Trends Microbiol 24, 928 (2016).
36. Lass-Florl, C., Dierich, M.P., Fuchs, D., Semenitz, E. & Ledochowski, M. Antifungal activity against Candida species of the selective serotonin-reuptake inhibitor, sertraline. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 33, E135-136 (2001).
37. Lass-Florl, C. et al. Antifungal properties of selective serotonin reuptake inhibitors against Aspergillus species in vitro. The Journal of antimicrobial chemotherapy 48, 775-779 (2001).
38. Zhai, B., Wu, C., Wang, L., Sachs, M.S. & Lin, X. The antidepressant sertraline provides a promising therapeutic option for neurotropic cryptococcal infections. Antimicrobial agents and chemotherapy 56, 3758-3766 (2012).
39. Rhein, J. et al. Efficacy of adjunctive sertraline for the treatment of HIV-associated cryptococcal meningitis: an open-label dose-ranging study. The Lancet. Infectious diseases 16, 809-818 (2016).
40. Lewis, R.J., Angier, M.K., Williamson, K.S. & Johnson, R.D. Analysis of sertraline in postmortem fluids and tissues in 11 aviation accident victims. J Anal Toxicol 37, 208-216 (2013).
41. Masaki, C. et al. Effects of the potential lithium-mimetic, ebselen, on brain neurochemistry: a magnetic resonance spectroscopy study at 7 tesla. Psychopharmacology (Berl) 233, 1097-1104 (2016).
42. Leitner, I. et al. The third-generation P-glycoprotein inhibitor tariquidar may overcome bacterial multidrug resistance by increasing intracellular drug concentration. The Journal of antimicrobial chemotherapy 66, 834-839 (2011).
43. Lemaire, S., Van Bambeke, F., Mingeot-Leclercq, M.P. & Tulkens, P.M. Modulation of the cellular accumulation and intracellular activity of daptomycin towards phagocytized Staphylococcus aureus by the P-glycoprotein (MDR1) efflux transporter in human THP-1 macrophages and madin-darby canine kidney cells. Antimicrobial agents and chemotherapy 51, 2748-2757 (2007).
44. Amin, M.L. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 7, 27-34 (2013).
45. Asok Kumar, K. et al. Evaluation of synergism between the aminoglycoside antibiotic streptomycin and the cardiovascular agent amlodipine. Biol Pharm Bull 27, 1116-1120 (2004).
46. Dutta, N.K., Mazumdar, K., DasGupta, A. & Dastidar, S.G. In vitro and in vivo efficacies of amlodipine against Listeria monocytogenes. European journal of clinical microbiology &

16
infectious diseases : official publication of the European Society of Clinical Microbiology 28, 849-853 (2009).
47. Mazumdar, K., Asok Kumar, K. & Dutta, N.K. Potential role of the cardiovascular non-antibiotic (helper compound) amlodipine in the treatment of microbial infections: scope and hope for the future. International journal of antimicrobial agents 36, 295-302 (2010).
48. Younis, W., Thangamani, S. & Seleem, M.N. Repurposing Non-Antimicrobial Drugs and Clinical Molecules to Treat Bacterial Infections. Curr Pharm Des 21, 4106-4111 (2015).
49. Ayaz, M. et al. Sertraline enhances the activity of antimicrobial agents against pathogens of clinical relevance. J Biol Res (Thessalon) 22, 4 (2015).
50. Forcade, N.A. et al. Prevalence, severity, and treatment of community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) skin and soft tissue infections in 10 medical clinics in Texas: a South Texas Ambulatory Research Network (STARNet) study. J Am Board Fam Med 24, 543-550 (2011).
51. Labreche, M.J. et al. Treatment failure and costs in patients with methicillin-resistant Staphylococcus aureus (MRSA) skin and soft tissue infections: a South Texas Ambulatory Research Network (STARNet) study. J Am Board Fam Med 26, 508-517 (2013).
52. Lee, G.C. et al. Comparative whole genome sequencing of community-associated methicillin-resistant Staphylococcus aureus sequence type 8 from primary care clinics in a Texas community. Pharmacotherapy 35, 220-228 (2015).
