Download - HPLC & MS - Bienvenue

HPLC & MS
A. Garnier 19 novembre 2014
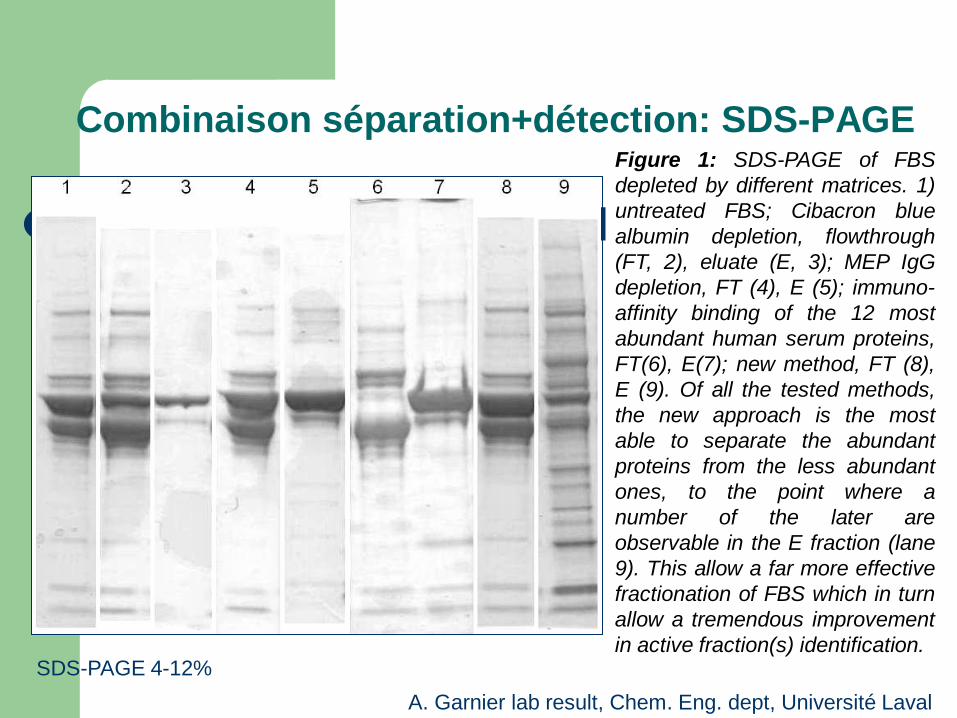
Combinaison séparation+détection: SDS-PAGE Figure 1: SDS-PAGE of FBS depleted by different matrices. 1) untreated FBS; Cibacron blue albumin depletion, flowthrough (FT, 2), eluate (E, 3); MEP IgG depletion, FT (4), E (5); immuno-affinity binding of the 12 most abundant human serum proteins, FT(6), E(7); new method, FT (8), E (9). Of all the tested methods, the new approach is the most able to separate the abundant proteins from the less abundant ones, to the point where a number of the later are observable in the E fraction (lane 9). This allow a far more effective fractionation of FBS which in turn allow a tremendous improvement in active fraction(s) identification.
SDS-PAGE 4-12% A. Garnier lab result, Chem. Eng. dept, Université Laval
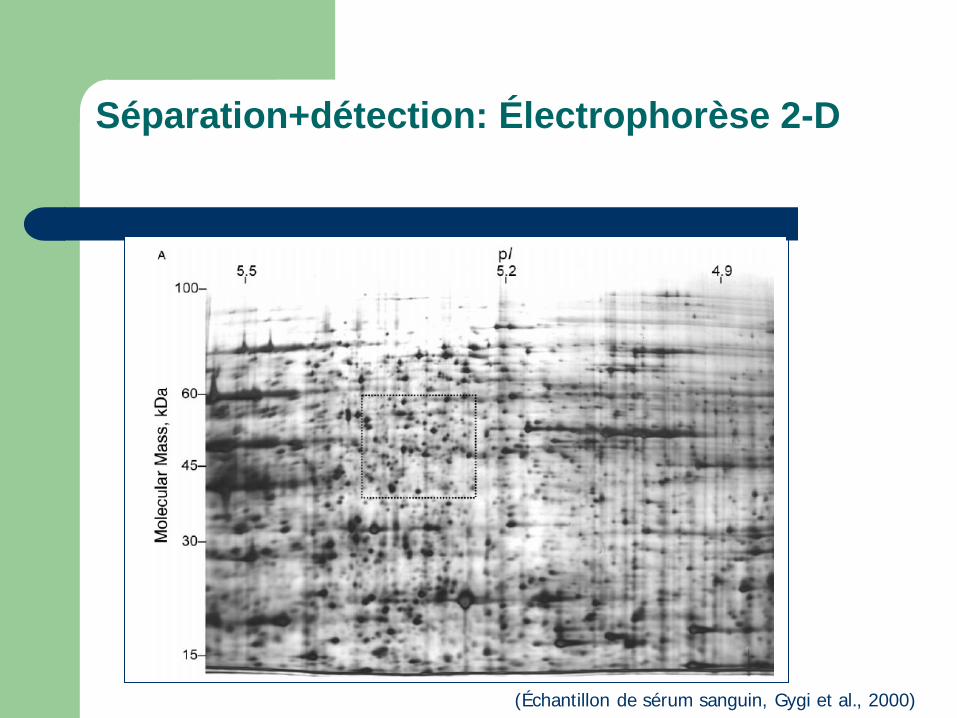
Séparation+détection: Électrophorèse 2-D
(Échantillon de sérum sanguin, Gygi et al., 2000)
2-DE des protéines soluble de levures

