dr. m. sofi md; frcp (london); frcpedin; frcsedin
TRANSCRIPT

MULTIPLE ENDOCRINE NEOPLASIA
Dr. M. Sofi MD; FRCP (London); FRCPEdin; FRCSEdin

1963 by Wermer, reported
MENsyndromes, consist of
rare, autosomal dominant
mutations in genes thatregulate cell growth.Current classification: Type I & type II MEN Type II divided into the
type IIA & type IIB MEN
Menin protein, produced by the MENIN gene, is a tumor suppressor.
Loss of this protein allows tumors to arise.
Ret protein, produced by the
RET gene , proto-oncogene,
can be constitutively activated,
causing abnormal cellProliferation.
MULTIPLE ENDOCRINE NEOPLASIA

Type 1 MEN: Hyper functioning of following tumors:
All 4 parathyroid glands Pancreatic islets ◦ gastrinoma◦ insulinoma ◦ glucagonoma◦ vasoactive intestinal
peptide tumor (VIPoma)◦ pancreatic polypeptide–
producing tumor (PPoma) Anterior pituitary ◦ prolactinoma ◦ somatotropinoma◦ corticotropinoma ◦ nonfunctioning tumors
Other associated tumors include:◦ lipomas, ◦ angiofibromas◦ located in the adrenal gland
cortex (rarely, in the adrenal medulla).
Hyperparathyroidism is the most common manifestation of type 1 MEN (80% of presentations)
Islet-cell tumors that secrete predominantly gastrin are called gastrinomas; these tumors frequently metastasize
Pheochromocytomas are reported in patients with type 1 MEN
Thymic and bronchial carcinoid tumors can also be associated with type 1 MEN
MULTIPLE ENDOCRINE NEOPLASIA

Type 2A MEN is defined by: Thyroid carcinoma Pheochromocytoma (50%
Hyperparathyroidism (20%)
However Familial MTC is hereditary MTC without other associated endocrinopathies
Type 2A MEN (Sipple syndrome) accounts for most cases of type 2 MEN.
Type 2 MEN affects about 1 in 40,000 individuals, and fewer than 1000 kindreds are known worldwide.
C-cell hyperplasia develops early in life and can be viewed as the precursor lesion for MTC, which often arises multifocally and bilaterally.
Pheochromocytomas are bilateral in 70% of cases and develop on the background of adrenomedullary hyperplasia secondary to an RET germline mutation.
Less than 25% of patients with type 2A MEN develop frank hyperparathyroidism
MULTIPLE ENDOCRINE NEOPLASIA
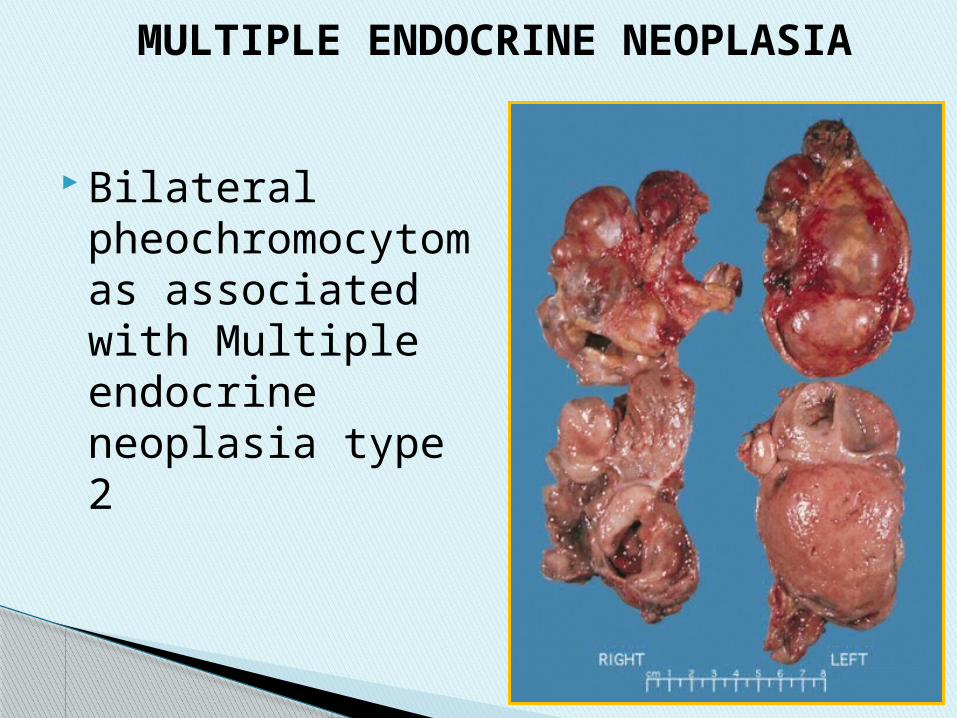
Bilateral pheochromocytomas associated with Multiple endocrine neoplasia type 2
MULTIPLE ENDOCRINE NEOPLASIA

