
1/26th/01 h:00 am/pm.
Inhibition of interactions and interconversions of prion protein isoforms by peptide fragments
from the C-terminal folded domain
Motohiro Horiuchi1,2, Gerald S. Baron1, Liang-Wen Xiong1 and Byron Caughey1
1Laboratory of Persistent Viral Diseases, Rocky Mountain Laboratories, NIAID, NIH, Hamilton,
Montana 59840 USA
2Department of Veterinary Public Health and the National Research Center for Protozoan
Diseases, Obihiro University of Agriculture and Veterinary Medicine, Obihiro Hokkaido 080-
8555 Japan
Running title: PrP peptide inhibitors of PrP-res formation
Correspondence to:Dr. Byron Caughey,Laboratory of Persistent Viral Diseases,NIAID, NIH, Rocky Mountain Laboratories,903 S 4th St.,Hamilton, MT 59840 USAPhone: 406-363-9264Fax: 406-363-9286e-mail: [email protected]
1
JBC Papers in Press. Published on February 1, 2001 as Manuscript M100288200 by guest on February 3, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Summary
The formation of protease-resistant prion protein (PrP-res or PrPSc) involves selective
interactions between PrP-res and its normal protease-sensitive counterpart, PrP-sen or PrPC.
Previous studies have shown that synthetic peptide fragments of the PrP sequence
corresponding to residues 119-136 of hamster PrP (Ha119-136) can selectively block PrP-res
formation in cell-free systems and scrapie-infected tissue culture cells. Here we show that two
other peptides corresponding to residues 166-179 (Ha166-179) and 200-223 (Ha200-223)
also potently inhibit the PrP-res induced cell-free conversion of PrP-sen to the protease-
resistant state. In contrast, Ha121-141, Ha180-199 and Ha218-232 were much less effective
as inhibitors. Mechanistic analyses indicated that Ha166-179, Ha200-223 and peptides
containing residues 119-136 inhibit primarily by binding to PrP-sen and blocking its binding to
PrP-res. Circular dichroism analyses indicated that Ha117-141 and Ha200-223, but not non-
inhibitory peptides, readily formed high beta sheet structures when placed under the conditions
of the conversion reaction. We conclude that these inhibitory peptides may mimic contact
surfaces between PrP-res and PrP-sen and thereby serve as models of potential therapeutic
agents for transmissible spongiform encephalopathies.
2
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Introduction
Transmissible spongiform encephalopathies (TSEs) are a group of fatal
neurodegenerative diseases that include scrapie in sheep and goats, bovine spongiform
encephalopathy in cattle, chronic wasting disease in deer and elk, and Creutzfeldt-Jakob
disease in humans. The preeminent neuropathological feature of TSEs is the accumulation of
the disease-specific, protease-resistant isoform of prion protein, designated as PrP-res or
PrPSc, in central nervous system. PrP-res is generated post-translationally from the normal,
protease-sensitive isoform of prion protein, PrP-sen or PrPC. Although several lines of
evidence suggest that PrP-res is a major component of the infectious TSE agent, the full
identity of the agent is still unclear.
Studies using transgenic mice, PrP-deficient mice and neuronal tissue grafts revealed
that an interaction between PrP-sen and PrP-res that leads to PrP-res accumulation plays a
central role in the propagation of infectivity and neurodegeneration (1-3). Furthermore, the
formation of PrP-res and propagation of TSE infectivity in animals and cultured cells require
PrP amino acid sequence compatibility between the recipient and donor of infectivity (4-6).
Cell-free experiments have shown that PrP-res can selectively bind PrP-sen (7,8) and
subsequently induce its conversion into a PrP-res-like protease-resistant molecule (9).
Further studies of this two-step process revealed that the direct binding of PrP-sen to PrP-res
is less dependent upon PrP amino acid compatibility than the conformational transformation to
the protease-resistant state (10,11). Although previous efforts have described several aspects
of the interaction between the two PrP isoforms, the molecular details of the interaction and
conversion mechanism remain to be elucidated.
Direct interactions of synthetic PrP peptides with PrP molecules indicated the possible
3
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

usefulness of PrP synthetic peptides for studying the mechanism of PrP-sen - PrP-res
interaction. For instance, Ha109-141 and Ha119-136 can inhibit PrP-res formation in cell-free
PrP-res-induced conversion reactions and in scrapie-infected cell cultures (12,13).
Stoichiometric excesses of a synthetic peptide corresponding to hamster PrP residues 90-145
(Ha90-145) can bind PrP-sen and protect it from proteolysis (14,15). The central region of PrP
containing residues 90-145 has been of prime interest because it is part of the usual C-terminal
protease-resistant core of PrP-res and it contains an amyloidogenic sequence AGAAAAGA
that appears critical for PrP-res formation (12,13,15-18).
Other data suggest that other regions in the C-terminal half of the molecule might be
important in PrP-sen - PrP-res interactions leading to PrP-res formation (8,19-21). In this
study, we have investigated this possibility further by testing the effects of various synthetic
peptides corresponding to portions of the C-terminal half of the PrP amino acid sequence.
Ha166-179 and Ha200-223 strongly inhibited the protease-resistant PrP formation in cell-free
conversion assays, while Ha180-199 showed much weaker inhibition. In addition, we have
addressed mechanistic aspects of the inhibition by various peptides, including Ha109-141, and
found that the peptides inhibit by binding to PrP-sen and blocking its binding to PrP-res.
Experimental Procedures
Cells and purification of PrP-sen
Hamster PrP-sen lacking the glycosylphosphatidylinositol (GPI) anchor was expressed in
PA317 and psi2 mouse fibroblasts (9). We refer to the PrP-sen lacking a GPI anchor as PrP-
sen(GPINEG). Metabolic labeling of the cells with 35S-methionine and purification of 35S-
4
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

labeled PrP-sen were performed as described previously (22) except for the elution of 35S-
labeled PrP-sen from protein A sepharose-immunocomplexes; PrP-sen was eluted with 0.1 N
acetic acid (pH 2.8) and kept at 4°C until use. In some experiments, culture supernatants of
35S-labeled cells were used as a source of 35S-PrP-sen(GPINEG).
Synthetic peptides
Synthetic peptides of hamster PrP corresponding to residues 109-141 (Ha109-141,
MKHMAGAAAAGAVVGGLGGYMLGSAMSRPMMHF), residues 117-141 (Ha117-141,
AAGAVVGGLGGYMLGSAMSRPMMHF), residues 121-141 (Ha121-141,
VVGGLGGYMLGSAMSRPMMHF) and residues 218-232 (Ha218-232,
YQKESQAYYDGRRSS) were described previously (12). In the experiments of the present
study, Ha109-141 and Ha117-141 were sometimes used as surrogates for one another since
they have been shown to be similarly inhibitory and both contain the core inhibitory sequence of
residues 119-136 (13). Synthetic peptides of hamster PrP corresponding to residues 166-179
(Ha166-179, VDQYNNQNNFVHDC), residues 180-199 (Ha180-199,
VNITIKQHTVTTTTKGENFTC) and residues 200-223 (Ha200-223,
ETDIKIMERVVEQMCTTQYQKESQ) were synthesized in this study (Fig. 1). Randomized
peptides of Ha166-179 (RHa166-179, VNFQNDVHNYQDNC) and Ha200-223 (RHa200-223a,
MIQETMKVTDQSIECQEVTKQRYE, and RHa200-223b, SQEKQYQTTCMQEVVREMIKIDTE)
were also synthesized. Synthetic peptides Ha109-141, Ha117-141, Ha121-141, Ha180-199,
Ha200-223 and Ha218-232 were dissolved with deionized water to make 2 mM stock solutions.
Due to poorer solubility in water, synthetic peptides Ha166-179, RHa166-179 and RHa200-
223 were first dissolved in dimethylsulfoxide (DMSO) to make 10 mM stock solutions. Aliquots
5
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