Séparation + détection = chromatographie
Chromatographie en phase gazeuse
Chromatographie sur papier buvard

Chromatographie gaz vs liquide
http://www.chem.agilent.com/Library/slidepresentation/Public/HPLC%20Separation%20Fundamentals%20-%20011409.pdf

Chromatographie en phase liquide à haute pression (HPLC)
Réservoir de Phase mobile Pompe
Échantillon
Colonne
Échantilloneur
Détecteur
Collecteur de fraction
•Échange d’ions •Tamis moléculaire •Interaction hydrophobe •Phase inverse •Électrophorèse
•En fonction de la colonne •Gradient
•UV/visible •Fluorescence •Infra-rouge •Indice de réfraction •Conductivité •Spectrométrie de masse
•Automatique •Réfrigéré

Exemple de HPLC (Agilent 1100)

Différentes phases stationnaires
Phase mobile
- - - - - -
+
+ +
++
NP
P
TP
NP
NP
P P
TP TP
TP
TP TP
P P
P
NP
NP
NP
Normale (hydrophobe) Inverse (hydrophile) Exclusion de taille Ionique P: polaire
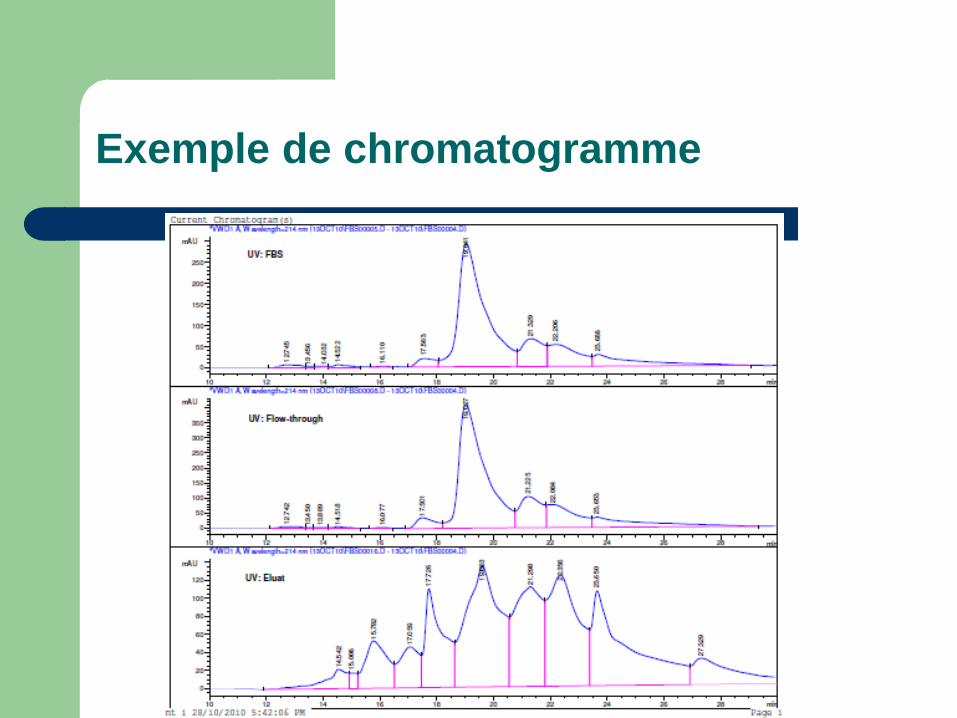
Exemple de chromatogramme
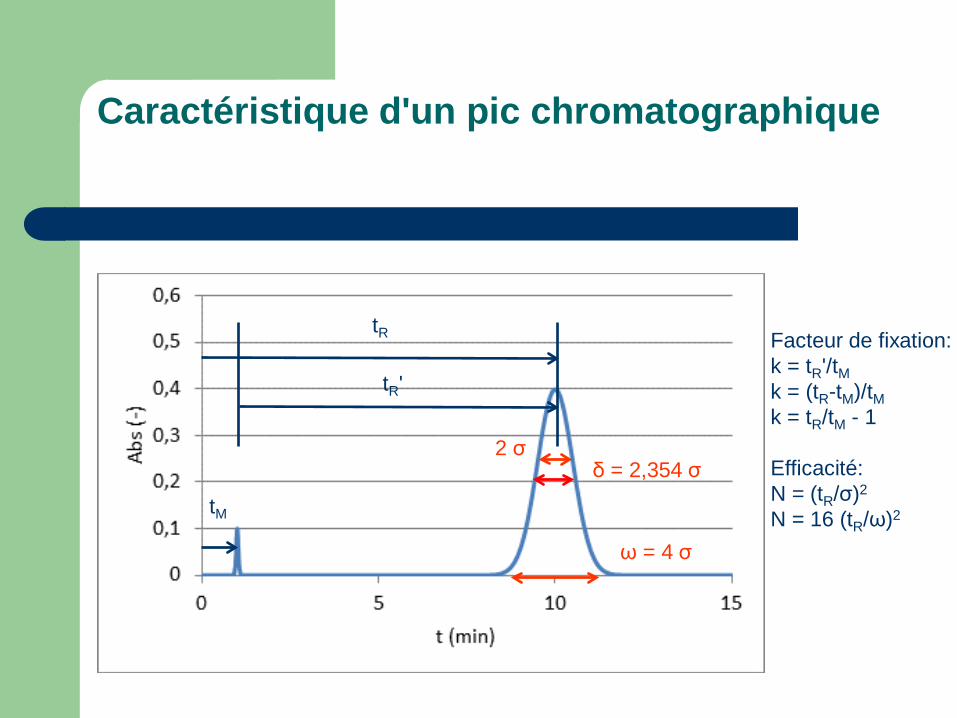
Caractéristique d'un pic chromatographique
tM
tR
tR'
ω = 4 σ
δ = 2,354 σ 2 σ
Facteur de fixation: k = tR'/tM k = (tR-tM)/tM k = tR/tM - 1 Efficacité: N = (tR/σ)2
N = 16 (tR/ω)2

