
Viral genome sequencing: applications to clinical management and public health
Professor Judy Breuer

Why do whole viral genome sequencing
Genome sequencing allows detection of multigenic resistance in widely spaced genes Genome sequencing may resolve nosocomial transmission events especially for DNA viruses where there is insuffient variation in small fragments Genome sequencing may identify outbreaks or patterns of spread
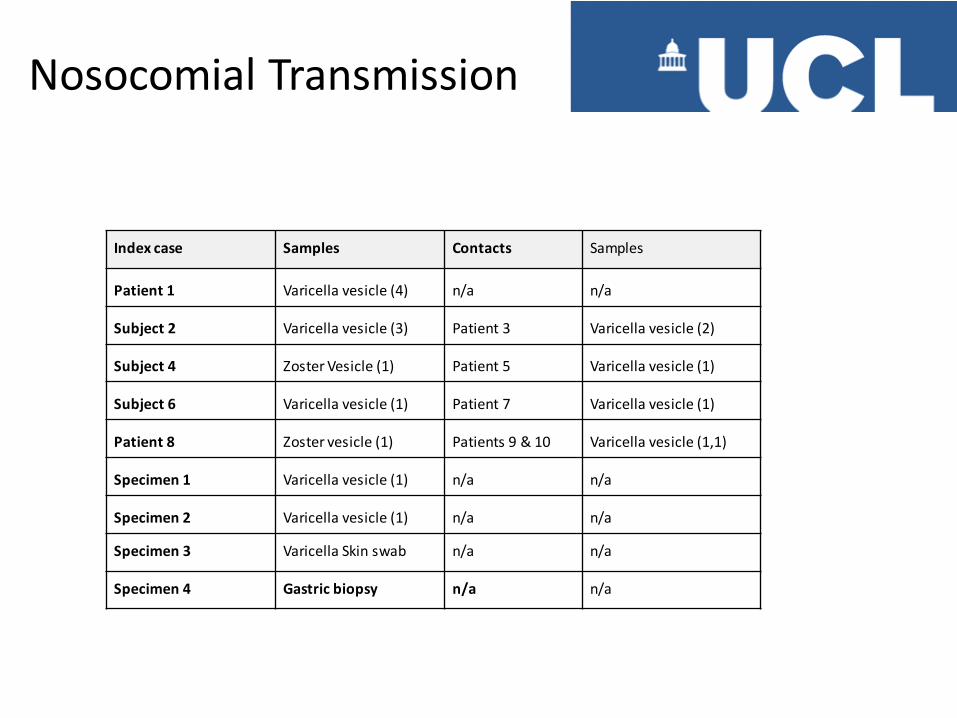
Index case Samples Contacts Samples
Patient 1 Varicella vesicle (4) n/a n/a
Subject 2 Varicella vesicle (3) Patient 3 Varicella vesicle (2)
Subject 4 Zoster Vesicle (1) Patient 5 Varicella vesicle (1)
Subject 6 Varicella vesicle (1) Patient 7 Varicella vesicle (1)
Patient 8 Zoster vesicle (1) Patients 9 & 10 Varicella vesicle (1,1)
Specimen 1 Varicella vesicle (1) n/a n/a
Specimen 2 Varicella vesicle (1) n/a n/a
Specimen 3 Varicella Skin swab n/a n/a
Specimen 4 Gastric biopsy n/a n/a
Nosocomial Transmission

