
Green synthesis of some…… Chapter 3
187
Green synthesis of some bioactive heterocyclic compounds from natural
precursors
Chemistry without catalysis, would be a sword without a handle, a light without brilliance,a bell without sound………………………………………………………………Alwin Mittasch
3.1. Introduction
Heterocyclic compounds, particularly those possessing N- or S- containing moieties, have
attracted significant interest due to their useful biological and pharmaceutical properties.
Many natural and synthetic drugs [Remington (2005)], dyes, pesticides are heterocyclic in
nature. Also, biological processes such as provision of energy, transmission of nerve
impulses, sight, metabolism and transfer of hereditary information are all based on chemical
reaction involving the participation of heterocyclic compounds, such as vitamins, enzymes,
coenzymes, ATP, DNA, RNA and serotonin. Heterocycles display intrinsic reactivity which
enables rich, versatile and productive transformations. Taking into cognizance, the
ubiquitous presence of heterocycles in natural products and drugs, the development of new,
fast and efficient preparative protocols for these structures remain an urgent task in
medicinal chemistry.
3.2. Dihydropyrimidin-2(1H)-ones
Dihydropyrimidinones (DHPMs) are important class of heterocyclic compounds due to their
wide range of bioactivities and their applications in the field of drug research. Out of the
five major bases in found in DNA and RNA, three are pyrimidinone derivatives which
comprises of Cytosine (1), Uracil (2) and Thymine (3) (Figure 1) and thus, have become
very important in the world of synthetic organic chemistry.
NH
N
OH2N NH
NH
OO NH
NH
OO
H3C
Cytosine (1) Thymine (3)Uracil (2)
Figure 1
The scope of this pharmacophore further widened with the identification of monastrol (4;
Figure 2) – an aryl-substituted 3,4-dihydropyrimidin-2(1H)-one – as a novel cell-permeable
molecule for the development of new anticancer drugs [Mayer et al. (1999)]. Monastrol is
known to affect the cell-division (mitosis) by a new mechanism which does not involve

Green synthesis of some…… Chapter 3
188
tubulin targeting and consists of the specific and reversible inhibition of the motility of the
mitotic kinesis, a motor protein required for spindle bipolarity [Mayer et al. (1999)].
NH
NH
S
OH
EtOOC
H3C NH
NH
S
OH
EtOOC
H3C
IC50 = 5 µM
vs
IC50 = 120 µM
Monastrol (mitotic kinase Eg5 inhibitor)
Figure 2
Over the years, several dihydropyrimidinone derivatives have been found as calcium
channel blocker (e.g. SQ 32926 (5) and SQ 32547 (6); Figure 3) [Atwal et al. (1991)],
alpha-1a-antagonist [Kappe et al. (1997)], neuropeptide Y antagonist [Sinder and Shi
(1993)], antiviral, antitumor, antibacterial, anti-inflammatory [Kappe (1993); (2000a);
(2000b)], antihypertensive [Jain et al. (2008)] and antimalarial [Chiang et al. (2009)]
agents.
NH
N
O2N
i-Pr OOC
H3C
CONH 2
O NH
NCOO
S
F3C
i-Pr OOC
H3C
SQ 32926 (5)(Antihypertensive agent)
N
FSQ 32547 (6)(Antihypertensive agent)
Figure 3
The most important examples of pharmaceutically active dihydropyrimidinone derivatives
are crambine (7) and batzelladine (8) alkaloids, which have been isolated from marine
sources and are potent HIV group-120-CD4 inhibitors (Figure 4) [Hojati et al. (2010)].
N
HNH2N
O
O
HN
(CH2)8CH3
NH2
NHNH2
H2NHN
O O
NHN
NH
O
O
NH
N
(CH2)6CH3H3C
HH
Batzelladine B (8) (Anti-HIV agent)Crambine (Anti HIV agent)
HO
3
6
7
3
Figure 4

Green synthesis of some…… Chapter 3
189
For these reasons, dihydropyrimidinones have not only attracted the attention of chemists to
synthesize, but also represent an interesting research challenge. In consequence, numerous
methods have been reported for the synthesis of this heterocyclic nucleus, some of which
are discussed as below:
3.3. Synthetic methodologies for dihydropyrimidinones
3.3.1. Classical method
The conventional method for the synthesis of DHPMs is the one-pot three-component
reaction of benzaldehyde, ethyl acetoacetate and urea in the presence of an acid catalyst
(Scheme 1). The product of this novel one-pot, three components synthesis that precipitated
on cooling of the reaction mixture was identified as 3,4-dihydropyrimidin-2(1H)-one and
this reaction came to be known as “Biginelli reaction”, or “Biginelli condensation”, or
“Biginelli dihydropyrimidine synthesis” after the name of its inventor “Pietro Biginelli”
[Biginelli (1893)].
CHO
C2H5OO CH3
OO
H2N NH2
O HCl
N
NHEtOOC
MeH
O
++EtOH, reflux
Scheme 1
Mechanism
Forty years after Biginelli’s initial report, the first mechanism for the synthesis of DHPMs
was conducted by Folkers and Johnson based on the reaction yields and visual observation
where N,N''-benzylidene bisurea, i.e. the primary bimolecular condensation product of
benzaldehyde and urea was suggested as the first intermediate in this reaction [Folkers and
Johnson (1933)]. In 1973, a second mechanistic proposal was suggested by Sweet and
Fissekis, which involved an aldol condensation between benzaldeyde and ethyl acetoacetate
to form a stabilized carbenium ion as the primary step [Sweet and Fissekis (1973)].
Kappe re-investigated the mechanism [Kappe (1997)] using 1H and 13C NMR spectroscopy
and established that the first step in the reaction involves the acid-catalyzed condensation
between aldehyde and urea, generating iminium ion 1. Interception of this iminium ion by
ethyl acetoacetate, possibly through its enol tautomer, produces an open-chain ureide 2
which subsequently cyclizes to dihydropyrimidine 3 by the removal of H2O [Kappe (1998)]
(Scheme 2).

Green synthesis of some…… Chapter 3
190
O
H2N NH 2
Ph-CHO
O
HN NH 2HO
Ph
H+
-H 2O O
HN NH 2HO
Ph
+
H3C
EtOOC
O
-H +
Ph
NHEtOOC
H3C O OH2N
-H2O
NH
NH
Ph
EtOOC
H3C O
1
23
Scheme 2
One major drawback of the classical Biginelli protocol is low to moderate yields of DHPMs
particularly, when substituted aromatic and aliphatic aldehydes are employed due to several
side reactions [Atwal et al. (1989); Barluenga et al. (1989)] besides harsh conditions and
high reaction times [Russowsky et al. (2004)]. This has led to the recent disclosure of
several improved reaction protocols for the synthesis of DHPMs, either by modification of
the classical one-pot Biginelli approach itself [Gupta et al. (1995); Dandia et al. (1998); Hu
et al. (1998); Lu and Ma (2000); Ma et al. (2000)] or by the development of novel, but more
complex multistep strategies [O’Reilly and Atwal (1987); Shutalev et al. (1998)].
3.3.2. Alternative multistep strategies
Apart from the traditional Biginelli condensation, there are only a few other synthetic
methods available that lead to DHPMs. One noticeable exception is the so-called “Atwal
modification” of the Biginelli reaction [Atwal et al. (1987); (1989); O’Reilly and Atwal
(1987)]. Here, an enone (a) is first condensed with a suitable protected urea or thiourea
derivative (b) under almost neutral conditions. Deprotection of the resulting 1,4-
dihydropyrimidine (c) with HCl or TFA leads to the desired DHPMs (Scheme 3).
NH 2
X NH
R
R1
O
O R2R3
+N
NH
R1
O
R2X
R
R3
HN
NH
R1
O
R2X
R
X= S, R3= 4-MeOC6H4CH2X= O, R3 = Me
NaHCO3, DMF
70oC
deprotection
a b cR1= CH3, C2H5R2 =CH3; R=CH3
Scheme 3
Another approach to DHPMs has been described by Shutalev et al. (1998). This synthesis is
based on the condensation of readily available R-tosyl-substituted (thio)ureas (a) with the

Green synthesis of some…… Chapter 3
191
(in situ prepared) enolates of acetoacetates or 1,3-dicarbonyl compounds. The resulting
hexahydropyrimidines (b) need not to be isolated and can be converted directly into
DHPMs (Scheme 4). This method works particularly well for aliphatic aldehydes and
thiourea and produces high overall yields of the desired target compounds.
NH2
X NH2HN
NH
R1
O
R2X
R
HN
NH
R1
O
R2X
R
X= O, S; Ts= p-toluenesulfonyl
R-CHO,TsH
ab
R
Ts NH
H2N XH2O
O
R1
R2 O
NaH,MeCN HTs OH
R1= CH3, C2H5etc.R= R2 =CH3 etc.
Scheme 4
In addition, several combinatorial approaches towards DHPMs have been advanced using
solid phase [Wipf and Cunningham (1995); Kappe (2000c); Valverde et al. (2001); Perez et
al. (2002)] or fluorous phase reaction conditions [Studer et al. (1997a); (1997b)]. In both the
solid-phase (Scheme 5) and fluorous-phase modifications (Scheme 6) of the Biginelli
condensations, the urea component is linked to the solid (or fluorous) support via the amide
nitrogen, which invariably leads to the formation of N1-functionalized DHPMs.
NH2.H Cl
HN S
R1
O H
EtO OC
R2 O
NH
NEtO OC
R1
R2 SNMPCs2CO3
90oC, 16 h
NH
NHEtO OCR1
R2 O
NH
NHEtO OCR1
R2 S
NH
NHEtO OCR1
R2 NH
NH4OAcMeCN
TFAEtSH
AcOHH2O
R1 =C6H5, CF3C6H4, NO2C6H4R2 = CH3
Scheme 5: Solid-Phase Synthesis
Substrate SubstrateF F + byproductsF reaction
Product
F Product
extraction
F Product+detachment
Productextraction
Scheme 6: Fluorous-Phase Synthesis
Though these alternate strategies lead to somewhat higher yields, yet lack the experimental
and conceptual simplicity of the Biginelli one-pot, one-step procedure, hence could not
compete with the original Biginelli MCR approach. Therefore, Biginelli reaction was

Green synthesis of some…… Chapter 3
192
reviewed and several modifications and improvements under classical reflux or solvent free
conditions, and microwave or ultrasound irradiation have been reported.
3.3.3. Modified Biginelli protocols
A number of improved variants employing Lewis acids or protic acids based new catalytic
systems, new solvents and new reagents have emerged allowing access to a large number of
multifunctionalized dihydropyrimidinone derivatives [Kappe and Roschger (1989); Kappe
(1993)].
For instance, with polyphosphate ester (PPE) as solvent in one-pot Biginelli's condensation,
a significant increase in the yields of DHPMs was observed, especially for systems that give
only moderate yields using traditional Biginelli conditions (Scheme 7) [Kappe and Falsone
(1998)].
H3C O
EtO OCNH 2
H2N O
O H
NH
NHEtOOC
H3C O
0.3 eq PPE
THF, reflux15 h
Scheme 7
In 2004, Tu and co-workers described an efficient synthesis of 3,4-dihydropryimidin-2(1H)-
one derivatives using potassium hydrogen sulfate as the promoter in glycol solution for the
Biginelli reaction (Scheme 8). The method was applicable not only to open-chained 1,3-
dicarbonyl compounds, but also to cyclic 1,3-dicarbonyl compounds. Bifunctional
compounds containing two dihydropyrimidinone units were also been synthesized using
isophthalaldehyde and terephthalaldehyde [Tu et al. (2004)].
O
R H
O
H3C R1
O O
H2N NH2 NH
NHR1
O
OH3C
R
KHSO 4+ +Ethylene glycol
R = C6H5, NO2C6H4, OCH3C6H4, ClC6H4 etc.; R1 = CH3, C2H5
Scheme 8
In another instance, catalytic behavior of a series of eleven transition metal
methanesulfonates [Mn(II), Fe(II), Co(II), Ni(II), Cu(II), Zn(II), La(III), Ce(III), Pr(III),
Nd(III), Yb(III)] in Biginelli condensation under reflux was investigated. The study
revealed that except for Mn(II), other ten methanesulfonates exhibited good catalytic effects
with Zn(II) methanesulfonate giving better results [Wang et al. (2005a); (2005b)].

Green synthesis of some…… Chapter 3
193
Similarly, a one-pot method for the synthesis of Biginelli-type 3,4-dihydropyrimidin-2(1H)-
ones using a recyclable and eco friendly heterogeneous Keggin-type heteropolyacid catalyst
(H3PMo12O40) has been reported. It was proposed that the catalytic effect probably arises
due to the acidity of H3PMo12O40 (Scheme 9) [Heravi et al. (2006)].
X
H2N NH2
O
R1 H
O
H3C OR2
O
NH
NHR2OOC
HR1
XH3C
H3PMo12O40 (2 mol%)
AcOH, reflux
X= O, S; R1 = C6H5, NO2C6H4, OCH3C6H4, ClC6H4 etc.; R2 = CH3, C2H5
+ +
Scheme 9
Furthermore, chlorotrimethylsilane (TMSCl) has been used as a catalyst for the synthesis of
N1-alkyl-, N1-aryl- and N1,N3-dialkyl-3,4-dihydropyrimidin-2(1H)-(thi)ones using N-
substituted urea and thiourea in Biginelli reaction (Scheme 10) [Ryabukhin et al. (2007)].
NH
YNH
R
R' Ar
H O
COO Et
O
N
NY
R
COO Et
Ar
R'4 eq. TMSCl
+
1.1-1.3 eqY = O, S; R = alkyl, aryl; R' = alkyl, H
+DMF, rt, 48 h
Scheme 10
Alike, a simple methodology has been reported for the synthesis of 3,4-dihydropyrimidin-
2(1H)-ones and thione analogs in moderate to good yields by the reaction of aldehydes, β-
ketoesters and urea or thiourea using copper(II) sulfamate as catalyst (Scheme 11) [Liu and
Wang (2009)].
O
R1 H
O
H3C OR2
O X
H2N NH 2NH
NHR2O
O
XH3C
R1
1m ol% C u(NH2SO3)2+ +AcOH, reflux
R1 =OCH3C6H4, ClC6H4, NO2C6H4 etc.; R2= CH3, C2H5; X = O, S
Scheme 11
Recently, first Brønsted base catalyzed Biginelli type condensations using potassium-tert-
butoxide (t-BuOK) has been reported for three-component Biginelli-type condensation of
aldehyde, 2-phenylacetophenone, and urea/thiourea (Scheme 12). It was proposed that
enone and bis-urea were intermediates for reactions involving thiourea and urea as
substrates, respectively [Shen et al. (2010)].

