
Universität Stuttgart
Institute of Polymer Chemistry
Laboratory Course
Polymer Chemistry
Contents:
page
Polymer Analogous Reactions 2
Polycondensation/Polyaddition 8
Radical Polymerization 22
Rheology 49
Polyinsertion and ROMP 70
Emulsion Polymerisation 88
Anionic Polymerization 99
Electropolymerization 107
Viscosimetry 126
Size Exclusion Chromatography 137
DSC 144

- 2 -
POLYMER ANALOGOUS REACTIONS
Assignment of tasks
1. Synthesis of a poly(vinylalcohol) through transesterification reaction of poly(vinyl acetate) using a
methanolic sodium hydroxide solution.
2. Investigations regarding the influence of H-bonding in terms of solubility of poly(vinylalcohol).
Literature
1. D. Braun, H. Cherdron, M. Rehahn, H. Ritter, B. Voit, Polymer Synthesis: Theory and
Practice, 4th Ed., Springer, 2005, page 333, 337.
2. H.-G. Elias, Makromolecules, Band 2, Wiley-VCH, 1. Auflage, 2007, page 276.
Content
1. Introduction
1.1. State of the reacting macromolecule
1.2. Degree of polymerization (end product)
1.2.1. Reactions maintaining the degree of polymerization
1.2.2. Reactions increasing the degree of polymerization
1.2.3. Reactions decreasing the degree of polymerization
1.3. Detail: poly(vinyl alcohol)
2. Experimental procedure
2.1. Safety
2.2. Experiment 1: Poly(vinylalcohol) through transesterification of poly(vinyl acetate)
2.3. Experiment 1: Intermolecular interactions in poly(vinyl acohol)
3. Questions

- 3 -
1 Introduction
Modifications of polymers via chemical reactions are used to modify the polymer and for
investigations regarding composition and constitution.
1.1 State of the reacting macromolecule
The nature of its state and/or its distribution is decisive for a macromolecule in solution for
responding to reactions.
In a good polymer solvent, a reaction of the macromolecule is only discriminable of the same
reaction of a low molecular weight macromolecule if:
1. "neighbouring group effects" are present, e.g., the functional group at the macromolecule is
different with regsards to the constitutional and stereochemical surrounding compared to the
functional group in a low molecular weight macromolecule.
2. side reactions occur. These side reactions, in low molecular chemistry, result in reduced
yields of the isolable main product. Using a macromolecule, chemically different products
result.
3. conversion is <<100%. The resulting macromolecular product is similar to a copolymer
("pseudo-copolymer").
In a poor polymer solvent, due to the high degree of cluster structure, intramolecular ring-closing
reactions occur. If the polymer is insoluble, the only possible reactions are reactions at the surface of
the cluster. If the polymer is swollen in the medium, the rate of reaction depends on the accessibility
of the functional groups in the solvent-swollen regions of the polymer matrix. The situation can
change if the swelling behavior changes or may complicate if insolubility occurs induced by newly
introduced groups. In partially crystalline polymers, reactions only occur in the amorphous regions,
because diffusion procedures are very slow (negligible) in crystalline regions.
1.2 The degree of polymerization of the reacted polymers
Reactions at macromolecules can be divided into three main groups of reactions:
1. reactions while maintaining,
2. while increasing
3. while decreasing the degree of polymerization.

- 4 -
1.2.1 Reactions maintaining the degree of polymerization are called "polymer analogous
reactions". Here, functional groups react in, on or at the end of the polymer chain with other
molecules intermolecularly or intramolecularly within the same chain. Technically important are the
conversion of cellulose to cellulose acetate, cellulose nitrate (collodion, gun cotton, films, coatings)
and cellulose xanthate (viscose). Ion exchange resins and Merrifield resins for peptide synthesis are
obtained by polymer-analogous reaction of functional side groups.
1.2.2 Reactions increasing the degree of polymerization are called “assembly reactions”. They
extend naturally only intermolecularly with functional groups in, on or at the end of the polymer
chain. Reactions in or on the main chain leading to monofunctional agents "graft" to the network
with multi-functional agents. Assembly reactions starting of the end of a polymer chain lead to
"block polymers".
1.2.3 Reactions decreasing the degree of polymerization are called “degradation reactions”.
These are targeted or untargeted reactions occurring chemical, photochemical, thermal or
mechanochemical in nature. These include the chemical and photochemical aging of polymers,
polymer degradation and analytic depolymerization reactions.
1.3 Detail: poly(vinylalcohol)
The saponification or transesterification of poly(vinyl acetate, PVAc) to poly(vinylalcohol, PVA)
represents a typical polymer-analogous reaction of polymer-side selection. Both products can have
the same average degree of polymerization after reaction, the obtained PVA can be re-acetylatet to
default PVAc. Technically, PVA is used for paints, fibers, as emulsifier and in form of protective
colloids. Atactic PVA dissolves very easily in water at any concentration. If such a solution is spun
through a spinneret into a coagulating bath (e.g., an alcoholic solution) and the resulting yarn is
stretched during the winding to a multiple of its original length, then by a parallel orientation of the
molecular chains, the intermolecular formation of hydrogen bridges over the OH groups is possible
along the PVA chains.
Fig. 1. Pattern of H-bondings in poly(vinylalcohol).

- 5 -
The resulting PVA fiber can no longer be dissolved even in warm water. Only at boiling temperature,
the thermal motion of the macromolecular chains is so strong that the parallel orientation of the
chains is disturbed. The hydrogen bonding between the chains is weakened and in its place water
molecules interact with the OH groups of PVA. The network is released, the molecular chain is
solvated and the PVA string dissolves.
2 Experimental Procedure
2.1 Safety: All persons being exposed to chemicals have to be instructed about the effects of
dangerous substances (toxicity, point of ignition, etc.) as well as about preventive measures. Before
the experiment is carried out, read the MSDS sheets for all of the chemicals used in this laboratory
and be familiar with their safe handling. The instructions of the teaching assistant must be followed
at all times. Especially the following points are relevant:
1. Wearing of suitable protective clothing (protective goggles, glove, laboratory coat, etc.)
2. Knowledge about the safety devices (e.g., laboratory hood, fire extinguisher, emergency
shower, first aid boxes, etc.) and exit.
3. Controlled disposal of toxic compounds in compliance with legal regulations.
4. Strict ban on eating/drinking/smoling in the laboratory.
2.2 Experiment 1: Poly(vinylalcohol) through transesterification of poly(vinyl acetate)
Chemicals: 25 mL methanolic NaOH-solution (1 wt.-%)
7.5 g poly(vinyl acetate, PVAc)
methanol, chloroform, tetrahydrofuran (THF), dimethyl formamide (DMF),
toluene, acetone
Equipment: 250-mL 3-neck flask
stirrer, stirring engine
reflux condenser
100-mL-dropping funnel with pressure balance
water bath, thermometer, porcelain suction filter
test tubes, glass rods, funnels
100 mL graduated cylinder
Procedure:
In a 250 mL three-necked flask with stirrer, reflux condenser and dropping funnel 25 mL of a l wt.-%
methanolic sodium hydroxide solution is heated in a water bath up to 50 °C. Under vigorous stirring

- 6 -
within 30 minutes, a solution of 7.5 g of PVAc in 80 mL of methanol is added dropwise. The
transesterification can be seen at the onset of the precipitation of PVA. After addition is complete,
stirring is continued for another 30 minutes, then the precipitate is filtered off, washed with methanol
and dried in vacuo free of alkali. The solubility of PVA is compared in test tubes with various cold
and warm (water bath!) organic solvents (methanol, chloroform, THF, DMF, toluene and acetone)
and in cold and warm water with the solubility of PVAc. Comment on the log of your observations.
With a solvent mixture of methanol / water 30/70, a 10 wt.-% PVA solution is prepared. This
solution is spread with a glass rod on a glass plate. After careful prolonged drying in an oven (PVA
is slightly hygroscopic!), a polymer film can be removed. Of these, an IR spectrum is made that you
should compare with an IR spectrum of PVAc provided.
2.3 Experiment 2: intermolecular interactions in PVA
Chemicals: different fibers made of poly(vinylalcohol)
dionized H2O
Equipment: 600 mL beaker, high form
piece weight
heating plate
thermometers
Procedure:
In an manual experiment, the solubility of three different PVA filaments under tension is
investigated. For this purpose, on each yarn made of PVA fibres a weight of about 100 g is attached
and slowly lowered into a beaker with water. Starting with yarn 1, the influence of the bath
temperature is examined for the solubility behavior. For examination, the water is slowly heated and
the dissolving of the respective threads is observed. What is observed, as long as the weight piece
hangs freely and is placed on the bottom of the cup-glass? Explain the observed behavior in the
protocol.

- 7 -
3 Question
1. What is the equation for the formation of PVAc?
2. Why can PVA not be produced by direct polymerization of the corresponding monomers?
3. Formulate the polymer-analogous reaction of PVA with aldehydes. Why are there no
networking products?
4. Give an example of an analytical polymer degradation and chemical aging of polymers.

- 8 -
POLYCONDENSATION and POLYADDITION
I Polycondensation
Assignment of tasks
1. Investigation of polycondensation kinetics via the reaction of succinic acid and 1,6-
hexandiol: In the acid-catalyzed polycondensation, the change of the carboxyl group‘s
concentration should be presented as a function of time. Further, the rate constant for this
condensation reaction should be determined.
2. Nylon-6, 10 thread should be made by the interfacial polycondensation of sebacoyl chloride
in cyclohexane and 1,6-hexamethylenediamine in water.
Literature
1. D. Braun, H. Cherdron, M. Rehahn, H. Ritter, B. Voit, Polymer Synthesis: Theory and
Practice, 4th Ed., Springer, 2005.
2. H.-G. Elias, Makromolecules Bd. 1, 1. Auflage, Wiley-VCH-Verlag, Weinheim, 2005.
3. P. J. Flory, J. Am. Chem. Soc. (1939), 61, 3334
Content
1. Introduction
1.1. Reactions by using different kinds of starting compounds with identical end groups
1.2. Reactions by using the same starting compounds with identical end groups
2. Experimental procedure
2.1. Safety
2.2. Experimental proceduce
2.3. Evaluation
2.4. Hands on experiment
3. Questions

- 9 -
1 Introduction
Condensation polymerizations are stepwise reactions between bifunctional or polyfunctional
components that entail the elimination of small molecules such as water, HCl, NaCl, NH3, HCN,
CH3OH etc. and the formation of macromolecular substances (step growth polymerization). For the
preparation of linear condensation polymers from bifunctional compounds, there are basically two
possibilities. One either starts from a monomer, which has two different groups suitable for
polycondensation (AB type), or one starts from two different monomers, each possessing a pair of
identical reactive groups that can reaction with each other (AA-BB type).
1.1 Reaction by using different kinds of monomers with identical end groups
An example of the AAB-B type polycondensation is the one of diols with dicarboxylic acids or
sebacic acid with hexamethylendiamine as follows:
n HO-(CH2)6-OH + n HOOC-(CH2)2-COOH HO-[(CH2)6-OOC-(CH2)2-COO]n-H +
(2n-1) H2O
1.2 Reaction by using monomers with two different end groups
Example: Nylon-6
n H2N-(CH2)5-COOH H2N-(CH2)5-CO-[NH-(CH2)5-CO]n-2-NH-(CH2)5-COOH +
(n-1) H2O
The formation of a condensation polymer is a stepwise process. Thus, the first step in the
polycondensation of both types of reactions mentioned above is the formation of a dimer that
possesses the same end groups as the initial monomer(s). The end group of this dimer can react in the
next step either with another dimer molecule or with the monomeric compound, and so on. In
addition, the exchange reactions must also be taken into account, which occur between free end
groups and any linking sites in the macromolecule, e.g., transesterification or transamidation:

- 10 -
In the polycondensation, the monomers, linear and cyclic oligomers and polymers are always in
equilibrium. However, the content of cyclic oligomers decreases with increasing molecular weight.
In contrast to chain growth polymerization, in which the polymerizations are typical chain-reactions
involving a starting step (initiation) followed by many identical chain-reaction steps (propagation),
each condensation step needs to be activated and requires the same activation energy.
Like the small-molecular organic chemistry reaction, the rate of H+-catalyzed esterification of a diol
with a dicarboxylic acid is proportional to the concentrations of alcohol and acid catalyst in the
polycondensation:
d COOH
dt
d OH
dtk K COOH OH (1)
If the initial concentrations of both reactive groups are equal (higher molecular weight polymers are
achieved only if this condition is fulfilled), then
COOH OH c (2)
It follows:
dc
dtk K c2 (3)
If the concentration of the catalyst is constant, integration will give:
1 1
c ck K t
t o (4)
If the number of such functional groups initially present is co, and at time t is ct, the extent p of
condensation is defined as the fraction of functional groups that have already reacted at that time:
pc c
co t
o
(5)
then
1
1
p
c
co
t (6)
Hence from eq. (4) and (6) one obtains:

- 11 -
1
11
pk K c to (7)
The plot of 1/(1-p) versus t should be linear with a slope of k.K.co from which k can be determined.
If the initial concentration of both bifunctional reactants is equal, the number of the functional
groups at any time is equal to the number of molecules originally present. The average degree of
polymerization is then defined as the ratio of the number of molecules originally present, co, to the
total number of unreacted molecules ct at the appropriate stage of the reaction. Hence from eq. (6)
one obtains:
pc
cP
t
on
1
1 (8)
(One could also define p as the ratio of functional groups that have already reacted at a certain time
to the number of functional groups in the beginning p = (N0-N)/N0 , because the number of those
functional groups is proportional to the molar concentration of reactants.)
The following graphic shows that a high degree of polymerization (or high molecular weight) can
only be achived, if the polycondensation reaction is carried out with high conversion and without
side reactions.
In order to obtain a polymer with a degree of polymerization 100, more than 99% of the functional
monomers should react and convert into product. It can only be obtained, if both functional
monomers are present in exact equimolar amounts. For some functional substances, e.g.,
hydroxycarbonyl acids or amino acids, the equivalent is automatic since they contain both groups.
Otherwise, a small excess of one component can heavily affect the molecular weight of the obtained
polymers.
In addition, the condensation is an equilibrium reaction, in order to obtain high conversion, it is
extremely important to remove as much of the low-molecular-weight reaction products as possible
(to draw the reaction toward product formation) usually by applying vacuo or azeotropic distillation.
Condensation reaction can be carried out in melting state, in solvent, in suspension or as interfacial
polycondensation.
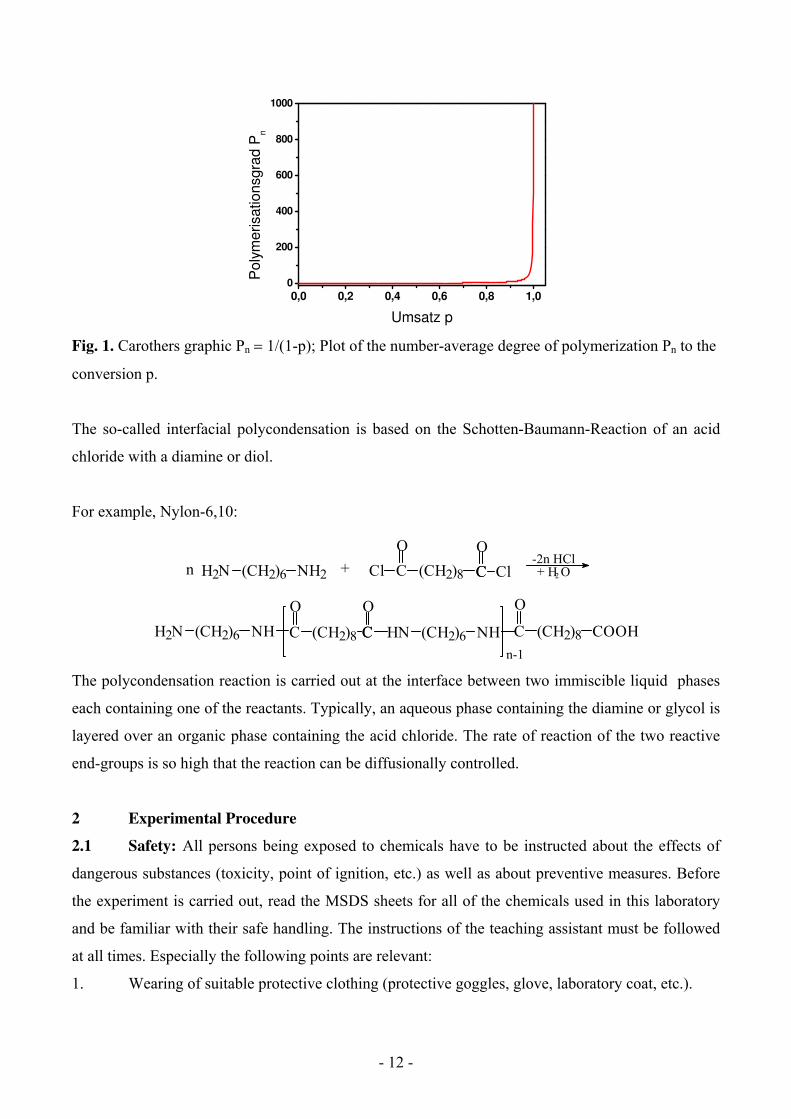
- 12 -
0,0 0,2 0,4 0,6 0,8 1,00
200
400
600
800
1000
Poly
merisationsgra
d P
n
Umsatz p
Fig. 1. Carothers graphic Pn 1/(1-p); Plot of the number-average degree of polymerization Pn to the
conversion p.
The so-called interfacial polycondensation is based on the Schotten-Baumann-Reaction of an acid
chloride with a diamine or diol.
For example, Nylon-6,10:
n-1
HN (CH2)6 NH
O
C (CH2)8 COOHC (CH2)8 C
O
C
O
H2N (CH2)6 NH
2+ H O -2n HCl
O
C
O
+H2N (CH2)6 NH2n C ClC (CH2)8Cl
The polycondensation reaction is carried out at the interface between two immiscible liquid phases
each containing one of the reactants. Typically, an aqueous phase containing the diamine or glycol is
layered over an organic phase containing the acid chloride. The rate of reaction of the two reactive
end-groups is so high that the reaction can be diffusionally controlled.
2 Experimental Procedure
2.1 Safety: All persons being exposed to chemicals have to be instructed about the effects of
dangerous substances (toxicity, point of ignition, etc.) as well as about preventive measures. Before
the experiment is carried out, read the MSDS sheets for all of the chemicals used in this laboratory
and be familiar with their safe handling. The instructions of the teaching assistant must be followed
at all times. Especially the following points are relevant:
1. Wearing of suitable protective clothing (protective goggles, glove, laboratory coat, etc.).

- 13 -
2. Knowledge about the safety devices (e.g., laboratory hood, fire extinguisher, emergency
shower, first aid boxes, etc.), exit.
3. Controlled disposal of toxic substances in compliance with legal regulations.
4. Strict ban on eating, drinking, smoking in the laboratory.
The polycondensation reaction is carried out with the following equipment:
Fig. 2. Circulation apparatus for preparation of condensation polymers via azeotropic esterification.
2.2 Experiment
Chemicals: p-toluic acid, 1,6-hexanediol, succinic acid, toluene, Mg(ClO4)2, NaOH,
phenolphthalein
Equipment: 500-mL flask, becker, buret
Procedure:
To 240 mL of toluene in a 500 mL round-bottomed flask are added 150 mg of p-toluenesulfonic
acid, 5.99 g (0.05 mol) of 1,6-hexandiol and 5.911 g (0.05 mol) of succinic acid. After addition of a
few boiling stones and opening of the cooling water, the heating mantle is switched to level two. An
extraction thimble filled with Mg(ClO4)2 is then inserted into the Ttube of the circulation apparatus.
Then the overflow nose is inserted into the top side of the extraction thimble. The obtained water can
be removed by opening tap 1 in the siphon. 30 min later a clear solution is obtained in the distillation
flask, at this point the distillation is interrupted briefly, 2 mL of the solution are removed by using a
pipette and titrated with n/100 NaOH solution. In order to determine the content of the carboxy end-
1 T

- 14 -
group, aiddtional samples will be taken at intervals of 30 min for a total reaction time of 5~6 hours
and then titrated with the above-mentioned NaOH solution.
2.3 Evaluation
After the beginning of the cycle there are exactly 100 mL of solution in the distillation flask.
Statistically every condensated molecule contains an average of one alcohol and one carboxyl end
group. From the consumption of n/100 NaOH solution the number of mole of carboxyl end groups
can be calculated. This number is plotted against time. By using equation (5) the validity of the
correlation in equation (7) is verified.
2.4 Hands on experiment
Synthesis of nylon-6, 10 via the interfacial polycondensation
Chemicals: 3 mL sebacoyl chloride
4,4 g hexamethylendiamine
100 mL cyclohexane
50 mL dist. water
sodium hydrogencarbonate, acetone,
phenolphthalein
Equiments: beakers (1x200 mL, 2x400 mL)
measuring zylinder (1x50 mL, 1x100 mL)
pipette 3 mL
stirrer, glas bar, glas funnel
A solution of 4.4 g (26 mmol) of hexamethylene diamine in 50 mL of water in a beaker is carefully
layered with a solution of 3 mL (14 mmol) of sebacic acid dichloride in 100 mL of cyclohexane by
means of a funnel. For a besser visualization of the separated phase a small amount of
phenolphthalein can be added into the aqueous solution. A thin film will form at the interface. Draw
a thread out of the interface by using a tweezer and place it on the glass rod in the stirrer motor. After
switching on the motor a fiber can be extracted continuously. The product is washed in a 400 mL
beaker first with sodium bicarbonate solution, then with water and finally with acetone and
eventually dried in a vacuum oven at 60 °C.

- 15 -
3 Questions
1. What kind of correlation of conversion p and reaction time t do you expect in case of the
auto-catalyzed polycondensation of a dicarboxylic acid and a diol? How can this be
demonstrated experimentally?
2. In principle what kind of possibilities are there to regulate the molecular weight of a
polycondensate and what has to be kept in mind regarding the end groups?
3. You are supposed to synthesize a polycondensate with a dicarboxylic acid and ethylene
glycol (Kp(760) = 198 oC). What difficulties do you expect when using an azeotropic
esterification and applying toluene Kp(760) = 110,6 oC) as hauler? How can this difficulty
be bypassed?
4. What kind of methods for the synthesis of polyamides do you know?

- 16 -
II Polyaddition
Assignment of tasks
Synthesis of polyurethane through a polyaddition reaction between a polyole and a diisocyanate.
Literature:
1. J. H. Saunders, W. Frisch, “Polyurethanes: Chemistry and Technology I: Chemistry”, in
“High Polymers” XVI, 1962, S. 63.
2. G. Oertel, Kunststoffbuch Bd. 7, Polyurethane, Hanser Verlag München 1983.
3. D. Braun, H. Cherdron, M. Rehahn, H. Ritter, B. Voit, Polymer Synthesis: Theory and
Practice, 4th Ed., Springer, 2005, page 333, 337.
4. H.-G. Elias, Makromolecules, Bd. 1, 1. Auflage, Wiley‐VCH‐Verlag, Weinheim, 2005.
Content
1. Introduction
2. Mechanism
2.1. Reaction in the absence of a catalyst
2.2. Reaction in the presence of a catalyst
3. Experimental procedure
3.1. Safety
3.2. Experiment
4. Questions

- 17 -
1 Introduction
The formation of synthetic polymers is a process, which occurs via chemical connection of many
hundreds up to many thousands of monomer molecules. As a result, macromolecular chains are
formed. They are, in general, linear, but can be branched, or crosslinked as well. The chemical
process of chain formation may be divided into two classes, depending on whether it proceeds as a
chain-growth or as a step-growth reaction. Condensation polymerizations (or step-growth
polymerization) comprise of the stepwise reaction between bifunctional or polyfunctional
components, with elimination of small molecules such as water, alcohol, or hydrogen and the
formation of macromolecular substances. Polymers such as polyamides and polyesters can be
prepared via condensation polymerization; this type of condensation polymerization is therefore
termed polycondensation. In contrast to condensation polymerization, addition polymerization (also
called polyaddition) involves a stepwise reaction of at least two bifunctional components, leading to
the formation of macromolecules; however, in course of this process no low-molecular-weight
compounds are eliminated. The coupling of the monomer units is a consequence of the migration of
a hydrogen atom. Like condensation polymerization, this kind of addition polymerization is also a
stepwise reaction, consisting of a sequence of independent individual reactions, so that the average
molecular weight of the resulting polymer steadily increases during the course of the reaction. The
oligomeric and polymeric products formed in the individual steps possess the same functional end
groups and the same reactivity as the starting materials; they can be isolated without losing their
reactivity. As all stepwise reactions, they are also governed by kinetic laws similar to those for
condensation polymerization.
The classical and important stepwise addition polymerization is the reaction of the di- or
polyfunctional isocyanate with di- or polyfunctional hydroxy compounds, or other compounds
having a plurality of active hydrogen atoms, to form the macromolecules in which the constitutional
repeating units are coupled with one another via urethane or urea groups (scheme 1).
Scheme 1. Polyurethane and polyurea formation.

- 18 -
The branched and crosslinked polyurethane (PU) can be prepared through the reaction of
diisocyanates with compounds that possess more than two hydroxyl groups per molecule, or the
reaction of linear oligourethanes, which possess either hydroxyl or isocyanate end group, with
suitable reactive compounds, followed by crosslinking reactions (Scheme 2). In the presence of
water, pairs of isocyanate end groups in the chain-extended polymer OCN-X-NCO first react with
one molecule of water; this results in a linear coupling through urea moieties, with simultaneous
elimination of CO2. The subsequent crosslinking probably occurs by the reaction of the hydrogen
atoms of the resulting urea groups with isocyanate groups still present in the starting polymer or the
chain-extended polymer, with the formation of biuret groups.
A key factor in the preparation of polyurethane is the activity of the isocyanate. Aromatic
diisocyanates are more reactive than aliphatic diisocyanates; primary isocyanates react faster than
secondary or tertiary isocyanates. The most important and commercially most accessible
diisocyanates are aliphatic and colorless hexamethylene-1,6-diisocyanate (HDI), isophorone
diisocyanate (IPDI), and aromatic, brownish colored diphenylmethane-4,4´-diisocyanate (MDI), 1,5-
naphthalenediisocyanate, and a 4:1 mixture of 2,4- and 2,6-toluenediisocyanates (TDI).
Scheme 2. The formation of biuret.
The addition of isocyanates to hydroxyl compounds is inhibited by acid compounds (e.g., hydrogen
chloride or p-toluenesulfonic acid ( blocked isocyanates), on the other hand, it can be accelerated
by basic compounds (e.g., tertiary amines like triethylamine, N,N-dimethylbenzylamine, and
espically 1,4-diazabicylco[2.2.2]octane and by certain metal salts or organometallic compounds (e.g.,
dibutyltin dilaurate, bismuth nitrate).