Clade 1
Clade 3
Clade 5

Clade 1
Clade 3
Clade 5
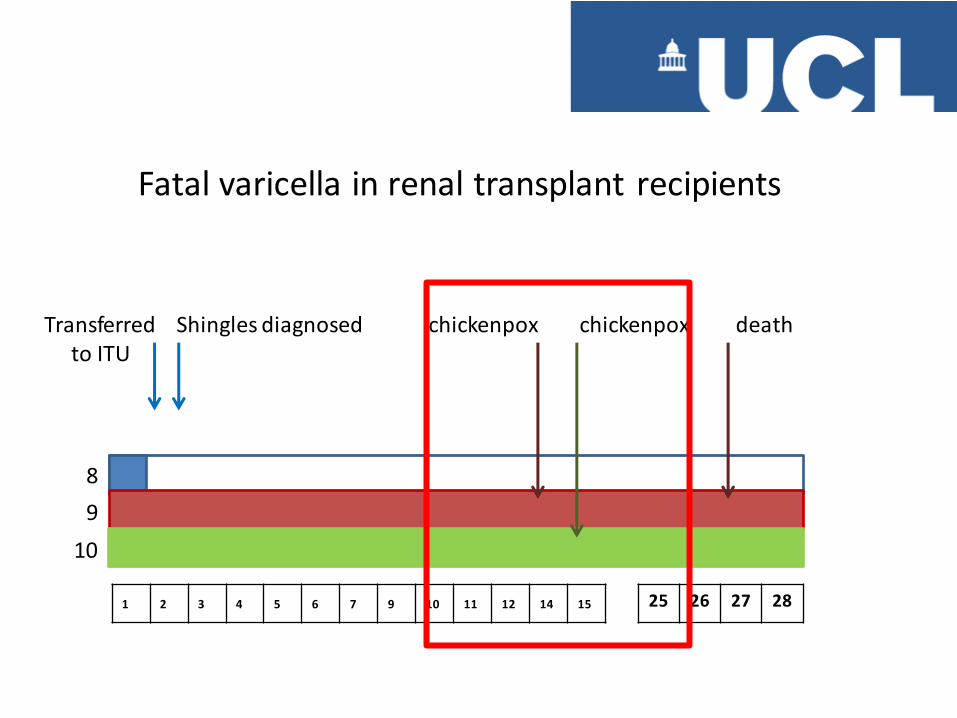
Transferred to ITU
8
9
10
1 2 3 4 5 6 7 9 10 11 12 14 15 25 26 27 28
Shingles diagnosed chickenpox chickenpox death
Fatal varicella in renal transplant recipients