Type 2B MEN is defined by: MTC Pheochromocytoma. Associated abnormalities
include: ◦ Mucosal neuromas ◦ Medullated corneal nerve
fibers◦ Marfanoid habitus
MTC is relatively aggressive and frequently occurs in childhood
Pheochromocytomas also occur earlier than in patients with type 2A MEN, and patients have the same features arising in the context of adrenomedullary hyperplasia, multifocality, and, often, bilateral involvement
Carney complex Carney complex is a
distinct, rare type of MEN characterized by:
Primary pigmented adrenocortical disease,
Pituitary adenoma, Sertoli-cell tumors, Thyroid nodules, and Additional nonendocrine
features. The most commonly
associated features are cardiac and skin myxomas, melanotic schwannomas, and lentigines.
MULTIPLE ENDOCRINE NEOPLASIA



Type 1 MEN The MENIN gene
responsible for type 1 MEN has been localized to chromosome band 11q13;
It produces a nuclear protein called menin, a tumor suppressor.
The MENIN gene is ubiquitously expressed and is localized to the nucleus of cells
There is increasing evidence that menin may act in DNA repair or synthesis.
Type 2 MEN The genetic mutation in type
2 MEN occurs in RET proto-oncogene, located on band 10q11.2
This gene defect has been observed in both MEN2A & MEN2B
Activation of RET leads to hyperplasia of target cells in vivo.
Subsequent secondary events then lead to tumor formation. RET is specifically expressed in neural crest–derived cells.
The presence or absence of RET expression in parathyroid tissue is unknown
Etiology

Type 1 MEN Hyperparathyroidism is most common initial clinical
manifestation of type 1 multiple endocrine neoplasia (MEN). Some patients may manifest findings of ZES before they have
hyperparathyroidism. Symptoms of gastrinoma may become clinically apparent
either with abdominal pain and diarrhea or with complications such as ulcer perforation or bleeding.
Type 2A MEN All patients develop MTC on the basis of C-cell hyperplasia. About 50% of patients with MTC manifest pheochromocytomas
(usually late in life), and 20% of patients have hyperparathyroidism.
Type 2B MEN Pheochromocytomas occur earlier than in patients with type
2A MEN.
Clinical presentation

The age of onset of endocrine tumors is usually in the teenage years, but symptoms from these tumors may not appear for several years, and the diagnosis is frequently delayed until the fourth decade of life.
Cutaneous tumors may develop prior to the manifestation of overt clinical symptoms resulting from endocrine tumors.
The earliest cutaneous tumors appear in the teenage years
Hyperparathyroidism: hypersecrete hormone causing hypercalcaemia and recurrent nephrolithiasis
Zollinger-Ellison syndrome (hypergastrinaemia)
hypoglycaemia (hyperinsulinaemia)
Prolactinoma: amenorrhoea (hyperprolactinaemia)
Acromegaly (excess growth hormone).
Tumors of the pituitary gland may cause symptoms by mass effects.
Angiofibromas, collagenomas, and lipomas do not typically cause symptoms, and they are mostly of cosmetic concern
Clinical presentation MEN Type 1