- 19 -
2 Mechanism
The reaction of an isocyanate with an active hydrogen compounds is carried out with or without a
catalyst. The self-addition reactions of isocyanates do usually not proceed as readily as reactions
with active hydrogen compounds.
2.1 Reaction in the absence of a catalyst
The active compound itself acts catalytically in the reaction as follows (Scheme 3).
Scheme 3. Isocyanate reaction in the absence of a catalyst.
As given in Scheme 3, in reactions proceeding in the absence of a catalyst, the electrophilic carbon
of the isocyanate is attacked by the nucleophilic centre of the active hydrogen compound; hydrogen
is added to the –NCO group. The reactivity of the –NCO groups is increased due to the presence of
the electron withdrawing groups, and decreases in the presence of electron donating groups. While
the aromatic isocyanates are more reactive than the aliphatic isocyanates, steric hindrance at the –
NCO or HXR’ groups reduce the reactivity. The order of reactivity of active hydrogen compounds
with isocyanates in uncatalyzed systems is as follows:
Aliphatic amines> aromatic amines> primary alcohols> water>secondary alcohol> tertiary alcohol>
phenol> carboxylic acid> ureas> amides>urethanes.
2.2 Reaction in the presence of a catalyst
The isocyanate reactions are also extremely susceptible to catalysis. The various isocyanate reactions
are influenced to different extents by different catalysts. Many commercial applications of
isocyanates utilize catalyzed reactions. Tertiary amines, metal compounds like tin compounds are
most widely used catalysts (Schemes 4). The mechanisms are similar to that of uncatalyzed reaction
(Scheme 3). Tertiary amines and metal salts catalyze the reaction as follows:

- 20 -
Scheme 4. Metal salt-catalyzed reaction.
3 Experimental procedure
3.1 Safety: All persons being exposed to chemicals have to be instructed about the effects of
dangerous substances (toxicity, point of ignition, etc.) as well as about preventive measures. Before
the experiment is carried out, read the MSDS sheets for all of the chemicals used in this laboratory
and be familiar with their safe handling. The instructions of the teaching assistant must be followed
at all times. Especially the following points are relevant:
1. Wearing of suitable protective clothing (protective goggles, gloves, laboratory coat, etc.)
2. Knowledge about the safety devices (e.g., laboratory hood, fire extinguisher, emergency
shower, first aid boxes, etc.), exit
3. Controlled disposal of toxic substances in compliance with legal regulations
4. Strict ban on eating in the laboratory
In addition, the handling and use of isocyanates should be undertaken with great care in order to
avoid any expose. Isocyanates are suspect carcinogens and cause irritation to the respiratory tract
(nose, throat, and lungs). Care should be excised in the handling of the amine or tin catalysts, polyols,
and blowing agents.
3.2 Experiment
Chemicals: Poly(ethyleneglycol), 1,4 butanediol, methylene diphenyldiisocyanate (MDI), DBTDL
catalyst, dry THF, methanol.
Equipment: 250-mL 3-necked flask
stirrer, stirring engine, reflux condenser
100-mL-dropping funnel with pressure balance
oil bath, thermometer, porcelain suction filter
test tubes, glass rods, funnels
100 mL graduated cylinder

- 21 -
Procedure:
Before starting the reaction, all monomers such as the macro-diol or 1,4-butanediol (BD) need to be
well dried in vacuo with appropriate temperature for removing residual moisture. The reaction is to
be performed by solution polymerization.
In a typical reaction, 1 equivalent of the macro-diol (3.0 g, 2.0 mmol ) and 2.2 equivalents of MDI (
1.1 g, 4.4 mmol) are mixed with 20mL of dry THF and taken in a dry 3 necked RB under dry
nitrogen atmosphere. Then the RB is placed on a magnetic stirrer and heated at 60°C. After complete
mixing of all monomers, 0.01% (0.003 g) of DBTDL catalyst (based on the weight of macro-diol ) is
added. 2 h later, 2 equivalents of 1,4 butanediol (0.1802 g, 2 mmol) are added and the mixture is
stirred again for 3-4 hr at reflux. The final mixture is then purified by precipitating the polymer from
methanol followed by repeated washings for removal of any unreacted monomer. The precipitate is
then dried in a vacuum oven at 60°C for 24 hrs.
4 Questions:
1. Formulate the reaction mechanism of the obtained polyurethane using a base as catalyst.
2. Why are neither primary nor secondary amines used as catalysts for the synthesis of
polyurethanes?
3. Why are low boiling tertiary amines not used as catalysts?
4. Formulate the equation for the formation of urea from the prepolymer containing isocyanate
groups and water.
5. Formulate the crosslinking equation with the formation of a biuret structure from the
starting polymer or the chain-extended polymer containing isocyanate groups in the
presence of water.
6. What will happen if the reaction of isocyanate and water is faster then the reaction of
isocyanate with polyol, and vice versa?
7. What are the side reactions occurring if some moisture is present in the reaction system?

- 22 -
RADICAL POLYMERIZATION
Literature
1. H. G. Elias, Bd. 1, 1. Auflage, Wiley-VCH-Verlag, Weinheim, 2005.
2. G. Odian, “Principles of Polymerization”, John Wiley & Sons, Inc., 3rd Ed., New York 1991, S. 198 ff
3. F. R. Mayo, F. M. Lewis, J. Am. Chem. Soc. (1944), 66, 1594
4. M. Fineman, S. D. Ross, J. Polym. Sci. (1959), 5, 259
Content
1. Introduction
2. General overview over the reaction mechanism
2.1. Initiation
2.2. Propagation
2.3. Termination
2.4. Dependence of the degree of polymerization on conversion
2.5. Reaction scheme and kinetics
3. Experimental verification of the rate law
3.1. Determination of the total rate
3.2. Dependence of the total rate on the concentration of the starting materials
4. Other concepts in the free-radical polymerization
4.1. Kinetic chain length and degree of polymerization
4.2. Chain transfer
4.3. The Trommsdorff-effect
5. Radical copolymerization
5.1. Copolymerization equation
5.2. Influence of the penultimate incorporated monomers on propagation
5.3. Discussion of the r1, r2 values
5.4. Experimental determination of the copolymerization parameters
5.5. Influence of the monomers constitution on the copolymerization parameters
6. Statistic of the polymer chain
6.1. Sequence distribution in copolymer
6.2. Calculation of the average sequence length ( I1 )
6.3. Definition and calculation of the run number ®
7. Experimental procedure
8. Questions

- 23 -
1 Introduction
Since all the essential principles of free-radical polymerization are known, this type of reaction is
particularly suited to practice and explains a number of definitions, concepts, methods and kinetics of
free-radical polymerization. The reaction of styrene with 2,2‘-azobisisobutylrontitrile (AIBN) has
been selected as a practical example:
n)(N C C
CH3
CH3
CH2 C
H
N
CH3
CH3
CCn H2C C
H
+N C C
CH3
CH3
N N C C
CH3
CH3
N
(1)
As has been shown, the total reaction consists of several sub-steps. It is the merits of G.V. Schulz, H.
Mark, J.W. Breitenbach and H.W. Melville to have solved this reaction mechanism. The below-
mentioned reaction mechanism evolved from a wealth of investigations, in which the measurement
of the rate of polymerization carried in this experiment played an important role.
2 General overview of the reaction mechanism
The polymerization, just like the well-known example of the chloride and hydrogen reaction at
school, proceeds according to a radical chain mechanism. The chain reaction can be divided into
three stages: initiation, propagation, and termination.
2.1 Initiation
In the initiation step the free radicals are formed from an initiator. Most of them are low-molecular
weight substances, which will decompose to radicals upon exposure to heat or light. Initiators can be
peroxides, persulfates and azo compounds. AIBN belongs to the mostly used initiators due to its easy
handling and clear decomposition. The decomposition of AIBN, as seen in the following equation,
strictly follows the first order rate law:
N2+2 N C C
CH3
CH3
N C C
CH3
CH3
N N C C
CH3
CH3
N
(2)
d AIBN
dt k AIBNz
(3)
d R *
dt2 k AIBNz
(4)

- 24 -
The radicals (R) so formed then react with the monomer (M) under opening of the double bond to
form a - bond between R and M. At the same time, a new radical is formed in the α-position of the
phenyl ring (why?!):
+N C C
CH3
CH3
H2C C
H
N C C
CH3
CH3
CH2 C
H
(5)
However, not all radicals generated by the initiators are capable of starting a polymer chain. Some of
the formed radicals recombine before they diffuse apart (see cage-effect). The initiator efficiency f is
defined as the ratio of the number of initiator molecules that start polymerization chain to the number
of initiator molecules decomposed under the given condition of the polymerization:
f can be experimentally determined by using C14-labelled AIBN in the experiment. The fAIBN for the
polymerization of styrene at 50°C is ca. 0.5.
2.2 Propagation step
Chain growth occurs by the addition of monomer to the monomer radicals formed in the initiation
step:
N C C
CH3
CH3
CH2 C
H H
CCH2N C C
CH3
CH3
CH2 C
H
+ H2C C
H
(6)
A growing radical, in which n monomer molecules have been added, is called polymer-radical Pn*.
In general, the successive additions can be formulated as follows:
Pn + M Pn+1* *
(7)
The rate of propagation is given by:
v k P * Mw w (8)
vw = rate of propagation; kw= rate constant of propagation. This reaction has all characteristics of a
chain reaction because a new polymer radical is formed with every step. The main difference
between this type of chain reaction and a low molecular weight chain reaction, as for example for the
explosive reaction of chlorine with hydrogen, is, that this reaction forms chemical bonds between the
different links of the chain.

- 25 -
2.3 Termination
The termination reaction will take place when two polymer radicals react with each other. In general,
there are two types of termination reactions. Either the two radicals combine with each other as
shown in the following (recombination reaction):
Pn + P Pn+mm* *
(9)
or a hydrogen atom of one chain is abstracted from the other, producing a terminal unsaturated group
and a polymer with a terminal saturated group (disproportion reaction).
P CH2 C
H
n-1+ PCH2C
H
m-1P CH C
H
n-1P CH2 C
H
Hm-1
+
(10)
Both types of termination reaction follow the same rate law. (The rate constant depends on the type
of termination reaction):
vab d Pn *
dt kab Pn * Pm *
vdis d Pn *
dt kdis Pn * Pm *
(11)
(11a)
vab = rate of the termination action; kab = rate constant of termination action.
2.4 Dependence of the degree of polymerization on conversion
In a radical polymerization reaction polymer molecules exist beside unreacted monomers, even at
very low conversions. The reason is that the propagation reaction (Eq. 7), compared with the
decomposition reaction of initiators, needs much lower activation energy. That is to say, the
decomposition reaction of initiators is the rate-determining step of the total polymerization. Once a
free radical is formed, the propagation reaction with the growth of the chain takes place in
milliseconds, until the termination occurs. The progress of the polymerization is therefore not an
increase of the molecular weight (in contrast to ionic polymerization and polyaddition), but an
increase of conversion. Plotting the average degree of polymerization Pn versus the conversion of
monomers, the following characteristic picture for the free-radical polymerization in the early stage
can be obtained:

- 26 -
Fig. 1: Dependence of the degree of polymerization on conversion for the radical polymerization.
2.5 Reaction scheme and kinetics
The rate law for the total reaction can be determined from the partial reaction and its rate law. The
total rate vBr is defined as the conversion of monomer to polymer per unit volume and per unit time:
vd M
dt
M
tBr
(12)
For clarity, all of the individual reactions are combined in a reaction scheme:
initiation:
AIBN k z
2 f R*
R* + M k st
P1*
propagation:
P1* + M k w1
P2*
P2* + M k w2
P3*
Pn-1* + M k wn-1
Pn*
termination:
Pn* + Pm* k ab
Pn+m
oder k ab
Pn + Pm
The kinetics of the ideal polymerization can then be derived with the help of the reaction scheme. In
order to be successful, we need the following assumptions:

- 27 -
1. All reactions are irreversible (this is reasonable).
2. We refer to a situation, where the concentration of the initiator radical R is constant; i.e. all
of the radical formed via the decomposition of the initiator should be consumed by the
following propagation reaction.
dR *
dt= 0 = 2 k f I - k R * Mz st
(13)
3. The concentration of initiator remains constant during the polymerization reaction. That is
to say, the concentration of the initiator [I] at time t is equal to the original concentration of
the initiator [I0].
4. The rate of the total reaction is approximately equal to that of the propagation reaction
v = -d M
dt= k P* M + k R * MBr w st
(14)
For high degree of polymerization, the consumption of the monomer at the starting reaction,
compared to the propagation reaction, is negligible.
This results in the following equation:
v v = k P * MBr w w (15)
5. Termination reaction occurs strictly by mutual deactivation of two polymer radicals.
6. The concentration of polymer radicals P is constant:
d P*
dt= k R * M k P*st ab
2 0, d.h. vst=vab, , kst [R*] [M]=kab [P*]2 (16)
The concentration of polymer radical is obtained from Eq. 16:
P * =k R * M
kst
ab
(17)
and the concentration of the initiator from Eq. 13:
R * =2 k f I
k Mz
st
(18)
substitution [R*] (Eq. 18) into Eq. 17, gives [P*] as:
P * =2 k f I
kz
ab
(19)
substitution [P*] into Eq.15 and considering [I] [I]0 give the equation for the total reaction rate:
v = k 2 k f
k I MBr w
z
ab0
(20)

- 28 -
3 The experimental verification of the rate law
3.1 Determination of the total rate
According to eq. 12, the total reaction rate or the rate of polymerization is defined as negative time
dependent change in the monomer concentration. All physical and chemical properties that will be
changed during the polymerization can be used for determining this change. One of the most
applicable methods is the dilatometric method. Its principal is based on the change of the specific
volume in the transition state of monomer to polymer. The term “dilatometry” is somehow
misleading, as the polymer has a higher density than the monomer and, therefore, only a contraction
and no dilatation can be observed during the polymerization. The conversion in % is then calculated
by using the following formula:
U =100 V
K V0
where K =
V V
Vsp(M) sp(Poly)
sp(M)
(21)
Vsp(M) = specific volume of the monomer
Vsp(Poly) = specific volume of the polymer
V0 = volume of the monomers used
V = volume change due to contraction
Here, K represents the relative change in volume at complete conversion and K = 0.167 for styrene at
50 °C. Usually, it is necessary to monitor the reaction over a long time, and then to measure the
conversion of the monomer at different time, finally to plot the so-called time-conversion curve.
According to equation 12, the rate of polymerization is then given as the slope of the time-
conversion-curve.
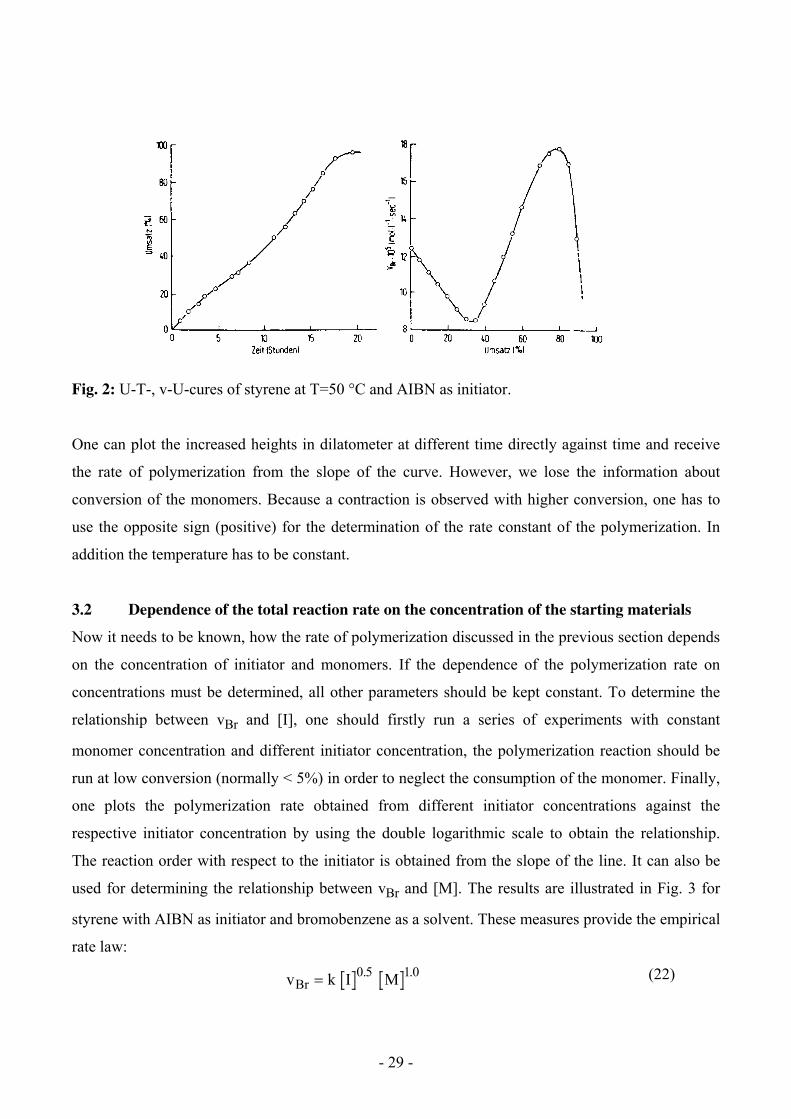
Fig. 2 shows such a time-conversion curve as well as the gross rates as function of the conversion for
the polymerization of styrene, which can be determined by graphical differentiation. This counts
only for bulk reactions (without addition of a solvent). The strange form of the curve is discussed in
5.3.
- 29 -
Fig. 2: U-T-, v-U-cures of styrene at T=50 °C and AIBN as initiator.
One can plot the increased heights in dilatometer at different time directly against time and receive
the rate of polymerization from the slope of the curve. However, we lose the information about
conversion of the monomers. Because a contraction is observed with higher conversion, one has to
use the opposite sign (positive) for the determination of the rate constant of the polymerization. In
addition the temperature has to be constant.
3.2 Dependence of the total reaction rate on the concentration of the starting materials
Now it needs to be known, how the rate of polymerization discussed in the previous section depends
on the concentration of initiator and monomers. If the dependence of the polymerization rate on
concentrations must be determined, all other parameters should be kept constant. To determine the
relationship between vBr and [I], one should firstly run a series of experiments with constant
monomer concentration and different initiator concentration, the polymerization reaction should be
run at low conversion (normally < 5%) in order to neglect the consumption of the monomer. Finally,
one plots the polymerization rate obtained from different initiator concentrations against the
respective initiator concentration by using the double logarithmic scale to obtain the relationship.
The reaction order with respect to the initiator is obtained from the slope of the line. It can also be
used for determining the relationship between vBr and [M]. The results are illustrated in Fig. 3 for
styrene with AIBN as initiator and bromobenzene as a solvent. These measures provide the empirical
rate law:
v k I MBr0.5 1.0 (22)

- 30 -
Fig. 3: Polymerization of styrene with AIBN in brombenzene (T=50°C), a) for [M] = const.; b) for
[I] = const.
4 Other criteria of free-radical polymerization
4.1 Kinetic chain length and degree of polymerization
The kinetic chain length indicates that how many monomer molecules are averagely deposited on
each active polymer radical before the termination takes place. Therefore, is defined as the ratio of
the probability of chain growth Ww to chain termination Wab. Since the probability is proportional to
the corresponding reaction rate, we can write
W
W
v
v
k P * M
k P *
w
ab
w
ab
w
ab2
(23)
Using the definition of the polymerization rate (eq. 15) then follows:
k M
k vw
2 2
ab Br
(24)
The prerequisite for the application of eq. 23 is that the initiator radical and also the growing chains
do not break. At low initiator concentration it can be taken as a good approximation. The degree of
polymerization and the corresponding molecular weights are closely related to the just defined
kinetic chain length. Assuming the validity of the reaction scheme discussed in section 3, the degree
of polymerization in chain termination is defined as follows for the combination:
Pn = 2 .
and for disproportionation:

- 31 -
Pn =
4.2 Chain transfer
One should distinguish between the chain as a term for a linear macromolecule and the chain as
reaction kinetics term; thus, the termination of the growing molecule does not also mean a
termination of the kinetic chain. The chain transfer reaction will occur when a growing chain radical
abstracts an atom from other molecules, for example, hydrogen, chlorine etc., at the same time, the
attacked molecule forms a new radical and initiates a new chain growth. The chain reaction proceeds
continuously, even though the chain growth of the first macromolecule is completed. Chain transfer
reactions can take place with initiator, polymer, monomer, solvent and the polymer radical itself, in
addition, the so-called regulator or chain transfer agent can be added for this purpose. Especially the
last three examples are of practical importance.
When such a chain transfer takes place in the polymerization reaction, an additional reaction should
be added into the reaction scheme, which [P*] is reduced without substantially affecting vBr. XQ is
generally referred to the chain transfer partner whose weakly bound atom X is transferred to the
polymer radical.
vd XQ
dtk P* XQÜ Ü
(25)
In analogy to the kinetic chain length , one defines `for the occurrence of chain transfer:
= v
v + vw
ab Ü
(26)
It includes all the monomers, which are connected by a sequence of chain growth and range from
chain starting or a transfer to the chain termination or a transfer. If no chain transfer takes place, then
´ = . For termination by disproportionation:
P n
For termination by combination it should be considered that two kinds of polymer molecules are
available:
a) Molecules, whose chain growth is terminated by chain transfer:
P n
b) Molecules, whose chain growth is terminated by combination:
P 2 n .

- 32 -
4.3 The Norrish-Trommsdorf-effect (NT-effect, gel-effect)
Following the polymerization to high conversion and assuming the validity of the rate law for
polymerization (eq. 20) we expect that, due to the reduction in monomer concentration, the overall
rate decreases linearly with conversion. The polymerization of styrene in a solvent can very well
explain these phenomena. However, the rate of the polymerization rises disproportionate, if the
polymerization is running in bulk. E. Trommsdorf interpreted this effect as follows:
During the polymerization reaction, the viscosity of the reaction mixture increases to such an extent
as a result of the formation of macromolecules that the mobility of the growing macro-radicals
becomes severely restricted and bimolecular termination is then hindered. However, the reactivity of
the chain ends remains unchanged and simultaneously the formation of the new radical via the
decomposition of initiator and the corresponding polymer radicals carry out continually; furthermore,
the unreacted monomer moves so relatively freely that the propagation reaction occurs continually,
which results in the extension of the kinetic chain. Before reaching 100% conversion, the rate of the
polymerization drops, due to the high viscosity, the monomer is also frozen, and the reaction solution
looks like a gel.
5 Radical copolymerization
By copolymerization we understand the mutual polymerization of two or more chemically different
monomers and the resulting copolymers containing repeat units of all the participating monomers.
The following discussion is limited to the copolymerization of two different monomers.
5.1 Copolymerization equation
In this section the derivation of the copolymerization equation via a kinetic approach is discussed.
For the derivation of the equation, the following assumption must be made:
1. Die polymerization is irreversible, that is to say, there are not reverse reaction in equations
28-31.
2. The total concentrations of monomers [M1] and [M2] are equal to the concentrations at the
reaction site.
3. The degree of polymerization is so high that the consumption of the monomers for
initiation, termination and transferring can be neglected.
4. The influence of the penultimate monomers incorporated in the polymer chain on the
activity of the polymer radicals is negligible.
5. The Bodenstein’ sche quasistationary state should be applied in the kinetic analysis.

- 33 -
The aim of the kinetic analysis of the copolymerization is to understand the molar incorporation ratio
m1/m2 of the monomers in the copolymer. This incorporation ratio is equal to the rate of decrease of
the monomers as a function of time:
1 11
2 2 2
d M / dt d Mm
m d M / dt d M
(27)
In the copolymerization of two monomers, there are two different polymer radicals, in which the
monomer can be deposited. It results in four possible chain growth reactions:
11k
1 1 1 1 11 11 1 1~ M M ~ M M v k ~ M M (28)
12k
1 2 1 2 12 12 1 2~ M M ~ M M v k ~ M M (29)
21k
2 1 2 1 21 21 21 1~ M M ~ M M v k ~ M M (30)
22k
2 2 2 2 22 22 2 2~ M M ~ M M v k ~ M M (31)
The properties of the polymer radicals are mainly determined by the last incorporated monomer (see
below for exception).
The concentration of monomer decreases according to eq. 28, 30 for M1 and 29, 31 for M2.
1
11 21 11 1 1 21 2 1
d Mv v k ~ M M k ~ M M
dt
(32)
2
12 22 12 1 2 22 2 2
d Mv v k ~ M M k ~ M M
dt
(33)
The concentration of the radicals is constant in the quasi stationary state (Bodenstein principle of
quasi stationarity):
1
21 12
d ~ Mv v 0
dt
(34)
12 1 2 21 2 1k ~ M M k ~ M M (35)

- 34 -
By using eq. 35, the concentration of the active species [~M2●] can be expressed via [~M1
●]. The
copolymerization equation (37) can be obtained by introduction of eq. 32, 33 and 35 into eq. 27,
concomitantly, r1, r2 are defined as copolymerization parameter.
11 221 2
12 21
k kr und r
k k (36)
1 1 1 21
2 2 2 2 1
M r M Mm
m M r M M
(37)
5.2 Influence of the penultimate incorporated monomers (penultimate effect)
If the penultimate incorporated monomer influences the reactivity of the growing chain end, the two
rate constants should be different:
111
211
k
1 1 1 1 1 1
k
2 1 1 2 1 1
~ M M M ~ M M M
~ M M M ~ M M M
That is to say, eight different propagation constants have to be considered for copolymerization of
two monomers. An impact is observed, when the last but one monomer has a strong inductive effect
on the added monomer (e.g., in the fumaronitrile/styrene system).
5.3 Discussion of the r1, r2 – value
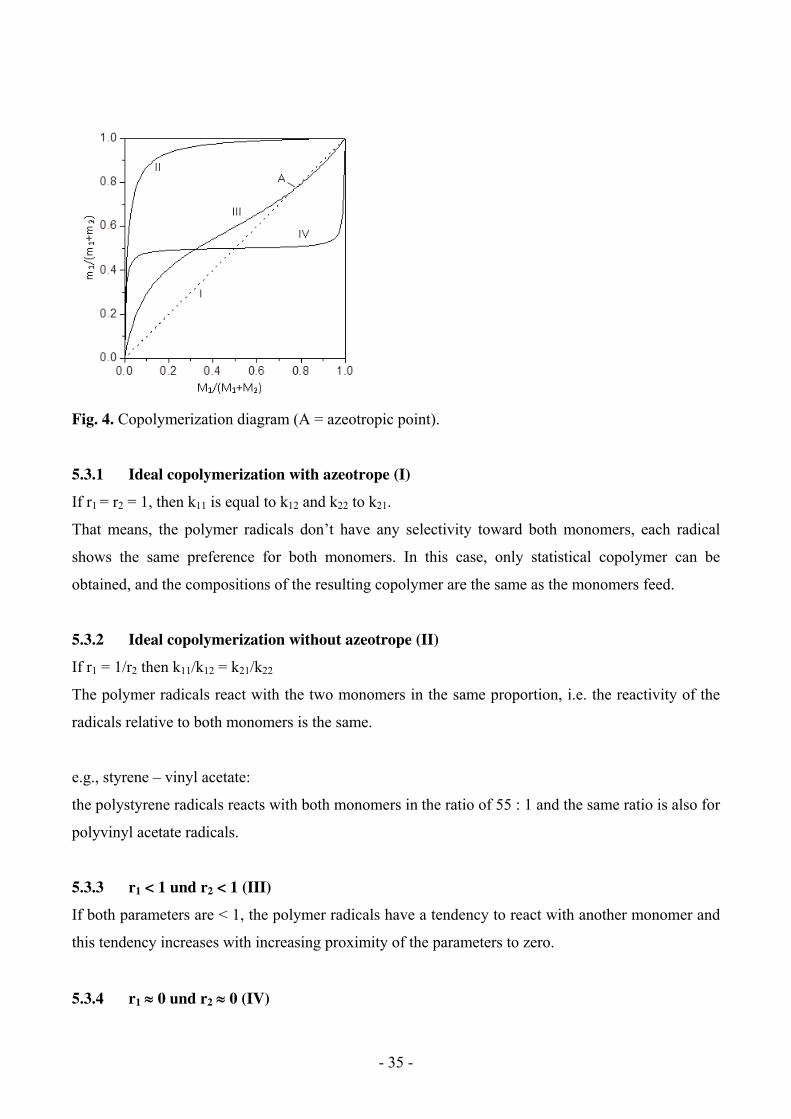
To plot the mole fraction m1/(m1 + m2) of one of the two monomeric units M1 (conversion < 5%) in
the resulting copolymer against the mole fraction of this monomer M1/(M1+M2) in the original
reaction mixture, the copolymerization diagram can be obtained as showed in Fig.4.
I Styrene r1= 1.0
p-Trimethylsilylstyrene r2= 1.0
II Styrene r1= 55 ± 10
Vinylacetate r2= 0.01 ± 0.01
III Styrene r1= 0.75 ± 0.03
Methylacrylate r2= 0.18 ± 0.02
IV Vinylether r1= 0.01
Maleic anhydride r2= 0.01
- 35 -
Fig. 4. Copolymerization diagram (A = azeotropic point).
5.3.1 Ideal copolymerization with azeotrope (I)
If r1 = r2 = 1, then k11 is equal to k12 and k22 to k21.
That means, the polymer radicals don’t have any selectivity toward both monomers, each radical
shows the same preference for both monomers. In this case, only statistical copolymer can be
obtained, and the compositions of the resulting copolymer are the same as the monomers feed.
5.3.2 Ideal copolymerization without azeotrope (II)
If r1 = 1/r2 then k11/k12 = k21/k22
The polymer radicals react with the two monomers in the same proportion, i.e. the reactivity of the
radicals relative to both monomers is the same.
e.g., styrene – vinyl acetate:
the polystyrene radicals reacts with both monomers in the ratio of 55 : 1 and the same ratio is also for
polyvinyl acetate radicals.
5.3.3 r1 < 1 und r2 < 1 (III)
If both parameters are < 1, the polymer radicals have a tendency to react with another monomer and
this tendency increases with increasing proximity of the parameters to zero.
5.3.4 r1 0 und r2 0 (IV)

- 36 -
Here the growing polymer radicals react only with the other monomer. This results in a polymer
chain, in which both monomers will be polymerized alternatively (alternating copolymer). The
polymerization usually ends if one of the monomer is completely used.
5.3.5 r1 > 1 and r2 > 1
The larger the parameter, the more easily the polymer radical reacts with its own monomers. For
very large r1 and r2 block copolymerization or simultaneous homopolymerization of both monomers
takes place. In the last case (though observed very rarely) a polymer blend is formed.
5.4 Experimental determination of the copolymerization parameters
For determining the copolymerization parameters r1 and r2, a monomer mixture of known
composition is polymerized at low conversion (<5%) in order to assume [M1] = [M1]0 and [M2] =
[M2]0. The composition of the obtained copolymer can then be determined by using analytical
methods, e.g., elemental analysis, UV-, NMR-, IR- spectroscopy, radiolabelled monomers or GC
analysis of the residual monomers. In principle, it is possible to calculate both r1 and r2 from the
composition of only two copolymers that have been obtained from two different mixtures of both
monomers. However, due to the uncertainty of the analytical methods, it is recommended to
determine the composition of the copolymers from several monomer mixtures and evaluate the
results by graphical methods.
5.4.1 Graphical determination of the copolymerization parameters according to Mayo and
Lewis
The linear relationship between r1 and r2 is obtained from the rearranged copolymerization equation
37:
2
2 21 11 2 2
2 1 21
M Mm mr r 1
m M mM
(38)
Slope and intercept of this equation are known, each copolymerization can then be characterized via
a linear relationship of r1 = f (r2) (Fig. 5). In practice, the lines for all copolymerization do not
intersect precisely at a point so that r1 and r2 are taken as the center of the smallest area that is cut or
touched by all the lines, the size of this area allows an estimate of the limits of error.

