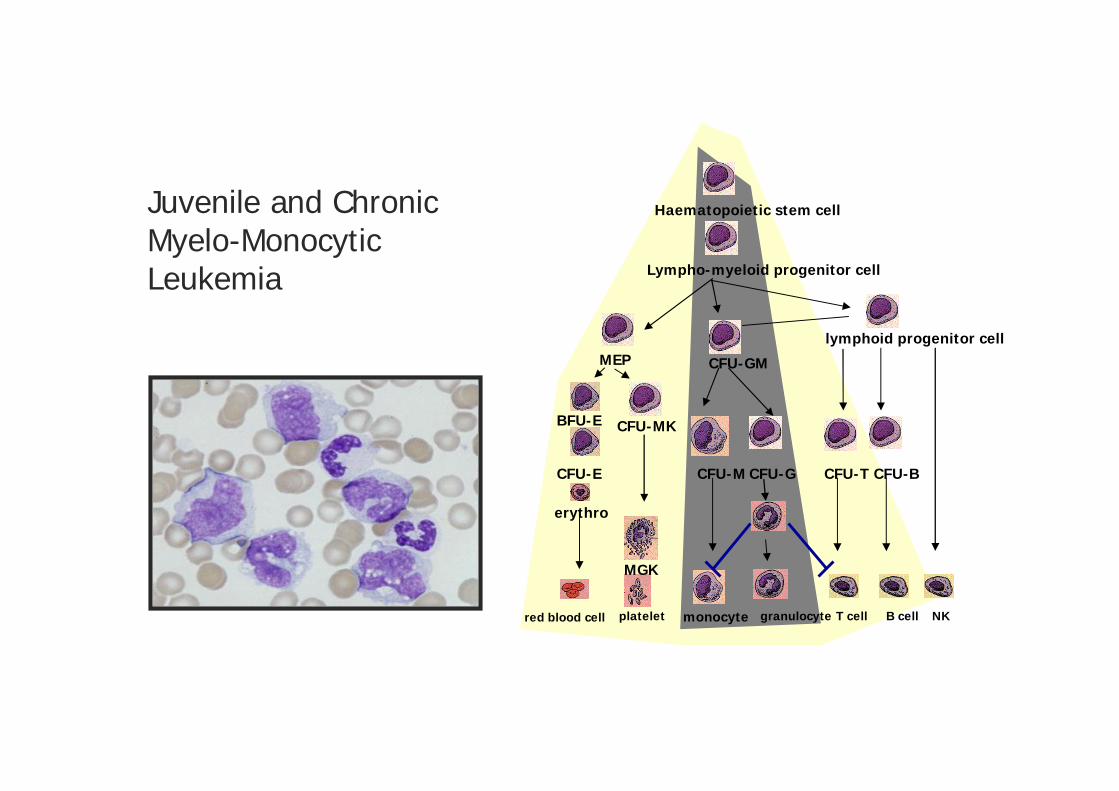
Juvenile and ChronicMyelo-MonocyticLeukemia
red blood cell
Haematopoietic stem cell
Lympho-myeloid progenitor cell
lymphoid progenitor cellMEP
BFU-E
CFU-E
CFU-MK
CFU-GM
CFU-M CFU-G
platelet granulocyte T cell B cell NK
erythro
MGK
CFU-BCFU-T
monocyte

MDS/MPN of the WHO classification
Clonal diseases of the HSC
Excluded in 2009 : PDGFBR rearrangement and eosinophilia
1. Chronic myelomonocytic leukemia
2. Juvenile myelomonocytic leukemia
3. Atypical chronic myeloid leukemia,
4. Refractory anemia with ring sideroblasts and thrombocytosis
5. MDS/MPN unclassifiable
No BCR-ABL or Ph1Bone marrow blast cells < 20%

JMML
An agressive myeloid malignancy of chilhood
MPN / MDS in the WHO classification
- Infant or young child (0-6), male > female, with fever and pallor, - Circulating WBC count > 10G/L; Peripheral monocytosis > 1 G/L- A few circulating myeloid precursor cells
- Sometimes : skin rash, cough, bloody stools
- Splenomegaly, hepatomegaly
- Lack of BCR-ABL- Less than 20% blast cells in the bone marrow
JMML
An agressive myeloid malignancy of chilhood
Cytogenetic abnormality (monosomy 7, 25%)
Hypersensitivity of myeloid progenitor cells to GM-CSF (not to IL-3 or G-CSF)
Increased HbF for age
Treatment : ABMT (EFS 52%)
AML4 / AML5 or spontaneous improvement