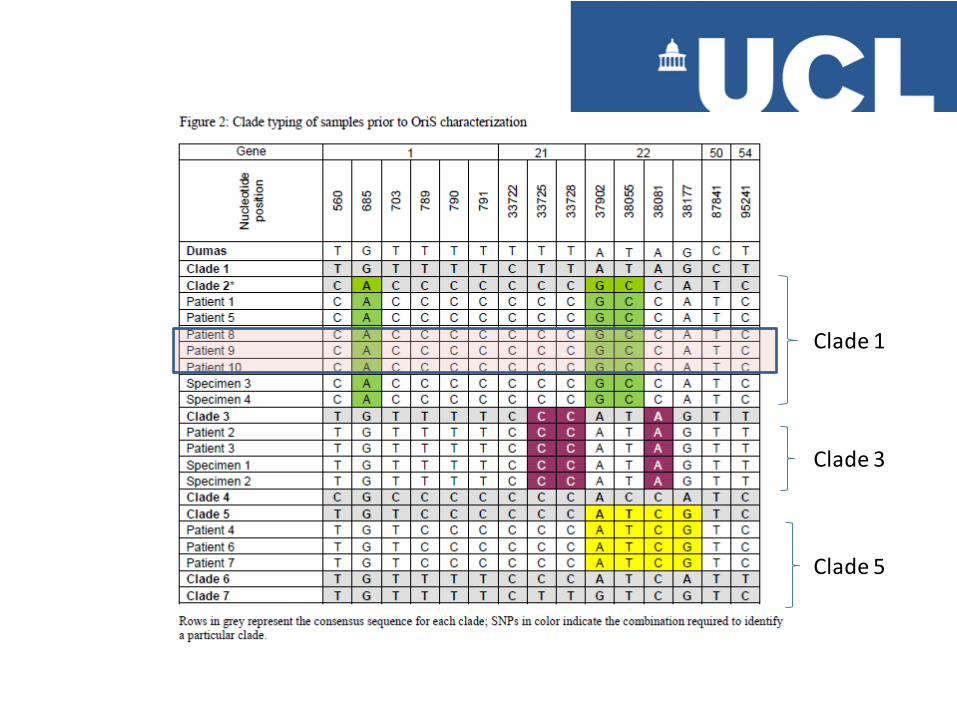
Clade 1
Clade 3
Clade 5
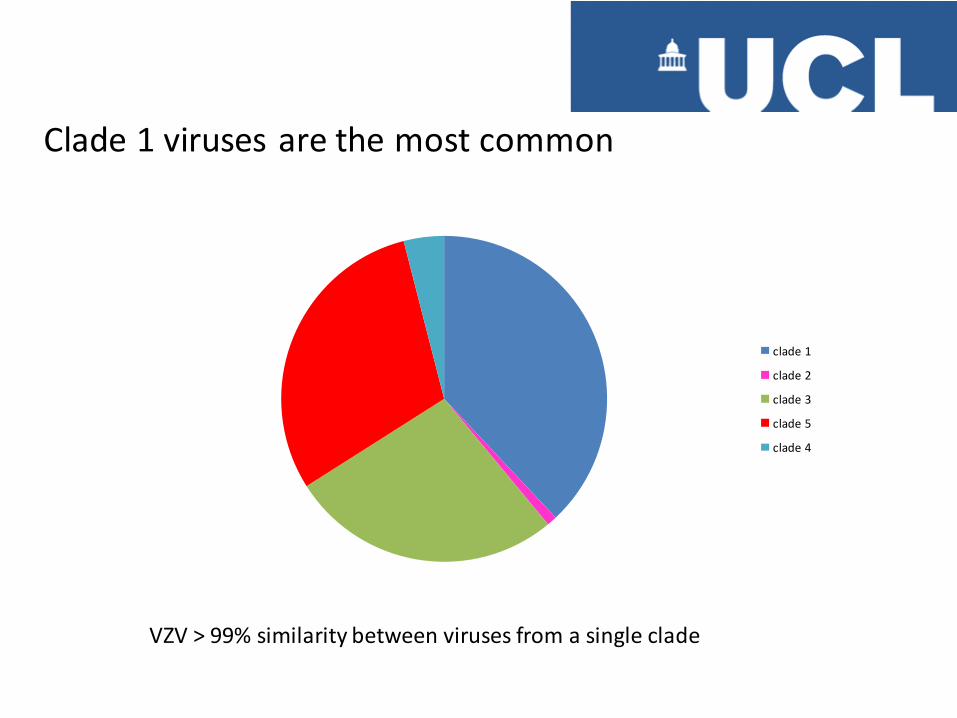
clade 1
clade 2
clade 3
clade 5
clade 4
Clade 1 viruses are the most common
VZV > 99% similarity between viruses from a single clade