- 37 -
Fig. 5. Graphical determination of the copolymerization parameters acc. to Mayo and Lewis.
5.4.2 Graphical determination of the copolymerization parameters according to Fineman
und Ross
Eq. 37 is rearranged to give eq. 39 according to Fineman und Ross so that r1 and r2 are, respectively,
the slope and intercept of a line:
2
1
2
2
2
2
11
1
2
2
1 rm
m
M
Mr
m
m1
M
M
(39)
Each copolymerization run is shown by the points of the plot of vs. . The
fit line provides r1 as slope and r2 as intercept.
5.5 Influence of the constitution of the monomers on the copolymerization parameters
A plausible method to correlate the reactivity of monomers is based on the assumption that under the
same conditions, the rate constant k11, e.g. for styrene, is the same for all copolymerization. Then, the
reciprocal value of r1 is a direct measurement of the relative reactivity of the monomer (M2) with
respect to styrene radical. The following Table is a compilation of the r1-1 values for different
monomers (M2):

- 38 -
Table 1: Relative reactivity (r1-1) of radicals (~ M1
●) against the monomer (M2).
2M 1~ M
styrene butadiene AN MMA
2-vinylpyridin 1.82 - 8.84 2.50
2-chlorostyrene 1.79 0.85 - 2.00
4-vinylpyridin 1.61 - 8.84 1.74
4-chlorstyrol 1.35 0.69 - 2.41
styrene 1.00 0.61 25.0 2.18
α-methylstyrene 0.85 - 16.7 2.00
The following effects are taken into account for the realization of the results:
5.5.1 Resonance stabilization of monomers and polymer radicals
The overall rate of polymerization for the homopolymerization depends on the resonance
stabilization of both in the monomer and the polymer. As shown in Table 2, the stabilization ability
of radical formed after the addition of a monomer has a huge influence on the total rate of
polymerization.
Table 2: Addition rate of a polymer radical to its own monomer and the resonance of the radical and
monomers.
Monomer relative rate of
addition
Resonance stabilization energy [kcal/mol]
double bond radical
vinyl acetate 23.0 1.7 4
MMA 7.05 4.2 23
styrene 1.45 4.2 24.5
butadiene 1 6.0 25
A decrease in resonance stabilization energy results in an increase of the reactivity of the radical. A
stable radical is not reactive enough for the reaction with a double bond (which would result in the
formation of a less stable radical). For copolymerization this means that only compounds with
similar radical stabilities can react with each other. The polystyrene radical (~M1*) is not reactive

- 39 -
enough to react with vinylacetate (M2) because this would form a less stable radical. So preferebly
another styrene monomer would react with the polystyrene radical.
5.5.2 Influence of the double bond polarity on the copolymerization parameters
The polarity of a double bond has a minor influence on the total rate of the homopolymerization
compared to the radical stability. However, the polarity of double bond plays a very important role in
the copolymerization, especially, on little stabilized radical types. Taken styrene as example, one can
see that the rate of addition of monomer M2 increases (i.e. r1 becomes smaller) if there are strong
electron-accepting substituents on M2. This tendency becomes predominant in case both monomers
yield similar resonance-stabilized radicals. Here, one should also assume that the polarity of the
obtained radical is the same as that of the monomer. If the polarity of the monomers is different
enough, they can copolymerize themselves, i.e. vinyl ether with styrene or maleic anhydride. There
are two possible interpretations for this behavior:
1. At the time of addition of monomers to the growing polymer radical, the monomer will
orient in such a way that the activation energy of the addition steps become very low, if
radical and monomers are oppositely polarized.
2. The monomers are pre-oriented like a charge-transfer complex (confirmed by charge-
transfer band in the UV). By partial charge transfer, the complex will obtain a “diradical”
character. This kind of polymerization reaction can, therefore, be thought of as
polycombination reaction (i.e. vinyl ether – maleic anhydride).
5.5.3 Steric effects
Sterically hindered monomers can also copolymerize, even if they do not tend to homopolymerize.
Table 3 shows the relative rates of addition of substituted ethylenes to radicals.
Table 3: Relative rate of addition reaction of steric hindered monomers on radicals.
monomer PAN● PVAc● PS●
vinyl chloride 1.0 1.0 1.0
vinylidene chloride 3.6 10 9.2
trichlorethylene 0.05 0.45 1.0
tetrachlorethylene 0.007 0.04 0.09
The increasing rate of addition reaction from vinyl chloride to vinylidene chloride to a polystyrene
radical can be explained with the increase in resonance stabilization. The addition reaction is not

- 40 -
hindered sterically with both monomers, but the increasing steric hindrance from trichloroethylene to
tetrachloroethylene causes a decrease of the relative reaction rate, although the stability of the radical
increases. (Fig. 6).
Fig. 6: Steric hindrance of the addition reaction for a 1,2-substituted ethylene.
5.5.4 Influences of the solvent, temperature and phase relationship on copolymerization
An influence of the solvent on the copolymerization of two monomers is to be expected when a
monomer associated or the solubility of both monomers is different in heterogeneous polymerization.
The processes of association on the polymer become particularly noticeable when the polymerization
is heterogeneous and the association of both monomers is obviously different. In addition, the
association of the polymers is strongly temperature dependent. For the styrene/MMA system, there is
no association effect, so the parameters approach the value 1 at a high temperature, which means, the
selectivity of the polymer radical decreases towards the monomers.
6 Statistics of the copolymer
Due to the nature of the formation process, the length and composition of the obtained polymer chain
follow different distribution functions, the measured properties on a copolymer sample are only an
average value and do not represent the structure of a single polymer chain. Of course, it is impossible
to detect the sequence of any length and any structure by analytical means using currently available
methods. With the help of nuclear magnetic resonance, the sequence of up to 5 repeating units can
now be identified quantitatively.
6.1 Sequence distribution in copolymers
The appearing frequency of sequences with one, two, three, etc. constitutional units of the same
monomer is determined by the probability of the addition of the monomer in question in the
copolymer chain. The probability p12 for the formation of M1-M2-sequence is defined by the ratio of
the rate of addition to the sum of all possible rate of addition, as shown in eq. 40:
12 1 212
12
11 12 11 1 2 12 1 2
k M Mvp
v v k M M k M M
(40)

- 41 -
Eq. 41 is obtained if eq. 40 is divided by k12 [M1*][M2] via elimination of the unknown concentration
M1● .
12
1
1
2
1p
M1 r
M
(41)
According to the above-mentioned rule, the probability of p21 and p22 can be formulated.
Furthermore, equation 42 should be fulfilled:
p11 + p12 + p22 = 1 (42)
By calculating the probability from the r-parameter and the initial concentration of the monomers,
the relationship between the formal kinetic of the copolymerization and the statistic structure of the
copolymer chain is made.
6.2 Calculation of the average sequence length ( I1)
The frequency of any sequence length from M1 and/or M2-unit can be calculated by using the above-
mentioned probability.
The average sequence length for monomer M1 is obtained as follows:
111
12
v1
v l (43)
or can be written as eq. 44 if the rate of reaction is substituted by eq. 28-31:
11 1 1 11 1
1
12 1 1 12 1
k M M k M1 1
k M M k M
l (44)
taken
111
12
kr
k (45)
then the average sequence length 1l of monomers M1 is expressed as:
1
1 1
2
Mr 1
M
l (46)
and similarly for the average sequence length 2l
2
2 2
1
Mr 1
M
l (47)

- 42 -
6.3 Definition and calculation of the run number
The chemical microstructure of linear copolymers can also be described by the run number
introduced by Harwood. The run number R is defined as the average number of the monomers
sequence (run numbers that are composed of only one type of constitutional unit) that occur in a
copolymer molecule per 100 monomer units. According to eq. 48, the run number R can be
calculated from the mole percent of the monomers rA and rB in copolymerization runs.
A B
200R
A B2 r r
B A
(48)
7 Experimentals
7.1 Safety
All persons being exposed to chemicals have to be instructed about the effects of dangerous
substances (toxicity, point of ignition, etc.) as well as about preventive measures. Before the
experiment is carried out, read the MSDS sheets for all of the chemicals used in this laboratory and
be familiar with their safe handling. The instructions of the teaching assistant must be followed at all
times.
Instruments:
Schlenk tube, inert gas system, 4 dilatometera), 1 volumetric flask 25 mL (for solvent 1),
3 volumetric flask 20 mL, Thermostat (T = 60°C), vacuum drying oven, bakers,
oil bath with magnetic stirrer, high vacuum pump, frits with bottle
a) V0(1) = 19,86 cm3, V0
(2) = 9,98 cm3, V0(3) = 10,16 cm3, V0(4) = 10,51 cm3
(V0 = volume of the flask)
Experiment 1:
Pure styrene is polymerized to a conversion of 5 % with AIBN as initiator at 60°C. Four solutions
with different concentrations of initiator are prepared: 0.15 x 10-2 M (solution 1), 0.6 x 10-2 M
(solution 2), 1.5 x 10-2 M (solution 3) und 2.4 x 10-2 M (solution 4).
The initiator is weighed into a small volume flask and then dissolved in styrene. (Noting the exact
sample weight, the above-mentioned concentration is an „ideal concentration“, which can be
achieved approximately). The volume flask is then filled with styrene to the mark (do not fill the cool
styrene to the calibration mark). The clear solution is introduced into the dilatometer above the

- 43 -
grinding. The slighly greased capillars (mL-scale) should be placed on top very carefully. The big
dilatometer is used for the polymerization with lowest initiator concentration (solution 1). After
inserting the dilatometer into the thermostat, which was previously temperatured at 60°C, the volume
is determined every 5 min after Kontraktion has started (every 2 min for the solution with the highest
initiator concentration, solution 4). The first three solutions can be read simultaneously. The
precalculated volume, which the desired conversion is assigned about 5%, is reached after 3 h for the
lowest initiator concentration and after 1.5 h for the other initiator concentrations. The dilatometer is
then removed from the thermostat and solution 4 is dropped slowly with stirring into cold methanol
(ca. 0°C). The precipitate is filtered off, washed with methanol, and then re-dissolved in a minimum
amount of chloroform. The polymer is then precipitated once again by adding its solution slowly into
the methanol. The precipitate is filtered off and dried in a vacuum oven at 60°C.
Evaluation:
1. Determine the order of the reaction with respect to the initiator concentration:
- Determination of the reaction rate vBr for different initiator concentration from the plot [Mt]
vs. t. For which value of [M] the four curves should be theoretically cut?
- Determine the order of the reaction from the plot vBr versus [I]. Why is the obtained value
not equal to 0.5?
2. The rate constant of reaction k is determined from the log-log plot of vBr against [I].
(Ordinate = log k + log [M])
Moreover, the rate constant of the reaction k1 (for initiator concentration I1) and the
determined exponent for [I] is calculated and compared with the constant k.
3. All of the obtained or measured results at the experiments should be filled into the table for
evaluation. The corresponding fit line equation for each obtained straight line should be
given.
4. The sample prepared with the highest concentration of initiator (solution 4) is purified by re-
precipitation and prepared for GPC measurement.
Determination of conversion:
The conversion using for the determination of the rate of polymerization can be obtained by many
methods:
1. Separation and weighing of the polymer.
2. Measurement of the decrease in monomer concentration (e.g., titration, IR- and UV
measurements)

- 44 -
3. Measurement of the refractive index
4. Measurement of the volume contract (dilatometer)
The most straighforward method for the determination of conversion is to observe the volume
contraction, which is based on the difference in density between the monomer and the polymer. The
volume contraction is for a 100% conversion at 25 °C, for example, 14.1 % for styrene, 26.8% for
vinyl acetate, 23.1 % for methyl methacrylate and 25.0% for isoprene. From experience, it can be
linearly interpolated for low conversion. In addition to the high sensitivity (conversion < 1%), the
application of the dilatometric method depends in particular on the fact that the density of a polymer
does not depend on the degree of polymerization and minor structural difference. The respective
monomer concentration [M]t can be calculated from the partial density of the monomer M and
polymer P in solution:
MV V
V
10
M
mol
ltt
t
3
M-1
P-1
M
(49)
MM= 104.14 g mol-1 M= 0.924 - 9.17 x 10-4 T
Vt= volume at t P= 1.087 - 7.00 x 10-4 T
V0= volume at t = 0 T= temperature in °C
V = m V 0
P
0 M
P
Experiment 2:
Monomers are weighed into the Schlenk flask according to the below-given mixing ratio and then
0.5 mol-% AIBN are added.
Table 4: Mixing ratio of both monomers.
styrene [mL] MMA [mL]
bottle 1 2 10
bottle 2 4 8
bottle 3 6 6
bottle 4 8 4
bottle 5 10 2

- 45 -
Important: the accurate information of the mixing rate (weighing!).
For degassing, the Schlenk flask is connected to the vacuum line, frozen by using liquid nitrogen,
evacuated, and then thawed with the tap closed. The flask is filled with nitrogen and then frozen
again. The process as described above is repeated twice. (Attention: vacuum grease disturbs the
spectroscopic investigation!)
The thawing can be considerably accelerated by immersing the flask in methanol and three flasks can
be degassed at the same time. The flask is then warmed to room temperature, removed from the inert
gas system under nitrogen, and then sealed with a glass stopper. The flask is put into an oil bath,
which is kept at 50 °C by using a thermostat. (note the time!). After one hour of polymerization
(corresponding to an approximate turnover of 5-10%), the flask is then removed from the oil bath
and the polymer is precipitated by adding the solution dropwise to 150 – 200 mL of methanol, the
precipitate is filtered off, washed carefully with methanol and dried in a vacuum oven at 40°C.
7.3 Determination of the copolymerization parameters via 1H-NMR-spectroscopy
For determining the monomer units in the copolymer, 1H-NMR-spectra are recorded from the
copolymer. Both monomers have different chemical shifts; the H atoms on the phenyl ring of the
styrene are used for characterization, while the signals of the methoxy group of MMA are used for
characterization. The obtained spectra are integrated; the integral is proportional to the number of the
H atoms. The ratio of the monomers in THE copolymer can be calculated according to the integral of
H-NMR.
7.4 Evaluation of the experiments
The r1- and r2-values of both monomers can be determined from the m1/m2 integrals of different
copolymers by NMR, if eq. 38 is substituted according to Mayo – Lewis and eq. 39 Fineman – Ross
The copolymerization diagram is then created with the obtained m1/m2 – values (see Fig.4).
- 46 -
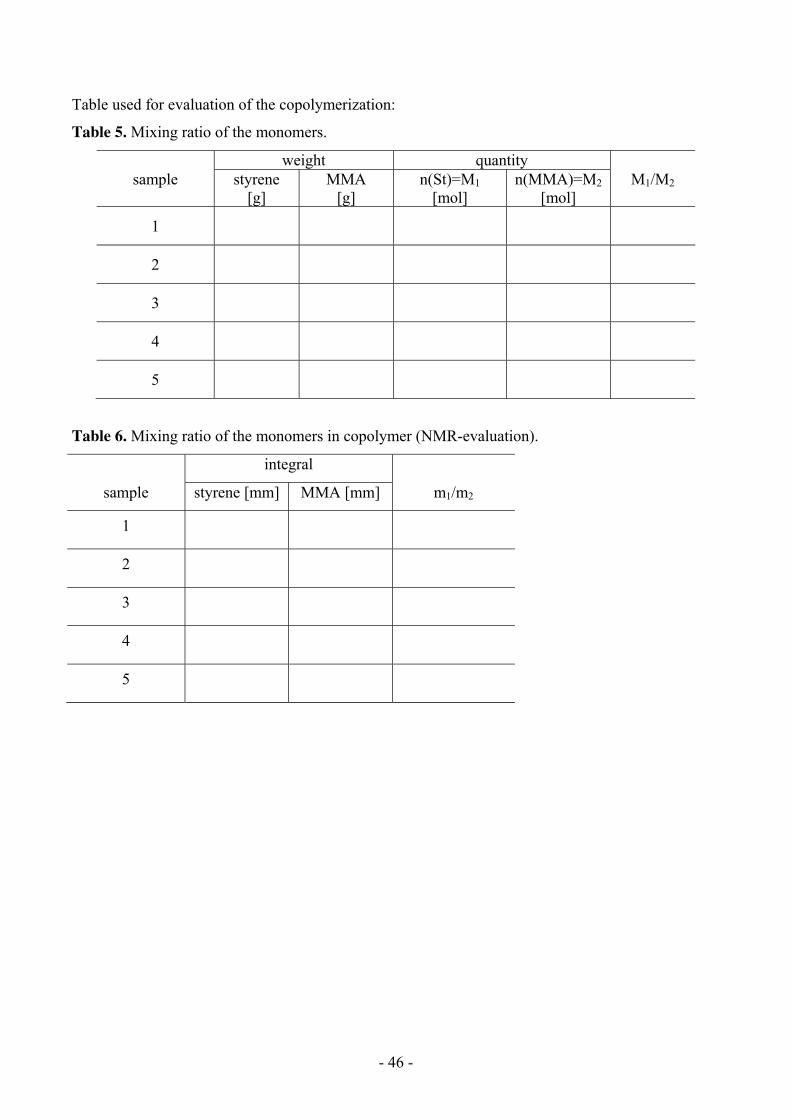
Table used for evaluation of the copolymerization:
Table 5. Mixing ratio of the monomers.
weight quantity sample styrene
[g] MMA
[g] n(St)=M1
[mol] n(MMA)=M2
[mol] M1/M2
1
2
3
4
5
Table 6. Mixing ratio of the monomers in copolymer (NMR-evaluation).
integral
sample styrene [mm] MMA [mm] m1/m2
1
2
3
4
5

- 47 -
8 Questions:
1. Discuss the presence of oxygen in the radical polymerization. Is it possible to use it as an
initiator?
2. Explain the term ceiling-temperature? Does a floor-temperature exist too?
3. Under which conditions is the relation vw [I]0.5 false?
4. Draw the diagrams for vBr against conversion and P against conversion (in solution). How
do the same diagrams for polycondensation and ionic polymerization look like? ( P against
conversion)
5. How does the termination rate change in the case of the NT-effect?
6. The activation energy of the decomposition of AIBN is ca. 30 kcal/mol. For the activation
energy the gross rate of the polymerization of styrene is ca. 20 kcal/mol. How does the gross
rate and the degree of polymerization change at low conversions if you decrease the
temperature from 40 to 20 °C (neglect side reactions)?
7. What is a block copolymer and graft copolymer and how would you synthesize them?
8. What is an „ideal“ copolymerization and how does the copolymerization diagram look like?
9. What consequences would you expect for the structure of a copolymer for the following r-
parameters:
r1 = 1.00 r2 > 1.00
r1 = 1.00 r2 < 1.00
r1 0 r2 0
r1 r2 .
10. What composition of a copolymer is expected for r1 0 and r2 0?
11. List methods for the experimental determination of the composition of copolymers?
12. Which values for r1 and r2 are needed for the formation of an azeotrope?
13. Are there structural differences between polymers made by radical or anionic
copolymerizations (at which r1 and r2)?
14. What extremes in the sequence distribution are possible in copolymers?
15. List requirements for a statistical derivation of the copolymerization equation?
16. How does the copolymerization diagram for r1 > 1 and r2 > 1 look like?
17. What is an alternating copolymerization?
18. Why do you observe several signals in the 1H-NMR spectrum for the methoxy group in a
copolymer (only 1 signal in the PMMA monomer spectrum)?

- 48 -
19. Determine the average sequence length and the number of blocks for the monomers styrene
and vinylidenechloride (r1=2.0, r2=0.14). Monomer concentrations are 30 mol% styrene and
70 mol% vinylidenechloride. Explain.

- 49 -
Rheology
1 Introduction .............................................................................................................................. 50
2 Theoretical foundation ................................................................................................................ 50
2.1 Fundamental terms in rheology............................................................................................ 50
2.2 Linear-viscoelastic behavior ............................................................................................... 52
2.3 Measuring technique ......................................................................................................... 55
2.3.1 Oscillation measurements ...................................................................................... 55
2.3.2 Rotational experiments .......................................................................................... 56
2.3.3 Tension experiment .............................................................................................. 57
2.3.4 Relaxation experiment ........................................................................................... 57
2.4 Borderline behavior of matter and rheological models .............................................................. 57
2.4.1 Elastic behavior ................................................................................................... 58
2.4.2 Plastic behavior .................................................................................................... 59
2.4.3 Viscous behavior .................................................................................................. 60
2.4.4 Viscoelastic behavior ............................................................................................ 61
2.4.5 Characterization by flow and viscosity curves ............................................................ 63
2.5 Time-dependent rheological behavior ................................................................................... 64
2.6 Rheometry (measuring technique) ........................................................................................ 65
3 Experimental ............................................................................................................................ 66
3.1 Conduction of rotational measurements ................................................................................. 66
3.2 Time-dependent change of viscosity ..................................................................................... 67
3.3 Oscillation measurements to determine the viscoelastic behavior ............................................... 67
4 Questions ................................................................................................................................. 68
5 Literature ................................................................................................................................. 68

- 50 -
1 Introduction
Rheology is the science which describes, explains, quantifies and applies the phenomena appearing
while bodies or liquids are deformed or while they flow. According to Ziabicki [1], the rheological
behavior is responsible for the drawability and thus of fundamental importance for the spinnability of
liquid systems. Further examples of applied rheology are found in various areas in natural science
and engineering. [2-7]
1. Colors and varnish (brushability, storage)
2. Polymer solutions and melts (polymer extrusion and spinning
3. Characterization of polymers (statement about molecular weights and molecular weight
distribution
4. Manufacture of high performance materials (ceramics)
5. Flow behavior of food (ketchup, convenience sauce)
6. Cosmetics and sanitary products (tooth paste, cream, shampoos)
7. Geo-rheology (simulation of volcanic flow)
8. Medicine (hemorheology)
9. Pharmaceutical products
10. Electronics
In the following paragraphs the basic principles of rheology, which are described in literature [1 –
20] are addressed.
2 Theory
2.1 Fundamental terms in rheology
To characterize the flow behavior of substances, they are subjected to defined forces and the
resulting deformations are described in detail in dependence of different parameters. Depending on
the direction of the affecting force, the relevant cases for rheometry are distinguished: elongation,
compression strain and shear strain. The effect occurring during the shear experiment of a liquid and
the associated fundamental terms in rheology can be explained with the aid of a two-plate-model
(Figure 1). In this model a liquid is located between two parallel plates of the area A. The upper plate
is moved relative to the lower static plate with a constant velocity. Thereby the power F needs to be
applied due to the internal friction. This shear strain causes a laminar flow of the velocity v which
linearly decreases from the moving to the static plate.

- 51 -
Fig.1: Two-plate-model. The resulting shear rate gradient is also called deformation velocity or shear velocity:
dt
d
dy
dx
dt
d
dt
dx
dy
d
dy
dv
(1)
velocity gradient = shear rate
Deformation: tandy
dx [-] (2)
Deformation velocity: dt
d [1/s] (3)
When the force applied during the shearing experiment is related to the plate area A, the shear stress
is obtained, which is connected with the deformation velocity by the shear viscosity .
Shear stress: A
F [Pa] (4)
Stationary shear viscosity:
[Pa·s] (5)
According to the dependence of the stationary shear viscosity on the shear rate, flow behavior can be
characterized as Newtonian, shear thinning and shear thickening (dilatant). Newtonian flow
behavior is present, when the viscosity is independent of the shear rate. If the viscosity decreases
with increasing shear rate, the behavior is called shear thinning, this is typical for polymer melts

- 52 -
and polymer solutions. When the viscosity increases with increasing shear rate the flow behavior is
referred to as shear thickening. Examples of viscosity curves are given in 2.4.5.
For a Newtonian liquid, a direct proportionality exists between shear stress and deformation rate,
whereas for an ideal Hook-type body a direct proportionality between shear stress and deformation is
present, i.e. Hook’s law is in force. The proportionality constant is the modulus of shear G:
Shear stress: G [Pa] (6)
It should be noted, that also elastic systems exist which to not apply to Hook’s law (e. g. rubbery-
elastic materials). A general definition of elastic behavior is given in 2.4.1.
2.2 Linear-viscoelastic behavior
Many materials do not show exclusively viscous or elastic behavior but a combination of these
characteristics. This is referred to as viscoelastic behavior. The theory of linear viscoelasticity
describes the rheological phenomena of polymer solutions and melts, which is connected to the
preservation of the resting structure/state of the temporary network of entangled molecular chains.
The theory of non-linear viscoelasticity describes viscoelastic phenomena, which are connected with
depletion of the temporary network structure. The rheological material value functions for describing
linear viscoelasticity are determined with the aid of oscillatory experiments. Thereby, the latent state
of the sample is not disturbed, which permits to separately display viscous and elastic behavior. At a
deformation-controlled oscillatory measurement, the sample, which is located in a gap between two
plates, is loaded by a periodic deformation (t) and thereby a periodic strain (t) is induced, which
shows a displacement of phase relative to the preset deformation.
preset deformation resulting strain (response of the system)
}cos{0 t }cos{0 t
radial frequency of the oscillation [rad/s]
strain amplitude [Pa] deformation amplitude [-]
Based on the displacement of phase δ, rheological behavior can be classified:
elastic behavior: 0

- 53 -
viscous behavior: 2
viscoelastic behavior: 2
0
Fig. 2: Course of strain and deformation during oscillatory measurements.
To simplify rheological calculations, complex values are introduced. The transition to complex
values is performed by extending strain and deformation with the respective imaginary part. This
allows the transition from trigonometric functions to the complex e-function, thus simplifying
calculations considerably. The following rheological values are defined:
Complex shear strain:
)}(exp{})sin{}(cos{* 00 titit (7)
Complex deformation:
}exp{})sin{}(cos{* 00 titit (8)
Complex deformation velocity:
}exp{*)(* 0 tiidt
d
(9) Viscoelastic behavior can be described by the complex modulus G*, which is defined as the quotient
of complex strain and complex deformation.

- 54 -
Complex modulus:
'''}exp{*
**
0
0 GiGiG
(10)
Here, G’ is the storage modulus and G’’ is the loss modulus. The storage modulus represents the
degree of elastic behavior, i.e. the energy, which is reversibly stored by restoring forces. The loss
modulus represents the amount of viscous behavior, i.e. the energy which irreversibly dissipated due
to viscous flow. For G’ and G’’ applies:
Storage modulus:
cos'0
0 G [Pa] (11)
Loss modulus:
sin''0
0 G [Pa] (12)
The ratio of loss and storage modulus is the dissipation factor :
Dissipation factor: '
"tan
G
G [-] (13)
The dissipation factor is used to estimate, whether the viscous or elastic behavior is dominating. The
following classification is used:
tan < 1: elastic behavior dominates
tan > 1: viscous behavior dominates
Based on equation (10), it accounts for the absolute value of the complex modulus:
0
022 '''*
GGG [Pa] (14)
The absolute value of the complex modulus corresponds to the ratio of stress amplitude and
deformation amplitude.
The viscoelastic behavior may also be described with help of the complex viscosity *, which is
defined as the quotient of complex sheer stress * and complex deformation velocity * .
Complex viscosity:

- 55 -
'''*
}exp{*
**
0
0
i
i
Gi
i (15)
’ is a measure for the viscose behavior, ’’ describes the elastic behavior.
For ’ and ’’ accounts:
'
cos''0
0 G
[Pa·s] (16)
''
sin'0
0 G
[Pa·s] (17)
It results for the absolute value of complex viscosity:
0
0
0
022
22 ''''''*
GG [Pa·s] (18)
Accordingly, the absolute value of complex viscosity corresponds to the ratio of stress amplitude and
deformation rate amplitude.
2.3 Measuring technique
2.3.1 Oscillation measurements
Oscillation measurements are used to study the linear-viscoelastic behavior. In the rheometers used,
the sample is located in a gap between an upper plate or cone and a lower plate. This setup is also
called plate-plate or cone-plate geometry. (In the following sections, measuring geometries will be
further explained.) In principle, both geometries are suitable for oscillation measurements.
2.3.1.1 Amplitude sweep (= strain sweep and stress sweep)
During the amplitude test, the frequency is held constant and the amplitude of the deformation signal
or the strain signal is varied, depending on whether the following frequency measurement is
supposed to be conducted by deformation control or by strain control. If the amplitude is not too
high, the rheological material value functions (i.e. G‘() and G‘‘()) do not show any dependence
on the amplitude. This measurement range is called region of linear viscoelasticity. In this range, the
idle state of the sample is not disturbed. Starting from a specific amplitude value, the rheological
material functions G‘() and G‘‘() decrease with increasing amplitude. In this range the laws of
non-linear viscoelasticity are applicable; the idle state of the sample is disturbed. For solutions and
melts of polymers a disentanglement of entangled (verhakt) molecular chains occurs.