Genetic syndromes that predispose to JMML
Neurofibromatosis, type I (NF1) : prone to JMML in the first decade
- Autosomal dominant disorder (or spontaneous)- At least 6 café au lait macules, - At least 2 neurofibromas or 1 plexiform neurofibroma,- Lisch nodules (iris hamartomas), axillary or inguinal freckling, and/or optic gliomas
- NF1 encodes neurofibromin protein, a GTPase activating protein for Ras- Enhances the hydrolysis of the active, GTP-bound conformation of Ras
- In the bone marrow of children with JMML: loss of the wild type allele
- Mouse models of nf1 conditional deletion reproduce the disease

Genetic syndromes that predispose to JMML
Noonan syndrome (NS)
- Autosomal dominant (or spontaneous) disorder- Facial dysmorphism, short stature, webbed neck, cardiac anomalies,- Varying levels of impaired cognition,- Self-resolving myeloproliferative disorder in infancy that resembles JMML
- Germine mutations in PTPN11 (former SHP2) in 50% of cases- Encodes a non-receptor protein tyrosine phosphatase (also SHP2)- That connects tyrosine kinase receptors to Ras
- Other Noonan : germline mutations in SOS1, a RAS-GEF (Guanine nucleotide-exchange factor) KRAS

Gene mutations in JMML
SHP2Yp
Grb2
SOS1
Ras-GDP
Ras-GTP
NF1
CBL
Raf
Mutually exclusive mutations in
PTPN11 35%NF1 25%N/KRAS 20%CBL 10%
Other in SOS1, FLT3, ASXL1
No TET2 mutation
AKT STAT5
ERK
MEK

CMML
a disease of the lederly
defined by only one positive criteria
MPN / MDS in the WHO classification
Clonal disease of HSC with monocytosis
- Persistant monocytosis (> 1 G/L)
- Lack of Ph1 or Bcr-Abl
- Blood and bone marrow blast cells < 20%
- Cell dysplasia, at least one cell line

Molecular abnormalities identified
in human CMML
1 – Cytogenetic abnormalities : 15-40%
2 – Uniparental disomy : ~50 %
3 – Mutations in
Epigenetic genes : TET2 ; ASXL1 ; AML1/RUNX1; IDH1/2; EZH2; UTX
Signalling : N/KRAS; CBL; FLT3-ITD, JAK2; NOTCH2 /NCST / MALM
Splicing : SRSF2; ZRSR2; J2AF35; SF3B1
4 – Deregulated expression of
TIF1, miRNA, CJUN, CFOS

IDH/TET2 pathway: 72% mutations
Molecular abnormalities identified
in human CMML (GFM 175 patients)

No specific mutationHigh frequency of TET2/IDH pathway mutations (> 70%)Combinations of mutations in signaling, epigenetic, splicingMore mutations to identify (NGS)
Molecular abnormalities identified
in human CMML

