
Characterization of Elongation factor I-A & I-Ba from
Methanobacterium thermoautotrophicum
by Nadeem Siddiqui
Department of Biochemistry
McGill University
Montreal, Quebec, Canada
August 2001
A thesis submitted ta the Faculty of Graduate Studies and Research in fulfillment of the
requirements for the degree of Master of Science
©Nadeem Siddiqui, 2001

National.Ubraryof Canada
Acquisitions andBibliographie Services
395 Wellington StreetOttawaON K1AON4Canada
Bibliothèque nationaledu Canada
Acquisitions etserVices bibliographiques
395, rueWellingtonOttawa ON KlA 0N4canada
Your file VOIre réIémnœ
Our file No/reréMrent:8
The author bas granted a nonexclusive licence allowing theNational Library ofCanada toreproduce,loan, distributeor.sencopies of this thesis inmicrofonn,paper or electronic.formats.
The author retains ownership ofthecopyright inthisthesis.N~ither thethesis nor substantial extractsfrom itmay be printed or otherWisereproduced withouttheauthor'spermission.
L'auteur a accordé une licence nonexclusive penne~t âlaBibliothèque nationale du Canada dereproduire, prêter,. distribuer ouvendre des copies de cette thèse sousla forme de microfiche/film, .dereproduction sur papier ou sur formatélectronique.
L'auteur conservela prqpriétéqudroit d'auteur qui protège.cette thèse.Ni la thèse. Rides extraits>substantielsde celle-Clne doivent être .imprimésou autrement reproduits sans sonautorisation.
0-612-78959-4
Canada

Abstract
A three-dimensional model of Methanobacterium thermoautotrophicum (M.t.) elongation factor
lA, (M.t.)EFI-A, was generated based on homologous crystal structures. The model shows that
structural elements essential for binding guanine nucleotides are conserved. (M.t.)EFI-A protein
was purified to homogeneity using a combination of immobilized metal ion affinity and HPLC
size exclusion chromatography. The protein specifically binds to guanine nucleotides and
exhibits nucleotide exchange rates that give values between 4.6 and 6.5 x 10-3S-l. The solution
structure of Methanobacterium thermoautotrophicum elongation factor-IBa, (M.t.)EFI-Ba, was
determined by NMR spectroscopy. Analysis of the structure reveals an a ~ superfold that is
commonly found in RNA binding proteins. The 3-D architecture of (M.t.)EFI-Ba is similar to
analogous domains catalyzing llucleotide exchange in human, yeast and bacterial proteins within
the translation elongation family.
2

Résumé
Un modèle tridimensionnel de la protéine EFI-A de Methanobacterium Thermoautotrophicum,
(M.t.)EFI-A, a étégénéré par homologie avec des structures crystallines connues. Ce modèle
montre que des éléments essentiels à la liaison avec des guanines sont conservés. (M.t.)EFI-A a
été purifiée en solution homogène par chromatographie d'affinité et gel-filtration sur HPLC. La
protéine se lie de manière spécifique aux guanines et montre une vitesse d'échange de nucléotides
de l'ordre de 4.6 à 6.5 x 10-3S-I. La structure en solution du facteur d'élongation I-Ba de
Methanobacterium Thermoautotrophicum, (M.t.)EFI-Ba, a été déterminée par spectroscopie
RMN. L'analyse de la structure révèle un repliement de type a~ communément trouvé dans les
protéines interagissant avec l'ARN. En outre, l'architecture tridimensionnelle de (M.t.)EF-lBa
est similaire aux domaines analogues catalysant l'échange de nucléotides chez les humains,
levures et bactéries.
3

Table of Contents
Abstract
Resume
Table of Contents
List of Figures and Tables
Abbreviations
Acknowledgements
Chapter 1: Introduction
1.1 Introduction to Archaeabacteria
1.2 Translation in Archaeabateria
1.2.1 Initiation
1.2.2 Elongation
1.2.3 Termination
1.3 Methanobacterium thermoautotrophicum
1.4 Rationale and Objective
Chapter 2: Homology Model and Characterization of EFI-A fromMethanobacterium thermoautotrophicum
2.1 Introduction to G-proteins and Translation Elongation Factors
2.1.1 Foreword
2.1.2 G-proteins and the Structure of G-Domains
2.2 Elongation Factors EF-Tu and EF1-A
2.2.1 Elongation Factor EF-Tu
2.2.2 Elongation Factor EF1-A
4
2
3
4
6
8
9
10
11
13
15
17
20
21
22
22
242424
31
31
33

2.3 Three-Dimensional Model of (M.t.)EF1-A 35
2.3.1 Method 352.3.2 Results 36
2.4 Expression and Purification of (M.t.)EF1-A 40
2.4.1 Materials and Methods 402.4.2 Results 43·
2.5 Nucleotide Exchange and Affinity Assays 442.5.1 Foreword 44
2.5.2 Materials 442.5.3 Nucleotide Exchange Assay 45
2.5.4 Nucleotide Affinity Assay 48
Chapter 3: Characterization and Solution Structure of Elongation 54Factor-lEa from Methanobacterium Thermoautotrophicum
3.1 Guanine Nucleotide Exchange Factors 55
3.2 Guanine Nucleotide Exchange Mechanism 583.2.1 GEF Mechanism in E. coli EF-Tu:Ts 583.2.2 GEF Mechanism in S. cerevisiae EFI-ABa 59
3.3 Expression, Purification and NMR spectrosopy of (M.t.)EFI-Ea 62
3.3.1 Materials and Methods 623.3.2 Purification Results 653.3.3 Solution Structure of (M.t.)EFI-Ba 66
Chapter 4: General Discussion 704.1.1 Summary of results 714.2.1 Insights into the mechanisms of guanine nucleotide exchange in 74
Archaeabacteria
Appendix 76
References 77
5

List of Figures and Tables
Chapter 1 10
Table 1
Chapter 2
Homology between archaeal Methanobacterium
thermoautotrophicum and eukaryotic Translation Factors.
14
23
Figure 2.1.1 A schematic diagram of a typical G-Domain.
Figure 2.1.2 The nucleotide binding site of a typical G~domain.
Figure 2.1.3 A comparison of G-domains from EF1-AJTu
from M. thermoautotrophicum, S. cerevisiae, T. aquatiqus
and E. coli.
Table 2.1 Residue conservation found in loop regions of the G-domain
in translation factors EF-Tu from E. coli, T. aquaticus,
and EF1-A from S. cerevisiae and M. thermoautotrophicum.
Figure 2.1.4 Schematic diagram showing hydrogen bonding interactions
between residues from EF-Tu with GDP cofactor and ligand
coordination around Mg2+.
Figure 2.2.1 Ribbon diagrams of CA) E. coli EF-Tu-GDP and
CB) T. aquaticus EF-Tu-GTP complex structures.
Figure 2.2.2 Ribbon diagram of S. cerevisiae EF1-A.
Figure 2.3.1 Ribbon representation of the homology model of
M. thermoautotrophicum EF1-A.
Figure 2.3.2 Ribbon representation of conserved loop regions involved
in guanine nucleotide binding and Mg2+ coordination from
S. cerevisiae, M. thermoautotrophicum and T. aquaticus.
6
26
27
28
28
30
32
33
37
38

Figure 2.4.1 (A) HPLC-gel filtration chromatogram of pooled fractions from 46immobilized metaI affinity chromatography.
(B) 12.5%SDS-PAGE of (M.t.)EFI-A purified by IMAC and elution
fractions after HPLC-gel filtration application.
Figure 2.5.1 (A) Guanine nucleotide exchange activity of (M. t.)EF1-A. 50(B,C) Nucleotide specificity of 47 and 30 kDa forms of(M.t.)EFI-A.
Chapter 3 54
Table 3.1 Sequence alignment of (M.t.)EFI-Ba with archaeal and 55
eukaryotic exchange f(ictors.
Figure 3.1 Comparisons of the nucleotide exchange domain from 56eukaryotes and prokaryotes (A) eEFI-Ba from Human
and S. cerevisiae (B) Cata1ytic domain of E. coli EF-Ts.
Figure 3.3.1 SDS-20% PAGE of (M.t.)EF1-Ba purification by immobilized 65
metal ion affinity chromatography.
Figure 304.1 Ribbon representation of the average solution structureof (M.t.)EF-lBa. from Methanobacterium thermoautotrophicum. 66
Figure 304.2 Structural comparison of guanine nucleotide exchange 68factors from S. cerevisiae EF1-Ba, M. thermoautotrophicum
EF1-Ba and E. coli EF-Ts.
Chapter 4
Table 4.4.1 Sequence alignment of the C-terminal region of archaealEFI-Ba and euk(lryotic exchange factors.
7
70
74

Abbreviations
Amino Acyl
Amino Acyl tRNA
Archaeal Elongation Factor
Escherichia coli
Electrospray Ionisation Mass Specrometry
Elongation Factor
Elongation Factor l-A
Elongation Factor I-Ba
Eukaryotic Elongation Factor
Guanine Nucleotide binding proteins
Guanine Nucleotide binding domain
Guanine Nucleotide binding loop
Guanine Nucleotide exchange factor
Guanine Nucleotide Triphosphate
Guanine Nucleotide Diphosphate
Immobolized Metal Ion Chromatography
Initiation Factors
Methanobaeterium thermoautotrophicum
Nuclear Magnetic Resonance Spectroscopy
Phosphate binding loop
Saccharomyces cerevisiae
Shine-Delgarno
Thermus aquaticus
Translation Factors
8
aa
aa-tRNA
aEF
E.c.
ESI-MS
EF
EFI-A
EFI-Ba
eEF
G-protein
G-Domain
G-Ioop
GEF
GTP
GDP
!MAC
IFs
M.t.
NMR
P-Ioop
S.e.
SD
T.a.
TFs

Acknowledgments
1 want to thank my supervisor, Dr. KaIle Gehring, for providing me with the opportunity to study
in his laboratory. 1 cannot thank him enough for his support, patience, freedom to grow and most
of aH his friendship.
1 want to acknowledge aIl my colleagues who have made this experience a most memorable one.
Their help, support, friendship and the goad times will always be remembered. Special thanks ta
N.B., T.S., A.N., D.C, P.L., R.C, N.C, S.C, M.B., A.D., J-F.T., P.G., and of course G.K. 1 also
want to thank Robert Laroque and Daniel Tessier for their many helpful discussions. Most of aH,
a very warm appreciation goes to Dr. Irena Eikel for her tutelage and everlasting enthusiasm.
FinaHy, 1 want to thank my family for their love and support and my friends for their continuous
encouragement. It is to my family and friends 1 dedicate this thesis.
9

Chapter 1
Introduction

1.1 Introduction to Archaeabacteria
Archaeabacteria are unique organisms that are found to inhabit sorne of the most extreme
environments in nature. These environments range from polar conditions to waste deposits,
alkaline or acidic waters, salt lakes and hot springs. Although once thought to occupy a lirnited
number of environments there is compelling evidence that show archaeabacteria to be globaIly
widespread (1).
Archaeabacteria, or archaea, were once classified as a part of the bacterial domain since both
shared many morphological features including similar ceIl sizes, absence of organelles, single
circular genomes, and the lack of a nuclear membrane. Although possessing clear physical
similarities to bacteria, molecular phylogenetic studies have revealed that archaeabacteria are
actuaUy different from bacteria. For example, their is significant primary sequence homology (>
45%) between transcription and translation factors from archaeabacteria and their eukaryotic
counterpart. From an evolutionary point of view, the evidence suggests that archaea are in fact
closer to eukaryotes than to bacteria (2). Furthermore, DNA sequence analyses of various
archaeal genomes have revealed an evolutionary distinction between the informational and the
metabolic features of the archaeal ceIl.
11

The informational aspects, principally transcription and translational machinery, of archaea are
essentially similar to eukaryotes whereas many metabolic aspects resemble those seen in bacteria
(3). It is generally accepted that eukaryotic and archaeal cells are fundamentally similar in their
informational design. However it is uncertain if this similarity extends to other cellular
components (4). Despite a wealth of research, from a molecular and biochemical standpoint,
archaeans are still the least understood out of the three domains of life.
The research presented here will examine translational machineries in archaea focusing on the
elongation cycle. For structural and perhaps biochemical studies of translation factors, archaea
present many advantages. Primary sequence analysis of archaeal genomes indicate that
translation factors are generally smaller but display high homology to their eukaryotic
counterparts (5). Given the similarities, one can assume that mechanisms for archaeal translation
factors may be similar to those seen in eukaryotes. More data now supports this idea and it is
suggested that what we are observing in the archaeal informational system is in fact a simpler
version of the eukaryotic system (6). Hence, generalizations made from archaeal informational
systems should yield more insight into eukaryotic systems rather than studies from bacteria.
12

1.2 Translation in Archaeabateria
Translation is the process of deciphering genetic infonnation stored in our DNA, via messenger
RNA (mRNA), to a functional polypeptide counterpart. This process occurs on the ribosome
with a series of accessory proteins, referred to as translation factors (TFs), to complete
polypeptide synthesis. In aU domains of life the process of translation is divided in main three
stages: initiation, elongation and termination.
Analysis of various archaeal genomes such as Methanococcus jannaschü, Methanobacterium
thermoautotrophicum, Sulfolobus solfactarious, Archaeoglobus fulgidus and Pyrococcus
horikoshii indicate that out of fifteen putative proteins involved in archaeal translation thirteen
share significant sequence homology to eukaryotic translation factors. For example, with the
exception of elongation factor I-Ba (EFI-Ba), the sequence homology between translation
factors from Methanobacterium thermoautotrophicum (M.t.) and their eukaryotic counterpart are
aU greater than 48% (Table 1). The assumption made is that if there is signifièant sequence
homology between TFs from both domains then they must possess similar function. The
foUowing section will describe the current model of archaeal translation including a comparison
of sorne features within the bacterial and eukaryotic systems. The model will be based on
research in archaeal systems in combination with functional data from homologous proteins in
eukaryotes.
13