Green synthesis of some…… Chapter 3
194
O
R1 H
O
R2R3
O
H2N NH2
S
H2N NH2
+ t-BuOK (20 mol%)
EtOH, 70oC
NH
NHR3
R2
R1
O
NH
NHR3
R2
R1
S
via
via
R1HN
HN
O
NH2
O
NH2
O
R2R3
R1
Scheme 12
However, some of these catalyzed conditions still have certain drawbacks, including the
cost of the catalyst, harsh reaction conditions such as need of strong acidic conditions,
anhydrous conditions, requiring inert atmosphere, long reaction times, high temperatures,
incompatibility with other functional groups on the benzene ring and unsatisfactory yields.
Moreover, in Brønsted acid conditions, acid sensitive aldehydes provided low yields
[Ghassamipour and Sardarian (2010)]. Consequently, there is scope for further
improvement towards milder reaction conditions, variations of constituents in all three
components and better yields.
3.3.3.1 Green Chemistry context
For the increasing environmental and economical concerns, development of non-hazardous
synthetic methodologies for reactions is one of the latest challenges to the organic chemists.
The areas of opportunity being exploited to engage green chemistry are: (i) choice of
solvent, (ii) the catalytic agent employed, (iii) solvent free conditions, and (iv) energy
efficient techniques etc.
In one instance, 3,4-Dihydropyrimidin-2(1H)-ones were synthesized in high yields in the
presence of room temperature ionic liquids such as 1-butyl-3-methylimidazolium
tetrafluoroborate ([BMIm]BF4) or hexafluorophosphorate ([BMIm]PF6) as catalysts under
solvent-free and neutral conditions (Scheme 13) [Peng and Deng (2001)].
RCH O +O
H2N NH 2
O
H3C R1
O
NH
NHR1
O R
OH3C
+ Ionic Liquid100oC, 0.5 h
R = C6H5, 4-OCH3-C6H4,4-Cl-C6H4, 4-NO2-C6H4, C5H11R1= OEt, CH3
Scheme 13In another modification, synergic effect of ultrasound and ionic liquid 1-butylimidazolium
tetrafluoroborate ([Hbim]BF4) has been employed to synthesize DHPMs in excellent yields

Green synthesis of some…… Chapter 3
195
within short reaction times and in the absence of any added catalyst (Scheme 14) [Gholap et
al. (2004)].
O
H3C OEt
O O
R H
Y
H2N NH2 NH
NHEtO
O R
H3C Y
+ + Ionic liquid,
30oC, 40-90minX = O, SR = alkyl, aryl
Scheme 14
A plausible mechanistic pathway was also postulated for illustrating the role of ionic liquid
in the above reaction (Scheme 15).
H
O H NN
BF4
Bu
O
O
O
H
H
H NN
Bu
BF4O
O
O
H NN
Bu
BF4
H
DHPM
d+
d +
Scheme 15
Furthermore, N-Bromosuccinimide (NBS) has also been reported as a neutral catalyst for
the synthesis of dihydropyrimidinones under microwave irradiation (Scheme 16)
[Hazarkhani and Karimi (2004)].
O
OEt
O O
R H
Y
H2N NH2 NH
NHEtO
O R
Y
+ + 0.2 eq. NBSEtOH, MW (600W)
3-6 minR = C6H5, 4-OCH3-C6H4,4-Cl-C6H4, 4-NO2-C6H4, C5H11; Y = O, S
Scheme 16
Similarly, benzyltriethylammonium chloride (TEBA) – a phase transfer catalyst has been
used as catalytic agent in Biginelli’s reaction under solvent-free conditions (Scheme 17).
The method presents an efficient route for the synthesis of important drug molecule
Monastrol [Bose et al. (2005)].

Green synthesis of some…… Chapter 3
196
R2OOC
OR1
R
H O
NH2
H2N X
NH
NHR2OOC
R1
R
X
+
Benzyltriethylammoniumchloride (10 mol%)
100oC, 30-90 min
R= OCH3C6H4, N(CH3)2C6H4, NO2C6H4, OHC6H4 etc.; X = O, SR1= CH3, C2H5; R2= CH3, C2H5, C(CH3)3
Scheme 17
Yet another modification involving titanium dioxide (TiO2) nanopowder catalyzed and
microwave induced protocol was reported with an endeavor to develop a rapid and greener
preparation of dihydropyrimidinone fused benzoquinolines (Scheme 18) [Naik et al.
(2009)].
N
N
HN NH
XO
ZCOOR2
Z
R2OOCO
H2N
NH2
X
+
TiO2 Nanopowder
MW/ EtOH
Z =Cl/SH/SeH; X= O/S; R2= CH3/C2H5
Scheme 18
In 2010, a simple protocol for Biginelli like reaction was reported under solvent-free
grinding method using catalytic amount of hydrated ferric nitrate or clayfen (Scheme 19).
The advantage of this protocol lies in the avoidance of organic solvent, high yield, energy
efficiency, variation of substrates, and use of inexpensive catalyst along with the recycling
of catalyst for more than three times [Phukan et al. (2010)].
CHO
R
CH3
O
H2N NH2
O HN NH
O
R
++
Fe(NO3)3.9H2O
rt, grinding
R = OCH3, CH3, Br, Cl, OH etc.
Scheme 19
Similarly, a green and environmentally benign water assisted protocol has been reported for
the synthesis of 3,4 DHMPs in excellent yields without using additional solvent/acid
catalyst under conventional heating, microwave irradiation/ultrasound. The presence of
water was found to be vital and the reactions were found to be faster under microwave
irradiation/ultrasound in comparison to conventional heating and afforded products in high
yields [Singhal et al. (2010)].
Recently, lemon juice has been employed as natural catalyst as a suitable replacement to
conventional acids in Biginelli condensation reaction (Scheme 20) [Patil et al. (2011)].

Green synthesis of some…… Chapter 3
197
RC 6H4CHO
H2N NH2
OR' NH
O R
ONH
+ + Lemon Juice
Stir, rt
R = H, Cl, NO2, OH etc; R' = OEt, Me
O
R'
O
Scheme 20
3.3.3.2. Organo- and biocatalytic protocols
In the past few years, several organocatalytic approaches have been developed for the
Biginelli reaction, mainly for the synthesis of asymmetric dihydropyrimidinones. The first
organocatalyst used in this multicomponent reaction was BINOL-derived phosphoric acid
developed by Gong and co-workers (Scheme 21) [Chen et al. (2006)].
CHO X
H2N NH2
O
R1O
O HN
HNX
COO R1
+ 10 mol% catalyst
CH2Cl2, 25oC,4 days
Catalyst =
+
R
R = H, Cl, NO2, CH3, OCH3, F, Br etc.; X = O, SR1 = Et, Me, i-Pr, t-Bu
R
OO
P OHO
Scheme 21
An enantioselective Biginelli reaction that proceeds by a dual-activation route has been
developed by using a combined catalyst of a readily available trans-4-hydroxyproline-
derived secondary amine and a Bronsted acid with an organic amino salt as additive
(Scheme 22). The corresponding dihydropyrimidinones were obtained in moderate-to-good
yields with up to 98% ee under mild conditions [Xin et al. (2008)]
CHO O
H2N NH2
O
EtO
OHN
HNO
COOEt
+5 mol% catalyst/5 mol%t-BuNH2.TFA5 mol% additive
1,4-dioxane/THF (2:8)rt, 36 h
HNCatalyst = Additive = t-BuNH2.HCl
+
R
R = H, Cl, NO2, CH3, OCH3 etc.
R
Scheme 22

Green synthesis of some…… Chapter 3
198
Similarly, three chiral bicyclic diamines [(1S,4S)-2,5-diazadicyclo[2.2.1]heptane derivatives]
were utilized as organocatalysts in the enantioselective Biginelli reaction. It was found that
(1S,4S)-2,5-diazabicyclo[2.2.1]heptane·2HBr and its N-methylated derivative effectively
catalyze the reaction between ethyl acetoacetate, representative aromatic aldehydes, and
urea to afford the expected DHPMs in good yields and moderate enantioselectivities (18-
37% ee), favoring the (S) enantiomer [González-Olvera et al. (2008)].
Of late, several chiral primary amines, mainly those derived from the cinchona alkaloids,
were evaluated as the organocatalysts for the asymmetric Biginelli reaction. With the
quinine-derived amine catalyst, DHPMs were obtained in moderate to good yields and 51–
78% ee from a three-component reaction of aryl and aliphatic aldehydes, urea, and
acetoacetate [Ding and Zhao (2010)].
In addition, simple amino acids like L-proline (Scheme 23) and its derivatives (e.g. L-
Proline methyl ester hydrochloride) have been reported as effective catalysts for assembling
(±)-dihydropyrimidinones under mild conditions [Yadav et al. (2004); Mabry and Ganem
(2006); Sohn et al. (2009)].
O
H2N NH2
O
EtO
H3C O
+
rt, 18 h
+ NHHN
O
COO CH 3
PhH3C
OH3CPhCHO L-Proline
MeOH NHHN
O
COOCH 3
PhH3C
Scheme 23
In comparison to above, relatively little attention has been paid to the synthesis of
heterocyclic Biginelli compounds through biocatalysis. Till date, only one enzymatic
system involving Baker yeast as catalyst has been reported for the synthesis of DHPMs. It
was observed that Biginelli compounds could be synthesized in good yields under
fermenting yeast conditions (Scheme 24) [Kumar and Maurya (2007)].
OR1
O O
H2N NH2
XRCH O
NH
NHR1OOC
R
X
+
Baker's YeastD-glucose
Phosphate buffer (pH 7)rt, 24 h
Scheme 24
However, the attempts to synthesize DHPMs by organo- and biocatalytic reactions are met
with certain limitations such as prolonged reaction time, need of additives, complex
synthesis of organocatalyst etc. Consequently, from the green chemistry perspective, there
is scope for further renovation towards milder and practical routes towards the synthesis of

Green synthesis of some…… Chapter 3
199
dihydropyrimidin-2(1H)-ones. Moreover, it would be doubly beneficial if such approaches
could be developed via utilization of abundantly available plant based feedstocks.
3.4. Results and discussion
In continuation of our recent forays into biocatalysis focusing on discovering new catalysts
for organic transformations [Kasana et al. (2007); Sharma et al. (2009); Sharma et al.
(2011a); (2011b)] and encouraged by the inspiring reports appearing on Brønsted base
[Shen et al. (2010)] and Baker’s yeast catalyzed Biginelli reaction [Kumar and Maurya
(2007)], we targeted the synthesis of chiral DHPMs using lipase as catalyst.
Initially, cyclocondensation of 0.25 mmol of benzaldehyde (1), ethyl acetoacetate (2) (1
equiv.) and urea (3) (3 equiv.) was attempted using various lipases in EtOH at 28oC for 6
days (Table 1; entries 1-6). Among all, Candida antarctica lipase-B (CAL-B) and porcine
pancreas lipase (PPL) provided better yields (28%; entries 1 and 3) with about 5%
enantioselectivity (% ee). In order to improve the yield and % ee some other solvents
reported for chiral DHPMs synthesis (Table 1; entries 7-13) were screened with CAL-B as
catalyst but without any success. Thereafter, the effect of temperature was probed to stir the
reaction in desired direction. It was observed that though increase in temperature from 28o
to 60oC brought about a linear change on the yield (from 28% to 62%), yet could not
exerted any positive effect on % ee (Table 1; entry 1 vs 14, 15). Moreover, significant yield
was observed in case of both denatured lipase (52%, Table 1; entry 16) and protein bovine
serum albumin (BSA) (80%, Table 1; entry 17). With these results in hand (lack of
enantioselectivity together with considerable yield observed in case of both denatured lipase
and BSA), we assumed that instead of involvement of any catalytic sites of lipase, it might
be the amino acid distribution on enzyme surface responsible for Biginelli reaction.
To confirm our hypothesis, catalytic influence of two neutral amino acids (glycine and L-
proline) was assessed for the cyclocondensation of 0.25 mmol of benzaldehyde (1) with 1
equiv. ethyl acetoacetate (2) and 3 equiv. urea (3) at 60oC (Table 2, entries 1-8) in terms of
reaction time. From the results, it was inferred that 48 h is the optimum time for formation
of 1b at 60oC. Moreover, it was established that there is not much difference in the yield of
the product obtained with both amino acids catalysts (Table 2, entries 1-8).