Yoshida Nature 2011
Molecular abnormalities identified
in human CMML
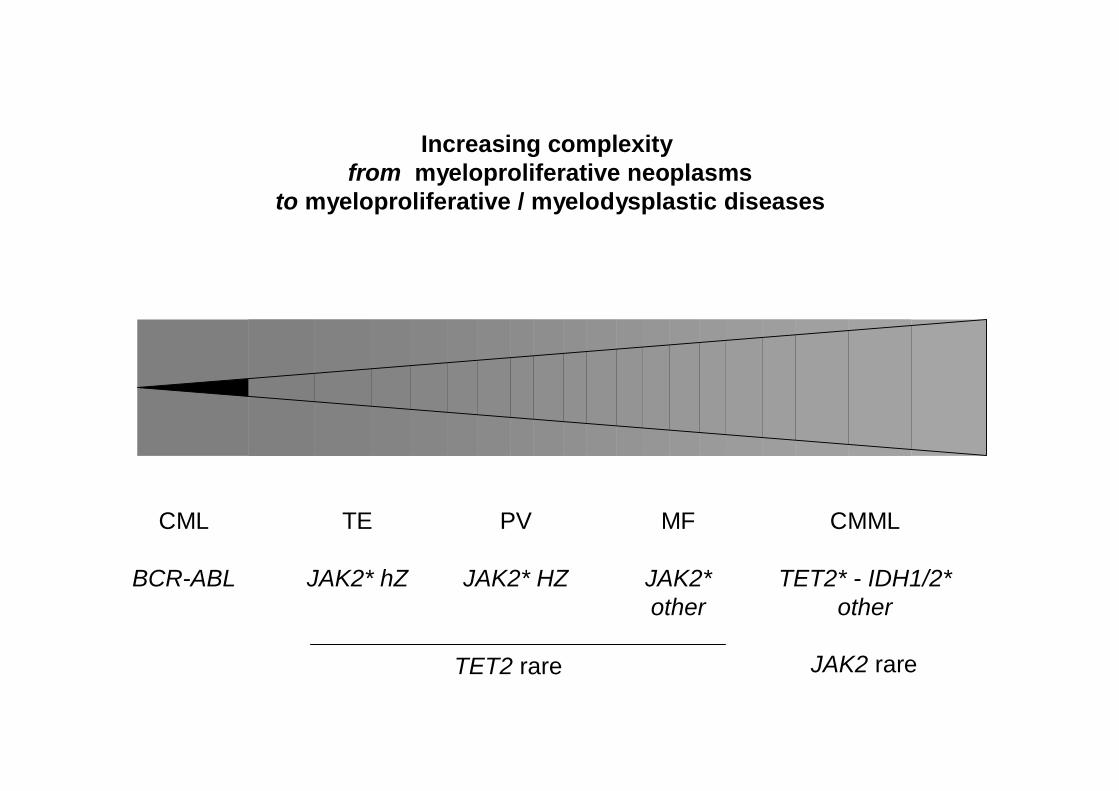
CML
BCR-ABL
TE
JAK2* hZ
PV
JAK2* HZ
MF
JAK2*other
CMML
TET2* - IDH1/2*other
TET2 rare JAK2 rare
Increasing complexityfrom myeloproliferative neoplasms
to myeloproliferative / myelodysplastic diseases

AML1 ASXL1 CBL JAK2 KRAS NRAS
29 MUTATIONS IN 17 PATIENTS
# CLONES
TET2
52 MUTATIONS IN 46 PATIENTS
Clonal expansion occurs in theCD34+/CD38- compartment
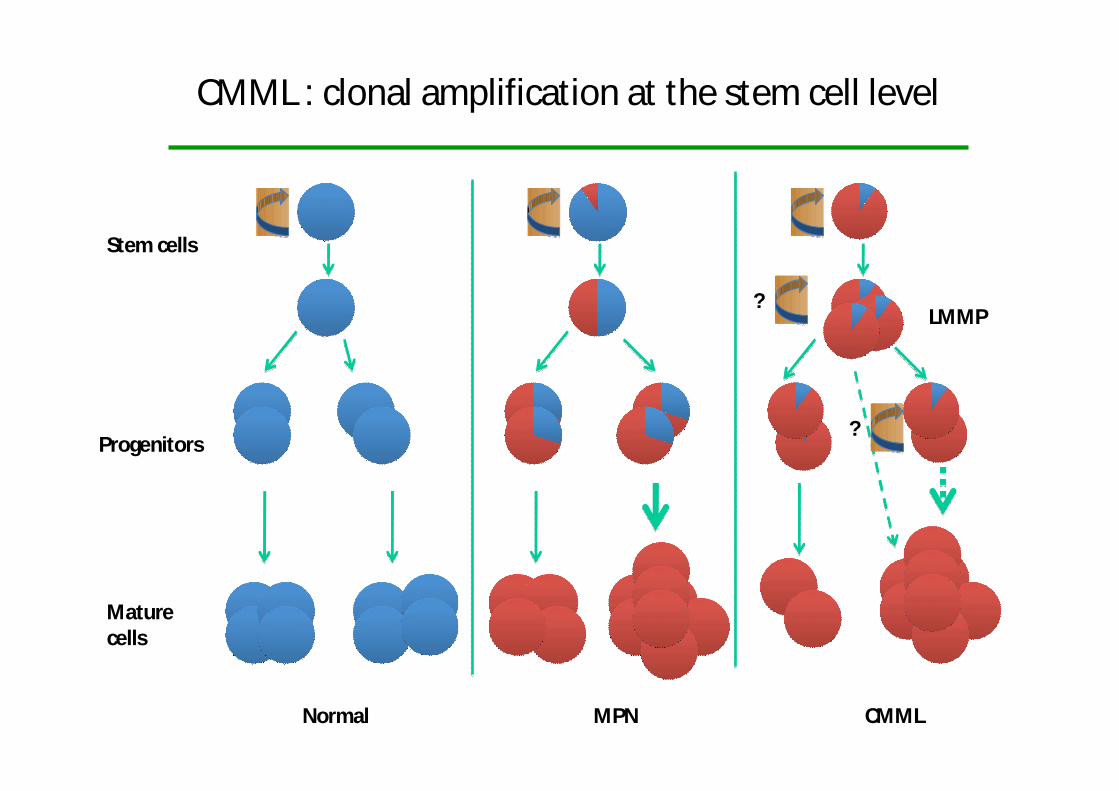
CMML : clonal amplification at the stem cell level
Stem cells
Progenitors
Maturecells
Normal MPN CMML
LMMP?
?