of each stock solution were stored at –20°C until use.
Cell-free conversion reactions
PrP-res was purified without proteinase K (PK) treatment from the brains of hamsters
infected with the 263K strain (23) as described previously (24). Cell-free conversion reactions
without the use of GdnHCl were performed as described elsewhere (8). Briefly, purified PrP-
res was diluted with deionized water and sonicated for 15 sec, and then 100 ng of PrP-res was
mixed with 20,000 cpm (~2 ng) of 35S-PrP-sen(GPINEG) in 20 µl of reaction mixture
containing 200 mM KCl, 1.25% Sarkosyl, 5 mM MgCl2, and 50 mM citrate buffer (pH 6.0). The
reaction mixtures were incubated at 37°C for 2 days. Nine-tenths of the reaction mixtures were
treated with 20 µg/ml of PK [50 mM Tris-HCl (pH 8.0), 150 mM NaCl in 100 µl] at 37°C for 30
min. PK digestion was stopped by adding Pefabloc (Boehringer Mannheim) to 2 mM,
thyroglobulin to 20 µg/ml as a carrier protein, and four volumes of methanol. The remaining
one-tenth of the reaction mixture was analyzed without PK treatment. The protein samples
collected by centrifugation were subjected to SDS-PAGE using Novex pre-cast acrylamide gels
and radioactive proteins were visualized and quantified with a PhosphorImager instrument
(Molecular Dynamics).
PrP-sen – PrP-res binding analysis and conversion reactions in multi-well plates
PrP-res was suspended in 2.5 M GdnHCl at 2.5 ng/µl and incubated at 37°C for 30 min.
Then 40 µl of the PrP-res solution was added to the round bottom 96-well plate (High bind,
Costar) and incubated at 37°C overnight. After adsorption of PrP-res, the wells were washed
once with PBS and then blocked with 0.5% skim milk in PBS at room temperature for 2 h. After
6
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

washing the wells once with PBS, 40 µl of 35S-PrP-sen(GPINEG) (40,000 cpm) solution
containing 200 mM KCl, 5 mM MgCl2, 1.25% Sarkosyl, and 0.1% fetal bovine serum in 50 mM
citrate buffer (pH 6.0) was added and the plates were incubated at 37°C for 2 days. For the
binding analysis, the wells were washed four times with a washing buffer containing 200 mM
KCl, 1.25% Sarkosyl and 50 mM citrate buffer (pH 6.0). For the conversion reactions, the wells
were washed once with 50 mM Tris-HCl (pH 8.0) and 150 mM NaCl, and then incubated with
50 µl of PK solution containing 20 µg/ml PK, 50 mM Tris-HCl (pH 8.0) and 150 mM NaCl at
37°C for 30 min. The PK digestion was terminated by adding 2 µl of 0.1 M Pefabloc and then the
wells were washed once with 50 mM Tris-HCl and 150 mM NaCl. Finally, 35S-PrP remaining
in the wells was eluted with 20 µl of 1x sample buffer (5% SDS, 4M urea, 62.5 mM Tris-HCl (pH
6.8), 3 mM EDTA, 5% glycerol, 5% 2-mercaptoethanol, 0.04% bromophenol blue) by heating
the plate at 80°C for 10 min and analyzed by SDS-PAGE as above.
Binding analysis between PrP-sen and PrP synthetic peptides
Peptides were diluted to various concentrations with 50 mM phosphate buffer either pH
8.5 or pH 7.0 containing 150 mM NaCl and 50 µl of the diluted peptide solutions were added to
the wells of a DNA-BIND 96-well plate (Costar). The wells were coated with a layer of reactive
N-oxysuccinimide esters so that peptides could be covalently crosslinked to the wells through a
primary amine group. After 90 min incubation at 37°C, the wells were washed once with the
corresponding buffer and blocked with 2% skim milk in the corresponding buffer. The wells
were washed once with PBS and then incubated with 40 µl of 35S-PrP-sen(GPINEG) (40,000
cpm) solution containing 200 mM KCl, 5 mM MgCl2, 1.25% Sarkosyl, and 0.2% fetal bovine
7
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

serum in 50 mM citrate buffer (pH 6.0) at 37°C overnight. After incubation, supernatants were
saved as the unbound fraction, and the wells were washed four times with washing buffer.
Finally, the 35S-PrP-sen bound to the wells (bound fraction) was eluted with 5% SDS and 4M
urea by heating the plate at 80°C for 10 min, and radioactivity in the bound and unbound
fractions was counted by liquid scintillation counter. To analyze the selectivity of the binding
between PrP-sen and peptides, the culture supernatants of 35S-labeled cells were used
instead of 35S-labeled purified PrP-sen. In this case, unbound and bound fractions were
analyzed by SDS-PAGE.
Immunoblotting
Transfer of the proteins from acrylamide gels to Immobilon-P membranes (Millipore) was
performed as described elsewhere (25) . PrP on the membrane was visualized by using anti-
PrP synthetic peptide (residues 89-103) antibodies and ECF western blotting reagents
(Amersham), and quantified with a PhosphorImager instrument.
Circular Dichroism Spectroscopy
Synthetic peptides were dissolved in deionized water to make 5 mg/ml stock solutions. Peptide
solutions (40 µL) were combined with 160 µL of conversion buffer (200 mM KCl, 5 mM MgCl2, 50 mM
citrate, pH 6.0) with or without 0.1% sarkosyl to give final peptide concentrations of 1 mg/ml . CD
measurements were performed using a 0.1 mm path length quartz cylindrical cell with an OLIS-16
DSM CD Spectrophotometer. The following parameters were used: 0.05 nm monochromator resolution,
1 nm bandwidth, dual beam mode, 400 kHz sampling rate, 2 V high volts criterion. Wavelength
8
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