Table 1 Homology between archaeal Methanobacterium thermoautotrophicum and eukaryotic
translation factors.
Translation Factor Eukarya Archaea (M.t.) % Homology todosest eukaryote
Initiation Factor 1 or (SUIl) eIF-1 aIF-l 61
Initiation Factor lA eIF-IA aIF-IA 61
Bacterial-type Initiation Factor 2 FUN121 FUN121bIF-2 55 2
(bIF-2 or FUN12)
Initiation Factor 2 alpha subunit eIF-2a aIF-2a 48
Initiation Factor 2 beta subunit eIF-2~ aIF-2~ 58
Initiation Factor 2 gamma subunit eIF-2y aIF-2y 58
Initiation Factor 2B EIF2-B a/~/8/Y/E aIF2-Ba3 52
Initiation Factor 5A eIF-5A aIF-5A 54
Initiation Factor 6 eIF-6 aIF-6 51
Elongation Factor 1A eEFI-A aEFI-A 65
Elongation Factor lB eEFI-D aEFI-Ba No significanteEFI-B similarity
Elongation Factor 2 eEF-2 aEF-2 48
Eukaryotic Peptide Chain eERF"1 aERF-l 51
ReleaseFactor subunit 1
1 Found in one eukaryotic lineage (60).
2 Percent homology to bacterial-type IF-2. Both eIF-2 and bIF-2 are found in ail sequenced archaeal genomes.
3 Only one subunit is found on the M.t. genome, however subunits a,~,() are found in other archaeal
genomes (7).
14

1.2.1 Initiation
One of the more obscure features in archaeal translation is the primary step in initiation. As
previously mentioned, archaeal translation factors show close homology to their eukaryotic
counterpart suggesting similar function. However, archaeal mRNAs, as in bacteria, are
polycistronic, employ the usage of aIl three start codons, posses no 5' cap, lack poly-A tails and
generally contain Shine-Delgarno (SD)-like sequences upstream from the start codon (4). In
addition, a complimentary SD-like sequence is also found on the. 3'-end of the 16S rRNA (8).
Since archaeans contain a bacterial-like initiation factor 2 (IF2), the evidenee suggests that they
utilize an initiation mechanism similar to bacteria (8).
However, bacterial-like IF2 and homologous eIF2 proteins are found in aH sequenced archaeal
genomes (9,10,11). IF2 and eIF2 perform essentially the same function in bacterial and
eukaryotic translation respectively. However, both proteins appear unrelated specifically in terms
of structure and number of subunits. IF2 functions as a single polypeptide chain where eIFs
consists of three subunits and require an additional multi-subunit factor, eIF2-B, for recycling
GTP. Archaeans also contain multi-subunit initiation factors, aIF2 and aIF2-B, that are
homologous toeukaryotic IFs. Henee, the question, is why are two types of IF2 factors present
in the archaeal system?
15

Archaeal genes within operons contain a Shine Delgarno (SD)-like sequence upstream from the
start codon while the first gene in the operon and monocistronic genes do not contain SD-like
sequences (12), It is suggested that in archaea, the initiation process involve alternative
mechanisms, For genes that are within operons, a bacterial-type initiation process is completed,
Given the absence of apoly-A tail, as expected, the only eukaryotic initiation factor missing in
archaea is an eIF4 homologue that is involved in mRNA S'-cap recognition, Thus, for the first
gene in an operon and monocistronic genes, start codon recognition is suggested to involve a cap-
independant 5'-3' scanning initiation mechanism found in sorne eukaryotes (13,14), Contrary to
recent suggestions (15), what we observe is not a hybrid between eukaryotic and bacterial
processes but two distinct mechanisms,
16

1.2.2 Elongation
In eukaryotes, archaea, and bacteria the synthesis of a polypeptide by the ribosome can be viewed
as a three-stage reaction cycle. Unlike some ambiguities surrounding the initiation stage, the
elongation cycle in archaea is clearly similar to that of eukaryotes (16). Three eukaryotic
homologues, central to the elongation cycle, are aIl found in archaeal systems: aEFI-A, aEFI-B,
and aEF-2. From comparative sequence and structural analysis of bacterial vs.
eukaryotic/archaeal elongation factors, EF-Tu, EF-Ts and EF-G are analogues to EFI-A, EFI-B,
and EF-2 respectively.
EFI-AlTu is the central translation factor involved in extending the growing polypeptide chain
one amino acid residue at atime. EFI-A, like IF-2, EF-Tu, EF-2 and EF~G, is a member of the
guanine nucleotide binding protein family. EFI-A is activated upon binding to guanine
nucleotide triphosphate (GTP) and subsequently forms a temary complex with amino acyl tRNA
(aa-tRNA). The first step of the elongation cycle commences when this complex enters the
ribosome's A-site aIlowing aa-tRNA to hybridize with mRNA through a codon-anticodon
dependant manner. Upon correct codon-anticodon recognition the ribosome stimulates
hydrolysis of GTP on EFI-A ensuing the release of an inactive EFI-A:GDP complex. The
second stage of the elongation cycle, transpeptidation, consists of lengthening the polypeptide
chain at its C-terminus by one residue. The formation of a new peptide bond between aa-tRNA
on the A- site and peptidyl-tRNA on the. P-site of the ribosome is catalyzed by peptidyl
17

transferase. After this process EF2/G translocates, or moves, the peptidyl-tRNA from the A-site
that is hybridized with rnRNA to the P-site of the ribosome. Upon GTP hydrolysis, EF2/G is
released and the A-site becomes available for a next cycle.
GDP interacts with EF1-B trs, a guanine nudeotide exchange factor (GEF), which induces the
release of its bound GDP for GTP. In the eukaryotic system, EFI-B4 is a complex that is
composed of subunits a, ~ and y where only a and ~ posses guanine nudeotide exchange activity
(18). This complex interacts with EF1-A and induces the exchange of GDP for GTP. In
prokaryotes only onYienzyme, EF-Ts, performs the same function. In the archaeal system only
one GEF is found - aEF1-Ba.. In eukaryotes, upon GTP binding, a ternary complex of EF1-A:
EF-1B:GTP is formed. This complex interacts with aa-tRNA synthase upon which aa-tRNA is
transferred. When aa-tRNA is bound, EF1-B disassociates and the active EF1-A:GTP:aa-tRNA
re-enters the elongation cycle. In contrast, during nucleotide exchange in bacteria, upon binding
to GTP, the GEF EF-Ts is ejected and a binary EF-Tu:GTP complex is formed. This binary
cornplex binds to aa-tRNA and enters the elongation cycle. How exactly tRNA is transferred on
to the EF1-A/Tu complex from aa-tRNA synthethases in both systems is not completely
understood and is under investigation.
4 EPI-Ba, EFI-B~ and EFI-Bywere farmerly referred ta as EF-I~, EPI-ô, EF-Iy(l7)
18

In contrast to EF-Tu:GTP, eEFI-A:GTP stimulates the activity of aa-tRNA synthethases (19). In
addition, aa-tRNA synthethases are known to associate with eEFI-A:IB:GTP through eEFl
B~(58). Therefore, since aa-tRNA is not found free in solution, theabove results suggest that
EFl~B may play a role in assisting aa-tRNA to the ribosome via EFI-A (17).
Given the homology between archaeal and eukaryotic elongation factors it is hypothesized that
the complexes formed at each step and specifie mechanisms of action would be sirnilar,however
there is insufficient data for conformation.
19

1.2.3 Termination
After completion of translating the mRNA transcript, polypeptide synthesis is terminated when
the termination codon in mRNA enters the A-site. Stop codons do not have a corresponding
tRNA; instead they are recognized by release factors (RFs). When a release factor binds to the
termination codon, ribosomal peptidyl transferase catalyses the transfer of a peptidyl group to
water instead of aa-tRNA releasing the polypeptide chain. Upon hydrolysis of GTP, the
uncharged. tRNA and RFs dissociate from the ribosome which prepares itself for another round
of translation. Prokaryotes have three release factors (RF): RF-l and RF-2 recognize UAA, UAG
and UAA, UGA respectively, while RF-3, a G-protein, stimulates the ribosomal binding of RF-}
and RF-2. Termination in eukaryotes and archaea is similar to prokaryotes but requires only one
release factor. Both eERF-l and aERF-l are G-proteins that show a high degree of homology to
each other and are functionally similar in that they recognize all three stop codons (20).
20

1.3 Methanobacterium thermoautotrophicum
The following study will investigate the structure and biochemical properties on two proteins in
the elongation cycle of translation: EFI-A and EFI-Ba from the archaeabacteria
Methanobacterium thermoautotrophicum (M.t.). M.t. is an anaerobic, autotrophic, methane
producing, thermophillic organism isolated from a municipal wastewater treatment facility. This
organism grows in surroundings of approximately 65 oc. Over the past 20 years, enzymes and
other methanogenic extracts from this organism have been purified and studied. A great deal of
our knowledge in biochemistry of the bioconversion of CO2 to CH4 is based on this organism.
Recently the entire genome of M.t., (-1751 Kbp, i.\H strain) was sequenced (la). Additional
information, sequence search tools and comparative analyses are available at the M.t. homepage
maintained by Ohio State University5. The following endeavor is part of a collaborative effort
(Structural Genomics Project) that aims at characterizing and solving three-dimensional
structures of M.t. proteins by nuclear magnetic resonance spectroscopy (NMR) and X-ray
crystallography.
5 Please refer to Appendix (Pg. 76)
21

1.4 Rationale and Objective
EF1-A belongs to the superfamily of guanine nucleotide binding proteins (G-proteins) and EFl-
Ba is its associated guanine nucleotide exchange factor (GEF). G-proteins are responsible for a
series of key regulatory functions in a eell ranging froID protein synthesis to signal transduction
(21). They act as binary IDolecular switches by alternating between an active GTP and inactive
GDP bound forms (27). One of the more intriguing aspects of G-proteins is how nucleotide
exchange is catalysed by their corresponding exchange factors. In general, G-proteins share
significant sequence identity with each other while their exchange factors do not. This is also
the case for G-proteins and GEFs found in translation. SpecificaIly, eukaryotic and archaeal
EF1-A, and EF-Tu show high sequence hOIDology while members of the EF1-B and EF-Ts
families share no significant sequence homology with each other. However, recent studies show
significant structural homology in the catalytic domain of translational exchange factors
(22,23,27,24). One would expect a protein that has a similar structure would employ similar
function with similar mechanisID. In this case, studies have demonstrated that GEFs do function
in a similar manner but despite the high structural similaritytheir mechanism differs considerably
(25,22,26). Most of the analyses arederived from prokaryotic and eukaryotic systems. Henee,
the objective of this thesis is to structurally and biochemically characterise both aEF1-A and
aEF1-Ba from the archaeon Methanobacterium thermoautotrophicum and offer insightinto the
mechanism of nucleotide exchange in the archaeal translation elongation .cycle.
22

Chapter 2
Homology ModeZ and Characterization ofEF1-Afrom
Methanobacterium thermoautotrophicum

2.1 Introduction to G-proteins and Translation Elongation Factors
2.1.1 Foreword
As previously mentioned, EFI-A belongs to the GTPase superfamily. This section introduces the
general structure of guanine nucleotide binding proteins and the elements involved in nucleotide
binding and Mi+ co-ordination. The following sections will focus on elongation factors from
prokaryotes (E. coli, T. aquaticus) and eukaryotes (S. cerevisiae).
2.1.2 G-proteins and the Structure ofG-Domains
G-proteins comprise several classes of proteins including initiation, elongation, termination
factors, Ras and Ras-related proteins, ADP-ribosylation facrtor-l and heterotrimeric membrane
proteins. These proteins are responsible for a variety of biochemical processes in the cell ranging
from protein synthesis to cell proliferation, secretion and hormone response (21). In all cases,
with the exception of heterotrimeric proteins (28), G-proteins binds as a complex with Mg2+ and
GTP or GDP and possess intrinsic GTPase activity (59). Structura11y, they a11 have a variation on
the phosphate binding motif of ATP (29) but are specifie for only GTP and GDP.
G-proteins act as molecular switches by alternating between GTP and GDP bound forms, which
represents active and inactive conformational states. When active, G-proteins causes target
proteins to perform their specifie function. The action is terminated upon hydrolysis of the GTP
24

to GDP which is accompanied by significant structural changes. Due to these structural changes,
GTP or GDP bound forms recognize different co-factors and binding partners thereby allowing
G-proteins to function as a molecular switches in the cell. Thus, the analysis of guanine
nucleotide dependent processes frequently involves studying the conformational switching
mechanisms between GTP and GDP bound forms.
Although G-proteins vary in size and number of domains, the unifying feature within this
superfamily is a common guanine nuc1eotide-binding core. This structural core is often referred
to as the G-domain. Revealed by crystallographic analyses from several G-proteins, a G-domain
consists of approximately 200 residues. Illustrated in Figure 2.1.1, similar to an G-proteins, a
typical nuc1eotide binding domain contains a central 6-stranded ~-sheet surrounded by a-helices
(21). In addition, all G-domains contain five highly conserved guanine nuc1eotide binding loops,
G-Ioops, that participate in nuc1eotide binding and Mg2+ co-ordination. The structural similarity
in G-domains is reflected in the significant sequence identity in an G-proteins suggesting a
common evolutionary origin for these proteins.
25