Résolution entre deux pics
Pic A Pic B
tM
tA
tB
ωA ωB
Facteur de sélectivité: α = tB'/tA' = kB/kA α est constant pour des conditions données Résolution: RS = Δt/ω
Δt
Cliquer ici pour accéder au fichier Excel
+
−
=B
BS k
kNR1
14 α
α
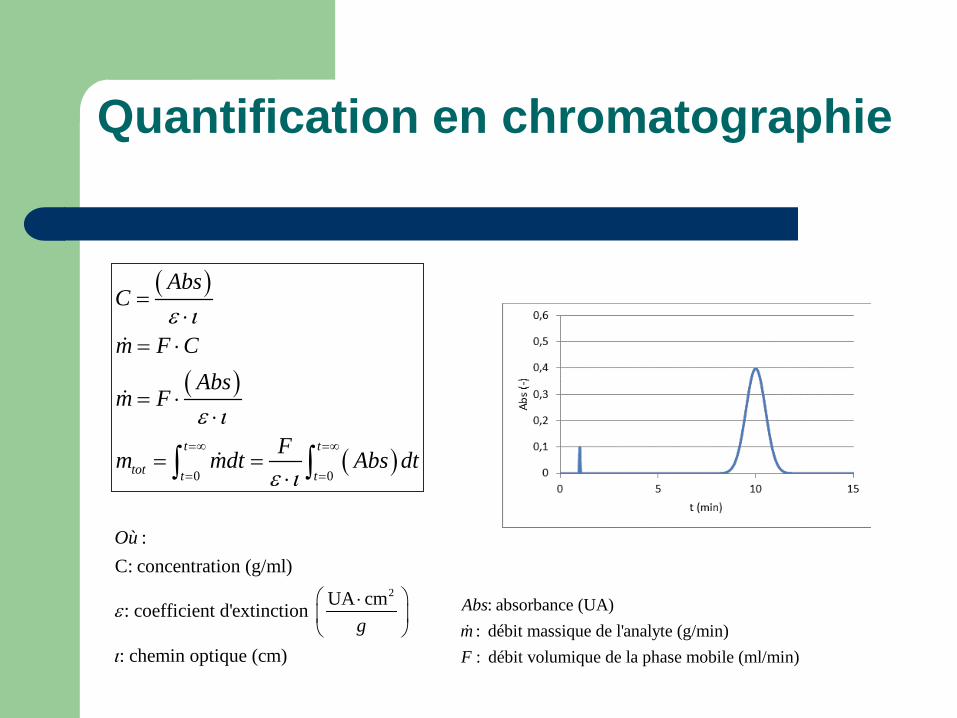
Quantification en chromatographie
( )
( )
( )0 0
t t
tot t t
AbsC
m F CAbs
m F
Fm mdt Abs dt
ε ι
ε ι
ε ι=∞ =∞
= =
=⋅
= ⋅
= ⋅⋅
= =⋅∫ ∫
2
:C: concentration (g/ml)
UA cm: coefficient d'extinction
: chemin optique (cm)
Où
gε
ι
⋅
: absorbance (UA): débit massique de l'analyte (g/min): débit volumique de la phase mobile (ml/min)