TIF1 involved in zebrafish erythropoiesis (severe anemia in « moonshine » mutant) human erythropoiesis (ex vivo differentiation of CD34+ cells)
TIF1 knock-out embryonic lethal in mice
TIF1 (Transcription Intermediary Factor 1 )
1127 AA1127 AARINGRING B1B1 B2B2 CoiledCoiled--coilcoil TSSTSS PHDPHD BromoBromo
RBCC ou TRIMRBCC ou TRIMTIF Signature TIF Signature SequenceSequence
FeaturesFeatures of of chromatinchromatinremodelingremodeling proteinprotein
MeMe33H3K4H3K4AcetylatedAcetylated
lysineslysines
TIF1- belongs to a group of four proteins (TIF1 ) - modulates the TGF- signaling pathway- is a transcriptional co-regulator (TAL1/PU1)

Ageing Tif1 / mice developa CMML-like disease
Peripheral blood
00
11
22
33
44 CtrlCtrl//
11--1313 1414--2626 2727--3939 ≥≥4040
Mon
ocyt
es (k
/mm
Mon
ocyt
es (k
/mm
33 ))
WeeksWeeks
n=11n=11n=23n=23
n=12n=12n=23n=23
n=12n=12n=20n=20
n=9n=9n=9n=9
**
****
Gr1Gr1--FITCFITC
Mac
1M
ac1 --
Ale
xa64
7A
lexa
647
CtrlCtrl //
44%44% 1%1%3%3% 51%51%
CtrlCtrl //
1 cm1 cm
Spleen

-139GGGAGGAYGT TYGTGYGTA YGTGYGYGTGT YGTAAT YGTTT TT TTTTAAA YGYGYGA YGYG
-139GGGAGGACGTCCGTGCGTACGTGCGCGTGCCGCAACCGCCCTCCTTCAAACGCGCGACGCG unconverted
non methylatedpartially methylatedtotally methylated
Control TIF1low
TIF1norm
alexpression (Subset# 1) (Part of subset# 2)
TIF1 gene promoter is methylatedin 35-50% of CMML
35%of patients

Months
Cum
ulat
ive
Pro
babi
lity
of S
urvi
val
0 6 12 18 24 30
0.0
0.2
0.4
0.6
0.8
1.0
17 15 11 7 2 0
18 14 11 9 3 1
low TIF1ghigh TIF1g
Low TIF1
Normal TIF1
1.0
0.8
0.6
0.4
0.2
0.0
0 6 12 18 24 30Months
Cum
ulat
ive
prob
ablil
ity
of s
urvi
val
TIF1 expression leveldoes not predict decitabine efficacy
0
50
100
150
200
250
300
mRN
Aex
pres
sion
(rel
ativ
e)
Cont
rol
Subs
et#
1
Subs
et#
2

Wild-type
Mutated
Months
19 15 11 7 219 17 14 12 5
0 6 12 18 24 300 6 12 18 24 30
Cum
ulat
ive
Prob
abili
tyof
Sur
viva
l
0.0
0.2
0.4
0.6
0.8
1.0
Mutated
Wild-type
TET2
13 10 9 7 325 22 16 12 4
Months
Cum
ulat
ive
Pro
babi
lity
of S
urvi
val
0 6 12 18 24 30
0.0
0.2
0.4
0.6
0.8
1.0
27 24 18 15 6 0
10 7 6 3 1 1
AML1 WTAML1 MUT
Mutated
Wild-type
0 6 12 18 24 30
10 7 6 3 1 Mutated28 24 18 15 6 Wild-type
AML1ASXL1
Gene mutations in TET2, ASXL1, AML1 do not affect survival