- 56 -
2.3.1.2 Frequency sweep
Within the frequency test, the radial frequency ω is varied, whereas the deformation amplitude 0 is
held constant in case of deformation-controlled oscillation experiments and the strain amplitude 0 is
held constant for strain-controlled experiments. Concerning an optimum in signal to noise ratio, the
highest possible deformation or strain amplitude is chosen from the rheograms of the previously
performed amplitude tests, which is just in the linear viscoelastic area. Usually, storage modulus
, loss modulus and the absolute value of complex viscosity I*I are measured and
drawn against radial frequency ω in a double logarithmic reference frame, because the rheological
material value functions change with radial frequency over several orders of magnitude.
2.3.1.3 Time sweep
Time sweep is carried out at constant amplitude 0 or 0, respectively and constant angular velocity.
The time-dependent behavior of the rheological material value functions is observed. Thus, changes
in the material properties over time can be recorded by using rheology. One example is the
thickening process in the manufacture of gels or the stability of polymer solutions against gelling.
2.3.1.4 Temperature sweep
A temperature ramp at constant angular frequency and deformation is applied. The temperature
dependent measurement is illustrated by a semi-logarithmic plot of storage and loss modulus as a
function of temperature. With the aid of a temperature sweep, for example, glass transition
temperature and crystallization temperature of polymers can be determined. This technique is often
called dynamic mechanical thermal analysis (DMTA).
2.3.2 Rotational experiments
Rotational experiments offer another possibility to either preset strain or deformation rate. In a
strain-controlled experiment, the resulting deformation rate is received as the answer of the system,
in deformation-controlled experiments, respectively, the resulting strain is obtained. Rotational
experiments are usually conducted with cone/plate geometry, because with this measuring system,
the deformation rate is independent of the distance r to the middle of the plate and thus constant.
Exclusively rotational experiments are performed to record flow curves (plot of against ) or
viscosity curves (plot of against ).

- 57 -
2.3.3 Tension experiment
This experiment is a rotational experiment in which the time-dependent behavior of rheological
quantities is recorded. Here, a constant deformation rate is preset and the resulting strain signal is
measured as a function of time. In the ideal case of the experiment, the angular frequency escalates
from the idle state to a constant value. Viscoelastic liquids exhibit a delayed increase in shear stress.
If the measured time-dependent shear stress is correlated to the preset constant deformation rate, the
time-dependent (transient) viscosity +(t) is obtained:
)()(
tt
[Pa·s] (19)
In the case of viscoelastic fluids the transient viscosity approximates a boundary value with
progressing measurement time. This boundary value is called stationary (= time-independent) shear
viscosity :
)(),(lim
tt
[Pa·s] (20)
2.3.4 Relaxation experiment
In the relaxation experiment, the sample is stressed by an escalating deformation o and the resulting
strain signal (t), which decreases with time, is measured. Due to its elastic restoring force a strain is
produced by the preset deformation. A relaxation process within the sample takes place by viscous
flow and the generated strain diminishes. If the strain signal is correlated with the preset
deformation, the nonlinear-viscoelastic relaxation modulus is obtained, with is dependent of time and
the existent deformation:
Relaxation modulus: 0
0
)(),(
t
tG [Pa]
(t): strain [Pa] o: deformation [-]
2.4 Borderline behavior of matter and rheological models
In rheometry material-specific dependencies of the above mentioned interactions of applied shear
stress and resulting shear deformation are obtained. Thereby, the material’s behavior is distinguished
by the following properties: viscous, elastic, viscoelastic and plastic behavior. These behaviors of
material are sketched in Figure 3. All liquids can be described with the aid of viscosity.
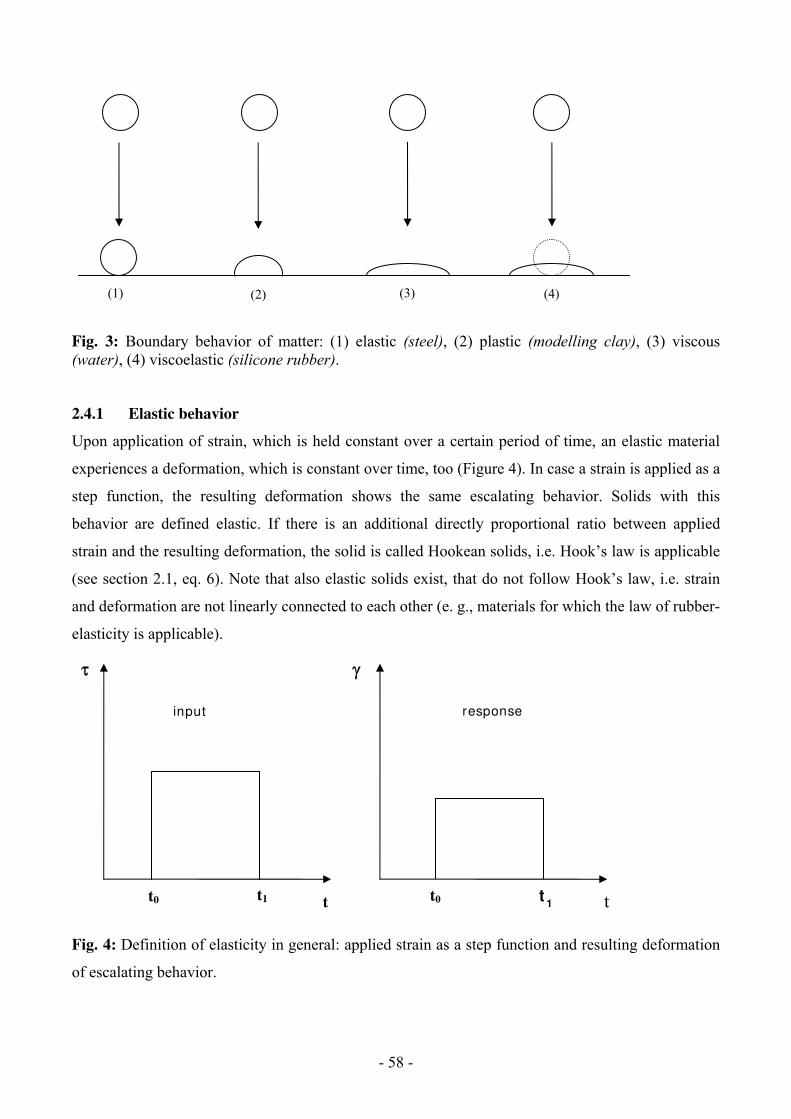
- 58 -
Fig. 3: Boundary behavior of matter: (1) elastic (steel), (2) plastic (modelling clay), (3) viscous (water), (4) viscoelastic (silicone rubber).
2.4.1 Elastic behavior
Upon application of strain, which is held constant over a certain period of time, an elastic material
experiences a deformation, which is constant over time, too (Figure 4). In case a strain is applied as a
step function, the resulting deformation shows the same escalating behavior. Solids with this
behavior are defined elastic. If there is an additional directly proportional ratio between applied
strain and the resulting deformation, the solid is called Hookean solids, i.e. Hook’s law is applicable
(see section 2.1, eq. 6). Note that also elastic solids exist, that do not follow Hook’s law, i.e. strain
and deformation are not linearly connected to each other (e. g., materials for which the law of rubber-
elasticity is applicable).
Fig. 4: Definition of elasticity in general: applied strain as a step function and resulting deformation
of escalating behavior.
t t0 t1
t t1
responseinput
(1) (2) (3) (4)
t0

- 59 -
The behavior of a Hookean solid is described by a spring for which Hook’s law is applicable.
Fig. 5: Spring as the mechanical model for elastic behavior.
2.4.2 Plastic behavior
Plastic behavior is characterized by a flow behavior with an existing flow limit. The flow limit is the
strain value under which no or only elastic deformation occurs. Above this limit a permanent
deformation appears. Plastic behavior is illustrated with the Saint Venant body. This mechanical
model consists of a slider, which only moves if the applied force overcomes the resistance of static
friction.
Fig. 6: Shearing of a plastic body as a consequence of strain; for deformation a certain shear stress is
necessary, then, shear stress is constant.
Fig. 7: Saint Venant body as a mechanical model for plastic behavior.
0

- 60 -
2.4.3 Viscous behavior
Viscous behavior is found for ideal liquids, the so-called Newtonian liquids. For the definition of the
stationary shear viscosity the previously described two plate model can be used, for which a
laminar flow is induced in the liquid between two plates as a consequence of the applied shear.
Because of the inner friction of the liquid, the layers only move partially against each other.
Assuming that a laminar flow is present, a velocity gradient is formed in the liquid between the two
plates. As described in section 2.1, for Newtonian liquids, there is a direct proportionality between
shear stress and shear rate, where the constant value is called shear viscosity.
Fig. 8: Applied strain as a step function and resulting deformation for Newtonian liquids.
The viscous flow behavior of a Newtonian liquid is described by the mechanical model of an
attenuator. At a constant affecting shear force, force and piston speed are proportional. The piston
immediately stops at the position where it was, when the force effect is finished. Therefore the
deformation of the liquid fully persists even when the force is relieved.
Fig. 9: Attenuator as a mechanical model for viscous flow behavior.
t t0 t1
t t1
response
t0
input

- 61 -
2.4.4 Viscoelastic behavior
As described in section 2.2 many materials do not show exclusively viscous or elastic behavior, but a
combination of both properties. Depending on the type of strain that is applied to the material, the
respective components of viscoelastic flow behavior appear more or less pronounced. Rheological
material value functions used for describing viscoelastic flow behavior are given in section 2.2.
Viscoelastic behavior can be described by mechanical models where spring and attenuator are
combined. For describing the behavior of viscoelastic liquids (polymer melts or concentrated
polymer solutions) one or several Maxwell elements are used. A Maxwell element is a series of
connected springs and attenuators. If a force is applied in form of a step function, the result is a
spontaneous deflection (elastic behavior). Then, the effect of the attenuator appears (viscous
behavior). The initial position is not reached again. This model describes the ideal behavior of
viscoelastic liquids.
Fig. 10: Applied strain as a step function and resulting deformation for a simple Maxwell element.
Fig. 11: Maxwell element as a simple mechanical model for viscoelastic liquids.
For many viscoelastic liquids this model does not describe the flow behavior adequately enough.
Only by parallel connection of several Maxwell elements viscoelastic liquids can be described with
satisfactory accuracy. This approach is called generalized Maxwell model.
t t0 t1
t t1 t
0
spring
at tenuator
regression of
the spring
input response

- 62 -
Fig. 12: Generalized Maxwell model for improved description of the behavior of viscoelastic liquids.
For viscoelastic solids, the viscoelastic behavior is described by the Kelvin Voigt model. Within this
model, spring and attenuator are connected in parallel. The deformation occurs as long as the
straining force is acting with constant intensity. Both components can only be deformed at the same
time and to the same degree, because they are connected by a fixed frame. The spring cannot be
deformed in the same spontaneous way as it would if it was a single spring with liberty of action,
because it is retarded by the attenuator. As a result of the strain period, deformation behavior is
observed as a curved, time-dependent e-function in the (t)-graph having deformation values rising
to a certain maximum value. Accordingly, the spring tends to move back to its initial state when the
force is released. This energy effects that both components reach their initial positions. However, this
happens after a certain time. Due to the presence of the attenuator, this is a time-delayed process, too.
Fig. 13: Strain input as a step function and resulting deformation for the Kelvin Voigt model.
t t0 t1
t t1 t
0
responseinput
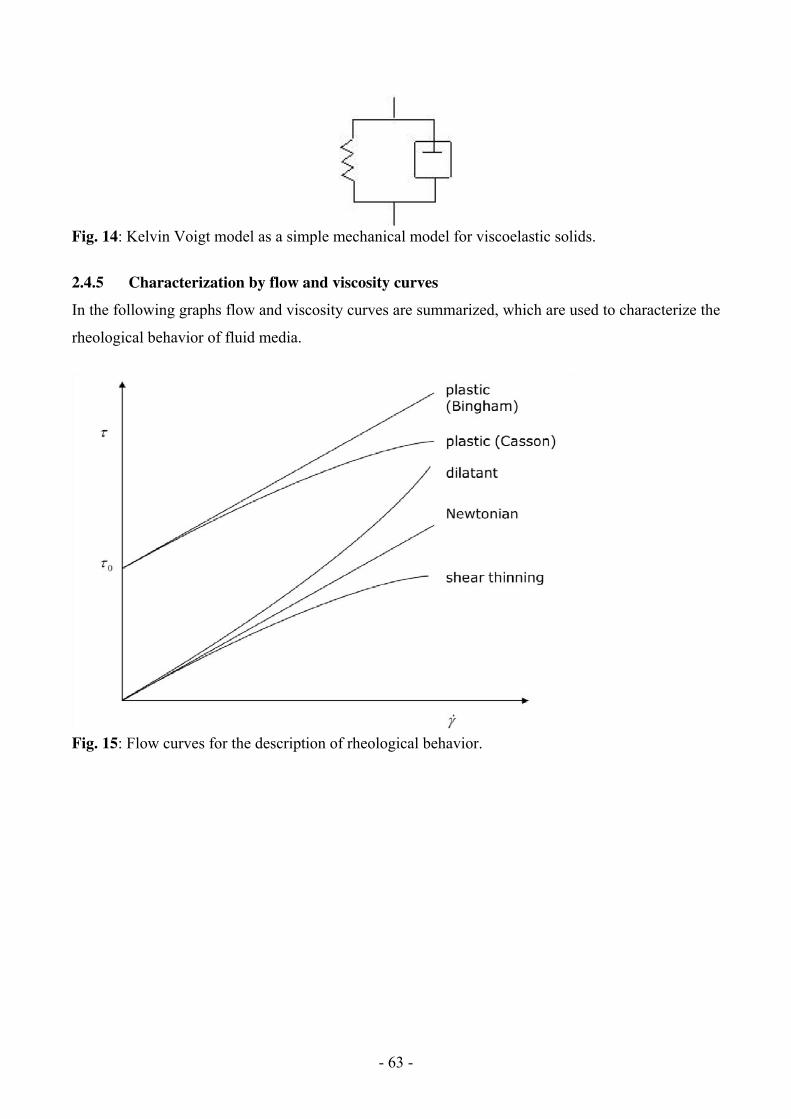
- 63 -
Fig. 14: Kelvin Voigt model as a simple mechanical model for viscoelastic solids.
2.4.5 Characterization by flow and viscosity curves
In the following graphs flow and viscosity curves are summarized, which are used to characterize the
rheological behavior of fluid media.
Fig. 15: Flow curves for the description of rheological behavior.

- 64 -
Fig. 16: Viscosity curves for the description of rheological behavior. For a shear thinning substance, viscosity is dependent on the extent of shear strain. With increasing
strain, the flow curve exhibits a negative slope, viscosity decreases, respectively. In the consequence
of shearing, a structural change is induced, which results in the decrease of viscosity. For the filled
systems, the arrangement of the particles is in favor of the lowest possible flow resistance. Thereby,
the arrangement significantly relies on the underlying structure of the deformed material.
Fig. 17: Effect of shearing on the structure of shear thinning materials.
2.5 Time-dependent rheological behavior
To study the time-dependency of structure disassembly and assembly, experiments are conducted
during which the deformation rate is preset as a step function and viscosity is measured in
dependence of time. By such measurements it is possible to distinguish between thixotropic

- 65 -
(structure disassembly) and rheopectic (structure amplification) behavior, whereat these notations
may only be used in case of fully reversible and isothermally occurring processes. If the disassembly
of structure is irreversible, the behavior is denoted non-thixotropic. These time-dependent
deformation phenomena are defined as follows:
Thixotropy:
Thixotropy describes the disassembly of a structure at constant shear strain and complete reassembly
of the structure after a certain period of time. This disassembly/reassembly cycle is a completely
reversible process. Well-known examples for materials with thixotropic behavior are, e.g.,
dispersions like paste, creams, ketchup, lacquer, etc.
Non-thixotropic behavior:
If the reassembly of structure is incomplete or not happening even after a long period of recovery,
the shear strain induces a permanent change in the structure. This effect is sometimes called “unreal”
or “incomplete” thixotropy. A very prominent example for this effect is mixing up yoghurt. After
mixing, the yoghurt is flowing much more than before, even after a long relaxation period.
Rheopecty:
Rheopecty means an increase in structural strength during shear strain, i.e. assembly of structure
during constant shearing and complete disassembly after relaxation. This assembly/disassembly
cycle is a completely reversible process.
2.6 Rheometry (measuring technique)
For conducting rheological studies, rotational rheometers of different geometries can be used. Each
measuring geometry is used for a different value of viscosity. The most common geometries are:
Plate-plate-geometry:
A plate-plate meassuring system consists of two even plates. Ususally the upper plate is the rotor and
thus the movable part of the meassuring geometry (“measuring plate”) und the lower plate is fixed on
the rheometer stand. The geometry is determined by the plate’s radius R. A disadvantage of this
geometry is, that even at a constant rotational speed, the deformation speed – viewed over the entire
plate gap – is not constant but depends on the distance r to the middle of the plate. That is, a radius-
dependent shear rate distribution exists. These non-uniform, non-constant shear conditions are seen
as a disadvantage for scientifically working rheologists, especially when performing exclusively

- 66 -
rotational experiments. These experiments should be conducted with cone-plate geometry for the
above mentioned reasons. However, this disadvantage is of low relevance when determining
rheological material value constants of linear viscoelasticity (that is G’, G’’ and I*I), for this reason,
the plate-plate geometry is often used for performing oscillation measurements, especially when the
sample is analyzed at different temperatures or, if the sample is filled with particles (e. g., filled
polymer melts or dispersions with relatively large particles). Plate-plate geometry allows for larger
gap positions, thus, phenomena which negatively affect the measured values, like thermal expansion
of the measuring tools or friction effects due to incorporated particles in filled systems could be
minimized.
Cone-plate geometry:
A cone-plate measuring system consists of a round measurement body with a slightly tilted, slightly
cone-shaped surface and a plate. Usually, the cone is the rotor and hence, the upper, moveable part of
the measuring geometry and the lower plate is unmovably fixed on the rheometer stand. The
dimensions of the conical surface are determined by the cone’s radius R and the cone angle α. A
major advantage of the cone-plate geometry is that with this measurement system, the deformation
rate is independent of the distance r to the middle of the plate. Because of this relationship, cone-
plate geometry is especially recommended for performing rotational experiments.
Coaxial cylinder geometry:
Coaxial cylinder measuring systems consist of a measurement body (inner cylinder) and a measuring
cup (outer cylinder). Coaxial means that both cylindrical components are located along one identical
rotationally symmetric axis when the system is in working position. These measuring systems are
especially used for studying low viscous liquids. According to the operating mode, two system types
are distinguished. In the Couette system, the outer cylinder is rotating whereas in the Searle system,
it is the inner cylinder.
3 Experimental
All experiments are conducted with a cone-system (Ø=50 mm, cone-plate-geometry). Temperature
control of the plate is conducted by a Peltier element. Unless stated otherwise, all experiments are
carried out at a temperature of T = 25 °C.
3.1 Conduction of rotational measurements

- 67 -
The following samples should be classified according to their characteristic flow behavior. The
respective flow phenomena and the underlying structure are to be discussed based on the measured
flow and viscosity curves.
Sample 1: Honey
Shear rate: = 10-1 to 103
Sample 2: 14 wt.-% PVP-solution (H2O) (Mw = 1,300,000 g/mol)
Temperature: T = 20°C
Shear rate: = 100 to 103
Sample 3: starch/H2O suspension (50 wt.-%)
Shear rate: = 10-1 to 102
3.2 Time-dependent change of viscosity
Time-dependent measurements at predetermined deformation rates are performed and the
characteristic evolution of the viscosity function depending on time is discussed. The observed effect
is to be discussed with regard to the underlying effects.
Sample 4: Ketchup
Measuring program: From t = 0 to 3 s: = 0.1
From t = 3 to 6 s: = 100
From t = 6 to 250 s: = 0.1
3.3 Oscillation measurements to determine the viscoelastic behavior
Two oscillation experiments are preformed and the measured curves are discussed. The highest
possible deformation amplitude is chosen from the viscoelastic region and used as a constant
parameter (default value) for the following frequency test.
3.3.1 Oscillation measurement by varying the deformation amplitude at a constant angular
frequency (amplitude test)
Sample 5: 20 wt.-% PVP-solution (H2O) (Mw = 360,000 g/mol)

- 68 -
Temperature: T = 20°C
Measuring program: angular frequency: ω = 10 rad/s
Deformation amplitude from = 1 % to 1000 %
Illustration of G’, G’’ and as a function of the deformation amplitude and choice of the
deformation amplitude from the measured curves for the subsequent oscillation measurement (from
linear viscoelastic to non-linear viscoelastic behavior).
3.3.2 Oscillation measurement by varying the angular frequency
Temperature: T = 20°C
Measuring program: Use of the deformation amplitude determined in 3.3.1 as a default
Angular frequency from ω = 100 to 0.1 rad/s
Illustration of G’, G’’ and as a function of angular frequency. Explain the rheological behavior
based on the depicted curves (viscosity, viscoelasticity and elasticity). Describe the structural events
at the transition between the borderline cases.
4 Questions
1. Term other applied examples for the determination of materials’ viscosity.
2. Sketch the most important flow and viscosity diagrams.
3. Describe the advantages and disadvantages of plate/plate and cone/plate geometry.
5 Literature
1. M. Dragoni, A. Tallarico, J. Volcanol. Geoth. Res. (1994), 59, 241
2. G. Miyamoto, S. Sasaki, Computers & Geosciences (1996), 23, 283
3. G. B. Thurston, Biophys. J. (1972), 12, 1205
4. W.P. Cox, E.H. Merz: J. Polym. Sci. (1958), 28, 619
5. G. Böhme, M. Stenger: Chem. Eng. Technol. (1988), 11, 199
6. G. V. Vinogradov, A. Y. Malkin, Y. G. Yanovsky, E. A. Dzyura V. F. Schumsky, V. G.
Kulichikhin: Rheol. Acta 8 (1969), 490-496
7. G. V. Vinogradov, N. V. Prozorovskaya: Rheol. Acta 3 (1964)
8. G. V. Vinogradov, A. Y. Malkin: J. Polym. Sci. A: Polym. Chem. (1964), 2, 2357
9. H. M. Laun: Progr. Coll. Pol. Sci. (1987), 75, 111

- 69 -
10. H. M. Laun: Rheol. Acta (1978), 17, 1
11. J. D. Ferry: Viscoelastic properties of polymers, John Wiley & Sons, Inc. (1970)
12. L. E. Nielsen: Polymer Rheology, Marcel Dekker, Inc., New York – Basel (1977)

- 70 -
POLYINSERTION and ROMP
I Polyinsertion
Assignment of tasks
Synthesis of isotactic polystyrene (it-PS) with the heterogeneous Ziegler-catalyst TiC14/(iBu)3A1 in
heptane as solvent.
Literature
1. H. G. Elias: Bd. 1, 1. Auflage, Wiley-VCH-Verlag, Weinheim, 2005.
2. Compr. Polym. Sci., Vol. 4, Part II, 1. Aufl., Pergamon Press GmbH 1989
Content
1. Theoretical basics
1.1. Ziegler-Natta catalysts
1.2. Mechanisms
1.3. Industrially produced polymers
2. Description of the experiment
2.1. Safety
2.2. Chemicals and equipment
2.3. Experimental procedure
3. Questions

- 71 -
1 Theoretical basics
1.1 Ziegler-Natta catalysts
In the last few years, transition metal-based catalysts have gained great importance, especially for the
polymerization of ethylene at low pressures, as well as for the synthesis of polypropylene.
Furthermore, they offer a wide range of applications in the preparation of stereoregular polymers.
Among the technically most important catalysts in this area are the so-called Ziegler-catalysts (also
named Ziegler-Natta-catalysts). Systems, which are made by mixing organometallic compounds of
transition group elements IV-VIII with metals alkyls or metals hydrides of the groups I – III of the
periodic system, are referred as Ziegler-catalyst. A typical Ziegler-catalyst is formed, for example,
by the reaction of TiCl4 with Et3Al. These catalysts is heterogeneous, because they precipitate as a
fine suspension in an organic solvent (for example from heptane). At the beginning of catalyst
research, this heterogeneity was assumed to be responsible for catalyst activity (“catalytic surfaces”),
however, Kaminsky et al. found that also soluble (homogeneous) systems display similar activities.
Such homogeneous Ziegler-catalysts can be generated through the combination of, e.g.,
bis(cyclopentadienyl)titanium(IV) dichloride (Cp2TiCl2, a metallocene catalyst) with
diethylaluminiumchloride (Et2AlCl).
A few further important catalytic systems:
Et2AlCl/TiCl3 heterogeneous
Et2AlCl/VCl4/anisole homogeneous
Et2AlCl/V(acac)3 homogeneous
Et2AlCl/Cr(acac)3 homogeneous
Cp2ZrMe2/alumoxane homogeneous
Besides the Ziegler-catalyst, the so-called "Phillips-catalyst" (CrO3/SiO2/A12O3) is of technical
importance, especially in the polymerization of ethylene.
1.2 Mechanisms
The polymerization occurs at the transition metal-carbon bond. The active species and the reaction
mechanism are, with the exception of Phillips-catalysts, mostly clarified. The growth step can
principally take place over a mono- or bimetallic mechanism.

- 72 -
Monometallic mechanism
In the so-called monometallic mechanism (Fig. 1) it is assumed that the olefin approaches first to a
vacant site of the transition metal with its π-bond, and then coordinates to it. The coordinated
monomer is then inserted into the metal-carbon bond via a four-membered transition state.
Fig. 1: Monometallic mechanism.
The main group metal complex is not involved into the chain growth reaction; it is only used for the
alkylation of the transition metal complex ("monometallic"). There are (idealized) separated ion
pairs. In fact, however, these ion pairs are in reality never completely separated from each other, and
the termination and transfer reactions can be easily explained via the following reaction scheme:
Termination caused by β-hydride elimination:
Transfer to the monomer:
Molecular weight control by H2:

- 73 -
Bimetallic mechanism
In the bimetallic mechanism (Fig. 2) both metal atoms are involved in the reaction. The π-electron of
the olefin interacts first with the orbitals of the transition metal, wherein an electron-deficient
compound is formed. The free rotation of the only partially resolved C-C double bond remains
rescinded, and there is also no free rotation around the metal-carbon bond. Therefore, the substituent
R remains fixed in the further reaction steps with respect to its position relative to the methyl group
of the last inserted monomer unit. By ring formation the free residual valences are saturated. The
overlap of electrons at C(1) and C(3) forms a σ-bond. At the same time the Al-C(3) bond is broken
and the starting complex can be formed again with an extended polymer chain.
Fig. 2: Bimetallic mechanism.
The general principles of such a mechanistic model can be applied to both homogeneous and
heterogeneous catalysis. The all-accepted fact is that, in the case of heterogeneous catalysis, the
catalytically active sites on the crystal surface of the heterogeneous contact are built from originally
defects presented in the crystal structure. If any α-olefin, e.g., styrene is used instead of ethylene as
monomer, there is the possibility of the formation of various stereoregular polymer chains (Fig. 3),
such as isotactic (it), syndiotactic (st) chains and atactic (at) for those that do not have
stereoregularity in the polymer chains. Chains, in which all the side-group carbon atoms have the
same absolute configuration (RRRR or SSSS) are called isotactic. In syndiotactic chains, the
configuration alternates along the chain (RSRS etc.). By means of the zig-zag-(also Natta-) projection
used in polymer chemistry, these regular and the non-regular (R and S are randomly distributed)

- 74 -
atactic forms can be described. The stereoregularity of polymers has a very strong influence on their
physical properties. Atactic polymers, for example, are generally amorphous, and their glass
temperatures are relatively low and extend as freeze-range over several degrees. However, the iso-
and syndiotactic polymers generally can be crystallized; their melting points Tm are high and well-
defined, also the solubility is greatly reduced due to the high crystallinity. The various forms can be
characterized by both IR- and NMR.
Fig 3: Natta-projection.
The following Table summarizes some of the properties (G. Natta, 1959).
Table 1. Comparison of the properties of stereoregular and atactic polymers.
product solid state Tm /
° C
Tg /
° C
density /
g·cm-3
solubility* in
diethyl ether
solubility* in
toluene
PP
at amorphous -20 0,85 s ss
it crystalline 158 - 160 -10 0,93 ds s
PB
at amorphous -24 0,87 s ss
it crystalline 126 - 128 0,91 ds s
PS
at amorphous 70 - 100 1,04 s ss
it crystalline 230 - 231 1,08 ds s
* s: soluble; ds: difficultly soluble; ss: slightly soluble.