calibration was done with (1S)-(+)-10–camphorsulfonic acid (Sigma). The temperature of sample
chamber was held at 37 °C. Data were collected from 260 to 90 nm with 1 datum/nm. The resulting
spectra were obtained by averaging 6 scans and subtracting spectra of the buffer with or without 0.1%
sarkosyl.
Results
Inhibition of protease-resistant PrP formation in cell-free reactions by synthetic PrP peptides.
Previous cell-free conversion studies have shown that synthetic peptides Ha106-128,
Ha109-141 and certain subunits thereof (e.g. Ha117-121) can inhibit PrP-res formation, while
peptides spanning most other portions of the PrP sequence do not (12,13). One portion of the
PrP amino acid sequence that was not addressed in previous analyses spanned residues 170-
218. Thus, we first examined whether PrP synthetic peptides from this region inhibit the PrP
conversion reaction. Cell-free PrP conversion reactions usually involve incubating
immunoprecipitated 35S-methionine-labeled PrP-sen with unlabeled PrP-res purified from
TSE-infected brain tissue and then assaying for the formation of partially proteinase K (PK)-
resistant 35S-PrP products (26). In the previous peptide inhibition studies, the cell-free
conversion reactions were performed using guanidine hydrochloride (GdnHCl) as a stimulant
(12,13). However, we recently established cell-free conversion reactions that occur under
much more physiologically compatible conditions without the use of chaotropic salts (8). Hence,
to better approximate in vivo PrP-res formation, we used these conditions in the experiments
that follow.
As reported previously using the GdnHCl-containing conditions (12), Ha117-141
9
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

inhibited the formation of the typical partially PK-resistant 35S-PrP conversion product. Like
brain-derived PrP-res itself (not shown), these 35S-PrP conversion product bands appear ~7
kDa lower in molecular mass than the full-length 35S-PrP-sen precursor after PK treatment
(Fig. 2a). The concentration of the peptide exhibiting 50% inhibition of the conversion reaction
(the IC50) was ~40 µM (Fig. 2g) compared to 80 µM observed under the GdnHCl-containing
conditions in the previous report (12). As in the previous study, the peptides Ha121-141 and
Ha218-232 did not inhibit the protease-resistant PrP formation at concentrations up to 500 µM
(Fig. 2b,f). Thus, other than the somewhat lower IC50 for Ha117-141, the effects of these
peptides were similar in GdnHCl-containing and GdnHCl-free cell-free conversion reaction
conditions.
Two newly synthesized peptides Ha166-179 and Ha200-223 inhibited protease-
resistant PrP formation with IC50 values of 10-15 µM (Figs. 2c,e,g). The inhibitory effects of
these peptides are at least comparable to that of the synthetic peptide Ha109-141 under the
present conditions (IC50= 15), which was the most efficient inhibitory peptide in previous studies
(data not shown). In contrast, the synthetic peptide Ha180-199 only partially inhibited
protease-resistant PrP formation at the highest concentration tested (500 µM) (Fig. 2d,g).
Since our data demonstrate the strongest inhibition by Ha166-179 and Ha200-223, we
wished to address the amino acid sequence specificity of the effects of these peptides. Thus,
we synthesized and examined peptides of the same amino acid composition, but randomized
sequence (RHa166-179, RHa200-223a and RHa200-223b, respectively) for effects on the
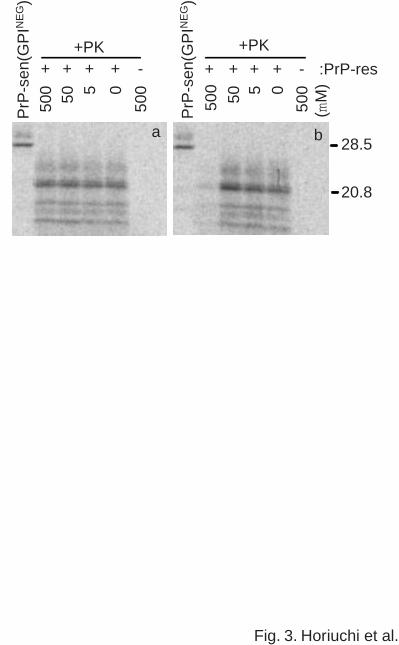
conversion reaction. RHa166-179 did not inhibit the reaction up to 500 µM, demonstrating the
amino acid sequence specificity of the Ha166-179 effect (Fig. 3a). However, two distinct
10
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

RHa200-223 peptides (data for only one is shown in Fig. 3b) substantially inhibited the reaction
at the highest concentration tested (500 µM) indicating that the effect of the Ha200-223 was not
completely dependent upon the full amino acid sequence of the peptide.
Inhibition of PrP-sen - PrP-res binding by synthetic peptides
Since some of the PrP synthetic peptides inhibited the formation of protease-resistant
35S-PrP, we examined whether this was due to the inhibition of the binding between PrP-sen and
PrP-res or the subsequent conversion of PrP-sen to the protease-resistant form. PrP-sen is
soluble and PrP-res is a readily pelletable aggregate. Thus, we first attempted to examine the
effect of the peptides on PrP-res binding by a sedimentation-based binding assay described
previously (8,11). However, 35S-PrP-sen was detected in the pellet in the presence of some
of the peptides without PrP-res (data not shown) thus hampering our ability to specifically
monitor binding to PrP-res. Therefore, we opted to monitor both the PrP-sen - PrP-res
binding and conversion reactions using a solid phase system with PrP-res adsorbed to a 96-
well plate.
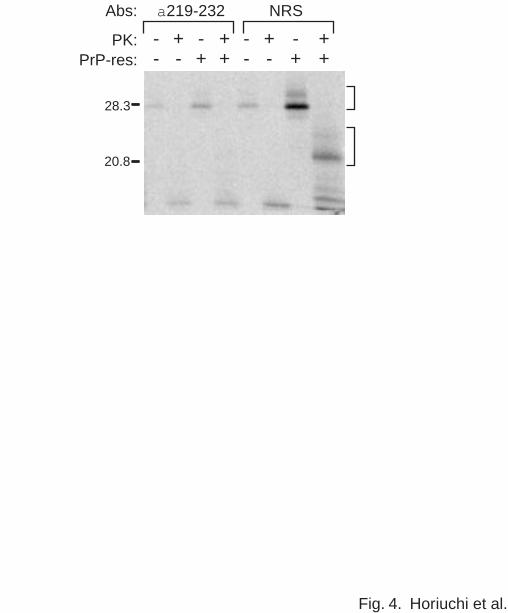
Fig. 4 shows the binding and the conversion reaction with the solid phase system. In the
absence of pre-adsorbed PrP-res, the non-specific binding of 35S-PrP-sen to the wells was
minimal and no protease-resistant 35S-PrP was detected after the PK digestion. However, in
the presence of PrP-res, both binding of 35S-PrP-sen and conversion to the PK-resistant form
was detected (Fig. 4, right side lane). Another indication of the specificity of the solid phase
binding and conversion reactions was inhibition by anti-PrP219-232, a rabbit antiserum known
to inhibit the binding of PrP-sen to PrP-res in suspension reactions (8), but not by normal rabbit
11
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

