Download - Quaternary structure / Protein interfaces

• Mostproteinsareoligomers
• Common3°structuredoesnotimplycommon4°structure• Mostoligomericproteinsarenotstableasmonomers
-thinkaboutthefreeenergyofthemonomer-canwedescribethesinglechainfoldingofaproteinwith
4°structurepurelyatthe3°level?
• Subunitinterfacesareoftenusedasfunctionalhotspots
• protein-protein interactions are a type of quaternary structure.
Quaternarystructure/Proteininterfaces

0.0
5.0
10.0
15.0
20.0
25.0
30.0
35.0
40.0
45.0
oligomericstate
heterooligomershomooligomers
Quaternary structure:More than 80% of proteins are oligomeric
1 2 3 4 5 6 7 8 9 10 11 12 <12
Goodsell and Olson Ann. Rev. Biomol. Struc.29, 105 (2000)

D-amino acid transferase KDGP aldolase neuraminidase
Lactate dehydrogenase Cholera toxin insulin

Molybdenum cofactor biosynthesis protein GroES co-chaperon Galactonate dehydratase
3-dehydroquinate dehydratase Rhino virus (soccer ball) proteosome

Examples of Point Group symmetries Chiralpointgroupsin3D
-theoperationdoesnotchangetheobject-rotationsonly(noreflectionsorinversions)-rotationsaxesintersectatapoint-resultsinrepeated,equivalentsurfacesbetween
chains-generates“closed”structures
Cyclic(Cn)n=2,3,4,...
rotationaboutasingleaxisDihedral(Dn)
rotationalsymmetryaboutoneaxis,andC2rotationsperpendiculartothen-foldaxis(resultsinnCnaxes)
Cubic
T:tetrahedral(C2andC3axes)O:Octahedral(C4,C3andC2axes)I:Icosahedral(C5,C3andC2axes)

Line Group symmetry (helical symmetry)• Combination of rotational and translational (1D) symmetries
Leads to fibrous structures.
• Combinerotationwithtranslationalongaxis(screwrotation)• Producessupermolecularhelices
e.g.Actin,microtubules,flagella,…
• Oftenusedinstructural/architecturalroles
• “open”structures-howtoterminatethechain?
needtocaptheends
Keratin
• rare(biologically):planegroup(2translations),spacegroup(3translations)

Advantages of oligomeric proteins
Evolutionary advantage - Symmetric oligomers are more “fit” Functional Genetic Physicochemical
Large size Larger proteins more stable against denaturation (smaller surf/vol) More efficient use of intracellular water Bigger is better - but limitations of protein synthesis
Oligomeric Error control Coding efficiency
Symmetrical Stability of association Multivalent binding / allostery Self assembly
Goodsell and Olson Ann. Rev. Biomol. Struc.29, 105 (2000)


• Generally multiple, weak contacts (large surface area).
• Shape complementarity (VdW)
• Additional stabilization: disulfides, metals, cofactors, ...
• Huge variety - cannot make many generalizations
• Range of affinities/exchange rates: “stable” (low Kd, generally has a hydrophobic character) vs. “transient” or “exchangeable” (higher Kd, usually more hydrophilic intermolecular surface).
• Individual exchange rates (kon, koff) do not necessarily correlate with affinity (Kd)
(but Kd is given by their ratio koff/kon)
• Stable: usually shielding many hydrophobic groups - Net result is a very strong association.
(i.e. these proteins typically exists exclusively in the oligomeric form)
These interfaces are often indistinguishable from the interior of proteins
• Transient or exchangeable: usually more polar, often contain bridging waters buried surface typically : 600-2000 Å**2 , polar, 0-10 H-bonds, bridging waters
Interfaces in oligomeric proteins“geometric and electrostatic complementarity”

The glycolytic enzymes PDB “molecule of the month” Feb 2004.
http://www.rcsb.org

Generation of Oligomers by 3D Domain Swapping
mechanism for the formation of new interfaces
results in higher order quaternary structures
requires only small changes in a “hinge loop”
Usually involves a C or N terminus

Examples of domain-swapped protein structures. Monomeric proteins are shown where available. A) α-IPMS (also called LeuA), pdbcode 1sr9,19 B) RNaseA N-terminal swapped dimer, pdbcode 1a2w,21 C) RNaseA C-terminal swapped dimer, pdbcode 1f0v,22 D) RNaseA cyclic C-terminal trimer, pdbcode 1js0,70 E) T7 helicase filament structure, pdbcode 1cr4,71 F) Designed 3-helix bundle, pdbcode 1g6u,28 G) suc1 monomer32 and H) domain-swapped dimer, pdbcode 1sce,33 I) wild-type GB1 monomer, pdbcode 1pgb,72 J) mutant GB1 domain-swapped dimer, pdbcode 1q1037 and K) mutant GB1 doubly domain-swapped tetramer, pdbcode 1mpe,38 L) Cystatin monomer, pdbcode 1gd3,73 M) Cystatin domain-swapped dimer, pdbcode 1g96,40 N) Cystatin amyloid-like domain-swapped dimer, pdbcode 1tij,74 O) Human prion protein monomer, pdbcode 1qm051 and P) domain-swapped dimer, pdbcode 1i4m.39
Rousseau et al., Madame Curie Bioscience Database