Months
Cum
ulat
ive
Pro
babi
lity
of S
urvi
val
0 6 12 18 24 300.
00.
20.
40.
60.
81.
0
17 16 13 12 5 0
18 13 9 4 1
low MYBhigh MYB
Months
Cum
ulat
ive
Pro
babi
lity
of S
urvi
val
0 6 12 18 24 30
0.0
0.2
0.4
0.6
0.8
1.0
16 15 14 10 2 0
19 14 8 6 3 1
low CJUNhigh CJUN
P = 0.06
Cum
ulat
ive
prob
ablil
ity
of s
urvi
val 1.0
0.8
0.6
0.4
0.2
0.0
Low
High
CJUN 1.0
0.8
0.6
0.4
0.2
0.0
CMYB
Low
HighP = 0.01
0 6 12 18 24 30 0 6 12 18 24 30Months Months
CJUN level correlates with responseCMYB level correlates with survival

Chronic myelomonocytic leukemia
Persistant monocytosis (> 1000/µL)
Why do these patients die?
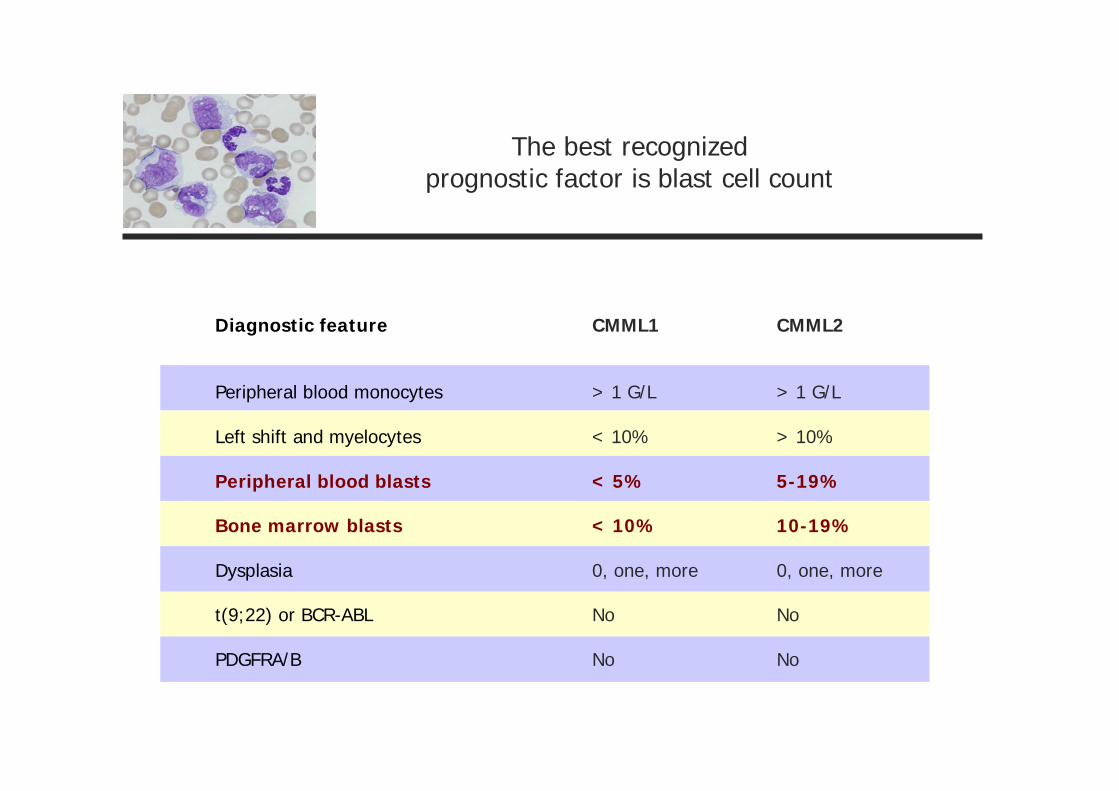
The best recognizedprognostic factor is blast cell count
Diagnostic feature
Peripheral blood monocytes
Left shift and myelocytes
Peripheral blood blasts
Bone marrow blasts
Dysplasia
t(9;22) or BCR-ABL
PDGFRA/B
CMML1
> 1 G/L
< 10%
< 5%
< 10%
0, one, more
No
No
CMML2
> 1 G/L
> 10%
5-19%
10-19%
0, one, more
No
No
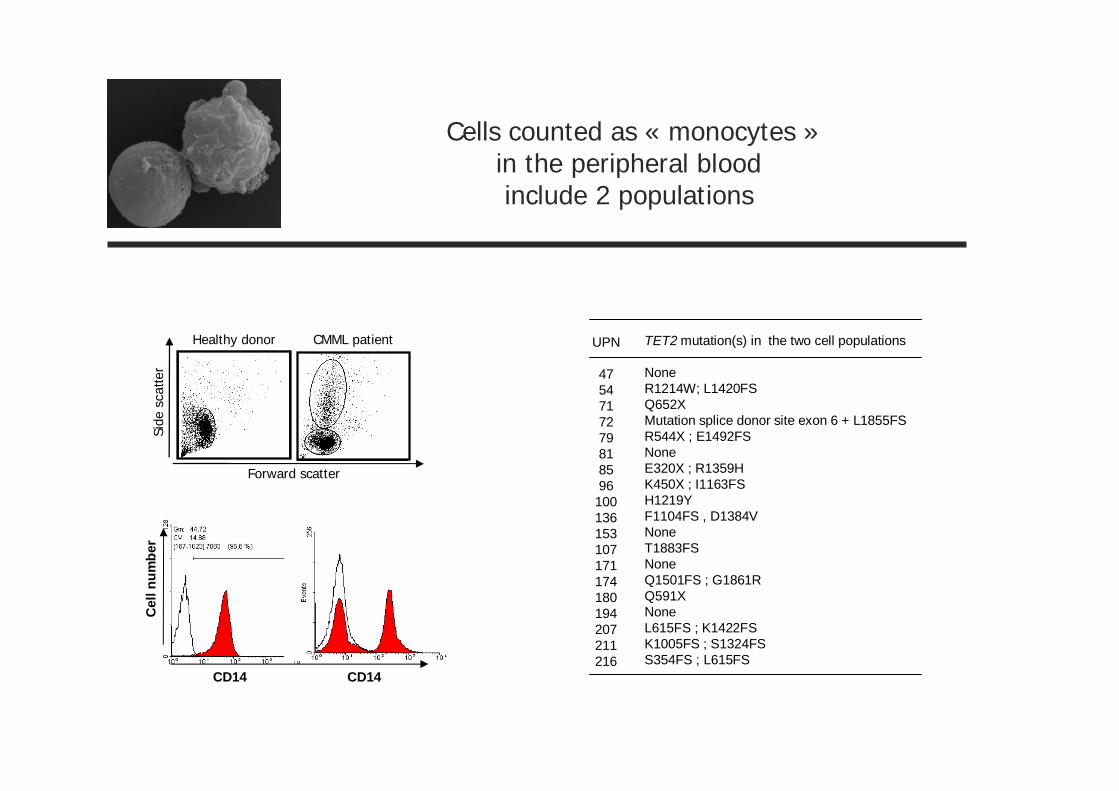
Forward scatter
