
Nitric oxide as neurotransmitter and neuromodulator Under supervision of : Dr. Bothaina Fouad
By : Hala Aly Hafez
Course : 1701802

2
Nitric oxide as neurotransmitter and neuromodulator
Abstract
Nitric oxide (NO), is a neurotransmitter and neuromodulator, was
discovered as an endothelium-derived relaxing factor more than two decades
ago. Since then, it has been shown to participate in many pathways. NO has
been described as a key mediator of different pathways in the central and
peripheral nervous system in both healthy and diseased processes.
Introduction
Nitric oxide (NO), a ubiquitous gaseous cellular messenger, plays
significant roles in a variety of neurobiological processes. Several functions of
this regulatory molecule have been identified in the nervous system, in the
process of endothelium-dependent vasodilatation, in neurotransmission, and in
host-defense mechanisms. The discovery that NO functions as a signalling
molecule in the nervous system had radically changed the concept of neural
communication. Indeed, the adoption of the term nitrergic for nerves whose
transmitter function depends on the release of NO or for transmission
mechanisms brought about by NO (Moncada et al., 1997) emphasizes the
specific characteristics of this mediator. The physical properties of NO prevent
its storage in lipid-lined vesicles and metabolism by hydrolytic degradatory
enzymes. Therefore, unlike established neurotransmitters, NO is synthesized on
demand and is neither stored in synaptic vesicles nor released by exocytosis, but
simply diffuses from nerve terminals. The distance of this NO diffusion (40 ±
300 mm in diameter) implies that structures in the vicinity of the producing cell,
both neuronal and non-neuronal, are influenced following its release. This
suggests that, as well as acting as a neurotransmitter, NO has a neuromodulatory
role (J Garthwaite et al., 1995). In addition, it diffuses into rather than binds
with protein receptors on adjacent cells, and most of its known actions are the
consequence of interplay with intracellular targets that would usually be
regarded as secondary messengers. The activity of conventional
neurotransmitters is terminated either by re-uptake mechanisms or enzymatic
degradation while inactivation of NO follows reaction with a substrate. There
are multiple points at which biological control can be exerted over the
production and activity of conventional neurotransmitters. However, control of
the synthesis of NO is the key to regulating its activity (Džoljić et al., 2015).

3
Several studies have demonstrated that NO, a freely diffusible gaseous
compound, has an important role in a variety of neurobiological processes
(Džoljić et al., 2015). Numerous functions of this regulatory molecule have
been identified in the CNS, in the process of endothelium-dependent
vasodilatation, in neurotransmission, and in host-defense mechanisms. NO is
produced from the oxidation of the terminal guanidine nitrogen of the amino
acid arginine. This reaction is catalyzed by the NADPH-dependent enzyme,
nitric oxide synthase (NOS). After its formation, NO diffuses outside the cell.
NO derived from eNOS maintains the CNS microcirculation by inhibiting
platelet aggregation and leukocyte adhesion and migration. NO derived from
nNOS is an important neurotransmitter related to neuronal plasticity, memory
formation, regulation of CNS blood flow, and neurotransmitter release (Toda et
al., 2003).
Nitric oxide synthesis
Much is known about the mechanism of NO synthesis from biochemical
studies of the purified NO synthase (NOS) enzymes. They are complex proteins
found constitutively in two isoforms, neuronal (nNOS) and endothelial (eNOS).
The third, inducible, type (iNOS) is rarely present normally but can be
expressed in numerous cell types (prototypically in macrophages, mainly in
microglia in the CNS) when subjected to immunological challenge. All three
isoforms generate NO from L-arginine but have distinct functional and
structural features. iNOS is usually linked with pathological situations (Stuehr
et al., 2004).
The three isoforms of nitric oxide synthase differ in their activity patterns
and expression in different cells. Neuronal nitric oxide synthase (nNOS) is
localized in synaptic spines, astrocytes, and the loose connective tissue
surrounding blood vessels in the brain; eNOS is present in both cerebral
vascular endothelial cells and motor neurons; and iNOS is induced in astrocytes
and microglia under pathological conditions. During physiological processes,
NO produced by eNOS/nNOS, respectively, controls blood flow activation, and
act as a messenger during long-term potentiation (LTP). However, under
pathological conditions, eNOS appears to be impaired, leading to a reduction in
blood flow and, consequently, low oxygen/metabolites delivery, efflux of
toxicological agents from the brain tissue and disturbance in the blood brain
barrier (BBB). The NO produced by iNOS in glial cells and nNOS, which
triggers the NMDA- excitotoxic pathway, combines with superoxide anion and