AbsmF
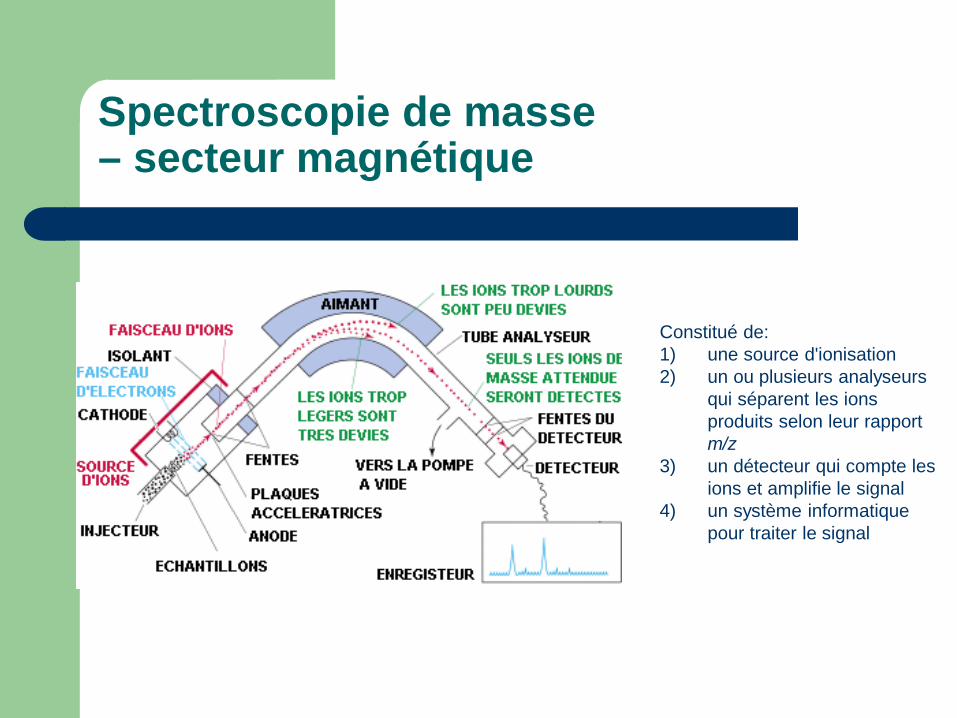
Spectroscopie de masse – secteur magnétique
Constitué de: 1) une source d'ionisation 2) un ou plusieurs analyseurs
qui séparent les ions produits selon leur rapport m/z
3) un détecteur qui compte les ions et amplifie le signal
4) un système informatique pour traiter le signal
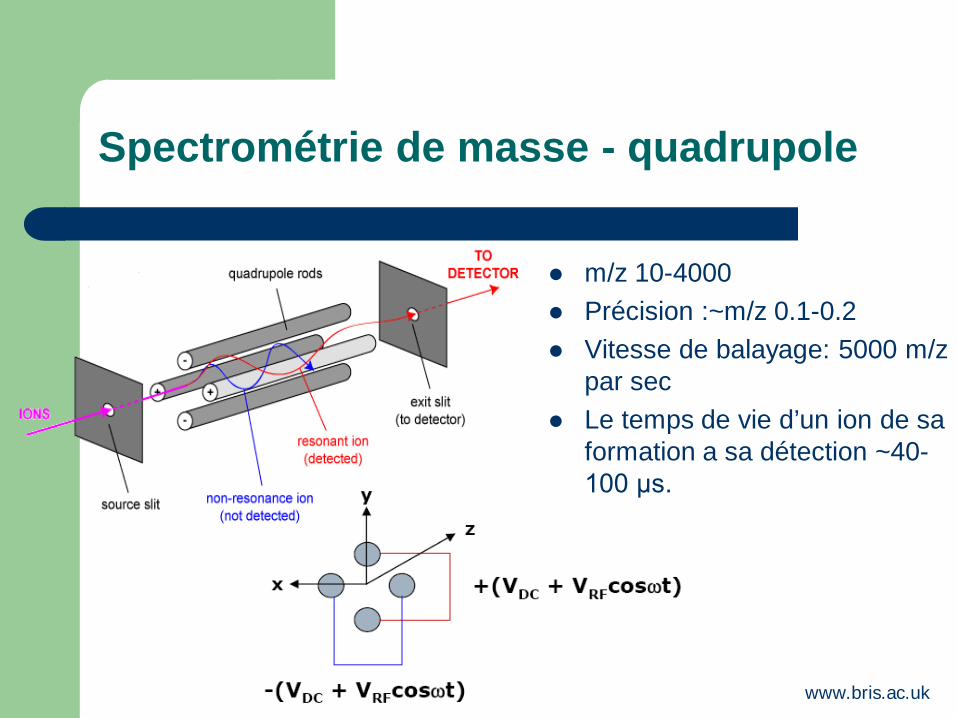
Spectrométrie de masse - quadrupole
m/z 10-4000 Précision :~m/z 0.1-0.2 Vitesse de balayage: 5000 m/z
par sec Le temps de vie d’un ion de sa
formation a sa détection ~40-100 μs.
www.bris.ac.uk