serum (NRS). Given these indications of the specificity of the solid phase system, we used it in
subsequent analyses of the effects of the peptide inhibitors.
Synthetic peptides Ha109-141, Ha166-179 and Ha200-223, which inhibited the
formation of protease-resistant 35S-PrP, also blocked the binding of 35S-PrP-sen to PrP-res
in a dose dependent manner (Fig. 5). In contrast, Ha121-141 and Ha218-232, which did not
inhibit the conversion reaction, did not block PrP-sen - PrP-res binding. Furthermore, Ha180-
199, which only partially inhibited the conversion reaction at 500 µM, did not significantly block
the binding up to 100 µM. Therefore, these results indicated that the inhibition of protease-
resistant PrP formation by Ha109-141, Ha166-179 and Ha200-223 was due primarily to a
blockade of the binding of PrP-sen to PrP-res.
Peptide inhibition by binding to PrP-sen
The preceding results raise the question of whether the inhibition of PrP-sen - PrP-res
binding by the peptide inhibitors is due to binding of the synthetic peptide to PrP-sen, PrP-res
or both. In a solid phase analysis, in which the synthetic peptides were covalently cross-linked
to a 96-well plate, binding of 35S-PrP-sen to Ha117-141, Ha166-179 and Ha180-199 was
observed (Fig. 6). In contrast, no binding of 35S-PrP-sen to Ha121-141, Ha200-223 and
Ha218-232 was detected. This experiment was performed with the multi-well plate coated with
N-oxysuccinimide that is reactive with primary amines (DNA-BindTM, Costar). Moreover, the
same results were obtained when the plate coated with a sulfhydryl-reactive maleimide group
(Reacti-BindTM, maleimide activated plate, Pierce) or a plate possessing a hydrophobic
surface (High bind, Costar) was used (data not shown). One apparent inconsistency in our data
12
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

was observed with Ha200-223; this peptide inhibited the binding of PrP-sen to PrP-res (Fig. 5)
but binding of PrP-sen to this peptide was not detected by the solid phase assay. However,
when sedimentation analysis was performed, 35S-PrP-sen was detected in the pellet with
Ha200-223 even without PrP-res (data not shown). We also observed by electron microscopy
that Ha200-223 forms fibrils under these buffer conditions (data not shown). These findings
suggest that PrP-sen was bound to Ha200-223 fibrils in suspension and was therefore pelleted
by centrifugation. Taken together, the solid phase and sedimentation experiments provide
evidence for PrP-sen binding to each of the inhibitory peptides.
Next we tested if the peptide can also inhibit the PrP-sen - PrP-res interaction by
binding to PrP-res. For this purpose, the synthetic peptides and 35S-PrP-sen were added
sequentially to the wells coated with PrP-res. The synthetic peptide was added first and
incubated for 1 day to allow binding to PrP-res. Then the peptide solution was removed and
35S-PrP-sen was added. No peptides inhibited binding between 35S-PrP-sen and PrP-res under
these circumstances (data not shown) indicating that the inhibitory peptides do not interact with
PrP-res in a manner which blocks PrP-sen binding after removal of the free peptide solution
and the addition of PrP-sen. Thus, the available evidence favors a mechanism in which the
inhibitory peptides act by binding to PrP-sen and blocking its binding to PrP-res.
Specificity of the binding between peptide inhibitors and PrP-sen
To examine the specificity of the binding between the peptide inhibitors and PrP-sen,
selectivity of the binding of PrP-sen versus other proteins was analyzed (Fig. 7). When
culture supernatants of [35S]methionine-labeled cells containing many different labeled
13
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

proteins besides PrP-sen(GPINEG) were incubated in wells coated with PrP-res, the binding
between PrP-sen and PrP-res was highly selective as described previously (8); the binding
of only the three major PrP-sen(GPINEG) bands was detected (indicated by arrowheads).
The identity of these bands as 35S- PrP-sen was confirmed by the ~80% decrease in
binding when PrP-sen(GPINEG) was first depleted from the culture supernatant by
immunoprecipitation. The binding of 35S-PrP-sen(GPINEG), as well as some other 35S-
labeled proteins, to PrP synthetic peptides was observed in wells coated with Ha109-141,
Ha166-179 or Ha180-199. This was consistent with the peptide-PrP-sen binding analysis
using purified 35S-PrP-sen (Fig. 6). The depletion of PrP-sen(GPINEG) reduced the
intensity of only the PrP-sen(GPINEG) bands (indicated by arrowheads), suggesting that the
other 35S-labeled bands were not SDS-stable oligomers of PrP-sen but other 35S-labeled
proteins in the culture supernatants. Although the binding between PrP-sen and PrP
synthetic peptides was not as selective as that between PrP-sen and PrP-res, this result
confirmed that Ha109-141, Ha166-179 and Ha180-199 bind PrP-sen in preference to a
large number of other proteins.
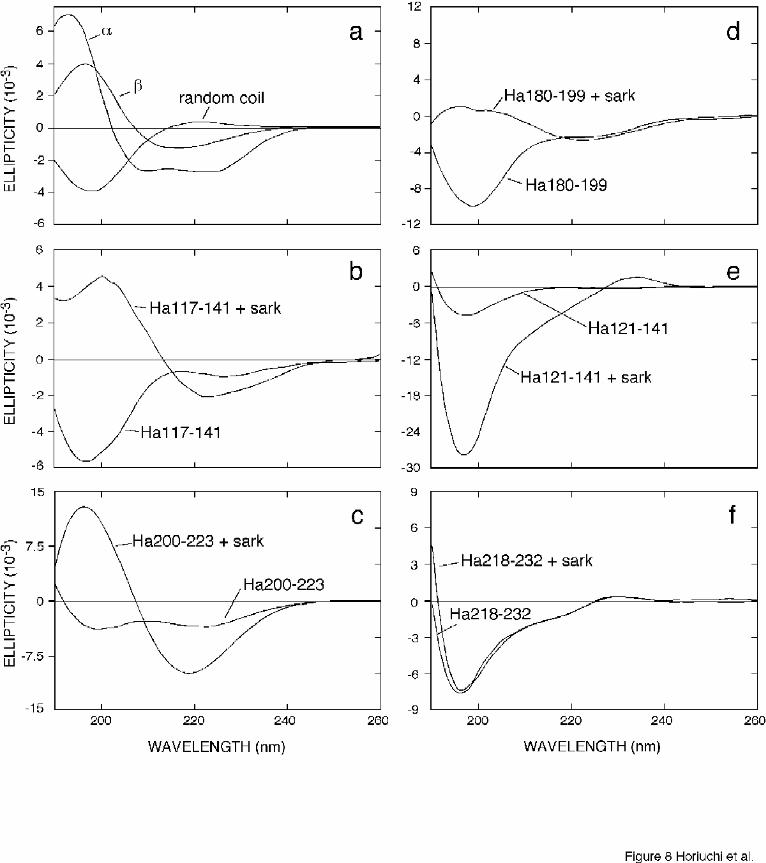
Secondary structures of the inhibitory peptides
Inhibitory peptides described by Chabry et al. (12) tended to form high β-sheet
aggregates. Thus, we analyzed the conformation of the synthetic peptides described in this
study to determine if they exhibited a similar tendency to form β-sheet structure. The
peptides were dissolved in water and then diluted in conversion buffer in the presence or
14
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