Parathyroid hyperplasia and adenomas
Hyperparathyroidism is the presenting feature of multiple endocrine neoplasia type 1 (MEN1) in about 80% of patients.
Patients present either with asymptomatic hypercalcaemia on biochemical screening or with the features of sporadic hyperparathyroidism.
All four glands are diffusely hyperplastic and there may be nodule formation.
Pancreatic endocrine tumors These occur in about 70% of
patients with MEN1. 60% of tumors are
gastrinomas and produce ZES PU account for most of the
MEN1 morbidity and mortality
About 30% are insulinomas. VIPoma (vasoactive intestinal
peptide and pancreatic polypeptide-secreting tumor)
Duodenal microgastrinoma is very common and probably accounts for almost half of all MEN1-associated gastrinomas.
They are usually multiple, with up to 15 separate tumors.
Clinical presentation MEN Type 1

Pituitary adenomas Present by screening in 30% of
patients, but is found at post-mortem in 50%.
Unlike the pancreas and parathyroid, there does not appear to be diffuse pituitary hyperplasia.
Prolactinoma producing hyperprolactinaemia occurs in about 30% of cases.
They tend to be more aggressive than sporadic cases.
Acromegaly, due to excessive human growth hormone (hGH) occurs in about 30%.
Adrenocorticotrophic hormone(ACTH) may produce Cushing's syndrome but other functioning tumors are rare.
Skin lesions Occur in nearly 90% of
patients, but they can be easily overlooked.
Benign tumors include multiple angiofibromas, collagenomas, and lipomas.
They should be sought because they can act as markers for this syndrome.
Other lesions Lesions in other tissues have
been reported, but their relationship remains controversial.
Carcinoid tumors of the foregut, midgut, and thymus occur in about 10%, and are often found in the pancreas, but they are rarely symptomatic.
Clinical presentation MEN Type 1

Screening of first- and second-degree relatives of patients with multiple endocrine neoplasia type 1 (MEN1)
Diagnosis of MEN1 depends on having a high level of suspicion in patients who present with multiple
Facial angiofibromas, Collagenomas, and
lipomas Features
hyperparathyroidism increased gastric acid
secretion.Investigations include: Hormone hypersecretion
blood tests Imaging studies to look for
the presence of tumors.
DNA testing is available and identifies a mutation in about 80% of patients with familial MEN1.
Mutation analysis may be used to confirm the clinical diagnosis
Screen asymptomatic family members.
Prenatal diagnosis for pregnancies at increased risk is possible if the disease-causing mutation in a family is known
Diagnosis MEN type 1

Bilateral, anteroposterior radiographic views of the hands in a patient with type 1 (MEN1) and primary hyperparathyroidism. These images show subperiosteal bone resorption along the radial aspects of the middle phalanges.
Imaging features MEN Syndrome

Computed tomography (CT) scan of the pancreas in a patient with multiple endocrine neoplasia syndrome type 1 (MEN1) and a gastrinoma. This image shows a pancreatic head mass (large, white arrow), as well as a low-attenuating lesion in the liver (small, black arrowhead) that indicates metastases. Calcifications of the right renal medullary pyramids (medullary nephrocalcinosis; black arrows) in this nonenhanced CT scan.
Imaging features MEN Syndrome

Computed tomography (CT) scan image with oral and intravenous contrast in a patient with biochemical evidence of insulinoma. The 3-cm contrast-enhancing neoplasm (arrow) is seen in the tail of the pancreas (P) posterior to the stomach (S)
Imaging features MEN Syndrome

6x5x5 cm adrenal mass arising from right adrenal
Imaging features MEN Syndrome

The screening of first- and second-degree relatives of patients with (MEN1) is aimed at early detection of parathyroid, pancreatic or pituitary lesions in gene carriers, to reduce the associated morbidity.
There is no evidence that screening reduces mortality
Identification of affected individuals in 'malignant kindred' with aggressive pancreatic disease may allow curative surgery which would be expected to prolong survival.
Screening lowers the age of detection of the syndrome by about 20 years.
Screening tests are serum calcium, fasting gastrin, and prolactin.
Sensitive markers of pancreatic disease are basal and test-meal stimulated pancreatic polypeptide and gastrin, and basal insulin and proinsulin.
80% of affected individuals will have been identified by the 5th decade.
Screening of sporadic pancreatic endocrine tumors for evidence of MEN1 is probably justified, especially for gastrinomas or insulinomas.
There is little evidence to support screening in those with sporadic pituitary tumors.
Screening tests MEN type 1