- 75 -
1.3 Industrially important polymers prepared via coordinative polymerization
HDPE, high density polyethylene
Hostalen (Hoechst), Marlex (Phillips)
vessels, bags, sheets, dishes, disposable products, cable sheathing, insulators, hip joints, ...
it-PP, it-polypropylen
Hostalen PP (Hoechst), Novolen (BASF)
temperature-resistant containers, vessels, automotive, upholstery, ...
it-PB, it-poly-l-butene
tear-resistant films, pipes, ...
poly-4-methyl-l-pentene
TPX (Mitsui)
electrical and lighting industry, temperature resistant (mp 240 ° C), medical applications
trans-1,4-polyisoprene
golf balls, medical applications (for example shoe inserts)
cis-1,4-polyisoprene und -butadiene
rubber and tire industry
2 Experimental produce
2.1 Safety: All persons being exposed to chemicals have to be instructed about the effects of
dangerous substances (toxicity, point of ignition, etc.) as well as about preventive measures. Before
the experiment is carried out, read the MSDS sheets for all the chemicals used in this laboratory and
be familiar with their safe handling. The instructions of the teaching assistant must be followed at all
times. Especially the following points are relevant:
1. Wearing of suitable protective clothing (protective goggles, glove, laboratory coat, etc.)
2. Knowledge about the safety devices (e.g., laboratory hood, fire extinguisher, emergency
shower, first aid boxes, etc.), exit
3. Controlled disposal of toxic substances in compliance with legal regulations
4. Strict ban on eating, drinking and fuming in the laboratory

- 76 -
The greatest possible care must be exercised when working with organoaluminum compounds, since
they ignite very easily on contact with air and water. All operations must, therefore, be carried out
with the complete exclusion of air and moisture and pipettes must be flushed with nitrogen.
Moreover, these substances cause wounds that are slow to heal, so that the wearing of safety goggles
is mandatory and all contact with the skin must be avoided. The pipettes are cleaned as follows: after
all organoaluminum have been run out, the pipette is filled with ether and allowed to drain again. It is
then washed with isopropanol/HCl solvents.
2.2 Chemicals and equipment
Chemicals: heptane abs., triisobutylaluminium (25% in hexane), TiCl4 (caution!), styrene (freshly
distilled), 2-propanol, methanolic HCl (ca. 10%), methanol, methyl ethyl ketone (MEK), toluene.
Equipment: Gas purification system, Schlenk line with pressure relief valve, 500 mL three-neck flask
with gas introduction, KPG-stirrer, dropping funnel with pressure balance and
three-way valve, thermometer, oil bath with temperature sensor, hotplate, extraction flask with reflux
condenser and heating mantle, heating gun, vacuum desiccator (heatable), vacuum pump, N2-
cylinder with pressure reducer.
2.3 Experiment
The apparatus is evacuated (vacuum pump, slightly heated (heat gun) and filled with N2. After
cooling down the apparatus, 50 mL of absolute heptane are added with the aid of a dropping funnel
to the flask. Under a nitrogen atmosphere 1.0 mL of TiCl4 is added by a syringe and then 25 mL of
the aluminumalkyl-solution are added drop wise over a period of 20 minutes. Since the reaction of
the two catalyst is highly exothermic, an external cooling may be necessary. After approximately 10
minutes “aging time” of the catalyst, 25 mL styrene are added to the solution at once. The reaction
mixture is heated up to 75 °C (inside temperature) for 3-4 hours. After the mixture is cooled to room
temperature, the polymerization is stopped by adding 20 mL of 2-propanol under N2 and then
100 mL of 2-propanolic HCl subsequently (thorough mixing by vigorous stirring required). The
polymer is filtered off and washed thoroughly with methanol. The crude product is boiled under
reflux with 80 mL of methyl ethyl ketone, and then portion wise 30 mL of toluene are added and
extracted for 5 hours. The residue is first decanted and then sucked off with a coarse frit. After
washing with methyl ethyl ketone, the polymer is dried at 60 ° C in a vacuum desiccator. Both the
yield of it polystyrene and the melting range are to be determinated.
3 Questions about coordinative polymerization

- 77 -
1. Why is the extraction with MEK and toluene necessary?
2. What are the molar concentrations of the catalyst components in the reaction mixture?
Calculate the ratio Ti:styrene!
3. What is the difference between Fischer-projection and Natta-(Zig-Zag-) projection?
4. What is the difference between low and high pressure PE? Physically? Chemically?
5. What is meant by a 3rd-generation Ziegler catalyst?
6. Why can vinyl ether not be coordinative polymerized by TiCl4/Et3Al?
7. Structures of HDPE, LDPE, LLDPE, it-PP, st-PP, PB, trans-1,4-polyisoprene,
poly(4-methyl-1-pentene), cis-1,4-polybutadiene?

- 78 -
II Ring-Opening Metathesis Polymerization (ROMP)
Task
Polymerization of norbornene derivatives using the metathesis reaction and a transition metal
alkylidene catalyst.
Literature
1. R. H. Grubbs (Ed.) Handbook of Metathesis, Wiley-VCH, Weinheim, (2003)
2. M. Chanda, Introduction to Polymer Science and Chemistry: A Problem Solving Approach, CRC Press Inc.,
(2006)
3. F. J. David, Polymer Chemistry: A Practical Approach, Oxford University Press, (2004)
4. P. W. N. M van Leeuwen, Homogeneous Catalysts: Activity, Stability, Deactivation, Wiley-VCH, Weinheim,
(2011)
5. M. R. Buchmeiser, Chem. Rev. (2000), 100, 1565
Content
1. Introduction
2. Mechanism
3. Experimental part
4. Chemicals, equipment and safety
5. Experiment
6. Questions

- 79 -
1 Introduction
Today catalytic olefin metathesis, in addition to palladium catalyzed coupling reactions, is one of the most important
chemical reactions for the formation of carbon-carbon bonds. It has had an undeniable impact on various areas of
chemistry. Olefin metathesis is the exchange of alkenyl groups of two olefins in the presence of a metal alkylidene
catalyst. The results is a thermodynamically equilibrated mixture of products consisting of cis- and trans-olefins (Figure
1).
Fig. 1: General olefin metathesis reaction.
Olefin metathesis opened the door for a wide range of new methods and research areas in both organic and polymer
chemistry. One important area is the research on organometallic compounds done by Schrock, Grubbs, Katz and many
others. The need for catalytically active species of these kinds is rising ever since.
Fig. 2: Catalytic cycle of olefin metathesis.
Outstanding contributions in this field were provided by Chauvin (postulation of the right mechanism), Schrock
(synthesis and characterization of the first highly active well defined metathesis catalysts) and Grubbs (synthesis of
broadly applicable and (air) stable catalysts). In 2005 they shared the Chemistry Nobel Prize.

- 80 -
In 1971, Y. Chauvin postulated that the olefin adds to the metal carbon double bond via a [2+2] cycloaddition. The
resulting intermediate is a metalla-cyclobutane species, which reacts to the product olefin and a new metal alkylidene via
a [2+2] cycloreversion. All steps in this reaction, in principle, are reversible. Thus, the product can be transformed back
to the starting material. Therefore, a driving force is needed to achieve high conversions and constantly shift the
equilibrium to the product side. This is illustrated in detail in Figure 2.
First observations of this kind of reactions were made in the 1950s when strange side products occured in the
polymerisation of ethylene in the presence of various metals. In early attempts, compounds like MoO3, WCl6, WOCl4 or
Re2O7 were used to selectively catalyze olefin metathesis. In addition, an alkylating agent had to be present (e.g., SnR4,
AlR3, etc.). These systems were highly active (but not selective). Another striking disadvantage was that they were
relatively short living and sensitive for impurities containing functional groups. In many cases, the real catalytically
active species remains unknown until today.
In 1964 Fischer synthesized the first metal-carbon double bond (carbene). The so called Fischer-carbenes are stabilized
by π-donors (Figure 3). Thus, these carbenes have a singlet configuration.
Fig. 3: Fischer-Carbene.
In 1974 Schrock prepaired the first metal-carbene without neighboring π-donors and with a transition metal in its highest
oxidation state. His alkylidenes (Figure 4) differ from Fischer-carbenes in that they possess a triplet configuration of the
metal carbon bond. Schrock synthesized complex 2 (Figure 4) by treating tantalum pentachloride with neopentyl lithium.
Fig. 4: First examples of a Schrock-carbene (alkylidene).
Through α-H-abstraction one equivalent of neopentane is lost and the new alkylidene forms. Schrock consequentially
recognized the unstable character of pentaalkyl substituted tantalum and proposed the formation of alkylidenes by this
mechanism. Over the years new and more active and stable catalysts were prepaired with great effort (Schrocks catalysts,
Figure 5). Grubbs made the invention of air (bench-top) stable rutheniumcarbenes (Figure 6), which set a new landmark
in the applicability of these catalysts. They are made by oxidation of Ru(II)-precursors. They complement to Schrocks
catalysts in their properties, because in general they are less active but also less sensitive. First and foremost they are
more stable to protic functional groups like alcohols or water. Schrock-alkylidenes in return have a better tolerance
towards functional groups containing sulfur, nitrogen or phosphorus.
- 81 -
Fig. 5: General structure of tungsten- or molybdenum-alkylidene catalysts.
RuR
Cl
Cl
L
L
Ru
P(Cy)3
P(Cy)3
Cl
Cl RuCl
ClP(Cy)3
NNMesMes
Grubbs I
Ru
P(Cy)3Cl
ClO Ru
Cl
ClO
NNMesMes
Grubbs-Hoveyda I
Grubbs II
Grubbs-Hoveyda IIRu
CF3COO
CF3COOO
NNMesMes
Buchmeiser-Grubbs-Hoveyda
Fig. 6: General structure and important examples of Grubbs-catalysts.

- 82 -
Olefin Metathesis Reactions
Some olefin metathesis reactions are shown in the following schemes.
nn
Rn
Rn
XX X
x yz
Ring Opening Metathesis Polymerization (ROMP)
1-Alkyne Polymerization
Cyclopolymerization
nn
n
+
Acyclic Diene Metathesis Polymerization (ADMET)
R
R'+ R
R'
+Crossmetathesis
R +R'
R R'
Cross-Ene-Yne-Metathesis
X
R
n
X Rn
RingclosingEne-yne-Metathesis
X
R +
X
R
Cross-Ene-Diyne-Metathesis
R +
RRing opening
Cross-metathesis
XTandem-
Ringopening CrossmetathesisX
X
Ringclosing metathesis X
+
Fig. 7: Examples for metathesis reactions.
One important aim of such catalytic reactions is the selective formation of E- and Z-double bonds. In general, E-double
bonds are thermodynamically favored. In the case of moderately controlled reactions a mixture of E- und Z- configured
products is formed. Another important factor is the control of tacticity in stereoregular polymers. Besides Mo- and W- ,

- 83 -
some highly selective Ru-catalysts have been reported. Normally, the driving forces for the reactions shown in Figure 7
are release of volatile molecules (ethylene, etc.), entropic effects, release of ring strain or the formation of highly
conjugated systems.
2 ROMP: Mechanisms
Ring opening metathesis polymerization (ROMP) is a reaction where mostly strained double bonds are opened by the
catalyst (better: initiator) and rearranged subsequently with other olefins to form chains. Basically, it follows a chain-
growth mechanism. Usually, a monocyclic (e.g., cyclopentene, cyclooctene, cyclooctadiene etc.) or polycyclic (e.g.,
norbornene, dicyclopentadiene etc.) monomer with higher ring strain is used. The general mechanism is shown in Figure
8.
Fig. 8: Mechanism of the ring opening metathesis polymerization.
Besides the mentioned growth and termination of a polymer chain, several side reactions are also of importance.
Particularly undesired terminations, which end the reaction in an unpredictable manner are responsible for a wide range
of products and a broad molecular weight distribution.
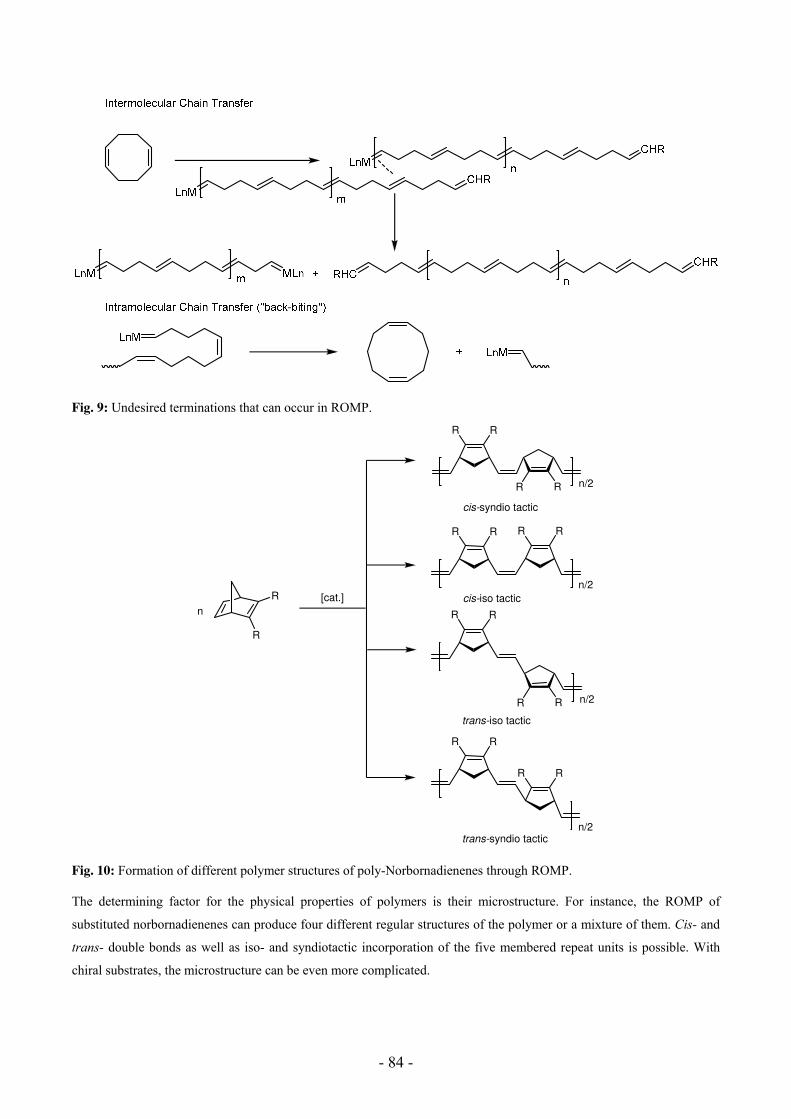
- 84 -
Fig. 9: Undesired terminations that can occur in ROMP.
RR
R R
n/2
R R
n/2
R R
RR
R R
n/2
R R
n/2
R R
R
R
[cat.]
n
cis-syndio tactic
cis-iso tactic
trans-iso tactic
trans-syndio tactic
Fig. 10: Formation of different polymer structures of poly-Norbornadienenes through ROMP.
The determining factor for the physical properties of polymers is their microstructure. For instance, the ROMP of
substituted norbornadienenes can produce four different regular structures of the polymer or a mixture of them. Cis- and
trans- double bonds as well as iso- and syndiotactic incorporation of the five membered repeat units is possible. With
chiral substrates, the microstructure can be even more complicated.

- 85 -
Whether regular or mixed structures are formed strongly depends on the applied catalyst system. The following schemes
show the addition of monomer to different catalysts and the consequences for the configuration of the double bond and
tacticity.
Fig. 11: Monomer addition in the polymerization of substituted norbornadienenes using a MAP-catalyst.
Fig. 12: Monomer addition to the 1st-generation Grubbs catalysts.
3 Chemicals, Equipment and Safety
Equipment:
Beaker 20 mL, Beaker 200 mL, heat gun, Schlenk flask 50 mL, suction filter, stirring bars, filter
paper, glas stopper, spatula, pipette, syringe, scintillation vials
Chemicals:
MW (g/mol)
Grubbs Catalyst 1st-gen. (RuCl2(PCy3)2CHPh) 822,96
5-Norbornen-2-yl-acetate 152,19
dichloromethane, methanol, ethyl vinyl ether, chloroform-d
3.1 Safety
Work under high vacuum bears the danger of imploding glasware and has to be done in closed fume
hoods. Ethyl vinyl ether as well as methanol are highly flamable and have to be protected from

- 86 -
ignition sources. The exposure of the chemicals used in this experiment to atmosphere must be
avoided. Dichloromethane and chloroform are suspected cancerogens. Methanol is toxic by any
exposure (skin, lung etc.). Norbornene and its derivates are flamable and have to be kept away from
ignition sources. Since other data for the used monomer is not available, it has to be considered as
highly dangerous. Prevent exposure and inhalation of the fumes. The applied ruthenium compound is
flamable and should be handled with great care. Avoid any contact with skin.
3.2 Experiment
Handling of the catalyst: The transition metal complex has to be stored in the fridge (2-8 °C).
Minimize its exposure to the atmosphere and/or humidity to prevent decomposition of this expensive
chemical. Before opening the bottle, it should reach room temperature to minimize water
condensation. Weighing should be carried out as quickly as possible and catalyst containers need to
be kept close when not used. Thus, dry glassware and solvents are required for this experiment. All
manipulations have to be conducted under a nitrogen atmosphere using Schlenk techniques.
The reaction vessel has be heated and evacuated to eliminate residual water and oxygen. 20 mL of
dry dichloromethane are added under a stream of nitrogen. The monomer (ca. 1.00 g, 3.29 mmol) is
added into the flask the same way. Use a scintillation vial for weighing and rinse thoroughly with dry
dichloromethane. Note the exact used amounts of catalyst and monomer. Now weigh in the catalyst
(ca. 54 mg, 0.0329 mmol, 1 mol-%) and dissolve in 2 mL dry dichloromethane. Transfer the solution
quickly and completely into a syringe. Add the catalyst solution as quickly as possible and in one
single shot into the heavily stirred reaction mixture. Close the flask with a stopper. The reaction is
being stirred for one hour at room temperature. After that time a small portion of the reaction mixture
(approx. 2 mL) is taken out by a syringe and added under heavy stirring to 20 mL of methanol. Note
your observations. To the rest of the reaction mixture 2 mL of ethyl vinyl ether is added. Stirr the
reaction for another 15 minutes. For isolation of the polymer, the reaction mixture has to be added
dropwise to heavily stirred 300 mL of methanol. The polymer should precipitate. After addition, stirr
for another 10 minutes and filter the product. Wash the polymer with some methanol. Residual
solvent is removed applying high vacuum. Use 10 mg of the dry polymer and dissolve it in 10 mL of
THF for GPC analysis. 20 mg of the dry polymer are dissolved in 0.6 mL of chloroform-d for NMR
analysis.

- 87 -
4 Questions
1. What was the visual difference you observed when precipitating the polymer for the first
and second time? Explain.
2. What is ethyl vinyl ether used for in this experiment and what is being formed after addition?
Write down the reaction equations for the reactions in this experiment and explain why
ethyl vinyl ether is a good candidate for the needed purpose. Would 1-pentene be an
alternative? Why/(not)?
3. Why has the catalyst solution to be added as quickly as possible? Clarify the role of
temperature, mixing and concentration of monomer solution in this regard.
4. Calculate the theoretical values for molecular weight of the synthesized polymer chains.
Compare them to the experimentally found values determined by GPC analysis. Explain any
deviations. Why, in general, can an experiment deliver much higher or smaller values
compared to the theoretical ones?
5. Do you see any possible way to reuse the catalyst? What is necessary to accomplish this?
6. Name some simple ways to prepare (in this experiment) polymers with higher solubility?
Name factors which play a crucial role.
7. Which influences can an applied catalyst system have on the reaction process and the
product? Why do expensive and sensitive systems still play an important role?
8. What is the striking structural feature of transition metal alkylidene complexes? What
significant difference would you expect from the NMR spectrum of a propagating species to
the parent catalyst?
9. Assign the observed signals you see in the 1H-NMR of the polymer and explain the
differences to the spectrum of the monomer.

88
Experiment 6: Emulsion Polymerization
Objective
Styrene is polymerized in an emulsion using dodecyl hydrogen sulfate sodium salt as an emulsifier and potassium persulfate as the initiator. Samples are drawn from the reactant solution in regular intervals in order to generate diagrams of the conversion as well as the reaction rate versus the reaction time.
References
1) H.‐G. Elias, Makromoleküle Bd. 1, Edition 6, Wiley‐VCH Verlag, Weinheim, 1999
2) P. J. Flory, Principles of Polymer Chemistry, Cornell, University Press, Ithaca, 1953
3) B. Tieke, Makromolekulare Chemie, Edition 2, Wiley‐VCH Verlag, Weinheim, 2005
4) W. V. Smith, H. Ewart, J. Chem. Phys. 16(6), 592 (1948)
5) W. D. Harkins, J. Am. Chem. Soc. 69, 1428 (1947)
Content
1. Theoretical Background
2. Experimental Part
3. Questions

89
1. Theoretical Background
The process of emulsion polymerization is of vital significance for the fabrication of various industrial
polymers such as polychloroprene, poly(vinyl acetate), polytetrafluoroethylene and poly(vinyl
chloride) as well as “cold rubber” (styrene/butadiene copolymers). The technological development
of this method started in the 1920s and has since gained ever‐increasing importance resulting in a
recent global production of several million tons per year.
The essential components of an emulsion polymerization are the following.
Monomer (insoluble in water)
Water
Surfactant
Initiator (radical forming agent, soluble in water)
In most cases anionic surfactants (e.g. salts of fatty acids, salts of alkyl hydrogen sulfates, salts of
alkyl sulfonic acids) are used, while cationic (quaternary ammonium salts, e.g. cetyltrimethyl
ammonium bromide) or non‐ionic surfactants (often high molecular weight) are rarely applied. All
types of surfactants comprise both hydrophilic and hydrophobic groups. When highly diluted, the
surfactants can be completely dissolved in water. However, at concentrations above the critical
micelle concentration (cmc), the molecules of the emulsifier aggregate into micelles. This process,
which is driven by thermodynamic principles, is accompanied by a significant drop in the surface
tension of the system. The emulsion polymerization is generally performed at surfactant
concentrations above the cmc.
Water‐soluble peroxo salts (e.g. potassium persulfate) are the classical choice for the initiator of the
polymerization reaction, even though organic hydroperoxides (e.g. cumene hydroperoxide) are also
frequently used. Redox initiators are of particular importance in industrial processes, since these
compounds can induce polymerization even at low temperatures.
The progress of a typical emulsion polymerization can be divided into three phases distinguished by
their characteristic profiles of the reaction rate over time (see Figure 1):
(I) increasing reaction rate
(II) constant reaction rate
(III) decreasing reaction rate.
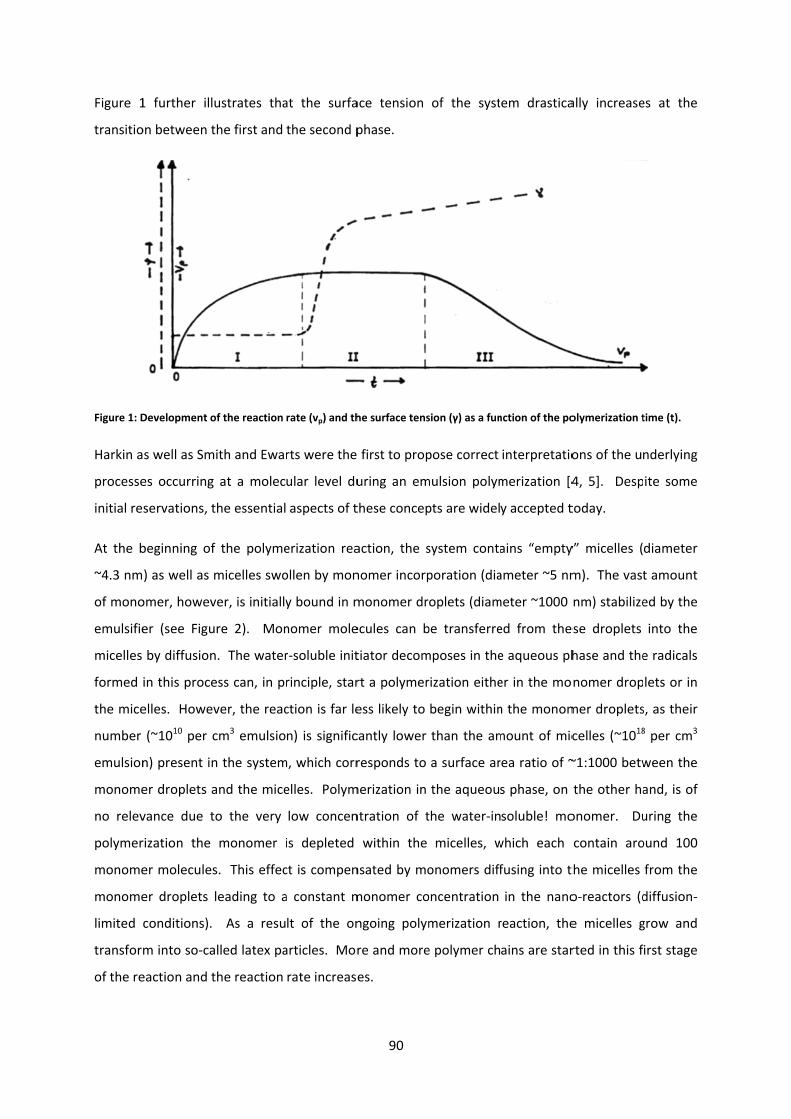
Figure 1
transitio
Figure 1: D
Harkin a
processe
initial re
At the b
~4.3 nm
of mono
emulsifie
micelles
formed
the mice
number
emulsion
monome
no relev
polymer
monome
monome
limited
transform
of the re
1 further ill
on between t
Development o
s well as Sm
es occurring
servations, t
beginning of
) as well as m
omer, howev
er (see Figu
by diffusion
in this proce
elles. Howev
(~1010 per c
n) present in
er droplets a
vance due t
rization the
er molecules
er droplets
conditions).
m into so‐ca
eaction and t
ustrates tha
the first and
of the reaction
ith and Ewa
at a molec
the essential
f the polyme
micelles swo
ver, is initially
ure 2). Mon
n. The water
ess can, in p
ver, the reac
cm3 emulsio
n the system
and the mice
o the very
monomer i
s. This effec
leading to a
As a resu
alled latex pa
the reaction
at the surfa
the second p
rate (vp) and th
rts were the
ular level du
aspects of t
erization rea
ollen by mon
y bound in m
nomer mole
r‐soluble init
rinciple, star
ction is far le
n) is signific
m, which corr
elles. Polym
low concen
is depleted
ct is compen
a constant m
lt of the on
articles. Mor
rate increase
90
ace tension
phase.
he surface tens
first to prop
uring an em
these concep
action, the s
nomer incorp
monomer dr
ecules can b
tiator decom
rt a polymer
ess likely to
cantly lower
responds to
merization in
ntration of t
within the
nsated by mo
monomer co
ngoing polym
re and more
es.
of the syst
sion (γ) as a fun
pose correct
mulsion polym
pts are widel
system conta
poration (dia
oplets (diam
be transferre
mposes in the
rization eithe
begin within
than the am
a surface ar
the aqueou
the water‐in
micelles, w
onomers diff
oncentration
merization r
e polymer ch
em drastica
nction of the po
interpretatio
merization [4
y accepted t
ains “empty
ameter ~5 nm
meter ~1000
ed from the
e aqueous ph
er in the mo
n the monom
mount of mic
rea ratio of ~
s phase, on
soluble! mo
which each
fusing into t
in the nano
reaction, the
ains are star
ally increase
olymerization t
ons of the un
4, 5]. Desp
today.
y” micelles (
m). The vast
nm) stabilize
ese droplets
hase and the
nomer drop
mer droplets
celles (~1018
~1:1000 betw
the other ha
onomer. Du
contain aro
the micelles
o‐reactors (d
e micelles g
rted in this f
es at the
time (t).
nderlying
ite some
diameter
t amount
ed by the
into the
e radicals
lets or in
s, as their 8 per cm3
ween the
and, is of
uring the
ound 100
from the
diffusion‐
grow and
irst stage

