
Doctoral dissertation
To be presented by permission of the Faculty of Medicine of the University of Kuopio
for public examination in Auditorium L22, Snellmania building, University of Kuopio,
on Friday 13th November 2009, at 12 noon
Institute of Clinical MedicineDepartment of Clinical Microbiology
University of Kuopio
JONNA EEVA
Receptor-Mediated Apoptosis in B Cells
From the Regulation of B Cell Survival to Potential Novel Approaches for Lymphoma Therapy
JOKAKUOPIO 2009
KUOPION YLIOPISTON JULKAISUJA D. LÄÄKETIEDE 463KUOPIO UNIVERSITY PUBLICATIONS D. MEDICAL SCIENCES 463

Distributor : Kuopio University Library P.O. Box 1627 FI-70211 KUOPIO FINLAND Tel. +358 40 355 3430 Fax +358 17 163 410 www.uku.fi/kirjasto/julkaisutoiminta/julkmyyn.shtml
Series Editors: Professor Raimo Sulkava, M.D., Ph.D. School of Public Health and Clinical Nutrition Professor Markku Tammi, M.D., Ph.D. Institute of Biomedicine, Department of Anatomy
Author´s address: Institute of Clinical Medicine Department of Clinical Microbiology University of Kuopio P.O. Box 1627 FI-70211 KUOPIO FINLAND E-mail : eeva@hytti .uku.fi Supervisors: Professor Jukka Pelkonen, M.D., Ph.D. Institute of Clinical Medicine Department of Clinical Microbiology University of Kuopio
Mine Eray, M.D., Ph.D. School of Medicine, University of Tampere Department of Pathology, Tampere University Hospital
Reviewers: Professor Marko Salmi, M.D., Ph.D. MediCity Research Laboratory University of Turku
Professor Mauno Vihinen, Ph.D. Institute of Medical Technology University of Tampere
Opponent: Professor Seppo Meri, M.D., Ph.D. Haartman Institute University of Helsinki
ISBN 978-951-27-1363-9ISBN 978-951-27-1380-6 (PDF)ISSN 1235-0303
KopijyväKuopio 2009Finland

Eeva, Jonna. Receptor-mediated apoptosis in B cells. From the regulation of B cell survival to
potential novel approaches for lymphoma therapy. Kuopio University Publications D. Medical
Sciences 463, 2009. 93 p.
ISBN 978-951-27-1363-9
ISBN 978-951-27-1380-6 (PDF)
ISSN 1235-0303
ABSTRACT Apoptosis or programmed cell death plays an important role in the regulation of B cell survival.
Failures in the apoptotic signaling can lead to autoimmune diseases or cancer. Moreover, apoptosis
is a major mode of cell death in cancer therapy, and resistance to therapy is often associated with
disturbances in the apoptotic signaling. The detailed knowledge of the apoptotic signaling pathways
can be used as an advantage in the development of new treatment modalities against B cell
malignancies. Two major signaling pathways leading to apoptosis have been described. The
extrinsic pathway is commonly triggered by activation of death receptors, such as Fas/CD95, and
involves the activation of the cysteine protease caspase-8. The intrinsic pathway is initiated by
various extracellular or intracellular stress signals, which induce changes in mitochondrial
membrane permeability and trigger the activation of the caspase-9. The initiator-caspases -8 or -9
activate the cascade of downstream caspases, which are responsible for organized degradation of
cellular organelles.
During adaptive immune response, B cells are selected based on their affinity for foreign antigens,
such as surface molecules of bacteria. The selection of antigen activated B cells takes place at the
germinal centers (GC) of lymphoid organs, and is regulated by surface receptors including B-cell
receptor (BCR), Fas/CD95 and CD40. In this study, BCR- and Fas -induced apoptosis of follicular
lymphoma cells was inhibited by CD40 stimulation. BCR-induced apoptosis involved the
mitochondrial breach and caspase-9 activation pathway, while in Fas-induced apoptosis caspases-8
and -3 were activated independently of mitochondria. The CD40-mediated protection against Fas-
induced apoptosis was associated with a rapid and NF-κB-dependent up-regulation of c-FLIP
proteins, natural inhibitors of caspase-8 activation. Based on our findings, BCR-induced apoptosis
may be involved in the deletion of self-reactive cells generated during the GC reaction, while Fas-
induced apoptosis may be involved in the deletion of low affinity B cells.
Follicular lymphoma is a cancer that originates from GC B cells. The second aim of this study was
to explore signaling pathways of apoptosis induced by rituximab, a chimeric monoclonal antibody
used for the treatment of B cell lymphomas. Rituximab-mediated apoptosis of follicular lymphoma
cells was dependent on the activation of caspase-9 and mitochondrial breach, while the death
receptor pathway was not involved. Moreover, the simultaneous triggering of Fas enhanced
significantly apoptosis induced by rituximab. Thus, the combined use of rituximab with drugs
designed to destabilize mitochondrial membranes, or with drugs that induce the activation of the
death receptor pathway, is warranted for further investigation in clinical settings.
National Library of Medicine Classification: QU 375, WH 200, WH 525, QZ 267
Medical Subject Headings: Apoptosis; Cell Death; Cell Survival; B-Lymphocytes; Signal
Transduction; Caspases; Caspase 8; Caspase 9; Mitochondria; Mitochondrial Membranes;
Receptors, Antigen, B-Cell; Antigens, CD95; Antigens, CD40; NF-kappa B; CASP8 and FADD-
Like Apoptosis Regulating Protein; Antibodies, Monoclonal; Receptors, Death Domain;
Lymphoma, Follicular/drug therapy


ACKNOWLEDGEMENTS
This study was carried out at the Department of Clinical Microbiology, Institute of Clinical
Medicine, University of Kuopio, during the years 2000-2009.
I express my deepest gratitude to my supervisor Professor Jukka Pelkonen, M.D., Ph.D. for his
positivism and encouraging support during this study. I want especially thank Jukka for his wide
expertise on the field of immunology and molecular biology, and always open-minded attitude for
new ideas. I owe my sincere thaks to my second supervisor Mine Eray M.D, Ph.D., for many
supporting discussions, and for her work contribution in the establishment and charachterization of
follicular lymphoma cell lines used in this study.
I address my warmest thanks to all my former and current colleagues at the Department of
Clinical Microbiology for their unforgettable companionship during all these years. Especially, I
want to thank my colleague and dear friend Ulla Nuutinen M.Sc. for her invaluable support, critical
review of the manuscripts and for fascinating discussions. I also wish to thank other members of our
“B cell group” Mikko Mättö Ph.D., Antti Ropponen M.Sc., Ville Postila M.D., and Anna-Riikka
Pietilä M.Sc. for their collaboration and invaluable help during these years. This study would not
exist without your contribution and expertise. My special thanks belong to Pia Keinänen, Päivi
Kivistö, Riitta Korhonen and Eila Pelkonen for their excellent technical assistance and friendship.
I am deeply grateful to official reviewers of this thesis, Professor Mauno Vihinen and Professor
Marko Salmi, for critical evaluation of this thesis and their most valuable comments.
I wish to thank all my friends for their support during the occasional disappointments during this
study, and for sharing many delight running, biking, skiing, paddling, skating and hiking moments
with me.
I owe my sincere thanks to my parents Päivikki and Hannu for giving me support and
encouragement during all these years, and providing me home where education was greatly
appreciated. I also express warm thanks to my sister Salla and her husband Renato, and my sister
Suvi-Tuuli and her life-companion Jaakko for sharing enjoyable moments during our holidays. I am
deeply indebted to Suvi-Tuuli for her precious contribution in the laboratory works, and caring for
Pihla when ever it was needed.
Finally, I dedicate my warmest thanks to Jarno for love, care and sharing the dark and delight
moments during this project. My most loving thanks goes to our little sunshine Pihla, for fulfilling
our days with joy of living, and for allowing me to accomplish this dissertation project during the
maternity leave.
This work was financially supported by Kuopio University Hospital (EVO-fund), Finnish Medical
Foundation, and KELA- the Social Insurance Institute of Finland.
Kuopio, October 2009
Jonna Eeva


ABBREVIATIONS
AIF apoptosis inducing factor
Bax Bcl-2 associated X protein
BAFF B-cell-activating factor of the tumor necrosis factor family
Bcl-2 B-cell lymphoma 2 protein
Bcl-xL Bcl-2 related gene (large variant)
BCR B-cell receptor
BH Bcl-2 homolgy
CD cluster of differentiation
CREB cAMP response element-binding protein,
DISC death-inducing signaling complex
DN dominant negative
ERK extracellular signal regulated kinase
FADD Fas associated death domain
FL follicular lymphoma
FLIP flice inhibitory protein
FLIPI follicular lymphoma prognostic index
GC germinal center
GFP green fluorescence protein
GSK3 glycogen synthase kinase-3
IL interleukine
IAP inhibitor of apoptosis protein
IMS intermembrane space
JNK c-Jun N-terminal kinase
kDa kilo Dalton
lpr lymphoproliferative
mAb monoclonal antibody
MAPK mitogen activated protein kinase
MOMP mitochondrial outer membrane permeabilization
NF-κB nuclear factor-κB
NF-AT nuclear factor of activated T cells
SHM somatic hypermutation
SMAC second mitochondria-derived activator of caspase/direct IAP-binding protein with
low PI
OM outer membrane
PI propidium iodide
PI3K phosphatidyl-inositol-3-kinase
PLC phospholipase-c
PKC protein kinase c
TNF tumor necrosis factor
TRAIL tumor necrosis factor-related apoptosis inducing ligand
TRAF tumor necrosis factor associating factor
zVAD benzyloxycarbonyl-Val-Ala-Asp
Ψm mitochondrial membrane potential


LIST OF ORIGINAL PUBLICATIONS
The thesis is based on the following original publications, which are referred to in the text by the
Roman numerals (I-IV).
I Eeva J *, Postila V*, Mättö M, Nuutinen U, Ropponen A, Eray M, Pelkonen J. Kinetics and
signaling requirements of CD40-mediated protection from B cell receptor-induced apoptosis.
Eur J Immunol 2003;33:2783-91.
II Eeva J, Ropponen A, Nuutinen U, Eeva ST, Mättö M, Eray M, Pelkonen J. The CD40-induced
protection against CD95-mediated apoptosis is associated with a rapid upregulation of anti-
apoptotic c-FLIP.
Mol Immunol 2007;44:1230-7
III Eeva J, Nuutinen U, Ropponen A, Mättö M, Eray M, Pellinen R, Wahlfors J, Pelkonen J.
Feedback regulation of mitochondria by caspase-9 in the B cell receptor-mediated apoptosis.
Accepted for publication. Scand J Immunol.(In press)
IV Eeva J, Nuutinen U, Ropponen A, Mättö M, Eray M, Pellinen R, Wahlfors J, Pelkonen
J. The involvement of mitochondria and the caspase-9 activation pathway in rituximab-
induced apoptosis in FL cells. Apoptosis 2009;14:687-98.
* Equal contributors
The original publications have been reproduced with permission of the copyright holders.
The thesis includes also previously unpublished data.


CONTENTS
1. INTRODUCTION ............................................................................................. 13
2. REVIEW OF THE LITERATURE ................................................................ 15
2.1. Apoptosis ..................................................................................................... 15 2.1.1 Different ways to die; apoptosis, autophagy and necrosis ............................................... 15 2.1.2 Mitochondrial changes during apoptosis ......................................................................... 17 2.1.3 Caspases – executioners of the apoptotic cell death ........................................................ 22
2.2. B cells and the humoral immune response .............................................. 24 2.2.1 An overview of the humoral immune response ............................................................... 24 2.2.2 The B cell antigen receptor; structure and activation ...................................................... 25 2.2.3. B cell maturation ............................................................................................................ 28 2.2.4 Life and death decisions of a B cell; regulation of B cell apoptosis ................................ 32 2.2.5 GC as an origin of B cell malignancies ........................................................................... 35
2.3. Monoclonal antibody therapy for cancer treatment ............................... 37
2.4. Follicular lymphoma .................................................................................. 40 2.4.1. Incidence and clinical characteristics ............................................................................. 40 2.4.2 Pathophysiology .............................................................................................................. 40 2.4.3 Current treatment of follicular lymphoma ....................................................................... 41
3. AIMS OF THE STUDY .................................................................................... 46
4. MATERIALS AND METHODS ..................................................................... 47
4.1. Cell line and culturing ............................................................................... 47
4.2. Establishment of overexpressing cell lines ............................................... 47
4.3. Cell treatment experiments ....................................................................... 48
4.4 Apoptosis detection ..................................................................................... 49 4.4.1 Quantification of fractional DNA content (I-IV) ............................................................ 49 4.4.2 Mitochondrial membrane potential depolarization (I-IV) ............................................... 50 4.4.3 Cytochrome c release (I-IV) ............................................................................................ 50 4.4.4 Caspase activation (I-IV) ................................................................................................ 50 4.4.5 Cell membrane permeabilization III ................................................................................ 51
4.5. Western blotting ......................................................................................... 51
4.6. Reverse transcriptional (RT)-polymerase chain reaction ...................... 52
4.7 Statistical analyses....................................................................................... 52
5. RESULTS .......................................................................................................... 53
5.1. The molecular mechanisms of B cell receptor-induced apoptosis (I, III)
............................................................................................................................. 53 5.1.1 The signal transduction pathways connected with B cell receptor-induced apoptosis .... 53 5.1.2 The role of caspase-9 and mitochondrion in B cell receptor-induced apoptosis (I, III) .. 54 5.1.3 The caspase-8 activation pathway showed only marginal role in B cell receptor-induced
apoptosis................................................................................................................................... 56 5.2. The molecular mechanisms of Fas/CD95-induced apoptosis (II, III, IV)
............................................................................................................................. 58
5.3. The molecular mechanisms of rituximab-induced apoptosis (IV) ......... 58

5.4. Molecular mechanisms of CD40-mediated protection against apoptosis
(I, II and IV) ...................................................................................................... 60 5.4.1 Kinetics and signaling requirements of CD40 mediated protection from B cell receptor-
mediated apoptosis (I) .............................................................................................................. 61 5.4.2 The CD40-induced protection against Fas-induced apoptosis was associated with a rapid
up-regulation of anti-apoptotic c-FLIP (II) .............................................................................. 61 6. DISCUSSION .................................................................................................... 63
6.1. FL cell lines as an experimental model for the study of receptor-
mediated apoptosis ............................................................................................ 63
6.2. Molecular mechanisms of receptor-mediated apoptosis ........................ 64 6.2.1 Fas-induced apoptosis proceed via caspase-8 activation pathway while B cell receptor-
and rituximab –induced apoptosis are dependent on mitochondrial pathway .......................... 64 6.2.2 Feedback regulation of mitochondria by caspase-9 during intrinsic apoptosis ............... 65 6.2.3. Caspase-9 –mediated apoptosis was poorly inhibited by pan-caspase-inhibitor z-VAD-
fmk ........................................................................................................................................... 67 6.2.4. What happens before mitochondrial breach during intrinsic apoptosis? ........................ 67
6.3. Molecular mechansims of CD40-mediated protection against apoptosis
(I, II and IV) ...................................................................................................... 68
6.4. A proposed model of the molecular interactions during the germinal
center negative selection ................................................................................... 71
7. CONCLUSIONS; Therapeutic applications and future directions ............. 73
7.1. Cell surface receptors in the regulation of life and death decisions in the
germinal center .................................................................................................. 73
7.2. CD40-CD40L interactions as potential targets for cancer therapy ....... 74
7.3. Novel approaches to enhance rituximab therapy.................................... 75
8. REFERENCES .................................................................................................. 77
APPENDIX: ORIGINAL PUBLICATIONS I-IV

13
1. INTRODUCTION
In our body, millions of cells are being produced every day. To maintain homeostasis, the
divisions of cells must be delicately balanced by deletion of unnecessary cells. Elegantly, these cells
are cleared by a tightly regulated process called apoptosis or programmed cell death.
Apoptosis is genetically regulated form of cell death, in which a single cell is sacrificed for the
benefit for the whole organisms. There are several circumstances in which multicellular organisms
benefit for the targeted deletion of cells. For example, cells irreversibly injured by virus infection,
radiation, growth factor withdrawal or other violating signals from outside of the cell are no longer
useful for the organism and thus eliminated. In addition, various stress signals originating from
inside of the cell, such as DNA lesions that have occurred during cell divisions, can induce
programmed cell death. Unfortunately, some of the cells which have gained abnormalities in genes
regulating apoptosis may escape from cell death and continue to proliferate. These cells are
potentially hazardous, since the further accumulation of growth promoting and apoptosis inhibiting
lesions can in rare circumstances lead to development of cancer, a disease characterized by
uncontrolled cell proliferation.
Follicular lymphoma (FL) is a heterogeneous cancer disease of B cell origin. In Finland, over two
hundred follicular lymphoma cases are diagnosed annually. Until these days, this disease has been
assumed as incurable, and thus only the patients with symptoms or with advanced disease are
treated with standard chemotherapy. However, immunotherapy with anti-CD20 antibody rituximab
has recently improved the overall survival and disease free time of FL patients when combined to
standard chemotherapy. When given to patients, rituximab depletes B cells by three overlapping
mechanisms; complement-mediated cytotoxicity, antibody-dependent cytotoxicity and apoptosis.
Thus far, the molecular mechanisms of rituximab-induced apoptosis are largely unsolved. The great
advantage of rituximab and also other therapies based on the use of immunotherapeutic antibodies is
their specificity for the cancer cells and thus milder side effects as compared to standard
chemotherapies. Inspired by rituximab, several immunotherapeutic drugs are currently under
clinical or preclinical trials for the treatment of B cell lymphomas and also for other cancer diseases.
Study of apoptotic signaling pathways is important, since these pathways are potential targets
when developing new treatment modalities for cancer. Commonly, cancer is associated with
dysregulation of apoptotic pathways, which offers targets for drug discovery. Majority of cancer
therapeutics used currently delete cancer cells by apoptosis. Thus, failures in apoptotic signaling can

14
result in the resistance against cancer therapy, which may be hindered by drugs promoting
apoptosis. This work was aimed to reveal the molecular mechanisms involved in the receptor-
mediated apoptosis of B cells. Apoptotic pathways induced by B cell receptor and Fas are important
regulators of B cell fate during B cell development and immune reactions. The molecular
mechanisms of CD20-induced cell death are currently unknown despite the wide use of anti-CD20
antibody rituximab in the immunotherapy. This work proposes signaling pathways involved in
BCR, Fas and CD20 –mediated apoptosis in a FL cell line. The detailed knowledge of the apoptotic
signaling pathways may be used in the future for the development of novel, apoptosis directed
treatment modalities against cancer diseases.

15
2. REVIEW OF THE LITERATURE
2.1. Apoptosis
2.1.1 Different ways to die; apoptosis, autophagy and necrosis
In the human body dazzling rate of continuous cell division must be compensated by cell death to
maintain tissue homeostasis. Either deficient or excessive cell death can lead to a variety of
pathologic conditions. For example, insufficient death of cells with deleterious genetic lesions may
result in the development of cancer. Conversely, ischemia induced cell death during stroke or
myocardial infarction incurs irreversible tissue damage.
Cell death can be classified according to morphological appearance, biochemical patterns and
functional aspects (programmed versus accidental) (Galluzzi et al., 2007). Mainly based on the
morphological features, three different forms of cell death can be classified; apoptosis, autophagy
and necrosis. However, in some instances the dying cell has overlapping features of different cell
death modalities making the exact classification unfeasible.
Apoptosis or programmed cell death (type I cell death) is genetically engineered, physiological
form of cell death where the single cell is sacrificed for the benefit for the whole organism. The
term apoptosis was first introduced in the 1970`s century by Kerr and Wyllie, who first described
the cell death with specific morphological features (Kerr et al., 1972). These well defined
morphological changes include cell shrinkage and nuclear condensation (pyknosis), nuclear
fragmentation (karyohexis) and plasma membrane blebbing (Galluzzi et al., 2007). Finally the intact
cytoplasmic organelles or fragments of the nucleus are cleanly packaged inside membrane bounded
vacuoles which are also called apoptotic bodies. Eventually, these apoptotic bodies are recognized
and engulfed by phagocytic cells, without evoking an inflammatory response which could be
harmful for the surrounding tissue. Apoptotic cell death is most often associated with typical
biochemical events including DNA fragmentation, cysteine protease activation and mitochondrial
membrane potential collapse, but these events are not definitive for apoptosis (Galluzzi et al., 2007).
Autophagy (type II cell death) is a process where parts of the cytoplasm are sequestered within
double-membrane vacuoles and finally digested by lysosomal hydrolases (Kroemer and Jäättelä,
2005). Autodigestion of cellular constituents may serve as a defense mechanism against nutrient
deficiency or sub-lethal cellular injury and therefore autophagy may even prevent the cell death in
some conditions. However, extensive autodigestion may lead to cell death especially in the cases

16
where apoptosis is somehow disturbed. The morphological hallmarks of autophagy include the
double-membrane autophagic vacuoles and the lack of chromatin condensation and other typical
features of apoptosis (Galluzzi et al., 2007).
Necrosis (type III cell death) is usually defined as a harmful event as it is often associated with
pathological conditions involving excessive cell loss, and the disadvantage of necrotic cells to
promote inflammatory response (Galluzzi et al., 2007). The necrotic cells undergo extensive
swelling and unorganized dismantling of cytoplasmic organelles which eventually leads to the
rupture of plasma membrane and spilling of the cell contents to surrounding extracellular space.
Necrosis lacks the typical morphological characteristics of apoptosis and massive vacuolization seen
during autophagic cell death. Necrosis is most often accompanied with the mitochondrial
dysfunction including extensive production of reactive oxygen species, mitochondrial membrane
permeabilization (MMP), ATP depletion and failure in Ca2+
homeostasis. The activation of calpain
and cathepsin proteases and the lysosomal rupture are typically observed during the necrotic cell
death. While sufficient ATP and glucose content are obligatory for apoptosis, their depletion favors
switching to necrosis. However, the view that apoptosis is the only physiological or advantageous
form of cell death whereas necrosis is always harmful and pathological process has recently gained
a momentum. Indeed, apoptotic cell death can be shifted to necrosis in the circumstances where the
activation of caspase-activation is artificially blocked (Goldstein et al., 2005). In addition, in some
instances necrosis can be programmed and triggered appropriately by specific plasma membrane
receptors, although these characteristics of cell death have been previously associated only with
apoptotic cell death (Galluzzi et al., 2007). Thus, there is marked overlap between the different cell
deaths, and in many cases features of different forms of cell death can be observed simultaneously.
The next chapters are focused on the detailed examination of apoptotic signaling pathways. In
general, there are two main apoptotic pathways, the extrinsic and intrinsic pathways, which differ in
their inducing agent and the involvement of mitochondria (Figure 1.). Both of these pathways lead
to the activation of a series of cysteine proteases called caspases, which are activated in a cascade in
which activated caspases cleave and activate downstream caspases (Boatright and Salvesen, 2003).
The extrinsic pathway is commonly triggered by activation of death receptors, such as Fas/CD95,
and involves the activation of the cysteine protease caspase-8. The intrinsic pathway is initiated by
various extracellular or intracellular stress signals, which induce changes in mitochondrial
membrane permeability and trigger the activation of the caspase-9. In both pathways, the initiator-
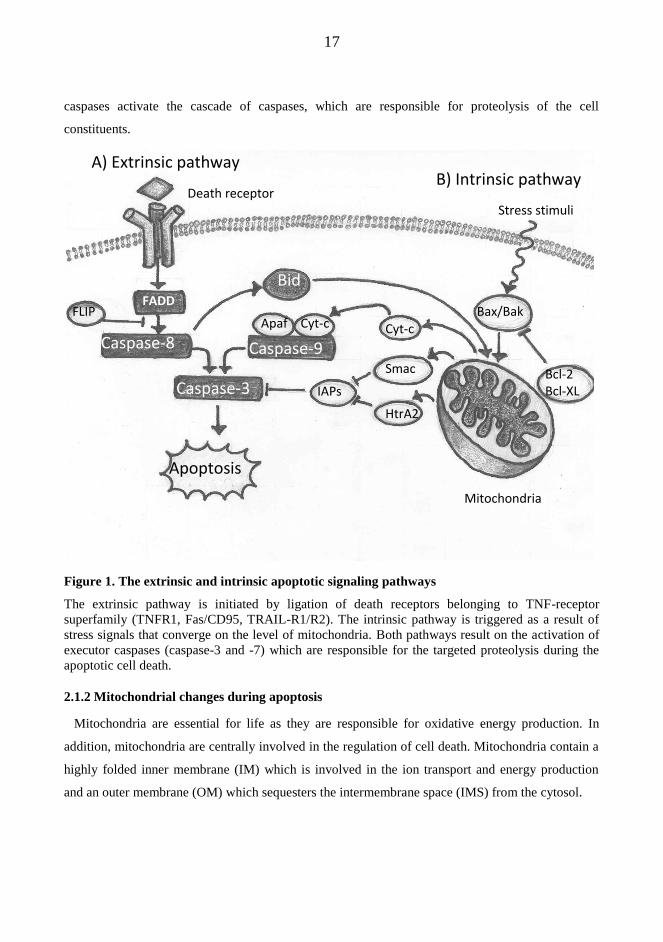
17
caspases activate the cascade of caspases, which are responsible for proteolysis of the cell
constituents.
Figure 1. The extrinsic and intrinsic apoptotic signaling pathways
The extrinsic pathway is initiated by ligation of death receptors belonging to TNF-receptor
superfamily (TNFR1, Fas/CD95, TRAIL-R1/R2). The intrinsic pathway is triggered as a result of
stress signals that converge on the level of mitochondria. Both pathways result on the activation of
executor caspases (caspase-3 and -7) which are responsible for the targeted proteolysis during the
apoptotic cell death.
2.1.2 Mitochondrial changes during apoptosis
Mitochondria are essential for life as they are responsible for oxidative energy production. In
addition, mitochondria are centrally involved in the regulation of cell death. Mitochondria contain a
highly folded inner membrane (IM) which is involved in the ion transport and energy production
and an outer membrane (OM) which sequesters the intermembrane space (IMS) from the cytosol.
Stress stimuli
Bax/Bak
Bcl-2 Bcl-XL
Mitochondria
Apoptosis
FLIP Apaf Cyt-c Cyt-c
Smac
HtrA2
IAPs
Death receptor
A) Extrinsic pathway B) Intrinsic pathway
FADD
Caspase-8
Bid
Caspase-3
Caspase-9