Switch II
Figure 2.1.1 A schematic diagram of a typical G-Domain. Switch l and il regions are shown in
dark grey. Secondary structrual elements and conserved guanine nucleotide binding loops are
labeled. GppCp is centered as a baIl and stick figure and Mg2+ ion is shown as a black sphere.
Secondary structural elements and conserved loop regions (G1 through G5) involved in
nucleotide binding and Mg2+ coordination are labeled (21).
As mentioned, G-proteins undergo global structural changes in response to altemate binding of
GDP and GTP. There are two key regions that undergo marked conformational changes when in
the GDP or GTP bound forms. These regions, designated as the effector regions switch 1 and
switch II, regulate these global structural changes. Two G-Ioops, G2 and G3, are within both
switch regions respectively and contain conserved residues that co-ordinate Mg2+. Figure 2.1.2
focuses on the loops (G-loops) that contribute in forming the nucleotide binding motifs and Mg2+
ion coordination site.
26

Figure 2.1.2 The nucleotide binding site of a typical G-domain. The side chaills of highly
conserved residues within the G-Ioops are in dark grey and labeled with one letter amino add
code. Centered is the GTP analogue, GppCp, complexed with a Mg2+ ion shown in light grey
(21).
Since this research focuses on elongation factors, the following sections will explore G-dorrtains
found in these proteins. Figure 2.1.3 illustrates the high degree of sequence conservation in G-
domain of translation factors. The high degree of conservation found in the loop regions is not
only observed in elongation factors but also in aH proteins containing G-domains. Table 2.1
denotes the conserved residues.that are found in the loop regions of elongation factors involved in
Mg2+ coordination and nucleotide binding.
The lst loop (G1), is referred as the phosphate binding loop (P-loop) because it contains a set of
residues that wrap around the a and ~ phosphates of the guanine nucleotide. In addition, the P-
loop contains an essential ThrlSer residue that is central in coordinatîng Mg2+.
27

G5 G5LVNYDEEKFn alkdEVAALI KTVGY----- ---------- -----KPSDV EFIPLSAFEG DNITSKSENT PWY------SVKWDESRFq eivkETSNFI KKVGY----- -----~---- -----NPKTV PFVPISGWNG DNMIEATTNA PWYkgweketMVDDpELLDL VEMEVRDLLN QYEFPGDEVP VIRGSALLAL EqrohrNPKTr ---------- -----RGENE WVD------MVDDEELLELVEMEVRELLS QYDFPGDDTP IVRGSÈiliKAL E--------- ---------- ------GDAE WEA-------
G3 G4HAKFETDKYE FTIV1J.':~PGHR DFVKNMITGA SQADAAVLVV AvdDGVMP-- -----QTKEH VFLSRTLGIN QLIVAINKMDLWKFETPKYQ VTVI2~PGHR DFIRb~ITGT SQADCAILII AGGVGefeag iskdgQTREH ALLAFTLGVR QLIVAV1QKMDHVEYETAKRH YSHVDCPGHA DYIKNMITGA AQMDGAILVV SAADGPMP-- -----QTREH ILLARQVGVP YIVVFMNKVDHVEYDTPtRH YAHVR~PGHA DrvKM~ITGA AQMDGAILVV AAtDGPMP-- -----QTREH ILLGRQVGVP YIIVFLNKCD
MT EFl-A 1SC EFI-A 1TA EF-Tu 1EC EF-Tu 1
MT EFl-A 67SC EFl-A 77TA EF-Tu 67EC EF-Tu 66
MT EFl-A 140SC EFl-A 157TA EF~Tu 140EC EF-Tu 139
GlMA----KEKE HMNLAFIGHV DHGKSTLVGH LLLQAGAlae q--------MG----KEKS HINVVVIGHV DSGKêITTGH LIYKCGGldk rtiekfekeaAKGEFIRTKP HVNVGTIGHV DHGKIILTAA LTFVTAaenp nvevkdygdSKEKFERTKP HVNVGTIGHV DHGK1~LTAA ITTVLAktyg gaarafdq--
G2
-QLAEGEDKF RFVt'TDRLSEE ~~i~~aELGKGSFKY AWVLDKLKAE---------- ----'vnhl~88
---------- ---IDNAPEE __~~~,~_
Figure 2.1.3 A comparison of G-domains from EFI-Affu from M. thermoautotrophicum, S.cerevisiae, T. aquatiqus and E. coli. Uppercase letters indicate conservation of residues. G-loopsare underlined and labeled. ltalicized residues shown in red designate switch 1 and II regions
respectively. Residues involved in: phosphate binding are labeled in purple, nudeotide binding inblue.and Mg2
+ co-ordination in green.
G l-Loop or Phosphate binding loop G2-1oop G3-1oop G4- Nucleotide G5-1oop
Mg2+ Mg2+ Binding Motif
His: Asp His Gly Lys Thr Thr Ile IAsp Asp C:,rs Pro Asn Lys Asp Ser ~a
EC EF-Tu 19 21 22 23 24 25 26 49 :JO ~;" B1 8? 135 136 138 173 74tJU
TA EF-Tu 19 21 22 23 24 25 26 50 r.:;. "1 81 82 83 136 137 139 174 1175v ~L
SC EFI-A 15 17 Ser 19 20 Ser21 22 L1;n;l, 61 91 92 93 b3 154 156 183 ~84
18 60MT EF1-A 15 17 18 19 20 Sez.:'21 22 Met. 51 81 82 83 136 137 139 1~<; ~.., r.. ~ ,':)
5D
Table 2.1 Residue conservation found in loop regions of the G-domain in translation factors EF
Tu. from E. coli, T. aquaticus, and EFI-A from S. cerevisiae and M. thermoautotrophicum. As in
Figure 2.1.3, residues involved in phosphate binding are labeled in purple, nucleotide binding inblue and Mg2
+ co-ordination in green.
28

Mutations of this residue reduce GTPase activity, and destabilize nucleotide and aa-tRNA
interaction (30). These results indicate the importance of Mi+ to ligand binding and GTP
hydrolase activity. The G2 loop is within the Switch 1 region and contains two residues that co
ordinate Mg2+ via water molecules. The G3 loop overlaps the switch II region and contains
residues that complete the co-ordination sphere of Mg2+. G4 contains a conserved NKXD motif
which is structurally important for binding and recognizing the guanine ring (31,32).
Furthermore, mutations in this motif reduce A-site fidelity on the ribosome which increases
misincorporation of amino acids in vitro (33). These results imply that this motif could also be
involved in ribosomal interaction. The G5 loop is not found on all G-proteins but does forms
part of the binding pocket for the guanine ring in translation factors. Figure 2.1.4 gives a
detailed examination of how residues from Table 2.1 interact with the nucleotide and Mg2+ co-
factor.
29
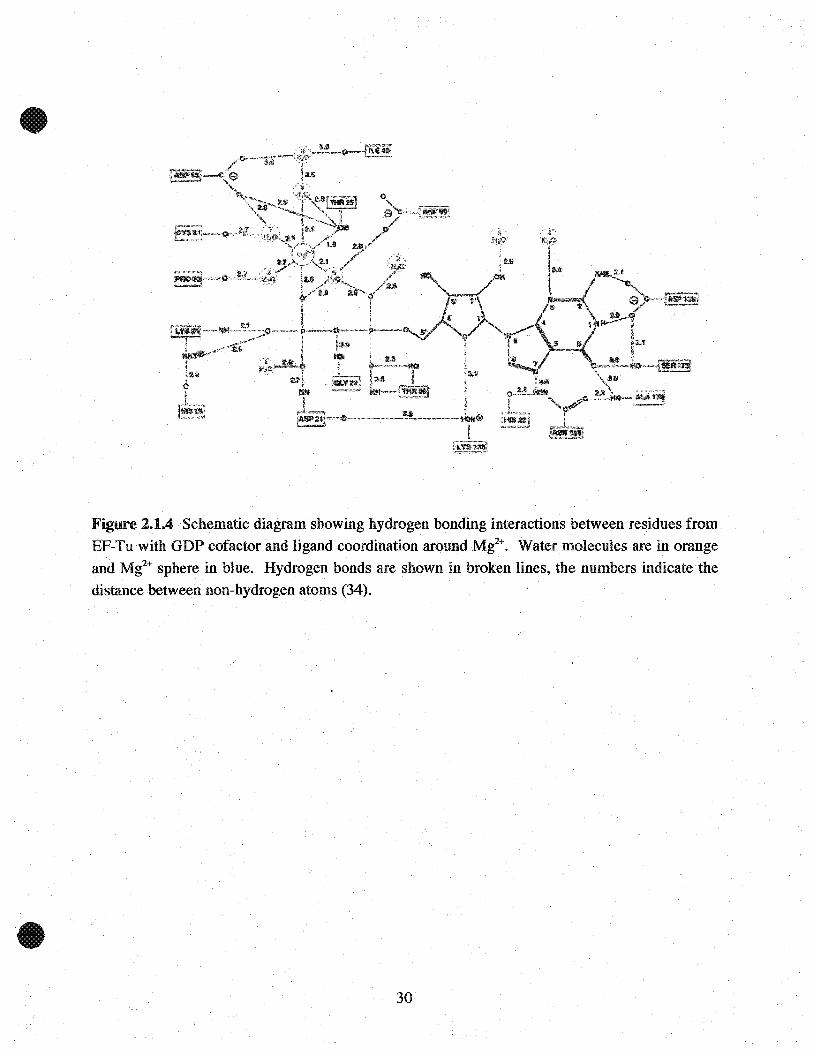
Figure 2.1.4 Schematic diagram showing hydrogen bonding interactions betwe~n residues fromEF-Tn with GDP cofactor and ligand coordination around Mt+. Water moIecuIes are in orangeand Mg2
+ sphere in bIne. Hydrogen bonds are shown in broken Hnes, the numbers indicatethedistance between non-hydrogen atoms (34).
30

2.2 Elongation Factors EF-Tu and EFI-A
The following section addresses how guanine nucleotide binding induces conformational changes
based on the structural information from crystal structures of EF-Tu (E. coli, T. aquaticus, T.
thermophilus) in their unbound and nucleotide bound forms and EF1-A (S. cerevisiae) in ils
complex form with EF-1Ba and nucleotides.
2.2.1 Elongation Factor EF-Tu
EF~Tu is a flexible complex whose structure is influenced by many external factors. Crystal
structures of EF-Tu (34, 35) show that il is comprised of two main structural units. The first unit
contains thefirst domain (Domain 1), located at the N-terminal, consisting of -200 residues and
has the structure of a prototypical G-domain. The second structural unit is composed of two
domains that are held together by strong interdomain interactions. Their close association allows
them to remain in the same relative orientation in the various conformational states of the protein
(36, 37, 38). Domain 2 and 3 are both - 100 residues and are both organized as antip.arallel B-
barrels. Domain 2 is involved in EF-Ts interaction (22, 25) and domain 3 is principally involved
in aa-tRNA recognition (39). Given that Domain 1 is the G-domain, herein lies the switch 1 and
switch II regions. When comparing the structure of EF-Tu in ils GDP (35,37) to GTP (36, 40)
bound forms, there are notable structural changes that occur within these regions including a
rearrangement of domains.
31

To illustrate, residues 54-59 (E. coli) in the switch 1 region form a l3-strand in the GDP form.
Upon binding to GTP these residues fold into a short a-helix. In addition, upon GTP binding, the
a-helix that foUows switch II is shifted by 4 residues. This unwinds one helical turn at the N-
terminus and forms another at the C-terminus. This changes the orientation of this helix which
induces domain 1 to rotate "-'90° relative to domain 2 and :3 (36). As seen in Figure 2.2.1, the
result is that the open pocket centered in the GDP form closes and the interaction between
domain 1 and 3 changes when GTP is bound.
Figure 2.2.1 Ribbon diagrarns of (A) E. coli EF-Tu-GDP (34) and (B) T. aquaticus EF-Tu-GTPcornplex structures. The polypeptide backbone for dornains 1,2 and 3 are shown in purple, lightblue and dark blue, respectively. The switch 1 region is shown in yeHow and switch Il in green.GDP and GTP rnolecules are in baU and stick model and Mt+ are shown as light blue spheres(36).
32

To summarize, the binding of GTP and its hydrolysis to GDP is accompanied by local changes in
switch 1 and il that contribute to a global conformational change wmch alters the relative
orientation of the nucleotide binding domain 1 to domain 2 and 3. It is these altemate switching
conformations that controis the overall affinity of EF-Tu for other molecules including the
nucleotide itself, the ribosome, aa-tRNA and EF-Ts.
2.2.2 Elongation Factor EFl-A
As expected elucidation of the crystal structure of S. cerevisiae (S.c.)EFI-A (17,21), Figure 2.2.2,
revealed a sirnilar structure to EF-Tu.
Figure 2.2.2 Ribbon diagram of S. cerevisiae EFI-A. Domain 1 contains u-helices in red and 13sheets in blue. Domain 2 and 3 are shown in purple and bIlle, respectively (17,41).
33