Figure 2:
from [3]).
~~~ = su
When t
simultan
system i
concentr
increase
beginnin
polymer
is fully c
kinetics)
Howeve
which al
of the re
for poly
leading t
yet anot
question
The rem
following
Schematic illu
Legend: S = m
urfactant
the reaction
neously more
ncreases an
ration of fre
e in the surf
ng of phase
rization withi
compensate
.
r, when the
l the monom
eaction only
merization.
to a charact
ther increas
ns).
markable ind
g advantage
stration of dif
micelle swollen
n progresse
e (but smalle
d a higher a
ee surfactan
face tension
II of the re
in the latex p
d by the dif
e emulsion p
mer molecule
those mono
From this p
teristic declin
e in the rea
dustrial relev
s:
fferent particle
with monome
s further, t
er) latex part
mount of em
t, which wil
is noticeab
eaction, whic
particles. At
ffusion of n
polymerizatio
es bound in t
mer molecu
point onward
ne in the rea
action rate c
vance of th
91
es/aggregates i
er, L = latex par
the amoun
ticles are for
mulsifier is b
ll eventually
ble and there
ch is charac
t this stage t
ew monom
on progress
the monome
les already lo
ds the numb
action rate (
can be obse
he emulsion
involved in an
rticle, M = mon
t of mono
rmed. Cons
ound. This
fall below t
e are no fre
cterized by a
he consump
er from the
es even furt
er droplets a
ocated withi
ber of these
(first order k
erved towar
polymeriza
emulsion poly
nomer droplet,
mer drople
equently, th
leads to a st
the cmc. A
ee micelles l
a constant r
tion of mono
e monomer
ther, a poin
re consumed
n the latex p
e reactants c
kinetics). In
ds the end
tion proces
ymerization (r
, = monomer
ets decrease
e surface ar
teady decrea
At this point
left. This m
reaction rate
omer by the
droplets (ze
nt will be re
d. In this th
particles are
constantly d
some cases
of the reac
ss is a resu
reproduced
r molecule,
es, while
ea of the
ase in the
a strong
marks the
e for the
e reaction
ero order
ached at
ird phase
available
decreases
, though,
tion (see
lt of the

92
a) The aqueous phase enables temperature regulation during the polymerization reaction.
b) Redox initiators can be applied, which enable polymerization reactions to proceed at comparably
low temperatures, yet keeping the reaction rate relatively high.
c) High degrees of polymerization can be achieved due to the small probability of chain termination.
This can be further regulated by the controlled addition of chain transfer agents.
d) Remaining monomer can be removed by steam distillation.
e) The obtained latex can be directly subjected to further applications (paints, glues, coatings).
On the downside, however, a frequently problematic extraction of the emulsifier as well as the
potentially high degree of branching of the polymer chains have to be taken into account as major
disadvantages of the method.
Kinetics and Mechanism
A hallmark of the emulsion polymerization process is that this method yields higher degrees of
polymerization than those accessible under comparable conditions in bulk or suspension
polymerization reactions. The kinetics and the mechanism of emulsion polymerization are,
therefore, distinct from other radical polymerization methods. The reaction rate vw(L) in an individual
latex particle is given by equation (1).
(1) [M][P*]k)(v Ww L
vw(L): reaction rate of the polymerization in the latex particle
kw: rate constant of the polymerization
[M]: concentration of monomer in the latex particle
[P*]: concentration of radicals in the latex particle
The measurable reaction rate vw of the entire reaction batch is represented by the sum of the
reaction rates in all isolated latex particles. Assuming a narrow size distribution of the latex particles
the reaction rate can be described as follows:

93
(2) A
wwN
nN[M]kv
vw: reaction rate of the polymerization
n: average number of free radicals per latex particle
N: number of latex particles per unit volume of the emulsion
NA: Avogadro constant
In equation (2), it is further assumed that the polymerization occurs exclusively within the latex
particles, i.e. there is no considerable amount of polymer forming either in the aqueous phase or in
the monomer droplets. For the determination of vW the factors included in equation (2) have to be
expressed as properties, which are experimentally accessible. This is particularly difficult in the case
of N and n. The most established quantitative model for the description of the kinetics and the
mechanism of the emulsion polymerization was derived by Smith and Ewart. It is particularly well‐
suited for systems showing a behavior consistent with the Harkin model.
The Harkin model is based on the following considerations: After a short phase of particle generation
(phase I) the reaction rate becomes constant in phase (II). Therefore, regarding the factors included
in equation (2), not only the monomer concentration [M] and the number of latex particles per unit
volume [N], but also the number of free radicals within the latex particles have to be constant.
However, according to the steady state approximation the number of these radicals will only remain
constant, when equal amounts of radicals are formed and consumed in a given period of time.
The following assumptions will be made: Once a radical is located within a latex particle, it cannot
leave this environment again. If a second radical enters the same latex particle, an immediate
recombination of the two will take place due to the small radius of the latex particle. An individual
latex particle, therefore, contains either one or no radical at any given time, corresponding to an
average number of radicals per latex particle of n = ½. With additional assumptions regarding the
number of latex particles equation (3) can be derived:
(3) 5
3
S5
2
A
ww [S])(a)N
σb(
2
1[M]kv
b: system constant in the range of 0.37‐0.53
σ: inflow/intake velocity of the radical into the latex particle
µ: constant volume increase of the latex particle

94
aS: surface area required by one surfactant molecule
[S]: concentration of the emulsifier
Combining all constant parameters included in equation (3) into a single constant K leads to a
simplified representation as given by equation (4):
(4) 5
3
w K[M]([S])v
On closer examination of equation (4) it becomes obvious, that the reaction rate of the emulsion
polymerization can be raised even without increasing the temperature, monomer concentration or
initiator concentration – simply by adding a higher amount of the surfactant.
The degree of polymerization Pn of a polymerization reaction is defined as the ratio between the sum
of all reaction rates associated with processes leading to an increase in the chain length and the sum
of reaction rates ascribed to those processes resulting in the termination of an individual chain. The
latter can occur either by direct termination or by chain transfer to monomer, solvent, or initiator
molecules (but not to polymer chains!).
(5) )v(v
vP
TA
wn
vA: sum of the reaction rates of all processes terminating the chain
vT: sum of the rates of all chain transfer reactions
As the concentration of the emulsifier directly influences vw, it also has an immediate effect on the
degree of polymerization. In contrast to all other radical polymerization processes, the
recombination of radical chain ends is inhibited in emulsion polymerization, since the chains exist in
isolated latex particles and are therefore effectively separated from each other.
Moreover, in its final stages the process of emulsion polymerization shows a behavior analogous to
the Norrish‐Trommsdorf effect, which is known from bulk polymerization. As a result, the reaction
rate and the degree of polymerization are comparably higher.

95
2. Experimental Part
The goal of this experiment is to prepare polystyrene by polymerization of styrene in emulsion. For
that purpose, dodecyl hydrogen sulfate sodium salt and potassium persulfate are used as the
surfactant and initiator, respectively. Plots of the conversion as well as the reaction rate versus the
reaction time should be obtained. The conversion is determined gravimetrically at various stages of
the reaction. The results shall be discussed with a special emphasis on the kinetics of the reaction.
Chemicals:
‐ Styrene (destabilized and freshly distilled)
‐ Water
‐ Dodecyl hydrogen sulfate sodium salt
‐ Potassium persulfate
‐ Hydroquinone (0.02% in water)
Equipment:
‐ Oil bath
‐ Round‐bottomed flask
‐ Hot‐plate magnetic‐stirrer device
‐ Reflux condenser
‐ Thermometer
‐ Magnetic stirring bars
‐ Sample dishes
‐ Glass vials
‐ Syringes and needles
‐ Plastic pipettes
‐ Analytical balance
‐ Vacuum drying oven
Procedure:
Water (50 mL), emulsifier (1 g) and styrene (5 g) are filled into a round‐bottomed flask of 100 mL
capacity. The mixture is heated to 70°C under stirring in an oil bath. After an equilibration period of
approximately 30 minutes the polymerization is started by the addition of the initiator dissolved in 4
96
mL water (t = 0 min). During the course of the next two hours, samples of 2 mL reactant solution
each are drawn in regular intervals according to the specifications in table 1.
Table 1. Sample identifier and reaction time at which the sample was taken.
Sample
identifier E1 E2 E3 E4 E5 E6 E7 E8 E9 E10 E11
t/min 2 4 6 10 15 20 30 45 60 90 120
These samples are immediately pipetted into glass vials containing 2 mL hydroquinone solution for
stabilization and are subsequently transferred to a vacuum drying oven to remove all volatile
components. Afterwards, the solid content is determined by gravimetric analysis. All relevant
masses and mass differences are calculated and documented according to table 2.
Table 2. Evaluation of the obtained sample weights and determination of the conversion.
Sample
identifier
m0
(dish)
m1
(dish +
hydroquinone)
m2
(dish +
hydroquinone
+ dispersion
before
drying)
m2‐m1
(msample)
m3
(dish +
dispersion
after drying)
m3‐m0
(msolid)
Conversion
E1
E2
E3
E4
E5
E6
E7
E8
E9
E10
E11
Evaluation:
Plots of conversion versus time.
1) Besides the polymer molecules the isolated solid additionally contains residual emulsifier. In order
to determine the conversion (conv) of the reaction accurately, the surfactant content has to be

97
subtracted from the total weight of the solid material. This calculation can be performed according
to the following equation:
conv = msolid
mM0
· 1
1+ mE0
mM0
· mtot
msample
·100%
msolid: weight of the solid
mE0: weight of emulsifier used in the reaction batch
mM0: weight of monomer used in the reaction batch
mtot: total weight of the reaction batch
msample: weight of the sample
2) Calculate conversions for all samples taken during the reaction and plot the results over the
reaction time.
Plots of reaction rate versus time.
3) Determine the reaction rates at the various time steps by constructing tangents on the conversion
vs. time curve. List the obtained reaction rates in a table, plot and discuss the results.

98
3. Questions
1) Which effects can be potentially caused by the presence of oxygen traces in the emulsion? What
could be an explanation for another increase in the reaction rate during the final stages of the
polymerization (Take into account that the reaction is a radical polymerization)?
2) Why is the coagulation of latex particles inhibited?
3) What is the average radical concentration in the latex particles under the following assumptions?
The radicals reach the latex particles by diffusion. This process determines the reaction rate.
Whenever two radicals are located in the same latex particle they will recombine
immediately.
4) Describe the principle of a redox initiator and list a number of examples.
5) Compare the characteristics of emulsion polymerization and suspension polymerization. Name
further examples for technologically relevant polymerization procedures.

-99-
Experiment 7
Anionic Polymerization
Assignment
The concept of living anionic vinyl polymerization will be demonstrated by the block
copolymerization of styrene and isoprene with sec-butyllithium in toluene.
References
1) H.-G. Elias, An Introduction to Polymer Science, 1. Auflage, Wiley-VCH-Verlag,
Weinheim, 1997
2) M. Morton, Anionic Polymerization: Principles and Practice, Academic Press, New
York, 1983
Contents
1. Introduction
1.1 General Remarks
1.2. Anionic Polymerization
1.2.1. Initiation Reaction
1.2.2. Propagation Reaction
2. Experimental Part
2.1. Anionic Polymerization of Styrene/Isoprene
2.2. Evaluation
3. Questions

1. Intr
1.1. Ge
The io
propag
1. In th
end (P
growth
Fig 1:
Ionic ch
The ch
For the
equilibr
species
solvent
These
the con
Fig. 2:
Differen
The rea
rate de
increas
towards
roduction
eneral Rem
onic vinyl
ation step
he case of
Pn+), where
mechanis
hain growt
arged term
e reactivity
ria with th
s, whose
t, the inter
species ca
nductivity.
nt forms of
activity of c
ecreases
sing solve
s free ions
n
marks
polymeriz
can be ei
f cationic g
eas the ch
m. In both
P Mn
h polymeri
minal group
of these c
he counte
abundanc
rmolecular
an be distin
f ion pairs
charged te
with decr
nt polarity
s (Fig. 2).
zation is
ther cation
growth the
hain end w
cases the
Pn 1
ization via
ps of propa
charged ter
erions in s
ce depend
ionic inter
nguished b
that are in
erminal gro
reasing io
y the equ
- 100 -
a chain-g
nic or anio
polymer c
will be neg
e propagati
or
macrocati
agating ch
rminal grou
solution. F
ds on vari
ractions an
by spectro
equilibrium
oups with t
onization o
uilibrium is
growth po
nic as illus
chain carri
gatively ch
ng chain a
P Mn
on and ma
hains do us
ups it is im
Fig. 2 sho
ous factor
nd the size
scopy as w
m with eac
he monom
of the co
s shifted f
lymerizatio
strated sch
ies a posit
harged (Pn
a macroion
Pn 1
acroanion.
sually not e
portant to
ows the d
rs, e.g. th
e of the me
well as by
ch other [1]
mer and the
ounterion/a
from aggr
on reactio
hematically
tive charge
n-) for an
n.
1
exist as fre
consider d
different e
he polarity
etallic cou
measurem
]
erefore the
anion bond
regated io
on. The
y in Fig.
e at the
anionic
ee ions.
dynamic
excisting
y of the
nterion.
ments of
e growth
d. With
n pairs

In con
polyme
externa
polyme
index.
synthes
1.2. An
1.2.1. I
The ini
Typical
lithium)
Fig. 3
butyl lit
Fig. 3:
Initiatio
The me
Consid
electro
form a
evolvin
step an
radical
trast to o
erization, t
al terminat
erizations a
Furthermo
sis of polym
nionic Poly
nitiation
tiators for
lly, alkali m
) and alkal
illustrates
thium, a fre
on step of a
echanism
ering spec
n tranfer r
naphthalid
g in this p
nother elec
anion (Fig
other chain
he ideal io
tion and t
and can b
ore, the livi
mers with a
ymerizatio
the anioni
metals, alky
i metalnap
the initiatio
equently us
a styrene p
of the initia
cifically the
reaction fr
de radical
process, th
ctron trans
g. 4), whic
n-growth p
onic polym
transfer re
be used to
ng charac
a well-defi
on
c polymeri
yl compoun
phthalides a
on step of
sed initiato
polymeriza
ation with
e system
om the m
anion (Fi
e color of
sfer reactio
ch sponta
- 101 -
polymeriza
merization
eactions. S
o synthesiz
cter of the
ned block
izations ca
nds of alka
are used.
f an anion
or.
tion with s
alkli metal
of naphth
etal to the
g. 4). As
the THF s
on occurs r
aneously d
ation react
is a poly
Such poly
ze polymer
polymeriza
copolymer
an be Brön
ali metals (
ic polymer
ec-butyl lit
naphthalid
alene with
e naphthal
a consequ
solution ch
resulting in
dimerizes i
tions such
reaction w
yreactions
rs with a l
ation allow
r structure.
nsted base
(e.g. cumy
rization of
hium
des is muc
h sodium m
lin molecu
uence of t
anges to g
n the forma
into a dist
h as free
without inte
are calle
low polydi
ws for a co
.
s or Lewis
yl potassium
styrene w
ch more co
metal in T
ule takes p
the radical
green. In t
ation of a
tyryl dianio
radical
ernal or
d living
spersity
ontrolled
s bases.
m, butyl
with sec-
omplex.
THF, an
place to
anions
the next
styrene
on. This

dianion
ends.
Fig. 4:
Reactio
sodium
1.2.2. P
The an
chracte
- t
-
-
In this
calcula
of mon
macroa
n starts the
on schem
m [1]
Propagatio
nionic cha
eristics of a
the rate co
(ki >> kp); t
in the prop
no termina
case the
ated from th
nomer con
anion, K =
e polymeriz
e for the
on
in-growth
an ideal liv
onstant of
the conseq
pagation st
ation and n
e number
he initial m
version an
2 for a ma
zation add
initiation
polymeriz
ing polyme
initiation (k
quence is t
tep only on
no chain tra
avarage
molar ratios
nd the fun
acrodianion
- 102 -
ding monom
of styren
ation is a
erization ar
ki) is much
that all cha
ne form of
anfer
of the de
s ([M]0/[I]0)
ctionality K
n).
mer molec
ne polyme
so-called
re:
h higher th
ains start a
ion pairs is
egree of p
of monom
K of the g
cules on bo
erization w
living po
an the pro
and grow si
s involved
polymerizat
mer and init
rowing ch
oth reactiv
with napht
lymerizatio
opagation r
imultaneou
tion (Pn)
tiator, the e
ains (K =
ve chain
thalene/
on. The
rate (kp)
usly
can be
extent p
1 for a

- 103 -
Kp
M**
0IP
0n
0M = initial monomer concentration
0I = initial initiator concentration
p = extent of monomer conversion
K = functionality of macroanion
For a kinetically controlled living polymerization with fast initiation the polydispersity
index (PDI) is given by the following equation:
1 1
wM = weight average molecular weight
nM = number average molecular weight
nP = number average of the degree of polymerization
With increasing degree of polymerization Pn the polydispersity index approaches 1.
For Pn = 500 the PDI is calculated as 1.002.

- 104 -
2. Experimental
2.1. Anionic Polymerization of Styrene/Isoprene
Reagents: toluene from the solvent purification system (SPS)
sec-butyllithium in cyclohexane (1.4 molar)
styrene (freshly distilled)
isoprene (freshly distilled)
methanol (technical grade)
Procedure:
40 ml toluene are added with a syringe through a septum into a completely dry,
argon-flushed 250 ml three necked-flask equipped with a magnetic stir bar. The three
necked-flask is kept under a slight argon stream. The toluene is then heated up to
50°C. Afterwards, 2 ml of styrene are added to the solvent and 0.3 ml of a sec-
butyllithium solution in cyclohexane (1.4 molar) will be injected fast. The reaction
mixture is then stirred for 60 minutes. Subsequently, 6.5 ml of Isoprene are added to
the reaction mixture at room temperature. The solution is stirred again for another 2
hours.
Isolation of the block copolymer
The dissolved polymer is precipitated by dropping the solution under stirring into a
beaker filled with 400 ml of methanol kept at 0°C. Subsequently, the polymer is
filtered by suction filtration, washed with methanol and dried at 50°C in a vacuum
oven.
2.1.2. Evaluation
1) The yield of polymerization is determined.
2) The molar mass averages as well as the molecular weight distribution are
determined by size exclusion chromatography (SEC).

- 105 -
3) Discuss the difference between the theoretically calculated and experimentally
determined values for the molecular weight.

- 106 -
3. Questions
1. How can you calculate the molar mass of the polymer using the monomer and
initiator concentration?
2. How much methanol do you theoretically need to stop the polymerization?
3. Give thereaction equation for the anionic polymerization of styrene with sec-
butyllithium (initiation, propagation and termination reactions).
4. Give the mechanism of the anionic ring opening polymerization of -caprolactam to
synthesize polyamide 6.

107
Versuch 6
Electropolymerization of Conducting Polymers
Assignment of tasks:
1. The theoretical and experimental aspects of the electrochemistry and electropolymerization of
conducting polymers shall be mediated.
2. Voltammetry is introduced as a tool for the synthesis of polythiophene films
3. The electrochromic behavior of the deposited films is studied and correlated to the redox-state.
4. A consistent analysis and evaluation of electrochemical data shall be performed to gain access to
the values of the HOMO level (HOMO = highest occupied molecular orbital)
References:
[1] György Inzelt, Conducting Polymers - A New Era in Electrochemistry, 2nd
Edition, Springer, 2012.
[2] Jürgen Heinze, Bernardo A. Frontana-Uribe, and Sabine Ludwigs, Electrochemistry of Conducting
Polymers - Persistent Models and New Concepts, Chem. Rev., 2010, 110, 4724–4771.
[3] Matthias Rehahn., Elektrisch Leitfähige Kunststoffe, Chemie unserer Zeit 2003, 37, 18-30.
[4] Alan MacDiarmid, Synthetische Metalle: eine neue Rolle für organische Polymere, Angew. Chem.,
2001, 113, 2649-2659.
[5] Jean-Luc Bredas and G.Brian Street, Polarons, bipolarons, and solitons in conducting polymers, Acc.
Chem. Res. 1985, 18, 309–315.
[6] Allen J. Bard and Larry R. Faulkner, Electrochemical Methods – Fundamentals and Applications, John
Wiley & Sons ; 2nd
Edition., 2001.
[7] Carl H. Hamann and Wolf Vielstich, Elektrochemie, Wiley-VCH Verlag GmbH & Co. KGaA, 4th
Edition,
2005.

108
1. Theoretical Background
1.1 Conducting polymers
In the year 2000 the scientists Alan G. MacDiarmid, Alan J. Heeger, and Hideki Shirakawa received the
Nobel Prize in chemistry for their discovery and development of conducting polymers (CPs). Their
discovery of the electrical conductivity of the organic material polyacetylene upon doping with iodine
was the key step in the modern development of conducting polymer science. From the late 1970s up to
now a variety of new CPs (Figure 1) has been introduced based on different monomers like pyrroles,
thiophenes, anilines or aromatic hydrocarbons.
Figure 1: Structures of common conducting polymers; polyacetylene 1, poly(paraphenylenevinylene) 2,
polythiophene 3, polypyrrole 4, polyaniline 5
The important features hi h all of the share are alter ati g - a d σ-bonds along the polymer
backbone resulting in a -conjugation. The electronic structure and the electronic properties of
conjugated polymers can be explained by the molecular orbital model. In ethylene two sp2 hybridized
carbons combine resulting in an occupied - and an unoccupied *-orbital, as shown in Figure 2.
Interaction with a uniform -bond results in a splitting of the orbitals. A polymer with n sp2 hybridized
carbons has likewise / - a d / *-orbitals, the distance in-between them is negligible and so they
can be described as bands. Also the distance between the highest occupied - (HOMO, also Valence
Band, VB, in analogy to the band theory) and the lowest u o upied *-orbital (LUMO, also Conduction
Band, CB) which is called band gap (Eg) decrease with increasing number of repeating units.

109
Figure 2: Schematic explanation of the formation of energy bands in conjugated polymers: Transition
from the atomic orbital of an individual sp2 carbon atom to the molecular orbitals of ethylene and finally
formation of the band structure in polyacetylene. [3]
As Eg is still so large, that electrons cannot easily be promoted from the Valence Band to the Conduction
band just by thermal energy at room temperature, conjugated polymers are semi-conductors or
insulators in the neutral state. However, upon oxidation (extraction of an electron, formation of a radical
cation, Figure 3, right) or reduction (uptake of an electron, formation of a radical anion, Figure 3 left) the
conductivity of such materials is dramatically increased. The oxidation and reduction process is often
called p- and n-doping process, respectively. Thus, conductive polymers are often called p-type (hole
transport) or n-type (electron transport) semi-conductors.
Figure 3: Structure of a neutral polythiophene (middle) as well as a radical cation (oxidized form, right)
and radical anion (reduced form, left).

110
During oxidation electrons are extracted from the HOMO. On the other hand, the reduction of the
polymer is coupled to an uptake of electrons by the LUMO. In both cases, i.e. upon reduction and
oxidation, charges are generated due to the uptake/extraction of electrons. Therefore, the reduction and
oxidation process is often called charging.
In the literature the term polaron is also found for the mono-ionic forms (radical cation and radical
anion) and bipolaron is used for di-ionic forms. These charged species (radical mono-anion and di-ion;
polaron and bipolaron) are stabilized through a delocalization of the charge/electron over the
o jugated -system of several monomer units of the polymer. The neutral form exhibits a benzoid-like
structure, whereas the charged states reveal a quinoid-like structure (Figure 4a). Thus, in the doped
polymer both structures are present. This change in the electronic structure is followed by a geometric
reorganization which causes a local upward shift Δε of the HOMO a d downward shift of LUMO levels,
respectively, see Figure 4b, which results in a decrease of the band gap. This may also serve as an
explanation for the increased conductivity.
Figure 4: a) chemical structure of the neutral state, polaron and bipolaron of polythiophene, b) change of
the electronic structure upon doping
As the color of a conducting polymer is defined by the band gap (absorption correlates to an excitation of
an electron from the HOMO to the LUMO), the change of the band gap will also evoke a color change.
Thus, conducting polymers are electrochromic materials.
Electrochromic materials are compounds that undergo a color change upon switching the redox-state of
the species. A variety of organic compounds show electrochromic behavior. For example, molecular
based materials that show electrochromism are viologens or metal complexes based on phthalocyanine
ligands. In particular polythiophenes and polyanilines are widely used as materials for electrochromic
devices due to their high stability and simple preparation as well as their well-established processability.

111
The optical properties of these materials can be tuned by the choice of appropriate thiophene or aniline
monomers.
Typical colors of a polythiophene film in the neutral and the p-doped state (oxidized form) are red and
blue/green, respectively. Polymers based on PEDOT (poly(3,4-ethylenedioxythiophene)) are of particular
interest because of their low oxidation potentials (high lying HOMO level) as well as their transparency
and high conductivity (hole transport) in the p-doped state. PEDOT polymers are often used in
electrochromic displays, namely in the form of the complex PEDOT:PSS (PSS = polystyrenesulfonate). In
this material PEDOT is present in its oxidized form. The polyanion PSS acts as stabilizer in the complex