18
Variety of lethal signals from the endoplasmic reticulum, cell surface receptors, nucleus or
extracellular environment converges on mitochondria to induce the mitochondrial outer membrane
permeabilization (MOMP). As a result of MOMP, caspase-activating molecules, such as
cytochrome c, and caspase-independent death effectors are released from the intermembrane space
of mitochondria to the cytosol (Figure 1.). The permeabilization of the outer mitochondrial
membrane is frequently associated with the permeabilization of the inner mitochondrial membrane,
which leads to the collapse of mitochondrial membrane potential (ΔΨm), metabolic failure and
eventually cell death.
The permeabilization of mitochondrial outer membrane is regulated by Bcl-2 family proteins and
is considered as “point of no return” in apoptosis (Garrido et al., 2006; Green and Kroemer, 2004).
The Bcl-2 family proteins are divided into two main groups according to their capacity to either
inhibit or promote apoptosis (Kuwana and Newmeyer, 2003) (Figure 2.). The pro-apoptotic family
members are further classified according to whether they have multiple Bcl-2 homology (BH)
domains or only one BH domain (BH3-only proteins). The common feature of Bcl-2 family proteins
is the formation of homo- or heterodimers with other family members thus neutralizing the action of
each other.
Several pieces of evidence demonstrate that MOMP is a central event during apoptosis, and that
the permeabilization process is precisely regulated by the Bcl-2 family proteins. Firstly, anti-
apoptotic Bcl-2 and Bcl-xL block the release of pro-apoptotic molecules from the intermembrane
space and thus inhibit apoptosis (Kluck et al., 1997; Kuwana et al., 2002; Yang et al., 1997).
Secondly, the pro-apoptotic Bcl-2 family members can permeabilize lipid bilayers, allowing the
release of macromolecules across the membrane. The first clues that the Bcl-2 family proteins can
permeabilize lipid membranes come from their structure, which has similarities with pore forming
bacterial toxins (Suzuki et al., 2000). Afterwards, it has been shown that oligomerized Bax can
form channels in plain liposomes through which cytochrome c could be released (Saito et al., 2000).
Moreover, it has been demonstrated that BH3 only protein Bid activates Bax to produce membrane
openings in mitochondrial outer membrane large enough to transmit mitochondrial IMS proteins
(Kuwana et al., 2002). This process could be inhibited by Bcl-xL and involved cardiolipin, an
abundant lipid in the mitochondrial membrane. These findings were reproduced in cell models since
it was found that cells lacking both Bax and Bak were completely resistant to cytochrome c release
and apoptosis induced by activated Bid (Wei et al., 2001). Thus, it is now widely accepted that

19
during apoptosis, Bax and Bak oligomerize and insert on the mitochondrial outer membrane
resulting in its permeabilization and the release of pro-apoptotic proteins from IMS to cytosol.
Mitochondrial outer membrane permeabilization (MOMP) is regulated by the Bcl-2 family
proteins
Figure 2. The Bcl-2 family of proteins
The Bcl-2 family of proteins is divided on three groups based on their composition of Bcl-2
homology (BH) domains. The anti-apoptotic members contain four domains (BH1-4). The pro-
apoptotic members are divided in to two groups; multidomain proteins (Bax, Bak) which contain
three domains (BH1-3), and the BH3-only proteins. Often, the BH3-only are subdivided into direct
activators (Bid, BIM) and de-repressors/ sensitizers. Each protein contains the hydrophobic
carboxyl terminal transmembrane domain (TM).
Inactive Bax exist in the cytosol as monomers whereas Bak is constitutively bound in
mitochondrial outer membranes (Hsu and Youle, 1998; Youle and Strasser, 2008). It is generally
believed that the activation of Bax/Bak involves the translocation of Bax to outer membrane and
oligomerization with Bak to form membrane spanning pores. The activation of Bax/Bak is induced
either directly or indirectly by “activator” BH3-only proteins, including Bid and Bim. According to
“direct activation model”, BH3-only proteins (i.e. Bid and Bim) bind directly to Bax/Bak leading to
their activation (Kim et al., 2006). According to an alternative “indirect activation model”, BH3-
Anti-apoptotic Bcl-2 proteins
Pro-apoptotic Bcl-2 proteins Multiple BH3 domains
BH3-only
Bcl-2, Bcl-w, Bcl-XL, Mcl-1, A1
Bax, Bak
Bid, Bim
Bad, Bik, Bmf, Hrk, Noxa, Puma
BH4 BH3 BH1 BH2
BH3 BH1 BH2
TM
TM
TM BH3

20
only proteins promote apoptosis exclusively by binding and neutralizing anti-apoptotic Bcl-2 family
members and thus unleashing Bax and Bak from their control. This model is supported by the
findings that Bax and Bak do not bind to “activating” BH3-only proteins, and apoptosis proceed
normally in cells deficient of Bim and Bid (Willis et al., 2007). The interactions between different
Bcl-2 family members are still not fully elucidated and different set of these proteins depending on
the cell type or an apoptosis inducing agent may be involved.
The mitochondrial permeability transition and the membrane potential collapse during
apoptosis
The mitochondrial permeability transition (MPT) denotes the increase of the permeability of the
mitochondrial inner membrane that leads to the collapse of ΔΨm, mitochondrial swelling, and
rupture of the outer mitochondrial membrane (Tsujimoto and Shimizu, 2007). It is generally thought
that MPT is induced as a result of channel formation (permeability transition pore, PTP) between
the inner and outer membrane. This channel is thought to consist of the voltage-dependent anion
channel (VDAC) in the outer membrane, the adenine nucleotide traslocator (ANT) in the inner
membrane and the cyclophilin D (Cyp D) in the matrix.
The role of MPT in apoptosis is controversially discussed. The hypothesis that MPT is involved in
apoptosis is largely based on findings that inhibitors of Cyp D or ANT, cyclosporine A and
Bonkrecik acid respectively, can inhibit apoptosis in some experimental models (Custodio et al.,
2001; Zamzami et al., 1996). In addition, MPT may result in the permeability changes of the outer
mitochondrial membrane leading to the cytochrome c release during apoptosis (Green and Kroemer,
2004). However, recently it has been demonstrated that cell lines isolated from Cyp D deficient
mouse undergo apoptosis normally in response to various stimuli, showing that MPT (or at least its
component Cyp D) is not involved in apoptosis (Nakagawa et al., 2005). In contrast, Cyp D and
MPT seem to be instrumental in some forms of necrotic cell death (Nakagawa et al., 2005).
However, the collapse of ΔΨm is frequently seen in apoptosis induced by variety of stimuli. Thus it
can be speculated that the permeabilization of the inner membrane participates also to apoptotic cell
death. The ΔΨm collapse leads to cessation of ATP synthesis, release of Ca2+ from the matrix,
production of reactive oxygen species (Skommer et al., 2007), and these changes may promote both
apoptotic and nectrotic cell death.

21
Cytochrome c and other pro-apoptotic molecules released from the IMS
Cytochrome c is a small heme-containing protein bounded to outer face of the IM and involved in
the electron transport and oxidative phosphorylation (Garrido et al., 2006). In addition to its vital
function in the respiratory chain, cytochrome c has distinct function as a regulator of apoptosis.
Upon apoptotic stimulus, cytochrome c is released through permeabilized mitochondrial outer
membrane in to the cytosol, where it binds to apoptotic protease-activating factor-1 (Apaf-1) and
induces ATP dependent formation of protein complex called the apoptosome (Riedl and Salvesen,
2007). In addition to cyt c, ATP and Apaf-1, cysteine protease caspase-9 is recruited to apoptosome
where it undergoes dimerization and activation. Caspase-9 functions as an initiator caspase of the
mitochondrial pathway of apoptosis and further activates the executioner caspases responsible for
the targeted proteolysis of cellular substrates (see later).
Recently, Hao et al. developed a mouse model in which mutated cytochrome c retained normal
respiratory function but lacked apoptotic function due to failure to oligomerize with Apaf-1 (Hao et
al., 2005). In these animals, apoptosis during brain- and lymphocyte development was severely
disrupted showing the importance of cytochrome-c mediated apoptosis in the organogenesis.
Although the fibroblasts of this cytocrome-c mutated mouse model were resistant to caspase
activation and apoptosis, the thymocytes were sensitive to apoptosis induced by variety of signals.
Thus the authors concluded that there may be cytochrome-c / apoptosome independent pathways of
caspase-activation. Indeed, in addition to cytochrome c, also other proapoptotic molecules are
released from the IMS during apoptosis. The SMAC/DIABLO and HtrA2/Omi released to the
cytosol bind and inactivate IAPs (Inhibitor of Apoptosis Proteins) and thus facilitate the activation
of caspases (Figure 1.) (Suzuki et al., 2001; Verhagen et al., 2000). Apoptosis-inducing factor (AIF)
is expelled from IMS due to apoptotic stimulus and translocates to nucleus where it contributes to
DNA fragmentation (Porter and Urbano, 2006). However, AIF is centrally involved in the oxidative
phosphorylation, and thus it has been speculated that the main mechanism of how AIF release
triggers cell death is related to failure in energy metabolism (Porter and Urbano, 2006). Endo G was
previously shown to cause chromatin condensation and DNA fragmentation independently of
caspase activation. However, the role of EndoG in apoptosis is recently questioned in the basis of
genetic studies (Irvine et al., 2005).

22
2.1.3 Caspases – executioners of the apoptotic cell death
Caspases are cysteine proteases which play an important role in the regulation and execution of
apoptotic cell death. The term caspase is derived from cysteine dependent, aspartatic acid specific
protease, which denotes that the cysteine is involved in the catalytic site of the protease which
cleaves its substrate proteins after the aspartatic acid residue (Boatright and Salvesen, 2003).
Recognition of at least four amino acids next to the cleavage site is involved for substrate binding
and cleavage (Thornberry and Lazebnik, 1998). This four amino acid recognition sequence differs
among the caspases and explains the differences in their action. The activation of caspases does not
lead to indiscriminate protein digestion, instead highly specific set of target proteins is cleaved
elegantly resulting in the loss or change in their function.
The caspases are synthesized and stored as inactive precursors (zymogens) which became
activated upon apoptosis induction (Boatright and Salvesen, 2003). The first caspases to be
activated in the proteolytic cascade are termed initiator caspases (caspases -2, -8, -9 and -10). The
zymogens of the initiator caspases exists within a cell as inactive monomers, which became
activated by dimerization at multiprotein activating complexes (Boatright and Salvesen, 2003;
Boatright et al., 2003; Donepudi et al., 2003). In general, two ways of initiator caspase-activation
have been described; the extrinsic pathway is initiated by ligation of death receptors belonging to
TNF-receptor superfamily (TNFR1, Fas/CD95, TRAIL-R1/R2) and the intrinsic pathway is
triggered by MOMP as a result of stress signals that converge on the level of mitochondria (Figure
1.).
The death receptor (extrinsic) pathway
The extrinsic pathway of apoptosis is triggered by binding of extracellular ligand (TNF, FAS-L,
TRAIL) to their specific transmembrane receptors (Ashkenazi and Dixit, 1999). However, in the
case of Fas/CD95 receptor, also ligand independent activation by receptor aggregation has been
described (Hennino et al., 2001). Upon activation, the death receptors form trimeric complexes
which bind and recruit the adaptor protein FADD (Fas-associated death domain). FADD in turn
binds caspase-8 zymogens resulting in the formation of the death inducing signaling complex
(DISC) (Medema et al., 1997), a multiprotein complex which promotes the dimerization of caspase-
8 leading to its activation (Donepudi et al., 2003). In addition to caspase-8, caspase-10 can be
recruited to DISC and work as an initiator caspase of the death receptor pathway at least in some
cell models (Kischkel et al., 2001).

23
Two types of cells which differ in their involvement of mitochondria in the death receptor
mediated apoptosis has been described (Scaffidi et al., 1998). In type I cells the amount of caspase-8
activated at the DISC is sufficient for the activation of the executioner caspases without the
involvement of mitochondria. In type II cells, small amount of activated caspase-8 cleaves and
activates Bid, a pro-apoptotic member of the Bcl-2 family, leading to its translocation to
mitochondria where it induces MOMP and engages the extrinsic pathway with the intrinsic pathway
of apoptosis (Li et al., 1998) (Figure 1.).
Besides functioning as an activation platform for caspase-activation, DISC is also involved in the
regulation of the initiator caspase activation by recruiting the caspase-8 homolog FLIP (FLICE-like
inhibitory protein) (Irmler et al., 1997; Scaffidi et al., 1999). FLIP has structural homology with the
caspase-8 and can thus replace it in the DISC. Generally, FLIP is considered as an apoptosis
inhibiting molecule as it can displace caspase-8 from the DISC while it does not itself have notable
proteolytic activity.
The mitochondria-dependent (intrinsic) pathway
The intrinsic or mitochondrial pathway of apoptosis is triggered by multiple cellular or
extracellular stress signals, such as growth factor withdrawal, DNA damage or radiation, which
converge on the level of mitochondria. The hallmark of the intrinsic pathway is the activation of
caspase-9 at the multiprotein activating complex called the apoptosome (Riedl and Salvesen, 2007).
As a result of mitochondrial permeability change, cytochrome c is released into the cytosol, where it
binds to an intracellular receptor molecule apoptosis-inducing factor-1 (Apaf-1) leading to its
oligomerization and ATP dependent conformational change. Apaf-1 in turn recruits caspase-9 via its
N-terminal caspase-activation recruitment domain (CARD). Activation of caspase-9 monomers at
the apoptosome is achieved by dimerization (Riedl and Salvesen, 2007). Moreover, it has been
suggested that the apoptosome increases the caspase-9 affinity for procaspase-3 (Yin et al., 2006)
Activation of the executioner caspases (caspases -3, -6 and -7) involves their proteolytic
processing by the initiator caspases (Thornberry and Lazebnik, 1998). The cleavage of the linker
domain of executioner caspases results in the formation of small and large caspase subunits, which
heterodimerize to form a catalytical unit. Executioner caspases contribute to apoptosis by cleaving
structural proteins of the nucleus (lamin), proteins involved in the regulation of cytoskeleton
(gelsolin, focal adhesion kinase) and proteins involved in the DNA repair (DNA-PKcs) or replication
(replication factor C) (Thornberry and Lazebnik, 1998). In addition, executioner caspases can

24
enhance mitochondrial dysfunction and hence cell death by cleavage of the respiratory chain
components (Ricci et al., 2004) or anti-apoptotic Bcl-2 family proteins (Chen et al., 2007; Cheng et
al., 1997).
The activity of executioner caspases is kept in check by Inhibitor of Apoptosis Proteins (IAPs) to
ensure that these proteases are not activated inappropriately (Verhagen et al., 2001). To date, several
members of the IAP-family have been characterized, including XIAP, cIAP1, cIAP2 and surviving
(reviewed in (D'Amelio et al., 2008)) The IAPs in turn can be inactivated by SMAC/DIABLO or
HtrA2/Omi released from IMS during the apoptotic insult (Figure 1.) (Suzuki et al., 2001; Verhagen
et al., 2000; Verhagen et al., 2001).
In addition of their death promoting effect during apoptosis, caspases may also function in the
regulation of cell survival, proliferation, differentiation and inflammation (Lamkanfi et al., 2007).
2.2. B cells and the humoral immune response
2.2.1 An overview of the humoral immune response
The main function of the immune system is to protect our body from invading infectious agents.
The adaptive immune response enables the specific destruction of invading micro-organisms such
as viruses and bacteria, whose foreign structures work as antigens. The immunological memory and
the adaptation to combat micro-organisms that are transforming all the time are instrumental
properties of the adaptive immunity.
Failures in the immune system are associated with several diseases. Disturbance in the
development of a specific immune response (immunodeficiency) leads to overwhelming or
recurrent infections. On the other hand, a normal immune response can be inappropriately directed
against harmless, non-infectious agents, such as in the case of allergy or autoimmune diseases.
T and B lymphocytes are greatly in charge for the development of a specific immune response. Each
individual lymphocyte bears a unique variant of an antigen receptor, so that lymphocytes
collectively have an enormous repertoire of antigen receptors which can bind highly diverse
antigens.
The humoral immune response is mediated by antibodies (immunoglobulins), which are secreted
by terminally differentiated B cells called plasma cells. The antibody is a soluble form of the B cell
surface antigen receptor, and recognizes the same antigen as the receptor. Antibodies secreted to
circulation delete micro-organisms by three main ways. Antibodies can bind to micro-organisms
25
and thus prevent their adherence to host cell (neutralization). Secondly, antibodies can bind to the
surface of pathogens and promote its phagosytosis by macrophages and other effector cells
(opsonization). Thirdly, antibody binding to the surface of micro-organism may trigger the
activation of complement system leading to pore formation on the target cell surface and cell lysis.
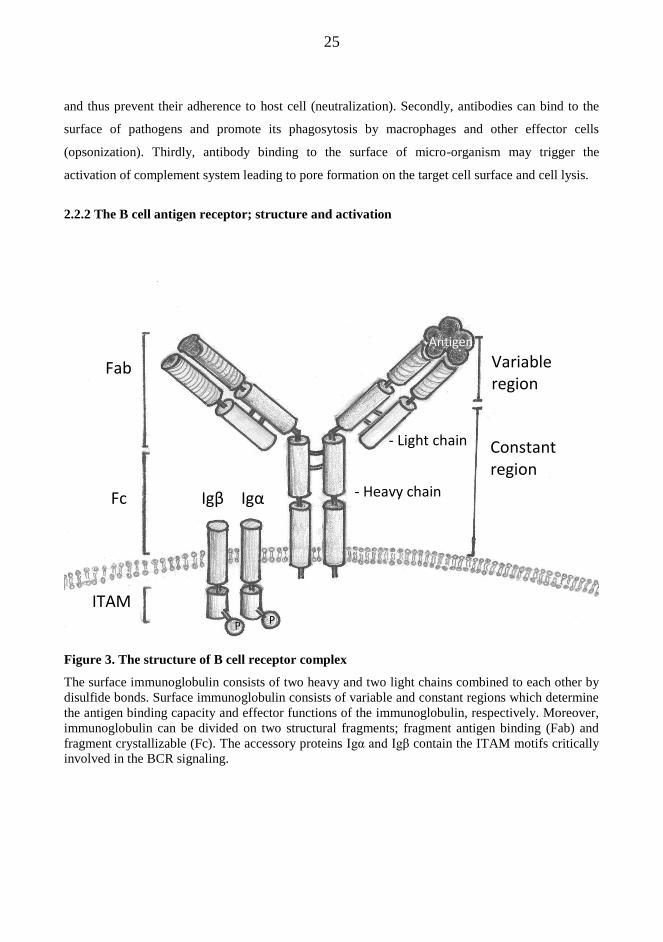
2.2.2 The B cell antigen receptor; structure and activation
Figure 3. The structure of B cell receptor complex
The surface immunoglobulin consists of two heavy and two light chains combined to each other by
disulfide bonds. Surface immunoglobulin consists of variable and constant regions which determine
the antigen binding capacity and effector functions of the immunoglobulin, respectively. Moreover,
immunoglobulin can be divided on two structural fragments; fragment antigen binding (Fab) and
fragment crystallizable (Fc). The accessory proteins Igα and Igβ contain the ITAM motifs critically
involved in the BCR signaling.
Variable region
Constant region
- Light chain
- Heavy chain
Antigen
Fab
Fc
ITAM P P
Igα Igβ

26
The main functions of the BCR are to recognize foreign antigens, to internalize antigens for
further processing and presentation to T-helper cells, and to transmit activating signals to the cells
interior. The activating signals initiated after BCR-stimulation have different outcomes depending
on the maturational stage of a B cell. In mature B cells, BCR triggering leads to proliferation and
survival, whereas in immature B cells BCR activation may lead to inactivation or apoptosis (Niiro
and Clark, 2002). The central question in the research concerning the BCR signaling is how the
signals initiated from the same receptor can evoke these different responses.
The B cell receptor complex is made up of the cell surface immunoglobulin combined with
accessory proteins Igα and Igβ (Figure 3.) (Reth and Wienands, 1997). The immunoglobulin part
consists of two heavy and two light chains combined to each other by disulfide bonds. The variable
regions of both light and heavy chains are unique in each B cell clone and responsible for antigen
binding specificity. Diversity of the antigen binding capacity is a result of immunoglobulin variable
region gene rearrangements during early B cell development (Busslinger, 2004). The surface
immunoglobulin has the same antigen specificity as the soluble immunoglobulin that the cell will
eventually produce as a plasma cell.
The surface immunoglobulin has a short cytoplasmic tail and can not transmit signals in to cells
interior. Accessory proteins Igα and Igβ provide the cytoplasmic domains crucial for BCR signaling
(Reth and Wienands, 1997). They contain amino acid sequences called immunoreceptor tyrosine-
based activation motifs (ITAMs), which become phosphorylated by Src-family protein tyrosine
kinases (PTKs), such as Lyn, after engagement of BCR by antigen (Niiro and Clark, 2002). The
phosphorylated ITAMs in turn recruit and facilitate the activation of protein kinases Syk and Btk,
which in turn activate the phopholipase Cγ2 (PLC γ2), phosphatidyl-inositol-3-kinase (PI3K) and
Ras-Raf-1-ERK downstream signaling pathways (Figure 4.) (Niiro and Clark, 2002). Adaptor
proteins such as B-cell linker (BLNK) and BAM 32 (B-lymphocyte adaptor molecule of 32 kD)
connect the initial kinases with downstream signaling molecules.
The activation of ERK1/2 (extracellular signal regulated kinase) has been associated with both
proliferation and apoptosis of B cells (Koncz et al., 2002; Lee and Koretzky, 1998). ERK1/2 are
activated by a cascade of upstream protein kinases Ras and Raf-1 (Niiro and Clark, 2002). In
addition to Ras-Raf-1, the PLC-γ2 signaling pathway has been shown to participate in the activation
of ERKs (Hashimoto et al., 1998). In mature B cells, sustained activation of ERK after BCR
stimulation was associated with the up-regulation CREB and Elk-1, transcription factors involved in
cell proliferation (Koncz et al., 2002). In line with this, the pharmacological inhibition of ERK

27
activation suppressed BCR-induced proliferation in mature B cells (Richards et al., 2001). In
contrast, it was shown in the murine immature B cell line that ERK2 plays an active role in anti-
IgM-mediated apoptosis (Lee and Koretzky, 1998). In human B lymphoma cell lines, the Ras-ERK
pathway activation was connected with up-regulation of Bim and induction of apoptosis (Stang et
al., 2009). Thus, the role of the Ras-Raf-1-ERK pathway in the regulation of B cell fate may depend
on the maturational stage of the B cell and the kinetics of ERK activation.
Figure 4. B cell receptor-induced signal-transduction pathways
Three main pathways are activated after B cell receptor activation; Ras, PLC and PI3-kinase
pathways. The most important downstream mediators include mitogen activated protein kinases
(MAPKs), NF-AT, cyclin D and NF-κB.
The PI3K mediates B cell survival by activating downstream kinase AKT which promotes cell
survival by directly inhibiting pro-apoptotic Bcl-2 family member BAD and by inducing the
RAS
Raf
PLC
PKC Ca2+
PI3K
AKT
GSK3
Cyclin D NF-κB NFAT CREB
ERK JNK P38
BCR
MAPKs
Src-family PTKs

28
expression of pro-survival proteins (Bcl- xL, Bcl-2, A1) through activation of NF-κB. In addition,
AKT inhibits GSK3 (glycogen synthase kinase-3) leading to stabilization of Cyclin D and Myc,
thereby facilitating cell proliferation (Niiro and Clark, 2002).
Activated PLCγ cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol 1,4,5-trisphosphate
(IP3) and diacylglyserol (DAG) (Campbell, 1999). IP3 binds to its receptors on endoplasmic
reticulum leading to the release of intracellular calcium stores, while DAG activates certain
isoforms of PKC. The release of intracellular Ca2+
and PKC activation are crucial for the activation
of mitogen activated protein kinases (MAPKs), such as ERK, JNK and P38 (Niiro and Clark, 2002).
In addition, elevated cytosolic Ca2+
level triggers the activation of calcineurin, a protein
phosphatase, which can activate target molecules including caspase-2, transcription factor NFATc2
or mitogen activated protein kinases (MAPKs) p38 and JNK (Chen et al., 1999; Graves et al., 1996;
Kondo et al., 2003). The activation of JNK and p38 have been associated with BCR apoptosis in
B104 cell line (Graves et al., 1996). In addition, JNK and p38 are activated after various stress
signals, such as radiation induced DNA damage (Chen et al., 1996).
2.2.3. B cell maturation
The selection of immature and transitional B cells; the negative selection during B cell
development in the bone marrow and periphery
During early B cell development in the bone marrow, the B cell precursors undergo recombination
of the immunoglobulin heavy (H) and light (L) chain genes which creates diversity to antigen
binding capacity. The recombination is accomplished by RAG1/2 proteins which rearrange first
variable (V), diversity (D) and joining (J) regions of the Ig heavy chain followed by VJ regions of
the light chain (Busslinger, 2004). If the rearrangement is completed successfully, the B cell
expresses functional surface IgM receptor combined with accessory proteins Igα and Igβ and its
development can continue.
The gene rearrangement process may probably produce B cells with self-reactive receptors which
have to be eliminated to maintain immune system self-tolerant. The mechanisms of this called
negative selection include deletion, receptor editing, anergy and inergy (Hartley et al., 1991).
Deletion of self-reactive immature B cells has been demonstrated in transgenic animal models
(Hartley et al., 1991; Nemazee and Burki, 1989). Moreover, in vitro, immature B cells die by
apoptosis after antigen receptor activation, and these cells have been used as a model of negative
selection (Norvell et al., 1995). Especially, strong signal generated by multivalent antigen drives

29
cells to deletion. However, the bone marrow developing B cells might be rescued by receptor
editing process, whereby self-reactive antigen receptor is replaced by newly generated non-self-
reactive antigen receptor after recurrent immunoglobulin gene-rearrangement (Melamed and
Nemazee, 1997).
Immature B cells which bind soluble self antigens are not deleted immediately in the bone
marrow, but instead they migrate to the periphery where they remain permanently unresponsive or
anergic to further antigenic stimulation (Duty et al., 2008). The anergic B cells die rapidly in the
periphery mostly because of the lack of survival signals from the antigen specific T cells. The fourth
potential fate of a self-reactive B cell is that they ignore the antigenic stimulus which is too weak to
activate antigen receptor. Alternatively, some self-antigens may not be presented in the bone
marrow, and B cells specific for these self-antigens are left unaffected and enter the periphery.
Contrary to anergy, ignorance can be later counteracted if the concentration of the soluble antigen
suddenly increases so that the antigen receptor activation is possible. Thus, ignorant B cells may be
thought as a “seeds” of the autoimmune diseases. However, normally these low-affinity self reactive
B cells are not activated because of the lack of proper T cell help.
Only the B cells, which are not negatively selected in the bone marrow, have the potential to
further develop to antibody secreting mature B cells. However, still majority of the cells that enter
peripheral B cell pool are short lived and die before maturation is completed (Thomas et al., 2006).
These transitional B cells are dependent on the survival signals which they get at the lymphoid
follicles of peripheral lymphoid tissues. Continuous expression of B cell receptor seems to be
instrumental for B cell survival, since gene deletion experiments have shown that B cells lacking the
functional BCR complex are rapidly eliminated from the peripheral B cell pool (Kraus et al., 2004;
Torres et al., 1996). It seems that instead of antigen dependent activation of the receptor, antigen
independent signaling from the BCR is sufficient for survival (Monroe, 2006). In addition,
maturating B cells of the periphery also need other survival signals for their survival, such has
BAFF receptor activation by BAFF, a ligand belonging to TNF receptor family (Stadanlick and
Cancro, 2008).