Problems with whole viral genome sequencing
Virus are intracellular organisms and unlike bacteria, sufficient viral nucleic acid for good quality NGS sequencing cannot easily be obtained from culture (human DNA overwhelms sequence) PCR can be used to amplify fragments for sequencing but this is not suitable for larger viruses or small volume clinical specimens (contain too little viral nucleic acid)

(6) Reference guided assembly using Burrows-Wheeler Aligner (mapped vs. VZV strain Dumas , pOka or vOKA)
(5) Quality control: Removes poor quality reads (QUASR)
(4) Sequencing (Illumina) generating paired-end reads (76bp) Incl 12-16 rounds PCR
(1) Total DNA extracted from clinical sample (± WGA)
(2) Fragmentation and library preparation ( Illumina-based protocol)
(3) Target DNA isolated by hybridisation with custom 120-mer RNA baits
Purifying viral DNA for NGS
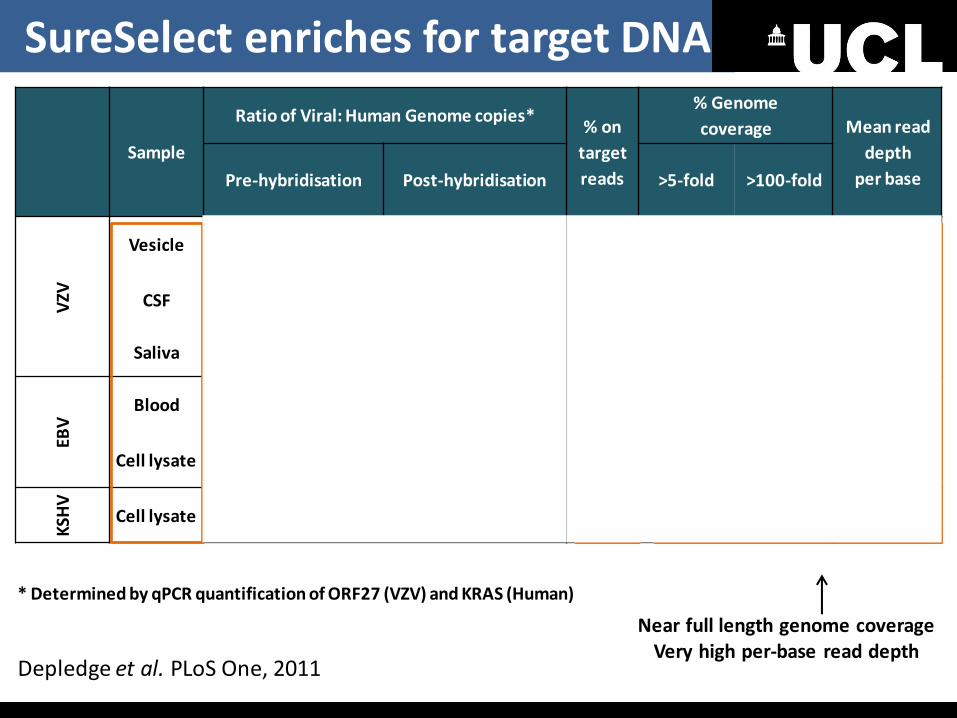
Sample
Ratio of Viral: Human Genome copies* % on
target
reads
% Genome
coverage Mean read
depth
per base Pre-hybridisation Post-hybridisation >5-fold >100-fold
VZV
Vesicle 10299 1157666 93.69 >99% >97% 3022
CSF 34976 1006398 64.47 >99% >97% 2416
Saliva 51987 9855143 42.61 100% >98% 1096
EBV
Blood
86.37 >99% >98% 2523
Cell lysate 52.84 >98% >97% 2599
KSH
V
Cell lysate 90.97 >98% >93% 1773
Near full length genome coverage Very high per-base read depth
* Determined by qPCR quantification of ORF27 (VZV) and KRAS (Human)
100-1000 fold enrichment
High % of on target reads
Depledge et al. PLoS One, 2011
SureSelect enriches for target DNA

Evolution and pathogenesis Sequencing viruses by NGS
A C A A T
Biallelic positions- important for minority variant resistance etc
Consensus
Multiple sequence reads for each base

Method Validated
1. Highly sensitive – can recover <1000 VZV genomes in < 10ng DNA Depledge 2011
2. Reproducible and representative of the original Depledge 2013
3. Less mutagenic than PCR (without loss of population structure due to culture)
Depledge 2011
Error<1%
Targeted enrichment

*
*
*
*
Viruses from transmission cluster
Kundu, Depledge unpub

* * *
*
*
* * * *
*
*
*
*
* *
* *
* * * *
*
* * *
*
Clade 1 Clade 3 Clade 5 Clade 4 Clade 2
Kundu, Depledge unpub

Outbreak monitoring
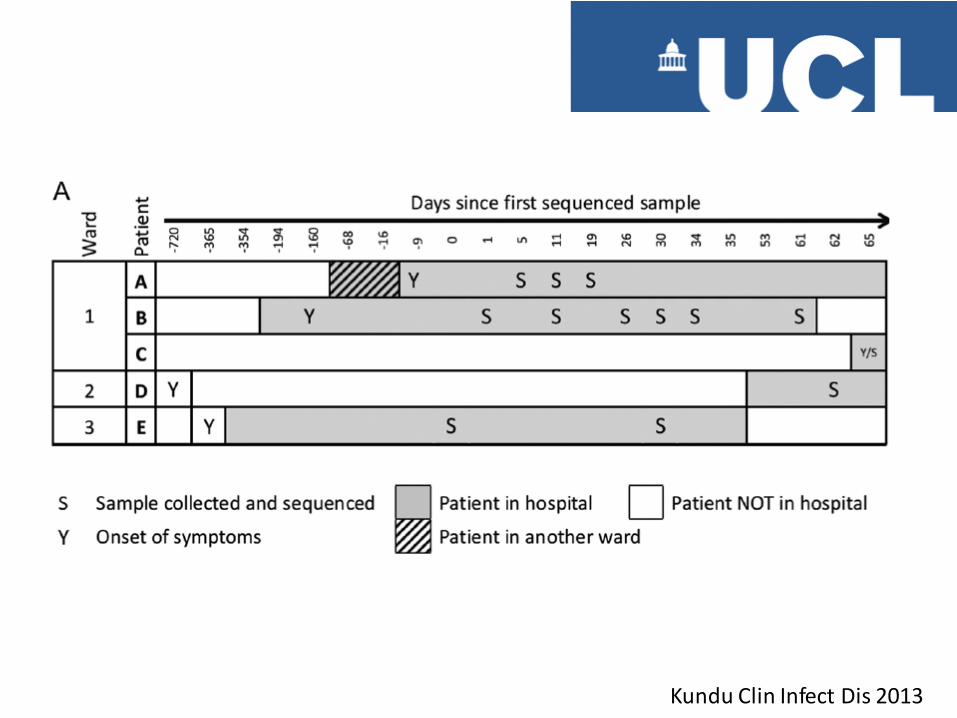
Kundu Clin Infect Dis 2013