Although there are some variants in the size and number of secondary structural elements as weIl
as the relative orientation between each domain, (S.c.)EFI-A still contains three distinct domains
that are similar in size and function. The only eukaryotic structures of EFI-A available are those
of S. cerevisiae complexed with the catalytic domain of its exchange factor EFI-Ba and
nucleotides GDP or GTP (26). Similar to EF-Tu, these structures show that switch 1 and II in the
G-domain are the two regions that influence conformational changes in the protein.
34

2.3 Three-Dimensional Model of (M.t.)EF1-A
A three-dimensional model of M. thermoautorophicum (M.t.)EFI-A was generated to observe the
overall topology of the protein, to identify the guanine nucleotide binding pocket and to compare
its foldwith existing structures of EFI-A and EF-Tu.
2.3.1 Method
A model of (M.t.)EFI-A was constructed using the following templates. Crystal structures of S..
cerevisiae (S.c.)EFI-A in complex with combinations of C-terminal (S.c.)EFl-Ba without
nucleotide (17), with GDP or the GTP analogue GDPNP (26) (PDB entries lF60, nIF, lUE &
lG7C) and T.aquaticus (T.a.)EF-Tu in complex with both GDPNP and Phe-tRNA (PDB entry
lTTT) (39). Homology model building was completedusing an automated modeling software
ProModII (Guex, N. and Peitsch, M.C.) found in SWISS-MODEL6 located on the ExPASy
molecular biology website. The final model was subject to Powell energy minimization using X
PLOR (Brunger, A.T.) software running on a SGI workstation (Silicon Graphie Inc,
Mountainview, CA, USA).
6 Please refer to Appendix
35

2.3.2 Results
Since the sequence identities between (M.t.)EFI-A with (S.c.)EFI-A and (T.a.)EF-Tu are 67%
and 51 % respectively, the homology model generated can be assumed to be reliable. The model
of (M.t.)EFI-A was based on template structures that are bound to a nucleotide and its
corresponding exchange factor. Rence, the homology structure presented is a hypothetical
conformation as it would be bound to its associated nucleotide exchange factor and guanine
nucleotide.
Figure 2.3.1 clearly shows three distinct domains that are comparable to other elongation factors
with known crystal structures (Figures 2.2.1 and 2.2.2). Domain 1 (residues 1-202) consists of
six (X-helices and a six-stranded ~-pleated sheet. This is the G-domain, which contains a
structural core common to aIl other G-nucleotide binding proteins (42). Within this structural
core are the highly conserved sequence elements that are involved in nucleotide binding and Mg2+
coordination (Figure 2.1.3). Given that this is a homology model, it is not possible to identify
with certainty which residues are involved in nucleotide binding and Mg2+coordination.
Rowever, since the loop regions contain aIl the significant conserved residues it can be assumed
that the binding apparatus is very similar (Table 2.1.1). As seen in Figure 2.3.2 the topology of
aIl the loops around the nucleotide are similar in aIl structures.
36

GDP-Mg2+
DomainI
DomainU Domain III
•
Figure 2.3.1 Ribbon representation of the hornology model of M. thermoautotrophicum EFI-A.
(M.t.)EFI-A is organized in three structural domains. From the N-terminus, domain 1 (GDomain) is shown in red u-helices and green (3-sheets. Imbedded is a multicolored GDP
complexed to Mg2+, which is shown as an orange sphere. Domain II and HI are shown in blue
and purple respectively.
37

klcp4
locp!
klcp :3
Figure 2.3.2 A ribbon representation of conserved loop regions involved in guanine nucleotidebinding and M~ coordination. EFI-A nucleotide binding pocket of S. cerevisiae (PDB lIJF,right), M. thermoautotrophicum (model, middle) and T.aquaticus (PDB ITTT, left). Loops 1
through 5 are shown in light purple, yellow, and dark purple, blue and orange respectively.
Guanine nucleotides are multicolored and Mi+ are shown as white spheres.
One noticeable difference is within the primary sequence of the 5th Ioop when comparing
eukaryotic/archaeal and prokaryotic elongation factors (Figure 2.1.3). Although there is Iow
sequence homology between the 5th loop,. the location of the 5th Ioop on the structure with
respect to the nucleotide is similar. In addition, within the 5th loop of both structures are a
conserved serine and alanine residues that participate in nucleotide binding is present.
Nevertheless, smaU differences in the primary sequence may cause subtle variations in the
binding apparams wmch could contribute to functionaI differences interms of recognition of both
nucleotides and other binding partners (42). Domain 2 of OW.t.)EFI-A (217-296) consists of 913-
38

sheets and is connected to domain 1 bya 16 residue long peptide (203-216). Domain 3 (307-410)
has 7 ~-sheets is connected to domain 2 by 10 residues (297-306). Both domains belong to the
tertiary structural class of antiparallel beta barrels typically seen in aH elongation factor
structures.
39

Chapter 2.4 Expression and Purification of (M.t. )EF1-A
The following section describes the purification procedures used to isolate a pure form of the
recombinant protein (M. t. )EF1-A.
2.4.1 Materials and Methods
Buffers usedfor protein purification
Base Buffer, 50mM HEPES (ICN), 500mM NaCI (ACS), 5% Glycerol (DDH), at pH 7.5. Wash
Buffer; base buffer with either 5mM or 30mM Imidazole (Boehringer Mannheim) at pH 7.5.
Resuspension Buffer, base Buffer with 100ug/rnllysozyme, 7mM ~-mercaptoethanol (Omnipur),
1mM MgCl2 (Boehringer Mannheim) and 1mM Phenylmethylsulfonyl fluoride (PMSF) at pH
7.5. Gel-Filtration Buffer, SOmM Tris-Hel (Gibco), 500mM NaCI, 10% Glyc~rol, lOmM EDTA
(Fischer Biotech) and lOrnM ~-mercaptoethanol at pH 7.3.
Elongation Factor-lA construct
The archaeal EF1-A construct from M. thermoautotrophicum (gene MT 1058) was provided by
Cheryl Arrowsmith and Aled Edwards from the Ontario Cancer Institute and University of
Toronto.
40

Vector and Expression strain
Fulllength (M.t.)EFl-A was subcloned into aN-terminal 6x-Histidine tag vector with a thrombin
cleavage site, pET15b (Novagen), at the BamHI and NdeI (NEB) restriction sites. The construct
was transformed into an E. coli BL21 Gold Magic (GM) strain for expression. This strain is a
variant of BL2l Gold (Stratagene) that carries an extra plasmid that encodes three rare transfer
RNAs (tRNA - Arg AGG, Arg AGA, Ile ATA) not commonly found in RcoU strains but are
more prevalent in eukaryotic organisms (43). The choice of this strain is based on the analyzing
the DNA sequence of the desired gene. If the sequence shows a high frequency of these rare
codons, this host is chosen to ensure high level expression. Ampicllin (Fischer) is used to select
pET15b, tetracycline (Fischer) is used to select the expression host and kanamycin (Fischer) is
used specifically to select the extra magic plasmid.
Sample Preparation and Purification
An overnight culture was prepared from a single colony of transformed BL2l-GM containing the
pET15b-MTl058 construct. 1 liter of lx Luria Broth (LB) (DIFCO) was supplemented with
lOOug/ml and 50ug/ml of ampicillin andkanamycin, respectively, and inoculated with 10 ml of
overnight culture. The culture wasvigorously shaken and grown at 37°C until OD600 = -0.8.
The growth temperature was reduced to 25°C and isopropyl-beta-D-thiogalactopyranoside
(IPTG) was added for a final concentration of 0.5 mM to induce protein expression. The culture
41

was left overnight for expressIOn of the fusion protein. The cens were harvested by
centrifugation and stored at -20°e.
For every gram (wet weight) of packed cells, 3 ml of resuspension butter was used. The cens
were lysed by sonication and centrifuged for 30 minutes in a Beckman JA-20 rotor at 30000g to
remove the insoluble fraction. The supernatant was then passed through a DEAE-sephacel
(Pharmacia) resin in O.S M NaCI to remove nucleic acids. The sample was subjected to heat
denaturation at 6S-70°C for 10 minutes followed by centrifugation in a 60Ti Rotor at 4S000g for
30 min to remove all precipitated proteins. The protein was purifiedusing irnrnobilized metal ion
affinity chromatography. NiS04 (Anachemia) is equilibrated with Chelating Sepaharose Fast
Flow resin (Pharrnacia) to immobilize the Ni2+ metal ion on to the resin. This technique exploits
histidine's (6x-His tag) affinity for chelated metals. The Ni2+ charged resin was poured into a
gravity column and equilibrated with binding buffer. Prior to column application, the supernatant
was passed through a Whatman (Fischer) filter to remove residual precipitates. After sample
application, the column was sequentially washed with SmM and 30mM imidazole wash buffer to
remove any non-specifically bound protein. The fusion protein was eluted with SOOmM
imidazole and 10 mM EDTA was subsequently added to the eluted fractions to prevent Ni2+
mediated aggregation of the histidine tag (44). Eluted samples were then prepared for sns-
PAGE to determine protein purity.
42

Gel-Filtration High Performance Liquid Chromatography (HR75 Superdex Prep)
The elution fractions from the previous step were pooled and dialyzed against Gel-filtration
buffer for HPLC column preparation and to remove imidazole, which absorbs UV. The sample is
concentrated down to 1 ml (1.5 üD at 280 nm) and applied to a HR-75 Prep Superdex
(Pharmacia) HPLC colurnn at a flow rate of 2 ml/min and monitored at 280 nm. 1 ml fractions
are collected and prepared for SDS-PAGE analysis. Integrating the area of the observed peaks on
the chromatogram gave 1.27 ûD at 280 nm for a recovery of 87% of total sample.
2.4.2 Results
To investigate the biochemical properties of (M.t.)EF1-A, the protein was overexpressed as an N
terminal 6x-Histidine tagged fusion protein and initially purified using irnrnobilized meta1 ion
affinity chromatography. This system has been successfully applied to the expression and
purification of other archaeal proteins (45,46). From SDS-PAGE analysis, there are a series of
proteins that was seen at the final elution fraction (Figure 2.4.1B, lane 2). Two proteins are
predominant in this fraction, one corresponding to the full length (M.t.)EF1-A ~47 kDa and
another at 30 kDa. In addition, multiple bands can be seen above and in between these two major
bands. A series of alternative conditions were attempted to improve expression and purification
conditions. These conditions included varying growth and induction temperature, heat shock
time, denaturing conditions, elution with imidazole gradient, addition of other reagents in
43

resuspension buffer including solubilzing agents such as 1% SDS and 2% Triton or a stabilizing
guanine nuc1eotide ligand. Unfortunately, none of the attempted modifications improved the
protein purity at the final elution step (data not shown). Given the series of bands observed in the
elution fraction another purification step was deemed to be required.
Further purification of (M.t.)EFl-A was completedusing HPLC-gel filtration column. From
SDS-PAGE analysis, this purification strategy proved to be successful in isolating a pure form of
(M.t.)EFl-A. Based on the HPLC chromatogram and SDS-PAGE analysis, Figure 2A.lA and B,
fraction l (tane 3) contains (M.t.)EFl-A. Fraction 1 was pooled together for further
characterization. The 47 kDa band was also seen in fractions 3 and 4 (Figure 2A.lB lanes 4 and
5, respectively) which eluted with a series of other proteins. Since the higher molecular weight
form was eluted at a later elution time, it was assumed that these proteins either non-specifically
interacted with the size-exc1usion resin or formed an aggregate and eluted together.
Subsequently, a variety of altemate conditions were used to optimize this purification procedure
inc1uding higher salt concentrations and solubilizing agents. None of the modifications resulted
in improving purification conditions (data not shown).
Immunoblotting analysis (data not shown) using an anti-6x-Histidine antibody indicated that both
47 kDa and 30 kDa band observed in the elution fraction (Figure 2A.lB, lane 2) reacted with the
antibody. Since proteins expressed from pET-15b contain an N-terrninal histidine tag, this infers
44