30
B cell activation by T cells
When the maturating B cells first encounter their specific antigen, they have to be activated by
BCR-stimulation to generate effective immune responses. As in the case of immature B cells, the
antigenic stimulus leads primarily to deletion process. However, when antigen is delivered with co-
stimulatory signals derived by the activated T cells, the outcome is B cell activation instead of
deletion.
Figure 5. Crosstalk between B cell and a helper-T cell
Two events are necessary for antigen dependent B cell activation. Firstly, antigen binding triggers
the B cell receptor activation, antigen internalization and antigen presentation as peptides on the
surface of MHCII molecule. Secondly, B cell receives activating CD40-ligand (CD40-L) and
cytokines from an antigen specific T cell. Abbreviations: BCR; B cell receptor, TCR; T cell
receptor, MHCII; major histocompatibility complex II.
Crosstalk between a B cell and a helper-T cell specific to the same antigen is mandatory for B cell
activation (Figure 5.). After the first encounter of an antigen, antigen specific T and B cells are
trapped in border between T-and B-cell zones in the spleen and lymph nodes, where antigen is
presented in the surface of antigen presenting cells (dendritic cells) (MacLennan et al., 1997). After
B cell T cell
CD40 CD40-L
MHCII TCR
BCR
Antigen
Cytokines

31
antigen binding by BCR, the antigen is internalized and presented as peptides on the surface of a B
cell in MHCII-antigen complex, which is recognized by a T cell specific to the same antigen.
Subsequently, the T cell delivers contact mediated signals, of which the most important are CD40-
ligand and cytokines (IL4, IL5 and IL-6), to the B cell leading to its activation (Van den Eertwegh
et al., 1993).
Affinity maturation in germinal centers
Figure 6. The germinal center reaction Antigen activated B cell differentiates into centroblasts
that undergo proliferation and somatic hypermutation (SHM) in the dark zone of the germinal center
(GC). Centroblasts then move in the light zone of the GC and differentiate into centrocytes which
have three fates: centrocytes with self-reactive or non-binding antigen receptors are deleted by
apoptosis, while high affinity centrocytes are selected for further differentiation to memory B cells
or antibody secreting plasma cells.
After B cell activation, some of the B cells directly differentiate to antibody secreting short lived
plasma cells, which are responsible for the primary IgM-mediated responses against an antigen.
This primary response offers the first line defense against the invading agent, but the immune
Antigen activated B-cell
BCR
SHM
Centroblast
Centrocyte
1. Self-reactive
2. High affinity → Selection
3. Non-binding
Memory B cell
Plasma cell
Ig
CD4+ T cell
FDC
Apoptosis
Apoptosis Dark zone
Light zone

32
response is further improved by the antibody affinity maturation and isotype switching, which
ensures that antibodies will have more effective binding capacity and can evoke different effector
functions.
The affinity maturation takes place in germinal centers, which are formed by rapidly proliferating
B cells termed centroblasts (MacLennan, 1994) (Figure 6.). The vigorous proliferation is associated
with a somatic hypermutation (SHM), a process which introduces non-random, single base point
mutations into IgV-gene regions that encode the antigen binding site. Centroblasts then differentiate
into centrocytes, which are subjected for selection on the basis of their antigen binding capacity
(Figure 6.). Most of the centrocytes have gained deleterious mutations on their Ig-genes leading to
expression of a low affinity or a non-functional antigen receptor. These centrocytes are eliminated
by apoptosis. In addition, the SHM may lead to generation of B cells with self-reactivity, and also
these cells are eliminated by apoptosis because of the lack of survival signals (CD40-ligand)
delivered by activated T cells. Only the centrocytes with a high affinity BCR are able to bind
antigen efficiently and present it to helper T cells and follicular dendritic cells (FDC) to gain the
appropriate signal for further differentiation to antibody secreting plasma cells or memory B cells.
Some of the B cells may go repeated rounds of proliferation, SHM and selection to further improve
the antigen binding capacity. A subset of centrocytes undergoes immunoglobulin glass –switch
recombination, which results in the production of different immunoglobulin subclasses by plasma
cells (MacLennan, 1994).
2.2.4 Life and death decisions of a B cell; regulation of B cell apoptosis
To maintain B cell homeostasis, the high generation rate of B cells must be counteracted by
elimination of deleterious or extraneous B cells. Mainly, these unwanted cells are deleted by
apoptosis during various check points of B cell maturation process (see B cell maturation).
Apoptosis plays an important role in the negative selection of bone marrow immature B cells and
GC B cells. In addition, excess B lymphocytes are eliminated by apoptosis after the antigenic
challenge. The fate of a B cell is regulated by both survival and death signals originating from
stromal cells of lymphoid organs and activated T cells. Here, I will focus on the regulation of B cell
apoptosis during the GC reaction, and the signaling through antigen-, Fas-, and CD40-receptors, the
main regulators of B cell apoptosis. Knockout models of Fas and CD40 receptors and their
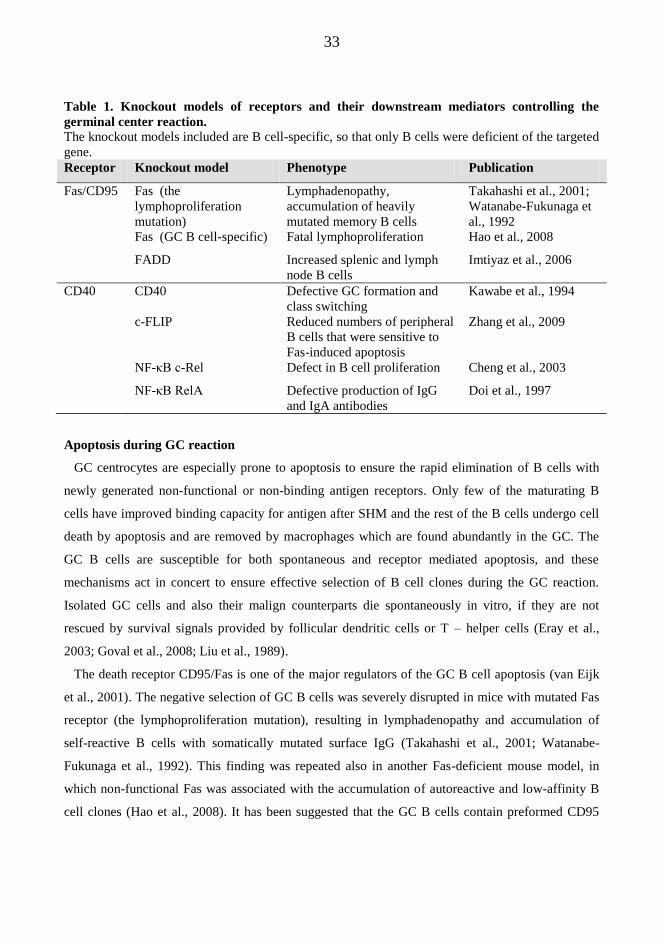
downstream mediators are developed to study their role in GC B cell development (table 1.).
33
Table 1. Knockout models of receptors and their downstream mediators controlling the
germinal center reaction.
The knockout models included are B cell-specific, so that only B cells were deficient of the targeted
gene.
Receptor Knockout model Phenotype Publication
Fas/CD95 Fas (the
lymphoproliferation
mutation)
Lymphadenopathy,
accumulation of heavily
mutated memory B cells
Takahashi et al., 2001;
Watanabe-Fukunaga et
al., 1992
Fas (GC B cell-specific) Fatal lymphoproliferation Hao et al., 2008
FADD Increased splenic and lymph
node B cells
Imtiyaz et al., 2006
CD40 CD40 Defective GC formation and
class switching
Kawabe et al., 1994
c-FLIP Reduced numbers of peripheral
B cells that were sensitive to
Fas-induced apoptosis
Zhang et al., 2009
NF-κB c-Rel Defect in B cell proliferation Cheng et al., 2003
NF-κB RelA Defective production of IgG
and IgA antibodies
Doi et al., 1997
Apoptosis during GC reaction
GC centrocytes are especially prone to apoptosis to ensure the rapid elimination of B cells with
newly generated non-functional or non-binding antigen receptors. Only few of the maturating B
cells have improved binding capacity for antigen after SHM and the rest of the B cells undergo cell
death by apoptosis and are removed by macrophages which are found abundantly in the GC. The
GC B cells are susceptible for both spontaneous and receptor mediated apoptosis, and these
mechanisms act in concert to ensure effective selection of B cell clones during the GC reaction.
Isolated GC cells and also their malign counterparts die spontaneously in vitro, if they are not
rescued by survival signals provided by follicular dendritic cells or T – helper cells (Eray et al.,
2003; Goval et al., 2008; Liu et al., 1989).
The death receptor CD95/Fas is one of the major regulators of the GC B cell apoptosis (van Eijk
et al., 2001). The negative selection of GC B cells was severely disrupted in mice with mutated Fas
receptor (the lymphoproliferation mutation), resulting in lymphadenopathy and accumulation of
self-reactive B cells with somatically mutated surface IgG (Takahashi et al., 2001; Watanabe-
Fukunaga et al., 1992). This finding was repeated also in another Fas-deficient mouse model, in
which non-functional Fas was associated with the accumulation of autoreactive and low-affinity B
cell clones (Hao et al., 2008). It has been suggested that the GC B cells contain preformed CD95

34
DISC, in which the caspase-8 can be activated spontaneously without the involvement of the Fas-
ligand (Hennino et al., 2001). In this model, the death receptor activation is inhibited by anti-
apoptotic c-FLIP (cellular FLICE inhibitory protein) which can interfere with caspase-8 in the pre-
formed DISC (Scaffidi et al., 1999). Without survival signals provided by CD40- or antigen
receptors or tropic factors from stromal cells, c-FLIP is rapidly degraded from the DISC leading to
caspase-8 activation and apoptosis (Hennino et al., 2001; van Eijk et al., 2001). However, Fas-
ligand expression at the mRNA level has been detected in GC T cells pointing out that the Fas
receptor activation might also be dependent on Fas ligand expressed on T-cells (Kondo et al., 1997).
The expression of Fas receptor is negatively correlated with the expression of anti-apoptotic Bcl-2
protein, since Bcl-2 protein level has been shown to drastically decrease during the GC reaction to
enhance Fas-mediated apoptosis (Kondo and Yoshino, 2007).
There is some controversy concerning the role B cell receptor stimulation in the selection process
of GC B cells. In the model of GC mediated selection presented above, BCR-mediated signal is
mainly proposed as a survival signal involved in the positive selection. However, according to
another hypothesis, signaling through the BCR is involved in the negative selection of somatically
mutated centrocytes with self-reactivity. This model is supported by findings that isolated BC B
cells and also their malign counterparts are susceptible for apoptosis induced by BCR triggering
(Billian et al., 1997; Eray et al., 2003). However, in both of the above mentioned models, activating
signals from CD4+ T cells (CD40L and IL-4), are critically involved in the positive selection of
high affinity, self tolerant centrocytes.
CD40-CD40L interactions in the regulation of B cell survival
CD40 is a 50 kDa transmembrane protein, which is expressed on B lymphocytes, monocytes and
dendritic cells and in addition several non-hematological tissues. It is also expressed by malignant
cells originating from these cells, including B and T cell lymphomas, multiple myeloma and
Hodgkin`s disease (Dallman et al., 2003). The ligand for CD40 (CD40L, CD154) is a
transmembrane protein expressed transiently on activated CD4+ T lymphocytes (van Kooten and
Banchereau, 2000; Younes and Kadin, 2003). In addition, CD40L expression has been documented
in activated B cells, natural killer cells, monocytes, basofils and dendritic cells. Constitutive
expression of CD40L has been demonstrated in a variety of B cell malignancies, including follicular
lymphoma, mantle cell lymphoma, diffuse large cell B cell lymphoma and chronic lymphoid
leukemia (Younes and Kadin, 2003).

35
The main function of CD40 is the regulation of T-cell mediated B-cell activation during humoral
immune response. The CD40 stimulation promotes B cell proliferation, immunoglobulin production
and class switching, GC formation and establishment of B cell memory (van Kooten and
Banchereau, 2000; Younes and Kadin, 2003).
The importance of CD40-CD40L interactions in the regulation of humoral immunity are
demonstrated in vivo in a human immunodeficiency disease X-linked hyper IgM syndrome in which
CD40 signaling is severely impaired due to mutations in the gene encoding CD40L. The disease is
characterized by accumulation of IgM type antibodies because of impaired Ig class switching, and
defects in the formation of GC and development of memory B cells (Korthauer et al., 1993). A
similar phenotype is presented also in CD40L or CD40 deficient mouse models (Dallman et al.,
2003).
Several in vitro studies have demonstrated the pivotal role of CD40 in the regulation of GC B cell
apoptosis. Isolated GC B cells die rapidly in vitro, unless they are not rescued by CD40L expressed
transiently on activated CD4+ T cells (Hennino et al., 2001). In addition, CD40 can counteract both
Fas and BCR -mediated apoptosis in both isolated GC B cells and in their malign counterparts
(Choe et al., 2000; Cleary et al., 1995; Eeva et al., 2003; Eeva et al., 2007).
CD40 contains a cytoplasmic tail, which can activate several intracellular signaling pathways by
binding adaptor molecules called TNF receptor associated factors (TRAFs) (van Kooten and
Banchereau, 2000). The activation of TRAF2 has been suggested as mediator of NF-κB activation
after CD40 ligation (Rothe et al., 1995). The NF-κB family includes the members p50, p65 (relA)
and c-Rel (Berberich et al., 1994), which are involved in the transcriptional control of several anti-
apoptotic genes including c-FLIP (Kreuz et al., 2001), survivin (Granziero et al., 2001), Bcl-2
(Holder et al., 1993), Bcl-xL and Bfl-1 (Lee et al., 1999). NF-κB activation is at least partially
mediated by a decrease in the half life of IκB-α and IκB-β proteins, which sequester NF-κB in the
cytosol in an inactive form (Schauer et al., 1996). Besides NF-κB activation, CD40 triggering leads
to the activation of other signaling pathways, including p38, JNK, ERK, JAK-STAT and NF-AT
pathways, but their importance in the regulation CD40 mediated survival is currently not well
characterized (van Kooten and Banchereau, 2000).
2.2.5 GC as an origin of B cell malignancies
The microenvironment that enhance vigorous cell proliferation, the SHM process, and a low
threshold for acquired mutations makes the GC B cells susceptible to malign transformation. The

36
hallmark of a malign transformation is the unregulated accumulation of single clone cells due to
overwhelming cell proliferation or inaccurate elimination by apoptosis. GC derived B cell
malignancies are most often associated with an aberrant Ig gene rearrangement, in which a highly
expressed Ig gene is joined next to genes that regulate cell growth or apoptosis. For example,
majority of the follicular lymphoma (FL) cells carry the chromosomal translocation t14:18 that
positions the Ig gene next to the antiapoptotic Bcl-2 gene leading to decreased apoptosis and
accumulation of long lived cells (Klein and Dalla-Favera, 2008). In Burkitt lymphoma cells the Myc
oncogene, involved in the control of cell cycle, is placed under the control of Ig loci leading to
enhanced cell proliferation (Klein and Dalla-Favera, 2008). Aberrant class switch recombination
leading to dysregulated expression of Bcl-6 may be the transforming event in the development of
DLBCL (diffuce large cell B cell lymphoma) (Klein and Dalla-Favera, 2008). Constitutive
expression of Bcl-6 results in the maintenance of proliferative, DNA-damage tolerant centroblastic
phenotype with subjects the cell for further genetic abnormalities.
The FL cells grow in a follicular pattern, and the same cellular components are found in the FL
follicles as are present in the normal germinal centers. It has been suggested that also the same
cellular interactions between FDCs, helper T cells and macrophages occur similarly in normal GC
B cells and FL cells (Kuppers, 2004). The dependence of FL cells as well as normal GC cells on the
growth support provided by their microenvironment is supported by the fact that the cells are
difficult to culture in vitro without survival signals provided by FDCs or CD40 (Eray et al., 2003;
Goval et al., 2008). Recently, a large scale gene-expression profiling study has demonstrated that
the clinical behavior of FL is not only determined by properties of tumor cells themselves, but also
by the molecular features of non-malignant tumor-infiltrating immune cells (Dave et al., 2004).
Another gene expression study demonstrated that especially the activation of CD4+ T-helper cells
play an important role in rapidly transforming FL with poor prognosis (Glas et al., 2007). In the
same study, it was found that the gene-expression profiles from of dendritic cells have an impact on
the growth properties and clinical behavior in FL. Further studies are needed for the better
understanding how the microenvironment specifically interacts with tumor cells, since this
knowledge can be used in the future for the development of new immunological treatment strategies
against FL.

37
2.3. Monoclonal antibody therapy for cancer treatment
In monoclonal antibody (mAb) therapy for cancer, the antibody against a specific tumor antigen is
given to patients as single therapy or in combination to chemotherapeutics to induce depletion of
cancer cells. In addition to cancer therapy, mAbs that target pro-inflammatory cytokines and their
receptors are currently used in the treatment of autoimmune diseases, such as rheumatoid arthritis,
systemic lupus erythematosus (SLE) and vasculitis (Bayry et al., 2007). The advantage of the mAb
therapy is the specificity of mAbs against tumor cells without a general toxicity associated with
conventional chemotherapeutics.
The target antigens currently used in the cancer therapy have several different biological
functions, such as regulation of cell death or proliferation, or conduction of growth promoting
signals inside the cell. Ideally, the target antigen should be expressed only in tumor cells, or in the
cell type from which the tumor cells arise. The expression of the antigen should not decrease upon
repeated mAb treatments, and the antigen should not detach from the cell surface upon antigen
binding.
Since the introduction of the hybridoma technique by Kohler and Milstein in 1975 (Kohler and
Milstein, 2005), specific mAbs have been available as large amounts needed for the therapy.
Hybridoma cells are produced by fusing a mouse myeloma cell with an antigen specific mouse B
cell extracted from a mouse immunized with the desired antigen. The resulting hybridoma cell line
is immortal and produces large amounts of the specific antibody. The pioneer study of mAb therapy
was conducted in the beginning of the 1980 century, when Nadler et. al applied intravenous mAb
against the tumor cell antigen to the lymphoma patient (Nadler et al., 1980). Although this pioneer
study showed no clinical response, it proofed for the first time that mAbs bind specifically to the
tumor cell antigen in vivo, and the therapy was not associated with a severe toxicity. In 1982, Levy
et al used an anti-idiotype antibody, an antibody specific to variable region of the B cell surface
immunoglobulin, to treat lymphoma patient (Miller et al., 1982). This clinical study showed that
mAb therapy can be directed specifically to lymphoma cells, since all lymphoma cells arise from a
single transformed cell and thus have uniform immunoglobulin variable regions (idiotype). The
lymphoma patient treated with anti-idiotype Ab had a complete response to the therapy showing the
curative potential of the treatment.
The first clinical studies of antibody therapy were conducted by using mouse antibodies, which
appeared to have several shortages in the clinical settings. The mouse antibodies are highly
immunogenic and thus induce the production of human-antimouse antibodies (HAMAs) as a result

38
of repeated dosing to the patients (Frodin et al., 1992). The mouse antibodies were associated with
mild adverse effects and the production of HAMAs resulted in too rapid clearance of the mAbs
from the circulation after their administration (Dillman et al., 1986). Later, the problems associated
with mouse antibodies were circumvented by the development of human-mouse chimeric antibodies
in which the Fab part of the immunoglobulin is mouse-derived and the Fc part of the antibody is
human specific (Figure 7.). More lately, the development of gene technology has enabled the
development of humanized monoclonal antibodies, in which only the six antigen binding
polypeptide loops (complementary determining regions (CDRs) are of non-human origin. For
example, the anti-CD52 antibody alemtuzumab, used for the treatment of chronic lymphocytic
leukemia, is produced by inserting mouse derived CDRs specific for CD52 into antibody
“framework” containing heavy and light chains of human origin (Crowe et al., 1992). Subsequently,
the humanized antibody, containing only CDRs of non-human origin, is expressed on immortalized
mouse cell line.
More recently, new technologies using phage display or transgenic mouse have emerged for the
generation of fully humanized antibodies (Lonberg, 2008). To date, two fully humanized
antibodies, adalimumab and panitumumab, are in the clinical use, and several antibodies in the
clinical trials. The anti-TNFα antibody adalimumab, used for the treatment of rheumatoid arthritis,
is produced by phage display technology. The antibody against epidermal growth factor receptor
(EGFR), panitumumab, is produced using “humanized” transgenic mice, which are able to produce
human immunoglobulin heavy and light chains after immunization. The antibody producing B cell
clone specific for EGFR is extracted from the transgenic mouse, and immortalized using Chinese
ovarian cell line.
The humanized and chimeric antibodies contain a human specific Fc part which can activate
leukocytes and complement system when applied to the human body. Indeed, antigen-dependent
cellular cytotoxicity (ADCC) and complement mediated cytotoxicity (CDC) are centrally involved
in the mAb-mediated tumor cell clearance in addition to the direct antigen specific growth-
inhibiting or apoptosis-inducing effect (Villamor et al., 2003) (Figure 8.). ADCC is mediated by
nonspecific cytotoxic cells such as NK cells, monocytes and macrophages which can recognize the
Fc part of a tumor-bound antibody by their Fcγ-receptors. The activation of Fcγ-receptors receptor
induces the secretion of lytic enzymes by the cytotoxic cells which results in the target cell lysis.
Complement dependent cytotoxicity is induced when antigen-antibody complex deposited on the
cell surface triggers the activation of classical complement pathway resulting in the formation of
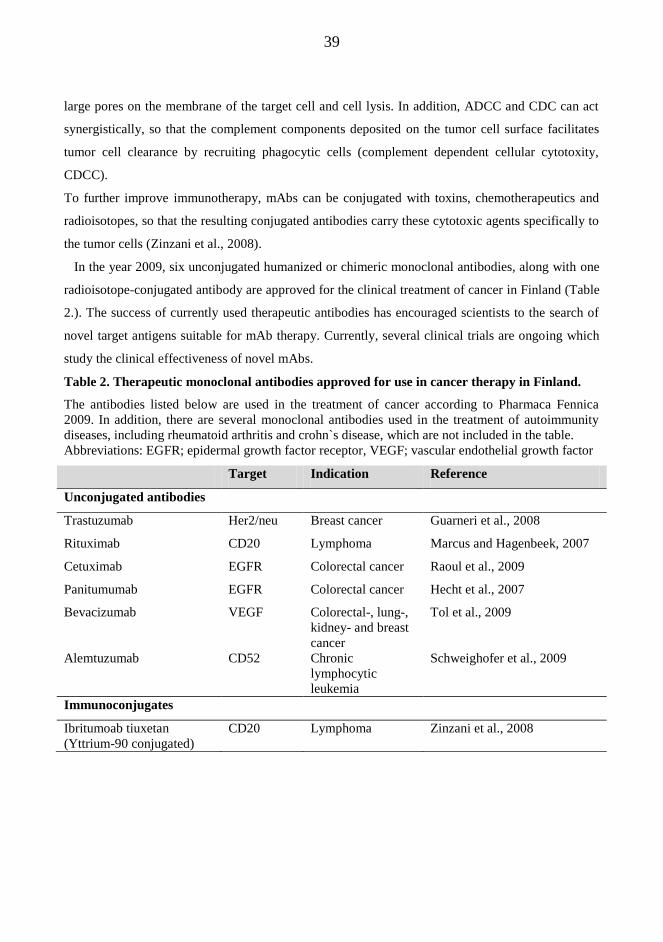
39
large pores on the membrane of the target cell and cell lysis. In addition, ADCC and CDC can act
synergistically, so that the complement components deposited on the tumor cell surface facilitates
tumor cell clearance by recruiting phagocytic cells (complement dependent cellular cytotoxity,
CDCC).
To further improve immunotherapy, mAbs can be conjugated with toxins, chemotherapeutics and
radioisotopes, so that the resulting conjugated antibodies carry these cytotoxic agents specifically to
the tumor cells (Zinzani et al., 2008).
In the year 2009, six unconjugated humanized or chimeric monoclonal antibodies, along with one
radioisotope-conjugated antibody are approved for the clinical treatment of cancer in Finland (Table
2.). The success of currently used therapeutic antibodies has encouraged scientists to the search of
novel target antigens suitable for mAb therapy. Currently, several clinical trials are ongoing which
study the clinical effectiveness of novel mAbs.
Table 2. Therapeutic monoclonal antibodies approved for use in cancer therapy in Finland.
The antibodies listed below are used in the treatment of cancer according to Pharmaca Fennica
2009. In addition, there are several monoclonal antibodies used in the treatment of autoimmunity
diseases, including rheumatoid arthritis and crohn`s disease, which are not included in the table.
Abbreviations: EGFR; epidermal growth factor receptor, VEGF; vascular endothelial growth factor
Target Indication Reference
Unconjugated antibodies
Trastuzumab Her2/neu Breast cancer Guarneri et al., 2008
Rituximab CD20 Lymphoma Marcus and Hagenbeek, 2007
Cetuximab EGFR Colorectal cancer Raoul et al., 2009
Panitumumab EGFR Colorectal cancer Hecht et al., 2007
Bevacizumab VEGF Colorectal-, lung-,
kidney- and breast
cancer
Tol et al., 2009
Alemtuzumab CD52 Chronic
lymphocytic
leukemia
Schweighofer et al., 2009
Immunoconjugates
Ibritumoab tiuxetan
(Yttrium-90 conjugated)
CD20 Lymphoma Zinzani et al., 2008