absence of sarkosyl to reproduce the manipulations used in the cell-free conversion
reactions and to evaluate the effect of sarkosyl on the conformation of the peptides. Circular
dichroism (CD) spectra of the Ha117-141, Ha121-141, Ha180-199, Ha200-223 and
Ha218-232 are shown in Fig. 8. The spectra indicate that sarkosyl has differential effects on
the CD spectra of the various peptides. The spectra of Ha117-141 and Ha200-223 indicated
changes in secondary structure from almost all random coil, which is characterized by a
negative ellipticity near 198 nm, to high β-sheet, characterized by strong positive ellipticity
near 198 nm and negative ellipticity near 220 nm (Fig. 8b,c). A similar, but much less
pronounced effect of sarkosyl was seen with Ha180-199. The spectrum of Ha218-232 was
initially indicative of random coil and was unaffected by sarkosyl. The spectrum of Ha121-
141 became more strongly indicative of random coil in the presence of sarkosyl. Interestingly,
the secondary structure changes in the peptides towards β-sheet by sarkosyl correlated with
the relative abilities of the peptides to inhibit the conversion of PrP-sen to PrP-res. The
peptides with strong tendencies to form increased β-sheet on addition of sarkosyl (Ha117-
141 and Ha 200-223) were the most efficient inhibitors of cell-free conversion of PrP-sen
(compare Fig. 8 to Fig. 2g). An intermediate effect was seen with the weaker inhibitor Ha180-
199 and no induction of β-sheet was seen for the non-inhibitory peptides Ha121-141 and
Ha218-232. Ha121-141 was previously reported to form β-sheet secondary structures (12)
but that observation was made at under different buffer and solubilization conditions
compared to the more physiologically compatible conditions used in the present study.
Discussion
15
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Since TSE pathogenesis and PrP-res formation involve precise interactions between PrP
isoforms, we have studied the effects of synthetic PrP peptides to gain insight into these
interactions. Previous studies revealed that Ha109-141 and shorter subfragments such as
Ha117-141 inhibited the transformation of PrP-sen to protease-resistant forms in vitro (12,13) .
In this study, we identify peptides from three other segments within the C-terminal third of the
PrP sequence that inhibit protease-resistant PrP formation. Ha166-179 and Ha200-223 have
potencies (IC50s) that are comparable to that of Ha109-141 and Ha117-141, while Ha180-199
was much less potent. There was strong amino acid sequence dependence to the inhibition by
Ha166-179, but this dependence was less pronounced for Ha200-223.
It is unclear why two different randomized permutations of Ha200-223 inhibited the PrP-
res formation to some extent. However, it is possible that a certain oligopeptide or structural
motif was maintained in the randomized sequences that allowed binding to PrP-sen or PrP-res
and inhibition of conversion. Although we could not discern any obviously conserved pattern of
residues on cursory examination, identification of such a motif may contribute to the
understanding of the molecular mechanism of PrP-res formation and possible therapeutics for
TSEs.
The PrP-res-induced transformation of PrP-sen into protease-resistant forms can be
separated kinetically and biochemically into two different steps; first, the binding between PrP-
sen and PrP-res, and second, the conversion of the bound PrP-sen to PrP-res (7,8,11) .
Thus, the generation of protease-resistant PrP can be impaired either at the binding or
conversion steps. Our data indicate that the inhibition of protease-resistant PrP formation by the
PrP peptides Ha109-141, Ha166-179, and Ha200-223 was due to interference with the initial
binding between the two PrP isoforms. Since Ha117-141 and Ha166-179 were able to bind
16
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

PrP-sen, but were not inhibitory when preincubated with PrP-res, it is likely that their inhibition
of conversion is due to binding to PrP-sen in a way that hinders the PrP-sen/PrP-res
interaction. Interestingly, Ha180-199 was more efficient than Ha200-223 at binding of PrP-sen
in the solid phase assay, but was 5-10 fold less potent as an inhibitor of conversion. Hence,
although we conclude that inhibition of PrP conversion by Ha109-141, Ha166-179 and Ha200-
223 is likely to be due to peptide binding to PrP-sen, the binding of peptides to PrP-sen is not
always sufficient to block conversion. Further analyses will be required to clarify whether or not
direct binding of the peptides to PrP-res occurs.
In the cases of Ha109-141, Ha117-141 and Ha166-179, where binding to PrP-sen
appears to be important for inhibition, at least a couple of different mechanisms can be
envisioned to account for these effects. First, the peptides, or polymers thereof, might bind to
PrP-sen and physically block the PrP-res binding site. Alternatively, the binding of the peptides
might over-stabilize PrP-sen or causes a change to a conformation that is incapable of binding
PrP-res.
For mechanistic reasons, it is important to consider the stoichiometry of the inhibitory
peptides relative to PrP-sen and PrP-res in the conversion reactions. The cell-free conversion
reactions contained ~2 nM PrP-sen(GPINEG) and ~150 nM PrP-res. Thus, the peptides with
IC50s of >10 µM required at least 60-fold stoichiometric excesses of peptide over PrP
molecules to exert their inhibitory effects. One possible explanation for the need for
stoichiometric excesses for inhibition by these peptides is that formation of high β-sheet
aggregates or polymers of the PrP peptides may be required to compete with PrP-res for
binding to PrP-sen. This would be consistent with the observations that peptides Ha109-141,
Ha117-141, and Ha 200-223 can form β-sheet rich structures according to FT-IR and/or CD
17
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

analyses [(12) and this study]. The secondary structure of Ha166-179 could not be assessed
with CD because of the presence of the highly far UV-absorbent solvent DMSO, which was
required for initial dissolution of this peptide. However, a similar synthetic peptide of human PrP
residues169-185 was reported to form an aggregate of rod-like structure and exhibit Congo red
birefringence (27), suggesting that Ha166-179 may form β-sheet rich structure. It is also
possible that monomers of the PrP peptides can bind to PrP-sen and inhibit conversion, but that
stoichiometric excesses of inhibitory peptides is required because of the relative affinities of the
peptides and PrP-sen for PrP-res. Although it remains to be elucidated which form of these
inhibitory peptides binds to PrP-sen, the binding itself suggests the peptides may serve as
useful probes of the sites of the PrP-sen - PrP-res interaction such as the regions on the PrP-
res molecule(s) which are involved in the interaction.
The direct binding of PrP-sen to Ha109-141, Ha166-179 and (apparently) Ha200-223,
but not numerous other PrP-derived peptides, suggests that the PrP-sen binding domain on
PrP-res may include residues contained in these inhibitory peptides. The lack of observed
binding between PrP-sen and Ha200-223 in the solid phase analysis may be due to a lack of
Ha200-223 fibril formation or some other artifactual interference with PrP-sen binding on the
solid phase support. It is noteworthy that Ha200-223, unlike the other peptides, has internal
lysine and cysteine residues that could react to the derivatized wells. This might restrict
interactions with PrP-sen more than binding solely via N- or C-terminal residues which is the
mode of attachment for the other peptides. Interestingly, a previous analysis has suggested that
a specific antibody that inhibits the binding and conversion reactions would likely sterically
hinder access to residues on PrP-sen that are contained in the peptides Ha109-141, Ha166-
179 and Ha200-223 (8). Thus, the available data suggest that residues contained in one or
18
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