with the p-doped PEDOT and ensures electroneutrality. In addition, the PSS part ensures the formation
of a stable aqueous dispersion of this complex which makes it an industrially applicable and solution-
processable form of PEDOT. Because of its high conductivity and transparency (transmission of light is
possible) PEDOT:PSS has found its application as printable electrode in organic solar cells for example.
Organic electrochromic materials have already been applied in anti-glare car rearview mirrors, in
controllable light reflective/transmissive devices, protective eyewear, smart windows (used in
automotive industry and buildings) and sunglasses. One interesting and promising application is the use
as long-term display of information due to the low energy demand of such types of displays.
1.2 Voltammetry / Electropolymerization
Voltammetry:
Voltammetry is one of the most common electroanalytical methods to study the redox-behavior of
electro-active compounds. In particular, cyclic voltammetry (also linear sweep voltammetry) has been
proven as a versatile tool for the characterization of electro-active species in aqueous and non-aqueous
solvents. The electro-active species can be either dissolved in the electrolyte or be present in the solid
state.
In a cyclic voltammetric experiment a linear potential scan with a constant scan rate (v) is performed in a
certain potential window. At the end of the potential window (Eλ) the sign of the scan direction is
inverted and the potential is linearly decreased (with the same v) until the start potential is reached
again (Figure 5 a : the pote tial le is losed . During this potential sweep, the current is recorded and
plotted vs. the applied potential. A typical I-E curve, a so called cyclic voltammogram, of a dissolved
redox-species is depicted in Figure 5 b).
From the corresponding I-E curves several characteristic data are obtained:
redox-potentials (thermodynamic parameters) and
peak currents (diffusion coefficients, kinetic parameters, etc.)
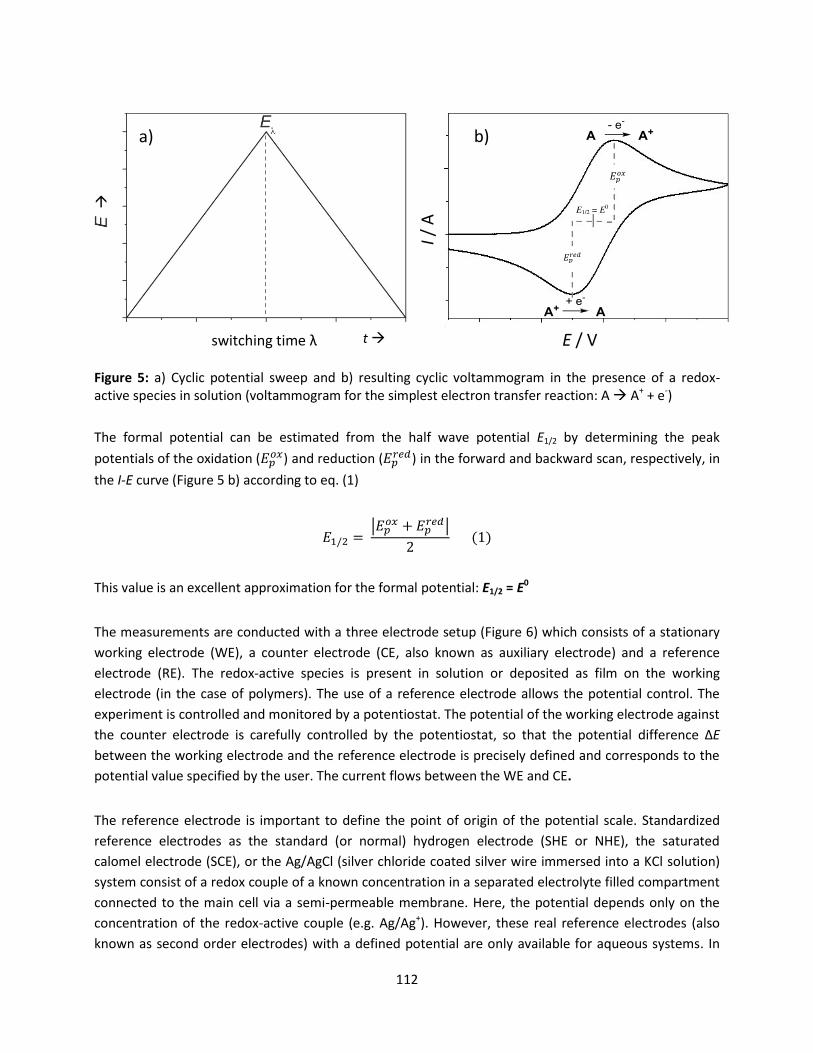
112
Figure 5: a) Cyclic potential sweep and b) resulting cyclic voltammogram in the presence of a redox-
active species in solution (voltammogram for the simplest electron transfer reaction: A A+ + e
-)
The formal potential can be estimated from the half wave potential E1/2 by determining the peak
potentials of the oxidation ( ) and reduction ( ) in the forward and backward scan, respectively, in
the I-E curve (Figure 5 b) according to eq. (1)
| |
This value is an excellent approximation for the formal potential: E1/2 = E0
The measurements are conducted with a three electrode setup (Figure 6) which consists of a stationary
working electrode (WE), a counter electrode (CE, also known as auxiliary electrode) and a reference
electrode (RE). The redox-active species is present in solution or deposited as film on the working
electrode (in the case of polymers). The use of a reference electrode allows the potential control. The
experiment is controlled and monitored by a potentiostat. The potential of the working electrode against
the counter electrode is carefully controlled by the potentiostat, so that the potential differe e ΔE
between the working electrode and the reference electrode is precisely defined and corresponds to the
potential value specified by the user. The current flows between the WE and CE.
The reference electrode is important to define the point of origin of the potential scale. Standardized
reference electrodes as the standard (or normal) hydrogen electrode (SHE or NHE), the saturated
calomel electrode (SCE), or the Ag/AgCl (silver chloride coated silver wire immersed into a KCl solution)
system consist of a redox couple of a known concentration in a separated electrolyte filled compartment
connected to the main cell via a semi-permeable membrane. Here, the potential depends only on the
concentration of the redox-active couple (e.g. Ag/Ag+). However, these real reference electrodes (also
known as second order electrodes) with a defined potential are only available for aqueous systems. In
t
s it hi g ti e λ
a
E / V
I /
A =
E1/2 = E0

113
organic solvents pseudo reference electrodes as an AgCl covered silver wire are commonly used. As the
potential of these is not well defined, IUPAC recommends for organic solvents is to measure an external
standard. Here the standard redox-couple ferrocene/ferrocenium (Fc/Fc+) is added, measured and all
potentials are reported against it (see, rit er . ta J. Recommendations on reporting electrode
potentials in nonaqueous solvents Pure Appl. Chem. 1984 − . This calibration allows the
comparison of different experiments conducted in different solvent systems and with different reference
electrodes.
Figure 6: Three electrode setup for cyclic voltammetric experiments.
To ensure charge transport in solution a supporting electrolyte is necessary. The electrolyte has to be
electrochemically inert for a broad potential window and should be chemically stable (reactions with
oxidized/reduced species must be avoided). Typically tetra-alkylammonium salts are used as cations. As
anions ClO4-, BF4
-, and PF6
- are commonly employed. Furthermore, the supporting electrolyte ensures the
electroneutrality in the diffusion layer in front of the working electrode (the diffusion layer corresponds
to the volume in which the oxidation/reduction takes place). In case of redox-active films, counter ions
are incorporated in the electro-active material during oxidation/reduction which also results in volume
changes of the films.
Electropolymerization:

114
Cyclic voltammetry is a powerful tool for the synthesis (electropolymerization) and characterization of
conducting polymers. A typical anodic electropolymerization takes place via a step-wise coupling of
radical cations. In particular, the electrodeposition of aromatic systems like thiophene or pyrrole has
been extensively studied. The oxidation of such monomers 1 (Figure 7) leads to the formation of radical
cations 2 followed by dimerization 3 and proton elimination. This leads to a neutral dimer 4. Lo ger -
conjugated systems are oxidized at lower potentials than the monomers, therefore the dimer 4 gets
oxidized immediately. Then the same reaction cascade is repeated: The oxidized dimer 5 reacts with
other radical cations and the formation of oligomers 6 takes place. Recent studies propose that the rate
determining step seems to be the elimination of protons rather than the dimerization step. This
mechanism is widely accepted in literature. One of the best models for the polymer growth mechanism
is the so called oligomer-approach. This mechanism proposes that dimerization rates of radical cations of
the same conjugation length are higher than those of coupling products of radical cations with different
conjugation lengths. Hence, the formation of dimers, tetramers and octamers is favored.
The larger the conjugation length the easier the oxidation and, thus, the lower the oxidation potential:
the polymer is oxidized at potentials smaller than the oxidation potential of the monomer unit (see
Figure 7). When the oligomers reach a certain length, the deposition onto the electrode surface takes
place. Further polymer growth includes now several steps under solid-state conditions. All these steps
are taking place as soon as the monomer oxidation potential is reached, and so the polymer oxidation
signal is observable already in the second cycle. Obviously, the peak current of the polymer increases
with increasing number of cycles: the polymer film grows. Voltammetric methods can be used to induce
and follow the polymer growth.
Figure 7: Electropolymerization of polythiophenes, a) cyclic voltammogram of the anodic polymerization
of a thiophene derivative under potentiodynamic conditions (first cycle in red), b) reaction mechanism of
the anodic polymerization of thiophene.
During the potentiodynamic growth of the film, the polymer is oxidized and reduced (to the neutral
form) in each potential cycle which means that the polymer can be obtained as neutral film (the
polymerization stops at a potential lower than the polymer oxidation) on the electrode.
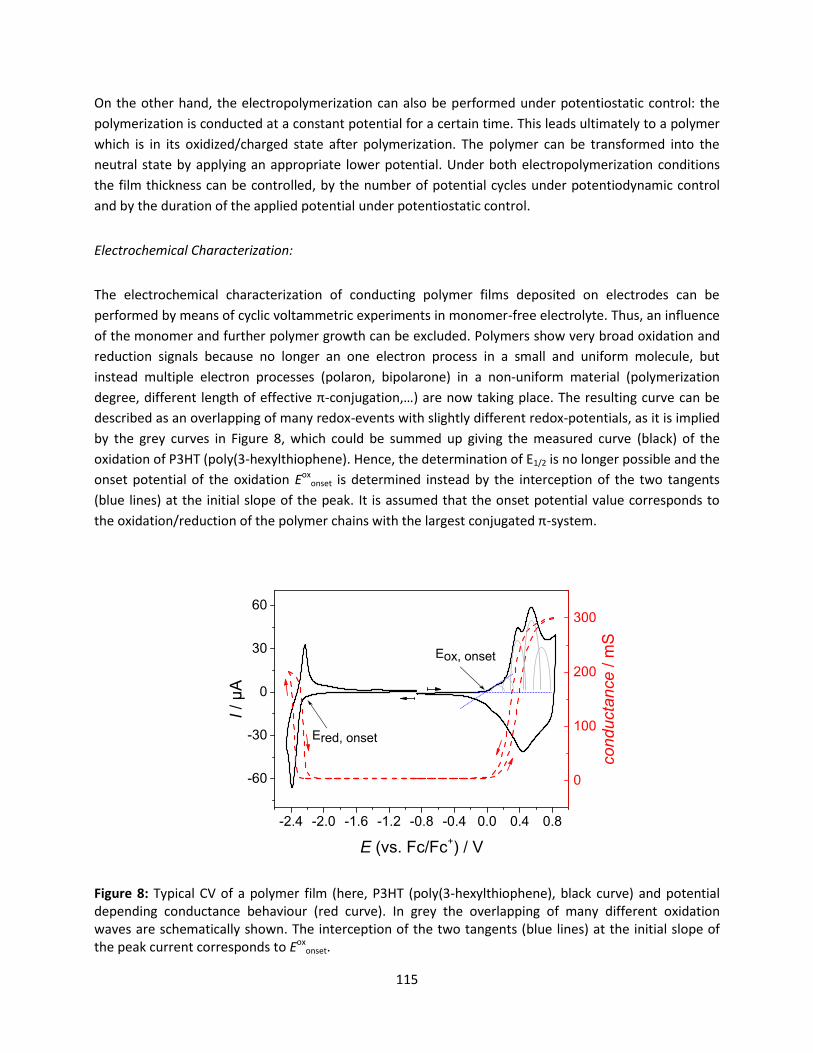
115
On the other hand, the electropolymerization can also be performed under potentiostatic control: the
polymerization is conducted at a constant potential for a certain time. This leads ultimately to a polymer
which is in its oxidized/charged state after polymerization. The polymer can be transformed into the
neutral state by applying an appropriate lower potential. Under both electropolymerization conditions
the film thickness can be controlled, by the number of potential cycles under potentiodynamic control
and by the duration of the applied potential under potentiostatic control.
Electrochemical Characterization:
The electrochemical characterization of conducting polymer films deposited on electrodes can be
performed by means of cyclic voltammetric experiments in monomer-free electrolyte. Thus, an influence
of the monomer and further polymer growth can be excluded. Polymers show very broad oxidation and
reduction signals because no longer an one electron process in a small and uniform molecule, but
instead multiple electron processes (polaron, bipolarone) in a non-uniform material (polymerization
degree differe t le gth of effe ti e -conjugation … are o taki g pla e. The resulti g ur e a e described as an overlapping of many redox-events with slightly different redox-potentials, as it is implied
by the grey curves in Figure 8, which could be summed up giving the measured curve (black) of the
oxidation of P3HT (poly(3-hexylthiophene). Hence, the determination of E1/2 is no longer possible and the
onset potential of the oxidation Eox
onset is determined instead by the interception of the two tangents
(blue lines) at the initial slope of the peak. It is assumed that the onset potential value corresponds to
the o idatio /redu tio of the pol er hai s ith the largest o jugated -system.
Figure 8: Typical CV of a polymer film (here, P3HT (poly(3-hexylthiophene), black curve) and potential
depending conductance behaviour (red curve). In grey the overlapping of many different oxidation
waves are schematically shown. The interception of the two tangents (blue lines) at the initial slope of
the peak current corresponds to Eox
onset.

116
From the onset potential values of the oxidation and reduction, the HOMO and LUMO levels,
respectively, of the polymer can be calculated according to equations (2) and (3) (see, C. M. Cardona et
al. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated
Polymers for Solar Cell Applications, Adv. Mater., 2011, 23, 2367–2371)
[ ]
( )[ ]
The value 5.1 corresponds to the formal potential of Fc/Fc+ in the Fermi scale. From the HOMO and
LUMO level, the electrochemical band gap can be estimated according to eq. (4), which is a critical
quantity for polymer based organic photovoltaics.
| |
For P3HT (poly(3-hexylthiophene), a typical electron donor material in organic photovoltaic devices, the
band gap is ≈ 2.3 eV.

117
2. Experiment
Equipment and General Methods:
For all experiments a PGSTAT101 potentiostat from Metrohm and a gas-tight full glass three electrode
cell containing an ITO covered glass slide as working electrode, a Pt plate as counter electrode and a AgCl
covered Ag-wire acting as the reference electrode are used. All manipulations are carried out under
nitrogen atmosphere using standard Schlenk techniques. Solvents are pre-dried and should be handled
under nitrogen atmosphere.
Chemicals:
10 ml of a 0.1 M NBu4PF6/MeCN solution (NBu4PF6: M = 387.43 g/mol),
3,4-ethylenedioxythiophene (EDOT) in a concentration of 0.02 M M = . 8 g/ ol = . g/mL),
CAUTION: EDOT is toxic!
pure acetonitrile, acetone
Electrodeposition:
Two electrochemical cells equipped with a reference electrode are filled with 10 ml of a 0.1 M
NBu4PF6/acetonitrile solution by means of a syringe under nitrogen atmosphere. The electrolyte solution
is deaerated by nitrogen bubbling for approximately 5 min. Then the calculated amount of monomer is
added via a syringe into one of the cells. The working and counter electrode are carefully placed in the
middle of the cell (Note: the working electrode should be placed near the reference electrode to
minimize the effect of an uncompensated IR-drop, an error that is based on the resistance in the
electrolyte solution). EDOT will be electropolymerized by potentiodynamic and potentiostatic control.
- For the potentiodynamic electropolymerization of EDOT, potential cycles in a specified range and scan
rate are performed. The precise values are given by the supervisor and need to be noted for the later
evaluation of the data. The peak maximum in the forward scan of the first cycle corresponds to the
oxidation potential Eox
of the monomer unit. After all cycles have been performed, the polymer covered
electrode is removed and washed gently with pure acetonitrile.
- For the potentiostatic electropolymerization of EDOT a new ITO electrode is mounted in the cell and a
constant potential is applied for a certain time. The polymer covered ITO electrode is removed and
washed gently with pure acetonitrile. The film - which is in its doped state - is stored for further
UV/Vis/NIR spectroscopic characterization. A second PEDOT film is prepared under identical conditions.
This second film is transformed into its neutral state after the deposition by applying a second constant
potential. The precise values are given by the supervisor and need to be noted for the later evaluation of

118
the data. The working electrode is removed again from the electrolyte solution and the film is washed
with pure acetonitrile. Also this film - which is in the neutral state - is stored for further UV/Vis/NIR
spectroscopic characterization.
After all electrochemical experiments have been performed the ferrocene standard is measured.
Therefore, a blank gold electrode is mounted in the cell and a small amount of ferrocene is added to the
electrolyte solution. For the determination of the formal potential of Fc/Fc+ the potential is cycled
between -0.1 to 0.7 V. For a consistent data set, at least three cycles (with stirring of the solution
between the single cycles) should be performed.
HOMO determination:
For the voltammetric characterization of the polythiophene film, cyclic voltammetry in monomer-free
solution is applied. The cell prepared above without monomer is used. The potentiodynamically
electropolymerized EDOT film is placed in the cell.
For the determination of the onset potential of the oxidation and thus the HOMO level potential three
cycles in a specified range and scan rate are performed. The precise values are given by the supervisor
and need to be noted for the later evaluation of the data. The first cycle is neglected to avoid artefacts
due to memory effects. The value for the onset potential is extracted of the 2nd
or 3rd
cycle.
Electrochromism:
Now, the electrochromic-behavior of PEDOT shall be analyzed. PEDOT is blue in its neutral and
transparent in its oxidized state. Here, the time which is needed for the film to be totally switched from
the one to the other state shall be determined. For that purpose a positive and a negative potential
(most suitable is the upper and lower switching potentials of the cyclic voltammetry performed before) is
repeatedly applied to the film and the current-time-slopes are recorded. As soon as the current stops
flowing the reaction in finished and the polymer film reaches the fully neutral or oxidized state. The
optical impression upon visual inspection (full conversion to the transparent state or not) should be
noted.
UV/Vis/NIR spectroscopic measurements of the charged and neutral PEDOT films deposited under
potentiostatic control shall be performed in transmission mode following the instructions of the
assistant.

119
3. Evaluation
3.1 Polymerizations
a) Determine the E1/2 of Fc/Fc+ of all measured cycles and build the average value out of this. For
evaluation of all your measured cyclic voltammograms substract E1/2 from the measured
potentials and plot all x-scales as E vs. Fc/Fc+.
b) Plot the I/E-curve of the potentiodynamic polymerization. Describe the curve and assign the
regions in the CV to processes happening. Draw and discuss in this context the mechanism of the
oxidative polymerization of EDOT.
3.2 Determination of the HOMO level
a) Estimate the onset potential value of the oxidation according to the protocol depicted in Figure
8. Give also a e pla atio h it’s ot possi le to deter i e E1/2 of conducting polymers.
b) Calculate the HOMO level according to equation (4).
3.3 Analysis of the optical properties of the charged and neutral PEDOT films
a) Describe the two polymer species obtained via the different electrostatic polymerizations,
compare the UV/Vis/NIR absorption spectra and discuss the differences.
b) Calculate the optical bandgap Eg from the UV/Vis/NIR absorption spectrum of the neutral PEDOT
film by determining the intersection of two tangents at the onset of the absorption, giving the
lowest energy which is absorbed by the polymer film.

120
4. Questions
a) Describe a chemical method for the synthesis of a polythiophene polymer based on a transition
metal complex catalyzed reaction (draw the reaction scheme in which the structure of the
monomer and the resulting polymer shall become clear as well as the catalytic cycle!).
b) Draw the molecular structure of PEDOT:PSS (poly(3,4-ethylenedioxythiophene):
polystyrenesulfonate) in its quinoid stabilized form. In which form is the polystyrenesulfonate
present? Name at least two applications of this conducting polymer.
c) Describe a detailed buildup of an electrochromic window.
d) Name two further electrochemically synthesized polymers and draw their structures.
e) The polymer P3HT is a common donor material in donor-acceptor based organic solar cells. The
acceptor material is often composed of a fullerene derivative (typically PCBM =[6,6]-Phenyl C61
butyric acid methyl ester). Draw a sketch of a polymer based organic solar cell. Draw an energy
diagram containing the HOMO and LUMO levels of the donor and acceptor material.

126
Experiment 10
Viscosimetry
Task
In this experiment the Staudinger index as well as the molar mass of a polystyrene
sample is to be determined by viscosimetry.
Literature
[1] M. Hoffmann, H. Krömer, R. Kuhn, Polymeranalytik, Bd. 1, Thieme Verlag
Stuttgart, 1977.
[2] D. Braun, H. Cherdron, H. Ritter, Praktikum Makromolekularer Stoffe, Wiley-
VCH-Verlag, Weinheim, 1999.
[3] H.-G. Elias, Makromoleküle, Bd. 2, Wiley-VCH-Verlag, 2001.
[4] W.-M. Kulicke, C. Clasen, Viscosimetry of Polymers and Polyelectrolytes,
Springer-Verlag, Berlin, 2004.
[5] B. Tieke, Makromolekulare Chemie, 2. Auflage, Wiley-VCH-Verlag, Weinheim
2005.
[6] Polymer Handbook, 2nd edition, John-Wiley & Sons, New York 1975.

127
Contents
1. Theoretical background
1.1. Determination of molar masses by relative methods
1.2. Viscosimetry
1.3. Viscosity of polymer solutions
1.4. Practical implementation of viscosimetry
2. Experiment
2.1. Procedure
2.2. Evaluation
3. Questions

1. Theo
1.1. De
Relativ
directly
curve i
chemis
chroma
on the
macro
the prin
polyme
concen
1.2. V
In a st
Starting
parabo
describ
F
Even w
conside
oretical ba
eterminati
e methods
y related pr
s always
stry are v
atography)
shape of
conformat
nciples tha
er solutions
ntration ser
iscosimet
treaming f
g from the
olic velocity
bed as a se
igure 1: Ve
when pres
erable incr
ackground
on of mol
s determin
roperty. Ho
required. T
iscosimetr
. The para
f the macr
tion. The v
at are valid
s deviation
ries as wel
try
fluid the g
wall of th
y profile is
equence of
elocity pro
sent in sm
rease in th
d
ar masses
ne the mol
owever, in
The most
y and ge
ameters ac
romolecule
validation o
in the idea
ns are obse
l as extrap
gross flow
e wall of th
built up. F
f fluid laye
ofile for lam
v =
mall conce
he viscosity
128
s by relati
lar mass o
order to va
frequently
el permeat
ccessible t
es in solut
of all absol
al dilute st
erved. Give
polation to
rate is in
he vessel,
For simplifi
rs with diff
minar flow i
= flow rate)
entrations,
y. For a w
ive metho
of a polym
alidate the
y applied r
tion chrom
through bo
tion which
lute and re
tate. Howe
en these li
infinite dilu
versely pr
through w
ication pur
ferent flow
in a capilla
).[4]
polymer
weighed po
ds
mer sample
obtained d
elative me
matography
oth method
is referre
elative met
ver, even
mitations,
ution are vi
roportional
which the l
rposes, this
rates.
ry (r = capi
molecules
ortion of a
e by meas
data, a cal
ethods in p
y (size ex
ds are dep
ed to by th
thods is ba
in strongly
measurem
ital.
l to the vi
iquid is flo
s gradient
illary radiu
s can lea
specific p
suring a
libration
polymer
xclusion
pendent
he term
ased on
y diluted
ments of
scosity.
owing, a
can be
us,
ad to a
polymer,

129
the increase of the viscosity caused by this polymer is not solely dependent on its
molar mass, but is also affected by the dimensions of the macromolecules.
Assuming a diameter of 2-10 Å for the solvent molecules and 500-1000 Å for a
macromolecular coil, it is clearly noticeable that one macromolecular coil can extend
over a number of solvent layers in a current gradient. This leads to an assimilation of
the flow rates. As a consequence, the gross flow rate decreases, which corresponds
to an increase of the macroscopic viscosity of the solution. It is important to note that
this increase in viscosity is not only dependent on the concentration of the polymer
but also on the spatial dimensions of the polymer coil in solution.
For statistically coiled macromolecules their macroconformation itself is dependent
on the solvent and also on the temperature of the apparatus. Usually, the efficacy of
solvents improves with increasing temperature. This results in bigger coils and thus
an increasing viscosity. Therefore, measurements of the viscosity always have to be
conducted at a constant temperature and in the same solvent when being used as a
relative method to determine molar masses.
1.3. Viscosity of polymer solutions
In connection with the investigation of the viscosity of polymer solutions different
kinds of viscosities are distinguished:
- Relative viscosity rel
- Specific viscosity sp rel 1
- Reduced viscosity red spc
- Inherent viscosity inh relc
Furthermore, the viscosity number (intrinsic viscosity, Staudinger index) is defined as
follows:
= Viscosity of the solvent
= Viscosity of the polymer solution
c = Concentration of the polymer solution

η rep
shear r
plottingηspc vers
temper
Fi
In mos
shear r
growing
neglect
the flow
1.4. P
resents the
rate of G
g ηspc versus
sus c for
rature of 25
gure 2: Re
molec
st cases it
rate. Even
g molecul
t this effec
wing mater
ractical im
e limit of th
= 0. It is p
s c. This of
polystyren
5 C.
duced visc
cular weigh
is forborne
though sta
ar weight
ct when vis
rial are use
mplementa
ηhe quotien
possible to
ften leads t
ne sample
cosities as
hts of polys
e to condu
atistical co
under the
scometers
ed.
ation of vi
130
limc→G→
nt ηspc at a c
o perform
to a linear
es exhibiti
a function
styrene (me
uct an extr
oils of poly
e influence
showing o
scosimetr
concentrati
a graphica
correlation
ing differe
n of the con
easured in
rapolation
mers tend
e of shea
only a sma
ry
on of c = 0
al extrapo
n. Figure 2
nt molecu
ncentration
toluene at
to c = 0 w
to increas
ar, it is us
all gradient
0 and in ca
olation to c
2 shows the
ular weigh
n for differe
t 25 C).
with respec
singly defo
sually pos
t in the ve
(1)
ase of a
c = 0 by
e plot of
ts at a
ent
ct to the
orm with
sible to
locity of

Measu
Ostwal
shows
Fig
Hagen-
determ
rements a
d- and the
a schemat
gure 3: Sch
-Poiseuille
ination of t
are perform
e Ubbelohd
tic illustrati
heme of an
’s law pro
the viscosi
r = capil
t = flow Δ = pre
l = capil
V = cap
= dens
h = heig
med in so-
de-viscome
ion of both
n Ostwald-
vides the
ity η of a p
llary radius
time
essure diffe
lary length
illary volum
sity of the
ght
131
-called cap
eter belong
h types of v
(left) and a
theoretica
olymer sol
∙ ∙with Δ
s
erence at t
h
me
solution
pillary visc
g to the mo
viscometer
an Ubbeloh
al basis for
lution:
∙ ∙∙
∙ g ∙ h.
the capillar
cometers, a
ost frequen
s.
hde-viscom
r the meas
ry
among wh
ntly used. F
meter (right
surement a
hich the
Figure 3
t).[5]
and the
(2)

132
The measurement itself is based on determining the time that a polymer solution
takes to travel a distinct distance in the capillary of the viscometer.
Considering Hagen-Poisseuille’s law the specific viscosity sp can be expressed in
the following way:
sp . (3)
As for small concentrations the density of the solution and the density of the
solvent are approximately the same, one can simplify (3) as follows:
sp . (4)
For linear, statistically coiled polymers the Staudinger index is linked to the molecular
weight via the Mark-Houwink equation, where represents the viscosity average
molar mass.
η K∙Mηa . (5)
The variables K and a are dependent on the geometrical form of the dissolved
polymer coil as well as on the type of solvent and the temperature. The value of a in
particular reflects the conformation of the polymer and is closely related to the
second virial coefficient. Usually the parameter a adopts values in the range between
0.6 and 0.9. In θ-solvents and at the corresponding θ-temperature a has a value of
0.5. In case of a non-perfused coil one obtains a = 0 and for a rigid rod a = 2. For
polymers with a known molar mass it is possible to extract the a and K values from a
double logarithmic plot of η versus resulting in a straight line where a and K are
represented by the slope and the y-intercept, respectively. Specific values for the
parameters a and K are documented in the literature for a variety of polymer-solvent-
temperature-systems.
In addition, the Staudinger index of a polydisperse substance without any interactions
can be expressed as an average weight (Philippoff-equation):

Consid
polyme
The mo
molar m
On exaM . Wi
in orde
is advi
method
ultrace
Finally,
determ
correla
than br
a sma
obtaine
ering this
er sample i
ost importa
mass and v
amination o
ith a decre
er to elimin
sable to c
d, which
ntrifuge).
, it shall als
ine the de
tion is the
ranched po
ller Staud
ed by meas
equation,
s given by
ant differen
viscosity m