40
2.4. Follicular lymphoma
Lymphoma is a cancer that originates from the lymphocytes of the immune system and presents as
a solid tumor of lymphoid cells. Traditionally, lymphomas are divided in two groups; hodgkin
lymphoma which was the first form of lymphoma ever described, and non-hodgkin lymphomas
(NHLs). More recently, lymphomas are grouped according to WHO classification that recognizes
over 40 different lymphoma types (Swerdlow et al., 2008).
2.4.1. Incidence and clinical characteristics
Non-Hodgin lymphomas (NHLs) represent a heterogeneous group of malignancies originating
mainly from B and T lymphocytes of the immune system. Follicular lymphoma (FL) is the second
most common NHL-subtype comprising 20% of the cases (Anderson et al., 1998). For yet unknown
reasons, the incidence of FL:s is increasing steadily (Chiu and Weisenburger, 2003). In Finland,
about 250 new cases of follicular lymphoma are diagnosed annually (Jyrkkiö et al., 2007).
The clinical course of FL is extremely variable. In some patients the disease proceed in an
indolent way over a period of many years whereas in other patients the disease transforms to diffuse
large cell B cell (DLBC) lymphoma with an aggressive course (Advani et al., 2004).
Patients with follicular lymphoma usually presents with an enlarged superficial lymph node.
Delays in diagnosis are not uncommon, since enlarged lymph nodes may be unnoticed for a
prolonged period of time. In some patients, lymph nodes grow in deep areas in the retroperitoneal,
iliac- or mesenteric regions. In those cases, patients may have atypical symptoms and tumor bulk
may be large at the time of diagnosis. Few patients have symptoms such as fatigue, weight loss and
night sweating at the time of diagnosis. The bone marrow is affected in about half of the cases.
Rarely, FL presents as extranodal tumor growth. (Salles, 2007)
2.4.2 Pathophysiology
FL originates from germinal center (GC) B cells of lymphoid tissue. GCs are the sites for antibody
affinity maturation, a process which enables the development effective immune response (Allen et
al., 2007). GC:s consist of rapidly dividing centroblasts undergoing somatic hypermutation and
centrocytes undergoing antigenic selection. Only the centrocytes with improved antigen binding
capacity escape from the selection and the rest of the centrocytes die by apoptosis. FL is thought to
arise from accumulation of inappropriately rescued long lived B cells due to resistance in apoptosis.
Classically, FL cells express anti-apoptotic Bcl-2 protein due to t14;18 translocation which places

41
the bcl-2 gene under the influence of the IgG gene enhancer (de Jong, 2005). However, the t14;18
translocation is not sufficient alone for lymphomagenesis but it enables the accumulation of
subsequent genetic lesions affecting proto-oncogenes and tumor-suppressor genes (Schwaenen et
al., 2009). Recently, gene expression studies have shown that tumor-infiltrating non-malignant
immune cells may influence the transformation risk and survival of FL patients (Dave et al., 2004).
The diagnosis of FL is based on the morphology and immunohistochemistry of an affected lymph
node. FL cells express the surface markers sIg (M, D, G, A), CD19, CD20, CD22 and CD79a which
can be detected by immunohistochemistry or by flow cytometry. In addition, germinal center B cell
antigens CD10 and Bcl-6 are expressed in FL cells (Marcus and Hagenbeek, 2007).
2.4.3 Current treatment of follicular lymphoma
The treatment and prognosis of FL has changed radically during the past years since the
introduction of monoclonal antibody therapies and stem cell transplantation. The standard
chemotherapies used at 1980-90s had no effect on overall survival rates and thus advanced stage FL
has been assumed as incurable disease. However, during the last years, the new therapies directed
against FL have markedly improved the life expectancy of FL patients.
The method of treatment is chosen on the basis of dissemination of a disease at the time of
diagnosis, which is stated by Ann Arbor classification (Table 3.) (Bendandi, 2008). In addition, the
age, symptoms and additional diseases of the patient contribute markedly to disease management.
The prognosis of FL is classified according to Follicular Lymphoma International Prognostic Index
(FLIPI) (Solal-Celigny et al., 2004) (Table 4.).
Table 3. The Ann Arbor classification
Stage Definition
I The cancer is located in a single region, usually one affected lymph node or one
extralymphatic organ
II Two or more affected lymph node regions, or local extralymphatic extension with lymph
nodes on the same side of the diaphgram
III Lymph node regions on both sides of the diaphgram, either alone or with local
extralymphatic extension
IV Diffuse involvement of one or more extralymphatic organs, including involvement of the
liver, bone marrow, or nodular involvement of the lungs

42
Table 4. Follicular lymphoma prognostic index (FLIPI)
Feature Good prognosis Adverse prognosis
Age ≤ 60 years > 60 years
Serum LDH level normal above normal
Ann Arbor stage I to II III to IV
Nodal sites involved ≤ 4 > 4
Hemoglobin level ≥ 120 g/l < 120 g/l
At most 20% of the patients have a regional disease without overall symptoms (Ann Arbor I-II) at
the time of diagnosis. In these cases radiotherapy of an affected lymph node is the most used
treatment modality. Radiotherapy can cure about half of the patients with stage I disease and quarter
of the patients with grade II FL (Wilder et al., 2001).
The majority of the patients have an advanced stage FL (Ann Arbor III-IV) at the time of
diagnosis. These patients are treated when disease related symptoms arise, FLIPI score is high or the
disease is morphologically aggressive (grade 3 which contains over 15 centroblasts per high-power
field) (Jyrkkiö et al., 2007). Elderly patients or patients with indolent disease and no symptoms
should first be managed by an active watch-and –wait approach because life expectancy of these
patients can not be improved by treatment (Ardeshna et al., 2003). Although a wide variety of
standard chemotherapeutics as single or combination regimen, with or without radiotherapy, have
been tested against FL, these treatment modalities have no effect on overall survival rates (Ardeshna
et al., 2003). However, at the last decade, the introduction of a monoclonal anti-CD20 antibody
rituximab has radically changed the treatment of B cell malignancies. Several large scale
randomized trials have proved that addition of rituximab to the standard chemotherapy significantly
improved clinical outcome (progression free survival and overall survival) in patients with
previously untreated advanced FL (Hiddemann et al., 2007). In addition, the use of rituximab as
maintenance therapy in relapsed FL responding to chemotherapy or immunotherapy has improved
progression free survival and overall survival rates (Marcus and Hagenbeek, 2007). The use of
rituximab maintenance therapy after first line therapy is currently being evaluated.
The rituximab therapy is usually well tolerated as compared to the standard chemotherapy which
is associated with considerable short and long term toxicity. Mild to moderate adverse effects, such
as hypotension, nausea and general weakness are relatively rarely reported after first infusion
(Kimby, 2005). Rituximab induces a rapid depletion of CD20 positive peripheral B cells, and the B

43
cell count remains low for the next 6 months (Kimby, 2005). Serum immunoglobulin levels remains
fairly stable, although a small reduction in serum IgM has been described during prolonged therapy.
Rituximab therapy has not associated with an increased risk to opportunist infections (Kimby,
2005), but further studies are needed to evaluate immunological adverse effects associated with the
long term use of rituximab.
In the future, the combination of anti-CD20 antibody with therapeutic radioisotopes may further
improve the survival of FL patients. Phase II study has demonstrated that 90-Ytrium-ibritumoab
(tiuxetan) either alone or as consolidation therapy followed by first line therapy improved response
rates in FL (Zinzani et al., 2008). Further studies are ongoing to establish the use of radiolabeled
anti-CD20 antibodies in FL treatment.
Myeloablative therapy followed by autologous stem cell transplantation (ASCT) and to a lesser
extend allogenic transplantation had improved long-term outcome of some patients with FL
(Hiddemann et al., 2007). These approaches are used not so commonly because of treatment related
toxicity and the risk of secondary malignancy, but may be considered in young patients with
relapsed FL with poor prognosis.
Rituximab- mechanism of action
Rituximab is a chimeric mouse/human monoclonal antibody targeted against human B cell surface
antigen CD20 (Figure 7.) (Reff et al., 1994). Several characteristics of rituximab make it excellent
tool for anticancer therapy. The expression pattern of CD20 on B cells is ideal, since it is expressed
on over 95% of B cell lymphomas (Mathas et al., 2000) and on normal B cells from the pre-B-cell
stage until the plasma cell differentiation (Reff et al., 1994). Thus, hematopoietic stem cells are left
unaffected by rituximab that enables the recovery of B cell pool after the therapy. In addition,
antibody secreting plasma cells do not express CD20, and thus the levels of antibodies are
maintained during the therapy. The humanized Fc part of the chimeric antibody enables the host
effector mechanism, which are important in rituximab induced cytotoxicity (see later). The
production of anti-chimeric antibodies is rare and not of clinical importance as compared to the
infusion of mouse mAbs which were associated with significant production of anti-mouse
antibodies (Kimby, 2005). Finally, the CD20 antigen is not internalized from the B cell surface
upon antibody binding (Reff et al., 1994).
Although the clinical results with rituximab have been promising, the exact mechanism how it
depletes B cells is controversial. Based on in vitro studies, rituximab can induce antibody-dependent

44
cell-mediated cytotoxicity (ADCC) (Reff et al., 1994), complement mediated cytotoxicity (CDC)
(Harjunpaa et al., 2000; Reff et al., 1994) and apoptosis (Mathas et al., 2000; Shan et al., 2000)
(Figure 8.). The relative contribution of these mechanisms to B cell depletion in vivo is not known,
and it is probable that synergistic action of these mechanisms is involved (Flieger et al., 2000). In
addition to direct induction of apoptosis, rituximab has been shown to sensitize lymphoma cells to
apoptosis induced by chemotherapeutics such as paclitaxel, fludarabine and cisplatin (Alas et al.,
2000; Alas et al., 2002; Jazirehi et al., 2003).
Figure 7. The structure of the rituximab antibody. The light- and heavy-chain variable regions are cloned from murine mAb 2B8 that specifically
recognizes CD20 molecule. The mouse derived genes encoding variable regions are linked with
genes encoding IgG1 heavy chain and kappa-light chain constant regions of human origin. After the
fusion of gene segments, the recombinant fusion antibody is produced in Chinese hamster ovarian
cells. (Reff et al., 1994). Abbreviations: FcγR; Fcγ-receptor
Variable region
Constant region
Binding region for FcγR
Light chain -
Heavy chain -
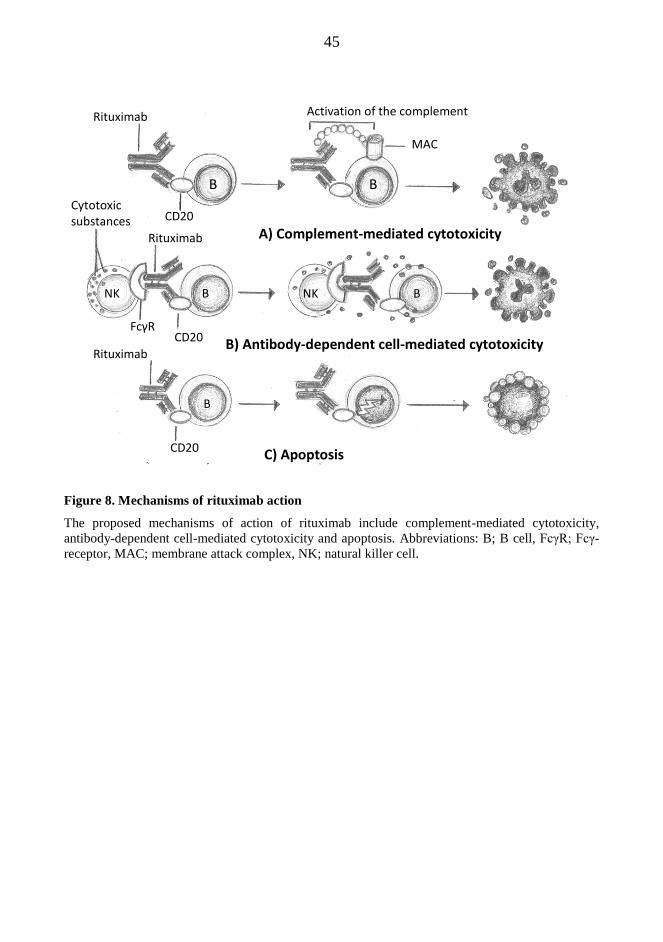
45
Figure 8. Mechanisms of rituximab action
The proposed mechanisms of action of rituximab include complement-mediated cytotoxicity,
antibody-dependent cell-mediated cytotoxicity and apoptosis. Abbreviations: B; B cell, FcγR; Fcγ-
receptor, MAC; membrane attack complex, NK; natural killer cell.
A) Complement-mediated cytotoxicity
B) Antibody-dependent cell-mediated cytotoxicity
C) Apoptosis
Rituximab
CD20
B B
Activation of the complement
MAC
Cytotoxic substances
FcγR CD20
Rituximab
CD20
Rituximab
NK NK B
B
B

46
3. AIMS OF THE STUDY
In general, the aim of the study was to examine the regulation of lymphoma cell apoptosis and
survival by cell surface receptors. The detailed knowledge of the apoptotic signaling pathways can
be used as an advantage in the development of new treatment modalities against B cell
malignancies.
Detailed aims were
1. to study the receptor-mediated regulation of B cell apoptosis during GC negative selection (I). To
get insight in the molecular mechanisms of B cell receptor – mediated apoptosis (I,III).
2. to study the molecular mechanisms of CD95/Fas –induced apoptosis (II, III). Could death
receptors serve as potential targets for cancer therapy?
3. to study the signaling pathways involved in the CD40-mediated protection against apoptosis (I,
II). Could CD40-CD40L interactions between lymphoma cells and the cells in their GC
microenvironment be responsible for resistance against cancer therapy? How the resistance
generated by anti-apoptotic function of CD40 can be counteracted?
4. to study the apoptotic signaling pathways induced by rituximab, and explore the means how
rituximab-induced cytotoxicity can be further enhanced (IV)

47
4. MATERIALS AND METHODS
4.1. Cell line and culturing
The studies presented in the original publications I-IV were performed in a human follicular
lymphoma (FL) cell line HF1A3. The cell line has been established from an enlarged lymph node of
a FL patient as described previously (Eray et al., 1994). The original patient sample from which the
cell line was established contained a nodular and diffuse centroblastic lymphoma according Kiel
classification (Eray et al., 2003). As a hallmark of a follicular lymphoma, HF1A3 cells contain the
t(14;18) translocation and overexpress the Bcl-2 protein. Based on the analysis of surface antigens
and responses against various physiological stimuli, the HF1A3 cells resemble germinal center
centrocytes undergoing negative selection. The cells were cultured as described in the original
publications I-IV.
4.2. Establishment of overexpressing cell lines
Transduction refers to the process where foreign DNA is introduced into cell by using a viral
vector. Commonly, the transduced gene coding for the particular protein is placed under the control
of a promoter that facilitates the transcription of the gene. In the publications III and IV, we used
transduced HF1A3 cell lines which overexpressed c-FLIP, Bcl-xL or dominat negative (DN) -
caspase-9 to study the signaling pathways essential for apoptosis. Transduction was performed by
using bicistronic lentivirus vector construct containing a gene of interest (DN-caspase-9, Bcl- xL,
FLIP-short or c-FLIP-long) followed by IRES (internal ribosomal entry site) and the GFP reporter
gene. Thus, the transduced cells always co-expressed the GFP reporter gene and the gene of interest.
After the transduction, the GFP-positive cells overexpressing the desired protein were sorted by
flow cytometry. Cells transduced with GFP reporter gene only were used as control cell line to
ensure that the cell transduction procedure in itself did not affect the viability of the cells, or made
them respond differently to apoptotic stimuli.
The establishment of DN-caspase-9 and Bcl-xL overexpressing cells has been described in detail
previously (Nuutinen et al., 2008). The establishment of c-FLIPlong/short overexpressing cells has
been described in the publication IV. Thus, only short overview of the cell transduction procedure is
given here.

48
Firstly, complementary DNA (cDNA) was synthesized by using the total mRNA extracted from
HF1A3 cells as a template. Thereafter, the sequences for caspase-9, Bcl-xL and c-FLIPshort / long were
amplificated by PCR using cDNA as template. Primer sequences for amplification contained SwaI-
sites which were designed to 5’ ends of the primers.
Dominant negative caspase-9 was generated by introducing a point mutation to catalytic cysteine,
Cys287Ser (TGT→AGT) by site-directed mutagenesis. This method uses oligonucleotide primer
containing the desired point mutation, and PCR to introduce the desired mutation to the gene of
interest.
The PCR fragments were isolated from agarose gel and inserted into a plasmid vector (pcr2.1-
TOPO-TA vector, Invitrogen, Carlsbad, CA). Subsequently, competent One Shot TOP10-E.coli
(Invitrogen) was transformed with the plasmid vector. Thereafter, the sequences were analysed to
avoid unwanted mutations. After sequencing inserts were cloned to SwaI site of the pWPI-IRES-
GFP (Tronolab, Switzerland) lentivirus-vector. In addition, pWPI-IRES-GFP lentivirus-vector was
produced and used for the establishment of control cell line. Subsequently, lentiviruses containing
the vector were produced following the protocol described earlier (Pellinen et al., 2004).
The HF1A3 cells were transduced with lentiviral vectors containing DN-caspase-9-IRES-GFP,
Bcl-xL-IRES-GFP, c-FLIPlong.-IRES-GFP and c-FLIPshort.-IRES-GFP. After three days, cells were
collected and cultured in fresh medium. The cultures were expanded and GFP positive cells were
enriched by using flow cytometer until over 95% of the cells were GFP positive. Overexpression of
Bcl-xL, DN-caspase-9, c-FLIPshort and c-FLIPlong was confirmed by Western blot.
4.3. Cell treatment experiments
In cell treatment experiments, apoptosis was induced by culturing cells in the presence of
antibodies directed against surface receptors. Physiologically, these receptors are activated by
binding of endogenous ligands that result in the clustering of adjacent receptors and activation of
intracellular signaling cascades. Similarly, antibodies used in the experiments cross-link adjacent
receptors resulting in their activation. The stimulating antibodies used in original publications are
introduced in table 5. Chemical inhibitors of signaling proteins were used to study importance of a
particular signaling pathway in apoptosis or survival. The inhibitors used are listed in the table 6.
The treatment of cells during the experiments is described in detail in the original publications.

49
Table 5. Antibodies used in cell treatment experiments. Abbreviations: κ; kappa, mAb;
monoclonal antibody, F(ab`)2; fragment antigen binding.
Antibody Type Antigen Publication
Anti-κ (anti-IgG) mAb, IgG1 κ-chain constant region of
human IgG
I, III
Anti-Fas (CH11) mAb, IgM Fas/ CD95 II, III, IV
AF.1.15 mAb, IgG4 CD40 I, II, IV
Rituximab chimeric, IgG1 CD20 IV
Anti-human
IgG(γ)
F(ab`)2 heavy chain constant region of
human IgG
IV
Table 6. Chemical inhibitors of signaling proteins
Inhibitor Target Publication
LY294002 phospahtidyl-inositol-3-kinase (PI3K) I
PD98059 mitogen-activated protein kinase kinase 1 (MEK1) I
Gö6850 protein kinase C (PKC) I
ZM336372 Raf I
PDTC NF- B II
z-IETD-fmk caspase-8 II
z-DEVD-fmk caspase-3 II, III
z-VAD-fmk caspases III, IV
4.4 Apoptosis detection
4.4.1 Quantification of fractional DNA content (I-IV)
Typical for apoptosis is the activation of endonucleases which cleave nuclear DNA at sites
between nucleosomes creating internucleosomal DNA fragments of about 200 base pairs and its
multiples. When DNA from apoptotic cells is analyzed by agarose gel electrophoresis, typical
“ladder” pattern of about 200 base pairs is detected. In the publications I-IV, flow cytometry and
the cell cycle analysis was used for the detection of cells with fragmented DNA. Before the
analysis, cells were fixed with ethanol and stained with a DNA binding dye propidium iodide.

50
As a result of DNA fragmentation, the amount of cells containing normal diploid chromosomal
content of DNA decreases, and a population of cells with decreased (hypodiploid) DNA content is
formed. The proportion of hypodiploid cells (sub-G1 fraction) can be quantified by flow cytometry.
4.4.2 Mitochondrial membrane potential depolarization (I-IV)
Mitochondrial membrane potential depolarization occurs as a result of changes in the
permeability of mitochondrial membranes during apoptosis. The depolarization can be analyzed
indirectly by using lipophilic cations which accumulate in the mitochondrial matrix driven by the
electrochemical gradient between the matrix and the intermembrane space. In publications II-IV,
loss of mitochondrial membrane potential ( Ψm) was assessed by using a cationic dye tetramethyl
rhodamine (TMRM, Molecular Probes) as previously described (Castedo et al., 2002). Cells lose
their capacity to retain TMRM stain when the electrochemical gradient between the matrix and
intermembrane space is collapsed. In the publication I, loss of mitochondrial membrane potential
( Ψm) was assessed by ApoAlert Mitochondrial Membrane Sensor Kit (Clontech Laboratories, Inc.,
California, USA). The kit contains a cationic dye MitoSensorTM
that fluoresces differently in
apoptotic and nonapoptotic cells, because of changes in mitochondrial membrane potential. After
staining, the cells were analyzed by flow cytometry. Only the early apoptotic cells with normal side-
and forward-scatter properties were included in the analysis.
4.4.3 Cytochrome c release (I-IV)
The analysis of cytochrome c release was based on the cell fractionation, which enabled the
separation of cell fraction containing mitochondria from the cytosol. The cell fractionation
procedure is described in detail in the publication I. After the fractionation, the amount of
cytochrome c in both mitochondrial and cytosolic fractions was detected by Western blotting.
Membranes were reprobed with an antibody against a mitochondrial protein cytochrome oxidase
subunit IV (COX-4) to ensure that the fractionation procedure was successful and mitochondrial
fractions contained equal amount of protein. Equal loading of cytosolic samples was assessed by
immunoblotting with polyclonal anti-actin antibodies.
4.4.4 Caspase activation (I-IV)
Commonly, more than one approach was used for the assay of caspase activity in original
publications. The activation of DEVD-specific caspases (i.e. caspase-3 and -7) was assessed by

51
employing fluorochrome substrate Ac-DEVD-AMC (Calbiochem, Ca, USA) which become
fluorescent upon cleavage by the caspase (publications I, II and IV). In addition to direct analysis of
caspase activity, activation of DEVD-specific caspases was assessed indirectly by testing whether
their specific inhibitor z-DEVD-fmk inhibited apoptosis (publications II and III). More specifically,
activation of caspase-3 was assayed by immunoblotting (publications II, III and IV). Upon
activation, full length caspase-3 is cleaved, and caspase activation can thus be detected by using an
antibody which recognizes both the cleavage product and the full length caspase-3.
The activation of caspase-8 was assayed indirectly by testing whether a chemical inhibitor z-
IETD-fmk, a specific inhibitor of caspase-8, inhibits apoptotic events (publication II). In addition,
cell lines expressing c-FLIP proteins, natural inhibitors of caspase-8 activation at the DISC, was
used to study the role of caspase-8 activation in apoptosis (publication III, IV). The activation of
caspase-8 was also assayed directly by immunoblotting as an increase of a cleavage product of
caspase-8 and a decrease of full length caspase-8. Recently, this method has been questioned since it
has been shown that merely dimerization, and not cleavage of caspase-8, is actually involved in its
activation (Pop et al., 2007). However, when caspase-8 activation was assessed in our experimental
model, we get similar results with immunoblotting and by inhibiting caspase-8 activation
chemically or by c-FLIP overexpression
The activation of caspase-9 was assayed by using cells stably overexpressing DN-caspase-9, in
which caspase-9 activity was blocked by single amino acid mutation at the catalytic site of the
protease (publications III, IV).
4.4.5 Cell membrane permeabilization III
During both necrotic and the late stages of apoptotic cell death, the cell membrane integrity is lost
and thus the cells can uptake stains such as propidium iodide, which can be detected by flow
cytometry. In the publication III, cell permeability assay was used as a cell viability analysis to
complement other assays of cell death. After staining with propidium iodide, cells were analyzed
with flow cytometry. The data were expressed as the proportion of living cells (the PI negative cells
with normal FCS properties) against the total cell count.
4.5. Western blotting
The samples containing equal amount of protein were run on SDS PAGE under reducing
conditions and transferred to nitrocellulose membranes. The membranes were pretreated with 3%

52
BSA buffer to reduce non-specific binding of antibodies. Following primary antibodies were used
for the immunoblotting: rabbit polyclonal anti-FLIPshort/long, rabbit polyclonal anti-caspase-3 H-277
and rabbit polyclonal anti-caspase-8 p20 (Santa Cruz Biotechnology, CA). Horseradish peroxidase-
conjugated goat anti-rabbit (Zymed Laboratories, CA) was used as a secondary antibody. ECL
chemiluminescence system (Amersham, Arlington Heights, IL) was used for detection.
4.6. Reverse transcriptional (RT)-polymerase chain reaction
In the publication II, C-FLIP mRNA expression was determined by quantitative reverse
transcription-polymerase chain reaction (RT-PCR). Total RNA extraction and PCR reaction were
performed as described in the publication II. The PCR products were analyzed on 2% agarose gel
and visualized by ethidium bromide staining.
4.7 Statistical analyses
In the publications I-II, student’s t-test was used to determine the statistical significance between
the selected groups. In the publication III, data were analyzed by repeated-measures
two-way ANOVA and Bonferroni post test. In the publication IV, Oneway ANOVA and Dunnett`s
post test was used for comparing selected group with other groups. Statistical analyses were
performed using GraphPad Prism version 4.0 for windows. p < 0.05 was considered significant.