Green synthesis of some…… Chapter 3
200
Table 1: Screening of different enzymes and solvents for Biginelli reaction
CHO
EtO CH 3
O O
H2N NH2
O
NH
NHEtO
O
O
EtOH
CH 2Cl2
C6H5CH3
EtOH
EtOH
EtOH
EtOH
EtOH
EtOH
EtOH
EtOHBS A
H2O
EtOH
ACN
THF
DMSO
MeOH
S. No. Enzyme TemperatureSolvent % Conversiona ee
1 CAL-B 28oC 28% 5%
7 CAL-B 28oC 24% 5%
8 CAL-B 28oC 19% 6%
14 CAL-B 40oC 44% 5%
2 CCL 28oC 24% <5%
3 PPL 28oC 28% <5%
4 MJL 28oC 12% 7%
5 TLL 28oC 15% <5%6 CRL 28oC 15% 6%
16 denatured CAL-Bb60oC 52% <5%
17 60oC 80% <5%
9 CAL-B 28oC 15% <5%
15 CAL-B 60oC 62% <5%
+ +
1a 2 3 4a6 days
10 CAL-B 28oC 18% 5%
11 CAL-B 28oC 10% 6%12 CAL-B 28oC 23% <5%
13 CAL-B 28oC 26% <5%
catalyst (50 mg)
Experimental conditions: 0.25 mmol 1a, 1 equiv. ethyl acetoacetate, 3 equiv. urea, 3 mL solvent;aon the basis of HPLC; CAL-B=Candida antarctica lipase B, CCL= C. cylindracea lipase, PPL=Porcine pancreas lipase, MJL= Mucor javanicus lipase; TLL= Thermomyces lanuginosus lipase,CRL= C. rugosa lipase; bPre-treated with urea and thiourea (7:1) at 100oC for 2 h.
In order to improve the efficiency of the condensation, thermal effects (reflux and
microwave (MW)) on the reaction were considered (Table 2, entries 9-12). It was found that
under reflux, optimum conversion was achieved in 8 h while the reaction occurred very fast
under microwave irradiation and was completed within 10 min (Table 2, entry 11).
However, reduced yield was noticed in both cases which were much prominent in case of L-
proline (Table 2, entries 10, 12). Out of above two catalyst, L-proline and its derivatives
have been well established for the synthesis of DHPMs [Yadav et al. (2004); Mabry and
Ganem (2006); Sohn et al. (2009)]. Recently, glycine was reported for the synthesis of
polyhydroquinolines in Hantzsch condensation under MW irradiation [Singh and Singh
(2010)]. However, there is no report on use of glycine as catalyst for Biginelli reaction thus;

Green synthesis of some…… Chapter 3
201
it was contemplated to exploit the potential of this organocatalyst for the synthesis of
DHPMs.
Table 2: Optimization of organo-catalyzed (glycine and L-proline) Biginelli reaction
CHO
EtO CH3
O O
H2N NH 2
OHN
NH
EtOO
O
MW b
MW b
EtOH
S. No. Catalyst TemperatureTime % Yielda
1 144 h 60oC 81%2 144 h 60oC 82%
3 96 h 60oC 80%
4 96 h 60oC 82 %
5 48 h 60oC 80%6 48 h 60oC 81%
9 reflux 71%10 reflux 61%
11 72%12 53%
1a 2 3 1b
7 24 h 60oC 64%8 24 h 60oC 68%
+
Catalyst
Temperature
+
8 h8 h10 min10 min
Experimental conditions: 0.25 mmol of benzaladehyde 1a, 1 equiv. ethylacetoacetate, 3 equiv urea, 3 equiv catalyst in 3 mL of solvent; aisolated yield ;bMW at P=100 W using CEM monomode microwave.
GlycineL-prolineGlycine
L-Proline
GlycineL-proline
GlycineL-proline
GlycineL-proline
GlycineL-proline
As indicated in Table 3, the
efficiency of glycine was
investigated by studying the
Biginelli condensation
reaction of 0.25 mmol of
benzaldehyde with different
amount of catalyst, ethyl
acetoacetate and urea under
focused MW irradiation at P
= 100 W for varying reaction
time. After several
permutations and
combinations (Table 3,
entries 1-8), the optimum
conditions selected for
conversion of benzaldehyde
CHO EtO CH3
O O
H2N NH2
O
NH
NHEtO
O
O
3 equiv.
EtOH, MW
2 equiv.4 equiv.4 equiv.3 equiv.
3 equiv.
S. No. Amount ofcatalyst
Time % Yielda
1 72%2 35%3 69%
1a
2
3 1b
+Glycine
10 min5 min
15 min
Experimental conditions: 0.25 mmol of benzaladehyde 1a, 1 equiv.dicarbonyl compound, 3 equiv urea, glycine in 3 mL of solvent underMW irradiation at P=100 W; aisolated yield ; b2 equiv. urea; c2 equiv.urea and 1.5 equiv. dicarbonyl compound
3 equiv.3 equiv.
4 63%10 min5 73%10 min6 71%15 min
7b 62%10 min8c 64%10 min
Table 3: Optimization of glycine catalyzed Biginelli
reaction

Green synthesis of some…… Chapter 3
202
(1a; 0.25 mmol) were: 1 equiv. of dicarbonyl compound (ethyl acetoacetate in this case), 3
equiv. of urea and 3 equiv. of glycine as catalyst under 10 min of microwave irradiation.
Products were easily isolated after pouring the reaction mixture in ice-cold water, filtration
and recrystallization from water-alcohol mixture.
In view of environmental friendly procedure, the reuse of a catalyst is quite preferable.
However, in our study recovery of amino acid (glycine) catalyst from water posed a
problem for its reusability. In the past few years, amino acids have emerged as a powerful
class of starting materials for the construction of ionic liquids (AAILs) [Bao et al. (2003);
Fukumoto et al. (2005); Tao et al. (2005); Branco et al. (2006); Fukumoto and Ohno
(2006); Guillen et al. (2006); Luo et al. (2006)]. AAILs are considered as “natural ILs” or
“bio-ILs” or ‘‘fully green ILs’’ due to their environment friendly nature [Tao et al. (2006)],
biodegradability [Gathergood et al. (2004); Garcia et al. (2005); Fukumoto and Ohno
(2006)], and lesser toxicity [Jastorff et al. (2003); Rosłonkiewicz et al. (2005); Pretti et al.
(2006)]. Moreover, low cost, easy preparation and property of amino acids to act as both
anions and cations are added advantage of these AAILs [Ohno and Fukumoto (2007)].
Accordingly, AAILs consisting of glycine as cation in combination with suitable inorganic
anions of the general formula
[AA]X (glycine nitrate;
GlyNO3, glycine sulphate;
GlySO4, and glycine chloride;
GlyCl) were synthesized as per
the previous reports [Tao et al.
(2005); (2006)]. Regarding the
anions, NO32-, SO4
2- and Cl- are
non-toxic, pharmaceutically
acceptable inorganic anions
[Swatloski et al. (2003)] and
the synthesis involves
acidification of glycine with
acid (HNO3, H2SO4, HCl),
which is an atom-economic
reaction where water is the reaction medium; thus making these ionic liquids ideal fully
green ones [Tao et al. (2005)]. To evaluate the ability of above AAILs as catalyst in
Biginelli reaction, the three component condensation reaction of benzaldehyde, ethyl
Table 4: Optimization of amino acid ionic liquid
(AAIL) catalyzed Biginelli reaction
CHOEtO CH3
O O
H2N NH2
O
NH
NHEtO
O
O
EtOH
EtOH
EtOHWater
MW,10m in
S. No. Catalyst Solvent % Conversiona
1a
2
3 1b
1 92% [90%]
+
Catalyst
Solvent
2 85% [70%]3 88% [80%]4 22%
Experimental conditions: 0.25 mmol of 1a, 1 equiv. ethyl acetoacetate, 3 equiv.urea, catalyst in 3 mL of solvent under MW at P=100 W; a% conversion on thebasis of HPLC, isolated yield in parenthesis
GlySO4
GlyNO3
GlyClGlyNO3
EtOH5 95%GlyNO3
EtOH6 98% [90%]GlyNO3
Catalystamount
1 equiv.
1 equiv.1 equiv.1 equiv.
0.5 equiv.0.4 equiv.
EtOH7 79%GlyNO3 0.3 equiv.

Green synthesis of some…… Chapter 3
203
acetoacetate and urea was performed at 1 equiv. of catalyst amount in EtOH (Table 4;
entries 1-3). From the results, it was noticed that all the three ionic liquids could promote
the reaction, with GlyNO3 (Table 4, entry 1) providing superior results. As a clean and
inexpensive solvent, water was also employed as reaction medium; however it failed to
produce any significant yield (Table 4; entry 4). Interestingly, in comparison with glycine,
the use of only 0.4 equiv. of GlyNO3 could make the yield reach 98% under the microwave
power (P) of 100 W and the irradiation time of 10 min (Table 4; entries 5-7).
Moreover, GlyNO3 was easily separated from the reaction medium by cooling the mixture
at 0oC and filtering the contents. The catalyst was regenerated by washing with water and
ethanol, followed by drying at room temperature. No appreciable loss of catalytic activity
was noticed for preparation of 1b up to ten cycles.
We next examined a variety of aromatic aldehydes obtained from abundantly available
phenylpropenes (Scheme 25) as per our previous reports [Sinha et al. (2003); Kasana et al.
(2007)] under the optimized reaction conditions to establish the catalytic importance of
GlyNO3 for this reaction.
R R
CHO
R= 2,4,5-OCH3 (3)beta-Asaronetrans-Anethole R= 4-OCH3 (2)
R
CH O
IsoeugenolR= 4-OH, 3-OCH3 (5)
Pseudomonas chlororaphisCDAE5
Isosafrole R= 3,4-(OCH2O)- (4)
NaIO4/OsO4
THF/H2OPTC, MW
Scheme 25
To expand the practical scope of developed method, some commercial benzaldehydes were
also subjected to Biginelli condensation. As shown in Table 5 (entries 1-19), yields of this
one-pot protocol reactions following recrystallization from ethanol were of the order 62-
90%, which is quite favorable. All the obtained products were characterized by NMR and
HRMS data. Another important aspect of the established protocol is the survival of a variety
of functional groups such as NO2, Cl, OH and OCH3 under the reaction conditions.
Thiourea was also used with similar success to provide the corresponding
dihydropyrimidine-(2H)-thiones, which are also of much interest with regard to the
biological activity (eg. monastrol 18, Table 5).

Green synthesis of some…… Chapter 3
204
Table 5: Substrate scope of GlyNO3 catalyzed Biginelli reaction
RCH O
R'O CH 3
O O
H2N NH2
X
R
NH
NHR'O
O
X
S. No. Time (min) Yield %a
10 98 [90]2 4-OMeC6H4 10 97 [72]3 2,4,5-(OMe)2C6H2 10 91 [64]4 10 90 [70]
6 4-OHC6H4
10 93 [83]
8 4-ClC6H4
910 96 [76]
103-BrC6H4 10 95 [78]
10 93 [85]
ab
7 3-OHC6H4
10 94 [65]10 91 [62]
GlyNO3 (0.4 equiv)
EtOH,
R
C6H5
3,4-(-OCH2O-)C6H3
R' X
4-NO2C6H4
13 4-MeC6H4
Et
EtEtEt
OOOO
O
O
O
OOO
Et
EtEt
EtEtEt
MW
16 20 96 [71]C6H5 Et S
1
Experimental conditions: 0.25 mmol of subsituted benzaladehyde a,1 equiv. dicarbonylcompound, 3 equiv. urea or thiourea, MW at P= 100 W in 5 mL of EtOH; a% Based onHPLC conversion, isolated yield (after recrystallization) in parenthesis
+ +X = O or S
11 10 97 [85]C10H7 OEt
17 20 91 [70]3-OMe, 4- OHC6H3 Et S
12 10 90 [74]4-N,N (CH3)2C6H4 OEt
15 10 93 [89]C6H5 t-Bu O
5 10 92 [70]3-OMe,4- OHC6H3 Et O
14 10 91 [84]C6H5 CH3 O
18 3-OHC6H4 20 85 [62]SEt19 4-ClC6H4 20 88 [73]SEt
Reaction of 2-hydroxy benzaldehyde, ethyl acetoacetate and urea provided a product (20c)
instead of the expected product (20b). The mass of both 20b and 20c was similar however;
the NMR spectral data was different from 20b (Scheme 26). Based on 1H NMR, 13C NMR
values and previous reports [Kumar and Maurya (2007); Abbas et al. (2008)], it was
confirmed that the structure of the compound 20c was oxygen-bridged instead of the
classical Biginelli structure (Scheme 26). The compound 20c was obtained in good yield
(75%) with high distereoselectivity as determined by 1H NMR.