Spectrométrie de masse – trappe ionique
Les ions sont capturés pour un certain intervalle de temps avant d’être soumis à l’analyse par un MS
Concentre les ions pour obtenir une détection significative
Sensible, peu dispendieux, mais faible précision massique (Aebersold et Mann, 2003)
www.rzuser.uni-heidelberg.de/

Spectrométrie de masse - temps de vol (TOF)
• Les ions sont accélérés dans un champ électrique et acquièrent ainsi une vélocité dépendante de leur masse.
• La mesure du temps nécessaire à l’ion (temps de vol) ainsi accéléré pour parcourir la distance le séparant du détecteur permet d’en déduire la masse

Spectrométrie de masse – résonance cyclotronique d’ion (FT-ICR)
http://www.chm.bris.ac.uk/ ; www.esi.umontreal.ca
En passant dans le champ B les ions subissent une mouvement circulaire perpendiculaire au champ B
La force de rotation des ions est fonction de leur m/z
Ils sont excités sur une orbite plus grande par induction d’un courant RF
La détection des ions est basée sur une trace de courant que l’ion laisse lors de son passage entre deux électrodes.
La fréquence de ce courant est la même que celle du courant RF et l’intensité du courant est proportionnel au nombre d’ions (Marshall et al., 1998)

Une fréquence peut être mesurée plus précisément que toutes autres lectures expérimentales
Avec seulement 100 ions d’un ratio m/z donné, le FT-ICR peut détecter un composé; détection de protéine jusqu’à des seuils de 450 amol (10-18)(Marshall
et al., 1998) Coûteux (> 1M $) et volumineux Fichiers LC-MS requierent espace mémoire de
plusieurs TB en mode continu
Spectrométrie de masse – résonance cyclotronique d’ion (FT-ICR)

Chromatographie en phase gazeuse - spectroscopie de masse (GCMS)

Spectroscopie de masse – diagramme d’application
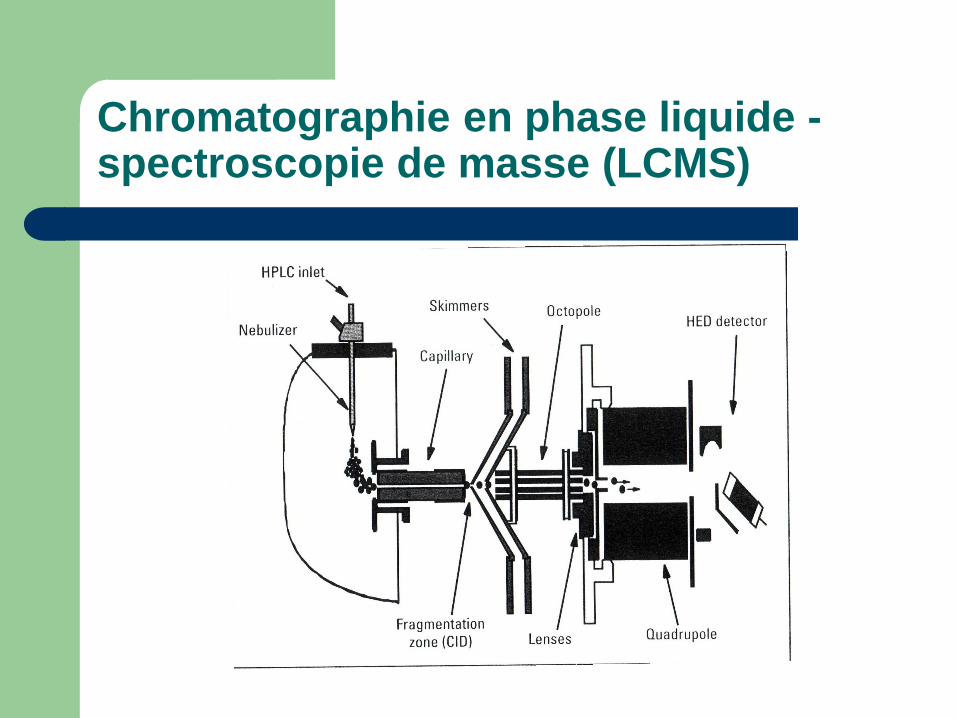
Chromatographie en phase liquide - spectroscopie de masse (LCMS)

Tout est dans l'interface LC-MS
L'electro-atomisation (electro-spray ionisation, ESI)
John Fenn, prix Nobel de chimie, 2002
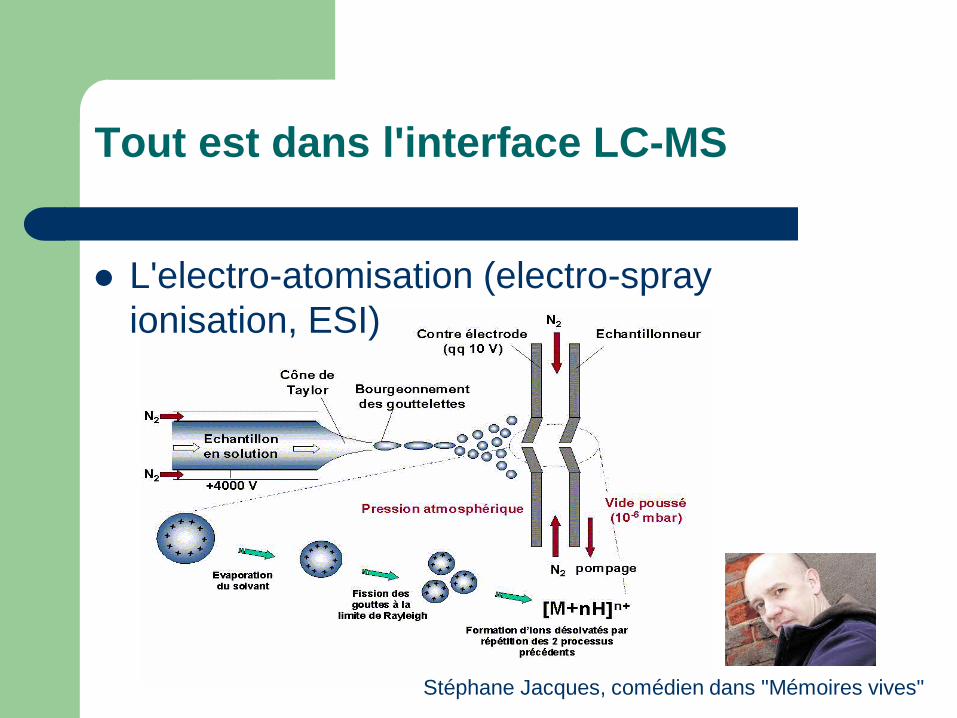
Tout est dans l'interface LC-MS
L'electro-atomisation (electro-spray ionisation, ESI)
Stéphane Jacques, comédien dans "Mémoires vives"