Side
scat
ter
Healthy donor CMML patient
CD14
Cel
lnum
ber
CD14
Cells counted as « monocytes »in the peripheral bloodinclude 2 populations
TET2 mutation(s) in the two cell populations
NoneR1214W; L1420FSQ652XMutation splice donor site exon 6 + L1855FSR544X ; E1492FSNoneE320X ; R1359HK450X ; I1163FSH1219YF1104FS , D1384VNoneT1883FSNoneQ1501FS ; G1861RQ591XNoneL615FS ; K1422FSK1005FS ; S1324FSS354FS ; L615FS
UPN
4754717279818596
100136153107171174180194207211216

-défensinesHNP1-3
CD14-/CD24+
Granuleux immatures
CD14+/CD24-
Monocytes
Lymphocyte
ExosomesIL-13
STAT3STAT6
Macrophage
M-CSF
The immunosuppressive propertiesof the CMML immature granulocytes

Take home messages
1 – JMML / CMML are distinct diseases
2 – JMML is a RAS pathway disease with hypersensitivity to GM-CSF
3 – CMML is a TET2 disease with many different mutations
4 – Epigenetic changes in addition to mutations (TIF1, miRNA)
5 – Immunosuppressive dysplastic granulocytes
To further explore the disease pathogenesis : [email protected]