53
5. RESULTS
5.1. The molecular mechanisms of B cell receptor-induced apoptosis (I, III)
The B cell receptor signaling has been a target of enthusiastic research during the past decades as
there are almost three thousand original articles published that concern BCR signaling. The BCR
has gained a lot of research interest because of its multiple and important functions in the B cell
development and adative immunity. As discussed earlier, the response to BCR-stimulation can be
proliferation, anergy or apoptosis depending on the maturational stage of a B cell and nature of
antigen binding (Niiro and Clark, 2002). This work studied the molecular mechanisms of BCR-
mediated apoptosis and how signals from the BCR regulate the fate of a germinal center B cell
coordinately with CD40 and Fas receptors.
5.1.1 The signal transduction pathways connected with B cell receptor-induced apoptosis
The signal transduction pathways involved in BCR-induced apoptosis are not yet clarified. Of the
major signaling pathways activated by BCR, the Ras-Raf-1-ERK and PLC-γ2 activation pathways
has been connected with apoptosis in some experimental models (Lee and Koretzky, 1998; Stang et
al., 2009) (Figure 9.). We used the specific inhibitors of Raf, ERK, PKC and p38 to study the
involvement of these kinases in BCR induced apoptosis (Figure 9.).
The Ras-Raf-1-ERK pathway was activated by BCR stimulation in HF1A3 cells, since we could
demonstrate a transient phosphorylation of ERK1/2 after BCR triggering analyzed by Western blot
(not shown). However, the selective inhibitors of Raf (ZM 336372) or ERK (PD 98059), showed
only slight reduction in the percentage of apoptotic cells after BCR triggering (Figure 10.). Pre-
treatment of cells with specific inhibitors of PKC (GÖ 6850) and p38 (SB 203580) showed only
minor effect on BCR-induced apoptosis (Figure 10.). Based on these results, we could not ascertain
the signal transduction pathway crucial for BCR-mediated apoptosis in HF1A3 cells.

54
Figure 9. The signal transduction pathways associated with B cell receptor-induced apoptosis. The specific inhibitors for the activation of Raf (ZM), PKC (GÖ), P38 (SB) and PI3K (LY) are
included in the picture.
5.1.2 The role of caspase-9 and mitochondrion in B cell receptor-induced apoptosis (I, III)
To get insight on the mechanism involved in BCR-induced apoptosis, we performed a careful
kinetic analysis of four sequential molecular events of apoptosis: activation of caspase-3,
fragmentation of nuclear DNA collapse of ΔΨm and release of cytochrome c (I). The activation of
caspase-3 was analyzed by a DEVD-specific protease assay and by Western blot analysis of the
caspase-3 cleavage product. The activation of caspase-3 started to increase 12 hours after BCR
triggering. Fragmentation of DNA, collapse of ΔΨm and release of cytochrome c followed similar
kinetics. BCR-induced apoptosis was dependent on the synthesis of new proteins, since the BCR-
induced DNA fragmentation and ΔΨm collapse were abrogated in the presence of the protein
synthesis inhibitor cycloheximide.
RAS
Raf
PLC
PKC Ca2+
PI3K
AKT
GSK3
Cyclin D NF-κB NFAT CREB
ERK JNK P38
BCR
ZM
BD
GÖ
MAPKs
LY
SB

55
Figure 10. The inhibitors of the signaling pathways connected with BCR show no effect on
apoptosis. HF1A3 cells were cultured in media containing ERK inhibitor PD 98059 (15 μM), Raf
inhibitor ZM 336372 (10 μM), PKC inhibitor GÖ 6850 (500nM) or p38 inhibitor SB 203580 (10
μM) for one hour followed by stimulation with anti-IgG antibodies for additional 23 hours.
Subsequently, cells were harvested for propidium iodide staining followed by analysis of cells with
fragmented DNA by flow cytometry. The data are presented as mean + SD from three experiments.
The data are previously unpublished.
It is well established that caspase-9 is the apical caspase in the intrinsic pathway of apoptosis. To
explore whether caspase-9 is involved in the BCR-mediated apoptosis, we generated a DN-caspase-
9 overexpressing cell line in which caspase-9 was inactivated by a single amino acid mutation at the
catalytic domain of the protease (III). The cells transduced with the empty IRES-GFP-vector were
used as a vector control cells. The overexpression of DN-caspase-9 could effectively inhibit the
BCR-induced fragmentation of nuclear DNA (Figure 11b.) and cleavage of caspase-3. However,
BCR-induced Ψm collapse (Figure 11a.) and cell death measured by PI exclusion assay were only
partly inhibited by DN-caspase-9 expression. Interestingly, the BCR-induced cytochrome c release
was almost totally inhibited in DN-caspase-9 overexpressing cells, although the cytochrome c
release has been previously suggested to be upstream of caspase-9 activation in the apoptotic
cascade (III).
The release of cytochrome c and collapse of Ψm are regulated by a balance between pro-and anti-
apoptotic members of the Bcl-2 family (Kim et al., 2006; Willis et al., 2007). In consistent with this,
0
10
20
30
40
50
control PD anti-IgG anti-IgG
+ PD
0
10
20
30
40
50
control GÖ anti-IgG anti-IgG
+ GÖ
0
10
20
30
40
50
control ZM anti-IgG anti-IgG
+ ZM
0
10
20
30
40
50
control SB anti-IgG anti-IgG
+ SB
% a
popto
tic c
ells
% a
popto
tic c
ells
% a
popto
tic c
ells
% a
popto
tic c
ells

56
overexpression of Bcl-xL prevented both BCR-induced cytochrome c release and collapse of Ψm in
HF1A3 cells. In addition, the BCR-induced DNA fragmentation, caspase-3 activation and cell death
was blocked by Bcl-xL overexpression. Thus, we next asked if BCR-induced apoptosis was
associated with the activation of pro-apoptotic members of the Bcl-2 family. We could not detect
any changes in the expression level or intracellular location of pro-apoptotic Bim, Bik , Bad or Bax
proteins after BCR triggering, although these proteins have been associated with BCR-induced
apoptosis previously (Eldering et al., 2004; Jiang and Clark, 2001; Malissein et al., 2003; Takada et
al., 2006).
5.1.3 The caspase-8 activation pathway showed only marginal role in B cell receptor-induced
apoptosis
The previous work has demonstrated that cross-linking of BCR induces an apoptotic pathway
involving caspase-8 activation (Besnault et al., 2001). To study the role of caspase-8 activation
pathway in BCR-induced apoptosis in our experimental model we generated a cell line
overexpressing the long splice variant of cellular FLICE inhibitory protein, c-FLIPlong. Both the long
and short splicing variant of c-FLIP interfere with caspase-8 activation at the death inducing
signaling complex (DISC) (Krueger et al., 2001). HF1A3 cell were transduced with wild type c-
FLIPlong in IRES-GFP-lentivector or with IRES-GFP-lentivectror only (vector control).
The overexpression of c-FLIPlong only marginally reduced the appearance of hypodiploid cells or
Ψm collapse after BCR stimulation (Figure 11). The poor effect of c-FLIP overexpression on BCR-
induced apoptosis could not be explained by inadequate expression of c-FLIP, since Fas-induced
DNA fragmentation and Ψm collapse were almost completely blocked in the same c-FLIPlong
overexpressing cell line (Figure 11.). In concordance, the specific inhibitor of caspase-8 (z-IETD-
fmk) failed to inhibit BCR-mediated apoptosis while it effectively rescued cells from Fas-mediated
apoptosis.
Figure 11a-b. The effect of Bcl-xL, DN-caspase-9 and c-FLIPlong overexpression on receptor
mediated apoptosis. The cells overexpressing Bcl-xL, DN-caspase-9 or c-FLIPlong were cultured
with anti-IgG (3 μg/ml), rituximab (10 μg/ml) or anti-Fas (10 ng/ml) antibodies for 24 hours. Cells
transduced with IRES-GFP-lentivector only were used as vector control cells. A) After culture, the
cells were harvested for TMRM staining and flow cytometric analysis of cells with collapsed
mitochondrial membrane potential (TMRM low cells). B) Cells were harvested for propidium
iodide staining followed by analysis of cells with fractional DNA content by flow cytometry
(hypodiploid cells).

57
A)
B)
0
5
10
15
20
25
30
35
HF
1A
3-G
FP
HF
1A
3-B
cl-
XL
HF
1A
3-D
N-c
asp
-9
HF
1A
3-F
LIP
-L
HF
1A
3-G
FP
HF
1A
3-B
cl-
XL
HF
1A
3-D
N-c
asp
-9
HF
1A
3-F
LIP
-L
HF
1A
3-G
FP
HF
1A
3-B
cl-
XL
HF
1A
3-D
N-c
asp
-9
HF
1A
3-F
LIP
-L
HF
1A
3-G
FP
HF
1A
3-B
cl-
XL
HF
1A
3-D
N-c
asp
-9
HF
1A
3-F
LIP
-L
Control Anti-IgG Rituximab Anti-Fas
% c
ells
with
co
llap
sed
ΔΨ
m
0
10
20
30
40
50
60
HF1
A3-G
FP
HF1A
3-B
cl-X
L
HF
1A
3-D
N-c
asp
-9
HF1A
3-F
LIP
-L
HF
1A
3-G
FP
HF1A
3-B
cl-X
L
HF
1A
3-D
N-c
asp
-9
HF
1A
3-F
LIP
-L
HF
1A
3-G
FP
HF
1A
3-B
cl-X
L
HF1A
3-D
N-c
asp-9
HF
1A
3-F
LIP
-L
HF1A
3-G
FP
HF
1A
3-B
cl-X
L
HF1
A3
-DN
-cas
p-9
HF
1A
3-F
LIP
-L
Control Anti-IgG Rituximab Anti-Fas
% H
yp
od
iplo
id c
ells

58
5.2. The molecular mechanisms of Fas/CD95-induced apoptosis (II, III, IV)
Fas triggering in HF1A3 cells induced the cleavage and activation of caspase-8, which could be
demonstrated by Western blot as an increase of the 18 kDa cleavage product and a decrease of the
57 kDa full length caspase-8 (II). The specific inhibitor of caspase-8 activation, z-IETD-fmk,
completely inhibited Fas-induced apoptosis showing the necessity of caspase-8 activation for the
process (II). In line with this, the overexpression of both short and long splicing variants of c-FLIP,
which interfere with caspase-8 activation at the DISC and thus inhibit caspase-8 activation, blocked
Fas-induced apoptosis (Figure 11.) (IV). The activation of caspase-8 was followed by the activation
of caspase-3 and fragmentation of nuclear DNA (II). The activation of caspase-9, the initiator
caspase of the mitochondrial pathway, was unnecessary for Fas-induced apoptosis, since the
overexpression of catalytically inactive DN-caspase-9 showed no effect on cell death (Figure 11.)
(III). Fas-stimulation induced a profound release of cytochrome c and collapse of Ψm which could
be inhibited by Bcl-xL overexpression (Figures 11.) (III). However, the overexpression of Bcl-xL
showed no effect on Fas-induced activation of caspases -8 and -3, showing that the caspase
activation was independent on the mitochondrial breach (III). In addition, the overexpression of Bcl-
xL only partly inhibited Fas-induced apoptosis as measured by DNA fragmentation (Figure 11.) (III)
5.3. The molecular mechanisms of rituximab-induced apoptosis (IV)
Rituximab, the chimeric human-mouse mAb against the CD20 antigen, has been widely used in
the therapy against follicular lymphoma. Rituximab has been shown to deplete malign B cells by
three different and overlapping mechanisms: CDC, ADCC and apoptosis (Manches et al., 2003;
Reff et al., 1994; Shan et al., 1998; van der Kolk et al., 2002). Binding of rituximab to the CD20
surface antigen induces apoptosis by yet unknown mechanisms. Detailed knowledge of CD20-
induced signaling pathways to apoptosis may allow the development of more effective treatment
modalities against B cell malignancies.
Propensity of various B cell lineages to undergo CD20-mediated apoptosis is highly variable
(Chan et al., 2003; Manches et al., 2003). Sensitivity to rituximab-induced apoptosis has been linked
with a maturational stage of a lymphoma progenitor cell (Mattila and Meri, 2008). This finding was
also reproduced in our FL cell lines representing different maturational stages, as only one (HF1A3)
of three available cell lines responded to rituximab treatment by apoptosis. The other cell lines,
HF28 and H4.9, were resistant to rituximab-mediated apoptosis even when rituximab bound to

59
CD20 antigen was cross-linked by secondary antibody (anti-Fc -F(ab2)) (not shown). Although the
use of cross-linking secondary antibodies against rituximab has augmented apoptosis in some
experimental models (Stel et al., 2007; van der Kolk et al., 2002), in HF1A3 cells rituximab-induced
apoptosis could not be enhanced by hyper-cross-linking. Instead, in HF1A3 cells, F(ab2) fragments
directed against the Fc part of antigen receptor induced apoptosis itself.
The involvement of caspases in the rituximab-induced apoptosis has been controversially
discussed. To explore the necessity of the caspase-9 and caspase-8 for rituximab-induced apoptosis,
we developed cell lines in which the activity of these caspases was artificially blocked. The
involvement of caspase-9, the initiator caspase of the intrinsic apoptosis pathway, was studied by
using DN-caspase-9 overexpressing cells, in which caspase-9 was inactivated by a single amino
acid mutation at the catalytic domain of the protease. The HF1A3 cells overexpressing FLIPshort and
FLIPlong, natural inhibitor proteins of caspase-8 activation, were used to study the involvement of
caspase-8 activation in the rituximab-induced apoptosis. According to our results, the caspase-9
activation pathway was necessary for the rituximab-induced apoptosis, since blocking of the
caspase-9 activation inhibited all apoptotic events induced by rituximab, including the activation of
caspase-3, fragmentation of DNA (Figure 11b.), collapse of Ψm (Figure 11a) and release
cytochrome c (IV). In contrast, the caspase-8 activation pathway was dispensable for apoptosis
induced by rituximab, since the overexpression of FLIP proteins showed no effect on rituximab-
induced apoptosis although in the same time Fas-induced apoptosis was totally abrogated by c-FLIP
overexpression (Figure 11.).
Overexpression of Bcl-xL almost completely blocked the cytochrome c release and collapse of
Ψm, showing that these changes were regulated by Bcl-2 family proteins (Figures 11.) (IV). In
addition, rituximab-induced DNA fragmentation (Figure 11b.), collapse of Ψm (Figure 11a.),
caspase-3 activation and cell death were also abrogated in Bcl-xL overexpressing cells.
The kinetics and magnitude of rituximab-induced apoptosis were substantially slower compared
with Fas-induced apoptosis in the same cell line. After four days culture with rituximab, only about
half of the cells were apoptotic. The slow induction of apoptosis may be partly explained by the fact
that rituximab induces only a subpopulation of cells to undergo apoptosis at a time whereas the
remainder of the cells may still proliferate and repopulate the culture.
The simultaneous culture with anti-Fas and rituximab antibodies showed an additive effect on
apoptosis. The percentage of apoptotic cells increased from 15.9% +/- 1.7 (rituximab) to 38.3% +/-
3.0 (rituximab combined with anti-Fas).
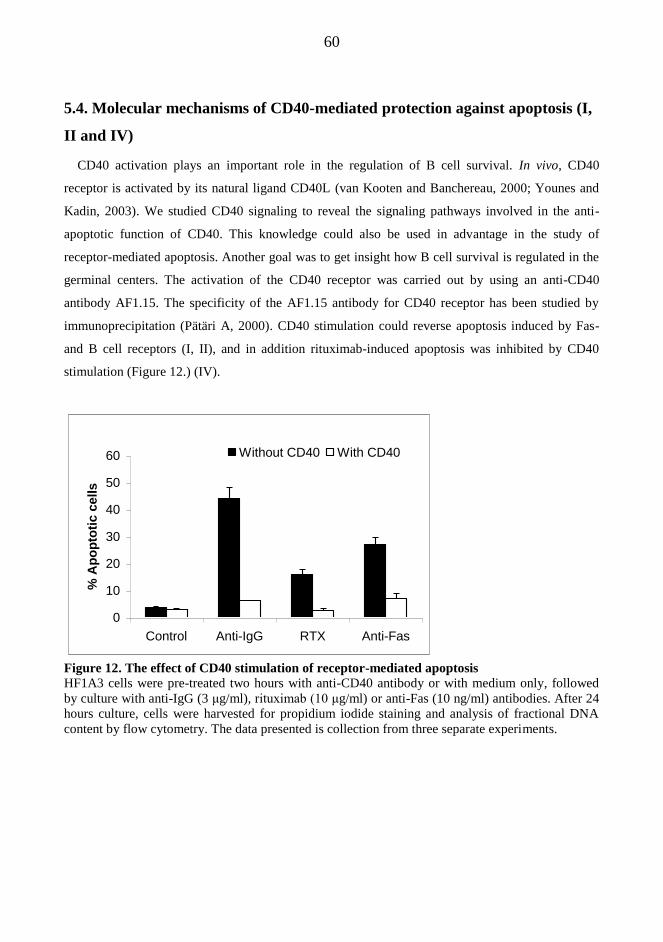
60
5.4. Molecular mechanisms of CD40-mediated protection against apoptosis (I,
II and IV)
CD40 activation plays an important role in the regulation of B cell survival. In vivo, CD40
receptor is activated by its natural ligand CD40L (van Kooten and Banchereau, 2000; Younes and
Kadin, 2003). We studied CD40 signaling to reveal the signaling pathways involved in the anti-
apoptotic function of CD40. This knowledge could also be used in advantage in the study of
receptor-mediated apoptosis. Another goal was to get insight how B cell survival is regulated in the
germinal centers. The activation of the CD40 receptor was carried out by using an anti-CD40
antibody AF1.15. The specificity of the AF1.15 antibody for CD40 receptor has been studied by
immunoprecipitation (Pätäri A, 2000). CD40 stimulation could reverse apoptosis induced by Fas-
and B cell receptors (I, II), and in addition rituximab-induced apoptosis was inhibited by CD40
stimulation (Figure 12.) (IV).
0
10
20
30
40
50
60
Control Anti-IgG RTX Anti-Fas
% A
po
pto
tic c
ells
Without CD40 With CD40
Figure 12. The effect of CD40 stimulation of receptor-mediated apoptosis
HF1A3 cells were pre-treated two hours with anti-CD40 antibody or with medium only, followed
by culture with anti-IgG (3 μg/ml), rituximab (10 μg/ml) or anti-Fas (10 ng/ml) antibodies. After 24
hours culture, cells were harvested for propidium iodide staining and analysis of fractional DNA
content by flow cytometry. The data presented is collection from three separate experiments.

61
5.4.1 Kinetics and signaling requirements of CD40 mediated protection from B cell receptor-
mediated apoptosis (I)
As presented above, BCR-induced apoptosis in HF1A3 cells was manifested by caspase-3
activation, DNA fragmentation, cytochrome c release and collapse of Ψm. All these apoptotic
events were completely blocked by CD40 stimulation. Interestingly, CD40 stimulation was able to
protect from BCR-induced apoptosis up to 10 h from the induction of apoptosis. When CD40
stimulation was done 12 h or later after the induction of apoptosis, its protective effect was
gradually lost.
CD40 stimulation has been connected with the activation of the PI3-kinase, and mitogen activated
protein kinases (MAPKs) P38 and ERK (van Kooten and Banchereau, 2000). In addition, the PKC
activation pathway has been connected with B cell survival (Kronfeld et al., 2002). We used
specific inhibitors of these pathways to study their role in CD40-mediated survival.
Pre-treatment of cells with PI3-kinase inhibitor LY294002 had no effect on CD40-mediated
protection from BCR-induced apoptosis. Similar results were also observed with specific ERK
inhibitor PD 98059, Raf inhibitor ZM 336372, P38 inhibitor SB 203580 and PKC inhibitor Gö 6850
(I).
5.4.2 The CD40-induced protection against Fas-induced apoptosis was associated with a rapid
up-regulation of anti-apoptotic c-FLIP (II)
As discussed above, activation of caspase-8 was indispensable for the induction of Fas-induced
apoptosis. The Fas-induced cleavage and activation of caspase-8 was completely blocked by CD40
stimulation, since the p18 active cleavage product of caspase-8 could not be detected when cells
were pre-treated with CD40 before Fas stimulation. In addition, Fas-induced caspase-3 activation,
DNA fragmentation, cytochrome c release and Ψm collapse were blocked by CD40 stimulation.
The specific inhibitor of NF-κB activation, PDTC, was used to study the involvement of NF-κB in
the anti-apoptotic signaling of CD40. When cells were pre-treated with PDTC before stimulation
with Fas and CD40 antibodies, CD40 stimulation could no longer protect the cells from Fas-induced
apoptosis. In addition, in the presence of PDTC, CD40 stimulation could not inhibit the Fas-induced
caspase-8 activation.
Among other targets, transcription factor NF-κB regulates the expression of c-FLIP proteins,
which can interfere with caspase-8 activation at the DISC (Krueger et al., 2001). Thus, we next
studied if CD40 stimulation could induce c-FLIP proteins in our experimental model. CD40

62
stimulation induced a rapid upregulation of both long and short isoforms of c-FLIP, which could be
seen at the mRNA within 2 hours and protein level within 4 hours after CD40 stimulation (II). The
CD40-induced upregulation of c-FLIPshort/long mRNA and protein were completely blocked by
PDTC, showing that the up-regulation of c-FLIP was dependent on the activation of NF-κB (II).

63
6. DISCUSSION
6.1. FL cell lines as an experimental model for the study of receptor-mediated
apoptosis
The cell line HF1A3 provides an excellent model for the study of receptor-mediated apoptosis of
GC B cells, since the cells have retained the same immunophenotype and responses against
physiological stimuli as their natural counterparts (Eray et al., 2003). However, although the cell
line HF1A3 mimics germinal center B cells, the cells have genetic abnormalities such as Bcl-2
overexpression, which may hinder cell death. We used the HF1A3 cell line to study receptor-
mediated selection of GC B cells. The findings presented here would be more easily generalized to
normal GC B cells if we could replicate the results with primary GC B cells. However, the natural
GC B cells die rapidly in vitro without growth support provided by their microenvironment, which
makes it difficult to study apoptosis by using GC derived primary B cells. In addition, the cell
transfection studies could only be conducted with stable cell lines, since large amount of cells is
needed for the experimental procedure.
The cell line and the original patient sample have been carefully diagnosed according to REAL
classification as follicular center cell lymphoma, a subtype of a follicular lymphoma (Eray et al.,
2003). Thus, the cell line offers a versatile tool for the pre-clinical study of FL directed cancer
therapeutics. We used HF1A3 cell line for the study of molecular mechanisms involved in
rituximab-induced apoptosis. Unfortunately, only one of our three FL cell lines was susceptible for
rituximab-mediated apoptosis, and the other two cell lines (HF28 and HF4.9) were resistant. Thus,
our findings concerning the molecular mechanism of rituximab-induced apoptosis, as well as BCR
and Fas –induced apoptosis, need to be replicated in other B cell lines to generalize the results to all
B cells. The establishment of FL cell lines has been demanding, and still over 10 years after their
establishment, suitable human FL cell lines are not common.

64
6.2. Molecular mechanisms of receptor-mediated apoptosis
6.2.1 Fas-induced apoptosis proceed via caspase-8 activation pathway while B cell receptor-
and rituximab –induced apoptosis are dependent on mitochondrial pathway
As discussed earlier, two major apoptosis pathways have been classified; the death receptor
(extrinsic) and mitochondria dependent (intrinsic) pathways. The death receptor pathway relies on
the activation of the initiator caspase-8 at the DISC, followed by the activation of effector caspase-3
(Medema et al., 1997). In mitochondria dependent (intrinsic) apoptosis, various apoptosis-inducing
stress signals converge on mitochondria, resulting in the permeabilization of MOM, release of pro-
apoptotic IMS proteins and collapse of ΔΨm. The release of cytochrome c from IMS triggers the
activation of caspase-9, an initiator caspase of the intrinsic pathway (Li et al., 1997). Commonly,
the extrinsic pathway is engaged with the mitochondria-dependent pathway of apoptosis. In these
cases, caspase-8 cleaves pro-apoptotic BH3-only protein Bid, and the truncated Bid translocates to
mitochondria where it can promote MOMP and apoptosis (Li et al., 1998).
We used cell lines stably overexpressing Bcl-xL, DN-caspase-9 and c-FLIP to explore the
involvement the death receptor (extrinsic) or mitochondrial (intrinsic) pathways in receptor-
mediated apoptosis. As expected, Fas-induced apoptosis of HF1A3 cells showed typical
characteristics of extrinsic apoptosis. Firstly, Fas-induced apoptosis was completely prevented when
caspase-8 activation was blocked by a specific caspase-8 inhibitor z-IETD-fmk, or overexpression
of either short or long splicing variants of c-FLIP. Secondly, Fas-induced apoptosis proceeded
normally in cells overexpressing catalytically inactive form of caspase-9, which is an initiator
caspase of the intrinsic apoptosis. Finally, Bcl-xL overexpression showed only 50% decrease in the
amount of apoptotic (hypodiploid) cells, while Fas-induced cytochrome c release and collapse of
ΔΨm were markedly inhibited in Bcl-xL overexpressing cells. In line with this, we showed that
caspases were mainly activated upstream of mitochondrial breach in Fas-induced apoptosis.
However, although mitochondria seem not be involved in the activation of caspases during Fas-
induced apoptosis, the mitochondrial breach may still enhance the cell death process. Loss of
mitochondrial normal function leads to unbalanced Ca2+
release from the matrix, uncoupling of
oxidative phosporylation leading to ATP depletion and superoxide production and acidification of
cytoplasm due to anaerobic lactate production (Skommer et al., 2007). These events may ensure that
the cell death is effectively completed.