4
results in peroxynitrite synthesis, a potent free radical that contributes to tissue
damage in the brain. Because these enzymes may have different sites of
expression and activation, they have a pivotal role in the divergent functions of
NO (Reis et al., 2017). All NOS isoforms have phosphorylation sites for
different protein kinases, including protein kinase A, B, and C, and calcium-
calmodulin kinase. NOS enzymes are very important for the maintenance of
physiological mechanisms within an organism, and the genetic ablation of NOS
in mice has been informative for establishing the functional roles of NOS-
generated NO in different systems (Cossenza et al., 2014).
Neurotransmission by Nitric oxide
Depending on the circuit, NO may be produced pre- or postsynaptically.
In many brain areas, the prototypic coupling with postsynaptic NMDA
receptors appears to apply. In others, NO may derive from presynaptic axon
terminals, much as it does in peripheral nitrergic nerves. In these nerves, the
stimulus for NO synthesis is usually the action potential-dependent opening of
presynaptic N-type and ⁄ or other Ca2+ channels, with the resulting NO
affecting smooth muscle relaxation (Toda et al., 2005).
Nitric oxide inactivation
It is often stated that NO does not need a specialised inactivation
mechanism because it is disposed of by virtue of its natural reactivity. However,
at the low nanomolar concentrations and below that are likely to be
physiological, NO is remarkably unreactive. Reaction with O2, for example, is
obvious at micromolar NO concentrations but negligible at low nanomolar
concentrations. Reaction with superoxide ions, giving peroxynitrite, is
extremely rapid but, over the presumed physiological NO concentration range,
superoxide dismutase is greatly in excess of NO so that superoxide ions are
removed too quickly to allow reaction with NO. Low NO concentrations will,
however, react avidly with lipid peroxyl radicals in a beneficial process that
stops lipid peroxidation. In physiological conditions, one pathway for NO
degradation will be through reaction with haemoglobin in circulating
erythrocytes, forming nitrate and methaemoglobin (John Garthwaite, 2008).
The main roles of nitric oxide in the brain
Nitric oxide binds to guanylyl cyclase, the cyclic guanosine-
monophosphate (cGMP)-producing enzyme which is a soluble NO receptor, and

5
through cGMP-mediated signaling cascades it expresses its modulating effects
either as a post- or a pre-synaptic retrograde messenger. As a retrograde
neurotransmitter, NO activates the cGMP-dependent protein kinase G (PKG)
pathway which phosphorylates synaptophysin, essential for fusion of glutamate
(Glu)-containing granules with the membrane of presynaptic nerve endings.
This potentiates and facilitates Glu-ergic neurotransmission, thus making NO
the neuromodulator of excitatory neurotransmission (Wang et al., 2005). NO
also acts on inhibitory gamma-aminobutyric acid (GABA)-ergic synaptic
transmission. Recent studies have demonstrated its actions, through cGMP-
dependent pathways, on ion channels and ion exchangers with directly
modulating effects on membrane excitability (Yamamoto et al., 2015).
Furthermore, other study had shown that NO signaling modulates synaptic
inhibition in the superior paraolivary nucleus via cGMP-dependent suppression
of a potassium/chloride co-transporter. Although, through this effect at post-
synaptic level, NO acts as a reducer of strength of inhibition, this in turn enables
fine tuning of information processing (Yassin et al., 2014).
Soon after its identification as an endothelium-derived relaxing factor and
neuromodulator, NO emerged as a possible mediator of neurovascular coupling.
Neurovascular coupling is an active mechanism through which vessel diameter
is enlarged in response to increasing metabolic demands imposed by neuronal
activity; it is of vital importance in preserving the structural and functional
integrity of the brain. NO, due to its peculiar properties – it is a potent
vasodilator, released during enhanced neuronal activity resulting from Glu-ergic
activation – is well suited to mediate the coupling between neuronal activity and
cerebral blood flow (Girouard et al., 2006).
Nitric oxide has been linked to the release of other neurotransmitters and
the effects which they produce, in particular acetylcholine, noradrenaline
dopamine, glutamate, g-aminobutyric acid (GABA), serotonin, adenosin
triphosphate (ATP), bombesin, carbon monoxide, opioids and endothelin. The
mechanisms responsible for these interactions are still not fully understood, but
direct S-nitrosylation of receptors, activation of cyclic GMP-dependent protein
phosphorylation cascades, regulation of neuronal energy dynamics and a
modulating effect on transporters are potentially involved. In addition, a
presynaptic modulation of NO release through the activation of a2-
adrenoceptors, nicotinic receptors, purinergic receptors etc. has also been
proposed. Finally, it has been suggested that NO modulates gene transcription