Three commonly used ways of classifying proteins into different groups: 1) By protein families
2) Using the domains they contain
3) Sequence features / motifs
Classificationofproteins

ProteinTaxonomies
•Allalpha•Allbeta b-sandwiches
b-propellers b-helices b-barrels Igfold ….
•Alpha/beta•Alpha+beta
Some of the projects that classify proteins: SCOP (Structural Classification of Proteins)
http://scop.mrc-lmb.cam.ac.uk/scop CATH (Class, Architecture, Topology and Homologous superfamily)
http://www.biochem.ucl.ac.uk/bsm/cath
TIM barrel Rossmann fold …

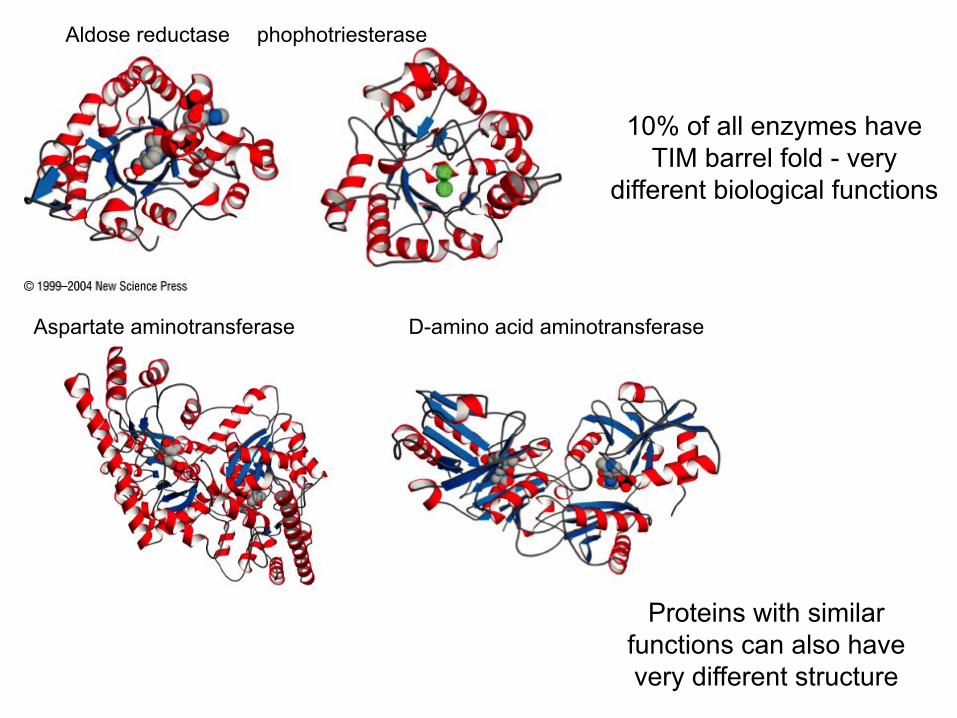
Proteins with similar functions can also have very different structure
Aldose reductase phophotriesterase
10% of all enzymes have TIM barrel fold - very
different biological functions
Aspartate aminotransferase D-amino acid aminotransferase

Many proteins are modular - this allows classification based on the domains they contain - common function for each domain

Evolutionarily linked set of proteins with related function, sequence and structure - Databases such as PFAM (http://pfam.sanger.ac.uk)
Proteinfamilies
http://www.ebi.ac.uk

Evolutionarily linked set of proteins with related function, sequence and structure
Proteinfamilies
http://www.ebi.ac.uk

Combiningfamilyanddomain
http://www.ebi.ac.uk

Sequenceelements- A set of amino acids in a specific pattern that confers a characteristic functional unit – metal binding site, signal sequence, catalytic group, etc.
H
D
S

Identical sequences ; identical function; identical structures. % identity
100
75
25
0
50 Similar structure - probably similar function.
Highly related sequences - high confidence that the two proteins have similar structure and function.
“twilight zone” : sequences with 25-35% sequence identity have a 50:50 chance of having similar structure.
In general, any two unrelated proteins (I.e. different sequence, structure and function) can be aligned to produce 10-25% identity).
But note that proteins with unrelated sequences may have very similar structures! (i.e. the two proteins may have diverged to the point that there is no detectable sequence signal - but the structures remain similar).
Hierarchy of divergence according to evolutionary distance
Sequence > Function > Structure.