65
Apoptosis induced by BCR triggering and rituximab showed typical characteristics of intrinsic
apoptosis since in the both forms of apoptosis mitochondria were centrally involved.
Overexpression of Bcl-xL could inhibit cytochrome c release, collapse of ΔΨm and apoptosis
induced by BCR triggering and rituximab, showing the importance of mitochondrial permeability
change in apoptosis induced by BCR and rituximab. In addition, cells with catalytically inactive
caspase-9, an initiator caspase of the intrinsic pathway, were resistant to DNA fragmentation
induced by BCR and rituximab. Rituximab-induced apoptosis seemed even more dependent on
caspase-9 activity when compared to BCR-induced apoptosis, since rituximab-induced cell death
was almost completely prevented by DN-caspase-9 overexpression, while BCR-induced cell death
was only partially inhibited. Our results are in line with a previous study showing that DN-caspase-
9 block completely BCR-mediated PARP cleavage, DNA laddering and caspase activation, while
the cell death measured by PI exclusion decreased only about 50% (Herold et al., 2002). Based on
our findings, we suggest that BCR-mediated cytochrome c release and collapse of ΔΨm may be
induced in two phases, and only the second phase is mediated by caspase activation (see Figure 8. in
the publication III).
Finally, overexpression of c-FLIP did not affect either BCR- or rituximab-induced apoptosis
showing that the caspase-8 activation pathway was not involved. These findings are in variance with
previous studies, which demonstrated that caspase-8 activation pathway was needed for BCR – and
rituximab –induced apoptosis (Besnault et al., 2001; Stel et al., 2007). However, in these studies
anti-IgM and rituximab were hyper-cross-linked by secondary antibodies, and it may be that in these
circumstances extensive receptor cross-linking induces DISC-formation and caspase-8 activation.
6.2.2 Feedback regulation of mitochondria by caspase-9 during intrinsic apoptosis
It has been widely accepted that during intrinsic apoptosis, MOMP allows the release of
cytochrome c to the cytosol where it binds to Apaf-1 leading to formation of the caspase-9
activating complex called the apoptosome (Riedl and Salvesen, 2007). Thus, it was rather
unexpected to find out that both BCR- and rituximab -induced cytochrome c release was inhibited in
DN-caspase-9 cells showing that caspase-9 regulated the release of cytochrome c from the
mitochondria. This finding is in contrary to well documented view that cytochrome c release
controls the caspase-9 activation (Zou et al., 1999), and not vice versa. Moreover, rituximab-
induced collapse of Ψm was completely dependent and BCR-induced collapse of Ψm was
partially dependent on caspase-9 activation. Similarly, caspase-9 was demonstrated to control the

66
dexamethasone-induced collapse of Ψm in another FL cell line HF28 (Nuutinen et al., 2009). Thus,
the caspase-9 mediated feedback regulation of mitochondrial breach seems to be a common
phenomenon during the intrinsic apoptosis independent of the apoptotic insult, and not restricted to
only one cell line. In line with this, a recent study showed that caspases-3 and -7 are involved in the
collapse of Ψm and release of pro-apoptotic proteins, such as AIF, from the intermembrane space
during intinsic apoptosis (Lakhani et al., 2006). In the same study cytochrome c release into cytosol
was delayed in caspase-9 -/- mouse embryonic fibroblasts. The view that caspases can mediate Ψm
collapse is supported by a finding that the respiratory chain enzymes are cleaved and inactivated by
caspases during apoptosis (Ricci et al., 2004). Moreover, caspases can cleave and inactivate anti-
apoptotic members of the Bcl-2 family, including Mcl-1 and Bcl-xL, which can enhance the
cytochrome c release and Ψm collapse during apoptosis (Chen et al., 2007). Thus, it is possible that
also in our experimental model, caspase-9 and downstream caspases feed back to mitochondria by
cleaving respiratory chain enzymes and anti-apoptotic Bcl-2 proteins. This feed back signaling to
mitochondria may enhance apoptosis and ensure that the cell death process is effectively completed.
The conflicting results concerning the temporal ordering of cytochrome c release and caspase
activation may be explained by the hypothesis that cytochrome c is released in biphasic fashion.
Indeed, a previous study has shown that cytochrome c can be released from mitochondria at two
distinct stages, the early cytochrome c activation being caspase-independent and late stage massive
cyt c release being dependent on caspase activation (Chen et al., 2000). Thus, the first wave of
cytochrome c release may be caspase-independent event which results from incomplete MOMP.
This initial release of cytochrome c may be enough to activate the cascade of caspases which in turn
may induce the complete release of cytochrome c. Indeed, it has been demonstrated that caspases
can cleave and inactivate anti-apoptotic Bcl-2 family proteins and thus favor the release of
cytochrome c to the cytosol (Chen et al., 2007; Cheng et al., 1997).
As a conclusion, caspases may have a more prominent role in the control of mitochondrial
changes during intrinsic apoptosis than we have previously thought. We suggest that caspases may
feedback on mitochondria to enhance mitochondrial deterioration during intrinsic apoptosis.

67
6.2.3. Caspase-9 –mediated apoptosis was poorly inhibited by pan-caspase-inhibitor z-VAD-
fmk
Interestingly, we found that the widely used pan-caspase-inhibitor z-VAD-fmk showed no effect
on rituximab-induced apoptosis although apoptosis could be completely abrogated in the cells
overexpressing DN-caspase-9. Similarly, BCR-induced DNA fragmentation was inhibited by DN-
caspase-9 overexpression but not by z-VAD-fmk. In the same time, z-VAD-fmk blocked
completely DNA fragmentation and apoptosis induced by Fas showing that the inhibitor was
functional. Thus, it seems that the pan-caspase-inhibitor z-VAD-fmk may not effectively inhibit
caspase-9 activation while it is capable inhibiting other caspases such as caspase-8. In line with this,
over-expressed caspase-9 has been shown to process caspase-3 even in the presence of z-VAD-fmk
(Eldering et al., 2004). The poor effect of caspase-inhibitors against caspase-9 may be partly
explained by the fact that the binding of the inhibitor may be hindered because the caspase-9 is
buried in the apoptosome complex (Riedl and Salvesen, 2007). Based on these findings, the
activation of caspases should not be assessed solely by using caspase-inhibitors.
6.2.4. What happens before mitochondrial breach during intrinsic apoptosis?
We and others have demonstrated the several hours delay between the triggering of cell surface
receptors and mitochondrial changes during intrinsic apoptosis (Mackus et al., 2002). What happens
during this delay and how the signals originating from different apoptotic insults are integrated at
the level of mitochondria are central questions of apoptotic research. It is widely accepted that pro-
apoptotic Bcl-2 family proteins Bax and Bak are centrally involved in MOMP and apoptosis, since
cells deficient of both Bax and Bak have lost their susceptibility to MOMP and apoptosis (Wei et
al., 2001). It is also generally accepted, that the BH3-only activator proteins can either directly or
indirectly activate Bax and Bak (Kim et al., 2006; Willis et al., 2007). However, it is not known
which pro-apoptotic Bcl-2 proteins are involved and how they are activated in apoptosis induced by
BCR or CD20 stimulation.
Previous studies with mouse immature B cell line have shown that BCR-induced apoptosis involves
upregulation of proapoptotic Bcl-2 protein Bik and changes in Bad phosphorylation (Jiang and
Clark, 2001; Malissein et al., 2003). In the other study, JNK dependent phosphorylation and
activation of Bim was required for anti-IgM-induced apoptosis in mouse immature B cell line
(Takada et al., 2006). In addition, BCR- induced apoptosis and deletion of autoreactive B cells was
inhibited in Bim deficient mouse knock out model (Enders et al., 2003). However, the involvement

68
of Bim in human cell lines is not established as there may be differences in the involvement of
apoptotic Bcl-2 proteins in mouse and men. Currently, there are no studies exploring the roles of
Bcl-2 proteins during rituximab induced apoptosis.
The delay between BCR triggering and irreversible changes of apoptosis (caspase activation and
mitochondrial breach) was several hours. Based on our studies with a protein synthesis inhibitor
cycloheximide, we suggest that the delay enables a synthesis of new protein(s) which are
responsible for the mitochondrial breach and induction of apoptosis. We took advantage of 2-D
electrophoresis and mass spectroscopy to reveal if there was any BCR stimulation –induced changes
in the protein composition of mitochondria. Unfortunately, we could not find out new apoptosis-
regulating proteins by using this method (unpublished). Neither could we ascertain the early signal
transduction pathways originating from the BCR responsible for apoptosis.
Gene expression profiling has offered a new insight how apoptosis related gene expression can be
studied. In a recent study, CLL cells responding to IgM triggering by proliferation showed increased
expression of CD40, NF-κB and TRAF, which are associated with apoptosis inhibition (Guarini et
al., 2008). In the contrary, CLL cells showing apoptotic response failed to express these survival
genes, and it was concluded that the loss of survival signals leads to apoptosis in these cells.
Induction of pro-apoptotic genes could not be detected in the same experiment. In another gene-
expression study specific genes related with BCR-induced proliferation and apoptosis was studied
in Ramos cell line (Ollila and Vihinen, 2007). The study showed no increase in the genes associated
with intrinsic pathway of apoptosis, while there was an induction of some genes involved in the
death receptor pathway of apoptosis. Nevertheless, it has been demonstrated by previous studies that
the death receptor (extrinsic) pathway is not involved in BCR apoptosis (Berard et al., 1999;
Bouchon et al., 2000). Also in our studies, c-FLIP overexpression or the specific inhibitor of
caspase-8 z-IETD-fmk failed to inhibit BCR-induced apoptosis, showing that the caspase-8
activation pathway was not involved in BCR-induced apoptosis. As a conclusion, the identification
of the proteins synthesized during apoptosis remains a central goal for future research.
6.3. Molecular mechansims of CD40-mediated protection against apoptosis (I,
II and IV)
The main function of CD40 is the regulation of T-cell mediated B-cell activation during humoral
immune responses. CD40 activation promotes B cell proliferation, immunoglobulin production and
class switching, GC formation and establishment of B cell memory (van Kooten and Banchereau,

69
2000; Younes and Kadin, 2003). Most importantly, CD40 activation is involved in the regulation of
survival of both normal GC B cells and lymphoma cells. According to previous studies and our
findings, CD40 stimulation can reverse apoptosis induced by Fas-, BCR-, CD20- and TRAIL-
receptors ((Cleary et al., 1995; Mackus et al., 2002; Merino et al., 1995; Metkar et al., 1998; Travert
et al., 2008). In addition to TRAIL-induced apoptosis, apoptosis induced by dexamethasone and
Doxorubisine was attenuated by CD40-stimulation in FL cells (Nuutinen et al. 2009, accepted for
publication in Scand, J Immunol).
The signaling pathways responsible for anti-apoptotic function of CD40 are not completely
understood. Among the earliest signaling events following CD40 stimulation are the activation of
PI3K, MAPK P38 and Ras-Raf-1-ERK signaling pathways (Craxton et al., 1998; Gulbins et al.,
1996; Ren et al., 1994). However, these signaling pathways may not be the ones responsible for
survival signaling of CD40. Based on our results, the specific inhibitors of PI3K, P38, Raf-1 and
ERK could not inhibit the CD40-mediated rescue against BCR-induced apoptosis (I). Similar results
were repeated in a recent study, where inhibitors of PI3K, MAPK, P38 and JNK could not block
CD40-mediated rescue against TRAIL-induced apoptosis (Travert et al., 2008).
Accumulating evidence suggest that CD40-mediated survival is dependent on the activation of
NF-κB family members p50, p65 (relA) and c-Rel (Andjelic et al., 2000; Berberich et al., 1994;
Kreuz et al., 2001; Lee et al., 1999). NF-κB activation is at least partially mediated by a decrease in
the half life of IκB-a and IκB-b proteins, which sequester NF-κB in cytosol in inactive form
(Schauer et al., 1996). NF-κB is involved in the transcriptional control of several anti-apoptotic
genes including c-FLIP (Kreuz et al., 2001), survivin (Granziero et al., 2001), Bcl-2 (Holder et al.,
1993), Bcl-xL and Bfl-1 (Lee et al., 1999). According to previous studies and our results, it seems
that contribution of these different molecules to the CD40-mediated survival depends on the
apoptotic insult. Whereas c-FLIP proteins may have a dominant role in the CD40-mediated
protection against death receptor -mediated (extrinsic) apoptosis, the up-regulation of anti-apoptotic
members of the Bcl-2 family plays substantial role in the CD40-mediated survival against
mitochondria dependent (intrinsic) apoptosis (Kreuz et al., 2001; Lee et al., 1999) (Figure 13.).
Based on our findings, the CD40-mediated protection against death receptor -induced apoptosis
was at least partly mediated by NF-κB dependent up-regulation of c-FLIP proteins. Firstly, the Fas-
induced apoptosis could be completely blocked by c-FLIP expression. Secondly, CD40 stimulation
induced a rapid up-regulation of both c-FLIP proteins, and the c-FLIP up-regulation as well as the
anti-apoptotic function was completely dependent on the activation of NF-κB. In concordance, c-

70
FLIP proteins mediated also the anti-apoptotic function of CD40 against TRAIL-mediated apoptosis
(Travert et al., 2008). However, in addition to c-FLIP, also other NF-κB dependent proteins, such as
Bcl-xL, may be partly involved in the anti-apoptotic CD40 signaling against death receptor induced
apoptosis. It seems likely that CD40 stimulation induces a large array of protective molecules
which can interfere with different steps in apoptotic pathways (figure 13).
Figure 13. Molecular mechansims of CD40-mediated protection against apoptosis
CD40 stimulation induces the upregulation of c-FLIP, which interfere with caspase-8 activation
during extrinsic apoptosis. Moreover, CD40 up-regulates anti-apoptotic Bcl-2 proteins, including
Bcl-2 and Bcl-xL, which can stabilize mitochondrial membranes Overexpression of c-FLIP proteins
could not block mitochondria dependent apoptosis induced by rituximab or BCR-stimulation.
Instead, the overexpression of pro-apoptotic Bcl-2 protein Bcl-xL potently inhibited both BCR and
rituximab induced apoptosis. The pro-survival members of Bcl-2 family seem potential effectors of
CD40-mediated action for several reasons. Anti-apoptotic Bcl-2 family proteins inhibit cytochrome
c release and Ψm collapse by controlling mitochondrial membrane permeability (Kim et al., 2006;
Kuwana and Newmeyer, 2003; Scorrano and Korsmeyer, 2003). In concordance, the overexpression
Stress stimuli
Bax/Bak
Bc l-2 Bc l-XL
Mitochondria
Apoptosis
FLIP Apaf Cyt-c Cyt-c
Smac
HtrA2
IAPs
Death receptor
A) Extrinsic pathway B) Intrinsic pathway
FADD
Caspase-8
Bid
Caspase-3
Caspase-9
CD40 →
CD40 ↓

71
of Bcl-xL could inhibit BCR-induced Ψm collapse, cytochrome-c release and apoptosis in our
experimental model. In addition, CD40 stimulation could increase the expression of Bcl-xL both in
mRNA and protein level through NF-κB activation (Nuutinen et. al 2009, accepted for publication
in Scand J Immunol). Bcl-xL may not be the only pro-survival Bcl-2 protein involved in the anti-
apoptotic action of CD40, and other proteins including Mcl-1 or Bcl-2 might also be involved.
However, Bcl-2 alone seems not a very potent inhibitor of apoptosis, since the FL cells
overexpressing Bcl-2 were still susceptible for apoptosis induced by different stimuli.
.
6.4. A proposed model of the molecular interactions during the germinal
center negative selection
We studied the mechanisms of receptor-mediated apoptosis in the follicular lymphoma cell line
HF1A3. Based on the morphology, immunophenotype and propensity to antigen-induced apoptosis,
the cell line represents GC centrocytes undergoing negative selection (Eray et al., 2003). As in the
case of HF1A3 cell line, cancer cells often retain the same immunophenotype and responses against
physiological stimuli as their founder cells. Thus, the cell line offers a good model to study the
signaling requirements of GC B cells undergoing the negative selection. On the other hand, the
knowledge of the regulation of cell survival and death in GC can be used as an advantage for the
development of new treatment modalities against B cell derived tumors, since normal GC B cells
and lymphoma cells are similarly influenced by their GC microenvironment (Kuppers, 2004).
After the somatic hypermutaion of their Ig variable regions, centrocytes are subjected for selection
on the basis of their antigen binding capacity (Figure 6.) The centrocytes with a high-affinity BCR
are able to present antigen derived peptides in the contexts of MHC class II molecules for CD4+
helper T cells. Subsequently, the CD40 ligand is expressed transiently on the surface of helper T
cells, and delivered for the centrocyte specific for the same antigen. The activating signal through
both BCR and CD40 receptor selects the centrocyte for proliferation and differentiation in to
antibody secreting cells. On the other hand, centrocytes that have became autoreactive during the
SHM process are eliminated by the lack of proper T cell help, and die by apoptosis. Our findings
support the previous view that BCR stimulus may be involved in the negative selection of self-
reactive cells (Billian et al., 1997), since the cells die by apoptosis after BCR stimulation without
CD40 stimulus delivered within substantial time frame. The kinetics of BCR-mediated apoptosis
strengthens this hypothesis, since there was over 12 hours delay between receptor activation and

72
irreversible changes of apoptosis. In addition, CD40 stimulation was able to reverse apoptosis up to
10 hours after the BCR triggering. The time frame for CD40 rescue may be physiologically relevant
because specific T cell help may not be immediately available after the BCR stimulus. Before the
expression of CD40L is induced on the T cell surface, T cells have to be activated by interactions
between the T cell and the centrocyte.
Since SHM is a random process, only few of the centrocytes improve their antigen binding
capacity, whereas the majority of the centrocytes gain non-binding or non-functional receptors.
Based on studies with Fas deficient mouse models (Hao et al., 2008; Takahashi et al., 2001), Fas-
induced apoptosis is centrally involved in the selection process of GC B cells. Thus, the centrocytes
which have gained a low affinity BCR after the SHM may be eliminated by Fas-mediated apoptosis,
which ensures a rapid deletion of these unnecessary cells. It has been suggested that GC B cells
have a preformed CD95 DISC, in which caspase-8 activation is interfered by c-FLIP (Scaffidi et al.,
1999). Without survival signals provided by CD40- or antigen receptors or tropic factors from
stromal cells, c-FLIP is rapidly degraded from the DISC leading to caspase-8 activation and
apoptosis (Hennino et al., 2001; van Eijk et al., 2001). Thus, c-FLIP seems to be an important
regulator of the positive selection in GCs. However, the B cells with improved antigen binding
capacity which are rescued from Fas-induced apoptosis by c-FLIPlong expression may be still
susceptible for BCR-induced apoptosis, which is implicated in the negative selection of self-reactive
B cells (Billian et al., 1997). This suggestion is based on our findings that apoptosis induced by
BCR triggering was only marginally inhibited by c-FLIPlong overexpression.
As a conclusion, the BCR stimulus may be involved in both negative and positive selection of
centrocytes, and the T cell signals in the form of CD40 ligand discriminates between these fates. In
addition, death receptor mediated apoptosis may be necessary for the elimination of low affinity
centrocytes. The finding that Fas- and BCR -induced apoptosis use different signaling pathways
may be physiologically relevant, since it enables that Fas- and BCR-induced apoptosis can be
regulated independently, and targeted to different subsets of centrocytes. However, the hypothesis
presented here need to be replicated in normal GC B cells, since the FL cell line used may have got
genetic abnormalities during the long in vitro culture time, and thus not respond as original GC B
cells.

73
7. CONCLUSIONS; Therapeutic applications and future directions
Cell surface receptors which regulate the live and death of lymphocytes are important in the
regulation of normal B lymphocyte homeostasis. For example, extensive B cell proliferation must
be balanced by elimination of extraneous or potentially harmful cells during the germinal center
reaction. In addition to their importance in physiology, the cell surface receptors regulating cell
death serve as attractive targets for cancer therapy. This work was devoted on the study of four cell
surface receptors which are important in the regulation of B cell survival; B cell receptor, Fas/CD95
and CD20 are receptors mediating apoptosis, and CD40 which regulates anti-apoptotic response
against variety of apoptotic signals.
7.1. Cell surface receptors in the regulation of life and death decisions in the
germinal center
The first aim of the study was to explore the receptor mediated regulation of B cell survival
during the germinal center reaction. The germinal center B cells die spontaneously in vitro without
the growth support provided by GC microenvironment, and thus the mechanisms of receptor-
induced cell death is hard to study with the normal B cells. To circumvent this problem, we used a
follicular lymphoma cell line HF1A3 as an experimental model. Based on the morphology,
immunophenotype and propensity to antigen-induced apoptosis, the cell line represents GC
centrocytes undergoing negative selection (Eray et al., 2003). As in the case of HF1A3 cell line, the
malign cells often retain the same immunophenotype and responses against physiological stimuli as
their founder cells. Thus, the cell line offers a good model to study the signaling requirements of GC
B cells undergoing the negative selection.
Based on the existing literature and our findings of receptor mediated apoptosis, the following
model of GC selection process can be depicted (see also Figure 6.). After the SHM, the centrocyte
may have three different fates based on the antigen binding capacity of its newly formed antigen
receptor. The few centrocytes with improved antigen binding capacity are positively selected as
they get the CD40 survival signal from the antigen specific T cell due to effective antigen capture
and presentation to the helper T cell. On the other hand, the centrocytes in which SHM has resulted
non-functional or non-binding antigen receptor are negatively selected by the lack of T cell survival

74
signal CD40, and these cells are effectively eliminated by Fas-mediated apoptosis. The third fate of
a centrocyte is to have an autoreactive antigen receptor. In these cells the activation of antigen
receptor without a co-stimulatory CD40 signal leads to apoptosis. Thus, the signal from the B cell
receptor is involved both in the positive and negative selection of a centrocyte, and the T cell help in
the form of CD40 ligand discriminates between these fates.
The knowledge of the regulation of cell survival and death in GC can be used as an advantage for
the development of new treatment modalities against B cell derived tumors, as the normal GC B
cells and their malign counterparts are similarly influenced by their GC microenvironment
(Kuppers, 2004). For example, the survival signals provided by activated T cells may counteract the
cytotoxic effect of various chemotherapeutics, which may lead to resistance against cancer therapy.
7.2. CD40-CD40L interactions as potential targets for cancer therapy
As demonstrated by our FL cell lines in vitro, CD40 activation counteracts apoptosis induced by
cell surface receptors (Fas, TRAIL, CD20, BCR) and by chemotherapeutic agents (such as
dexamethasone and doxorubisin). Similarly, CD40 ligation may counteract in vivo the action of
various chemotherapeutics given to lymphoma patients, and thus generate resistance against cancer
therapy. This resistance problem might be circumvented by development of CD40 blocking agents,
which inhibit CD40-induced survival signaling. These CD40 blocking agents may function on the
receptor level, such as antibodies which bind to CD40L blocking its activation. Another approach is
to inhibit the downstream mediators of CD40 signaling. As demonstrated in this work, the most
important mediators of CD40 signaling include the up-regulation of c-FLIP and Bcl-xL proteins
which inhibit the death receptor-mediated and mitochondria-dependent apoptosis respectively.
Attractive approach might be blocking of these proteins by siRNA method, in which the mRNA of
these proteins is bound and cleaved specifically. Another approach might be the development of
Bcl-xL small molecule inhibitors. Indeed, several small molecule inhibitors against another survival
protein Bcl-2 are already developed and currently under clinical trials (Zeitlin et al., 2008).
However, it should be mentioned that in addition to its pro-survival function in several low grade
lymphomas, CD40 activation may be apoptotic signal in some more aggressive lymphomas and in
these circumstances CD40 agonistic antibody is more practical choice.

75
7.3. Novel approaches to enhance rituximab therapy
Until these days, FL has been suggested as incurable disease as the therapies used showed no
effect on the survival of FL patients. Introduction of rituximab has radically changed this view as its
use in combination with chemotherapy has improved the live time expectancy of FL patients.
However, still majority of the patients relapse after first line treatment. Thus efforts are being made
to develop more efficient therapy against FL.
One of the aims of this work was to study the molecular mechanism of rituximab induced
apoptosis. We demonstrated here the critical signaling events of rituximab-induced apoptosis in a
FL cell, which is currently one of the most important targets of rituximab therapy in the clinics. The
apoptosis induced by rituximab was dependent on the activation of caspase-9 and mitochondrial
breach, while the death receptor pathway was not involved. Thus, drugs designed to destabilize
mitochondrial membranes, such as BH3 mimetics or the antagonists of anti-apoptotic Bcl-2
proteins, may be used in the future to enhance rituximab-induced apoptosis.
We showed also evidence, that the simultaneous triggering of the death receptor pathway by Fas
enhanced significantly apoptosis induced by rituximab. The additive effect of simultaneous Fas and
rituximab treatment on apoptosis may be a result of simultaneous activation of both the extrinsic
(caspase-8-mediated) pathway by Fas and intrinsic (mitochondria dependent) pathway by rituximab.
The death receptors appear as ideal targets for cancer therapy, as their activation directly leads to
caspase-8 activation and initiation of the cell suicide program. The resistance against cancer therapy
is most often associated with mutation of the tumor suppressor gene p53, and this resistance could
be elegantly circumvented by death receptor mediated apoptosis, which can proceed independently
of p53 activation. However, the use of FAS-ligand or Fas agonistic antibodies is not possible in the
clinical settings due to the expression of Fas in hepatocytes and extreme hepatotoxicity
demonstrated in mouse models (reviewed in (Yonehara, 2002)). To circumvent this problem, small
molecules which can induce self-aggregation and ligand-independent activation of Fas receptor
complex have been introduced. Of these molecules, edelfosine (ET-18-OCH) and perifosine can
induce apoptosis in primary multiple myeloma cell line by recruitment of Fas receptor and
downstream apoptotic signaling molecules into lipid rafts, leading to activation of the extrinsic
pathway of apoptosis (Gajate and Mollinedo, 2007). Edelfosine is selectively taken up by cancer
cells, and its therapeutic potential against B cell malignancies is currently being addressed in a
phase II clinical study (Feller et al., 2005). In the future, it might be worth of studying the effect of
edelfosine – rituximab combination therapy against FL in the clinical settings.