Exemple d’analyse – myoglobine humaine
AN: P02144, séquence: MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASEDLKKHGATVLTALGGILKKKGHHEAEIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKHPGDFGADAQGAMNKALELFRKDMASNYKELGFQG 154 acides aminés MM = 17184 Da # R (Arg, pKa= 12,1) = 2 # K (Lys, pKa= 10,6) = 20 # H (His, pKa= 6,0) = 9 # E (glu, pKa= 4,2) = 14 # D (asp, pKa= 3,7) = 8
Informations obtenues sur UniProtKB

Exemple de résultat - myoglobine
• Myoglobine • Le résultat obtenu est un
spectre de masse représentant les rapports m/z des ions détectés sur l'abscisse et l'abondance relative de ces ions sur l'ordonnée
• Le spectre est le résultat de la distribution de charges sur la population moléculaire
• Permet d'analyser qualitativement et quantitativement la protéine d'intérêt
• ΔMM = 233 ≈ 2 ac. aminés
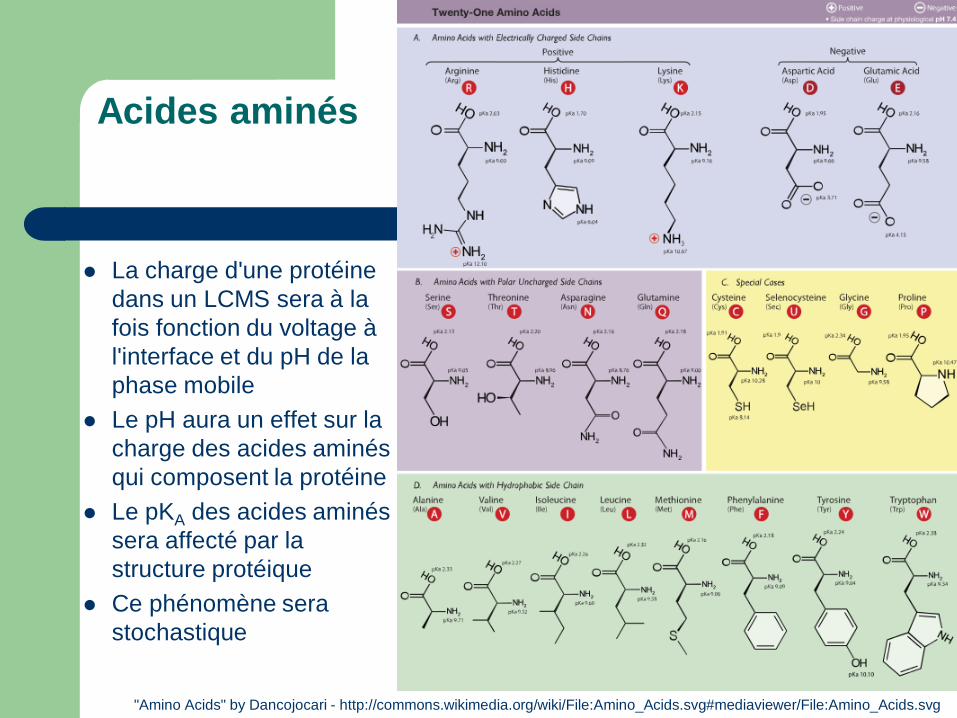
Acides aminés
La charge d'une protéine dans un LCMS sera à la fois fonction du voltage à l'interface et du pH de la phase mobile
Le pH aura un effet sur la charge des acides aminés qui composent la protéine
Le pKA des acides aminés sera affecté par la structure protéique
Ce phénomène sera stochastique
"Amino Acids" by Dancojocari - http://commons.wikimedia.org/wiki/File:Amino_Acids.svg#mediaviewer/File:Amino_Acids.svg

Exemple de calcul de la distribution de charge
Séquence: AAAAHHHHAAAA On suppose un pKA commun = 6 (pour His) à un pH = 6 Chaque His a une probabilité de 50% d'être chargé
Cliquer ici pour accéder au fichier
Excel