If the condition is confirmed, then genetic counseling is required
Pharmacological Diazoxide can be used to
inhibit release of insulin, especially in tumors that are beyond surgery.
High-dose proton pump inhibitors are required for gastrin-secreting tumors.
After surgery to the pituitary, hormone replacement may be required
Surgical Skin tumors may be removed The surgical approach to
pancreatic endocrine tumors in MEN1 is controversial:◦ Surgical cure is best
achieved by removing the pancreas and duodenum with adjacent lymph nodes. There is still a high rate of recurrence but the overall mortality remains low
Pituitary tumors: Same as for sporadic pituitary tumors.
Parathyroidectomy, subtotal or complete, is practiced for MEN1 but long-term follow-up reveals a high rate of recurrence in MEN1.
The treatment of metastatic disease is the same as in sporadic cases.
Management

The average age of death in individuals with multiple endocrine neoplasia type 1 (MEN1) is significantly lower (55.4 years for men and 46.8 years for women) than that of the general population.
Pancreatic endocrine tumors, particularly gastrinomas, become malignant in about 50% of patients with MEN1.
Untreated, patients may die from peptic ulcer disease, metastatic endocrine pancreatic carcinoma, or foregut carcinoid malignancy.
Pancreatic endocrine tumors associated with MEN1 are less malignant than sporadic tumors and carry a better prognosis, with a median survival of 15 years compared to 5 years for patients with sporadic tumors.
This may reflect more indolent disease or earlier diagnosis.
Prognosis MEN type 1

Patients may present with symptoms related to:◦ Medullary thyroid
carcinoma◦ Hyperparathyroidism◦ Pheochromocytoma.
Virtually all patients have MCT symptoms may include:◦ hypertension ◦ episodic sweating ◦ diarrhoea ◦ pruritic skin lesions◦ lump in the neck(which
may cause compressive symptoms).
Hypercalcaemia may lead to:◦ constipation,◦ polyuria,◦ polydipsia,◦ memory problems◦ depression, ◦ nephrolithiasis, ◦ glucose intolerance, ◦ gastro-oesophageal reflux,
and◦ fatigue
Chronic constipation.Cutaneous lichen amyloidosis
in multiple endocrine neoplasia type 2A (MEN2A) presents with
multiple pruritic, hyperpigmented, lichenoid papules in the scapular area of the back.
Clinical presentation MEN Type 2A

Cutaneous lichen amyloidosis

Present earlier than MEN2A. Neuromas usually predate
MTC and phaeochromocytoma.
Almost all patients have a Marfan's-like habitus.
Neuromas appear as:◦ Glistening bumps around
the lips, tongue, and lining of the mouth.
◦ Bumps on the eyelids, which are often thickened - neuromas may also appear on the cornea and conjunctiva.
Intestinal ganglioneuromatosis affects about 75% of cases.
Neuromas involve the autonomic nerves of both the myenteric and submucosal plexi and can cause poor suckling, with failure to thrive, constipation, diarrhea, recurrent pseudo-obstruction, toxic megacolon.
Patients also often develop spinal abnormalities. These features, along with thickened lips and eyelids are associated with Marfanoid habitus.
Involvement of peripheral motor and sensory nerves can cause a peroneal muscular atrophy (Charcot-Marie-Tooth syndrome)
Delayed puberty is a common feature.
Clinical presentation MEN Type 2B

MULTIPLE ENDOCRINE NEOPLASIA
Mucosal neuromas

Screening for pheochromocytoma is 24 hours urine for elevated catecholamines and catecholamine metabolites, especially vanillyl-mandelic acid (VMA).
Clinical suspicion or elevated urinary catecholamine values demand an abdominal MRI scan. A metaiodobenzylguanidine (MIBG) scan is useful for localizing pheochromocytomas
Thyroid tumors can be investigated initially by ultrasound and fine-needle aspiration.
Medullary thyroid carcinoma (MTC) is suspected with an elevated plasma calcitonin concentration.
This is a specific and sensitive marker.
In provocative testing, calcitonin concentration is measured before and 2 and 5 minutes after intravenous administration of calcium.
Parathyroid abnormalities are diagnosed when there are simultaneously elevated serum calcium and parathyroid hormone levels with an elevated urinary calcium to creatinine ratio.
Diagnosis of multiple endocrine neoplasia type 2