Green synthesis of some…… Chapter 3
205
CHO
OHOC2H5
OO
H2N NH 2
O+ +
NH
NH
OH3C
C2H5OOC
OH
NH
HN
OCH3C2H5OOC
O
H
Isolated in 75% yield
20b
20c
20a
Scheme 26
To increase the applicability of the developed method, the above conversion of 1a into 1b
with glycine nitrate as catalyst and the combination of benzaldehyde/ ethyl acetoacetate/
urea (1: 1: 3) was also performed under a multimode domestic microwave (due to its easy
access in almost every laboratory). The reaction was repeated thrice providing almost
similar yield each time (89%) which implied that our method worked efficiently in both
monomode and multimode microwave system.
Further, the applicability of the current method for the large scale synthesis was examined
by carrying out the reaction of 1 mol benzaldehyde with 1 mol ethyl acetoacetate, 3 mol
urea and 0.3 equiv GlyNO3. The product 1b was isolated in 85% yield after 10 minutes.
3.5. Conclusion
While exploring an enzymatic system for the synthesis of chiral DHPMs, it was found that
above reaction is protein catalyzed without any significant enantioselectivity. Further
exploration led to a novel amino acid ionic liquid (GlyNO3) based green approach for the
synthesis of bioactive 3,4-dihydropyrimidin-2(1H)-ones. The developed method not only
preserved the simplicity of Biginelli’s one-pot condensation but also provided good yields
of dihydropyrimidinones in shorter reaction times (10 min) besides good recyclability of the
catalyst.
3.6. Experimental
3.6.1. Materials and instrumentation
All reagents were obtained from commercial sources (Merck or Acros or HiMedia). The
phenylpropenes were purified from natural sources following the reported procedure [Sinha
et al. (2003)]. The solvents used for isolation/purification of compounds were obtained from
Merck and used without further purification. Melting points were obtained manually by

Green synthesis of some…… Chapter 3
206
capillary methods and are uncorrected. 1H (300 MHz) and 13C (75.4 MHz) NMR spectra
were recorded on a Bruker Avance-300 spectrometer. TMS was used as internal reference
for NMR. HRMS-ESI spectra were determined using Micromass Q-TOF Ultima
spectrometer. Column chromatography was done on silica gel (60-100 mesh). Thin layer
chromatography (TLC) was performed on silica TLC plates and compounds visualized in
iodine or under UV lamp. CEM Discover© focused microwave (2450 MHz, 300W) was
used wherever mentioned. The temperature of reactions in microwave experiments was
measured by an inbuilt infrared temperature probe that determined the temperature on the
surface of reaction flask. The sensor is attached in a feedback loop with an on-board
microprocessor to control the temperature rise rate. In the case of conventional heating, the
temperature of reaction mixture was monitored by thermometer.
HPLC analysis was performed using a Shimadzu HPLC (Model LC-20AT pump, DGU-
20A5 degasser) equipped with auto sampler (SIL-20AC), photo diode array detector (CBM-
20A; Shimadzu, Kyoto, Japan) and interfaced with IBM Pentium 4 personal computer. The
separation was performed on a Purospher star RP-18e column (150 x 4.6 mm id, 5 µM;
Merck) at 30oC. The mobile phase consisted of (A) 0.05% TFA (Trifluoroacetic acid) in
H2O and (B) methanol/acetonitrile (in 70:30; v/v) with gradient elution (0-5 min, 40-70% B;
5-10 min, 70-100% B; 10-12 min, 100-40% B; 12-20 min, 40% B) with a flow rate of 1
mL/min. Analysis wavelength was set at 280 nm. The quantification was performed using
external standard method.
3.6.2. Optimization of reaction conditions
3.6.2.1. Condensation of benzaldehyde (1a) with ethyl acetoacetate and urea in ethanol
using various lipases [Candida antartica lipase (CAL-B), C. cylindracea lipase (CCL);
Porcine pancreas lipase (PPL), Mucor javanicus lipase (MJL), Thermomyces
lanuginosus lipase (TLL); C. rugosa lipase (CRL)] (Table 1, entries 1-6)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and lipase (50
mg) was taken in 3 mL ethanol in a round bottom flask and the reaction mixture was
incubated at 28oC for 6 days. Thereafter, ice cold water was added to the reaction mixture
resulting in formation of precipitates which were collected by filtration and air-drying. The
product was analyzed by HPLC (section 3.6.1) in comparison to a reference standard
showing a conversion yield of 12-28% (Table 1, entries 1-6).

Green synthesis of some…… Chapter 3
207
3.6.2.2. Effect of various solvents on the condensation of benzaldehyde (1a) with ethyl
acetoacetate and urea using CAL-B (Table 1, entries 7-13)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and CAL-B
(50 mg) was taken in 3 mL dichloromethane or toluene or water or acetonitrile or
tetrahydrofuran or dimethyl sulfoxide or methanol in a round bottom flask and the reaction
mixture was incubated at 28oC for 6 days. After the completion of reaction, the reaction
mixture was worked up and analyzed with the HPLC as described above (section 3.6.1).
The conversion yield of 1b was in the range of 10-26% (Table 1, entries 7-13).
3.6.2.3. Effect of reaction temperature (40oC and 60oC) on the condensation of
benzaldehyde (1a) with ethyl acetoacetate and urea using CAL-B (Table 1, entries 14,
15)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and CAL-B
(50 mg) was taken in 3 mL ethanol in a round bottom flask and the reaction mixture was
incubated at 40oC or 60oC for 6 days. After the completion of reaction, the reaction mixture
was worked up and analyzed with the HPLC as described above (section 3.6.1). The
conversion yield of 1b was found to be 44% and 62%, respectively (Table 1, entries 14, 15).
3.6.2.4. Experiments with denatured CAL-B and bovine serum albumin at 60oC
(Table 1, entries 16, 17)
To study the exact role of enzyme in the above process, experiments with denatured lipase
(pretreated with urea and thiourea (7:1) at 100oC for 2 hours; Table 1, entry 16) or bovine
serum albumin (BSA) in place of lipase (Table 1, entry 17) were carried out. The
conversion yield of 1b was found to be 52% and 80%, respectively hinting at the role of
amino acid distribution on the enzyme surface for Biginelli condensation reaction.
3.6.2.5. Condensation of benzaldehyde (1a) with ethyl acetoacetate and urea in ethanol
using amino acid [glycine or L-proline] at different time intervals at 60oC (Table 2,
entries 1-8)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and amino acid
(glycine or L-proline; 3 equiv.) was taken in 3 mL ethanol in a round bottom flask and the
reaction mixture was incubated at 60oC for 24 h to 144 h. After the completion of reaction,
the reaction mixture was worked up and analyzed with the HPLC as described above
(section 3.6.1). From the results, 48 h of reaction time was found sufficient for conversion
of 1a to 1b with 81% yield in case of glycine and 82% yield in case of L-proline.

Green synthesis of some…… Chapter 3
208
3.6.2.6. Effect of refluxing temperature on the condensation of benzaldehyde (1a)
with ethyl acetoacetate and urea in ethanol using amino acid [glycine or L-proline] as
catalyst (Table 2, entries 9, 10)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and amino acid
(glycine or L-proline; 3 equiv.) was taken in 3 mL ethanol in a round bottom flask and the
reaction mixture was refluxed for 8-10 h till completion of the reaction (indication by TLC).
Afterwards, the reaction mixture was worked up and analyzed with the HPLC as described
above (section 3.6.1). From the results, it was observed that 8 h of reaction time was
required for condensation reaction of 1a (Table 2, entries 9, 10).
3.6.2.7. Effect of microwave irradiation on the condensation of benzaldehyde (1a)
with ethyl acetoacetate and urea in ethanol using amino acid [glycine or L-proline] as
catalyst (Table 2, entries 11, 12)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and amino acid
(glycine or L-proline; 3 equiv.) was taken in 3 mL ethanol in a round bottom flask and the
reaction mixture was subjected to microwave irradiation using CEM monomode microwave
at Power = 100 W for 10 min. Afterwards, the reaction mixture was worked up and
analyzed with the HPLC as described above (section 3.6.1). From the results, it was
observed that, out of two catalyst, glycine provided better yield (72%; Table 2, entry 11) as
compared to L-Proline (53%; Table 2, entry 12).
3.6.2.8. Effect of microwave irradiation time on % yield of 1a in Biginelli condensation
with glycine as catalyst (Table 3, entries 1-3)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and glycine (3
equiv.) was taken in 3 mL ethanol in a round bottom flask and the reaction mixture was
subjected to microwave irradiation using CEM monomode microwave at Power = 100 W
for 5-15 min. Afterwards, the reaction mixture was worked up and analyzed with the HPLC
as described above (section 3.6.1). From the results, it was observed that 10 min of reaction
time was sufficient for condensation of 1a providing 72% yield (Table 3, entry 1).
3.6.2.9. Effect of increase or decrease in glycine amount on Biginelli condensation of 1a
(Table 3, entries 4-6)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and 2 equiv. or
4 equiv. glycine (instead of 3 equiv) was taken in 3 mL ethanol in a round bottom flask and
the reaction mixture was subjected to microwave irradiation using CEM monomode
microwave at Power = 100 W for 10-15 min. Afterwards, the reaction mixture was worked

Green synthesis of some…… Chapter 3
209
up and analyzed with the HPLC as described above (section 3.6.1). The conversion yield of
1b was found to be 63% and 73%, respectively (Table 3, entries 4, 5).
3.6.2.10. Effect of decrease in urea amount on condensation of 1a (Table 3, entry 7)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), 2 equiv. urea (instead of 3
equiv.) and glycine (3 equiv.) was taken in 3 mL ethanol in a round bottom flask and the
reaction mixture was subjected to microwave irradiation using CEM monomode microwave
at Power = 100 W for 10 min. Afterwards, the reaction mixture was worked up and
analyzed with the HPLC as described above (section 3.6.1). Reduced yield of 1b (62%) was
observed (Table 3, entry 7).
3.6.2.11. Effect of change in ethyl acetoacetate and urea amount on condensation of 1a
(Table 3, entries 7, 8)
Benzaldehyde (1a, 0.25 mmol), 1.5 equiv. ethyl acetoacetate (instead of 1 equiv.), 2 equiv
urea (instead of 3 equiv.) and glycine (3 equiv.) was taken in 3 mL ethanol in a round
bottom flask and the reaction mixture was subjected to microwave irradiation using CEM
monomode microwave at Power = 100 W for 10 min. Afterwards, the reaction mixture was
worked up and analyzed with the HPLC as described above (section 3.6.1). Reduced yield
of 1b (64%) was observed (Table 3, entry 8).
3.6.3. Preparation of amino acid ionic liquids (glycine nitrate (GlyNO3), glycine
sulphate (GlySO4) and glycine chloride (GlyCl))
7.5 g (0.1 mol) of glycine was dissolved in 20 mL water. One mole equivalent of nitric acid
or hydrochloric acid or 0.5 mol equivalent of sulfuric acid was added drop wise. The
reaction mixture was then warmed to 60oC for 24 h. After evaporating in vacuo at 60oC and
lyophilization, the resulting white solid was collected and recrystallized from
methanol/ether. 1H and 13C NMR spectra were recorded and matched with reported values
[Tao et al. (2005)].COOH
H3N HH
X X = NO3-, Cl-, 1/2 SO4
2-
Glycine nitrate (GlyNO3)1H NMR (DMSO-d6, 300 MHz): δ 8.11 (3H, s), 3.68 (2H, s); 13C NMR (DMSO-d6, 75.4
MHz): δ 169.1, 39.7.

Green synthesis of some…… Chapter 3
210
Glycine sulphate (GlySO4)1H NMR (DMSO-d6, 300 MHz): δ 8.08 (3H, s), 3.64 (2H, s); 13C NMR (DMSO-d6, 75.4
MHz): δ 168.7, 38.8.
Glycine chloride (GlyCl)1H NMR (DMSO-d6, 300 MHz): δ 8.12 (3H, s), 3.67 (2H, s); 13C NMR (DMSO-d6, 75.4
MHz): δ 169.0, 39.6.
3.6.3.1. Biginelli condensation of benzaldehyde (1a) using amino acid ionic liquid
[glycine nitrate or glycine sulphate or glycine chloride] as catalyst in ethanol (Table 4,
entries 1-3)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and amino acid
ionic liquid glycine nitrate or glycine sulphate or glycine chloride (1 equiv.) was taken in 3
mL ethanol in a round bottom flask and the reaction mixture was subjected to microwave
irradiation using CEM monomode microwave at Power = 100 W for 10 min. Afterwards,
the reaction mixture was worked up and analyzed with the HPLC (section 3.6.1) showing a
conversion yield 85-92% Further, the crude product was purified by recrystallization from
water-ethanol mixture giving an isolated yield of 1b in the range of 70-90%. 1H and 13C
NMR spectra were recorded and matched with reported values and further confirmed by
HRMS/MS.
Ethyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate
(compound 1b, Table 4)
NHHN
O
CH3
COOC2H5 White solid (Yield 90%) m.p. 206-207°C, 1H NMR (DMSO-d6, 300
MHz): 9.21 (1H, s), 7.75 (1H, s), 7.35-7.23 (5H, m), 5.16 (1H, d, J = 3.08 Hz ), 4.02 (2H,
q, J = 7.07 Hz), 2.25 (3H, s), 1.12 (3H, t, J = 7.06 Hz ); 13C NMR (DMSO-d6, 75.4 MHz):
166.2, 153.0, 149.2, 145.7, 129.2, 128.1, 127.1, 100.1, 60.0, 54.8, 18.6 and 14.9. HRMS-
ESI: m/z [M+H]+ for C14H16N2O3, calculated 261.1370; observed 261.1374. The spectral
data matched well with the reported values [Karade et al. (2007)].
3.6.3.2. Biginelli condensation of benzaldehyde (1a) using glycine nitrate as catalyst in
water under microwave irradiation (Table 4, entry 4)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and glycine
nitrate (1 equiv.) was taken in 3 mL water (instead of ethanol) in a round bottom flask and