6
and translation in neurones and glia (Peunova et al., 1995). However, these
effects would seem to be indirect since there is little evidence of the existence of
DNA elements within the promotor regions of eukaryotic cells that respond
directly to NO (Morris, 1995).
Effects of centrally released nitric oxide
Modulation of synaptic plasticity
Nitric oxide has been proposed as the retrograde messenger which co-
ordinates the enhancement of both pre- and post-synaptic mechanisms involved
in two forms of synaptic plasticity; namely long-term potentiation (LTP) and
long-term depression (LTD). LTP is a property of many central excitatory
synapses characterized by a prolonged enhancement of synaptic transmission, or
an activity-dependent increase in synaptic strength, lasting from hours to weeks
or even longer. The process by which LTP is induced is not completely clear,
but it involves glutamate acting on amino-3-hydroxy-5-methylisooxazole-4-
propionic acid (AMPA) or NMDA-receptors. This activates a series of events in
which Ca2+/calmodulin-dependent protein kinase II, NOS and protein tyrosine
kinases are implicated. LTP is thought to be a synaptic correlate of learning and
memory, and is most pronounced in higher brain centres involved in cognitive
functions, particularly in the cerebral cortex and hippocampus. Guanylate
cyclase seems to be the main effector of NO in the induction of LTP (Figure 1)
(Reis et al., 2017), however, ADP-ribosylation and activation of calmodulin-
dependent kinases have also been implicated. LTD is characterized by a long
lasting depression of parallel fibre synapses, which follows repeated excitation
of the climbing fibres of Purkinje cells. The reduction in synaptic strength
appears to result from a diminished sensitization of postsynaptic AMPA
receptors which is mediated by activation of protein kinases C and G and of the
NO-cyclic GMP signalling pathway. LTD can be observed in higher regions of
the brain, although it has been particularly well studied in the cerebellum where
it has been proposed as a model for the learning of motor movements (Hölscher,
1997).
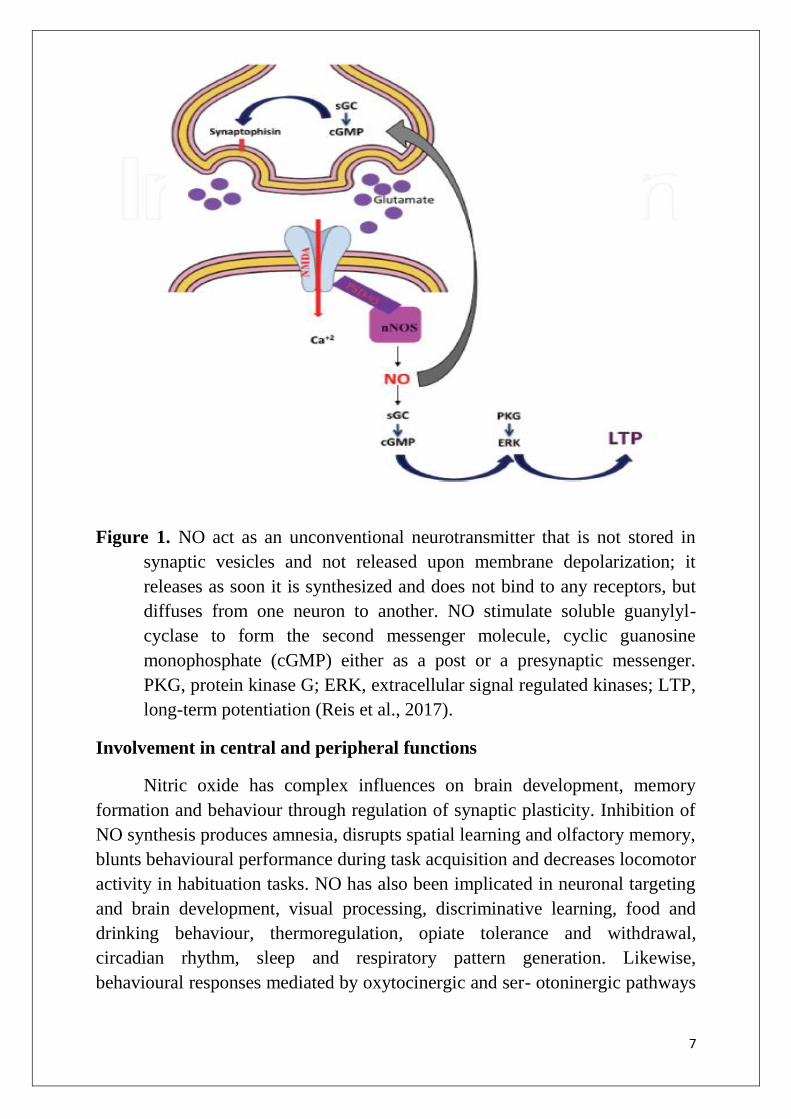
7
Figure 1. NO act as an unconventional neurotransmitter that is not stored in
synaptic vesicles and not released upon membrane depolarization; it
releases as soon it is synthesized and does not bind to any receptors, but
diffuses from one neuron to another. NO stimulate soluble guanylyl-
cyclase to form the second messenger molecule, cyclic guanosine
monophosphate (cGMP) either as a post or a presynaptic messenger.
PKG, protein kinase G; ERK, extracellular signal regulated kinases; LTP,
long-term potentiation (Reis et al., 2017).
Involvement in central and peripheral functions
Nitric oxide has complex influences on brain development, memory
formation and behaviour through regulation of synaptic plasticity. Inhibition of
NO synthesis produces amnesia, disrupts spatial learning and olfactory memory,
blunts behavioural performance during task acquisition and decreases locomotor
activity in habituation tasks. NO has also been implicated in neuronal targeting
and brain development, visual processing, discriminative learning, food and
drinking behaviour, thermoregulation, opiate tolerance and withdrawal,
circadian rhythm, sleep and respiratory pattern generation. Likewise,
behavioural responses mediated by oxytocinergic and ser- otoninergic pathways