The two types of moleculardiagnosis for MEN2 are
mutationanalysis and linkage analysis
ofthe RET proto-oncogene(chromosomal locus 10q11) Genetic linkage analysis has 98-
99% predictive accuracy Screening, to identify affectedindividuals and for early
detectionof thyroid, parathyroid andadrenal disease, reduces bothmorbidity and mortality in MEN2: Annual 24-hour urine
collections for catecholamine concentrations to detect pheochromocytoma at the earliest age possible.
Recurrence of medullary thyroid carcinoma (MTC) should be monitored with◦ Calcitonin ◦ Carcinoembryonic antigen
(CEA) ◦ Provocative calcitonin
testing with the pentagastrin stimulation test, with serial measurements of serum calcitonin measured.
False-positive and false-negative results have been reported
Screening for multiple endocrine neoplasia type 2

General principles In type 2 (MEN2) the goals
are: Identify individuals with
germline RET-disease-causing mutations associated with MEN2 before symptoms develop.
Reduce morbidity and mortality in the highest-risk individuals through either:
Prophylactic thyroidectomy Screening for medullary
thyroid carcinoma (MTC) Screening
phaeochromocytoma and parathyroid disease before symptoms develop.
Counseling of patients with MEN that they know what to expect and why continued follow-up
MEN2A The treatment for adrenal
medullary hyperplasia or phaeochromocytoma is bilateral adrenalectomy.
If an adrenal lesion is identified at the same time as MTC, the adrenalectomy performed first.
Total thyroidectomy as young as 3 years for MEN2A if they contain the genetic mutation.
Hyperparathyroidism: subtotal parathyroidectomy is advised, along with cervical thymectomy because of the increased risk of supernumerary parathyroid glands.
Recurrent hyperparathyroidism is less likely to occur in MEN2A patients than in MEN1 patients
Management MEN type 2

In MEN 2B, thyroidectomy with lymph node clearance should be performed at the earliest possible age.
MTC is biologically aggressive in these patients and has been reported as early as 15/12 age, with metastases by the age of 3 years.
Patients with the genetic mutation for MEN2B, total thyroidectomy is recommended in infancy.
Patients not identified by screening, thyroidectomy should still be performed.
In all patients with palpable tumors, central lymph node dissection should also be performed.
Useful markers in the follow-up of MTC are plasma calcitonin and carcinoembryonic antigen (CEA).
Prognosis Patients with MEN2B tend to
do worse than those with MEN2A, as the MTC is more aggressive.
The prognosis is poor in this group, with recurrent disease in about 20% of patients with clinically occult but macroscopic MTC
Over 60% with palpable MTC. It is particularly poor in
individuals with MEN2B who present with clinically apparent MTC.
Their 10-year survival is about 50%, and death from metastatic disease in the mid-twenties is common
Management MEN type 2B

Initial thyroid lesion in MEN2 is C-cell hyperplasia, which has been found as early as the age of 3 years in MEN2A and may be present at birth in MEN2B.
Over 5 to 10 years microscopic MTC develops and finally gross tumors become apparent.
MTC typically presents as a neck mass at about age 15 to 20 years.
More than 50% already have cervical lymph node metastases.
MTC may occur alone without other features of MEN2.
MTC is a tumor of the C cells which secrete calcitonin, and this acts as a tumor marker
Inherited MTC accounts for 25% of cases in association with MEN types 2A and 2B
It may present with a thyroid lump diarrhea may also occur.
Metastases may occur, to the lung, liver and bone.
Paraneoplastic syndromes are rare Cushing's or carcinoid may occur
Investigations include ultrasound scan, calcitonin levels, FNA, and DNA testing for familial cases.
Adverse prognostic indicators include older age, higher-grade lesions and incomplete surgical resection of the lesion
Medullary cell carcinoma of the thyroid

Phaeochromocytoma occurs in 50% of MEN2.
In patients with MEN2 are usually found in the adrenals
About 70% are bilateral, almost all are benign.
Produce excessive adrenaline secretion leading to tachycardia, palpitations, hypertension and headache.
Investigations include plasma concentrations of free metanephrines, and CT or MRI
Positron emission tomography (PET) is also used for diagnosis.
Treatment surgical, laparoscopic surgery is increasingly used.
Overall, half of the patients with malignant pheochromocytomas remain alive for five years.
Pheochromocytoma