Green synthesis of some…… Chapter 3
211
the reaction mixture was subjected to microwave irradiation using CEM monomode
microwave at Power = 100 W for 10 min. Afterwards, the reaction mixture was worked up
and analyzed with the HPLC (section 3.6.1) showing a conversion yield of 22%.
3.6.3.3. Effect of glycine nitrate amount on the Biginelli condensation of 1a (Table 4,
entries 5-7)
Benzaldehyde (1a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and glycine
nitrate (0.3 equiv. or 0.4 equiv. or 0.5 equiv.) was taken in 3 mL ethanol in a round bottom
flask and the reaction mixture was subjected to microwave irradiation using CEM
monomode microwave at Power = 100 W for 10 min. Afterwards, the reaction mixture was
worked up and analyzed with the HPLC as described above (section 3.6.1.). The conversion
yield of 1b was from 79-98% (Table 4, entries 5-7) with 0.4 equiv. glycine nitrate providing
better results (98%; Table 4, entry 6).
3.6.4. Synthesis of methoxylated benzaldehydes (2a-4a) from abundantly available
natural phenylpropenes (trans-anethole, β-asarone and isosafrole) (Table 5)
A mixture of phenylpropene derivative (1.5 mmol), OsO4 (1.5 mmol), NaIO4 (1.5 mmol)
and benzyltriethylammonium chloride (0.01 g) were dissolved in H2O-THF (3 mL, 4:1 v/v)
and irradiated under microwave. After completion of reaction, the mixture was filtered and
washed with dichloromethane. Evaporation of the solvent under reduced pressure gave a
crude product, which was purified on silica gel column with hexane: ethyl acetate to obtain
the corresponding benzaldehyde [Sinha et al. (2003)].
4-methoxybenzaldehyde (Compound 2a, Table 5) (obtained from trans-anethole)CHO
H3CO Colorless liquid (Yield 86%), 1H NMR (CDCl3, 300 MHz): 9.84 (1H,
s), 7.68 (2H, m), 6.94 (2H, m), 3.70 (3H, s); 13C-NMR (CDCl3, 75.4 MHz): 190.3, 164.4,
131.3, 129.2, 113.8 and 55.2. The spectral data matched well with the reported values
[Sinha et al. (2003)].
2,4,5-trimethoxybenzaldehyde (Compound 3a, Table 5) (obtained from β-asarone)OC H3
OC H3
H3CO
CHO
White needles (Yield 83%), 1H NMR (CDCl3, 300 MHz): δ 10.31 (1H,
s), 7.36 (1H, s), 6.52 (1H, s), 3.98 (3H, s), 3.92 (3H, s), 3.86 (3H, s); 13C NMR (CDCl3, 75.4

Green synthesis of some…… Chapter 3
212
MHz): δ 187.7, 158.4, 155.8, 143.4, 117.1, 109.0, 98.3 and 56.2. The spectral data matched
well with the reported values [Sinha et al. (2003)].
3,4,-dioxymethylenebenzaldehyde (Compound 4a, Table 5) (obtained from isosafrole)CHOO
O Yellow oil (Yield 76%), 1H NMR (CDCl3, 300 MHz): δ 9.85 (1H, s),
7.25 (1H, d), 7.18 (1H, s), 6.84 (1H, d), 5.87 (2H, s); 13C NMR (CDCl3, 75.4 MHz): δ 190.2,
153.4, 147.9, 130.1, 123.4, 116.2, 115.5 and 91.2. The spectral data matched well with the
reported values [Sinha et al. (2003)].
3.6.4.1. Synthesis of vanillin (5a) from isoeugenol (Table 5)
To 2 g of isoeugenol taken in a round bottom flask, 100 mL of whole cell culture of
Pseudomonas chlororaphis CDAE5 was added and the reaction mixture incubated at 25oC
and 180 rpm for 24 h [Kasana et al. (2007)]. Thereafter, the mixture was extracted three
times with dichloromethane. Evaporation of the solvent under reduced pressure after drying
over anhydrous sodium sulphate gave a crude product, which was purified on silica gel
column with hexane: ethyl acetate to obtain the corresponding benzaldehyde 5a.
4-Hydroxy-3-methoxybenzaldehyde (Compound 5a, Table 5)CH O
OH
OC H3
Creamish white crystals. m.p. 81-83°C, 1H NMR (CDCl3, 300 MHz): 9.87
(1H, s), 7.41-7.43 (2H, m), 6.98 (1H, d, J = 8.15 Hz), 6.46 (1H, s), 3.94 (3H, s); 13C NMR
(CDCl3, 75.4 MHz): 190.5, 151.3, 147.2, 129.2, 127.1, 114.4, 108.3 and 56.1. HRMS-ESI:
m/z [M+H]+ for C8H8O3, calculated 153.0684; observed 153.0678.
3.6.4.2. Synthesis of 3,4-dihydropyrimidin-2(1H)-ones (1b-13b) from substituted
benzaldehydes (Table 5, entries 1-13)
Substituted benzaldehyde (1a-13a, 0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.)
and glycine nitrate (0.4 equiv.) was taken in 3 mL ethanol in a round bottom flask and the
reaction mixture was subjected to microwave irradiation using CEM monomode microwave
at Power = 100 W for 10 min. Afterwards, the reaction mixture was worked up and
analyzed with the HPLC (see section 3.6.1.) with a conversion yield of 90-98%. Further, the
crude product was purified by recrystallization from water-ethanol mixture and subjected
for spectral analysis (NMR and mass spectrometry).

Green synthesis of some…… Chapter 3
213
Ethyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate
(compound 1b, Table 5)
NHHN
O
CH3
COO C2H5 White solid (Yield 90%), The NMR spectra matched well with that
obtained in section 3.6.3.1.
Ethyl 4-(4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-
carboxylate (Compound 2b Table 5)
NHHN
O
CH3
COOC2H5H3CO White solid (Yield 72%) m.p. 201-202oC, 1H NMR (DMSO-d6,
300 MHz): 9.17 (1H, s), 7.68 (1H, s), 7.17 (2H, d, J = 8.66 Hz), 6.89 (2H, d, J = 8.69 Hz),
5.10 (1H, d, J = 3.17 Hz), 4.01 (2H, q, J = 7.07 Hz), 3.72 (3H, s), 2.24 (3H, s), 1.13 (3H, t, J
= 7.07 Hz); 13C NMR (DMSO-d6, 75.4 MHz): 166.2, 159.3, 153.0, 148.9, 137.9, 128.3,
114.6, 100.4, 60.0, 55.9, 54.2, 18.6 and 14.9. HRMS-ESI: m/z [M+H]+ for C15H18N2O4,
calculated 291.1520; observed 291.1528. The spectral data matched well with the reported
values [Karade et al. (2007)].
Ethyl 6-methyl-2-oxo-4-(2,4,5-trimethoxyphenyl)-1,2,3,4-tetrahydropyrimidine-5-
carboxylate (Compound 3b, Table 5)
NHHN
O
CH3
COO C2H5
OCH 3
H3CO
OC H3
White solid (Yield 64%) m.p. 207-209°C, 1H NMR (DMSO-d6,
300 MHz): 8.66 (1H, s), 6.64 (1H, s), 6.52 (1H, s), 5.78 (1H, s), 5.68 (1H, s), 4.10 (2H, q,
J = 7.05 Hz), 3.88 (3H, s), 3.85 (3H, s), 3.76 (3H, s), 2.42 (3H, s), 1.14 (3H, t, J = 7.09 Hz);13C NMR (DMSO-d6, 75.4 MHz); 166.3, 154.2, 151.7, 149.9, 148.4, 143.2, 122.3, 112.1,
98.9, 97.8, 60.2, 57.3, 56.6, 50.3, 18.8 and 14.6. HRMS-ESI: m/z [M+H]+ for C17H22N2O6,
calculated 351.1820; observed 351.1829. The spectral data matched well with the reported
values [Beşoluk et al. (2010)].

Green synthesis of some…… Chapter 3
214
Ethyl 4-(1,3-benzodioxol-5-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-
carboxylate (Compound 4b, Table 5)
NHHN
O
CH3
COOC2H5
O
O White solid (Yield 70%) m.p. 180-181°C, 1H NMR (DMSO-d6, 300
MHz): 9.18 (1H, s), 7.69 (1H, s), 6.86-6.69 (3H, m), 5.98 (2H, s), 5.08 (1H d, J = 2.85
Hz), 4.03 (2H, q, J = 7.01 Hz), 2.25 (3H, s), 1.13 (3H, t, J = 7.05 Hz); 13C NMR (DMSO-d6,
75.4 MHz): 166.2, 152.9, 149.1, 148.1, 147.2, 139.7, 120.2, 108.9, 107.5, 101.8, 100.2,
60.1, 54.5, 18.6 and 14.9. HRMS-ESI: m/z [M+H]+ for C15H16N2O5, calculated 305.1358;
observed 305.1361.
Ethyl 4-(4-hydroxy-3-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-
carboxylate (Compound 5b, Table 5)
NHHN
O
CH3
COOC2H5
OCH3
HO
White solid (Yield 70%) m.p. 146-148°C, 1H NMR (DMSO-d6, 300
MHz): 9.11 (1H, s), 8.91 (1H, s), 7.64 (1H, d, J = 2.66 Hz), 6.80 (1H, d, J = 8.09 Hz ),
6.63-6.60 (2H, m), 5.07 (1H, d, J = 3.19 Hz), 4.03 (2H, q, J = 7.07 Hz ), 3.72 (3H, s), 2.23
(3H, s), 1.14 (3H, t, J = 7.21 Hz); 13C NMR (DMSO-d6, 75.4 MHz): 166.3, 153.1, 148.7,
148.1, 146.7, 136.8, 119.2, 116.1, 11.8, 100.4, 59.9, 56.9, 54.4, 18.6 and 15.0. HRMS-ESI:
m/z [M+H]+ for C15H18N2O5, calculated 307.1514; observed 307.1521.
Ethyl 4-(4-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-
carboxylate (Compound 6b, Table 5)
NHHN
O
CH3
COOC2H5HO White solid (Yield 65%) m.p. 232-234°C, 1H NMR (DMSO-d6,
300 MHz): 9.34 (1H, s), 9.12 (1H, s), 7.63 (1H, s), 7.04 (2H, d, J = 7.99 Hz), 6.70 (2H, d,
J = 7.97 Hz), 5.05 (1H, s), 4.01 (2H, q, J = 6.71 Hz), 2.23 (3H, s), 1.12 (3H, t, J = 6.92 Hz );13C NMR (DMSO-d6, 75.4 MHz): 165.4, 156.5, 152.2, 147.7, 135.4, 127.4, 114.9, 99.8,

Green synthesis of some…… Chapter 3
215
59.1, 53.4, 17.7 and 14.1. HRMS-ESI: m/z [M+H]+ for C14H16N2O4, calculated 277.1364;
observed 277.1374.
Ethyl 4-(3-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-
carboxylate (Compound 7b, Table 5)
NHHN
O
CH3
COO C2H5
OH White solid (Yield 62%) m.p. 179-182°C, 1H NMR (DMSO-d6, 300
MHz): 8.46 (1H, s), 8.23 (1H, s), 6.76 (1H, s), 6.18 (1H, t, J = 8.12 Hz), 5.77-5.72 (3H,
m), 4.15 (1H, d, J = 3.17 Hz), 3.09 (2H, q, J = 7.10 Hz), 1.32 (3H, s), 0.22 (3H, t, J = 7.12
Hz); 13C NMR (DMSO-d6, 75.4 MHz): 166.3, 158.2, 153.1, 148.9, 147.1, 130.1, 117.8,
115.0, 113.9, 100.3, 60.1, 54.7, 18.6 and 14.9. HRMS-ESI: m/z [M+H]+ for C14H16N2O4,
calculated 277.1364; observed 277.1372. The spectral data matched well with the reported
values [Xin et al. (2008)].
Ethyl 4-(4-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate
(Compound 8b, Table 5)
NHHN
O
CH3
COOC2H5Cl White solid (Yield 76%) m.p. 213-214°C, 1H NMR (DMSO-d6,
300 MHz): 9.25 (1H, s), 7.78 (1H, s), 7.41 (2H, d, J = 8.41 Hz), 7.26 (2H, d, J = 8.43 Hz),
5.15 (1H, s), 4.01 (2H, q, J = 7.05 Hz), 2.25 (3H, s), 1.12 (3H, t, J = 7.09 Hz); 13C NMR
(DMSO-d6, 75.4 MHz): 165.2, 151.9, 148.7, 143.8, 131.8, 128.4, 128.2, 98.8, 59.2, 53.4,
17.8 and 14.0. HRMS-ESI: m/z [M+H]+ for C14H15ClN2O3, calculated 295.5819; observed
295.58.23. The spectral data matched well with the reported values [Karade et al. (2007)].