more of these three inhibitory peptides may be important in the binding sites on both PrP-sen
and PrP-res. Dimerization of PrP molecules is proposed to generate a PrP-res specific epitope
(28), suggesting the possibility that a conformational domain comprising several regions of one
PrP-res molecule or several regions of different PrP-res molecules is involved in binding to
PrP-sen and inducing its conformational transformation. It is conceivable these regions on the
PrP-res molecule correspond to the inhibitory peptides identified in this and previous studies.
The binding of PrP-sen to the individual PrP peptide inhibitors was not adequate to form
protease-resistant PrP even at the highest concentration tested (Fig. 2), consistent with the
idea that multiple regions on PrP-res are involved in causing the conformational transformation
of PrP-sen. As noted above, a previous study showed that vast stoichiometric excesses of
Ha90-145 could cause PrP-sen to gain some PK-resistance, however, this was not the
characteristic partial PK-resistance of TSE-associated PrP-res (14). Thus, it is not yet
apparent that any synthetic peptide can induce the conversion of PrP-sen to bona fide PrP-res.
The conformation of the peptide inhibitors may have similarity but not identity to the
corresponding PrP-sen binding domain on PrP-res polymers. Thus incomplete reconstitution
of the PrP-sen binding domain by individual peptides may also account for lower selectivity than
PrP-res in the binding of PrP-sen versus other proteins (Fig. 8). Perhaps a combination of
different PrP synthetic peptides in a given spatial distribution will be required to more closely
mimic PrP-sen - PrP-res interactions. However, initial attempts at combining Ha166-179 and
Ha200-223 at 2.5 µM and 5 µM, respectively, (i.e. slightly below their IC50 values) did not
indicate either additive or synergistic effects of these two peptide inhibitors (data not shown).
Our data suggest that the region including residues 166-179, which forms a loop structure
between the second β-strand and second α-helix (29) plays an important role in PrP-res
19
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

formation. By contrast, a PrP mutant lacking residues 23-88 and 141-176 (PrP106) was able
to form protease-resistant PrP (30,31). In addition, an amber mutation of human PrP at
residue 145 caused Gerstmann-Straussler-Scheinker syndrome with PrP plaques consisting of
mutant PrP (32). These findings suggest that the region corresponding to Ha166-179 may not
be essential for PrP-res formation. However, this is apparently not generally true for PrP-res
formation in TSE diseases of infectious origin. For instance, it is well known that sheep
homozygous for Arg/Arg at residues 171 of PrP are resistant to scrapie [reviewed in (33)]. In
addition, substitutions of PrP residues 167 and 171 prevented PrP-res formation in scrapie-
infected cell cultures (21). Taken together, these observations indicate that there are multiple
types of PrP-res formed and that the region corresponding to residues 166-179 is involved in
the formation of PrP-res from full-length PrP-sen precursors.
Compounds that can facilitate the clearance of accumulated PrP-res (34), stabilize PrP-
sen, or prevent interactions between the two PrP isoforms, are possible candidates for TSE
therapeutics (12,13,35-37) . In addition, compounds indirectly affecting PrP-res formation may
also have therapeutic value. The combined usage of compounds with different mechanisms for
inhibiting of PrP-res accumulation may have synergistic effects in TSE therapeutics. The list of
compounds which inhibit PrP-res formation and/or prolong the incubation periods of scrapie in
rodents is growing (35,37-40). It is important to know the mechanism of action of these
compounds. The experimental procedures used in this study may contribute to the analysis of
such mechanisms. In addition, the assays of the direct interaction between PrP-sen and PrP-
res and/or PrP synthetic peptides in multi-well plates may provide high throughput screens for
compounds that inhibit those interactions.
20
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Acknowledgements
We thank Gregory Raymond for technical assistance and Gary Hettrick and Anita Golden
for graphics assistance. G.S.B. was supported in part by a post-doctoral fellowship from the
Natural Sciences and Engineering Research Council of Canada.
FOOTNOTES
a Abbreviations: TSE, transmissible spongiform encephalopathy; PrP-res, proteinase-resistant
prion protein; PrP-sen, proteinase-sensitive prion protein; PK, proteinase K; PBS, phosphate
buffered saline; GPI, glycosylphosphatidylinositol; GPINEG, glycosylphosphatidylinositol-
negative; GdnHCl, guanidine hydrochloride. CD, circular dichroism.
References
1. Scott, M., Foster, D., Mirenda, C., Serban, D., Coufal, F., Walchli, M., Torchia, M., Groth, D.,
Carlson, G., DeArmond, S. J., Westaway, D., and Prusiner, S. B. (1989) Cell 59, 847-857
2. Bueler, H., Fischer, M., Lang, Y., Bluethmann, H., Lipp, H.-P., DeArmond, S. J., Prusiner, S.
B., Aguet, M., and Weissmann, C. (1992) Nature 356, 577-582
3. Brandner, S., Isenmann, S., Raeber, A., Fischer, M., Sailer, A., Kobayashi, Y., Marino, S.,
Weissmann, C., and Aguzzi, A. (1996) Nature 379, 339-343
4. Prusiner, S. B., Scott, M., Foster, D., Pan, K. M., Groth, D., Mirenda, C., Torchia, M., Yang,
S. L., Serban, D., Carlson, G. A., Hoppe, P. C., Westaway, D., and DeArmond, S. J. (1990) Cell
63, 673-686
5. Scott, M. R., Kohler, R., Foster, D., and Prusiner, S. B. (1992) Protein Sci. 1, 986-997
6. Priola, S. A., Caughey, B., Race, R. E., and Chesebro, B. (1994) J. Virol. 68, 4873-4878
21
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