53. Clinical and Laboratory Standards Institute (CLSI) in CLSI document M07-A10, Edn. 10th (Wayne, PA; 2015).
54. Bhavnani, S.M., Ambrose, P.G. & Jones, R.N. Pharmacodynamics in the evaluation of drug regimens. Ann Pharmacother 36, 530-532 (2002).
55. Ambrose, P.G. et al. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 44, 79-86 (2007).
56. Clinical and Laboratory Standards Institute (CLSI) in CLSI document M26-A (Wayne, Pennsylvania; 1999).
57. Couto, I., Costa, S.S., Viveiros, M., Martins, M. & Amaral, L. Efflux-mediated response of Staphylococcus aureus exposed to ethidium bromide. The Journal of antimicrobial chemotherapy 62, 504-513 (2008).
58. Gibbons, S. & Udo, E.E. The effect of reserpine, a modulator of multidrug efflux pumps, on the in vitro activity of tetracycline against clinical isolates of methicillin resistant Staphylococcus aureus (MRSA) possessing the tet(K) determinant. Phytother Res 14, 139-140 (2000).
59. Marquez, B. Bacterial efflux systems and efflux pumps inhibitors. Biochimie 87, 1137-1147 (2005).
60. Pankey, G.A. & Sabath, L.D. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 38, 864-870 (2004).
61. Costa, S.S., Viveiros, M., Amaral, L. & Couto, I. Multidrug Efflux Pumps in Staphylococcus aureus: an Update. The open microbiology journal 7, 59-71 (2013).
62. Jang, S. Multidrug efflux pumps in Staphylococcus aureus and their clinical implications. J Microbiol 54, 1-8 (2016).
63. Van Bambeke, F., Pages, J.M. & Lee, V.J. Inhibitors of bacterial efflux pumps as adjuvants in antibiotic treatments and diagnostic tools for detection of resistance by efflux. Recent Pat Antiinfect Drug Discov 1, 157-175 (2006).
17
APPENDICES A. FDA Expedited Review Programs
* Fast track designation was amended by the FDA Safety and Innovation Act (FDASIA) in 2012 to include the Generating Antibiotics Incentives Now Act (GAIN Act)
Appendix B. Examples of ‘Rescued’ Drugs26
Program
name
Year instituted
Criteria for qualifying products Key characteristics
Accelerated approval
1992 - Treatment of serious condition - Evidence suggests reasonable advantage over existing therapies
- Can be approved on basis of surrogate endpoint - Approval granted on a conditional basis (Phase IV confirmatory trials are required)
Priority review
1992
- Treatment of serious condition or a drug designed as a Qualified Infectious Disease Product (QIDP) - Improvement in safety or effectiveness over existing therapies
Shorter FDA review timeline (ten vs six months)
Fast track 1997; 2012*
- Treatment of serious condition or a drug designed as a QIDP - Preclinical or clinical evidence demonstrating potential to address unmet medical need.