8
are thought to involve NO generation or stimulation of central nitrergic
neurones (Cossenza et al., 2014).
There is evidence implicating central NO in the regulation of blood
pressure, heart rate, stimulated renal sympathetic nerve activity, gastric acid
secretion and motility and motor disruption associated with alcohol abuse.
Likewise, NO in the CNS appears to be involved in reflexes leading to a
diminished sympathetic output to the periphery and the modulation of various
neuroendocrine responses, including the production of oxytocin, luteinizing
hormone-releasing hormone, osmoregulator peptide corticotropin-releasing
hormone and adrenocorticotrophic hormone (Cossenza et al., 2014).
Neuronal damage and protection
The neuronal damage that accompanies cerebral ischaemia involves an
excessive release of glutamate and a subsequent activation of NMDA receptors
that, if maintained for a sufficient period of time, induces a massive influx of
Ca2+ into the postsynaptic neurone which, in turn, triggers the activation of
nNOS and overproduction of NO. In contrast, NO produced by activation of
eNOS, and even NMDA receptors, plays a protective role in brain ischaemia by
maintaining regional cerebral blood flow. The first indications that NO could
mediate neurotoxic effects came with the discovery that inhibition of NOS
attenuates glutamate toxicity in primary neuronal cultures from the rat cerebral
cortex and induces neuroprotection in animal models of stroke (Nowicki et al.,
1991).
The interactions and signalling mechanisms involved in these NO-related
effects are complex. Vascular protection is linked to cyclic GMP-mediated
mechanisms. In addition, s-nitrosylation of glutathione by NO has been
implicated in the antioxidative neuronal defence system, while NO is thought to
scavenge reactive oxygen species and partially offset ischaemia induced
oxidative damage. Likewise NO could be directly neuroprotective by interacting
with a specific site of the NMDA-receptor channel, resulting in a decreased
binding of glutamate or a diminished flow of Ca2+ through the channel after
activation (Džoljić et al., 2015).
Generation of peroxynitrite seems to be the leading cytotoxic mediator in
glutamate-induced damage. Damage caused to DNA by NO and peroxynitrite
appears to be an important neurotoxic mechanism. This is due to the subsequent