7. DebBurman, S. K., Raymond, G. J., Caughey, B., and Lindquist, S. (1997) Proc. Natl. Acad.
Sci. USA 94, 13938-13943
8. Horiuchi, M., Chabry, J., and Caughey, B. (1999) EMBO J. 18, 3193-3203
9. Kocisko, D. A., Come, J. H., Priola, S. A., Chesebro, B., Raymond, G. J., Lansbury, P. T.,
and Caughey, B. (1994) Nature 370, 471-474
10. Kocisko, D. A., Priola, S. A., Raymond, G. J., Chesebro, B., Lansbury, P. T.,Jr., and
Caughey, B. (1995) Proc. Natl. Acad. Sci. USA 92, 3923-3927
11. Horiuchi, M., Priola, S. A., Chabry, J., and Caughey, B. (2000) Proc. Natl. Acad. Sci. U. S.
A. 97, 5836-5841
12. Chabry, J., Caughey, B., and Chesebro, B. (1998) J. Biol. Chem. 273, 13203-13207
13. Chabry, J., Priola, S. A., Wehrly, K., Nishio, J., Hope, J., and Chesebro, B. (1999) J. Virol.
73, 6245-6250
14. Kaneko, K., Peretz, D., Pan, K., Blockberger, T. C., Wille, H., Gabizon, R., Griffith, O. H.,
Cohen, F. E., Baldwin, M. A., and Prusiner, S. B. (1995) Proc. Natl. Acad. Sci. 92, 11160-11164
15. Kaneko, K., Wille, H., Mehlhorn, I., Zhang, H., Ball, H., Cohen, F. E., Baldwin, M. A., and
Prusiner, S. B. (1997) J. Mol. Biol. 270, 574-586
16. Giaccone, G., Verga, L., Bugiani, O., Frangione, B., Serban, D., Prusiner, S. B., Farlow, M.
R., Ghetti, B., and Tagliavini, F. (1992) Proc. Natl. Acad. Sci. U. S. A. 89, 9349-9353
17. Zhang, H., Kaneko, K., Nguyen, J. T., Livshits, T. L., Baldwin, M. A., Cohen, F. E., James,
T. L., and Prusiner, S. B. (1995) J. Mol. Biol. 250, 514-526
18. Holscher, C., Delius, J., and Burkle, A. (1998) J. Virol. 72, 1153-1159
19. Muramoto, T., Kitamoto, T., Hoque, M. Z., Tateishi, J., and Goto, I. (1993) J. Virol. 67,
22
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

6808-6810
20. Raymond, G. J., Hope, J., Kocisko, D. A., Priola, S. A., Raymond, L. D., Bossers, A.,
Ironside, J., Will, R. G., Chen, S. G., Petersen, R. B., Gambetti, P., Rubenstein, R., Smits, M. A.,
Lansbury, P. T.,Jr., and Caughey, B. (1997) Nature 388, 285-288
21. Kaneko, K., Vey, M., Scott, M., Pilkuhn, S., Cohen, F. E., and Prusiner, S. B. (1997) Proc.
Natl. Acad. Sci. U. S. A. 94, 2333-2338
22. Caughey, B., Kocisko, D. A., Raymond, G. J., and Lansbury, P. T. (1995) Chem. & Biol. 2,
807-817
23. Kimberlin, R. H. and Walker, C. A. (1978) J. Gen. Virol. 39, 487-496
24. Caughey, B. W., Dong, A., Bhat, K. S., Ernst, D., Hayes, S. F., and Caughey, W. S. (1991)
Biochemistry 30, 7672-7680
25. Caughey, B. and Raymond, G. J. (1991) J. Biol. Chem. 266, 18217-18223
26. Caughey, B., Raymond, G. J., Priola, S. A., Kocisko, D. A., Race, R. E., Bessen, R. A.,
Lansbury, P. T.,Jr., and Chesebro, B. (1999) Mol. Biotech. 13, 45-55
27. Goldfarb, L. G., Brown, P., Haltia, M., Ghiso, J., Frangione, B., and Gajdusek, D. C. (1993)
Proc. Natl. Acad. Sci. USA 90, 4451-4454
28. Korth, C., Stierli, B., Streit, P., Moser, M., Schaller, O., Fischer, R., Schulz-Schaeffer, W.,
Kretzschmar, H., Raeber, A., Braun, U., Ehrensperger, F., Hornemann, S., Glockshuber, R.,
Riek, R., Billeter, M., Wuthrich, K., and Oesch, B. (1997) Nature 390, 74-77
29. Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshuber, R., and Wuthrich, K. (1996)
Nature 382, 180-182
30. Muramoto, T., Scott, M., Cohen, F. E., and Prusiner, S. B. (1996) Proc. Natl. Acad. Sci. U.
S. A. 93, 15457-15462
23
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