MembraneProteinsSomegeneralrulesforunderstandingmembraneproteinstructure:-Satisfythemain-chainhydrogenbonds
-transmembranealpha-helices-betabarrels-NounsatisfiedHbondinggroupsexposedtolipid
-Matchthehydrophobicpropertiesofthesidechainsandlipids- alignproteinsurfaceswiththehydrocarbonchains
Exceptionsoccur!

Structure of biological membranes

Hydrocarbon region 30 Å
15 Å Headgroup region
15 Å Headgroup region
Bulk solvent
Bulk solvent
The membrane presents a very complex environment - and there are many types of membranes!

Membrane proteins
• Low dielectric constant within the bilayer
• No water in the middle of the bilayer • Main chain fully H-bonded (2° structure)
- α-helices: local (i, i+4) H-bonding - β-barrels: H-bonding between widely separated parts of the chain
• TM region: Simpler topologies (3° structure) than soluble proteins
α-helix bundles ; β-barrels
• Side chains point outward from helices or barrels
• energetics and folding pathways are very different than for soluble proteins
• Many MPs have both a membrane domain and a soluble domain

Contributions to the free energy of folding for membrane proteins are different (and often more complex) than for soluble proteins
• The “cost” of unformed H-bond in a membrane is very high
• No competing water in the bilayer
• MPs largely limited to TM helices and beta-barrels. Simpler topologies
• The dielectric constant changes from 2 - 4 within the bilayer to ~80 on the
outside surface.
• charged and H-bonding groups are driven away from the lipid phase.
• There is essentially no water in the middle of the bilayer
- no “hydrophobic force” to drive 3° structure!
- Helices pack in a hydrophobic environment What holds them together?
MP structure is not driven by the effects of water - van der Waals (packing) term is important

Alpha-helical bundles • Single or Multiple “passes” through the membrane
• Bundles with ~ aligned helices
• Commonly find cofactors
Most membrane proteins can be classified into one of two structural classes Very few exceptions known so far, but there are many variations to the theme. Proteins are complicated!
Beta-barrels • antiparallel beta-sheet
• the “last”” strand is H-bonded to the “first” strand to close the barrel.
This solves the “edge” problem and satisfies all main chain H bonding.

CompositionofTMhelices
• Richinhydrophobicaminoacids(Leu,Val,...).
• TrpandPheoftenobservedattheinterfacialregion.
• Glycine,prolineandcysteinearefairlycommon.But“no”disulfidebonds
• Sometimesfindpolarresidues(H-bondingbetweenTMhelices)
• Saltbridgescanoccur.
• Channels,pores,transporters,etc.oftenhavepolarinteriors.
• PolaraminoacidsinaTMhelixoftenpointin.Functionallyimportant.

Prediction of transmembrane segments based on hydrophobicity

Example of an α-helical membrane protein: ABC transporter
α-helical membrane proteins occur in: - eukaryotes: plasma membrane, most organelle membranes, inner
membrane of the mitochondria - bacteria: cell membrane (Gram positive); inner membrane (Gram negative)

some beta-barrel structures:
n=8 n=22 n=12 n=16
n=3x4=12
Present in: - outer membrane of Gram -ve bacteria - outer membrane of mitochondria

Effect of interfacial residues on TM domain folding
! Trp and Lys residues are often found at the ends of membrane-spanning segments – they help to ‘anchor’ the helices in place at the acyl chain-lipid headgroup interface.

àAnalysis of TM helix packing in high-resolution structures reveals tight packing at interhelical interfaces à This suggests that close van der Waals contacts may play an important role in TM protein folding à frequently mediated by amino acid motifs such as G X X X G which form a groove in one helix, allowing packing of an adjacent helix

à Due to the hydrophobic environment (lack of water within the bilayer), the formation of backbone hydrogen bonds (i.e. in helix/sheet) is much more favorable (2-10 kcal/mol). à This may contribute significantly to stage 1 of folding

à See both sidechain-sidechain and sidechain-backbone hydrogen bonds. The free energy associated with this is much more favorable than in solution.

à Loops may be important to constrain the protein structure in some cases, locking helices into position. à However, many proteins (eg. rhodopsin) can be assembled from individual helical peptides, without the loops

Chain length influences the thermodynamics of membrane protein folding. à This is primarily due to ‘hydrophobic mismatch’, a phenomenon in which the hydrophobic region of the protein is too long or too short for the hydrophobic thickness of the membrane.