Exemple de résultat - myoglobine
• Myoglobine • Le résultat obtenu est un
spectre de masse représentant les rapports m/z des ions détectés sur l'abscisse et l'abondance relative de ces ions sur l'ordonnée
• Le spectre est le résultat de la distribution de charges sur la population moléculaire
• Permet d'analyser qualitativement et quantitativement la protéine d'intérêt
• ΔMM = 233 ≈ 2 ac. aminés
Cliquer ici pour accéder au fichier Excel Cliquer ici pour un exemple de calcul des pKA via PROPKA
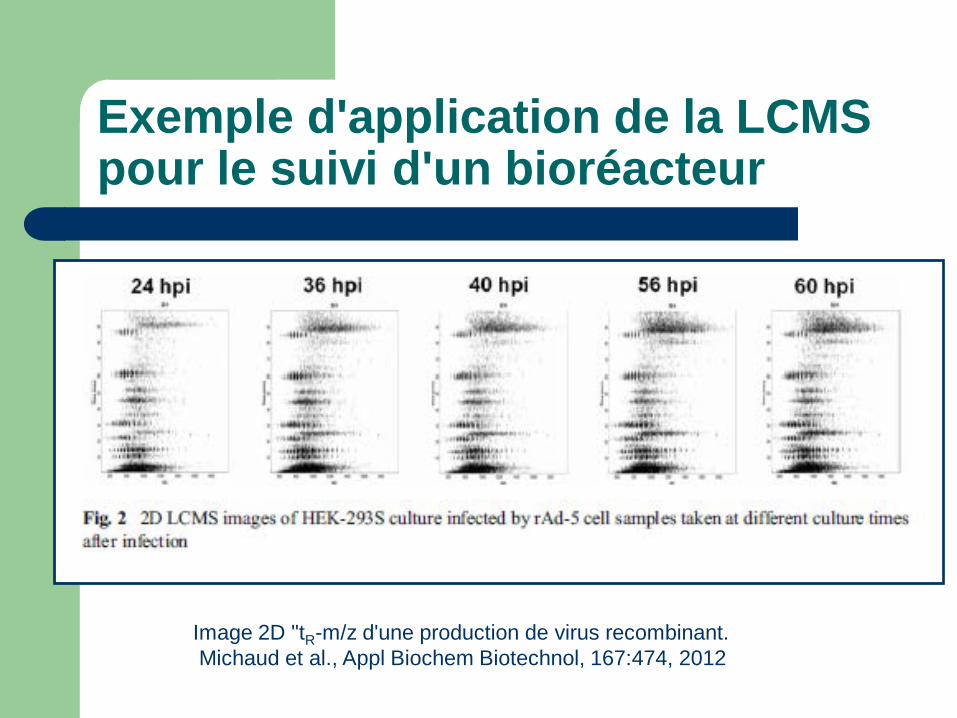
Exemple d'application de la LCMS pour le suivi d'un bioréacteur
Image 2D "tR-m/z d'une production de virus recombinant. Michaud et al., Appl Biochem Biotechnol, 167:474, 2012

Spectrométrie de masse en tandem (MSMS) – analyse de protéines
(Yates, 1998; Westermeier et Naven, 2002; Graves, 2002)
MS spectrum
MS/MS ion spectrum
m/z
m/z
Dissociation nomenclature fragment proposée par Roepstorff, 1984

Spectrométrie de Masse, Analyseurs: Tandem MS (MS/MS)
MS/MS, 2 étapes : – Détection et sélection d’un composé – Décomposition en fragments fournissant de l’information
structurelle. Cette fragmentation est effectuée par "Collision
Induced Dissociation" (CID). MS/MS, 2 types de configurations :
– Analyseurs en séries (tandem dans l’espace) triples quadrupole, hybride quadrupole-TOF,
– Analyseurs employant les méthodes de trappes ionique (tandem en temps): quadrupole-trappe ionique (trappe ionique linéaire) ou
directement un appareil de type FT-ICR (Westermeier et Naven, 2002).

Exemple d'analyse MSMS
Albumine
Protéine majeure du sérum sanguin (20-30 g/L), souvent utilisé en milieu de culture de cellules de mammifères
BSA, numéro d'accès P02769 sur UniProt
MM: 68kDa

MS de BSA
Michaud, Garnier, Lemieux, Duchesne, Proteomics, 9:512, 2009

Exemple d'analyse MSMS
Albumine
Informations obtenues sur Expasy, outil PeptideMass: http://web.expasy.org/peptide_mass/
>P02769|ALBU_BOVIN Serum albumin - Bos taurus (Bovine). MKWVTFISLLLLFSSAYSRGVFRRDTHKSEIAHRFKDLGEEHFKGLVLIAFSQYLQQCPF DEHVKLVNELTEFAKTCVADESHAGCEKSLHTLFGDELCKVASLRETYGDMADCCEKQEP ERNECFLSHKDDSPDLPKLKPDPNTLCDEFKADEKKFWGKYLYEIARRHPYFYAPELLYY ANKYNGVFQECCQAEDKGACLLPKIETMREKVLASSARQRLRCASIQKFGERALKAWSVA RLSQKFPKAEFVEVTKLVTDLTKVHKECCHGDLLECADDRADLAKYICDNQDTISSKLKE CCDKPLLEKSHCIAEVEKDAIPENLPPLTADFAEDKDVCKNYQEAKDAFLGSFLYEYSRR HPEYAVSVLLRLAKEYEATLEECCAKDDPHACYSTVFDKLKHLVDEPQNLIKQNCDQFEK LGEYGFQNALIVRYTRKVPQVSTPTLVEVSRSLGKVGTRCCTKPESERMPCTEDYLSLIL NRLCVLHEKTPVSEKVTKCCTESLVNRRPCFSALTPDETYVPKAFDEKLFTFHADICTLP DTEKQIKKQTALVELLKHKPKATEEQLKTVMENFVAFVDKCCAADDKEACFAVEGPKLVV STQTALA
Number of amino acids: 594 Molecular weight: 68026.9 Theoretical pI: 5.77