9
activation of the nuclear repair enzyme polyADP-ribose synthase which is
capable of triggering massive energy depletion resulting in cellular death if the
DNA damage is severe. In addition, inhibition of mitochondrial respiratory
chain enzymes exacerbates the depletion of neuronal energy stores. High local
concentrations of NO may also reduce cellular viability by nitrosylating several
enzymes, including phosphokinases C and glyceraldehyde-3-phosphate
dehydrogenase, or by interacting with the iron present in haem or non- haem
complexes associated with enzymes such as cytochrome P450 or aconitases
(Moncada et al., 2002).
Role in central disorders
Nitric oxide is also an important mediator under pathological conditions.
For instance, in brain ischemia-reperfusion injury, NO formation is initially
increased and has a protective function by inducing collateral perfusion as a
result of its powerful stimulatory effect on vasodilatation and angiogenesis. NO
donors induce neuroprotective effects. NO can exist in distinct
oxidation/reduction states and present dual biological actions as either a
neuroprotective or a neurotoxic molecule. Under physiological conditions,
nNOS produces hydrogen peroxide (H2O2) and superoxide (O2 •−) in addition
to NO (Costa et al., 2016).
There is growing evidence to support a role for NO in the aetiology of
neurologic conditions, including autoimmune and chronic neurodegenerative
diseases. Concentrations of NO present in inflamed tissue cause reversible
conduction block in normal, demyelinated and early remyelinated axons. Thus,
diseases such as multiple sclerosis and Guillain-Barre syndrome, characterized
by widespread loss of myelin, may see their neuronal symptoms exacerbated via
the release of NO that accompanies the severe inflammation in the central and
peripheral nervous system occurring in these conditions. NO may also be
important in secondary neuronal cell death following trauma. In the spinal cord,
nNOS expression precedes the death of motorneurones, which follows avulsion
of spinal nerve roots, and pre-treatment with nNOS inhibitors substantially
increases the number of surviving neurones. NOS neurones are resistant to
NMDA and NO neurotoxicity although the protective mechanism responsible is
still not fully known. Likewise, the excessive release of both glutamate and NO,
coupled with mitochondrial dysfunction and oxidative stress, has been
implicated in a number of neurodegenerative diseases. This highlights a