that the 30kDa fragment is a truncated form of (M.t.)EFI-A. From sequence analysis of
(M.t.)EFI-A, the 30kDa fragment corresponds to the N-terminal region of the protein that
contains the G-domain. Renee, fraction 5 (Figure 2A.IB, lane 7) was also collected for further
analysis. The pooled fractions were analyzed by UV spectroscopy at 280nm and a Bio-Rad assay
to determine protein concentration. After purification procedures approximately 0.5 mg of full
lengthand the N-terminal truncated form of (M.t.)EFI-A was isolated from 500 ml of culture.
45
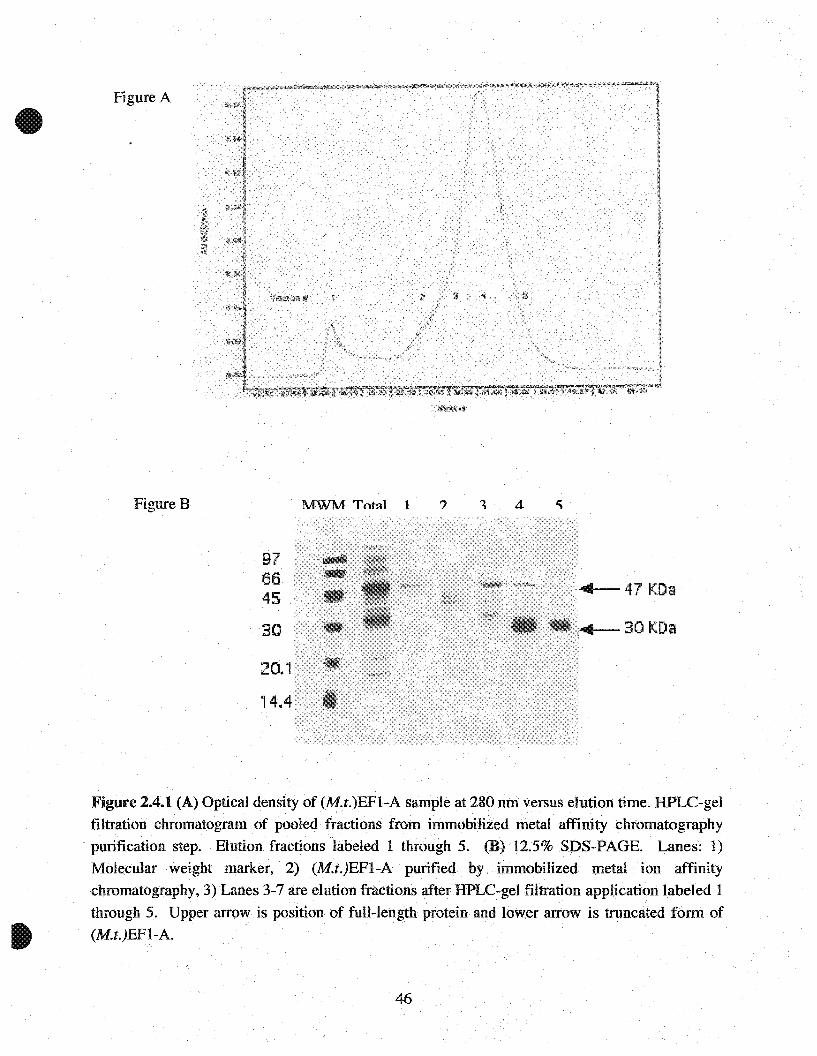
Figure A
Figure B
976645
30
20.1
14.4
MWM Tot~J 1
4-47 KDa
4-3QKDa
Figure 2.4.1 (A) Optical density of (M.t.)EFI-A sample at 280 nm versus elution time. HPLC-gelfiltration chromatogramof pooled fractions from immobilized metal affinity chromatography
. purification step. Elution fractions labeled 1 through 5.(B) 12.5% SDS-PAGE. Lanes: 1)
Molecular weight marker, 2) (M.t.)EFI-A purified by. immobilized metal ion affinitychromatography, 3) Lanes 3-7 are elution fractions afterHPLC-gel filtration application labeled 1
through 5. Upper arrow is .position of fuH-length protein and lowerarrow is truncated form of(M.t.)EFI-A.
46

2.5 Nucleotide Exchange and Affinity assays
2.5.1 Foreword
(M.t.)EFI-A was assayed based on its ability to bind 3H-GDP and form a binary complex
(M. t.)EF1-A-3H-GDP. The following experiments were comp1eted to determine the rate of
guanine nucleotide exchange in both forms of (M.t.)EFI-A as a representation of enzymatic
activity. In addition, a nucleotide competition experiment was completed to determine nucleotide
specificity for (M.t.)EFI-A and relative affinity for GDP versus GTP. The experimental method
was based on a filter binding assay as described in previous literature (47), (48).
2.5.2 Materials
[8'-3H]-Guanosine-5'-diphosphate-NH4 salt (Sp.Act. 10.1 Ci/mmol) was purchased from
Amersham Life Sciences. A filter binding apparatus and 0.45 um filters were purchased from
Fischer Scientific and Millipore respectively. Nucleotides, GMP, GDP, GTP, ATP and CTP
were purchased from ICN laboratories. The following assay buffers were used. Loading buffer:
50mM Tris-Hel, 150mM NaCl, 10mM MgCl2, 10% Glycerol, 5mM ~-mercaptoethanolat pH 7.3
and Wash buffer: 50mM Tris-Hel, 150mM NaCI, lOmM MgClz at pH 7.3.
47

2.5.3 Nucleotide Exchange Assay
Method
100 pmols of fulliength and truncated (M.t.)EFI-A was incubated for 1 hour at 37°C with 300
pmols oeE-GDP (Sp. Activity 7000 cpm/pmol) in 250 ul of loading buffer. After incubation,
lml of loading buffer was added and the mixture cooled to room temperature. The guanine
nucleotide. exchange reaction was initiated by adding a 100 fold excess of unlabelled GDP
(30000 pmol). 150ul aliquots were withdrawn at fixed time intervals and the reaction was
immediately stopped by filtration through a 0.45 mm nitrocellulose filter and quickly rinsed three
times with lml of cold (4° C) wash buffer. The filters were subsequently dried and prepared for
scintillation counting. Radioactivity was determined in liquid scintillation counting fluid (ACS,
Amersham) using a 1219 Rackbeta liquid scintillation counter (LKB Wallace).
Results and Discussion
The disassociation of 3H-GDP from (M.t.)EFI-A was followed by diluting the binary complex
with anexcess of unlabelled GDP. Under these conditions rebinding of 3H-GDP to (M.t.)EFI-A
became negligible and the time dependent decrease in the amount of labeled complex was
followed by first-order kinetics. The observed exchange activity for both full length and
truncated (M.t.)EFI-A was determined to be 6.5 x 10 -3 S-l and 4.6 x 10-3s'l, respectively (Figure
2.5.1A). These values are within an order of magnitude to those of other archaeal elongation
factors such as Sulfolobus solfactarius (M.t.)EFI-A (48) and analogues such as EF-Tu (49). The
48

similar exchange rates for both forms of (M.t.)EFI-A imply that the 30 kDa fragment contain the
apparatus neccesary to bind the guanine nucleotide GDP. This was expected given that the 30
kDa from should contain the elements that comprise the G-domain (residues 1-216,24 kDa) of
(M.t.)EFI-A. Whether or not the fragment possess GTPase activity was notaddressed; however,
truncated forms of aEFI-A that contain only the G-domain have both exchange and hydrolase
activity (50).
2.5.4 Nucleotide Affinity Assay
Methods
150 pmols of fulliength and truncated (M.t.)EFI-A of the was incubated for 1 hour at 37°C with
500 pmols ot3H-GDP (Sp. Activity 7000 cpm/pmol 3H-GDP) in 400ulloading buffer. After
incubation, 800 ul of loading buffer was added, mixed and then aliquoted to 5 separate tubes
containing a 100 fold excess of the following unlabelled purine nucleotides GDP, GTP, GMP,
ATP and the pyrimidine CTP. Each sample tube was equilibrated for 1 hour at room
temperature. Samples are passed through the filtration apparatus and washed three times with
cold (4°C) wash buffer. The filters were dried and prepared for scintillation counting.
Determination of radioactivity is described as above.
49

Fig A0 .....- .....---........--...,...----.
400300
y=-0.0065xR2 =0.9747
200
Time (sec)
y=-0.0046xR2 =0.9762
100-2.5 .....------------......1
o
-2
-0.5
~ -1;f..5-1.5
Fig B 30KDa
'1) 80....,.10'... 60...
CI
'-? 40T
"";;lii: 20
03H- GDP
GDPGTP GMP ATP CTP
Nucleotide
Fig C 47KDa100
-g 80.,.01
:60CI
'-?4OT
"";;lii: 20
o3H- GDP GTP GMP ATP CTP
GDPNucleotide
Figur~ 2.5.1 (A) Guanine nucleotide exchange activity of (M.t.)EFI-A. Activity was determined
by. the exchange. of bound 3H-GDP with an exce.ss of unIabeled GDP as a function of time. Theobserved rate of exchange. (Kobs) was determined assuming 1:1 binding and first order kinetics.Kobs for full length and truncated torm of (M.t.)EFI-A was 6.5 x 10-3 and 4.6 x 10-3 S-1,
respectively. (8, C) Nucleotide specificity ofhoth forms was determinedby displace.ment of 3H_
GDP by an excess of unlabeled. nucleotides GDP, GTP, GMP, ATP and CfP. Relativespecificity was determined by the. displacement of bound 3H-GDP by unlabeled nucleotides.
50

Results and Discussion
Relative affinity is defined as the capacity for a nuc1eotide to displace bound 3H-GDP from the
EFl-A-CH]-GDP complex. Figures 2.5.1 Band C illustrates that GDP and GTP are successfully
able to displace [3H]-GDP from the binary complex, implying that both forms of(M.t)EF1-A are
able to bind these nucleotides. As found with aIl other elongation factors, (M.t)EF1-A is specifie
for GDP and GTP but not for other nucleotides including GMP. Furthermore, GTP has a lower
relative affinity for (M.t)EFI-A than GDP; another feature previously observed in other EF
TuilA studies. Further comparisons of Figures 2.5.1 Band C, also indicates that the full form of
(M.t)EF1-A has a stronger affinity for guanine nuc1eotides than the 30kDa truncated protein.
The observed rate constant kobs ' as determined in section 2.5.3, is the off rate of the nuc1eotide
from the enzyme. A protein's affinity for a ligand, determined in terms of its equilibrium
dissacociation constant, is defined as Kd =Kor/Kon' If the off rate, Korr, is the same in both forms
but the affinity of guanine nuceotides for thefuIl (M.t.)EF1-A protein is higher, then the on rate
(Kon) of the 30 kDa form must be greater than that of the 47 kDa form. OveraIl, since the full
length (M.t)EFI-A has a higher relative affinity for GTP and GDP, this suggests that the full
length forms a more stable complex with gunaine nucleotides than the 30 kDa form.
As discussed, the nucleotide binding core is a weIl conserved structural unit found in G-proteins.
In addtion, amino acid residues that contribute to hydrogen bonding interactions between GDP
51

and Mg2+are highly conserved in aIl EF-Tu structures. Given that (M.t)EFI-A also possess these
residues in the primary sequence (please refer to Figure 2.1.3 and Table 2.1), it is assumed that
interaction with the guanine nucleotide and Mg2+coordination occurs in a similar manner. With
exception to heterotrimeric G-proteins, Mg2+is required to form a tight [GDP~Mg2+]-EF1-Tu!A
complex. In the absence of Mg2+, the affinity for the guanine nucleotides is significantly lowered
(32). Elongation factors posesses intrinsic nucleotide exchange activity when Mg2+is removed.
Kobs values were also determined for (M.t.)EFI-A in the absence of Mg2+(data not shown). The
K obs values were found to be the same in presence or absence of Mg2+. This result implys that
Mg2+may not be required for nucleotide binidng as seen in heterotrimeric G-proteins.
Figure 2.1.4 illustrates the hydrogen bonding interaction between amino acid residues from (E.
coli) EF-Tu with GDP and ligand coordination around Mg2+. With reference to Table 2.1 looking
at residues from E.coli, the G1-loop containsessential residues involved in binding the a and ~
phosphates of the nucleotide. The a-phosphate forms hydrogen bonds with Gly~23 and Thr-26
while the ~-phosphate interactswith three residues His-19, Asp-21 and Thr-25. In addition, the
~-phosphate directly coordinates Mg2+ and forms hydrogen bonds with surrounding water
molecules. The sum of these interactions contributes to the protein's affinity for the nucleotide.
In the absence of Mg2+, coordination between the metal with the a and ~ phosphates of
nucleotide and residues of the protein with stabilizing water molecules cannot occur; thus
explaining reduced nucleotide affinity. Given how a guanine nucleotide interacts with the
52

binding pocket of EF-Tu, it is evident why GMP binds weakly. Without the ~-phosphate, there
are only two hydrogen bond interactions that can occur between the nucleotide (from the a...
phosphate) and residues from the protein. Although the a-phosphate indirectly interacts with
Mg2+ via a H20 molecule, the presence of Mg2
+ would not contribute to GMP stability sincethe
~-phosphateis required for direct coordination of Mg2+.
The relative lack of affinity for ATP and other purine nucleotides can be explained by looking at
the structure and composition of their bases. It has been previously reported that GTP binds with
greater affinity than ATP by six orders of magnitude in EF-Tu. (49). In comparison to GTP, the
adenine base of ATP contains an amino instead of a carboxyl group at position 6 and lacks an
amino group at position 2. Without NH2 at position 2, hydrogen bonding to (E.c.)Asp-138
cannot occur. Furthermore the presence of NH2 at position 6 results in an unfavorable interaction
with the amino group of (E.c.)Ala-174 since both are hydrogen-bond donating groups. These
features allow G-proteins to discriminate against adenosine nucleotides. G-proteins discriminate
against pyrimidine nucleotides mainly due to its size. Pyrimidine bases only contain one six
membered ring and, due to its position in the binding pocket, hydrogen bond formation between
the pyrimidine moiety and residues that are involved in nucleotide bindingcannot occur.
53

Chapter 3
Characterization and Solution Structure of
Elongation Factor l-Bafrom Methanobacterium Thermoautotrophicum