Green synthesis of some…… Chapter 3
216
Ethyl 4-(3-bromophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate
(Compound 9b, Table 5)
NHHN
O
CH3
COOC2H5
Br White solid (Yield 78%) m.p. 187-189°C, 1H NMR (DMSO-d6, 300
MHz): 9.27 (1H, s), 7.80 (1H, s), 7.47-7.23 (4H, m), 5.15 (1H, d, J = 2.85 Hz), 4.02 (2H,
q, J = 7.07 Hz), 2.26 (3H, s), 1.13 (3H, t, J = 7.01 Hz); 13C NMR (DMSO-d6, 75.4 MHz):
166.0, 152.8, 149.8, 148.3, 131.7, 130.9, 130.0, 126.1, 122.4, 99.5, 60.2, 54.5, 18.7 and
14.9. HRMS-ESI: m/z [M+H]+ for C14H15N2O3Br, calculated 340.0332; observed 340.0338.
The spectral data matched well with the reported values [Karade et al. (2007)].
Ethyl 6-methyl-4-(4-nitrophenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate
(Compound 10b, Table 5)
NHHN
O
CH3
COOC2H5O2N Creamish solid (Yield 85%) m.p. 220-225°C, 1H NMR (DMSO-d6,
300 MHz): 9.38 (1H, s), 8.15 (2H, d, J = 8.31 Hz), 7.91 (1H, s), 7.72 (2H, d, J = 8.38 Hz),
5.31 (1H, d, J = 2.89 Hz), 4.05 (2H, q, J = 7.18 Hz), 2.28 (3H, s), 1.13 (3H, t, J = 7.13 Hz);13C NMR (DMSO-d6, 75.4 MHz): 165.9, 152.6, 150.3, 148.6, 147.9, 133.8, 131.1, 123.2,
121.9, 99.2, 60.2, 54.4, 18.7 and 14.9. HRMS-ESI: m/z [M+H]+ for C14H15N3O5, calculated
306.1411; observed 306.1417. The spectral data matched well with the reported values
[Karade et al. (2007)].
Ethyl 6-methyl-4-(naphthalen-2-yl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate
(Compound 11b, Table 5)
NHHN
O
CH3
COOC2H5 White solid (Yield 85%) m.p. 257-258°C, 1H NMR (DMSO-d6,
300 MHz): 9.24 (1H, s), 7.93 (1H, s), 7.86 (1H, d, J = 7.88 Hz), 7.79 (1H, s), 7.56-7.43
(5H, m), 6.07 (1H, d, J = 3.01 Hz), 3.83 (2H, q, J = 7.59 Hz), 2.37 (3H, s), 0.84 (3H, t, J =

Green synthesis of some…… Chapter 3
217
7.07 Hz); 13C NMR (DMSO-d6, 75.4 MHz): 165.6, 151.9, 148.9, 140.7, 133.7, 130.3,
128.7, 126.3, 126.0., 125.9, 124.5, 123.9, 99.4, 59.3, 50.1, 18.0 and 14.1. HRMS-ESI: m/z
[M+H]+ for C18H18N2O3, calculated 311.1526; observed 311.1534. The spectral data
matched well with the reported values [Xin et al. (2008)].
Ethyl 4-[4-(dimethylamino)phenyl]-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-
carboxylate (Compound 12b, Table 5)
NHHN
O
CH3
COOC2H5N
Creamish solid (Yield 74%) m.p. 252-255°C, 1H NMR (DMSO-d6,
300 MHz): 9.09 (1H, s), 7.60 (1H, s), 7.05 (2H, d, J = 8.38 Hz), 6.67 (2H, d, J = 8.42 Hz),
5.04 (1H, d, J = 2.18 Hz), 4.01 (2H, q, J = 6.86 Hz), 2.85 (6H, s), 2.23 (3H, s), 1.14 (3H, t, J
= 7.02 Hz); 13C NMR (DMSO-d6, 75.4 MHz): 165.2, 151.9, 149.4, 147.2, 132.3, 126.6,
111.9, 99.6, 58.8, 53.0, 17.39 and 13.8. HRMS-ESI: m/z [M+H]+ for C16H21N3O3,
calculated 304.1791; observed 304.1796. The spectral data matched well with the reported
values [Yadav et al. (2001)].
Ethyl 6-methyl-4-(4-methylphenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate
(Compound 13b, Table 5)
NHHN
O
CH3
COOC2H5H3C White solid (Yield 83%) m.p. 206-208oC, 1H NMR (DMSO-d6,
300 MHz): 9.12 (1H, s), 7.67 (1H, s), 7.11 (3H, s), 5.12 (1H, d, J = 2.67 Hz), 3.98 (2H, d,
J = 7.07 Hz), 2.25 (6H, s), 1.11 (3H, t, J = 7.07 Hz); 13C NMR (DMSO-d6, 75.4 MHz):
166.3, 153.2, 148.9, 142.7, 137.3, 129.8, 126.9, 100.4, 60.0, 54.5, 21.4, 18.6 and 14.9.
HRMS-ESI: m/z [M+H]+ for C15H18N2O3, calculated 275.1526; observed 275.1533. The
spectral data matched well with the reported values [Xin et al. (2008)].
3.6.4.3. Synthesis of 3,4-dihydropyrimidin-2(1H)-one (14b) from benzaldehyde with
methyl acetoacetate and urea (Table 5, entry 14)
Benzaldehyde (0.25 mmol), methyl acetoacetate (1 equiv.), urea (3 equiv.) and glycine
nitrate (0.4 equiv.) was taken in 3 mL ethanol in a round bottom flask and the reaction
mixture was subjected to microwave irradiation using CEM monomode microwave at
Power = 100 W for 10 min. Afterwards, the reaction mixture was worked up and analyzed

Green synthesis of some…… Chapter 3
218
with the HPLC (see section 3.6.1.) with a conversion yield of 91%. Further, the crude
product was purified by recrystallization from water-ethanol mixture and isolated yield
calculated. Structure of the compound was confirmed through NMR and mass spectrometry.
Methyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate
(Compound 14b, Table 5)
NHHN
O
CH3
COOCH3 White solid (Yield 84%) m.p. 205-210°C, 1H NMR (DMSO-d6, 300
MHz): 9.22 (1H, s), 7.76 (1H, s), 7.35-7.30 (2H, m), 7.26-7.23 (3H, m), 5.16 (1H, d, J =
3.28 Hz), 3.53 (3H, s), 2.26 (3H, s); 13C NMR (DMSO-d6, 75.4 MHz): 166.7, 153.0,
149.5, 145.5, 129.3, 128.1, 127.0, 99.9, 54.7, 51.6 and 18.7. HRMS-ESI: m/z [M+H]+ for
C13H14N2O3, calculated 247.0714; observed 247.0725. The spectral data matched well with
the reported values [Karade et al. (2007)].
3.6.4.4. Synthesis of 3,4-dihydropyrimidin-2(1H)-one (15b) from benzaldehyde with
tert-butyl acetoacetate and urea (Table 5, entry 15)
Benzaldehyde (0.25 mmol), tert-butyl acetoacetate (1 equiv.), urea (3 equiv.) and glycine
nitrate (0.4 equiv.) was taken in 3 mL ethanol in a round bottom flask and the reaction
mixture was subjected to microwave irradiation using CEM monomode microwave at
Power = 100 W for 10 min. Afterwards, the reaction mixture was worked up and analyzed
with the HPLC (see section 3.6.1.) with a conversion yield of 93%. Further, the crude
product was purified by recrystallization from water-ethanol mixture and isolated yield
calculated. Structure of the compound was confirmed through NMR and mass spectrometry.
tert-Butyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate
(Compound 15b, Table 5)
NHHN
O
CH3
COOC(CH3)3 White solid (Yield 89%) m.p. 220-222°C, 1H NMR (DMSO-d6, 300
MHz): 9.04 (1H, s), 7.64 (1H, s), 7.34-7.30 (2H, m), 7.27 (3H, d, J = 7.49 Hz), 5.10 (1H,
d, J = 2.67 Hz), 2.22 (3H, s), 1.28 (9H, s); 13C NMR (DMSO-d6, 75.4 MHz): 165.7, 153.0,
148.1, 145.8, 129.1, 128.1, 127.1, 101.5, 55.2, 49.4, 28.6 and 18.5. HRMS-ESI: m/z

Green synthesis of some…… Chapter 3
219
[M+H]+ for C16H20N2O3, calculated 289.1682; observed 289.1694. The spectral data
matched well with the reported values [Falsone and Kappe (2001)].
3.6.4.5. Synthesis of 3,4-dihydropyrimidin-2(1H)-thiones (16b-19b) from substituted
benzaldehydes (Table 5, entries 16-19)
Substituted benzaldehyde (0.25 mmol), ethyl acetoacetate (1 equiv.), thiourea (3 equiv.) and
glycine nitrate (0.4 equiv.) was taken in 3 mL ethanol in a round bottom flask and the
reaction mixture was subjected to microwave irradiation using CEM monomode microwave
at Power = 100 W for 20 min. Afterwards, the reaction mixture was worked up and
analyzed with the HPLC (see section 3.6.1.) with a conversion yield of 85-96%. Further, the
crude product was purified by recrystallization from water-ethanol mixture and isolated
yield calculated. Structure of the compounds was confirmed through NMR and mass
spectrometry.
Ethyl 6-methyl-4-phenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate
(Compound 16b, Table 5)
NHHN
S
CH3
COOC2H5 White solid (Yield 71%) m.p. 202-206°C, 1H NMR (DMSO-d6, 300
MHz): 9.61 (1H, s), 8.93 (1H, s), 6.67-6.53 (5H, m), 4.50 (1H, d, J = 3.60 Hz), 3.35 (2H,
q, J = 7.10 Hz), 1.60 (3H, s), 0.43 (3H, t, J = 7.03 Hz); 13C NMR (DMSO-d6, 75.4 MHz):
175.2, 165.9, 145.8, 144.3, 129.3, 128.5, 127.2, 101.6, 60.4, 54.9, 17.9 and 14.7. HRMS-
ESI: m/z [M+H]+ for C14H16N2O2S, calculated 277.2036; observed 277.2047. The spectral
data matched well with the reported values [Karade et al. (2007)].
Ethyl 4-(4-hydroxy-3-methoxyphenyl)-6-methyl-2-thioxo-1,2,3,4
tetrahydropyrimidine-5-carboxylate (Compound 17b, Table 5)
NHHN
S
CH3
COOC2H5
OC H3
HO
White solid (Yield 70%) m.p. 203°C, 1H NMR (DMSO-d6, 300
MHz): 10.25 (1H, s), 9.56 (1H, d, J = 1.74 Hz), 9.01 (1H, s), 6.63-6.60 (3H, m), 5.09 (1H,
d, J = 3.57 Hz), 4.04 (2H, q, J = 7.03 Hz ), 3.73 (3H, s), 2.28 (3H, s), 1.15 (3H, t, J = 7.08
Hz); 13C NMR (DMSO-d6, 75.4 MHz): 174.9, 166.1, 148.2, 147.8, 145.4, 153.4, 119.4,

Green synthesis of some…… Chapter 3
220
116.3, 111.8, 101.9, 60.4, 56.4, 54.5, 17.9 and 14.9. HRMS-ESI: m/z [M+H]+ for
C15H18N2O4S, calculated 323.2180; observed 323.2194.
Ethyl 4-(3-hydroxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-
carboxylate (Compound 18b, Table 5)
NHHN
S
C H 3
C OO C 2H 5
OH White solid (Yield 62%) m.p. 179-182°C, 1H NMR (DMSO-d6,
300 MHz): 9.37 (1H, s), 9.17 (1H, s), 7.70 (1H, s), 7.12 (1H, t, J = 7.92 Hz), 6.68-6.61
(3H, m), 5.06 (1H, s), 4.03 (2H, q, J = 6.97 Hz), 2.24 (3H, s), 1.19 (3H, t, J = 7.09 Hz ); 13C
NMR (DMSO-d6, 75.4 MHz): 165.4, 157.3, 152.2, 148.1, 146.3, 129.3, 116.9, 114.2,
113.1, 99.4, 59.2, 53.8, 17.8 and 14.1. HRMS-ESI: m/z [M+H]+ for C14H16N2O3S,
calculated 293.2030; observed 293.2051. The spectral data matched well with the reported
values [Dallinger and Kappe (2007)].
Ethyl 4-(4-chlorophenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-
carboxylate (Compound 19b, Table 5)
NHHN
S
CH3
COOC2H5Cl White solid (Yield 71%) m.p. 172-178°C, 1H NMR (DMSO-d6,
300 MHz): 10.38 (1H, s), 9.66 (1H, s), 7.44 (2H, d, J = 8.43 Hz), 7.24 (2H, d, J = 8.43
Hz), 5.18 (1H, d, J = 3.60 Hz), 4.04 (2H, q, J = 7.02 Hz), 2.29 (3H, s), 1.12 (3H, t, J = 7.08
Hz); 13C NMR (DMSO-d6, 75.4 MHz); 175.1, 165.9, 146.2, 143.2, 133.1, 129.4, 129.2,
101.2, 60.5, 54.3, 18.0 and 14.9. HRMS-ESI: m/z [M+H]+ for C14H15N2O2SCl, calculated
311.6485; observed 311.6491. The spectral data matched well with the reported values
[Dallinger and Kappe (2007)].
3.6.4.5. Synthesis of 3,4-dihydropyrimidin-2(1H)-thiones (20c) from 2-hydroxy
benzaldehyde (Scheme 26)
2-hydroxy benzaldehyde (0.25 mmol), ethyl acetoacetate (1 equiv.), urea (3 equiv.) and
glycine nitrate (1 equiv.) was taken in 3 mL ethanol in a round bottom flask and the reaction
mixture was subjected to microwave irradiation using CEM monomode microwave at

Green synthesis of some…… Chapter 3
221
Power = 100 W for 20 min. Afterwards, the reaction mixture was worked up and analyzed
with the HPLC (see section 3.6.1.) with a conversion yield of 88%. Further, the crude
product was purified by recrystallization from water-ethanol mixture and isolated yield
calculated. Structure of the compound was confirmed through NMR and mass spectrometry.
Ethyl 9-methyl-11-oxo-8-oxa-10,12-diazatricyclo[7.3.1.02,7]trideca-2,4,6-triene-13-
carboxylate (Compound 20c, Scheme 26)
NH
HN
OCH3C2H5OOC
O
H
White solid (Yield 75%) m.p. 202-205°C, 1H NMR (DMSO-d6,
300 MHz): 7.60 (1H, s), 7.24-7.17 (3H, m), 6.96-6.88 (1H, m), 6.80 (1H, d, J = 8.35 Hz),
4.49 (1H, m), 4.19 (2H, q, J = 7.31 Hz ), 3.26 (1H, s), 1.74 (3H, s), 1.26 (3H, t, J = 7.11 Hz
); 13C NMR (DMSO-d6, 75.4 MHz): 168.7, 154.8, 150.9, 129.6, 128.9, 125.7, 120.7,
116.8, 83.4, 60.8, 44.2, 40.6, 24.2 and 14.3. HRMS-ESI: m/z [M+H]+ for C14H16N2O4,
calculated 277.1364; observed 276.1369. The spectral data matched well with the reported
values [Kumar and Maurya (2007)].
3.7. References
Abbas, E.M.H, Abdallah, S.M., Abdoh, M.H., Tawfik, H.A. and El-Hamouly, W.S. (2008).
Behaviour of salicylaldehyde and some of its derivatives in the Biginelli reaction for the
preparation of aryl tetrahydropyrimidines. Turkish Journal of Chemistry 32: 297-304.
Atwal, K.S., O'Reilly, B.C., Gougoutas, J.Z. and Malley, M.F. (1987). Synthesis of
substituted 1,2,3,4-tetrahydro-6-methyl-2-thioxo-5-pyrimidinecarboxylic acid esters.
Heterocycles 26: 1189-92.
Atwal, K.S., Rovnyak, G.C., O’Reilly, B.C. and Schwartz, J. (1989). Synthesis of
selectivity functionalized 2-hetero-1,4-dihydropyrimidines. The Journal of Organic
Chemistry 54: 5898-907.
Atwal, K.S., Swanson, B.N., Unger, S.E., Floyd, D.M., Moreland, S., Hedberg, A. and
O’Reilly, B.C. (1991). 3-Carbamoyl-4-aryl-1,2,3,4-tetrahydro-6-methyl-5-
pyrimidinecarboxylic acid esters as orally effective antihypertensive agents. Journal of
Medicinal Chemistry 34: 806-11.