01
potential therapeutic role for specific NOS inhibitors in their pharmacological
control (Hobbs et al., 1999).
The gaseous signaling molecule NO has a variety of cellular functions,
including neurotransmission, regulation of blood‐vessel tone, and immunity.
Under pathological conditions, free radicals may deplete NO produced by
eNOS through the formation of ONOO − , thus decreasing the vascular
bioavailability of NO, which results in BBB dysfunction. This ultimately results
in endothelial damage, edema development, and hypoxia. Furthermore, the NO
produced by iNOS in glial cells or by nNOS under excitotoxic process can form
with free radicals (particularly O2−) ONOO− and produce several deleterious
effects on tissue, such as through tyrosine nitration and cysteine oxidation in
various proteins. These free radicals can further decompose into highly toxic‐
free radicals, such as NO2 • and •OH (Figure 2) (Reis et al., 2017).
The influence of peripheral systemic inflammatory conditions on the
expression of central NOS isoforms is still controversial, however,
overproduction of NO by iNOS is known to play a pathological role in acute
inflammatory disorders of the CNS. There are increased levels of NO
production in viral and bacterial infections such as meningitis, and a role for NO
has been clearly indicated in the disruption of the blood-brain barrier during
inflammatory conditions (Esplugues, 2002).

00
Figure 2. Different steps in the NO signaling cascade under
physiological/pathological conditions in the brain. During long‐ term
potentiation, NOS1 or neuronal NOS (nNOS) catalyze the NO synthesis
after the activation of the NMDA receptor by Ca2+. Under excitotoxic
conditions, excessive Ca2+ leads to nNOS hyperactivity, whereas
excessive NO production can combine with superoxide to form
peroxynitrite, which is responsible for tissue damage due to several
biological effects, including blockage of the eNOS pathway and BBB
impairment. NO is synthesized following the transcriptional expression of
a Ca2+‐independent NOS2 or iNOS isoform in glial cells (astrocytes
and microglia) after cytokine exposure, thereby contributing to
neuroinflammation and tissue damage in the brain. Intracellular Ca2+
activates NOS3 or eNOS to release NO from brain microvessels. This NO
binds to soluble guanylyl‐cyclase (sGC) receptors, which triggers a
cGMP‐dependent pathway and interacts with its downstream mediators
of the physiological regulation of vasodilation and vascular resistance,
platelet adhesion and aggregation, leukocyte‐ endothelial interaction,
and BBB integrity maintenance (Reis et al., 2017).

02
Nitric oxide has been implicated as a potential mediator of microglia-
dependent primary demyelination, a hallmark of multiple sclerosis, and iNOS
induction has been noted in the brains of patients with this autoimmune disease.
NO may also be involved in the pathogenesis of sporadic amyotrophic lateral
sclerosis and that of AIDS dementia. In the latter condition, neurotoxicity
induced by certain HIV coat proteins is partially mediated by activation of
NOS, whereas HIV patients who develop severe dementia exhibit a substantial
increase in cortical iNOS (Esplugues, 2002).
Nitric oxide in the peripheral nervous system
It is now well established that NO has a leading role as an inhibitory
neurotransmitter of peripheral non-adrenergic, non-cholinergic (NANC) nerves.
Peripheral nitrergic nerves have a widespread distribution, and are particularly
important in that they produce relaxation of smooth muscle in the
gastrointestinal, respiratory, vascular and urogenital systems. It is generally
assumed that free NO is the transmitter substance released by nitrergic nerves.
However, several controversial experimental observations have pointed to the
possibility of obtaining a closely related redox product from NO, and also
suggest that the inhibitory nitrergic transmitter is a NO-releasing molecule
(Barbier et al., 1992; Lefebvre, 1995).
Effects of peripherally released nitric oxide
Gastrointestinal system
In the gastrointestinal tract the majority of NOS positive fibres are
intrinsic, with smooth muscle cells containing sCG next to axon varicosities
containing NOS. This neuronally produced NO is implicated in many
physiological and pathophysiological reflexes in which changes in
gastrointestinal muscle relaxation are noted. Dysfunction of the inhibitory
NANC nerves in the lower oesophageal sphincter results in the motility
disorder, achalasia, and is probably involved in oesophageal spasms and related
primary motor disorders in the oesophageal body (Barrachina et al., 2001;
Calatayud et al., 2001).
Pulmonary system
The density of extrinsic NOS-containing fibres increases progressively
from the top of the trachea to the primary bronchi, and then diminishes as the