3.1 Guanine Nucleotide Exchange Factors
As discussed in Chapter 1.2.2, EF-Ts and EF1-B regulates the activity of EF-Tu and (M.t.)EFI-A
respectiveIy, by catalyzing the exchange of GDP for GTP thereby regenerating their active forms.
The primary sequences of (M.t)EFI-A and EF-Tu are highly homologous, possess similar
structures and have a common function in the elongation cycle. On the other hand, the
corresponding exchange factors aEF-IBa., eEF-lB and EF-Ts affect a common function but do
not share any significant sequence homology. Comparative sequence analysis of arcbaeal l'lnd
eukaryotic exchange factors indicate the presence of a conserved P+AFG+ motif (Table 3.1).
M.thermoHalobaC.P.abyssiA.pernixS.solifaA.fulgidT.acidopT.volcanS.cerivD.melanHuman
1818205118181821191720
DLEALKiŒIQ ERIPEGTELH KIDEDLDGLEADLS ESLPDGAEIN GTDTNLDELEEKLK AVIPEKYGLA KVEDPKTLAERIK SKLPSGYEVL AEGENLDNLYTDIS SKLPKEYKIIRKETDLNEVLEKIK NIQMEGVEIR DSAIDIKSIEDQVK RNIEGLCSIN RMDDTSVMESEVK EKLNGVCKIN NIEENLEEMVANVK AIEMEGLTWG AHQFDMKEMENNVR TIEMDGLLWG ASKLDMAKLEECVR SIQADGLVWG SSKL
ilAL.NVl!!fVV'V -GDAEG--GTEALLTTVIV -PDDSG-GTEALKFYVLG -KDEEGySFD
KLVIAM NEDTEG-GTEALILYVQM PEQTEG-GTD
IVLMAVM -PDMEG~IGD
YIKLEIIV -QDKEG-EIDIHLEVIV -EDKEG-EVD
KLQINCVV -EDDKV-SLDKLQIMCVI -EDDKV~SID
KKLQIQCVV -EDDKV-GTDP+AFG +
(100)(54)(75)
(48)
(51)(51)(52)(49)(N)
(N)(N)
Table 3.1 Sequence alignment of (M.t.)EF1-Ba. witharchaeal and eukaryotic exchange factors.
Above the lineare sequences of other archaean species andbtHow are sequences of eukaryotic
species. Percent homology is in brackets. (N) isno significant homology found. ConservedP+AFG+ motif is highlighted in blue and Pheffyr residue implicated in nucleotide exchange
in purple.
55
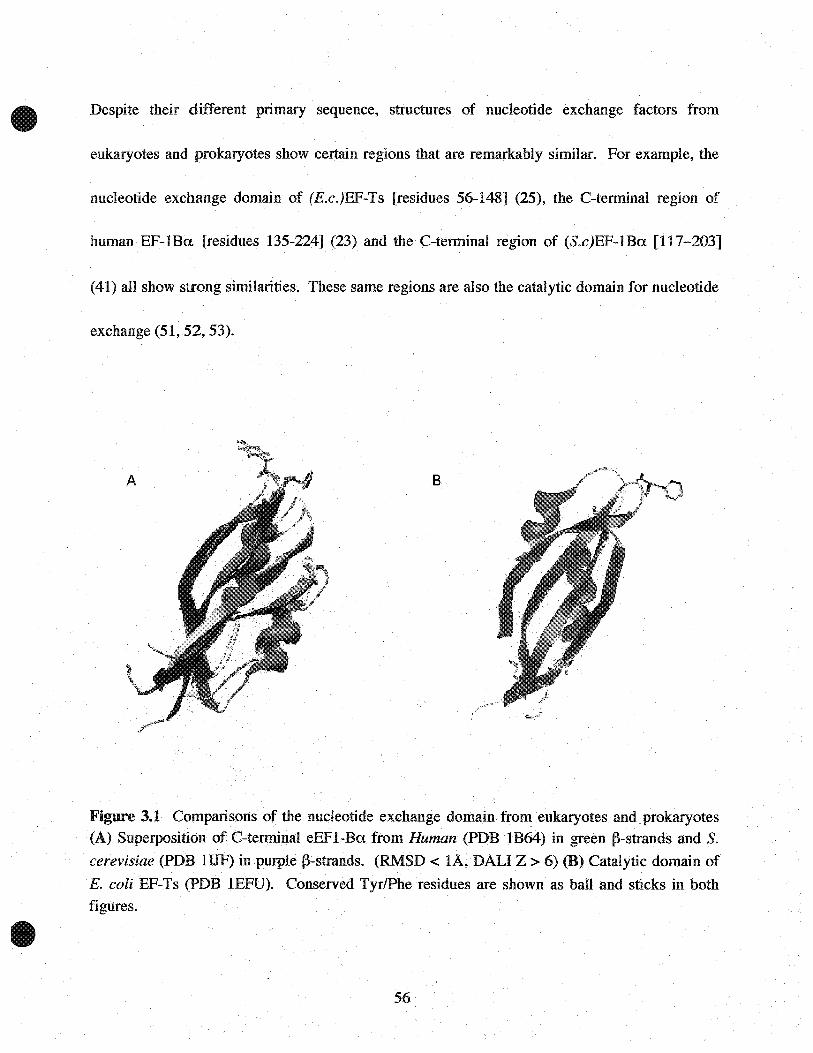
Despite their different primary sequence, structures of nucleotide exchange factors from
eukaryotes and prokaryotes show certain regions that are remarkably similar. For example, the
nucleotide exchange domain of (E.c.)EF-Ts [residues 56-148] (25), the C-terminal region of
human EF-lBa [residues 135-224J (23) and the C-terminal region of (S.c)EF-lBa [117-203]
(41) all show strong similarities. These same regions are also the catalytic domain for nucleotide
exchange (51, 52, 53).
B
Figure 3.1 COtIlparisons of the nucleotide exchange domain. from eukaryotes andprokaryotes(A) Superposition ofC-tenninal eEFI-Ba from Human (PDB IB64) in green j}-strands and S.
cerevisiae (PDB lIJF) in purplej}-strands. (RMSD < lA, DALI Z > 6) (B) Catalytic domain of
E. coli EF-Ts (PDB .1EFU). Conserved TyrlPhe residues are shown as baIl and sticks in bothfigures.
56

Analysis of the complex structure of (E.c.)EF-Tu:Ts reveals that Phe-81 of (E.c.)EF-Ts is
responsible for inducing nucleotide exchange (25) (22). Phe-81 lies within a sequence motif 78
QTDFV-82 that is highly conserved between prokaryotes and rnitochondrial EF-Ts proteins. As
seen in Figure 3.1, the phenylalanine ring is found on a loop region exposed to the solvent.
Superposition of the guanine nucleotide catalytic domains of eEF1-Ba and and EF-Ts results in
an RMSD of < lA (23). Comparing Figure 3.1 A and B, shows that the conserved P+AFG+
motif is also found on a loop region at a similar position. Due to their apparent structural
similarity, it was proposed that the phenylalanine ring plays an analogous role to Phe-81 of
(E.c.)EF-Ts (23). However, the recent structure elucidation of S.cerevisiae EFI-A in complex
with its exchange factor EF1-A:Ba (26) reveals a completely different mechanism for nucleotide
exchange. The following chapter reviews and compares the mechanism of guanine. nucleotide
exchange found in prokaryotes and eukaryotes.
57

3.2 Guanine NudeotideExchange Mechanism
3.2.1 GEF Mechanism in E.Coli EF-Tu:Ts
In the prokaryotic system, EF-Ts recognizes domain 1 and 3 of the EF-Tu-GDP complex (22,25).
sPhe-Sl? is inserted into a hydrophobie pocket of EF-Tu between His-S5 located on the G3-loop
within switch II of the 2nd u-helix and His-llS on the 3rd u-helix. Upon insertion of sPhe-SI
three significant events occur. First, the phenylalanine ring disrupts the Mg2+ binding site by
displacing the G3-loop that contains three residues (Asp-80, Cys-81, and Pro-82)8 involved in
stabilizing water molecules that coordinate Mg2+. Secondly, EF-Ts induces a conformational
change in the G I-phosphate binding loop that results in a flip of the Val-20 and Asp-21 peptide
bond. This disrupts the hydrogen bond between the ~-phosphate of GDP and the amino group of
Asp-21. In addition, the carboxyl group of Val-20 is placed in a position that overlaps the
negatively charged ~-phosphate and therefore sterically and electrostatically displaces that part of
GDP.. Finally, EF-Ts shifts the G4-loop (l35-NXLD-13S ) which contains two residues, Lys-136
and Asp-138, involved in stabilizing the guanine and ribose rings respectively. The sum of these
occurrences, which is disruption of the Mg2+ coordination sphere and weakening the grip on the
guanine base and phosphate groups, allow GDP to diffuse away from EF-Tu. Upon re-entry of
GTP, the y-phosphate occupies the same region as sPhe-81. Since the peptide flip of Val-20/Asn-
7 The prefix s to EF-Ts residues. No prefix are EF-Tu œsidues.
8 Please œfer to Figures 2.1.3, 2.1.4 and Table 2.1.1.
58

21 only destabilizes the binding of GDP, but not GTP (22) and a ternary complex of EF-Tu:Ts
GTP is not found. It is proposed that y-phosphate is suffièient to force Phe81 out of the binding
pocket.
3.2.2 GEF mechanism in S.cerevisiae EF1-A:Ba
As mentioned in Section 2.2.3, the structures of S.cerevisiae EFI-A complexed with the catalytic
domain of its exchange factor EFI-Ba and nucleotides GDP or GTP are known (26). Since the
structures of eukaryotic EF-IA-GDP and -GTP are not known, it is not possible to identify with
certainty which residues participate in nucleotide binding and Mg2+ coordination. However, the
structures of the GDP (37,41) and GTP (38) bound states for EF-Tu are known. The high
sequence and structural conservation in the nucleotide binding apparatus between EF-Tu and
eukaryotic EFI-A allows a description of the mechanism of nucleotide exchange.
In the eukaryotic system, the crystal structure of (S.c)EFI-ABa (17) shows that in addition to
domain 1, EFI-Ba recognizes domain 2 of (S.c.)EFI-A which is very different from the
recognition by EF-Ts of domain 3 in EF-Tu. Furthermore, the catalytic site for nucleotide
exchange in (S.c.)EFI-Ba is located on the C-terminal end in three amino acids 2ü4-QKL-2ü6.
This motif is conserved in aU eukaryotic EFI-Ba as -NKl- located at the extreme C-terminus.
59

Upon interaction of 204-QKL-206 with the G-domain of (S.c.)EFI-A, a series of events occur
which contribute to the release of GDP.
First, the insertion of the positively charged bLys-2059 in effect disrupts the Mg2+binding site by
forming a hydrogenbond with aSer-21, which is expected by homologylO (conserved as Thr-25 in
E. coli) to act as one of the six ligands for Mg2+. Secondly, bGln-204is inserted between the
switch I and II regions of the G-domain which shifts aAsp-91 in the third loop (G3). This residue
is homologous to Asp-80 in EF-Tu, which forms a hydrogen bond to Thr-25 (aSer-21) and water
molecules involved in Mg2+ coordination. Although the hydrogen bond to aSer-21 remains,
aAsp-91 forms a salt bridge to aLys-20, which is homologous to Lys-24 of EF-Tu. In both
systems, lysine is located on the P-Ioop and binds the ~-phosphate of GDP.
The sum of these interactions invariably causes a re-organization of switch II that contains
residues that coordinate Mg2+. Hence, similar to EF-Tu, the disruption of the Mg2+binding site
and elimination of residue-phosphate interaction lowers EFI-Als affinity for GDP. Without the
nucleotide exchange factor, eEFI-A's affinity for GDP is higher than GTP. Upon catalysis the
affinity is lowered and since the cellular concentration of GTP is -10 times higher than GDP,
9 The prefix a and b refer to residues in (S.c.)EFI-A and (S.c.)EFI-Ba respectiveIy. No prefix indicates EF-Tu
residues.
10 Please refer to Figures 2.1.3, 2.1.4 and Table 2.1.1
60

formation of EFI-A-GTP is favored over EFI-A-GDP. Unlike EF-Tu:Ts, GTP is not sufficient
for displacing EF1-Ba from EF1~A. Instead a ternary EF1-A:Ba-GTP complex is formed (18).
As mentioned, (S.c)EF1-Ba interacts with both the G-domain and domain 2 of EF1-A, a feature
that is also distinct from EF-Tu:Ts recognition. Specifically, the C-terminal region of EF1-Ba,
containing the conserved catalytic -QKL- motif, interacts with domain 1. While on the opposite
pole of the protein, the conserved phenylalanine in the loop motif 160-P+AFG+-164 interacts
with domain 2 which is proposed to be the binding site of aa-tRNA. The placement of
phenylalanine (Phe-163) in the aa-tRNA binding pocket of domain 2 suggests a competition
between EFI-Baand aa-tRNA. It is hypothesized that when the ternary EFI-A:Ba-GTP
complex interacts with aa-tRNA synthethase; aa-tRNA triggers the release of EFI-Ba to form a
EFI-A-GTP-aa-tRNA complex.
61

Chapter 3.3 Expression, Purification and NMR spectroscopy of Elongation Factorl-Ba
from Methanobacterium thermoautotrophicum
3.3.1 Materials and Methods
Reagents
The following reagents were used for preparation of isotopically labeled protein: I5NH4Cl, 13C_
glucose (Cambridge Isotopes Laboratory). Minimal media contains glucose, NH4Cl, vitamin BI'
FeS04, and CaClz. Buffers used for purification are as described in Chapter 2.4.
Elongation Factor lBa construct
The archaeal EF1-Ba construct from M.thermoautotrophicum (gene MT 1699) was provided by
Cheryl Arrowsmith and Aled Edwards from the Ontario Cancer Institute and University of
Toronto.
Vector and Expression strain
M.thermoautotrophicum EF1-Ba was subcloned into an N-terminal 6x-histidine tag vector, pET-
15b (Novagen), at the BamHI and NdeI restriction sites. The construct was transformed into
E.coli BL21 Gold (Stratagene) for expression purposes.
62