Green synthesis of some…… Chapter 3
222
Bao, W., Wang, Z. and Li, Y. (2003). Synthesis of chiral ionic liquids from natural amino
acids. The Journal of Organic Chemistry 68: 591-93.
Barluenga, J., Tomas, M., Ballesteros, A. and Lopez, L.A. (1989). 1,4-Cycloaddition of 1,3-
diazabutadienes with enamines: An efficient route to the pyrimidine ring. Tetrahedron
Letters 30: 4573-76.
Beşoluk, Ş., Küçükislamoğlu, M., Zengin M., Arslan, M. and Nebioğlu, M. (2010). An
efficient one-pot synthesis of dihydropyrimidinones catalyzed by zirconium hydrogen
phosphate under solvent-free conditions. Turkish Journal of Chemistry 34: 411-16.
Biginelli, P. (1893). Aldehyde-urea derivatives of aceto- and oxaloacetic acids. Gazzetta
Chimica Italiana 23: 360-413.
Bose, D.S., Sudharshan, M. and Chavhan, S.W. (2005). New protocol for Biginelli reaction-
a practical synthesis of Monastrol. Arkivoc (iii): 228-36.
Branco, L.C., Gois, P.M.P., Lourenço, N.M.T., Kurteva, V.B. and Afonso, C.A.M. (2006).
Simple transformation of crystalline chiral natural anions to liquid medium and their use
to induce chirality. Chemical Communications 2006: 2371-72.
Chen, X.H., Xu, X.Y., Liu, H., Cun, L.F. and Gong, L.Z. (2006). Highly enantioselective
organocatalytic Biginelli reaction. Journal of the American Chemical Society 128:
14802-03.
Chiang, A.N., Valderramos, J.C., Balachandran, R., Chovatiya, R.J., Mead, B.P., Schneider,
C., Bell, S.L., Klein, M.G., Huryn, D.M., Chen, X.S., Day, B.W., Fidock, D.A., Wipf, P.
and Brodsky, J.L. (2009). Select pyrimidinones inhibit the propagation of the malarial
parasite, Plasmodium falciparum. Bioorganic and Medicinal Chemistry 17: 1527-33.
Dallinger, D. and Kappe, C.O. (2007). Rapid preparation of the mitotic kinesin Eg5
inhibitor Monastrol using controlled microwave-assisted synthesis. Nature Protocols 2:
317-21.
Dandia, A., Saha, M. and Taneja, H. (1998). Synthesis of fluorinated ethyl 4-aryl-6-methyl-
1,2,3,4-tetrahydropyrimidin-2-one/thione-5-carboxylates under microwave irradiation.
Journal of Fluorine Chemistry 90: 17-21.
Ding, D. and Zhao, C.G. (2010). Primary amine-catalyzed Biginelli reaction for the
enantioselective synthesis of 3,4-dihydropyrimidin-2(1H)-ones. European Journal of
Organic Chemistry 20: 3802-05.
Falsone, S.F. and Kappe, C.O. (2001). The Biginelli dihydropyrimidone synthesis using
polyphosphate ester as a mild and efficient cyclocondensation/dehydration reagent.
Arkivoc (ii): 122-34.

Green synthesis of some…… Chapter 3
223
Folkers, K. and Johnson, T.B. (1933). Researches on pyrimidines. CXXXVI. The
mechanism of formation of tetrahydropyrimidines by the Biginelli reaction. Journal of
the American Chemical Society 55: 3784-91.
Fukumoto, K. and Ohno, H. (2006). Design and synthesis of hydrophobic and chiral anions
from amino acids as precursor for functional ionic liquids. Chemical Communications
2006: 3081-83.
Fukumoto, K., Yoshizawa, M. and Ohno, H. (2005). Room temperature ionic liquids from
20 natural amino acids. Journal of the American Chemical Society 127: 2398-99.
Garcia, M.T., Gathergood, N. and Scammells, P.J. (2005). Biodegradable ionic liquids Part
II. Effect of the anion and toxicology. Green Chemistry 7: 9-14.
Gathergood, N., Garcia, M.T. and Scammells, P.J. (2004). Biodegradable ionic liquids: Part
I. Concept, preliminary targets and evaluation. Green Chemistry 6: 166-75.
Ghassamipour, S. and Sardarian, A.R. (2010). One-pot synthesis of dihydropyrimidinones
by dodecylphosphonic acid as solid Brønsted acid catalyst under solvent-free conditions
via Biginelli condensation. Journal of Iranian Chemical Society 7: 237-42.
Gholap, A.R., Venkatesan, K., Daniel, T., Lahoti, R.J. and Srinivasan, K.V. (2004). Ionic
liquid promoted novel and efficient one pot synthesis of 3,4-dihydropyrimidin-2(1H)-
ones at ambient temperature under ultrasound irradiation. Green Chemistry 6: 147-50.
González-Olvera, R., Demare, P., Regla, I. and Juaristi, E. (2008). Application of (1S,4S)-
2,5-diazabicyclo[2.2.1]heptane derivatives in asymmetric organocatalysis: the Biginelli
reaction. Arkivoc (vi): 61-72.
Guillen, F., Brégeon, D. and Plaquevent, J.C. (2006). (S)-Histidine: the ideal precursor for a
novel family of chiral aminoacid and peptidic ionic liquids. Tetrahedron Letters 47:
1245-48.
Hazarkhani, H. and Karimi, B. (2004). N-Bromosuccinimide as an almost neutral catalyst
for efficient synthesis of dihydropyrimidinones under microwave irradiation. Synthesis
2004: 1239-42.
Heravi, M.M., Bakhtiari, K. and Bamoharram, F.F. (2006). 12-Molybdophosphoric acid: A
recyclable catalyst for the synthesis of Biginelli-type 3,4-dihydropyrimidine-2(1H)-
ones. Catalysis Communications 7: 373-76.
Hojati, S.F., Gholizadeh, M., Haghdoust, M. and Shafiezadeh, F. (2010). 1,3-Dichloro-5,5-
dimethylhydantoin as a novel and efficient homogeneous catalyst in Biginelli reaction.
Bulletin of Korean Chemical Society 31: 3238-40.

Green synthesis of some…… Chapter 3
224
Hu, E.H., Sidler, D.R. and Dolling, U.H. (1998). Unprecedented catalytic three component
one-pot condensation reaction: An efficient synthesis of 5-alkoxycarbonyl-4-aryl-3,4-
dihydropyrimidin-2(1H)-ones. The Journal of Organic Chemistry 63: 3454-57.
Jain, K.S., Bariwal, J.B., Kathiravan, M.K., Phoujdar, M.S., Sahne, R.S., Chauhan, B.S.,
Shah, A.K. and Yadav, M.R. (2008). Recent advances in selective α1-adrenoreceptor
antagonists as antihypertensive agents. Bioorganic and Medicinal Chemistry 16: 4759-
800.
Jastorff, B., Störmann, R., Ranke, J., Mölter, K., Stock, F., Oberheitmann, B., Hoffmann,
W., Hoffmann, J., Nüchter, M., Ondrushka, B. and Filser, J. (2003). How hazardous are
ionic liquids? Structure–activity relationships and biological testing as important
elements for sustainability evaluation. Green Chemistry 5: 136-42.
Kappe, C.O. (1993). 100 years of the Biginelli dihydropyrimidine synthesis. Tetrahedron
49: 6937-63.
Kappe, C.O. (1997). A re-examination of the mechanism of the Biginelli dihydropyrimidine
synthesis. Support for an N-acyliminium ion intermediate. The Journal of Organic
Chemistry 62: 7201-04.
Kappe, C.O. (1998). 4-Aryldihydropyrimidines via the Biginelli condensation: Aza-analogs
of nifedipine-type calcium channel modulators. Molecules 3: 1-9.
Kappe, C.O. (2000a). Recent advances in the Biginelli dihydropyrimidine synthesis. New
tricks from an old dog. Accounts of Chemical Research 33: 879-88.
Kappe, C.O. (2000b). Biologically active dihydropyrimidines of the Biginelli type-a
literature survey. European Journal of Medicinal Chemistry 35: 1043-52.
Kappe, C.O. (2000c). Highly versatile solid-phase synthesis of biofunctional 4-aryl-3,4-
dihyropyrimidines using resin-bound isothiourea building blocks and multidirectional
resin cleavage. Bioorganic and Medicinal Chemistry Letters 10: 49-51.
Kappe, C.O., Fabian, W.M.F. and Semones, M.A. (1997). Conformational analysis of 4-
aryl-dihydropyrimidine calcium channel modulators. A comparison of ab-initio,
semiempirical and X-ray crystallographic studies. Tetrahedron 53: 2803-16.
Kappe, C.O. and Falsone, S.F. (1998). Polyphosphate ester-mediated synthesis of
dihydropyrimidines. Improved conditions for the Biginelli reaction. Synlett 7: 718-20.
Kappe, C.O. and Roschger, P. (1989). Synthesis and reactions of Biginelli-compounds.
Journal of Heterocyclic Chemistry 26: 55-64.

Green synthesis of some…… Chapter 3
225
Karade, H.N., Sathe, M. and Kaushik, M.P. (2007). Synthesis of 4-aryl substituted 3,4-
dihydropyrimidinones using silica-chloride under solvent free conditions. Molecules 12:
1341-51.
Kasana, R.C., Sharma, U.K., Sharma, N. and Sinha, A.K. (2007). Isolation and
identification of a novel strain of Pseudomonas chlororaphis capable of transforming
isoeugenol to vanillin. Current Microbiology 54: 457-61.
Kumar, A. and Maurya, R.A. (2007). An efficient bakers’ yeast catalyzed synthesis of 3,4-
dihydropyrimidin-2-(1H)-ones. Tetrahedron Letters 48: 4569-71.
Liu, C.J. and Wang, J.D. (2009). Copper (II) sulfamate: An efficient catalyst for the one-pot
synthesis of 3,4-dihydropyrimidine-2(1H)-ones and thiones. Molecules 14: 763-70.
Lu, J. and Ma, H. (2000). Iron(III)-catalyzed synthesis of dihydropyrimidinones. Improved
conditions for the Biginelli reaction. Synlett 2000: 63-64.
Luo, S., Zu, D., Yue, H., Wang, L.,Yang, W. and Xu, Z. (2006). Synthesis and properties of
novel chiral-amine-functionalized ionic liquids. Tetrahedron: Asymmetry 17: 2028-33.
Ma, Y., Qian, C., Wang, L. and Yang, M. (2000). Lanthanide triflate catalyzed Biginelli
reaction. One-pot synthesis of dihydropyrimidinones under solvent-free conditions. The
Journal of Organic Chemistry 65: 3864-68.
Mabry, J. and Ganem, B. (2006). Studies on the Biginelli reaction: a mild and selective
route to 3,4-dihydropyrimidin-2(1H)-ones via enamine intermediates. Tetrahedron
Letters 47: 55-56.
Mayer, T.U., Kapoor, T.M., Haggarty, S.J., King, R.W., Schreiber, S.L. and Mitchison, T.J.
(1999). Small-molecule inhibitor of mitotic spindle bipolarity indentified in a
phenotype-based screen. Science 286: 971-74.
Naik, H.R.P., Naik, H.S.B. and Aravinda, T. (2009). Nano-Titanium dioxide (TiO2)
mediated simple and efficient modification to Biginelli reaction. African Journal of
Pure and Applied Chemistry 3: 202-07.
O’Reilly, B.C. and Atwal, K.S. (1987). Synthesis of substituted 1,2,3,4-tetrahydro-6-
methyl-2-oxo-5-pyrimidinecarboxylic acid esters: The Biginelli condensation revisited.
Heterocycles 26: 1185-88.
Ohno, H. and Fukumoto, K. (2007). Amino acid ionic liquids. Accounts of Chemical
Research 40: 1122-29.
Patil, S., Jadhav, S.D. and Deshmukh, M.B. (2011). Natural acid catalyzed multi-component
reactions as a green approach. Archives of Applied Science Research 3: 203-08.