Kundu Clin Infect Dis 2013
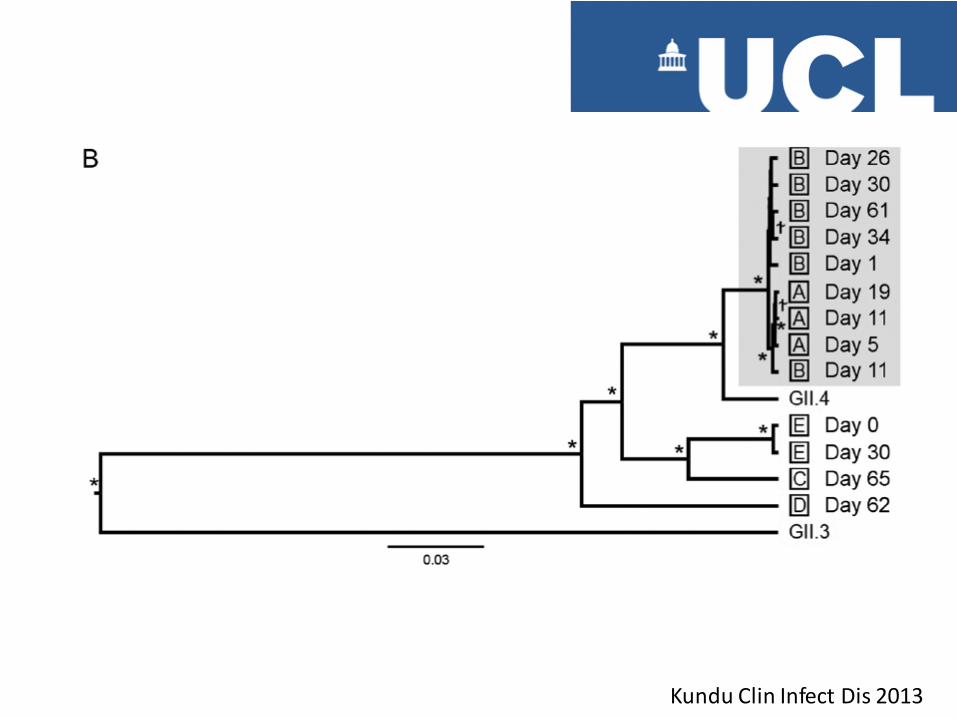
Kundu Clin Infect Dis 2013

Kundu Clin Infect Dis 2013

Substitutions between A and B
Kundu Clin Infect Dis 2013
20 SNPs, (6 coding for aa substitution) 9 Informative ie biallelic

Direction of Transmission
Minority variant in B becomes fixed in A following transmission bottleneck Confirms direction of transmission is B to A
Kundu Clin Infect Dis 2013

What does this study tell us 1. Whole genome sequencing identifies nosocomial
transmission(even where patients are isolated)
2. Norovirus shed by immunocompromised is infectious (A became infected only after moving to same ward as B)
3. Norovirus infection triggers chronic diarrhoea in immunocompromised A developed diarrhoea only after becoming infected with norovirus
4. Deep sequencing data can be used to identify direction of spread

Breuer group UCL
Viral genome sequencing Dan Depledge Samit Kundu Julianne Lockwood Eleanor Grey Fanny Wegner Rachel Williams Helena Tutil Mark Quinlivan Mette Christiansson
Cambridge University
Ian Goodfellow Lucy Thorne
GOSH John Hartley
UCL Genomics Mike Hubank
PHE Portsmouth Gill Underhill