Can be approved after one phase-II study
Breakthrough therapy
2012
- Treatment of serious condition - Demonstrates substantial improvement over existing therapies on one or more clinically important endpoints
Intensive FDA guidance throughout development to
generate additional safety and efficacy data
Drug Intended Indication “Rescue” Indication
Allopurinol Antineoplastic Gout
Atomoxetine Antidepressant Attention-deficit/hyperactivity disorder
Azidothymidine (AZT)
Cancer HIV/AIDS
Eflornithine Cancer Hirsutism
Gemcitabine Antiviral Anticancer agent
Lomitapide Common lipidemia Familial hypercholesterolemia
Miltefosine Cutaneous metastases of breast cancer
Visceral leishmaniasis
Thalidomide Nausea, morning sickness Leprosy, multiple myeloma
18
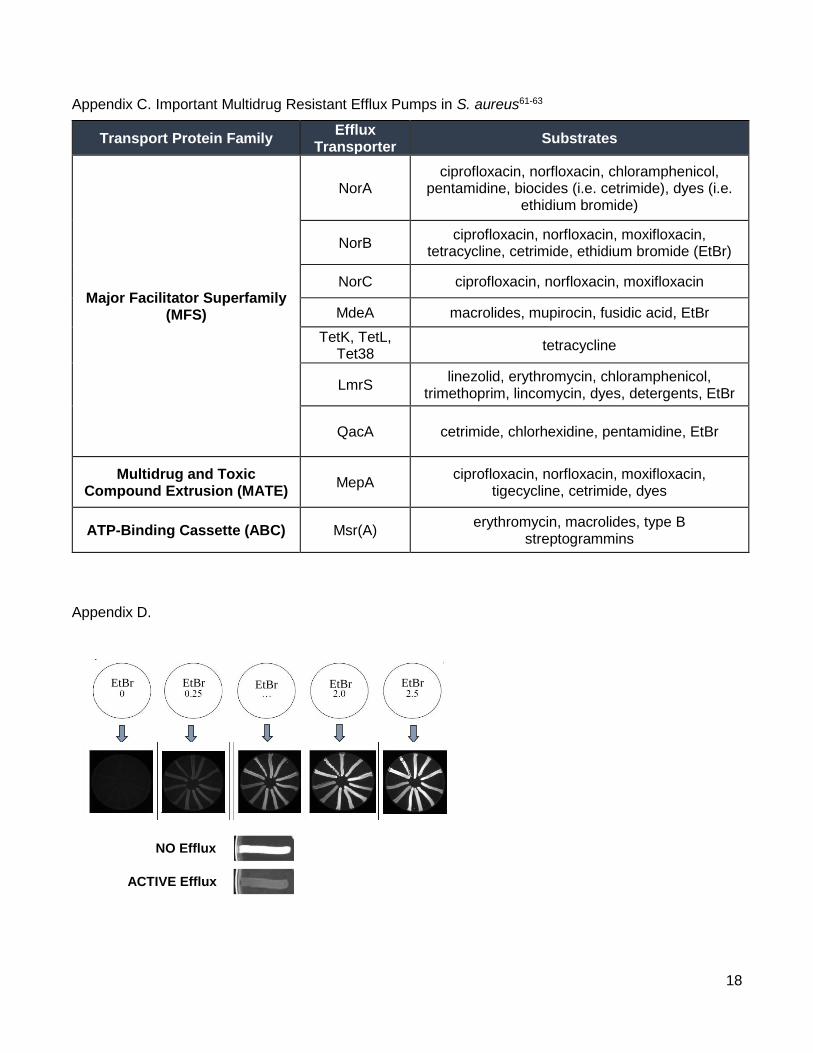
Appendix C. Important Multidrug Resistant Efflux Pumps in S. aureus61-63
Appendix D.
Transport Protein Family Efflux
Transporter Substrates
Major Facilitator Superfamily (MFS)
NorA ciprofloxacin, norfloxacin, chloramphenicol,
pentamidine, biocides (i.e. cetrimide), dyes (i.e. ethidium bromide)
NorB ciprofloxacin, norfloxacin, moxifloxacin,
tetracycline, cetrimide, ethidium bromide (EtBr)
NorC ciprofloxacin, norfloxacin, moxifloxacin
MdeA macrolides, mupirocin, fusidic acid, EtBr
TetK, TetL, Tet38
tetracycline
LmrS linezolid, erythromycin, chloramphenicol,
trimethoprim, lincomycin, dyes, detergents, EtBr
QacA cetrimide, chlorhexidine, pentamidine, EtBr
Multidrug and Toxic Compound Extrusion (MATE)
MepA ciprofloxacin, norfloxacin, moxifloxacin,
tigecycline, cetrimide, dyes
ATP-Binding Cassette (ABC) Msr(A) erythromycin, macrolides, type B
streptogrammins
NO Efflux
ACTIVE Efflux
EtBr EtBr EtBr EtBr EtBr
EtBr EtBr EtBr EtBr EtBr