76
In vitro studies have shown that rituximab-induced apoptosis was enhanced also by simultaneous
activation of the death receptor TRAIL (Daniel et al., 2007). Activation of death receptors TRAIL-
R1 or -R2 by their ligands or agonistic antibodies induces death receptor pathway in cancer cells of
different origin, including lymphoma cells (Younes and Kadin, 2003). TRAIL receptors are
expressed predominantly on tumor cells making them excellent targets for cancer therapy. Several
clinical trials investigating TRAIL as a potential anti-cancer drug are currently ongoing (Younes
and Kadin, 2003). The efficacy and safety of TRAIL and rituximab combination therapy for NHL
should be addressed in the clinical trial in the future.
Current trend in immunotherapy is the development of humanized antibodies which can be used in
the treatment of hematological malignancies and inflammatory diseases. The infusion of humanized
antibodies is associated with lesser side-effects caused by human anti-mouse-antibodies compared
with chimeric antibodies. The progress in gene technology has enabled the production of humanized
antibodies engineered to induce more efficient B cell depletion. For example, the humanized CD20
antibody ofatumumab is more effective than rituximab in promoting the activation of complement
dependent cell death (Pawluczkowycz et al., 2009). Another humanized CD20 antibody
ocrelizumab is associated with higher antibody dependent cytotoxicity as compared with rituximab
(Sikder and Friedberg, 2008). The tissue penetration of antibodies has been enhanced by the
development of “small-weight antibodies”, which consist of only single heavy- and light -chain
variable regions of human origin combined with engineered human IgG constant regions (Sikder
and Friedberg, 2008). Whether the novel humanized antibodies will supplant rituximab for
treatment of hematological malignancies is currently being assessed by clinical trials.

77
8. REFERENCES
Advani, R., Rosenberg, S.A., and Horning, S.J. (2004). Stage I and II follicular non-Hodgkin's
lymphoma: long-term follow-up of no initial therapy. J. Clin. Oncol. 22, 1454-1459.
Alas, S., Bonavida, B., and Emmanouilides, C. (2000). Potentiation of fludarabine cytotoxicity on
non-Hodgkin's lymphoma by pentoxifylline and rituximab. Anticancer Res. 20, 2961-2966.
Alas, S., Ng, C.P., and Bonavida, B. (2002). Rituximab modifies the cisplatin-mitochondrial
signaling pathway, resulting in apoptosis in cisplatin-resistant non-Hodgkin's lymphoma. Clin.
Cancer Res. 8, 836-845.
Allen, C.D., Okada, T., and Cyster, J.G. (2007). Germinal-center organization and cellular
dynamics. Immunity 27, 190-202.
Anderson, J.R., Armitage, J.O., and Weisenburger, D.D. (1998). Epidemiology of the non-
Hodgkin's lymphomas: distributions of the major subtypes differ by geographic locations. Non-
Hodgkin's Lymphoma Classification Project. Ann. Oncol. 9, 717-720.
Andjelic, S., Hsia, C., Suzuki, H., Kadowaki, T., Koyasu, S., and Liou, H.C. (2000).
Phosphatidylinositol 3-kinase and NF-κB/Rel are at the divergence of CD40-mediated proliferation
and survival pathways. J. Immunol. 165, 3860-7.
Ardeshna, K.M., Smith, P., Norton, A., Hancock, B.W., Hoskin, P.J., MacLennan, K.A., Marcus,
R.E., Jelliffe, A., Vaughan, G., Hudson, et al. (2003). Long-term effect of a watch and wait policy
versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: a
randomised controlled trial. Lancet 362, 516-522.
Ashkenazi, A., and Dixit, V.M. (1999). Apoptosis control by death and decoy receptors. Curr. Opin.
Cell Biol. 11, 255-260.
Bayry, J., Lacroix-Desmazes, S., Kazatchkine, M.D., and Kaveri, S.V. (2007). Monoclonal antibody
and intravenous immunoglobulin therapy for rheumatic diseases: rationale and mechanisms of
action. Nat. Clin. Pract. Rheumatol. 3, 262-272.
Bendandi, M. (2008). Aiming at a curative strategy for follicular lymphoma. CA Cancer. J. Clin. 58,
305-317.
Berard, M., Mondiere, P., Casamayor-Palleja, M., Hennino, A., Bella, C., and Defrance, T. (1999).
Mitochondria connects the antigen receptor to effector caspases during B cell receptor-induced
apoptosis in normal human B cells. J. Immunol. 163, 4655-62.
Berberich, I., Shu, G.L., and Clark, E.A. (1994). Cross-linking CD40 on B cells rapidly activates
nuclear factor-κB. J. Immunol. 153, 4357-4366.
Besnault, L., Schrantz, N., Auffredou, M.T., Leca, G., Bourgeade, M.F., and Vazquez, A. (2001). B
cell receptor cross-linking triggers a caspase-8-dependent apoptotic pathway that is independent of
the death effector domain of Fas-associated death domain protein. J. Immunol. 167, 733-740.

78
Billian, G., Mondiere, P., Berard, M., Bella, C., and Defrance, T. (1997). Antigen receptor-induced
apoptosis of human germinal center B cells is targeted to a centrocytic subset. Eur. J. Immunol. 27,
405-14.
Boatright, K.M., Renatus, M., Scott, F.L., Sperandio, S., Shin, H., Pedersen, I.M., Ricci, J.E., Edris,
W.A., Sutherlin, D.P., Green, D.R., et al. (2003). A unified model for apical caspase activation.
Mol. Cell 11, 529-541.
Boatright, K.M., and Salvesen, G.S. (2003). Mechanisms of caspase activation. Curr. Opin. Cell
Biol. 15, 725-731.
Bouchon, A., Krammer, P.H., and Walczak, H. (2000). Critical role for mitochondria in B cell
receptor-mediated apoptosis. Eur. J. Immunol. 30, 69-77.
Busslinger, M. (2004). Transcriptional control of early B cell development. Annu. Rev. Immunol.
22, 55-79.
Campbell, K.S. (1999). Signal transduction from the B cell antigen-receptor. Curr. Opin. Immunol.
11, 256-264.
Castedo, M., Ferri, K., Roumier, T., Metivier, D., Zamzami, N., and Kroemer, G. (2002).
Quantitation of mitochondrial alterations associated with apoptosis. J. Immunol. Methods 265, 39-
47.
Chan, H.T., Hughes, D., French, R.R., Tutt, A.L., Walshe, C.A., Teeling, J.L., Glennie, M.J., and
Cragg, M.S. (2003). CD20-induced lymphoma cell death is independent of both caspases and its
redistribution into triton X-100 insoluble membrane rafts. Cancer Res. 63, 5480-5489.
Chen, M., Guerrero, A.D., Huang, L., Shabier, Z., Pan, M., Tan, T.H., and Wang, J. (2007).
Caspase-9-induced mitochondrial disruption through cleavage of anti-apoptotic BCL-2 family
members. J. Biol. Chem.
Chen, Q., Gong, B., and Almasan, A. (2000). Distinct stages of cytochrome c release from
mitochondria: evidence for a feedback amplification loop linking caspase activation to
mitochondrial dysfunction in genotoxic stress induced apoptosis. Cell Death Differ. 7, 227-233.
Chen, W., Wang, H.G., Srinivasula, S.M., Alnemri, E.S., and Cooper, N.R. (1999). B cell apoptosis
triggered by antigen receptor ligation proceeds via a novel caspase-dependent pathway. J. Immunol.
163, 2483-2491.
Chen, Y.R., Wang, X., Templeton, D., Davis, R.J., and Tan, T.H. (1996). The role of c-Jun N-
terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK
activation may determine cell death and proliferation. J. Biol. Chem. 271, 31929-31936.
Cheng, E.H., Kirsch, D.G., Clem, R.J., Ravi, R., Kastan, M.B., Bedi, A., Ueno, K., and Hardwick,
J.M. (1997). Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 278, 1966-1968.

79
Cheng, S., Hsia, C.Y., Leone, G., and Liou, H.C. (2003). Cyclin E and Bcl-xL cooperatively induce
cell cycle progression in c-Rel-/- B cells. Oncogene 22, 8472-8486.
Chiu, B.C., and Weisenburger, D.D. (2003). An update of the epidemiology of non-Hodgkin's
lymphoma. Clin. Lymphoma 4, 161-168.
Choe, J., Li, L., Zhang, X., Gregory, C.D., and Choi, Y.S. (2000). Distinct role of follicular
dendritic cells and T cells in the proliferation, differentiation, and apoptosis of a centroblast cell
line, L3055. J. Immunol. 164, 56-63.
Cleary, A.M., Fortune, S.M., Yellin, M.J., Chess, L., and Lederman, S. (1995). Opposing roles of
CD95 (Fas/APO-1) and CD40 in the death and rescue of human low density tonsillar B cells. J.
Immunol. 155, 3329-37.
Craxton, A., Shu, G., Graves, J.D., Saklatvala, J., Krebs, E.G., and Clark, E.A. (1998). p38 MAPK
is required for CD40-induced gene expression and proliferation in B lymphocytes. J. Immunol. 161,
3225-36.
Crowe, J.S., Hall, V.S., Smith, M.A., Cooper, H.J., and Tite, J.P. (1992). Humanized monoclonal
antibody CAMPATH-1H: myeloma cell expression of genomic constructs, nucleotide sequence of
cDNA constructs and comparison of effector mechanisms of myeloma and Chinese hamster ovary
cell-derived material. Clin. Exp. Immunol. 87, 105-110.
Custodio, J.B., Cardoso, C.M., Madeira, V.M., and Almeida, L.M. (2001). Mitochondrial
permeability transition induced by the anticancer drug etoposide. Toxicol. in. Vitro. 15, 265-270.
Dallman, C., Johnson, P.W., and Packham, G. (2003). Differential regulation of cell survival by
CD40. Apoptosis 8, 45-53.
D'Amelio, M., Tino, E., and Cecconi, F. (2008). The apoptosome: emerging insights and new
potential targets for drug design. Pharm. Res. 25, 740-751.
Daniel, D., Yang, B., Lawrence, D.A., Totpal, K., Balter, I., Lee, W.P., Gogineni, A., Cole, M.J.,
Yee, S.F., Ross, S., et al. (2007). Cooperation of the proapoptotic receptor agonist rhApo2L/TRAIL
with the CD20 antibody rituximab against non-Hodgkin lymphoma xenografts. Blood 110, 4037-
4046.
Dave, S.S., Wright, G., Tan, B., Rosenwald, A., Gascoyne, R.D., Chan, W.C., Fisher, R.I., Braziel,
R.M., Rimsza, L.M., Grogan, T.M., et al. (2004). Prediction of survival in follicular lymphoma
based on molecular features of tumor-infiltrating immune cells. N. Engl. J. Med. 351, 2159-2169.
de Jong, D. (2005). Molecular pathogenesis of follicular lymphoma: a cross talk of genetic and
immunologic factors. J. Clin. Oncol. 23, 6358-6363.
Dillman, R.O., Beauregard, J.C., Halpern, S.E., and Clutter, M. (1986). Toxicities and side effects
associated with intravenous infusions of murine monoclonal antibodies. J. Biol. Response Mod. 5,
73-84.

80
Doi, T.S., Takahashi, T., Taguchi, O., Azuma, T., and Obata, Y. (1997). NF-κB RelA-deficient
lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and
reduced proliferative responses. J. Exp. Med. 185, 953-961.
Donepudi, M., Mac Sweeney, A., Briand, C., and Grutter, M.G. (2003). Insights into the regulatory
mechanism for caspase-8 activation. Mol. Cell 11, 543-549.
Duty, J.A., Szodoray, P., Zheng, N.Y., Koelsch, K.A., Zhang, Q., Swiatkowski, M., Mathias, M.,
Garman, L., Helms, C., Nakken, B., et al. (2008). Functional anergy in a subpopulation of naive B
cells from healthy humans that express autoreactive immunoglobulin receptors. J. Exp. Med. 206,
139-151.
Eeva, J., Postila, V., Mätto, M., Nuutinen, U., Ropponen, A., Eray, M., and Pelkonen, J. (2003).
Kinetics and signaling requirements of CD40-mediated protection from B cell receptor-induced
apoptosis. Eur. J. Immunol. 33, 2783-2791.
Eeva, J., Ropponen, A., Nuutinen, U., Eeva, S.T., Mätto, M., Eray, M., and Pelkonen, J. (2007). The
CD40-induced protection against CD95-mediated apoptosis is associated with a rapid upregulation
of anti-apoptotic c-FLIP. Mol. Immunol. 44, 1230-1237.
Eldering, E., Mackus, W.J., Derks, I.A., Evers, L.M., Beuling, E., Teeling, P., Lens, S.M., van Oers,
M.H., and van Lier, R.A. (2004). Apoptosis via the B cell antigen receptor requires Bax
translocation and involves mitochondrial depolarization, cytochrome C release, and caspase-9
activation. Eur. J. Immunol. 34, 1950-1960.
Enders, A., Bouillet, P., Puthalakath, H., Xu, Y., Tarlinton, D.M., and Strasser, A. (2003). Loss of
the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis
and deletion of autoreactive B cells. J. Exp. Med. 198, 1119-1126.
Eray, M., Postila, V., Eeva, J., Ripatti, A., Karjalainen-Lindsberg, M.L., Knuutila, S., Andersson,
L.C., and Pelkonen, J. (2003). Follicular lymphoma cell lines, an in vitro model for antigenic
selection and cytokine-mediated growth regulation of germinal centre B cells. Scand. J. Immunol.
57, 545-555.
Eray, M., Tuomikoski, T., Wu, H., Nordstrom, T., Andersson, L.C., Knuutila, S., and Kaartinen, M.
(1994). Cross-linking of surface IgG induces apoptosis in a bcl-2 expressing human follicular
lymphoma line of mature B cell phenotype. Int. Immunol. 6, 1817-27.
Feller, N., Jansen-van der Weide, M.C., van der Pol, M.A., Westra, G.A., Ossenkoppele, G.J., and
Schuurhuis, G.J. (2005). Purging of peripheral blood stem cell transplants in AML: a predictive
model based on minimal residual disease burden. Exp. Hematol. 33, 120-130.
Flieger, D., Renoth, S., Beier, I., Sauerbruch, T., and Schmidt-Wolf, I. (2000). Mechanism of
cytotoxicity induced by chimeric mouse human monoclonal antibody IDEC-C2B8 in CD20-
expressing lymphoma cell lines. Cell. Immunol. 204, 55-63.
Frodin, J.E., Lefvert, A.K., and Mellstedt, H. (1992). The clinical significance of HAMA in patients
treated with mouse monoclonal antibodies. Cell Biophys. 21, 153-165.

81
Gajate, C., and Mollinedo, F. (2007). Edelfosine and perifosine induce selective apoptosis in
multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid
rafts. Blood 109, 711-719.
Galluzzi, L., Maiuri, M.C., Vitale, I., Zischka, H., Castedo, M., Zitvogel, L., and Kroemer, G.
(2007). Cell death modalities: classification and pathophysiological implications. Cell Death Differ.
14, 1237-1243.
Garrido, C., Galluzzi, L., Brunet, M., Puig, P.E., Didelot, C., and Kroemer, G. (2006). Mechanisms
of cytochrome c release from mitochondria. Cell Death Differ. 13, 1423-1433.
Glas, A.M., Knoops, L., Delahaye, L., Kersten, M.J., Kibbelaar, R.E., Wessels, L.A., van Laar, R.,
van Krieken, J.H., Baars, J.W., Raemaekers, J., et al. (2007). Gene-expression and
immunohistochemical study of specific T-cell subsets and accessory cell types in the transformation
and prognosis of follicular lymphoma. J. Clin. Oncol. 25, 390-398.
Goldstein, J.C., Munoz-Pinedo, C., Ricci, J.E., Adams, S.R., Kelekar, A., Schuler, M., Tsien, R.Y.,
and Green, D.R. (2005). Cytochrome c is released in a single step during apoptosis. Cell Death
Differ. 12, 453-462.
Goval, J.J., Thielen, C., Bourguignon, C., Greimers, R., Dejardin, E., Choi, Y.S., Boniver, J., and de
Leval, L. (2008). The prevention of spontaneous apoptosis of follicular lymphoma B cells by a
follicular dendritic cell line: involvement of caspase-3, caspase-8 and c-FLIP. Haematologica 93,
1169-1177.
Granziero, L., Ghia, P., Circosta, P., Gottardi, D., Strola, G., Geuna, M., Montagna, L., Piccoli, P.,
Chilosi, M., and Caligaris-Cappio, F. (2001). Survivin is expressed on CD40 stimulation and
interfaces proliferation and apoptosis in B-cell chronic lymphocytic leukemia. Blood 97, 2777-
2783.
Graves, J.D., Draves, K.E., Craxton, A., Saklatvala, J., Krebs, E.G., and Clark, E.A. (1996).
Involvement of stress-activated protein kinase and p38 mitogen-activated protein kinase in mIgM-
induced apoptosis of human B lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 93, 13814-13818.
Green, D.R., and Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science
305, 626-629.
Guarini, A., Chiaretti, S., Tavolaro, S., Maggio, R., Peragine, N., Citarella, F., Ricciardi, M.R.,
Santangelo, S., Marinelli, M., De Propris, M.S., et al. (2008). BCR ligation induced by IgM
stimulation results in gene expression and functional changes only in IgV H unmutated chronic
lymphocytic leukemia (CLL) cells. Blood 112, 782-792.
Guarneri, V., Frassoldati, A., Bruzzi, P., D'Amico, R., Belfiglio, M., Molino, A., Bertetto, O.,
Cascinu, S., Cognetti, F., Di Leo, A., et al. (2008). Multicentric, randomized phase III trial of two
different adjuvant chemotherapy regimens plus three versus twelve months of trastuzumab in
patients with HER2- positive breast cancer (Short-HER Trial; NCT00629278). Clin. Breast Cancer.
8, 453-456.

82
Gulbins, E., Brenner, B., Schlottmann, K., Koppenhoefer, U., Linderkamp, O., Coggeshall, K.M.,
and Lang, F. (1996). Activation of the Ras signaling pathway by the CD40 receptor. J. Immunol.
157, 2844-50.
Hao, Z., Duncan, G.S., Chang, C.C., Elia, A., Fang, M., Wakeham, A., Okada, H., Calzascia, T.,
Jang, Y., You-Ten, A., et al. (2005). Specific ablation of the apoptotic functions of cytochrome C
reveals a differential requirement for cytochrome C and Apaf-1 in apoptosis. Cell 121, 579-591.
Hao, Z., Duncan, G.S., Seagal, J., Su, Y.W., Hong, C., Haight, J., Chen, N.J., Elia, A., Wakeham,
A., Li, W.Y., et al. (2008). Fas receptor expression in germinal-center B cells is essential for T and
B lymphocyte homeostasis. Immunity 29, 615-627.
Harjunpää, A., Junnikkala, S., and Meri, S. (2000). Rituximab (anti-CD20) therapy of B-cell
lymphomas: direct complement killing is superior to cellular effector mechanisms. Scand. J.
Immunol. 51, 634-641.
Hartley, S.B., Crosbie, J., Brink, R., Kantor, A.B., Basten, A., and Goodnow, C.C. (1991).
Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing
membrane-bound antigens. Nature 353, 765-769.
Hashimoto, A., Okada, H., Jiang, A., Kurosaki, M., Greenberg, S., Clark, E.A., and Kurosaki, T.
(1998). Involvement of guanosine triphosphatases and phospholipase C-γ2 in extracellular signal-
regulated kinase, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase activation
by the B cell antigen receptor. J. Exp. Med. 188, 1287-1295.
Hecht, J.R., Patnaik, A., Berlin, J., Venook, A., Malik, I., Tchekmedyian, S., Navale, L., Amado,
R.G., and Meropol, N.J. (2007). Panitumumab monotherapy in patients with previously treated
metastatic colorectal cancer. Cancer 110, 980-988.
Hennino, A., Berard, M., Krammer, P.H., and Defrance, T. (2001). FLICE-inhibitory protein is a
key regulator of germinal center B cell apoptosis. J. Exp. Med. 193, 447-458.
Herold, M.J., Kuss, A.W., Kraus, C., and Berberich, I. (2002). Mitochondria-dependent caspase-9
activation is necessary for antigen receptor-mediated effector caspase activation and apoptosis in
WEHI 231 lymphoma cells. J. Immunol. 168, 3902-9.
Hiddemann, W., Buske, C., Dreyling, M., Weigert, O., Lenz, G., and Unterhalt, M. (2007). Current
management of follicular lymphomas. Br. J. Haematol. 136, 191-202.
Holder, M.J., Wang, H., Milner, A.E., Casamayor, M., Armitage, R., Spriggs, M.K., Fanslow, W.C.,
MacLennan, I.C., Gregory, C.D., and Gordon, J. (1993). Suppression of apoptosis in normal and
neoplastic human B lymphocytes by CD40 ligand is independent of Bc1-2 induction. Eur. J.
Immunol. 23, 2368-2371.
Hsu, Y.T., and Youle, R.J. (1998). Bax in murine thymus is a soluble monomeric protein that
displays differential detergent-induced conformations. J. Biol. Chem. 273, 10777-10783.

83
Imtiyaz, H.Z., Rosenberg, S., Zhang, Y., Rahman, Z.S., Hou, Y.J., Manser, T., and Zhang, J. (2006).
The Fas-associated death domain protein is required in apoptosis and TLR-induced proliferative
responses in B cells. J. Immunol. 176, 6852-6861.
Irmler, M., Thome, M., Hahne, M., Schneider, P., Hofmann, K., Steiner, V., Bodmer, J.L., Schroter,
M., Burns, K., Mattmann, C., et al. (1997). Inhibition of death receptor signals by cellular FLIP.
Nature 388, 190-5.
Irvine, R.A., Adachi, N., Shibata, D.K., Cassell, G.D., Yu, K., Karanjawala, Z.E., Hsieh, C.L., and
Lieber, M.R. (2005). Generation and characterization of endonuclease G null mice. Mol. Cell. Biol.
25, 294-302.
Jazirehi, A.R., Gan, X.H., De Vos, S., Emmanouilides, C., and Bonavida, B. (2003). Rituximab
(anti-CD20) selectively modifies Bcl-xL and apoptosis protease activating factor-1 (Apaf-1)
expression and sensitizes human non-Hodgkin's lymphoma B cell lines to paclitaxel-induced
apoptosis. Mol. Cancer. Ther. 2, 1183-1193.
Jiang, A., and Clark, E.A. (2001). Involvement of Bik, a proapoptotic member of the Bcl-2 family,
in surface IgM-mediated B cell apoptosis. J. Immunol. 166, 6025-6033.
Jyrkkiö, S., Jantunen, E., Keskinen, L., Kuittinen, O., Leppä, S., Vasala, K., Vornanen, M., and
Lehtinen, T. (2007). Follikulaarisen lymfooman hoito. Lääkärilehti 62, 1729-1733.
Kawabe, T., Naka, T., Yoshida, K., Tanaka, T., Fujiwara, H., Suematsu, S., Yoshida, N., Kishimoto,
T., and Kikutani, H. (1994). The immune responses in CD40-deficient mice: impaired
immunoglobulin class switching and germinal center formation. Immunity 1, 167-178.
Kerr, J.F., Wyllie, A.H., and Currie, A.R. (1972). Apoptosis: a basic biological phenomenon with
wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239-257.
Kim, H., Rafiuddin-Shah, M., Tu, H.C., Jeffers, J.R., Zambetti, G.P., Hsieh, J.J., and Cheng, E.H.
(2006). Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat.
Cell Biol. 8, 1348-1358.
Kimby, E. (2005). Tolerability and safety of rituximab (MabThera). Cancer Treat. Rev. 31, 456-
473.
Kischkel, F.C., Lawrence, D.A., Tinel, A., LeBlanc, H., Virmani, A., Schow, P., Gazdar, A., Blenis,
J., Arnott, D., and Ashkenazi, A. (2001). Death receptor recruitment of endogenous caspase-10 and
apoptosis initiation in the absence of caspase-8. J. Biol. Chem. 276, 46639-46646.
Klein, U., and Dalla-Favera, R. (2008). Germinal centres: role in B-cell physiology and malignancy.
Nat. Rev. Immunol. 8, 22-33.
Kluck, R.M., Bossy-Wetzel, E., Green, D.R., and Newmeyer, D.D. (1997). The release of
cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275,
1132-1136.

84
Kohler, G., and Milstein, C. (2005). Continuous cultures of fused cells secreting antibody of
predefined specificity. 1975. J. Immunol. 174, 2453-2455.
Koncz, G., Bodor, C., Kovesdi, D., Gati, R., and Sarmay, G. (2002). BCR mediated signal
transduction in immature and mature B cells. Immunol. Lett. 82, 41-49.
Kondo, E., Harashima, A., Takabatake, T., Takahashi, H., Matsuo, Y., Yoshino, T., Orita, K., and
Akagi, T. (2003). NF-ATc2 induces apoptosis in Burkitt's lymphoma cells through signaling via the
B cell antigen receptor. Eur. J. Immunol. 33, 1-11.
Kondo, E., and Yoshino, T. (2007). Expression of apoptosis regulators in germinal centers and
germinal center-derived B-cell lymphomas: insight into B-cell lymphomagenesis. Pathol. Int. 57,
391-397.
Kondo, E., Yoshino, T., Nishiuchi, R., Sakuma, I., Nishizaki, K., Kayagaki, N., Yagita, H., and
Akagi, T. (1997). Expression of Fas ligand mRNA in germinal centres of the human tonsil. J.
Pathol. 183, 75-79.
Korthauer, U., Graf, D., Mages, H.W., Briere, F., Padayachee, M., Malcolm, S., Ugazio, A.G.,
Notarangelo, L.D., Levinsky, R.J., and Kroczek, R.A. (1993). Defective expression of T-cell CD40
ligand causes X-linked immunodeficiency with hyper-IgM. Nature 361, 539-541.
Kraus, M., Alimzhanov, M.B., Rajewsky, N., and Rajewsky, K. (2004). Survival of resting mature
B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell 117, 787-800.
Kreuz, S., Siegmund, D., Scheurich, P., and Wajant, H. (2001). NF-κB inducers upregulate cFLIP, a
cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell. Biol. 21, 3964-3973.
Kroemer, G., and Jäättelä, M. (2005). Lysosomes and autophagy in cell death control. Nat. Rev.
Cancer. 5, 886-897.
Kronfeld, I., Kazimirsky, G., Gelfand, E.W., and Brodie, C. (2002). NGF rescues human B
lymphocytes from anti-IgM induced apoptosis by activation of PKCzeta. Eur. J. Immunol. 32, 136-
43.
Krueger, A., Schmitz, I., Baumann, S., Krammer, P.H., and Kirchhoff, S. (2001). Cellular FLICE-
inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-
inducing signaling complex. J. Biol. Chem. 276, 20633-40.
Kuppers, R. (2004). Prognosis in follicular lymphoma--it's in the microenvironment. N. Engl. J.
Med. 351, 2152-2153.
Kuwana, T., Mackey, M.R., Perkins, G., Ellisman, M.H., Latterich, M., Schneiter, R., Green, D.R.,
and Newmeyer, D.D. (2002). Bid, Bax, and lipids cooperate to form supramolecular openings in the
outer mitochondrial membrane. Cell 111, 331-342.
Kuwana, T., and Newmeyer, D.D. (2003). Bcl-2-family proteins and the role of mitochondria in
apoptosis. Curr. Opin. Cell Biol. 15, 691-699.