Sample Preparation and Purification
Preparation and purification of unlabeled and isotopically labeled protein for NMR analysis was
accomplished using theprotocol as described in Chapter 2.4.1. The following modifications
were applied: only ampicillin (100 ug/ml) was supplemented in lx LB for plasmid selection since
BL21 Gold does not harbor the "Magic" plasmid which requires kanamycin. In addition, only 3
hours of growth at 25°C was required for maximum expression of fusion protein after induction
with IPTG.
For isotopically labeled protein, samples are prepared by growing transformed cells in M9
minimal media supplemented with a final concentraion of 0.2% 13C-Glucose, 2mM MgS04,
O.lmM CaClz, luM FeS04, 1ug/ml Vitamin BI and 0.05% 15NH4Cl. After purification of fusion
protein, the N-terminal 6x-histidine tag was cleaved by treating the sample with thrombin
protease (Pharmacia) at l unit/mg of fusion protein overnight at 25°C. The sample was then
treated with p-aminobenzamidine resin (Sigma) and Niz+ charged sepharose to remove thrombin
andthe c1eaved histidine tag respectively.
Mass Spectrometry
Molecular weight determination of (M.t.)EF1-Ba was accomplished by using electrospray
ionization mass spectrometry (ESI-MS) (Perkin Elmer, API III). Prior toapplication, -0.5 mg/ml
of both samples were dialyzed against 10% acetic acid until complete exchange occured.
63

NMR spectroscopy and structure calculations
Backbone assignments of (M.t)EF1-Ba were done by Dr. I.Eikel and Dr. N.Beglova and final
solution structure was completed by Dr. G.Kozlov. Experiments and procedures were as
described in Kozlov, G., et al., Journal ofBiomolecular NMR, (0); 1-8,2000.
64

3.3.2 Purification Results
(M.t.)EF1-Ba was purified to investigate its biochemical properties and to determine its 3-
dimensional structure. IMAC purification was sufficient in obtaining a pure sample of protein
(Figure 3.3.1). On SDS-PAGE, the elution fraction (Lane 5) gave a single band migrating at 14
kDa. Digestion of sample with thrombin protease was successful as only one band is seen below
the 14 kDa mark. Further characterization of (M.t.)EF-1Ba was done using ESI-MS, which gave
a molecular weight of 9675 Da correlating with its theoretical molecular weight.
KDa
94674330
20.114.4
1 234 5 6
11.6 KDa
9.6KDa
Figure 3.3.1 SDS-20% PAGE of (M.t.)EF1-Ba purification by immobilized metal ion affinity
chromatography (IMAC). Lanes: 1) Molecular Weight Marker 2) Total extract 3) Sample afterheat denaturation 4) After Ni2
+ column application 5) Purified fusion protein (upper arrow) 6)MtEF1-Ba after cleavage with thrombin (lower arrow).
65

3.3.3 Structure of(M.t.)EFl-Ba
From the N-terminus, (M.t.)EFI-Ba is eomposed of four anti-parallelj3-sheets that are loeated on
one side of the domain and two a-helices loeated on the other. The secondary structural elements
are sequentially conneeted in the order j31-al-j32-j33-a2-j34, and form a split j3-a-j3 fold.
This aj3 sandwich supeIfold is similar to folds found in many funetionally unrelated and non-
homologous proteins (54). In partieular they are found on many RNA binding proteins (55-57).
One unusual feature of this protein is a loop, between 132 and 133, composed of hydrophobie
residues (Ile44, Ala45, Phe-46, and Leu-48) that is exposed to the solvent.
N-terID
JI
-QKL-motif
Phe-163
Figure 3.4.1 Ribbon representation of the average M thermoautotrophicum EFI-Ba solution
structure. a-heliees, j3-strands and loop regions are shown in red, blue and yellow, respectively.
Residues proposed to be involved in guanine nudeotide exchange are labeled and shown as baUand stick figures.
66

The only aromatic residue in this protein, Phe-46, is found in this loop and points out directly into
the solvent. üther features of this protein include a highly negatively charged surface close to the
N- and C-terminal region, in between al and ~2, and ~l and ~4.
Discussion
Figure 3.4.2 compares (M.t.)EF-lBa with the catalytic regions other GEFs in theelongation
family with known structures. Using a DALI1 structure comparisontool, overlaying (M.t.)EFl-
Ba with (S.c.)EFl-Ba and (E.c)EF-Ts, respectively, results in a Z score of 6.9 and 0.3
respectively. These scores suggest that the (M.t.)EF-lBa structure is more similar to its
eukaryotic counterpart. This is not surprising since the pattern of secondary structural elements is
different between (M.t.)EFl-Ba and (E.c.)EF-Ts. Superposition of (M.t.)EFl-Ba with the
catalytic domains of (S.c.)EFl-Ba and (E.c.)EF-Tu, respectively, results in an RMSD of < lÀ
for the Ca atoms suggesting that their 3-D architecture is remarkably similar despite low
sequence homology.
Primary sequence analysis of (M.t.)EFl-Ba with other GEFs show very few homologous
sequences and none outside of the archaeal domain (Table 3.1). However, as found in other
archaeal and eukaryotic EF-lBa proteins, (M.t.)EFI-Ba contains the conserved P+AfG+ motif.
1 Please see Appendix (Pg. 76)
67

Figure 3.4.2: Structural comparison of guanine nucleotide exchange factors from the translationelongation family. A ribbon representation of structures from C-terminal S. cerevisiae EFI-Ba.
(left, PDB lUE), (M.t.)EF-lBa (middle, PDB IGH8) and central region of E. coli EF-Ts (right,
PDB lEFU). Residues proposed to be involved in guanine nucleotide exchange are shown as
bans and sticks.
Similar to (S.c.)EFI-Ba, this motif (M.t.)-42-PIAFG-47 is found on the loop region and is
solvent exposed. At the same time, superposition of (M.t.)EFI-Ba with EF-Ts also shows the
loop région to be in a similar position. As discussed in Chapter 3.1.1, the loop region (E.c.)EF-
Tu 78-QTDFV-82 and Phe-81, in particular, from E. coli is implicated as the catalytic site for
nucleotide exchange. On the basis of structural similarity and the pattern ofamino acid sequence
conservation, we could propose that the mechanism for nucleotide exchange in archaea is similar
to bacteria where (M.t.)Phe-46 plays an analogous role to Phe~81 of E. coli.
68

Prior to the complex structure of (S.c.)EFI-A:Ba, a similar proposition was made when the
structure of the catalytic domain of human EFI-Ba was completed (23). However, the (S.c.)EFl-
A:Ba complex demonstrated a different mechanism to that seen in bacteria. As discussed in
Chapter 3.2.1, the catalytic site for nucleotide exchange in (S. c. )EFI-Ba is located on the C
terminal end in three amino acids 204-QKL-206. Comparison of the (M.t.)EFI-Ba and (S.e.
)EFI-Ba structures shows that (M.t.)EFI-Ba contains a three residue sequence (86-RRL-88) at
the opposite pole of the protein to the P+AFG+ loop analogous to the nucleotide exchange
catalyitic site (204-QKL-206) of (S.e. )EFI-Ba.
As discussed in Chapter 3.2.2, the positively charged lysine, (S.c.)Lys-205, forms a hydrogen
bond with the side chain oxygen of Ser-21 (homologous to (E.c.)Thr- 25) which contributes to
the disruption of Mg2+ coordination in (S.c.)EFI-A.Furthermore, (S.c.)Gln-204 is inserted in a
region of ~-strands that shifts the switch regions of (S.c.)EFI-A. Since (M.t.)Arg-87 is also
positively charged, it is plausible that it plays an analogous role to (S.c.)Lys-205. Although
(M.t.)Arg-86 contains a larger side chain than (S.c.)Gln-204, this residue could be sufficient in
displacing the switch regions of (M. t. )EF1-A. On the basis of this sequence and structural
similarity, we could propose that the mechanism for nucleotide exchange is similar in archaea
and eukaryotes. Furthermore, the conserved P+AFG+ motif could have analogous roles in both
archaeal and eukaryotic systems.
69

Chapter 4
General Discussion

4.1.1 Summary ofResults
A three-dimensional model ofM. thermoautorophicum (M.t.)EFI-A was generated to observe the
overall topology of the protein and to identify the guanine nucleotide binding pocket. (M.t.)EFl
A was constructed using the crystal strucutures of S.cerevisiae (S.c.)EFI-A (17,26) and
T.aquaticus (T.a. )EF-Tu (39) as templates. The sequence identity between (M.t.)EFI-A with
(S.c.)EFI-A and (T.a.)EF-Tu are 67% and 51 %, respectively. The model of (M.t.)EFI-A clearly
shows three distinct domains that are comparable to other elongation factors with known crystal
structures. Since high sequence identity correlates to strong structural similarity, it is not
surprising that the model was similar to its its homologues. From comparative sequence and
structural allalysis, the first domain was identified as the guanine nucleotide binding domain (G
domain). The G-domain contained highly conserved sequence elements found within its loop
regions that are implicated in nucleotide binding and Mg2+ ion coordination. The topology of the
loops surrounding the nucleotide and Mg2+ ion in (M.t.)EFI-A are highly similar to homologous
structures suggesting that the binding apparatus is very similar.
To investigate the biochemical properties of (M.t.)EFI-A, the protein was over expressed as an
N-terminal 6x-histidine tagged fusion protein. Purification of (M.t.)EFI-A was completed using
a combination of immobilized metal ion affinity and HPLC size exclusion chromatography.
Further characterization was accomplished by SnS-PAGE analysis which revealed a band
71

migrating above the 43 kDa mark which corresponds to its theoretical molecular weight of 47
kDa. Along with the fulliength protein, a -30 kDa truncated form of (M.t.)EFI-A was isolated
and determined to be part of the N'-terminal region of the protein that contains the G-domain.
After aH purification procedures 0.5 mg of (M.t.)EFI-A was isolated from 500 ml of lx LB
culture. (M.t.)EFI-A was further characterized by its ability to bind on to guanine nuc1eotides.
Using a filter binding apparatus technique, (M.t.)EFI-A was treated with radioactive guanine
nuc1eotide eH] ...GDP and its dissociation was followed by diluting the binary complex with an
excess of unlabelled GDP. The observed exchange activity (off rates) for both fulliength and
truncated (M.t. )EFI-A was determined to be 6.5 x 10 -3 S-I and 4.6 x 10-3S-I respectively. These
values are within an order of magnitude to those of other homologous elongation factors. In
addition, a nuc1eotide competition experiment was done to determine nuc1eotide specificity for
(M.t.)EFI-A. Both GDP and GTP were able to displace eH]-GDP from the binary complex,
implying that the both forms of (M.t.)EFI-A are able to bind these nuc1eotides. As found with
allother elongation factors, (M.t.)EFI-A is specifie for GDP and GTP but not for other
nuc1eotides inc1uding GMP. Although similar off rates were found for both forms, fuHlength
(M.t.)EFI-A had a higher affinity for both GDP and GTP suggesting that fulliength (M.t.)EFI-A
is more stable than the truncated form.
(M.t.)EFI-Ba was purified to homogeneity using immobilized metal ion affinity
chromatography. Further characterization of (M.t.)EFI-Ba was done using ESI-MS, which gave
72

a molecular weight of 9675 Da correlating with its theoretical molecular weight. The solution
structure of (M. t. )EFI-Ba was determined by heteronuc1ear and multi-dimensional NMR
spectroscopy. The structure was composed of two a-helices overlaying a four stranded anti-
parallel ~-sheets. The overall structure contains a split ~-a-~ fold that is found in many
functionally unrelated and non-homologous proteins and in particular on many RNA binding
proteins.
73

4.2 Insights into the mechanisms ofGuanine nucleotide exchange in Archaeabacteria
In section 3.3.3 two propositions were made: either aEFI-Ba. catalyses nucleotide exchange
using a mechanism analogous to its prokaryotic or its eukaryotic counterpart. aEFI-Ba. is more
similar to (S.c.)EFI-Ba. in the foHowing manner. They both possess the same sequential pattern
of secondary structural elements (a split f3-a.-f3 fold), an RMSD < lÀ and residues that are
located at the C-terminus in the same structural pattern. This evidently suggests that the
mechanism for nucleotide exchange in archaea should be similar to eukaryotes. However,
sequence comparison of the C-terminal end between other archaeans and eukaryotic exchange
factors (Table 4.4.1) show that archaea do not possess the highly conserved -NKI- motif as seen
in eukaryotic homologues.
M. thermo 66Halobé3.cter 66P.abyssi 69A.pernix 100S.solifact 67A. fulgidus 66T.acidophi 66T.volcaniu 69S.cerivisi 67D.melanoga 65Human 68
AAEESLSGIE G-VSNIEVTDAVEDAFGAVD D-VESVSVEEEVAEKFKEVE N-VESAEVETEVEQLLKNIE G-VQEVEVENNLEEVVNNIQ G-VSHAEVVG lRYIEEIGKIE G-VESVEIEDRVENSISSIE G-VGEISTENRIEKVLSMVK G-VGEINTENDLQQSIEEDE DHVQSTDlAALLQEKIEEFE DFVQSVDlAAMLEEQITAFE DYVQSMDVAA
Table 4.4.1 Sequence alignment of the C-terminal region of EFI-Ba. with archaeal and
eukaryotic exchange factors. Above the line are sequences of otherarchaean species and beloware sequences of eukaryotic species. Percent homology is in brackets. Highlighted in red are thehigWy conserved -(N/Q)-KI- motif responsible for GNE activity in eukaryotes. In yeHow are the
archaeal sequences at similar positions.
74