position mass peptide sequence position mass peptide sequence
25-28 500,2463 DTHK 300-309 1177,5591 ECCDKPLLEK
29-34 712,3736 SEIAHR 310-318 1015,4877 SHCIAEVEK
37-44 974,4577 DLGEEHFK 319-336 1955,9596 DAIPENLPPLTADFAEDK
45-65 2435,2427 GLVLIAFSQYLQQCPFDEHVK 341-346 752,3573 NYQEAK
66-75 1163,6306 LVNELTEFAK 347-359 1567,7427 DAFLGSFLYEYSR
76-88 1349,546 TCVADESHAGCEK 361-371 1283,7106 HPEYAVSVLLR
89-100 1362,6722 SLHTLFGDELCK 375-386 1388,5708 EYEATLEECCAK
101-105 545,3405 VASLR 387-399 1497,6314 DDPHACYSTVFDK
106-117 1364,4803 ETYGDMADCCEK 402-412 1305,7161 HLVDEPQNLIK
118-122 658,3155 QEPER 413-420 1011,42 QNCDQFEK
123-130 977,4509 NECFLSHK 421-433 1479,7954 LGEYGFQNALIVR
131-138 886,4152 DDSPDLPK 438-451 1511,8427 VPQVSTPTLVEVSR
139-151 1519,7461 LKPDPNTLCDEFK 460-468 1052,4499 CCTKPESER
157-160 537,282 FWGK 469-482 1667,8131 MPCTEDYLSLILNR
161-167 927,4934 YLYEIAR 483-489 841,46 LCVLHEK
169-183 1888,9268 HPYFYAPELLYYANK 490-495 660,3563 TPVSEK
184-197 1633,6621 YNGVFQECCQAEDK 499-507 1024,455 CCTESLVNR
198-204 701,4014 GACLLPK 508-523 1823,8996 RPCFSALTPDETYVPK
205-209 649,3338 IETMR 524-528 609,2878 AFDEK
212-218 703,4097 VLASSAR 529-544 1850,8993 LFTFHADICTLPDTEK
223-228 649,3338 CASIQK 549-557 1014,6193 QTALVELLK
229-232 508,2514 FGER 558-561 509,3194 HKPK
236-241 689,3729 AWSVAR 562-568 818,4254 ATEEQLK
249-256 922,488 AEFVEVTK 569-580 1399,6926 TVMENFVAFVDK
257-263 789,4716 LVTDLTK 581-587 725,2593 CCAADDK
267-280 1578,5981 ECCHGDLLECADDR 588-597 1050,4924 EACFAVEGPK
281-285 517,298 ADLAK 598-607 1002,583 LVVSTQTALA
286-297 1386,6206 YICDNQDTISSK
Albumine: fragments trypsiques

Analyse LC et MS1 de l'albumine trypsique
ANALYSE MS2, cliquez ici
Identification de protéine, cliquez ici (Logiciel Mascot, Matrix Science)
Pour en savoir plus, suivez ce lien (cliquer ici)

Spectrométrie de Masse - quantification
Un problème majeur lors de l’analyse de peptide et/ou protéines en MS est la variation d’ionisation due à des paramètres non contrôlable.
L’intensité du signal d’un peptide ne reflète pas directement la quantité de ce peptide (Lu et
al., 2004) Solution: implémentation d’un standard
interne; un isotope stable

Spectrométrie de Masse - ICAT
Isotope coded affinity tagging (ICAT)
Réactif sous 2 formes réagissant avec les groupements thiol des cystéines des protéines permettant d’avoir un groupement d’isotopes sur les peptides
– Différence de 8 unités de masse entre l’échantillon vs un standard (Gygi et al., 1999)
(Graves, 2002)

Spectrométrie de Masse - quantification: autres méthodes
ICAT: réactif coûteux (25$ /nmol) Se lie seulement aux cystéines des peptides
(Goshe et Smith, 2003) SILAC: “stable isotope labeling by amino
acids in cell culture” (0.2$ /nmol) (Ong et al., 2002)
Autres méthodes: iTRAQ et AQUA

Protéomique du sérum sanguin
Tirumalai et al., 2003
•60 à 80 g/L de protéine (Tirumalai et al., 2003) •10 000 protéines différentes •22 protéines = 99% de la quantité protéinique du sérum (Zhang et al., 2005) •12 log de variation de concentration (Anderson et Anderson, 2002; Zhang et al., 2005)

Protéomique du sérum sanguin
Anderson et Anderson, 2002