Green synthesis of some…… Chapter 3
226
Peng, J. and Deng, Y. (2001). Ionic liquids catalyzed Biginelli reaction under solvent-free
conditions. Tetrahedron Letters 42: 5917-19.
Perez, R.; Beryozkina, T., Zbruyev, O.I., Haas, W. and Kappe, C.O. (2002). Traceless solid-
phase synthesis of bicyclic dihydropyrimidones using multidirectional cyclization
cleavage. Journal of Combitorial Chemistry 4: 501-10.
Phukan, M., Kalita M.K., and Borah, R. (2010). A new protocol for Biginelli (or like)
reaction under solvent-free grinding method using Fe (NO3)3.9H2O as catalyst. Green
Chemistry Letters and Reviews 3: 329-334.
Pretti, C., Chiappe, C., Pieraccini, D., Gregori, M., Abramo, F., Monni, G. and Intorre, L.
(2006). Acute toxicity of ionic liquids to the zebrafish. Green Chemistry 8: 238-40.
Remington. (2005). The science and practice of pharmacy. Troy, D.B. (ed). 21st edn. Vol 2.
Lippincott Williams and Wilkens, Philadelphia.
Rosłonkiewicz, A.C., Pernak, J., Feder, J.K., Ramani, A., Robertson, A.J. and Seddon, K.R.
(2005). Synthesis, anti-microbial activities and anti-electrostatic properties of
phosphonium-based ionic liquids. Green Chemistry 7: 855-62.
Roy, D.K. and Bordoloi, M. (2006). Synthesis of some substituted 2-oxo-1,2,3,4-
tetrahydropyrimidines(3,4-dihydropyrimidin-2(1)-ones) and 2-thioxo-1,2,3,4-
tetrahydropyrimidines catalyzed by tin(II) chloride dihydrate and tin(II) iodide under
microwave irradiation. Indian Journal of Chemistry B 45: 1067-71.
Russowsky, D., Lopesa, F.A., da Silvaa, V.S.S., Cantoa, K.F.S., Montes D’Ocab, M.G. and
Godoi, M.N. (2004). Multicomponent Biginelli’s synthesis of 3,4-dihydropyrimidin-
2(1H)-ones promoted by SnCl2.2H2O. Journal of Brazilian Chemical Society 15: 165-
69.
Ryabukhin, S.V., Plaskon, A.S., Ostapchuk, E.N., Volochnyuk, D.M. and Tolmachev, A.A.
(2007). N-Substituted ureas and thioureas in Biginelli reaction promoted by
chlorotrimethylsilane: Convenient synthesis of N1-alkyl-, N1-aryl-, and N1, N3-dialkyl-
3,4-dihydropyrimidin-2(1H)-(thi)ones. Synthesis 2007: 417-27.
Sharma, N., Sharma, U.K., Kumar, R., Katoch, N., Kumar, R. and Sinha, A.K. (2011a).
First bovine serum albumin promoted synthesis of enones, cinnamic acids and
coumarins in ionic liquid: An insight into the role of protein impurities in porcine
pancreas lipase for olefinic bond formations. Advanced Synthesis and Catalysis 353:
871-78.
Sharma, N., Sharma, U.K., Salwan, R., Kasana, R.C. and Sinha, A.K. (2011b). A synergic
blend of newly isolated Pseudomonas mandelii KJLPB5 and [hmim]Br for

Green synthesis of some…… Chapter 3
227
chemoselective 2o aryl alcohol oxidation in H2O2: Synthesis of aryl ketone or aldehydes
via sequential dehydration-oxidative C=C cleavage. Catalysis Letters 141: 616-22.
Sharma, U.K., Sharma, N., Kumar, R., Kumar, R. and Sinha, A.K. (2009). Biocatalytic
promiscuity of lipase in chemoselective oxidation of aryl alcohols/acetates: A unique
synergism of CAL-B and [hmim]Br for the metal-free H2O2 activation. Organic Letters
11: 4846-48.
Shen, Z.L., Xu, X.P. and Ji, S.J. (2010). Brønsted base-catalyzed one-pot three-component
Biginelli-type reaction: An efficient synthesis of 4,5,6-triaryl-3,4-dihydropyrimidin-
2(1H)-one and mechanistic study. 75: 1162-67.
Shutalev, A.D., Kishko, E.A., Sivova, N. and Kuznetsov, A.Y. (1998). A new convenient
synthesis of 5-acyl-1,2,3,4-tetrahydropyrimidine-2-thiones/ones. Molecules 3: 100-06.
Sinder, B.B. and Shi, Z. (1993). Biomimetic synthesis of (+-)-crambines A, B, C1, and C2.
Revision of the structure of crambines B and C1. The Journal of Organic Chemistry 58:
3828-39.
Singh, S.K. and Singh, K.N. (2010). Glycine-catalyzed easy and efficient one-pot synthesis
of polyhydroquinolines through Hantzsch multicomponent condensation under
controlled microwave. Journal of Heterocyclic Chemistry 47: 194-98.
Singhal, S., Joseph, J.K., Jain, S.L. and Sain, B. (2010). Synthesis of 3,4-
dihydropyrimidinones in the presence of water under solvent free conditions using
conventional heating, microwave irradiation/ultrasound. Green Chemistry Letters and
Reviews 3: 23-26.
Sinha, A.K., Acharya, R. and Joshi, B.P. (2002). A mild and convenient procedure for the
conversion of toxic beta-asarone into rare phenylpropanoids: 2,4,5-
trimethoxycinnamaldehyde and gamma-asarone. Journal of Natural Products 65: 764-
65.
Sinha, A.K., Joshi, B.P. and Acharya, R. (2003). Microwave-promoted synthesis of
methoxylated benzaldehydes from natural cis-phenylpropenes using NaIO4/OsO4 (cat.).
Chemistry Letters 32: 780-81.
Sohn, J.H., Choi, H.M., Lee, S., Joung, S. and Lee, H.Y. (2009). Probing the mode of
asymmetric induction of Biginelli reaction using proline ester salts. European Journal of
Organic Chemistry 2009: 3858-62.
Studer, A., Hadida, S., Ferritto, R., Kim, S.Y., Jeger, P., Wipf, P. and Curran, D.P. (1997a).
Fluorous synthesis: A fluorous-phase strategy for improving separation efficiency in
organic synthesis. Science 275: 823-26.

Green synthesis of some…… Chapter 3
228
Studer, A., Jeger, P., Wipf, P., Curran, D.P. (1997b). Fluorous synthesis: Fluorous protocols
for the Ugi and Biginelli multicomponent condensations. The Journal of Organic
Chemistry 62: 2917-24.
Swatloski, R.P., Holbrey, J.D. and Rogers, R.D. (2003). Ionic liquids are not always green:
hydrolysis of 1-butyl-3-methylimidazolium hexafluorophosphate. Green Chemistry 5:
361-63.
Sweet, F. and Fissekis, J.D. (1973). On the synthesis of 3,4-dihydro-2(1H)-pyrimidones and
the mechanism of the Biginelli reaction. Journal of the American Chemical Society 95:
8741-49.
Tao, G.H., He, L., Sun, N. and Kou, Y. (2005). New generation ionic liquids: cations
derived from amino acids. Chemical Communications 2005: 3562-64.
Tao, G.H., He, L., Liu, W.S., Xu, L., Xiong, W., Wang, T. and Kou, Y. (2006). Preparation,
characterization and application of amino acid-based green ionic liquids. Green
Chemistry 8: 639-46.
Tu, S., Fang, F., Zhu, S., Li, T., Zhang, X. and Zhuang, Q. (2004). A new Biginelli reaction
procedure using potassium hydrogen sulfate as the promoter for an efficient synthesis of
3,4-dihydropyrimidin-2(1H)-one. Synlett 2004: 537-39.
Valverde, M.G., Dallinger, D. and Kappe, C.O. (2001). Solid-phase synthesis of
dihydropyrimidones via N-acyliminium ion-based α-ureidoalkylations. Synlett 2001:
741-44.
Wang, M., Wang, Z.C., Sun, Z.L. and Jiang, H. (2005a). Synthesis and characterization of
transition metal methanesulfonates and their catalytic behavior in Biginelli reactions.
Transition Metal Chemistry 30: 792-96.
Wang, M., Jiang, H. and Wang, Z. (2005b). Biginelli condensation of aliphatic aldehydes
catalysed by zinc methanesulfonate. Journal of Chemical Research 2005: 691-93.
Wipf, P. and Cunningham, A. (1995). A solid-phase protocol of the Biginelli
dihydropyrimidine synthesis suitable for combinatorial chemistry. Tetrahedron Letters
36: 7819-22.
Xin, J., Chang, L., Hou, Z., Shang, D., Liu, X. and Feng, X. (2008). An enantioselective
Biginelli reaction catalyzed by a simple chiral secondary amine and achiral Brønsted
acid by a dual-activation route. Chemistry- A European Journal 14: 3177-81.
Yadav, J.S., Kumar, S.P., Kondaji, G., Rao, R.S. and Nagaiah, K. (2004). A novel L-Proline
catalyzed Biginelli reaction: One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones
under solvent-free conditions. Chemistry Letters 33: 1168-69.

Green synthesis of some…… Chapter 3
229
Yadav, J.S., Reddy, B.V.S., Reddy, K.B., Raj, K.S. and Prasad, A.R. (2001). Ultrasound-
accelerated synthesis of 3,4-dihydropyrimidin-2(1H)-ones with ceric ammonium nitrate.
Journal of Chemical Society, Perkin Transactions 1: 1939-41.


Green synthesis of some…… Chapter 3
i
NMR spectra of some compounds
NHHN
O
CH3
COOCH3
9 8 7 6 5 4 3 2 1 ppm1H NMR (in DMSO-d6) spectrum of Methyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-
5-carboxylate (14b, Table 5)
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 ppm
13C NMR (in DMSO-d6) spectrum of Methyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-
tetrahydropyrimidine-5-carboxylate (14b, Table 5)

Green synthesis of some…… Chapter 3
ii
NHHN
S
CH3
COOC2H5
OCH3
HO
10 9 8 7 6 5 4 3 2 1 ppm1HNMR (in DMSO-d6) spectrum of Ethyl 4-(4-hydroxy-3-methoxyphenyl)-6-methyl-2-thioxo-
1,2,3,4 tetrahydropyrimidine-5-carboxylate (17b, Table 5)
180 160 140 120 100 80 60 40 20 ppm
13CNMR (in DMSO-d6) spectrum of Ethyl 4-(4-hydroxy-3-methoxyphenyl)-6-methyl-2-thioxo-
1,2,3,4 tetrahydropyrimidine-5-carboxylate (17b, Table 5)
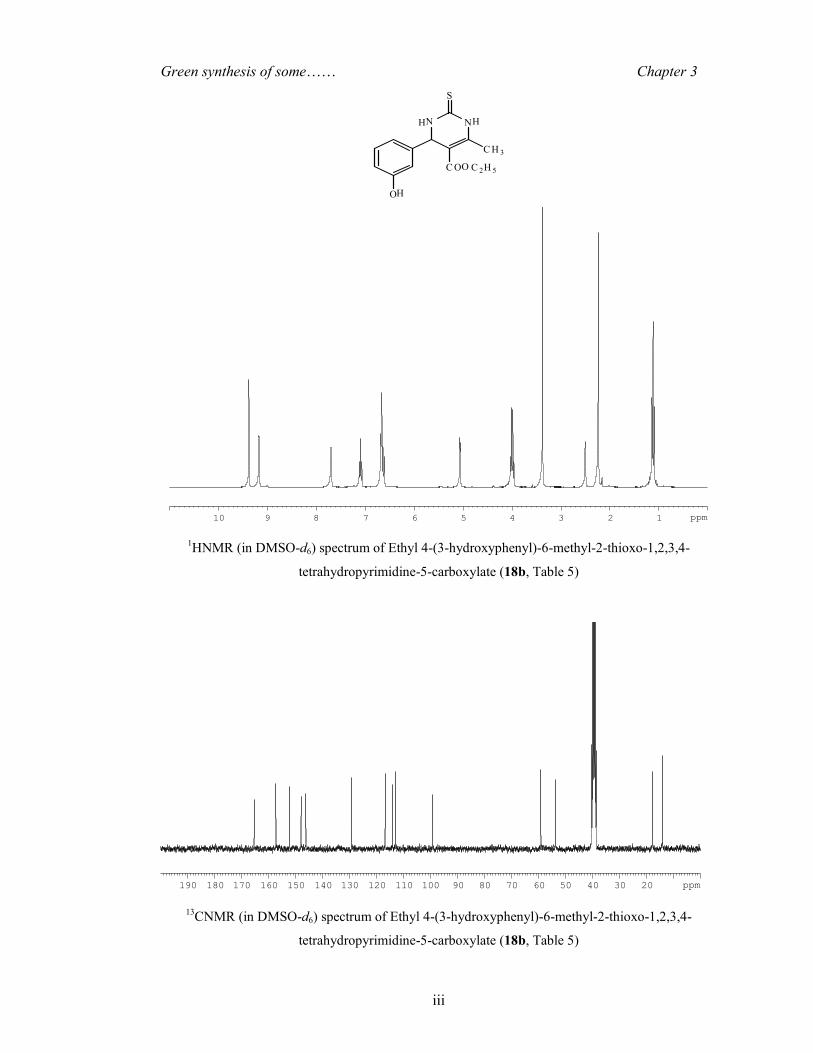
Green synthesis of some…… Chapter 3
iii
NHHN
S
C H 3
C OO C 2H 5
OH
10 9 8 7 6 5 4 3 2 1 ppm
1HNMR (in DMSO-d6) spectrum of Ethyl 4-(3-hydroxyphenyl)-6-methyl-2-thioxo-1,2,3,4-
tetrahydropyrimidine-5-carboxylate (18b, Table 5)
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 ppm
13CNMR (in DMSO-d6) spectrum of Ethyl 4-(3-hydroxyphenyl)-6-methyl-2-thioxo-1,2,3,4-
tetrahydropyrimidine-5-carboxylate (18b, Table 5)

Green synthesis of some…… Chapter 3
iv
NH
HN
OCH3C2H5OOC
O
H
9 8 7 6 5 4 3 2 1 ppm
1HNMR (in DMSO-d6) spectrum of Ethyl 9-methyl-11-oxo-8-oxa-10,12-
diazatricyclo[7.3.1.02,7]trideca-2,4,6-triene-13-carboxylate (20c, Scheme 26)
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 ppm
13CNMR (in DMSO-d6) spectrum of Ethyl 9-methyl-11-oxo-8-oxa-10,12-
diazatricyclo[7.3.1.02,7]trideca-2,4,6-triene-13-carboxylate (20c, Scheme 26)