The key residue in the -NKI- motif is lysine (Lys-20S in S. cerevisiae) which is lethal when
mutated (26). Lys-20S is an essential residue involved in re-organizing the switch II region of the
G-domain in eEFl ~A. Without re-organization of switch II, nucleotide exchange will not occur
and translation elongation will be halted. In sorne archaeans, Lys-20S is conserved as an arginine
residue and could function in an analogous manner. However, this residue is not conserved in
other archaeans. Although it is not shown that asparagine (conserved as Gln-204 in S.e.) is lethal
when mutated, its position in the (S.e.)EFI-A:Ba complex suggests that its structure is important
in displacing the switch regions in EFI-A. This residue is poorly conserved among archaeans.
AU archaeans do however posses the conserved phenylalanine-Ioop region that is solvent
exposed, which could suggest that archeans utilize a mechanism for nucleotide exchange that is
similar to bacteria. OveraU, it is not evident what type of nucleotide exchange mechanism
archaeans utilize. Given the variety of exchange mechanisms seen in other G-protein/GEF
complexes (21), it is plausible that archaeans utilize a unique mechanism for nucleotide
exchange.
75

Appendix
http://www.biosci.ohio-state.edu/-genomes/mthermo/
http://www.expasy.org/swissmod/SWISS-MüDEL.html
DaliZ
76

Appendix
Methanobacterium thermoautotrophicum Home Page
http://www.biosci.ohio-state.edu/-genomes/mthermo/
Sequence Alignment Tools
Primary amino acid sequence alignment (as found in Tables 2.1.3, 3.1 and 4.4.1) from various
species were compared and percent homology was calculated using the DIALIGN 2.1 software
found on: http://bibiserv.techfak.uni-bielefeld.de/dialign/
Homology Modeling
Homology model of Elongation Factor 1-A fromM. Thermoautotrophicum was completed using
an automated comparative protein modeling software found on:
http://www.expasy.org/swissmod/SWISS-MODEL.html
Stru~tural Comparison Tools
The structures of the catalytic domain from Elongation Factor 1-B from S. cerevisiae, M.
thermoautotrophicum and EF-Tu from E. coli was compared using The DALI Server 3D
structural comparison tool found on: http://www.ebi.ac.ukJdali/
Upon structural comparisons of two proteins using this software, the significant output parameter
is the Z-score. The strength of similarity between two proteins is based on this score. Z scoreS
are calculated based on sequence similarity, pattern of secondary structural elements, structural
similarity, and functional similarity. Z scores less than 2 indicate the compared proteins to be
structurally dissimilar.
76

References
1. Stein, J. L., and Simon, M. I. (1996) Pme NatlAead Sei USA 93(13), 6228-30.
2. Woese, C. R, Kandler, O., and Wheelis, M. L. (1990) Pme Natl Aead Sei USA 87(12),
4576-9.
3. Keeling, P. J., Charlebois, R L., and Doolittle, W. F. (1994) Curr Opin Genet Dev 4(6),
816-22.
4. Brown, J. R, and Doolittle, W. F. (1997) Mierobiol Mol Biol Rev 61(4), 456-502.
5. Dennis, P. P. (1997) Ce1l89(7), 1007-10.
6. Kyrpides, N. C., and Woese, C. R (1998) Pme Natl Aead Sei USA 95(1), 224-8.
7. Kyrpides, N. c., Olsen, G. J., Klenk, H. P., White, O., and Woese, C. R (1996) Microb
Comp Genotnies 1(4), 329-38
8. Keeling, P. J., and Doolittle, W. F. (1995) Pme NatlAead Sei USA 92(13), 5761-4.
9. Bult, C. J., White, O., Olsen, G. l, Zhou, L., Fleischmann, RD., Sutton, G. G., Blake, J.
A, FitzGerald, L. M., Clayton, R A, Gocayne, l D., Kerlavage, A R, Dougherty, B. A,Tomb, J. F., Adams, M. D., Reich, C. I., Overbeek, R, Kirkness, E. F., Weinstock, K. G.,
Merrick, J. M., Glodek, A, Scott, J. L., Geoghagen, N. S., and Venter, J. C. (1996)
Seience 273(5278), 1058-73.
10. Smith, D. R, Doucette-Stamm, L. A, Deloughery, C., Lee, H., Dubois, J., Aldredge, T.,
Bashirzadeh, R, Blakely, D., Cook, R., Gilbert, K, Harrison, D., Hoang, L., Keagle, P.,
Lumm, W., Pothier, B., Qiu, D., Spadafora, R, Vicaire, R, Wang, Y, Wierzbowski, J.,
Gibson, R, Jiwani, N., Caruso, A, Bush, D., Reeve, J. N., and et al. (1997) J Baeteriol
179(22), 7135-55.
11. Klenk, H. P., Clayton, R A, Tomb, J. F., White, O., Nelson, K E., Ketchum, KA,Dodson, R J., Gwinn, M., Hickey, E. K, Peterson, J. D., Richardson, D. L., Kerlavage, AR, Graham, D. E., Kyrpides, N. C., Fleischmann, RD., Quackenbush, J., Lee, N. H.,
Sutton, G. G., Gill, S., Kirkness, E. F., Dougherty, B. A., McKenney, K., Adams, M. D.,
Loftus, B., Venter, J. C., and et al. (1997) Nature 390(6658), 364-70.
12. Toistrup, N., Sensen, C. W., Garrett, R A., and Clausen, I. G. (2000) Extremophiles 4(3),
175-9.
13. Pelletier, J., and Sonenberg, N. (1988) Nature 334(6180),320-5.
14. Sonenberg, N., and Pelletier, J. (1989) Bioessays 11(5), 128-32.
15. Bell, S. D., and Jackson, S. P. (1998) Trends MicrobioI6(6), 222-8.
16. Bocchini, V., Adinolfi, B. S., Arcari, P., Arcucci, A, Dello Russo, A, De Vendittis, E.,Ianniciello, G., Masullo, M., and Raimo, G. (1998) Biochimie 80(11), 895-8.
17. Andersen, G. R, Pedersen, L., Valente, L., Chatterjee, 1. 1., Kinzy, T. G., Kjeldgaard, M.,
and Nyborg, J. (2000) Mol Ce1l6(5), 1261-6.
18. Janssen, G. M., and Moller, W. (1988) J Biol Chem 263(4), 1773-8.
77

19. Negrutskii, B. S., Budkevich, T. V.,~halak, V. F., Turkovskaya, G. V., and El'Skaya, A.
V. (1996) FEBS Lett 382(1 ..2), 18-20.
20. Dontsova, M., Frolova, L., Vassilieva, J., Piendl, W., Kisselev, L., and Garber, M. (2000)
FEBS Lett 472(2-3),213-6.
21. Sprang, S. R (1997) Annu Rev Biochem 66, 639-78
22. Wang, Y., Jiang, Y., Meyering-Voss, M., Sprinzl, M., and Sigler, P. B. (1997) Nat Struct
Biol 4(8), 650-6.
23. Perez, J. M., Siegal, G., Kriek, J., K, H. r., Dijk, J., Canters, G. W., and Moller, W. (1999)
Structure FoldDes 7(2),217-26.
24. Kozlov, G., Ekiel, 1, Beglova, N., Yee, A, Dharamsi, A, Engel, A., Siddiqui, N., Nong,
A, 'and Gehring, K. (2000) J Siomol NMR 17(3), 187-94.
25. Kawashima, T., Berthet-Colominas, c., Wulff, M., Cusack, S., and Leberman, R (1996)
Nature 379(6565), 511-'8.
26. Andersen, G. R., Valente, L., Pedersen, L., Kinzy, T. G., and Nyborg, J. (2001) Nat Struct
Biol 8(6), 531-4.
27. Boume, H. R, Sanders, D. A, and McCormick, F. (1991) Nature 349(6305),117-27.
28. Higashijima, T., Ferguson, K. M., Stemweis, P. c., Smigel, M. D., and Gilman, A G.
(1987) J Biol Chem 262(2), 762-6.
29. Saraste, M., Sibbald,P. R, and Wittinghofer, A (1990) Trends Biochem Sei 15(11),430
4.
30. Krab, I. M., and Parmeggiani,A (1999) J Biol Chem 274(16), 11132-8.
31. Hwang, Y. W., Sanchez, A, and Miller, D. L.(1989) J Biol Chem 264(14),8304-9.
32. Hwa.ng, Y. W., McCabe, P. G., Innis, M. A, and Miller, D. L. (1989) Arch BiochemBiophys 274(2), 394-403.
33. Carr-Schmid, A., Durko, N., Cavallius, J., Merrick, W. C., and Kinzy, T. G. (1999) J Biol
Chetn 274(42), 30297-302.
34. Song,H.,Parsons,M. R, Rowsel1, S., Leonard, G., andPhillips, S. E. (1999) J Mol Biol
285(3), 1245-56.
35. Kjeldgaard, M., and Nyborg, J. (1992) JMol Biol 223(3), 721-42.
36. Kjeldgaard, M., Nissen, P., Thirup, S., and Nyborg, J. (1993) Structure 1(1), 35-50.
37. Polekhina, G., Thirup, S., Kjeldgaard, M .., Nissen, P., Lippmann, C., and Nyborg, J.
(1996) Structure 4(10), 1141-51.
38. Nissen, P., Thirup, S., Kjeldgaard, M., and Nyborg, J. (1999) Structure Fold Des 7(2),
143-56.
39. Nissen, P., Kjeldgaard, M., Thirup, S., Clark, B. F., and Nyborg, J. (1996) Biochimie
78(11-12),921-33
40. Berchtold, H., Reshetnikova, L., Reiser, C. O., Schirmer, N. K., Sprinzl, M., and
Hilgenfeld, R. (1993) Nature 365(6442), 126-32.
78

41. Andersen, G. R., Thirup, S., Spremulli, L. L., and Nyborg, 1. (2000) J Mol Biol 297(2),
421-36.
42. Kjeldgaard, M., Nyborg, J., and Clark, B. F. (1996) Faseb J 10(12), 1347-68.
43. Brinkmann, D., Mattes, R. E.,and Buckel, P. (1989) Gene 85(1),109-14.
44. Sprules, T., Green, N., Featherstone, M., and Gehring, K. (1998) Biotechniques 25(1), 20
2.
45. Christendat, D., Yee, A, Dharamsi, A, Kluger, Y., Gerstein, M., Arrowsmith, C. H., and
Edwards, A. M. (2000) Prog Biophys Mol Biol 73(5), 339-45
46. Christendat, D., Yee, A, Dharamsi, A, Kluger, Y., Savchenko, A, Cort, J. R., Booth, V.,
Mackereth, C. D., Saridakis, V., Ekiel, 1., Kozlov, G., Maxwell, K. L., Wu, N., McIntosh,
L. P., Gehring, K., Kennedy, M. A, Davidson, A R., Pai, E. F., Gerstein, M., Edwards,
A. M., and Arrowsmith, C. H. (2000) Nat Struct Biol 7(10), 903-9.
47. Perez, J. M., Kriek, J., Dijk, J., Canters, G. W., and Moller, W. (1998) Protein Expr Purif
13(2), 259-67.
48. Masullo, M., Raimo, G., Parente, A, Gambacorta, A, De Rosa, M., and Bocchini, V.
(1991) Eur J Biochem 199(3), 529-37.
49. Wagner, A., Simon, I.,Sprinzl, M., and Goody, R. S. (1995) Biochemistry 34(39), 12535
42.~
50. Masullo, M., Ianniciello, G., Arcari, P., and Bocchini, V. (1997) Eur J Biochem 243(1-2),
468-73.
51. Raimo, G., Masullo, M., and Bocchini, V. (1999) FEBS Lett 451(2), 109-12.
52. van Damme, H.,Amons, R., Janssen, G., and Moller, W. (1991) Eur J Biochem 197(2),
505-11.
53. Arai, K. (1977) Seikagaku 49(7), 509-49
54. Orengo, C. A, and Thornton, J. M. (1993) Structure 1(2), 105-20.
55. Nagai, K., Oubridge, C., Jessen, T. H., Li, J., and Evans, P. R. (1990) Nature 348(6301),
515-20.
56. Garcia, C., Fortier, P. L., Blanquet, S., Lallemand, 1. Y., and Dardel,F. (1995) J Mol Biol
254(2),247-59.
57. Lindahl, M., Svensson, L. A, Liljas, A, Sedelnikova, S. E., Eliseikina,I. A, Fomenkova,
N. P., Nevskaya, N., Nikonov, S. V., Garber, M. B., Muranova, T. A., and et al. (1994)
Embo J 13(6), 1249-54.
58. Bec, G., Kerjan, P., Waller,J.P. (1994) J Biol Chem 269(3),2086-92
59. Gillman, AG. (1987) Annu Rev Biochem (56), 615-49
60. Keeling P.J. (1996) Syst. Appl. Microbiology (19),32-321
79