03
bronchial diameter decreases. Nitrergic nerves are believed to represent the
main nervous bronchodilator pathway in humans, and dysfunction of this
system may be implicated in the increased tone and hyper-responsiveness
observed in asthma. Furthermore, inhalation of NO has become an important
therapeutic tool in the treatment of diseases such as acute respiratory distress
syndrome, hypoxic respiratory failure, high pulmonary artery pressure, lung
transplantation, sickle cell disease and specifically paediatric conditions such as
neonatal pulmonary hypertension (Weinberger et al., 1999).
Vascular system
Neuronal NOS is found in the perivascular nerves of various blood
vessels and appears to constitute an alternative regional control mechanism for
blood flow, independent of eNOS. This neuronally produced NO seems to be
particularly relevant in the regulation of cerebral blood flow. High levels of
nNOS are present in vasodilator nerves in cerebral blood vessels, although, in
most cases, nNOS is co-localized with different vasoactive neurotransmitters. In
the brain, activity-dependent activation of nNOS is associated with a local
increase in blood blow, and this response is prevented by inhibitors of NOS
(Toda et al., 2003).
Urogenital system
Neuronal NOS is most prominent in the para-sympathetic postganglionic
innervation of the urethra. Like-wise, stimulation of bladder afferent nerves
leads to the release of NO and chronic irritation of the bladder augments nNOS
expression in dorsal root ganglion cells. Finally, bladder hyperactivity provoked
by intravesical irritants can be moderated by inhibition of NO synthesis, thus
suggesting a role for spinal cord NO in the micturition reflex pathway (Sciarra
et al., 2000).
Recent years have seen a major focus on the pharmacological modulation
of the NO released by the endothelium and nitrergic nerves and which is
involved in penile erection. nNOS neurones innervate the corpus cavernosum
and blood vessels of the penis and nerve stimulation leads to erection, which
involves sGC stimulation and is blocked by NOS inhibitors. Nitrergic neurones
are also implicated in the effects of sexual hormones. For instance, nNOS levels
in the penis decrease substantially after castration but return to normal levels

04
following testosterone replacement. Levels of nNOS diminish with age, and this
decrease correlates with impaired erectile responses (Sciarra et al., 2000).

05
REFERENCES
Barbier, A. J., & Lefebvre, R. A. (1992). Effect of LY 83583 on relaxation
induced by non-adrenergic non-cholinergic nerve stimulation and
exogenous nitric oxide in the rat gastric fundus. European journal of
pharmacology, 219(2), 331-334.
Barrachina, M., Panes, J., & Esplugues, J. (2001). Role of nitric oxide in
gastrointestinal inflammatory and ulcerative diseases: perspective for
drugs development. Current pharmaceutical design, 7(1), 31-48.
Calatayud, S., Barrachina, D., & Esplugues, J. V. (2001). Nitric oxide: relation
to integrity, injury, and healing of the gastric mucosa. Microscopy
research and technique, 53(5), 325-335.
Cossenza, M., Socodato, R., Portugal, C. C., Domith, I. C., Gladulich, L. F.,
Encarnação, T. G., . . . Paes-de-Carvalho, R. (2014). Nitric oxide in the
nervous system: biochemical, developmental, and neurobiological aspects
Vitamins & Hormones (Vol. 96, pp. 79-125): Elsevier.
Costa, E. D., Rezende, B. A., Cortes, S. F., & Lemos, V. S. (2016). Neuronal
nitric oxide synthase in vascular physiology and diseases. Frontiers in
physiology, 7, 206.
Džoljić, E., Grbatinić, I., & Kostić, V. (2015). Why is nitric oxide important for
our brain? Functional neurology, 30(3), 159-163.
doi:10.11138/fneur/2015.30.3.159
Esplugues, J. V. (2002). NO as a signalling molecule in the nervous system.
British journal of pharmacology, 135(5), 1079-1095.
Garthwaite, J. (2008). Concepts of neural nitric oxide‐mediated transmission.
European Journal of Neuroscience, 27(11), 2783-2802.
Garthwaite, J., & Boulton, C. (1995). Nitric oxide signaling in the central
nervous system. Annual review of physiology, 57(1), 683-706.
Girouard, H., & Iadecola, C. (2006). Neurovascular coupling in the normal
brain and in hypertension, stroke, and Alzheimer disease. Journal of
applied physiology, 100(1), 328-335.
Hobbs, A. J., Higgs, A., & Moncada, S. (1999). Inhibition of nitric oxide
synthase as a potential therapeutic target. Annual review of pharmacology
and toxicology, 39(1), 191-220.