of equatio
easing valu
ate the inf
conduct th
is avera
so be men
egree of br
e fact that
olymers of
dinger inde
suring a lin
, the visco
y (7).
nce betwe
molar mass
n (7) it is e
ue of M th
fluence of t
e calibrati
ging in
ntioned tha
ranching fo
linear pol
the same
ex. Figure
near and a
133
η =∑ wi∑ wi
osity aver
M ∑en numbe
s is the fact
easily see
his parame
the polydis
ion of the
a similar
at viscosim
or branche
lymers ge
molecular
e 4 shows
branched
i[η]iwi
.
rage mola
∑∑ .
r average
t that M d
n that in t
eter becom
spersity of
Staudinge
way (e.
metry additi
ed macrom
nerally sho
weight, an
s an exam
polymer.
r mass fo
molar mas
epends on
he case of
es smaller
the invest
er index u
g. static
onally prov
molecules.
ow smalle
nd consequ
mple of c
or a polyd
ss, mass a
n the expon
f a = 1 Mr than M .
tigated pol
using an a
light sca
vides an o
The basis
er coil dim
uently also
calibration
(6)
disperse
(7)
average
nent a.
equals
Hence,
ymer, it
absolute
attering,
option to
for this
ensions
o exhibit
curves

Fi
poly
2. Ex
2.1. P
Initially
polysty
then fil
syringe
polyme
polyme
mark.
Prior to
the sto
heating
pressin
automa
about 2
measu
igure 4: Int
yethylene (
periment
rocedure
, portions
yrene samp
led with c
e attachme
er, the flas
er is comp
Figure 5:
o the meas
rage vesse
g bath and
ng the Sta
atic tempe
2 cm abov
re the pas
trinsic visc
(HDPE) and
s of about
ple are we
circa 18 mL
ent filters be
sks are sw
pletely diss
Scheme of
surement,
el (4) throu
d connect
art-button
ring the li
ve the char
ss-through
cosity as a
d long-chai
tetra
t 200 mg,
eighed out
L of toluen
efore usag
wung in t
solved, the
f the Ubbel
the solutio
ugh the wid
ted to the
the meas
quid is pu
racteristic
time of th
134
function o
in branche
ralin at 120
, 100 mg,
t into 20 m
ne (Note: A
ge). In orde
he heating
e flasks ar
lohde-visco
ons as we
de tube (3)
pressure
surement
ushed thro
mark (M1)
he solution
of the molec
ed polyethy
C.[4]
50 mg a
mL-graduat
All toluene
er to achiev
g bath of
re filled up
ometer use
ll as the p
). The visc
hoses at
is initiated
ugh a cap
). The prog
between
cular weigh
ylene (LDPE
and 25 mg
ed flasks.
e has to b
ve a faster
the visco
p with solv
ed in the ex
ure toluen
ometer is t
t positions
d. After a
pillary (7)
gram will t
the marks
hts for line
E) measure
g of the
These fla
be filtered
r dissolutio
ometer. On
vent to the
xperiment.
ne are pou
then place
s (1) and
a few min
up to a he
then autom
s (M1) and
ear
ed in
existing
sks are
through
on of the
nce the
e 20 mL
red into
ed in the
(2). By
utes of
eight of
matically
(M2) 5

135
times in succession. If these 5 measurements do not lead to reproducible results,
another set of 5 measurements should be performed. Important: Every time you start
a new set of measurements, all values of the previous measurement are deleted.
Therefore, it is crucial to copy the results manually. Between the measurements the
viscometers are thoroughly rinsed with toluene (3x) and acetone (2x) and
subsequently dried with compressed air.
2.2. Evaluation
All measured values are summarized in one table. The values that are important for
the following calculations are the corrected average values. According to equation
(4) sp is determined. The values thus obtained are then used to calculate sp by
dividing sp by the respective concentrations. Plotting sp versus c (g/mL) and
extrapolation to c = 0 gives the Staudinger index η , from which the molar mass of
the polystyrene sample can be calculated according to equation (5).
K- and a-values for polystyrene in toluene at 30 °C: K = 0.012 mL/g, a = 0.71.[6]
3. Questions
1) How and why do the Staudinger indices of polystyrene, polystyrene-lithium
and sulfonated polystyrene differ?
2) Which factors for the determination of the Staudinger index are no longer
negligible at concentrations below 2 · 10-2 g/mL?
3) What is the difference between Ostwald- and Ubbelohde-viscometers?
4) Which solution exhibits the bigger Staudinger index – it-PMMA/3-heptanone or
at-PMMA/3-heptanone? (same molecular weights and measuring conditions)

Experiment 9 Size Exclusion Chromatography
‐137‐
Size exclusion Chromatography (SEC)
Shorttaskdescription
The objective of this laboratory course is to analyze the characteristic column parameters for
a given SEC system (calibration curve, number of theoretical plates) and to determine the
molecular weight as well as the polydispersity index (PDI) for two polystyrene‐based
samples: a homopolymer prepared by radical polymerization and a polystyrene‐block‐
polyisoprene block copolymer synthesized by ionic polymerization.
Theoreticalbackground
Generalinformation
Size Exclusion Chromatography (SEC) has evolved into a modern routine method for the
determination of the average molecular weight as well as the molecular weight distribution
of a polymer sample.
As a general principle of this separation technique, macromolecules are separated on a
chromatographic column according to their size, or more precisely, their hydrodynamic
volume in a given solvent. The hydrodynamic volume determines the degree of permeation
of the macromolecules into the porous material of the stationary phase and depends mainly
on the molecular weight, but also on the chemical and physical nature of the polymer, its
constitution and conformation in the solvent as well as the temperature.
In common with other chromatographic techniques, the column setup, on which the
polymer samples are separated, consists of a mobile phase moving with respect to a
stationary phase. In the specific case of SEC, the chromatographic column is filled with beads
of a porous gel, which are surrounded and swollen by the solvent. The small solvent‐filled
pores of the gel form the stationary phase (Figure 1), while the mobile phase is represented
by the solvent outside of the pores.

Experim
Figure 1
gel phas
Driven b
macrom
position
When t
pores, m
this beh
can per
case eit
the upp
on the m
(1)
(2)
KD repre
accessib
on the h
affected
to deriv
ent 9
1. Schemati
se, V0: volum
by the conc
molecules di
n of the equ
he hydrody
molecules c
havior is cal
meate into
ther, which
per and the
molecular w
esents the s
ble pores. T
hydrodynam
d by multipl
ve a direct c
Po
c illustratio
me of the m
centration g
iffuse into t
uilibrium de
ynamic volu
annot be se
led upper li
all pores. C
is referred
lower limit,
weight of th
separation c
The separati
mic volume
le paramete
correlation b
lymer solut
Size Exclu
n showing a
mobile phas
gradient bet
the pores of
pends on th
me of the p
eparated ac
mit (UL). O
Consequent
to as the lo
, the elution
e analyzed
coefficient,
ion coefficie
of the com
ers such as
between KD
ion
usion Chrom
‐138‐
a cross sect
se, VT: total
tween the m
f the gel un
he hydrody
polymer cha
ccording to
n the other
tly, effective
ower limit (L
n volume Ve
compound
which can
ent can ado
mpound. How
the temper
D and the m
atography
tion of the S
volume.
mobile and
til the equi
ynamic volu
ains is too b
their molec
r extreme, v
e separation
LL). For hydr
e is a charac
.
be regarded
opt values b
wever, as th
rature, the s
olecular we
SEC column
stationary p
librium is re
me of the m
big for them
cular weight
very small m
n cannot be
rodynamic v
cteristic par
d as the pro
etween 0 a
he hydrodyn
solvent etc.
eight.
. Vx: volume
phase the
eached. The
macromolec
m to enter an
t. The thres
macromolec
e achieved i
volumes be
rameter dep
oportion of
and 1 and de
namic volum
., it is not po
e of the
e
cules.
ny
shold for
cules
n this
etween
pending
epends
me is
ossible

Experim
Figure 2
derived
a polym
For that
individu
molecu
a functi
directly
shape c
polyme
(3)
ent 9
2. The relati
by the mea
mer sample(
t reason, ea
ual calibratio
lar weight d
on of the e
proportion
an be descr
r with the e
ionship betw
asurement
bottom) are
ach SEC syst
on perform
determined
lution volum
nal to Ve in t
ribed by a li
elution volul
Up
Molecular w
eight /
gmol‐1
Signal
Size Exclu
ween the e
of standard
e illustrated
tem (colum
ed by meas
by absolut
me Ve on a l
the region b
inear regres
me Ve accolog
pper limit
Elu
usion Chrom
‐139‐
lution volum
d samples (t
d.
n, polymer,
suring polym
e methods.
logarithmic
between the
ssion line co
ording to eq
ution volume V
Elution vo
Polymer s
atography
me Ve and l
top) as well
, solvent an
mer standar
. When the
scale, it be
e upper and
orrelating th
uation (3).
Lower l
Ve
olume Ve
sample
og (M) (cali
as a typica
d temperat
rds with a w
molecular w
comes obvi
d the lower
he molecula
imit
ibration cur
l chromatog
ture) requir
well‐defined
weight is pl
ious that lo
r limit. The c
ar weight o
rve)
gram of
res an
d
otted as
g (M) is
curve
f the

Experim
Experi
Figure 3
In gene
1. S
g
w
s
s
2. T
T
s
c
3. T
S
Injec
Sa
ent 9
imentals
3. Schemati
ral, the SEC
Solvent pum
Due to the
generated b
which is fre
sample volu
solvent.
The column
The actual s
In order to
several colu
in made fro
is micropor
resistance.
chloroform
The detecto
olvent
ctor
mple
etup
c illustratio
C system can
mp + inject
high flow re
by a high pr
ee of pulsati
ume into th
n
separation
cover a bro
umns with d
om polystyr
rous glass, w
Typical solv
.
or
Pump
Porous particles
Size Exclu
n showing a
n be divided
ion
esistance of
ressure pum
ion. An app
he SEC‐syste
of the comp
oader range
different po
ene crosslin
which is par
vents used f
Co
s
usion Chrom
‐140‐
a typical set
d into three
f the SEC sy
mp is requir
propriate inj
em without
ponents tak
e of molecul
ore sizes in s
nked by dive
rticularly po
for SEC are
olumn
atography
tup of a SEC
e main comp
ystem a con
ed to provid
jector enab
disturbing
kes place on
lar weights,
series. In m
enyl benzen
opular due t
tetrahydro
D
D
C apparatus
ponents:
stant press
de a consta
les the inse
the constan
n a chromat
it is also po
ost cases th
ne. Another
o its relativ
furan (THF)
Detector 1
Detector 2
s.
ure of 5 to
ant solvent f
ertion of a d
nt flow rate
tographic co
ossible to co
he column m
r common m
vely low flow
), toluene a
500 atü
flow
efined
e of the
olumn.
onnect
material
material
w
nd

Experim
c
v
These
Figure 4
shows a
The sma
be sepa
technica
contribu
benzene
system,
signal, c
(4)
(5)
The leng
the heig
frequen
ent 9
In order to
concentrati
positioned
viscosity or
eparation
4. Detector
a Gaussian s
aller the slo
arated on th
al broadeni
ution can be
e. As a quan
, the numbe
can be estim
gth of the c
ght equivale
ntly used to
achieve a c
ion, a UV‐V
at the end o
light scatte
performa
signal S plo
shape with
ope betwee
he column. D
ng of the G
e derived fr
ntitative pa
er of theore
mated accor
column norm
ent of theor
characteriz
Size Exclu
ontinuous q
is absorptio
of the chrom
ering detect
ance
otted as a fu
standard de
n the upper
Due to the f
aussian pea
rom measur
rameter rep
etical plates
rding to equ
/ /malized for
retical plate
ze the perfo
usion Chrom
‐141‐
quantitative
on or refrac
matograph
tors can be
unction of th
eviation σ.
r‐ and the lo
flow profile
ak recorded
rements of
presenting
s (N), which
uation (4).
∙/
the numbe
es (HETP). B
ormance of
T
atography
e determina
tive index (
ic column. A
applied.
he elution v
ower limit t
e on the col
d on the det
a monomo
the separat
is related t
/
er of theore
Both parame
a SEC syste
Turning Points
ation of the
RI) detecto
Alternativel
volume Ve. T
the better th
umn there w
tector. This
lecular subs
tion perform
o the broad
tical plates
eters, N and
em.
s
e polymer
r is typically
ly, fluoresce
The elution
he molecul
will always
instrument
stance such
mance of th
dening of th
is referred
d HETP, are
y
ence,
profile
es will
be a
tal
h as
he
he
to as

Experiment 9 Size Exclusion Chromatography
‐142‐
Dataanalysis
Synthetic polymers usually comprise a mixture of molecular weights. Therefore, the
characteristic elution profile of a polymer sample represents the sum of all components.
Even though the separation of all the different molecular weight fractions into completely
distinct signals proofs to be impossible in many cases, the shape of the elution curve
provides information about the molecular weight distribution of the polymer.
From the distribution profile of the molecular weight, the number‐averaged molecular
weight Mn as well as the weight‐averaged molecular weight Mw can be derived as
characteristic parameters for the description of a polymer sample. These properties are
defined as follows:
(6) ∑∑ ∑∑ ∑∑
(7) ∑∑ ∑∑ ∑∑
To each value of Ve in the elusion diagram a molecular weight Mi can be assigned, while the
abundance of molecule i is related to the detector intensity Hi.
Experimentaldetails
The aim of this practical course is to use the SEC method to determine the average
molecular weight values Mn and Mw as well as the molecular weight distribution for two
polymer samples, the first of which being a polystyrene homopolymer synthesized by radical
polymerization (sample from experiment 2), while the second one represents a polystyrene‐
block‐polyisoprene copolymer prepared by ionic polymerization (sample from experiment 7).
The calibration curve for the specific SEC system used in the experiments will be provided by
the demonstrator.
Prior to the analysis, the polymer samples have to be dissolved in THF (approximately 5%
w/w) followed by filtering through a syringe filter with 450 µm pore diameter. Subsequently,
the samples are applied onto the SEC system to record the chromatogram. The number of
theoretical plates and the HETP value should be determined by the evaluation of the o‐
dichlorobenzene signal.

Experiment 9 Size Exclusion Chromatography
‐143‐
Requirementsfordataanalysisandreport
Introduction including the theoretical background of the method and a description of the experimental setup.
Comparison of the different elution curves and detailed description of all signals. Analysis of the molecular weight distribution and averaged molecular weights Detailed discussion and comparison of the results.
Questions
1. How is the column volume defined?
2. Which absolute methods for the measurement of the molecular weight do you know
and how can you determine the molecular weight distribution?
3. How can continuous degassing of the solvent be achieved?
4. Which parameters influence the separation performance of the SEC‐system?
5. Which requirements do the pump and detectors have to fulfill?
6. Explain the setup of a differential refractometer!

144
Experiment 9
Thermal Analysis of Polymers by Means of Differential Scanning Calorimetry (DSC)
Task
Different thermal phase transitions of amorphous and semi-crystalline polymers
should be investigated by means of DSC.
Literature
1) H.-G. Elias, Makromoleküle, Bd. 2: Physikalische Strukturen und Eigenschaften, Wiley-VCH
Weinheim, 2001
2) W.F. Hemminger, H.K. Cammenga, Methoden der thermischen Analyse, Springer-Verlag, Berlin
1989
3) G.W.H. Höhne, W.F. Hemminger, H.-J. Flammersheim, Differential Scanning Calorimetry, Springer-
Verlag, Berlin 2003
Content
1. Theoretical Background
1.1 Polymers in the Solid State
1.2 Thermal Phase Transitions
1.3 Analysis of Thermal Phase Transitions
1.3.1 Measuring Principle of DSC
1.3.2 Thermal Phase Transitions - Examples and Interpretation
2. Experimental Section
3. Evaluation
4. Questions

145
1. Theoretical Background
1.1 Polymers in the Solid State
a) Amorphous Polymers
In the melt state, polymers exist as random coils (undisturbed dimensions, -state),
which interpenetrate each other. If the polymer exhibits a sufficiently high molecular
weight, this effect leads to a rubber-elastic melt, where the elasticity is based on
entanglements between the chains.
Upon temperature reduction, the viscosity of the melt increases strongly until a glassy
solidification occurs. The glass shows a similar microstructure as the melt and the
characteristic transition temperature is referred to as the glass temperature Tg. At the
molecular level the mobility of the chains becomes strongly reduced during the
transition from melt to glass. Further cooling leads to a reduced mobility of the main
chains and thus the material becomes brittle.
The formation of a glass, which does not have a possibility for crystallization, requires
an irregular structure of the polymer chains. A frequently used approach to obtain
amorphous structures of polymers that are otherwise capable of crystallization, is the
disturbance of the crystallization process by the introduction of comonomers or by
rapid quenching from melt.

b) Sem
Macrom
polyoxy
chain
arrange
Slow c
below t
crystall
crystall
crystall
treatme
only be
from hi
There
polyme
chain fo
semi-cr
caused
paralle
lamella
either t
chains
mi-crystallin
molecules
ymethylene
structure
ement.
ooling of s
the melting
ites due to
ization. Ch
ization deg
ent. It is g
een prove
ghly dilute
are two m
ers, the frin
olded crys
rystalline p
d by chain
l with thei
ae. The am
the same
are stretch
ne Polymer
with linea
e, PTFE)
enables
such polym
g point are
o kinetic a
hain foldin
gree α dep
enerally ve
n possible
solutions)
models us
nged micel
tal (figure
polymers
folding. In
r longitudi
morphous re
or the ad
hed or sho
a)
rs
ar and sy
are partic
s a we
mers from
e reached.
nd thermo
g occurs a
pends on
ery difficul
e for a few
).
sually app
le, where t
1b). X-ray
revealed
n these cry
nal axes a
egions bet
djacent lam
w a helica
146
ymmetrical
cularly pro
ell-ordered
melt leads
Usually th
odynamic c
and the th
the polym
lt to obtain
w polymer
plied to de
the chains
analysis a
a lamellar
ystalline re
aligned pe
tween the
mella. Usu
l arrangem
ly constru
one to crys
, close-p
s to crysta
he chains a
constrains,
us created
er structur
n single cr
rs under w
escribe th
are fully e
and electro
r structure
gions the
erpendicula
lamellae c
ually within
ment.
ucted chain
stallization
packed, t
llization wh
are not full
which hin
d folds are
re and the
ystals of p
well-defined
e semi-cr
extended (f
on microsco
e of the c
polymer ch
ar to the to
onsist of c
n the lame
b)
ns (polyet
n, as their
three-dime
hen tempe
ly extende
nder the co
e amorpho
previous
polymers a
d condition
rystalline s
figure 1a)
copy perfor
crystalline
hains are
op surface
chains whic
ellae the p
thylene,
simple
ensional
eratures
d in the
omplete
us. The
thermal
and has
ns (e.g.
state of
and the
rmed on
regions
all lying
e of the
ch enter
polymer

147
Fig. 1:
Schematic illustration of fringed micelle (a) und chain folded crystal with two different
kinds of chain folding (b).
1.2 Thermal Phase Transitions
Low molecular weight molecules show sharp changes in their physical properties
(melting point, ceiling point) at precisely defined temperatures. Polymers undergo
such thermal phase transitions as well, but usually these processes cover a broader
temperature range. Notably, a glass transition region and a melting region can be
identified.
Thermal phase transitions can be categorized into first and second order phase
transitions. First order transitions show jumps in the first derivation of the Gibbs free
energy G with respect to pressure or temperature (H, S and V). Hence, jumps appear
in the second derivation of G as well. Melting of polymers represents a first order
phase transition.
Second order phase transitions show a jump in the second derivation of G with
respect to pressure or temperature (, Cp oder but not in thefirstWhile the glass
transition indeed shows such jumps in or Cp, the value of the glass transition
temperature Tg is path-dependent (heating/ cooling rate). This type of transition is
called pseudo second order.
1.3 Analysis of Thermal Phase Transitions
Thermal phase transitions are accompanied by characteristic changes in V, H and S
of the sample. Thus, these properties were used to investigate phase transitions. In
this context, calorimetrical methods are of great importance. Due to the huge
experimental effort (adiabatic calorimetry) associated with a direct determination of
the enthalpy, differential calorimeters are frequently applied.
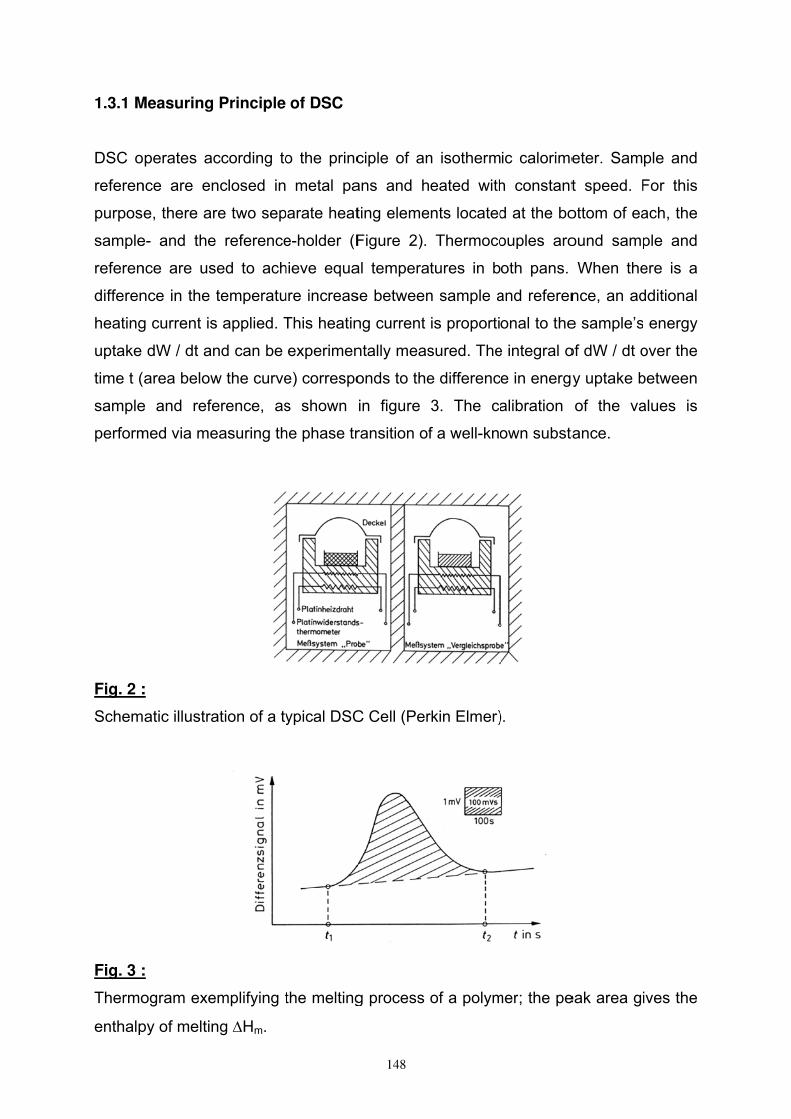
1.3.1 M
DSC o
referen
purpos
sample
referen
differen
heating
uptake
time t (
sample
perform
Fig. 2 :
Schem
Fig. 3 :
Thermo
enthalp
Measuring
operates ac
nce are en
e, there ar
e- and the
nce are us
nce in the
g current is
dW / dt an
area below
e and refe
med via me
:
atic illustra
:
ogram exe
py of meltin
Principle
ccording to
nclosed in
re two sep
e reference
sed to ach
temperatu
s applied. T
nd can be
w the curve
erence, as
easuring th
ation of a ty
emplifying t
ng Hm.
of DSC
o the princ
metal pa
parate heat
e-holder (F
hieve equa
ure increas
This heatin
experimen
e) correspo
s shown
he phase tr
ypical DSC
the melting
148
ciple of an
ans and h
ting eleme
Figure 2).
al tempera
se between
ng current
ntally meas
onds to the
in figure
ransition of
C Cell (Per
g process
n isotherm
eated with
ents located
Thermoco
tures in b
n sample a
is proporti
sured. The
e differenc
3. The ca
f a well-kno
rkin Elmer)
of a polym
ic calorime
h constant
d at the bo
ouples aro
oth pans.
and referen
onal to the
e integral o
e in energy
alibration
own substa
).
mer; the pe
eter. Sam
t speed. F
ottom of ea
ound samp
When the
nce, an ad
e sample’s
of dW / dt o
y uptake b
of the va
ance.
eak area gi
ple and
For this
ach, the
ple and
ere is a
dditional
energy
over the
between
alues is
ives the

Hm
1.3.2 T
a) Glas
Heating
of the
state. T
which i
Fig. 4 :
Thermo
Blockco
glass tr
b) Mel
= HProbe
Thermal Ph
ss transiti
g an amor
linked cha
This chang
s reflected
:
ogram of a
opolymers
ransitions.
lting of se
e HR
hase Tran
on in amo
rphous poly
ain segme
ge in mobi
d in a prono
an amorpho
consisting
emi-crysta
Referenz
sitions - E
orphous p
ymer to th
nts (30-50
lity is acco
ounced ste
ous polyme
g of two a
lline polym
149
Examples
olymers
e glass te
0 chain lin
ompanied
ep in the th
er.
amorphous
mers
and Interp
mperature
ks), which
by a chan
hermogram
s blocks c
pretation
leads to i
h were froz
nge in the
m as shown
can show
ncreased
zen in the
heat capa
n in figure 4
two indep
mobility
e glassy
acity Cp,
4.
pendent
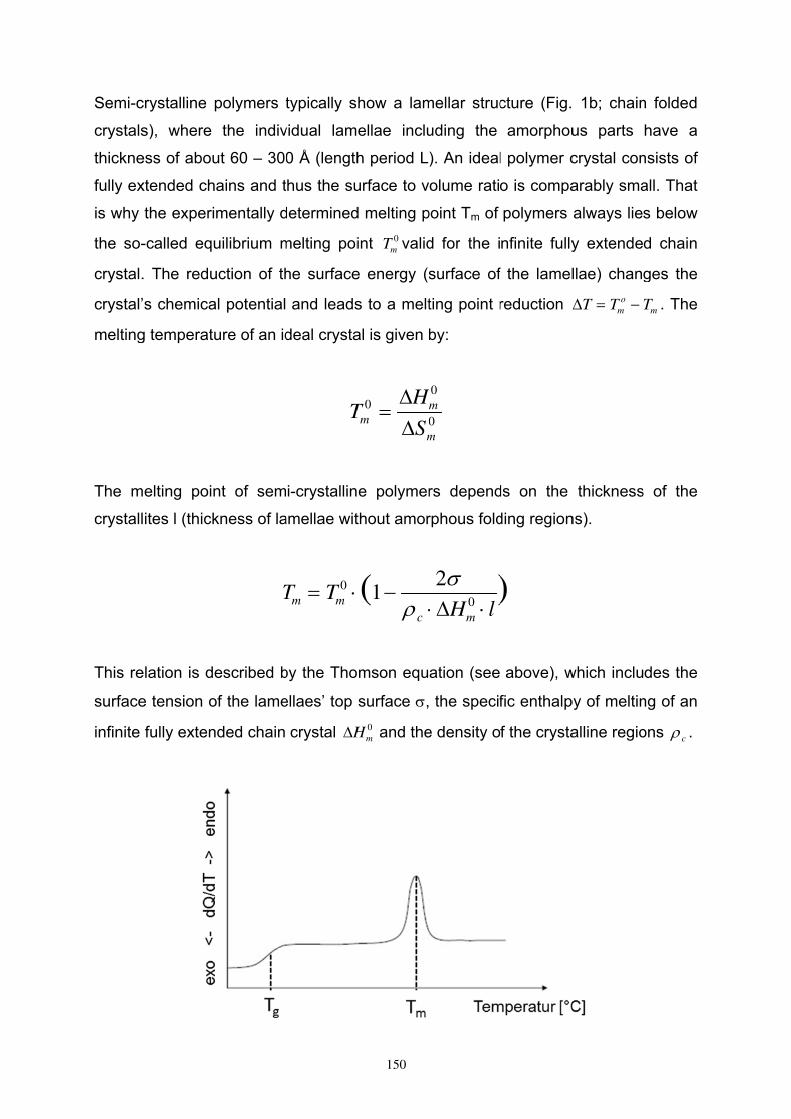
Semi-c
crystals
thickne
fully ex
is why
the so-
crystal.
crystal’
melting
The m
crystall
This re
surface
infinite
crystalline
s), where
ess of abou
xtended ch
the experi
-called equ
. The redu
s chemica
g temperatu
elting poin
ites l (thick
elation is d
e tension o
fully exten
polymers t
the indiv
ut 60 – 300
hains and t
mentally d
uilibrium m
uction of th
al potential
ure of an id
nt of sem
kness of la
T
escribed b
of the lame
nded chain
typically s
vidual lam
0 Å (length
thus the su
determined
melting poi
he surface
and leads
deal crysta
T
i-crystallin
amellae wit
T Tm m 0
by the Tho
ellaes’ top
crystal H
150
how a lam
ellae inclu
h period L
urface to v
d melting p
int Tm
0 valid
e energy (s
s to a melt
al is given
TH
Sm
m
0
e polymer
thout amor
c
1(
mson equ
surface ,
Hm
0 and the
mellar struc
uding the
). An ideal
olume rati
point Tm of
d for the i
surface of
ting point r
by:
Hm
m
0
0
rs depend
rphous fold
H lm
0
2 )
ation (see
the specif
e density o
cture (Fig.
amorphou
l polymer c
o is compa
polymers
nfinite fully
f the lamel
reduction
ds on the
ding region
)
above), w
fic enthalp
f the crysta
1b; chain
us parts
crystal con
arably sma
always lie
y extende
llae) chan
T Tm
o
thickness
ns).
which inclu
py of meltin
alline regio
n folded
have a
nsists of
all. That
s below
d chain
ges the
Tm
. The
s of the
des the
ng of an
ons c.

151
Fig. 5 : Thermogram of a semi-crystalline polymer.
Considering the Thomson equation, it can be derived that “thicker” crystalline regions
generally lead to a higher melting point of the polymer (figure 5). As there is typically
a distribution of crystallite sizes in a polymer sample, a broad melting region is
usually observed.
With this in mind, the melting point of a polymer is defined as the temperature where
the peak associated with the endothermic phase transition in the dW / dt curve
reaches a maximum, i.e. most of the crystallites melt at this temperature.
c) Determination of the degree of crystallization from the melting enthalpy
DSC is a simple method to determine the degree of crystallization of a semi-
crystalline polymer. The parameter can be calculated according to the following
equation, where Hm
denotes the area underneath the melting curve and 0
mH
represents the melting enthalpy of an ideal crystal.
H
H
m
m
0 ,
In order to calibrate the area underneath the melting curve, a sample with known
Hm
has to be measured. Due to the lack of ideal polymer crystals, 0
mH has to be
extrapolated to the ideal conditions. As an example: for PE the melting enthalpy of
linear alkanes CH3-(CH2)n-CH3 was measured and extrapolated to n = (plotting
H versus 1/n)
152
2. Experimental Section
Table 1: Measurement procedure for the investigated samples:
Measurement
No.
Sample Amount Starting
temperature
Final
temperature
Heating rate
[mg] [°C] [°C] [K/min]
1 Indium 5.525 130 170 10
2 it - PS 50
270
50
270
50
270
10
10
10
3 PET 30 300 20
300
30
30
300
10
10
Enthalpies of melting Hm
0 :
Indium: 28.2 J/g
it - PS: 86.8 J/g
Tonset (Indium) = 156.6 °C

153
3. Data Evaluation
1) Display the experimental parameters (see table 1) and values obtained from the
different measurements (melting and glass transition temperatures, transition
enthalpies) in a table. Correct the temperatures by subtracting the indium onset.
Correct the enthalpies by multiplying with the factor (theoretical / measured) derived
from the indium measurement. Display the corrected values in a table.
2) Discuss the profile of the indium heating curve.
3) Discuss the it – PS measurement.
Determine the degree of crystallization of the it – PS sample.
4) Discuss the PET measurement.
Determine the degree of crystallization of the PET sample (search for the
corresponding Hm
0 value in the literature).

154
4. Questions
1) Isotactic polypropylene crystallizes in a „31 - helix“ structure. What is the
meaning of the coefficents? (Polybutene: 85 - helix)
2) Describe synthesis routes to isotactic polypropylene and isotactic polystyrene!
3) How does the degree of crystallinity affect the mechanical properties of the
polymers?
4) Describe possible solid state structures of blockcopolymers comprising
crystallizable and non-crystallizable blocks (e.g. PE - PS - PE)?
5) Rapid quenching of a crystallizable polymer from melt leads to a polymer
glass. By which methods can the glass be transformed into the semi-
crystalline state?