85
Lakhani, S.A., Masud, A., Kuida, K., Porter, G.A.,Jr, Booth, C.J., Mehal, W.Z., Inayat, I., and
Flavell, R.A. (2006). Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science
311, 847-851.
Lamkanfi, M., Festjens, N., Declercq, W., Vanden Berghe, T., and Vandenabeele, P. (2007).
Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 14, 44-55.
Lee, H.H., Dadgostar, H., Cheng, Q., Shu, J., and Cheng, G. (1999). NF-κB-mediated up-regulation
of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc. Natl. Acad.
Sci. U. S. A. 96, 9136-41.
Lee, J.R., and Koretzky, G.A. (1998). Extracellular signal-regulated kinase-2, but not c-Jun NH2-
terminal kinase, activation correlates with surface IgM-mediated apoptosis in the WEHI 231 B cell
line. J. Immunol. 161, 1637-44.
Li, H., Zhu, H., Xu, C.J., and Yuan, J. (1998). Cleavage of BID by caspase 8 mediates the
mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491-501.
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S.M., Ahmad, M., Alnemri, E.S., and Wang, X.
(1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell 91, 479-89.
Liu, Y.J., Joshua, D.E., Williams, G.T., Smith, C.A., Gordon, J., and MacLennan, I.C. (1989).
Mechanism of antigen-driven selection in germinal centres. Nature 342, 929-31.
Lonberg, N. (2008). Fully human antibodies from transgenic mouse and phage display platforms.
Curr. Opin. Immunol. 20, 450-459.
Mackus, W.J., Lens, S.M., Medema, R.H., Kwakkenbos, M.J., Evers, L.M., Oers, M.H., Lier, R.A.,
and Eldering, E. (2002). Prevention of B cell antigen receptor-induced apoptosis by ligation of
CD40 occurs downstream of cell cycle regulation. Int. Immunol. 14, 973-82.
MacLennan, I.C. (1994). Germinal centers. Annu. Rev. Immunol. 12, 117-39.
MacLennan, I.C., Gulbranson-Judge, A., Toellner, K.M., Casamayor-Palleja, M., Chan, E., Sze,
D.M., Luther, S.A., and Orbea, H.A. (1997). The changing preference of T and B cells for partners
as T-dependent antibody responses develop. Immunol. Rev. 156, 53-66.
Malissein, E., Verdier, M., Ratinaud, M.H., and Troutaud, D. (2003). Changes in bad
phosphorylation are correlated with BCR-induced apoptosis of WEHI-231 immature B cells.
Biochimie 85, 733-740.
Manches, O., Lui, G., Chaperot, L., Gressin, R., Molens, J.P., Jacob, M.C., Sotto, J.J., Leroux, D.,
Bensa, J.C., and Plumas, J. (2003). In vitro mechanisms of action of rituximab on primary non-
Hodgkin lymphomas. Blood 101, 949-954.
Marcus, R., and Hagenbeek, A. (2007). The therapeutic use of rituximab in non-Hodgkin's
lymphoma. Eur. J. Haematol. Suppl. (67), 5-14.

86
Mathas, S., Rickers, A., Bommert, K., Dorken, B., and Mapara, M.Y. (2000). Anti-CD20- and B-
cell receptor-mediated apoptosis: evidence for shared intracellular signaling pathways. Cancer Res.
60, 7170-7176.
Mattila, A.M., and Meri, S. (2008). Responses to rituximab vary among follicular lymphoma B cells
of different maturation stages. Scand. J. Immunol. 68, 159-168.
Medema, J.P., Scaffidi, C., Kischkel, F.C., Shevchenko, A., Mann, M., Krammer, P.H., and Peter,
M.E. (1997). FLICE is activated by association with the CD95 death-inducing signaling complex
(DISC). EMBO J. 16, 2794-804.
Melamed, D., and Nemazee, D. (1997). Self-antigen does not accelerate immature B cell apoptosis,
but stimulates receptor editing as a consequence of developmental arrest. Proc. Natl. Acad. Sci. U.
S. A. 94, 9267-9272.
Merino, R., Grillot, D.A., Simonian, P.L., Muthukkumar, S., Fanslow, W.C., Bondada, S., and
Nunez, G. (1995). Modulation of anti-IgM-induced B cell apoptosis by Bcl-xL and CD40 in WEHI-
231 cells. Dissociation from cell cycle arrest and dependence on the avidity of the antibody-IgM
receptor interaction. J. Immunol. 155, 3830-3838.
Metkar, S.S., Naresh, K.N., Redkar, A.A., and Nadkarni, J.J. (1998). CD40-ligation-mediated
protection from apoptosis of a Fas-sensitive Hodgkin's-disease-derived cell line. Cancer Immunol.
Immunother. 47, 104-12.
Miller, R.A., Maloney, D.G., Warnke, R., and Levy, R. (1982). Treatment of B-cell lymphoma with
monoclonal anti-idiotype antibody. N. Engl. J. Med. 306, 517-522.
Monroe, J.G. (2006). ITAM-mediated tonic signalling through pre-BCR and BCR complexes. Nat.
Rev. Immunol. 6, 283-294.
Nadler, L.M., Stashenko, P., Hardy, R., Kaplan, W.D., Button, L.N., Kufe, D.W., Antman, K.H.,
and Schlossman, S.F. (1980). Serotherapy of a patient with a monoclonal antibody directed against
a human lymphoma-associated antigen. Cancer Res. 40, 3147-3154.
Nakagawa, T., Shimizu, S., Watanabe, T., Yamaguchi, O., Otsu, K., Yamagata, H., Inohara, H.,
Kubo, T., and Tsujimoto, Y. (2005). Cyclophilin D-dependent mitochondrial permeability transition
regulates some necrotic but not apoptotic cell death. Nature 434, 652-658.
Nemazee, D.A., and Burki, K. (1989). Clonal deletion of B lymphocytes in a transgenic mouse
bearing anti-MHC class I antibody genes. Nature 337, 562-566.
Niiro, H., and Clark, E.A. (2002). Regulation of B-cell fate by antigen-receptor signals. Nat. Rev.
Immunol. 2, 945-956.
Norvell, A., Mandik, L., and Monroe, J.G. (1995). Engagement of the antigen-receptor on immature
murine B lymphocytes results in death by apoptosis. J. Immunol. 154, 4404-4413.

87
Nuutinen, U., Ropponen, A., Suoranta, S., Eeva, J., Eray, M., Pellinen, R., Wahlfors, J., and
Pelkonen, J. (2009). Dexamethasone-induced apoptosis and up-regulation of Bim is dependent on
glycogen synthase kinase-3. Leuk. Res. 33, 1714-1717.
Nuutinen, U., Simelius, N., Ropponen, A., Eeva, J., Mätto, M., Eray, M., Pellinen, R., Wahlfors, J.,
and Pelkonen, J. (2008). PDTC enables type I TRAIL signaling in type II follicular lymphoma cells.
Leuk. Res.
Ollila, J., and Vihinen, M. (2007). Immunological systems biology: gene expression analysis of B-
cell development in Ramos B-cells. Mol. Immunol. 44, 3537-3551.
Pätäri A, K.M.,Anderson L. (2000). Abstracts of the SSi 31st annual meeting; (B14)
Characterization of a novel monoclonal antibody with a strong proliferating effect on human
peripheral blood B cells and an inhibitory effect on follicular lymphoma cells. Scand. J. Immunol.
52, 325.
Pawluczkowycz, A.W., Beurskens, F.J., Beum, P.V., Lindorfer, M.A., van de Winkel, J.G., Parren,
P.W., and Taylor, R.P. (2009). Binding of submaximal C1q promotes complement-dependent
cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab
(RTX): considerably higher levels of CDC are induced by OFA than by RTX. J. Immunol. 183,
749-758.
Pellinen, R., Hakkarainen, T., Wahlfors, T., Tulimaki, K., Ketola, A., Tenhunen, A., Salonen, T.,
and Wahlfors, J. (2004). Cancer cells as targets for lentivirus-mediated gene transfer and gene
therapy. Int. J. Oncol. 25, 1753-1762.
Pop, C., Fitzgerald, P., Green, D.R., and Salvesen, G.S. (2007). Role of proteolysis in caspase-8
activation and stabilization. Biochemistry 46, 4398-4407.
Porter, A.G., and Urbano, A.G. (2006). Does apoptosis-inducing factor (AIF) have both life and
death functions in cells? Bioessays 28, 834-843.
Raoul, J.L., Van Laethem, J.L., Peeters, M., Brezault, C., Husseini, F., Cals, L., Nippgen, J., Loos,
A.H., and Rougier, P. (2009). Cetuximab in combination with irinotecan/5-fluorouracil/folinic acid
(FOLFIRI) in the initial treatment of metastatic colorectal cancer: a multicentre two-part phase I/II
study. BMC Cancer 9, 112.
Reff, M.E., Carner, K., Chambers, K.S., Chinn, P.C., Leonard, J.E., Raab, R., Newman, R.A.,
Hanna, N., and Anderson, D.R. (1994). Depletion of B cells in vivo by a chimeric mouse human
monoclonal antibody to CD20. Blood 83, 435-445.
Ren, C.L., Morio, T., Fu, S.M., and Geha, R.S. (1994). Signal transduction via CD40 involves
activation of lyn kinase and phosphatidylinositol-3-kinase, and phosphorylation of phospholipase C
γ 2. J. Exp. Med. 179, 673-80.
Reth, M., and Wienands, J. (1997). Initiation and processing of signals from the B cell antigen
receptor. Annu. Rev. Immunol. 15, 453-479.

88
Ricci, J.E., Munoz-Pinedo, C., Fitzgerald, P., Bailly-Maitre, B., Perkins, G.A., Yadava, N.,
Scheffler, I.E., Ellisman, M.H., and Green, D.R. (2004). Disruption of mitochondrial function
during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron
transport chain. Cell 117, 773-786.
Richards, J.D., Dave, S.H., Chou, C.H., Mamchak, A.A., and DeFranco, A.L. (2001). Inhibition of
the MEK/ERK signaling pathway blocks a subset of B cell responses to antigen. J. Immunol. 166,
3855-3864.
Riedl, S.J., and Salvesen, G.S. (2007). The apoptosome: signalling platform of cell death. Nat. Rev.
Mol. Cell Biol. 8, 405-413.
Rothe, M., Sarma, V., Dixit, V.M., and Goeddel, D.V. (1995). TRAF2-mediated activation of NF-
κB by TNF receptor 2 and CD40. Science 269, 1424-1427.
Saito, M., Korsmeyer, S.J., and Schlesinger, P.H. (2000). BAX-dependent transport of cytochrome c
reconstituted in pure liposomes. Nat. Cell Biol. 2, 553-555.
Salles, G.A. (2007). Clinical features, prognosis and treatment of follicular lymphoma. Hematology
Am. Soc. Hematol. Educ. Program. 2007, 216-225.
Scaffidi, C., Fulda, S., Srinivasan, A., Friesen, C., Li, F., Tomaselli, K.J., Debatin, K.M., Krammer,
P.H., and Peter, M.E. (1998). Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17, 1675-87.
Scaffidi, C., Schmitz, I., Krammer, P.H., and Peter, M.E. (1999). The role of c-FLIP in modulation
of CD95-induced apoptosis. J. Biol. Chem. 274, 1541-8.
Schauer, S.L., Wang, Z., Sonenshein, G.E., and Rothstein, T.L. (1996). Maintenance of nuclear
factor-κB/Rel and c-myc expression during CD40 ligand rescue of WEHI 231 early B cells from
receptor-mediated apoptosis through modulation of IκB proteins. J. Immunol. 157, 81-6.
Schwaenen, C., Viardot, A., Berger, H., Barth, T.F., Bentink, S., Dohner, H., Enz, M., Feller, A.C.,
Hansmann, M.L., Hummel, M., et al. (2009). Microarray-based genomic profiling reveals novel
genomic aberrations in follicular lymphoma which associate with patient survival and gene
expression status. Genes Chromosomes Cancer 48, 39-54.
Schweighofer, C.D., Ritgen, M., Eichhorst, B.F., Busch, R., Abenhardt, W., Kneba, M., Hallek, M.,
and Wendtner, C.M. (2009). Consolidation with alemtuzumab improves progression-free survival in
patients with chronic lymphocytic leukaemia (CLL) in first remission: long-term follow-up of a
randomized phase III trial of the German CLL Study Group (GCLLSG). Br. J. Haematol. 144, 95-
98.
Scorrano, L., and Korsmeyer, S.J. (2003). Mechanisms of cytochrome c release by proapoptotic
BCL-2 family members. Biochem. Biophys. Res. Commun. 304, 437-444.
Shan, D., Ledbetter, J.A., and Press, O.W. (2000). Signaling events involved in anti-CD20-induced
apoptosis of malignant human B cells. Cancer Immunol. Immunother. 48, 673-683.

89
Shan, D., Ledbetter, J.A., and Press, O.W. (1998). Apoptosis of malignant human B cells by ligation
of CD20 with monoclonal antibodies. Blood 91, 1644-1652.
Sikder, M.A., and Friedberg, J.W. (2008). Beyond rituximab: the future of monoclonal antibodies in
B-cell non-Hodgkin lymphoma. Curr. Oncol. Rep. 10, 420-426.
Skommer, J., Wlodkowic, D., and Deptala, A. (2007). Larger than life: Mitochondria and the Bcl-2
family. Leuk. Res. 31, 277-286.
Solal-Celigny, P., Roy, P., Colombat, P., White, J., Armitage, J.O., Arranz-Saez, R., Au, W.Y.,
Bellei, M., Brice, P., Caballero, D., et al. (2004). Follicular lymphoma international prognostic
index. Blood 104, 1258-1265.
Stadanlick, J.E., and Cancro, M.P. (2008). BAFF and the plasticity of peripheral B cell tolerance.
Curr. Opin. Immunol. 20, 158-161.
Stang, S.L., Lopez-Campistrous, A., Song, X., Dower, N.A., Blumberg, P.M., Wender, P.A., and
Stone, J.C. (2009). A proapoptotic signaling pathway involving RasGRP, Erk, and Bim in B cells.
Exp. Hematol. 37, 122-134.
Stel, A.J., Ten Cate, B., Jacobs, S., Kok, J.W., Spierings, D.C., Dondorff, M., Helfrich, W., Kluin-
Nelemans, H.C., de Leij, L.F., Withoff, S., et al. (2007). Fas receptor clustering and involvement of
the death receptor pathway in rituximab-mediated apoptosis with concomitant sensitization of
lymphoma B cells to fas-induced apoptosis. J. Immunol. 178, 2287-2295.
Suzuki, M., Youle, R.J., and Tjandra, N. (2000). Structure of Bax: coregulation of dimer formation
and intracellular localization. Cell 103, 645-654.
Suzuki, Y., Imai, Y., Nakayama, H., Takahashi, K., Takio, K., and Takahashi, R. (2001). A serine
protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death.
Mol. Cell 8, 613-621.
Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., and
Vardiman, J.W. (2008). In WHO Classification of Tumours of Haematopoietic and Lymphoid
Tissues, Fourth Edition , (Lyon: IARC)
Takada, E., Hata, K., and Mizuguchi, J. (2006). Requirement for JNK-dependent upregulation of
BimL in anti-IgM-induced apoptosis in murine B lymphoma cell lines WEHI-231 and CH31. Exp.
Cell Res. 312, 3728-3738.
Takahashi, Y., Ohta, H., and Takemori, T. (2001). Fas is required for clonal selection in germinal
centers and the subsequent establishment of the memory B cell repertoire. Immunity 14, 181-92.
Thomas, M.D., Srivastava, B., and Allman, D. (2006). Regulation of peripheral B cell maturation.
Cell. Immunol. 239, 92-102.
Thornberry, N.A., and Lazebnik, Y. (1998). Caspases: enemies within. Science 281, 1312-1316.

90
Tol, J., Koopman, M., Cats, A., Rodenburg, C.J., Creemers, G.J., Schrama, J.G., Erdkamp, F.L.,
Vos, A.H., van Groeningen, C.J., Sinnige, H.A., et al. (2009). Chemotherapy, bevacizumab, and
cetuximab in metastatic colorectal cancer. N. Engl. J. Med. 360, 563-572.
Torres, R.M., Flaswinkel, H., Reth, M., and Rajewsky, K. (1996). Aberrant B cell development and
immune response in mice with a compromised BCR complex. Science 272, 1804-1808.
Travert, M., Ame-Thomas, P., Pangault, C., Morizot, A., Micheau, O., Semana, G., Lamy, T., Fest,
T., Tarte, K., and Guillaudeux, T. (2008). CD40 ligand protects from TRAIL-induced apoptosis in
follicular lymphomas through NF-κB activation and up-regulation of c-FLIP and Bcl-xL. J.
Immunol. 181, 1001-1011.
Tsujimoto, Y., and Shimizu, S. (2007). Role of the mitochondrial membrane permeability transition
in cell death. Apoptosis 12, 835-840.
Van den Eertwegh, A.J., Noelle, R.J., Roy, M., Shepherd, D.M., Aruffo, A., Ledbetter, J.A.,
Boersma, W.J., and Claassen, E. (1993). In vivo CD40-gp39 interactions are essential for thymus-
dependent humoral immunity. I. In vivo expression of CD40 ligand, cytokines, and antibody
production delineates sites of cognate T-B cell interactions. J. Exp. Med. 178, 1555-1565.
van der Kolk, L.E., Evers, L.M., Omene, C., Lens, S.M., Lederman, S., van Lier, R.A., van Oers,
M.H., and Eldering, E. (2002). CD20-induced B cell death can bypass mitochondria and caspase
activation. Leukemia 16, 1735-1744.
van Eijk, M., Defrance, T., Hennino, A., and de Groot, C. (2001). Death-receptor contribution to the
germinal-center reaction. Trends Immunol. 22, 677-82.
van Kooten, C., and Banchereau, J. (2000). CD40-CD40 ligand. J. Leukoc. Biol. 67, 2-17.
Verhagen, A.M., Coulson, E.J., and Vaux, D.L. (2001). Inhibitor of apoptosis proteins and their
relatives: IAPs and other BIRPs. Genome Biol. 2, REVIEWS3009.
Verhagen, A.M., Ekert, P.G., Pakusch, M., Silke, J., Connolly, L.M., Reid, G.E., Moritz, R.L.,
Simpson, R.J., and Vaux, D.L. (2000). Identification of DIABLO, a mammalian protein that
promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102, 43-53.
Villamor, N., Montserrat, E., and Colomer, D. (2003). Mechanism of action and resistance to
monoclonal antibody therapy. Semin. Oncol. 30, 424-433.
Watanabe-Fukunaga, R., Brannan, C.I., Copeland, N.G., Jenkins, N.A., and Nagata, S. (1992).
Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis.
Nature 356, 314-317.
Wei, M.C., Zong, W.X., Cheng, E.H., Lindsten, T., Panoutsakopoulou, V., Ross, A.J., Roth, K.A.,
MacGregor, G.R., Thompson, C.B., and Korsmeyer, S.J. (2001). Proapoptotic BAX and BAK: a
requisite gateway to mitochondrial dysfunction and death. Science 292, 727-730.

91
Wilder, R.B., Jones, D., Tucker, S.L., Fuller, L.M., Ha, C.S., McLaughlin, P., Hess, M.A.,
Cabanillas, F., and Cox, J.D. (2001). Long-term results with radiotherapy for Stage I-II follicular
lymphomas. Int. J. Radiat. Oncol. Biol. Phys. 51, 1219-1227.
Willis, S.N., Fletcher, J.I., Kaufmann, T., van Delft, M.F., Chen, L., Czabotar, P.E., Ierino, H., Lee,
E.F., Fairlie, W.D., Bouillet, P., et al. (2007). Apoptosis initiated when BH3 ligands engage
multiple Bcl-2 homologs, not Bax or Bak. Science 315, 856-859.
Yang, J., Liu, X., Bhalla, K., Kim, C.N., Ibrado, A.M., Cai, J., Peng, T.I., Jones, D.P., and Wang, X.
(1997). Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked.
Science 275, 1129-1132.
Yin, Q., Park, H.H., Chung, J.Y., Lin, S.C., Lo, Y.C., da Graca, L.S., Jiang, X., and Wu, H. (2006).
Caspase-9 holoenzyme is a specific and optimal procaspase-3 processing machine. Mol. Cell 22,
259-268.
Yonehara, S. (2002). Death receptor Fas and autoimmune disease: from the original generation to
therapeutic application of agonistic anti-Fas monoclonal antibody. Cytokine Growth Factor Rev. 13,
393-402.
Youle, R.J., and Strasser, A. (2008). The BCL-2 protein family: opposing activities that mediate cell
death. Nat. Rev. Mol. Cell Biol. 9, 47-59.
Younes, A., and Kadin, M.E. (2003). Emerging applications of the tumor necrosis factor family of
ligands and receptors in cancer therapy. J. Clin. Oncol. 21, 3526-3534.
Zamzami, N., Susin, S.A., Marchetti, P., Hirsch, T., Gomez-Monterrey, I., Castedo, M., and
Kroemer, G. (1996). Mitochondrial control of nuclear apoptosis. J. Exp. Med. 183, 1533-1544.
Zeitlin, B.D., Zeitlin, I.J., and Nor, J.E. (2008). Expanding circle of inhibition: small-molecule
inhibitors of Bcl-2 as anticancer cell and antiangiogenic agents. J. Clin. Oncol. 26, 4180-4188.
Zhang, H., Rosenberg, S., Coffey, F.J., He, Y.W., Manser, T., Hardy, R.R., and Zhang, J. (2009). A
role for cFLIP in B cell proliferation and stress MAPK regulation. J. Immunol. 182, 207-215.
Zinzani, P.L., d'Amore, F., Bombardieri, E., Brammer, C., Codina, J.G., Illidge, T., Jurczak, W.,
Linkesch, W., Morschhauser, F., Vandenberghe, E., et al. (2008). Consensus conference:
implementing treatment recommendations on yttrium-90 immunotherapy in clinical practice - report
of a European workshop. Eur. J. Cancer 44, 366-373.
Zou, H., Li, Y., Liu, X., and Wang, X. (1999). An APAF-1.cytochrome c multimeric complex is a
functional apoptosome that activates procaspase-9. J. Biol. Chem. 274, 11549-11556.

Kuopio University Publications D. Medical Sciences D 434. Hassinen, Maija. Predictors and consequences of the metabolic syndrome: population-based studies in aging men and women. 2008. Acad. Diss. D 435. Saltevo, Juha. Low-grade inflammation and adiponectin in the metabolic syndrome. 2008. 109 p. Acad. Diss. D 436. Ervasti, Mari. Evaluation of Iron Status Using Methods Based on the Features of Red Blood Cells and Reticulocytes. 2008. 104 p. Acad. Diss. D 437. Muukka, Eija. Luomun tie päiväkotiin: luomuruokailun toteutettavuus ja ravitsemuksellinen merkitys päiväkotilapsille. 2008. 168 p. Acad. Diss. D 438. Sörensen, Lars. Work ability and health-related quality of life in middle-aged men: the role of physical activity and fitness. 2008. 83 p. Acad. Diss. D 439. Maaranen, Päivi. Dissociation in the finnish general population. 2008. 97 p. Acad. Diss. D 440. Hyvönen, Juha. Suomen psykiatrinen hoitojärjestelmä 1990-luvulla historian jatkumon näkökulmasta. 2008. 279 p. Acad. Diss. D 441. Mäkinen, Heidi. Disease activity and remission in rheumatoid arthritis: comparison of available disease activity measures and development of a novel disease sctivity indes: the mean overall index for rheumatoid arthritis (MOI-RA). 2008. 129 p. Acad. Diss. D 442. Kousa, Anne. The regional association of the hardness in well waters and the incidence of acute myocardial infarction in rural Finland. 2008. 92 p. Acad. Diss. D 443. Olkku, Anu. Glucocorticoid-induced changes in osteoblastic cells: cross-talk with wnt and glutamate signalling pathways. 2009. 118 p. Acad. Diss. D 444. Mattila, Riikka. Effectiveness of a multidisciplinary lifestyle intervention on hypertension, cardiovascular risk factors and musculoskeletal symptoms. 2009. 92 p. Acad. Diss. D 445. Hartmann-Petersen, Susanna. Hyaluronan and CD44 in epidermis with special reference to growth factors and malignant transformation. 2009. 103 p. Acad. Diss. D 446. Tolppanen, Anna-Maija. Genetic association of the tenomodulin gene (TNMD) with obesity- and inflammation-related phenotypes. 2009. 111 p. Acad. Diss. D 447. Lehto, Soili Marianne. Biological findings in major depressive disorder with special reference to the atypical features subtype. 2009. 115 p. Acad. Diss. D 448. Nieminen, Jyrki. Effect of functional loading on remodelling in canine, and normal and collagen type II transgenic murine bone. 2009. 107 p. Acad. Diss. D 449. Torpström, Jaana. Yliopistokoulutus ravitsemusasiantuntijuuden kehittäjänä. 2009. 164 p. Acad. Diss.