06
Hölscher, C. (1997). Nitric oxide, the enigmatic neuronal messenger: its role in
synaptic plasticity. Trends in neurosciences, 20(7), 298-303.
Lefebvre, R. A. (1995). Nitric oxide in the peripheral nervous system. Annals of
medicine, 27(3), 379-388.
Moncada, S., & Erusalimsky, J. D. (2002). Does nitric oxide modulate
mitochondrial energy generation and apoptosis? Nature Reviews
Molecular Cell Biology, 3(3), 214-220.
Moncada, S., Higgs, A., & Furchgott, R. (1997). XIV. International union of
pharmacology nomenclature in nitric oxide research. Pharmacological
reviews, 49(2), 137-142.
Morris, B. J. (1995). Stimulation of immediate early gene expression in striatal
neurons by nitric oxide. Journal of Biological Chemistry, 270(42), 24740-
24744.
Nowicki, J., Duval, D., Poignet, H., & Scatton, B. (1991). Nitric oxide mediates
neuronal death after focal cerebral ischemia in the mouse. European
journal of pharmacology, 204(3), 339-340.
Peunova, N., & Enikolopov, G. (1995). Nitric oxide triggers a switch to growth
arrest during differentiation of neuronal cells. Nature, 375(6526), 68-73.
Reis, P. A., de Albuquerque, C. F. G., Gutierrez, T., Silva, A. R., & de Castro
Faria Neto, H. (2017). Role of nitric oxide synthase in the function of the
central nervous system under normal and infectious conditions. Nitric
Oxide Synthase–Simple Enzyme-Complex Roles. London: InTech, 55-70.
Sciarra, A., Voria, G., Gentile, V., Pastore, A., Di, C. C., Loreto, A., & Di, F. S.
(2000). Role of nitric oxide in the urogenital system: physiology and
pathology. Minerva urologica e nefrologica= The Italian journal of
urology and nephrology, 52(4), 201-206.
Stuehr, D. J., Santolini, J., Wang, Z.-Q., Wei, C.-C., & Adak, S. (2004). Update
on mechanism and catalytic regulation in the NO synthases. Journal of
Biological Chemistry, 279(35), 36167-36170.
Toda, N., & Herman, A. G. (2005). Gastrointestinal function regulation by
nitrergic efferent nerves. Pharmacological reviews, 57(3), 315-338.
Toda, N., & Okamura, T. (2003). The pharmacology of nitric oxide in the
peripheral nervous system of blood vessels. Pharmacological reviews,
55(2), 271-324.

07
Wang, H.-G., Lu, F.-M., Jin, I., Udo, H., Kandel, E. R., De Vente, J., . . .
Antonova, I. (2005). Presynaptic and postsynaptic roles of NO, cGK, and
RhoA in long-lasting potentiation and aggregation of synaptic proteins.
Neuron, 45(3), 389-403.
Weinberger, B., Heck, D. E., Laskin, D. L., & Laskin, J. D. (1999). Nitric oxide
in the lung: therapeutic and cellular mechanisms of action. Pharmacology
& therapeutics, 84(3), 401-411.
Yamamoto, K., Takei, H., Koyanagi, Y., Koshikawa, N., & Kobayashi, M.
(2015). Presynaptic cell type-dependent regulation of GABAergic
synaptic transmission by nitric oxide in rat insular cortex. Neuroscience,
284, 65-77.
Yassin, L., Radtke-Schuller, S., Asraf, H., Grothe, B., Hershfinkel, M.,
Forsythe, I. D., & Kopp-Scheinpflug, C. (2014). Nitric oxide signaling
modulates synaptic inhibition in the superior paraolivary nucleus (SPN)
via cGMP-dependent suppression of KCC2. Frontiers in neural circuits,
8, 65.