31. Supattapone, S., Bosque, P., Muramoto, T., Wille, H., Aagaard, C., Peretz, D., Nguyen, H.
O., Heinrich, C., Torchia, M., Safar, J., Cohen, F. E., DeArmond, S. J., Prusiner, S. B., and
Scott, M. (1999) Cell 96, 869-878
32. Kitamoto, T., Iizuka, R., and Tateishi, J. (1993) Biochem. Biophys. Res. Commun. 192,
525-531
33. Bossers, A., Schreuder, B. E., Muileman, I. H., Belt, P. B., and Smits, M. A. (1996) J. Gen.
Virol. 77, 2669-2673
34. Soto, C., Kascsak, R. J., Saborio, G. P., Aucouturier, P., Wisniewski, T., Prelli, F., Kascsak,
R., Mendez, E., Harris, D. A., Ironside, J., Tagliavini, F., Carp, R. I., and Frangione, B. (2000)
Lancet 355, 192-197
35. Caughey, W. S., Raymond, L. D., Horiuchi, M., and Caughey, B. (1998) Proc. Natl. Acad.
Sci. U. S. A. 95, 12117-12122
36. Demaimay, R., Chesebro, B., and Caughey, B. (2000) Arch. Virol. (in press)
37. Perrier, V., Wallace, A. C., Kaneko, K., Safar, J., Prusiner, S. B., and Cohen, F. E. (2000)
Proc. Natl. Acad. Sci. U. S. A. 97, 6073-6078
38. Priola, S. A., Raines, A., and Caughey, W. S. (2000) Science 287, 1503-1506
39. Supattapone, S., Nguyen, H. O., Cohen, F. E., Prusiner, S. B., and Scott, M. R. (1999)
Proc. Natl. Acad. Sci. U. S. A. 96, 14529-14534
40. Doh-ura, K., Iwaki, T., and Caughey, B. (2000) J. Virol. 74, 4894-4897
41. Donne, D. G., Viles, J. H., Groth, D., Mehlhorn, I., James, T. L., Cohen, F. E., Prusiner, S.
B., Wright, P. E., and Dyson, H. J. (1997) Proc. Natl. Acad. Sci. U. S. A. 94, 13452-13457
24
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Figure legends
Fig. 1. Schematic representation of the location of PrP synthetic peptides. The top line with
boxes indicates HaPrP23-232 which corresponds to the sequence of the mature PrP-sen
which lacks the N- and C-terminal signal sequences. Open boxes indicate regions forming β-
strands (β1 and β2) and hatched boxes indicate regions forming α-helix (α1, α2, and α3)
according to NMR studies (29,41). The thick bars with associated residue numbers indicate the
regions spanned by synthetic peptides used in this study. In some experiments a peptide
corresponding to residues 117-141 (not illustrated) was also used.
Fig. 2. The effect of PrP synthetic peptides on the cell-free formation of protease-resistant PrP.
(a) Ha117-141. (b) Ha121-141. (c) Ha166-179. (d) Ha180-199. (e) Ha200-223. (f) Ha218-
232. The lanes labeled “PrP-sen(GPINEG)” show one-tenth equivalent of 35S-labeled PrP-
sen(GPINEG) used for the cell-free conversion reactions. Peptide concentrations (µM) are
indicated above the gel images. +PK indicates the samples treated with PK. The presence and
absence of PrP-res in the reaction are indicated by + and -, respectively. The upper and lower
brackets on the left indicate PK-untreated 35S-PrP-sen(GPINEG) and PK-resistant 35S-PrP,
respectively, that were used in the phosphor autoradiographic quantitation shown in (g).
Conversion efficiencies [mean % conversion +/- SD (n = 3-5)] were calculated relative to a
control reaction without peptide as described previously (8). Molecular mass markers (kDa) are
on the right.
25
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Fig. 3. Effects of randomized permutations of Ha166-179 (RHa166-179) (a) and Ha200-223
(RHa200-223a) (b) on the formation of protease-resistant PrP. Details other than the peptides
used are the same as described in the legend of Fig. 2.
Fig. 4. Solid phase binding and conversion reactions. 35S-PrP-sen(GPINEG) was incubated
for 2 d in 96-well plates with or without precoating with PrP-res. The wells were washed and
incubated with (+) or without (-) PK. 35S-PrP species bound to the wells were solubilized and
analyzed by SDS-PAGE and phosphor autoradiography. Specificity of the reaction was tested
by comparing the effects of inclusion of α219-232, an antiserum known to inhibit binding of
35S-PrP-sen(GPINEG) to PrP-res, and normal rabbit serum (NRS). The brackets are as
described in legend to Figure 2.
Fig. 5. Effects of synthetic PrP peptides on binding of 35S-PrP-sen(GPINEG) to immobilized
PrP-res. Using the solid phase binding assay used in Figure 4, peptides were added in
increasing concentrations to the binding buffer with 35S-PrP-sen(GPINEG). After incubation
for 2 d, the wells were washed and assayed for bound 35S-GPI(-)PrP. The data points indicate
means +/- SD (n = 3-4) normalized to radioactivity bound in the absence of any peptide. The
graphs are labeled with the span of HaPrP residues to which the peptides correspond.
Fig. 6. Binding of 35S-PrP-sen(GPINEG) to covalently immobilized PrP peptides. 35S-PrP-
sen(GPINEG) (40,000 cpm) was added to wells pre-coated with PrP synthetic peptides as
26
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

described in Experimental Procedures. Supernatants were saved as the unbound fraction and
the 35S-GPI(-)PrP eluted from the wells with 5% SDS and 4 M Urea by heating at 80°C was
designated the bound fraction. The graph shows means +/- SD (n=3) of the percentage of the
bound 35S-GPI(-)PrP against the total 35S-GPI(-)PrP in the reaction (the sum of the unbound
and bound fractions).
Fig. 7. Selectivity of the binding between PrP-sen and PrP synthetic peptides. Culture medium
of metabolically labeled cells secreting PrP-sen(GPINEG) and other proteins were precleared
by centrifugation at 10,000 rpm for 10 min. The culture supernatants were added to wells pre-
coated with the designated synthetic PrP peptides (200 µM, 50 µl/well) or PrP-res (100 ng/40
µl/well). After incubation for 24 hr, the cells were washed four times and bound proteins were
eluted with SDS-PAGE sample buffer with heating. Half of each eluate volume was subjected
to SDS-PAGE and phosphor autoradiography. To confirm the identity PrP bands in the bound
fractions, we also used culture supernatants depleted of PrP-sen by single round of
immunoprecipitation with anti-HaPrP219-232 rabbit serum and protein A sepharose (indicated
by “D”). The lane labeled “PrP-sen(GPINEG)” shows 35S-PrP-sen(GPINEG) purified from
metabolically-labeled culture supernatants by immunoprecipitation with the same antiserum.
Lanes labeled “Culture sup.” contain one-tenth equivalent of the culture supernatants added to
the binding reactions. Arrowheads indicate a major three bands of 35S-PrP-sen(GPINEG)
bound to PrP synthetic peptides or PrP-res. The lowest arrowhead marks an N-terminally
truncated fragment that is especially prominent in culture supernatants as described previously
(8).
27
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Fig. 8. Circular dichroism spectra of synthetic PrP peptides in the presence and absence of
0.1% sarkosyl. (a) Reference CD spectra of all-alpha, all-beta, and all-random (Redrawn from:
Brahms, s., Brahms, J., J. Mol. Biol., 1980, 138: 149-178); (b) Ha117-141. (c) Ha200-223. (d)
Ha180-199. (e) Ha121-141. (f) Ha218-232. The final peptide concentrations were 1 mg/ml
peptide with or without 0.1% sarkosyl in buffer containing 160 mM KCl, 4 mM MgCl2, 40 mM
citrate, pH 6.0, 37°C.
28
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

PrP
-sen
(GP
INE
G)
500 50 5 0
500
+PK
PrP
-sen
(GP
INE
G)
500 50 5 0
500
PrP
-sen
(GP
INE
G)
500 50 5 0
500
+PK +PK+ + + + - + + + + - + + + + -
a b c
d e f
28.3
20.5
28.3
20.5
(mM
)
:PrP-res
Fig. 2a-f. Horiuchi et al.
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from

50050 5 0
500
PrP
-sen
(GP
INE
G)
+PK
28.5
20.8
+ -+++
50050 5 0
500
PrP
-sen
(GP
INE
G)
+PK
+ -+++ :PrP-res
(mM
)
a b
Fig. 3. Horiuchi et al.
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from
+- ++++
+ + +- ----
- -PrP-res:
PK:
a219-232 NRSAbs:
28.3
20.8
Fig. 4. Horiuchi et al.
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from



DD
DD
DD
DD
D
Culture sup.
None
aa109-141
aa121-141
aa166-179
aa180-199
aa200-223
aa218-223
PrP-res
PrP-sen(GPINEG)
Bound to P
rP synthetic peptide or P
rP-res
39.8
30.0
16.6
Fig. 7. H
oriuchi et al.
by guest on February 3, 2018 http://www.jbc.org/ Downloaded from

Motohiro Horiuchi, Gerald S. Baron, Liang-Wen Xiong and Byron Caugheyfragments from the C-terminal folded domain
Inhibition of interactions and interconversions of prion protein isoforms by peptide
published online February 1, 2001J. Biol. Chem.
10.1074/jbc.M100288200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on February 3, 2018http://w
ww
.jbc.org/D
ownloaded from







